 Weronika Bielska1,2†
Weronika Bielska1,2† Igor Jaszczyszyn1,3†
Igor Jaszczyszyn1,3† Pawel Dudzic1
Pawel Dudzic1 Bartosz Janusz1
Bartosz Janusz1 Dawid Chomicz1Sonia Wrobel1
Dawid Chomicz1Sonia Wrobel1 Victor Greiff4,5
Victor Greiff4,5 Ryan Feehan6Jared Adolf-Bryfogle6
Ryan Feehan6Jared Adolf-Bryfogle6 Konrad Krawczyk1*
Konrad Krawczyk1*- 1NaturalAntibody, Szczecin, Poland
- 2Medical University of Lodz, Lodz, Poland
- 3Medical University of Warsaw, Warsaw, Poland
- 4Department of Immunology, University of Oslo, Oslo, Norway
- 5Imprint Labs, LLC., New York, NY, United States
- 6Janssen Pharmaceuticals, Titusville, NJ, United States
Machine learning applications in protein sciences have ushered in a new era for designing molecules in silico. Antibodies, which currently form the largest group of biologics in clinical use, stand to benefit greatly from this shift. Despite the proliferation of these protein design tools, their direct application to antibodies is often limited by the unique structural biology of these molecules. We note that multiple methods attempting antibody design focus on the discovery of an antigen-specific antibody. Here, we review the current computational methods for antibody design, focusing on binder discovery, contextualizing their role in the drug discovery process.
Introduction
Antibodies represent the largest class of biotherapeutics (1), demonstrating significant versatility and efficacy in treating a wide array of diseases, including cancer, autoimmune disorders, and infectious diseases. These Y-shaped proteins, also known as immunoglobulins, possess the unique ability to specifically bind to antigens, thereby marking them for destruction or neutralization by the immune system. The specificity and affinity of antibodies make them invaluable tools in both therapeutic and diagnostic applications.
Traditionally, the discovery and development of therapeutic antibodies have relied on two experimental paradigms: immunization and display technologies (2). The immunization approach involves the administration of an antigen into a host animal, such as mice or rabbits, to elicit an immune response. This process leads to the generation of polyclonal antibodies, from which monoclonal antibodies can be derived through hybridoma technology. Köhler and Milstein’s pioneering work in the 1970s on hybridoma technology revolutionized antibody production by enabling the creation of monoclonal antibodies with defined specificity and uniform characteristics (3).
In contrast, display technologies, such as phage display, yeast display, ribosome display and mammalian display (4), have emerged as powerful tools for antibody discovery without the need for immunization. These methods involve the presentation of vast libraries of antibody variants on the surface of bacteriophages, yeast cells, ribosomes or mammalian cells, respectively. Through iterative rounds of selection and amplification, antibodies with high affinity and specificity for a target antigen can be isolated. Phage display, in particular, has been instrumental in the discovery of several clinically approved antibodies, with Smith’s 1985 innovation marking a significant milestone in this field (5). However, this technology has inherent limitations. Because bacterial folding machinery does not readily support the production of full-length antibodies, phage display is often limited to smaller constructs such as single-chain variable fragments (scFvs). Moreover, controlling post-translational modifications in microbial expression systems is challenging, an issue resolved by using mammalian display systems (6). While display technologies help circumvent the need for direct immunization (and in many cases can be used in tandem for affinity or specificity improvement), it is worth noting that in vivo approaches—such as immunization—also allow receptor editing processes that can reduce the likelihood of autoreactivity.
Together, these traditional methods have laid a robust foundation for antibody discovery. However, they also present limitations, such as time-consuming processes and dependence on the host immune response or large library sizes. To address these challenges, computational antibody design has emerged as a promising complementary approach. It leverages advances in computational biology, structural bioinformatics, and artificial intelligence to expedite and enhance antibody development (7).
At its core, Computational Antibody Design is a sub-problem of the more generalistic Computational Protein Design (CPD), that aims to engineer novel proteins with desired functions and properties. CPD involves the prediction and optimization of protein structures and sequences to achieve specific functional outcomes. Key early methods in CPD include de novo design, homology modeling, and molecular dynamics simulations. De novo design involves creating novel protein structures from scratch, guided by principles of protein folding and stability (8). Homology modeling, on the other hand, predicts structures based on the alignment with known homologs, facilitating the design of proteins with altered functions while maintaining structural integrity (9). Molecular dynamics simulations provide insights into the dynamic behavior of proteins, allowing for the refinement of models and prediction of their stability and interactions under physiological conditions (10). Because of reliance on structural information, such early design methods mostly used structural fragments, energy functions and statistical potentials to design new structures and sequences (11–14).
Recent advancements in machine learning-based structure (15, 16) and sequence prediction (16) have given a major boost to CPD. Thanks to advancements in structure prediction spearheaded by AlphaFold2 (15), three-dimensional structures have become much more accessible (17). Merging learnings from machine learning on natural language with protein sequences resulted in large language models such as ESM that can accurately model the distribution of natural sequences, to generate new ones. Specifically, the shift towards a ‘generative’ paradigm in protein and thus antibody design is the most prominent. As much as earlier methods relied on assembling fragments of known proteins, novel tools such as RFDiffusion (18, 19), ProteinMPNN (20) or ESM-IF (21) can generate novel structures/sequences that incorporate specific features found in natural antibodies, yet represent entirely new designs not observed in nature. Such methods are increasingly being applied to antibodies (22–24), and their focus is development of novel binders. Here, we provide a review of such novel binder design methods and a perspective, contextualized to other tasks associated with antibody discovery such as developability.
Computational protein design primer
Computational protein design is crucial for developing novel biotechnological applications such as new therapeutics or industrial enzymes (19, 25). Computational protein design predominantly uses methods that leverage physicochemical calculations or machine learning to perform tasks ranging from single point mutations with increased activity to de novo design of highly thermostable proteins. Computational protein design strategies can be loosely categorized into three overlapping groups, template based protein design given structure, sequence optimization given sequence or structure and finally de novo design.
Template-based protein design relies on using existing protein structures as starting points to guide the design process - for both sequence and backbone redesign. Since protein structure determines function, this approach is particularly effective for designing proteins with new functions or enhancing existing ones. An instrumental piece of software in this sphere is Rosetta (26). Rosetta is a software suite for molecular modeling and design with a wide range of applications that are centered around the use of protein structure and a scoring function, made up of empirical and physicochemical terms. The simplest form of computational design with Rosetta (27) is optimizing a protein’s function by identifying mutations that improve its energy score.
Historically, template-based design has been limited to proteins with solved structures of closely related homologs. Recent developments in methods using ML have significantly expanded the number of use cases for computational protein design that leverage protein structures as input. Previously, starting points for designs were limited to proteins with experimentally solved structures in the PDB (28), or close homologs that could be modeled from those structures. The ability to make high-quality, computationally generated protein structures increases the number of starting structures from ~200,000 available proteins in the PDB to 200 million known protein structures in the AlphaFold database (17). Moreover, the predicted structures of designed sequences can be used to filter out poor designs using the predicted structure’s confidence metrics or by aligning the predicted structure to the designed structure. It should be noted that co-folding the interaction between two proteins to use as starting templates using tools such as Alphafold-Multimer (29) is still a very difficult challenge and even more difficult for antibody-antigen interactions.
Such large numbers of predicted structures improve the power of sequence optimization algorithms. Here, given a structural template, one is tasked with developing a sequence that would ‘fit’ into it (i.e. maximize the probability of sequence given structure). Current sequence optimization strategies typically take the form of inverse folding, where algorithms such as ESM-IF (21) or ProteinMPNN (20) trained on millions of predicted structures are tasked with returning the original sequences. Both ESM-IF and ProteinMPNN use a graph architecture to turn information about residues in the local neighborhood of a specific position into features for that position (20), (21). Using a message-passing neural network (MPNN) in an iterative fashion allows features at each residue position to encode information about the microenvironment of the neighboring residues. A decoder uses the structure-based embedding to generate a protein sequence that is likely to successfully fold into the input protein structure. A common evaluation for protein design tools is to calculate the sequence recovery rate, which is the percent of generated residues that match the native amino acid at that position. ESM-IF achieves 51% sequence recovery (21), while ProteinMPNN achieved 53% sequence recovery rate (20). That is a significant improvement over Rosetta’s 33% sequence recovery rate for the same proteins. Moreover, experimental validation was used to show ProteinMPNN can successfully rescue previous failed designs, increase stability, increase solubility, and even redesign membrane proteins to be available in solution (30).
In contrast to template-based and sequence-optimization methods that require the existence of a basis structure or starting sequence, de novo protein design involves creating entirely new folds from scratch. Traditional approaches, grounded in physics-based modeling, use atomistic representations and energy functions to optimize sequences for a defined protein backbone (31). These methods rely on iterative cycles of structure generation and sequence optimization, as exemplified in early successes like the first de novo protein design of Top7 (32). Advancements in methods using diffusion models have further expanded the potential for computational protein design by generating protein backbones that are different (but inspired by) those found in nature. For instance, RFDiffusion (18) learned to sample the large conformational landscape of protein structure by training to recover solved protein structures corrupted with noise. During inference, unconstrained predictions transform random noise into proteins that can have little overall structural similarity to any known protein structure. Additionally, RFDiffusion can be constrained with a given active site, motif, or binding partner, which enabled successful computational designs of de novo protein binders with higher rates of success than previous methods. These tools emphasize modularity, tunability, and precision; facilitating the design of proteins with programmable behaviors for applications in catalysis, molecular recognition, and synthetic biology (33, 34).
Computational protein design is currently undergoing an exciting transition from predominantly energy-based methods to those using machine learning. The recent developments and success of the field have been emphasized by the Nobel Prize in Chemistry 2024 awarded for computational protein design and structure prediction to David Baker, John Jumper, and Demis Hassabis (The Nobel Prize in Chemistry 2024, 35). A large area of interest for protein design is the development and optimization of protein therapeutics. While many protein families can act as drugs, such as enzymes and cytokines, antibodies are the most widely used class of biologics owing to their quasi-programmable nature (36). The convergence of generic protein design methods with therapeutic antibody discovery presents a promising avenue for translating advancements in protein design into therapeutic applications.
Specifics of antibody structure and function for protein design
Antibodies are proteins of the immune system that have evolved in jawed vertebrates to recognize foreign pathogens and facilitate their expulsion from the organism. They are the actuators of the adaptive immunity, as opposed to innate immunity mediated mostly by T-cell receptors. Though they are versatile binders, they are much more structurally constrained than general proteins (Figures 1A, B), which introduces nuances in the way that protein design methods addressing them need to be adjusted.
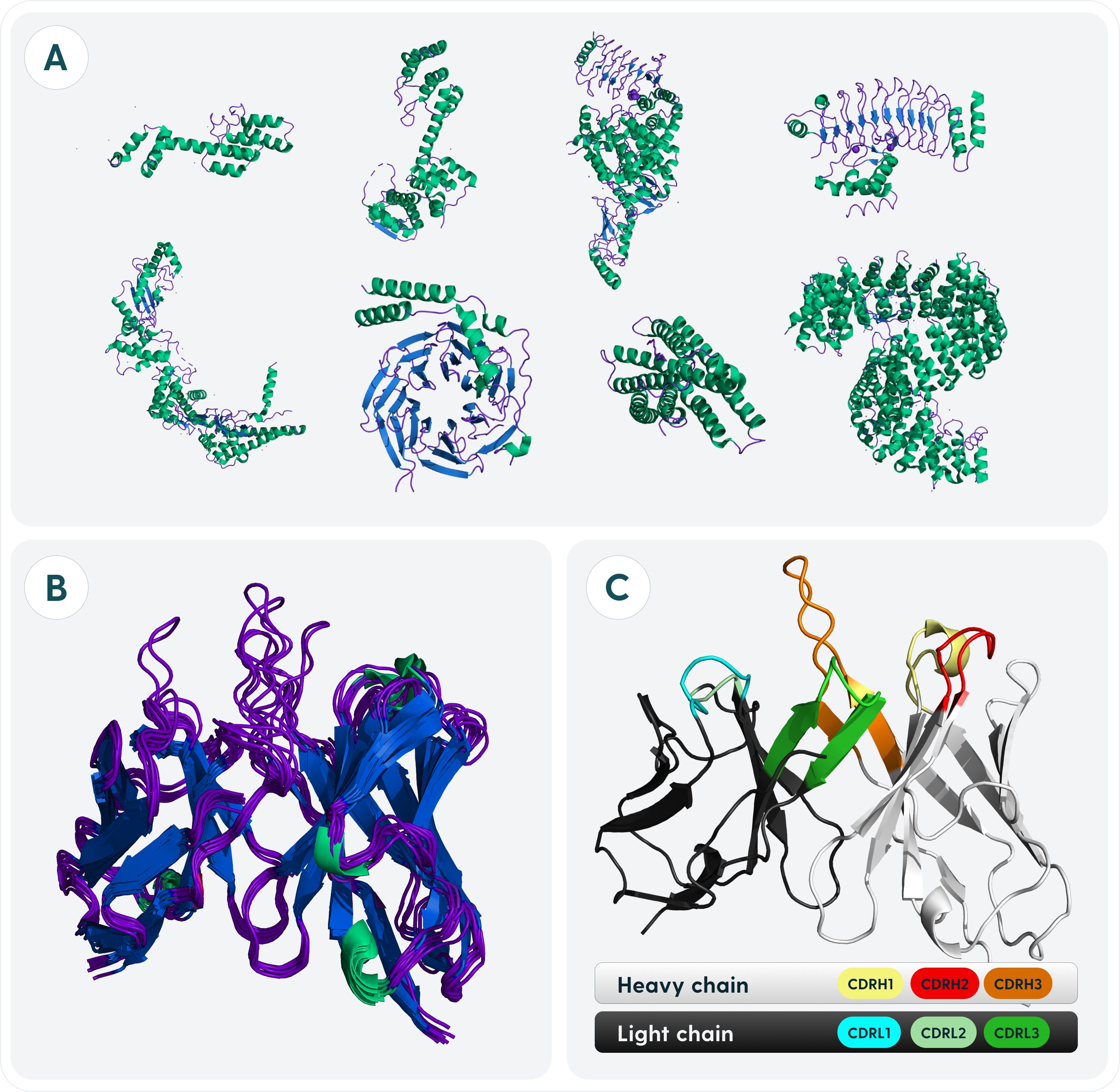
Figure 1. Specifics of antibody structure relating to its designability as opposed to other proteins. (A) Structural heterogeneity of proteins. Proteins in general adopt a variety of conformations. Relations between folds can be drawn on an evolutionary level from sequences alone. (B) Structural homogeneity of antibodies. Antibodies have a very conserved fold with a framework housing a diverse binding site. The differences between any two antibodies cannot be explained evolutionarily as it is the case with most proteins. (C) Regions of antibodies responsible for antigen-recognition. Antibodies are divided into a heavy chain, light chain. Each chain is composed of three Complementarity Determining Regions (CDRs) and four Framework Regions (FR).
Each organism has millions of distinct antibodies that collectively represent molecular diversity that should be capable of weakly binding a non-self antigen to start an immune response (37). The ability of antibodies to recognize virtually limitless amounts of antigens is the key to their success and of interest for protein design. Nature evolved antibodies to have their binding site composed of six complementarity determining regions, housed in a largely invariant framework (Figure 1C). Minute changes between CDRs can radically alter the binding affinity and specificity (38, 39). For this reason, whilst general de novo protein design might focus on building the entire scaffold that could interact with a binding partner, in case of antibodies, roughly 80% of the sequence should be known a priori because of the relative invariability of the framework.
Much of the focus for antibody redesign is devoted to the CDR-H3, since it is the most variable and in many cases, confers most of the binding affinity and specificity (40–42). The structural uniqueness of this loop eludes even the best models, such as the AlphaFold series (43). Accurate modeling of CDR-H3 is known to be a blocker to effective antibody design (44).Though most antibody design is focused on the CDR regions, it is known that the framework also has some influence on the binding ability (23, 24, 45). For humanization, one needs to replace the murine framework with a human one (Figure 2A), whilst maintaining high-affinity binding (46). This becomes a reverse design problem to focusing on CDRs alone, as one seeks to find a human framework that would be most structurally suitable to house the novel CDRs. Typically, ‘universal frameworks’ originating from certain germlines were preferred, because of empirical evidence, however, computational methods now allow us to select others that might be more suitable rather than going with such a safe choice (47).
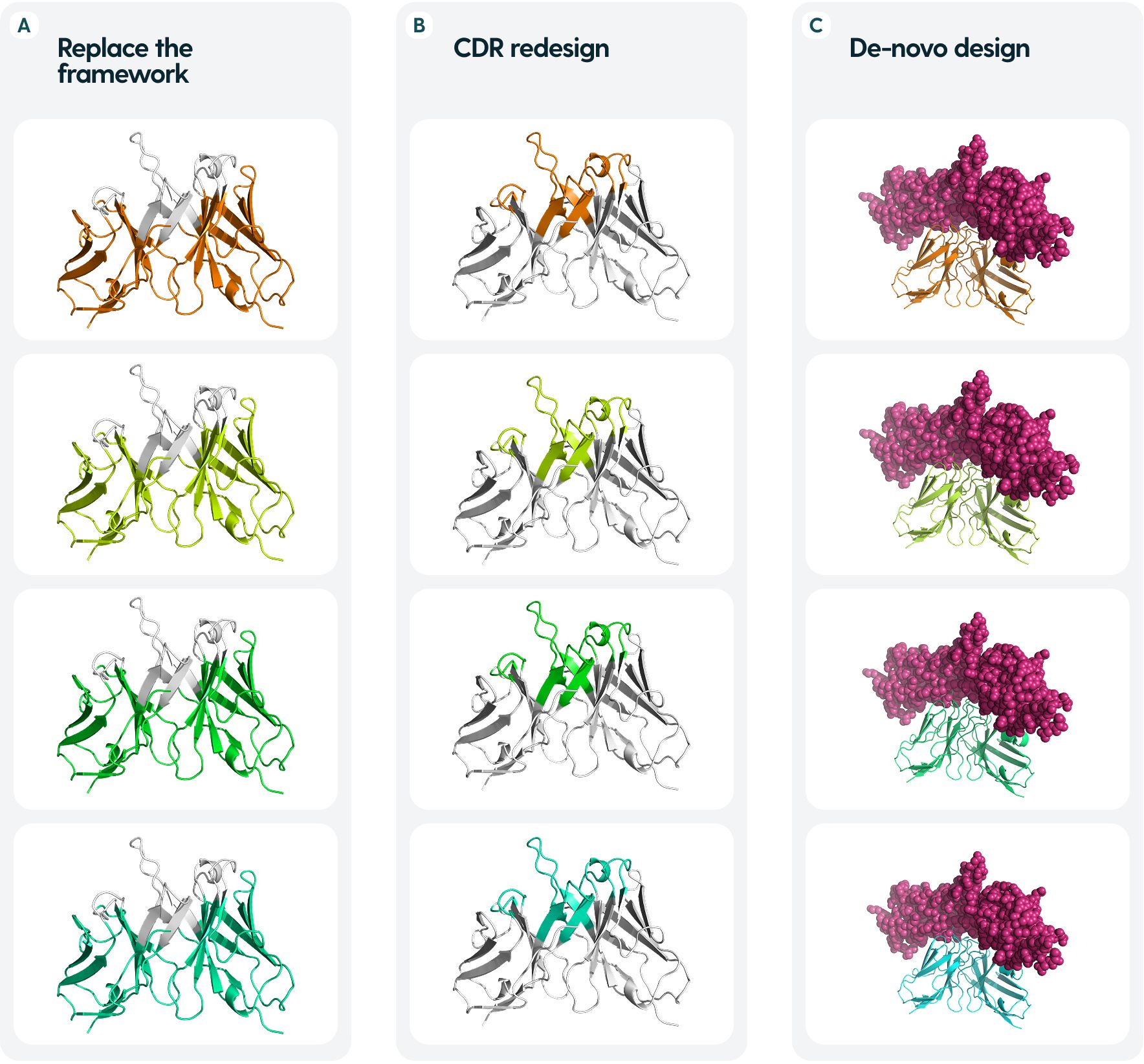
Figure 2. Common tasks in antibody design. (A) Re-designing frameworks aim to maintain the binding of the CDRs, whilst optimizing for properties such as stability or smaller immunogenicity. (B) Re-designing CDRs is chiefly aimed at modulating the binding abilities - specificity and affinity, usually starting from a known binder (C). De-novo design aims to create a novel antibody molecule from the ground up, given an antigen and/or an epitope site to be targeted.
Because of the relative invariability of the framework, one can often start from an existing binder and re-design the CDRs in one-shot fashion (Figure 2B). Arguably, the more difficult task is de novo design, when given a target antigen and epitope, one needs to create a whole new antibody that binds specifically to this epitope (Figure 2C).
Though a large unsolved part of antibody design is developing a binder, much of the preclinical work in antibody discovery is spent not on finding the right binder but on tuning the overall properties of the antibody to be more favorable as a therapeutic. These properties are commonly referred to as developability properties (48; 49; 50). This is an umbrella term encompassing multiple biophysical properties that ensure that an antibody can be economically produced in necessary quantities, can be stored for a defined period of time, and has a non-risky profile from pk/pd, specificity, and toxicity point of view, before eventually moving to clinical trials. Here, optimization takes multiple forms, with both CDRs and frameworks are becoming the targets for re-design. Nevertheless, in the most widely used meaning of the term antibody design we mean developing or redeveloping a binder towards a specific antigen first, modulating developability properties second.
To introduce a level of ontology into the antibody design field, we divided the methods into a number of categories, depicted in Figure 3 with details in Tables 1–8. Methods are broadly categorized based on their inputs, antibody/antigen specific focus, and the role they play in the design pipeline (end-to-end or just providing sequence for backbone). In terms of benchmarking, we indicate the extent of experimental validation. Otherwise, there appears to be no single metric of success amongst the methods. The list is not comprehensive as it is intended to demonstrate the methods associated with their respective categories.
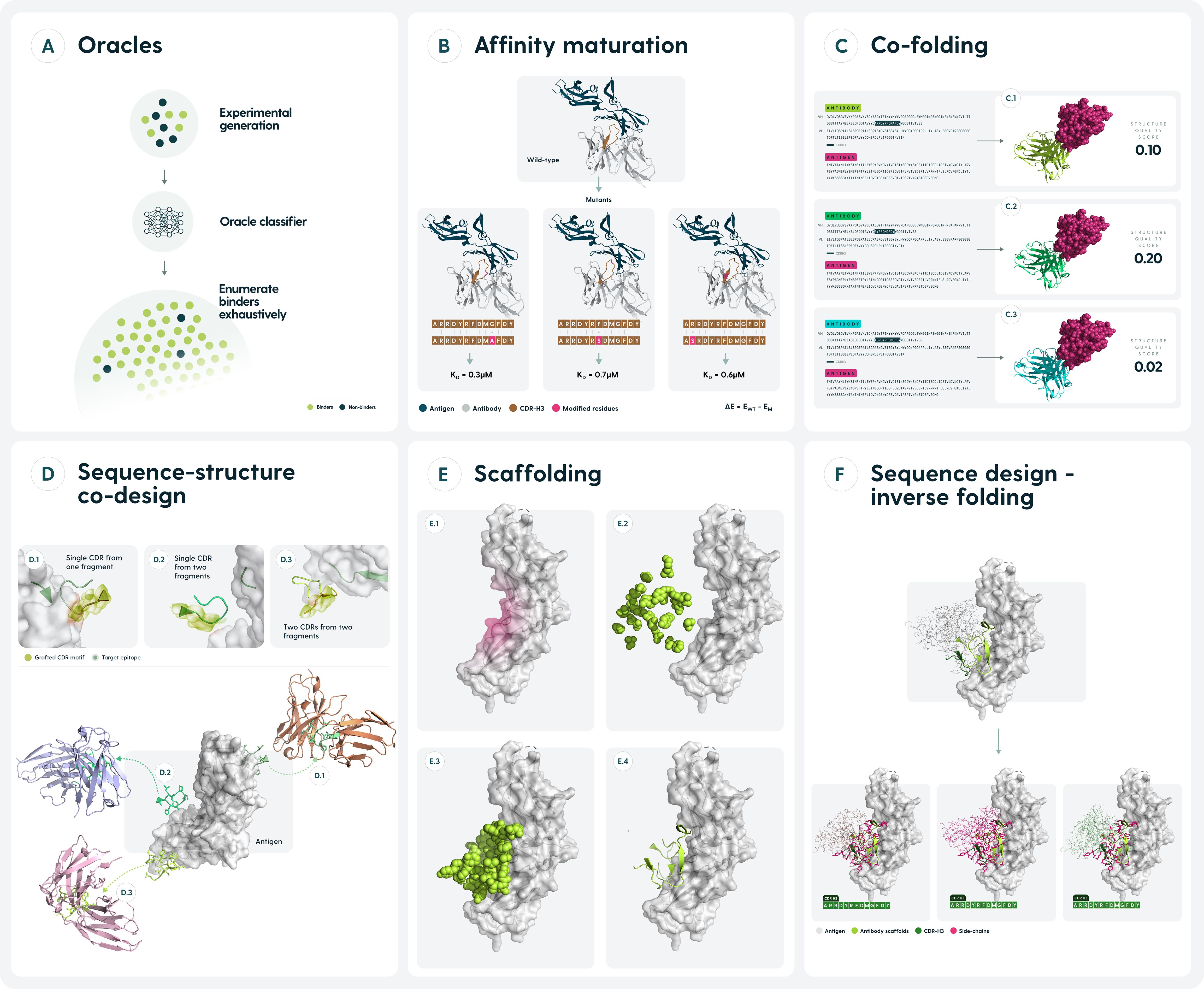
Figure 3. Current approaches to designing antibody binders computationally. (A). Binders and non-binders against a target are generated experimentally. Subsequent prediction of the binder/non-binder class allows for much more comprehensive sampling of the entire design space. (B) Affinity maturation approaches predict the free energy changes of mutations, chiefly to the CDRs to obtain a larger set of binding antibodies. (C) Current co-folding structure predictors provide confidence scores for the model of the entire complex which can be used as a proxy to gauge whether the two molecules would interact. (D) Sequence & structure can be co-designed from pre-existing elements, such as CDR fragments or entire canonical CDRs. (E). Backbone structure of an antibody binding a target epitope can be obtained, typically by diffusion approaches. Such predictions require a follow-up in the form of inverse folding. (F) Given a structure of an antibody, predict a sequence that could fold into it. Applicable as a follow up to scaffolding approaches or to obtain larger sets of potential binders that have the same structure.
Table 1. Methods in antibody design.
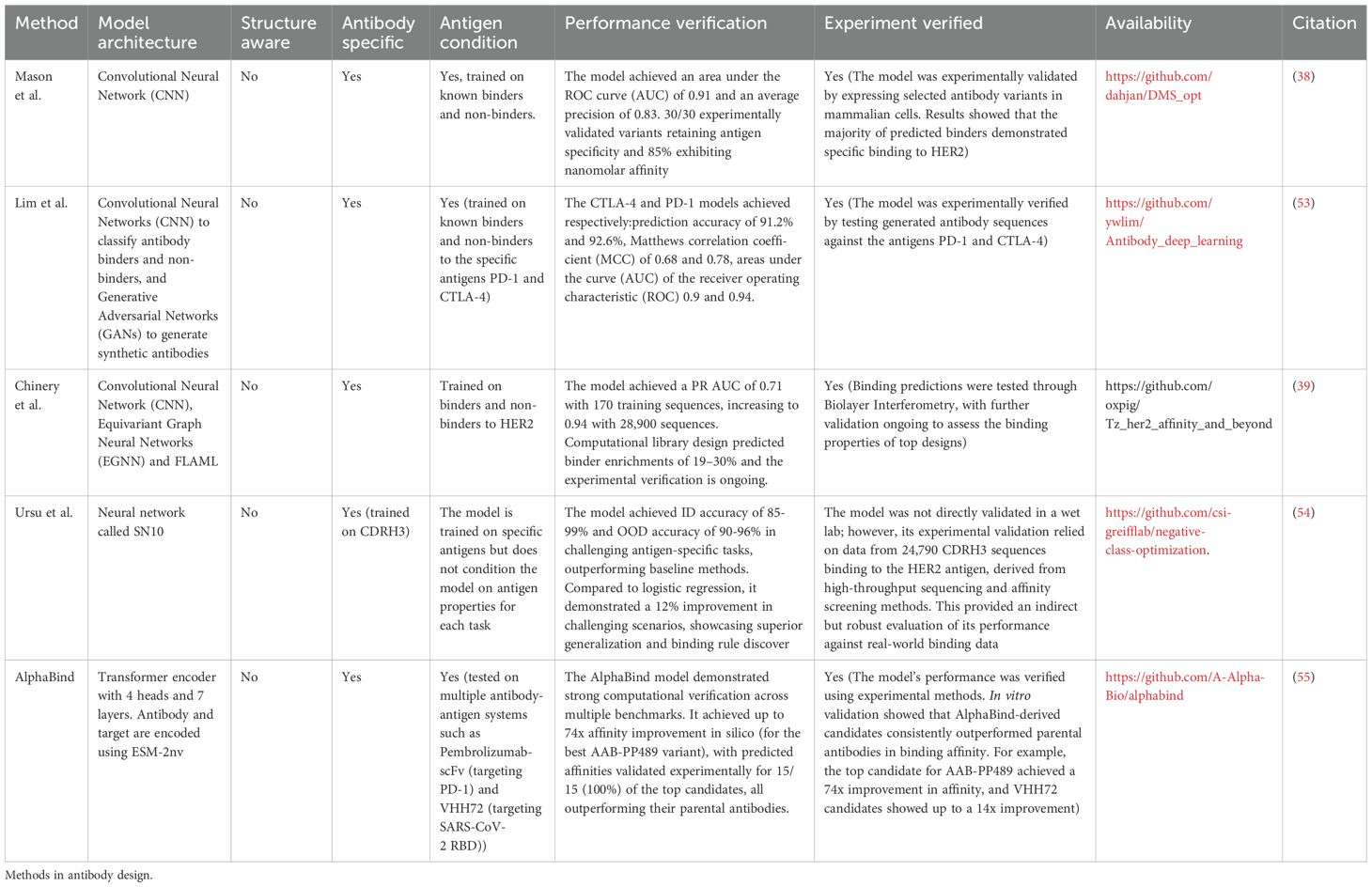
Table 2. Methods presented in this table focus on Oracle models.
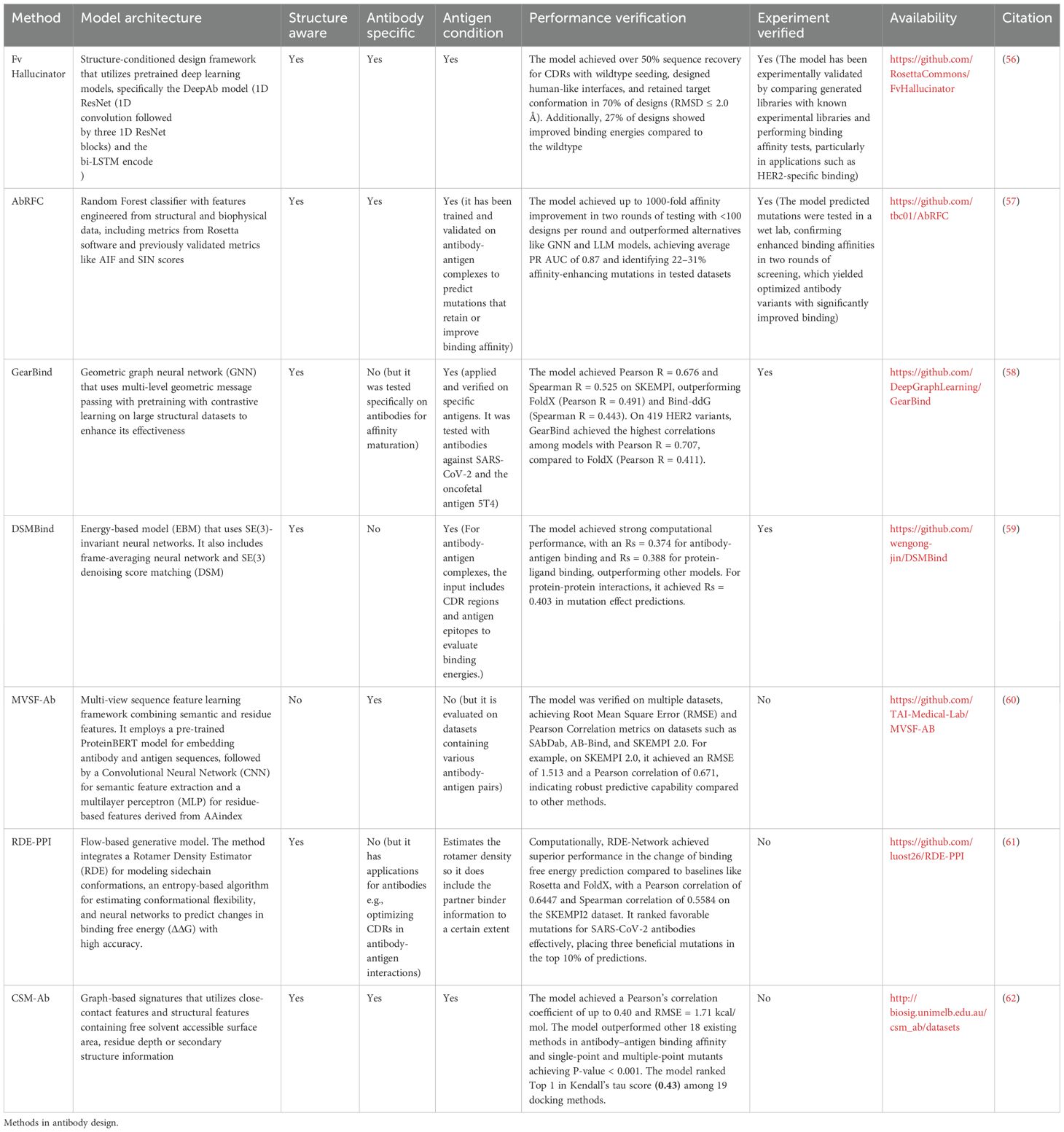
Table 3. Methods presented in this table focus on affinity maturation. Methods in antibody design.
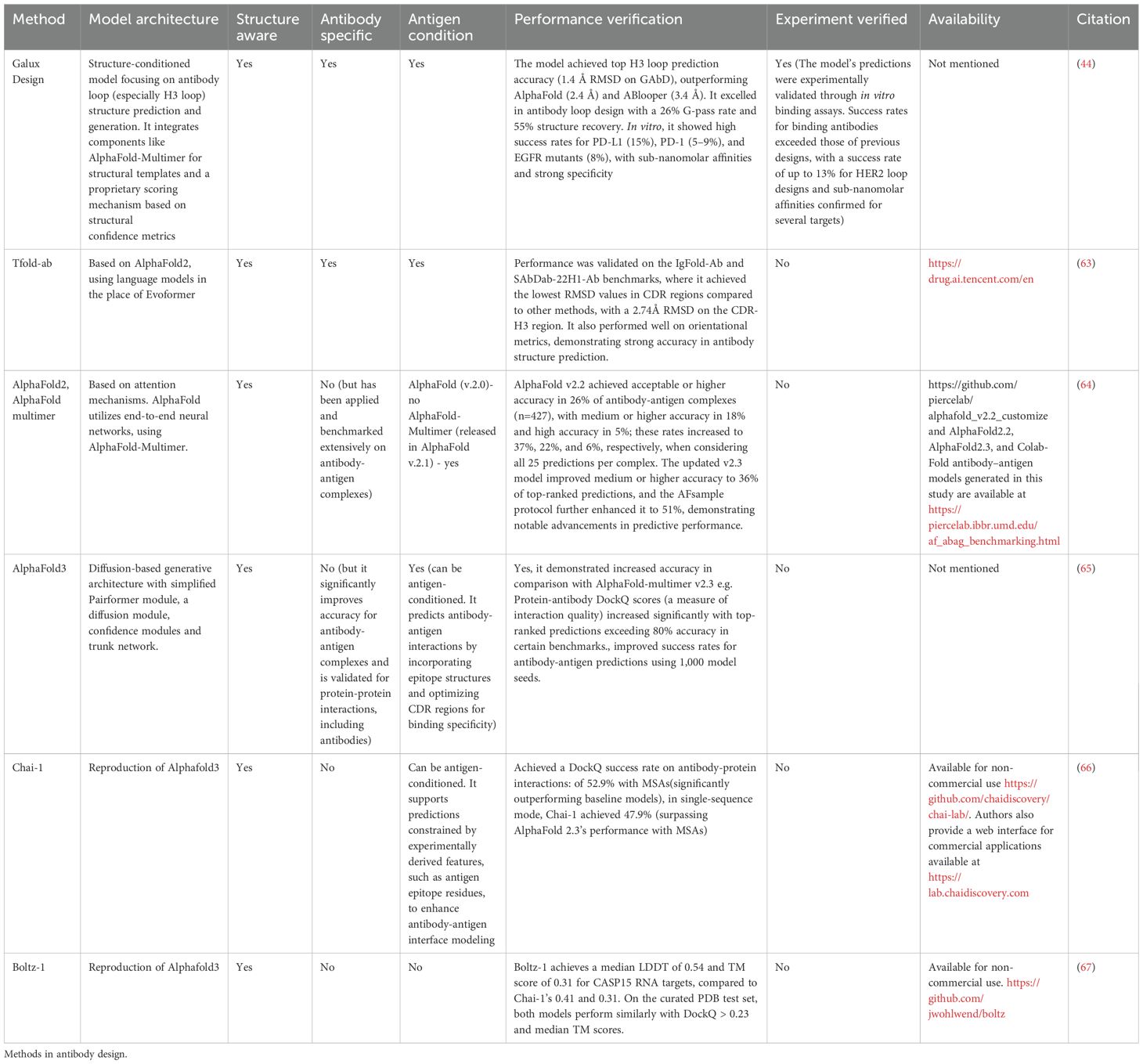
Table 4. Methods presented in this table focus on co-folding. Methods in antibody design.
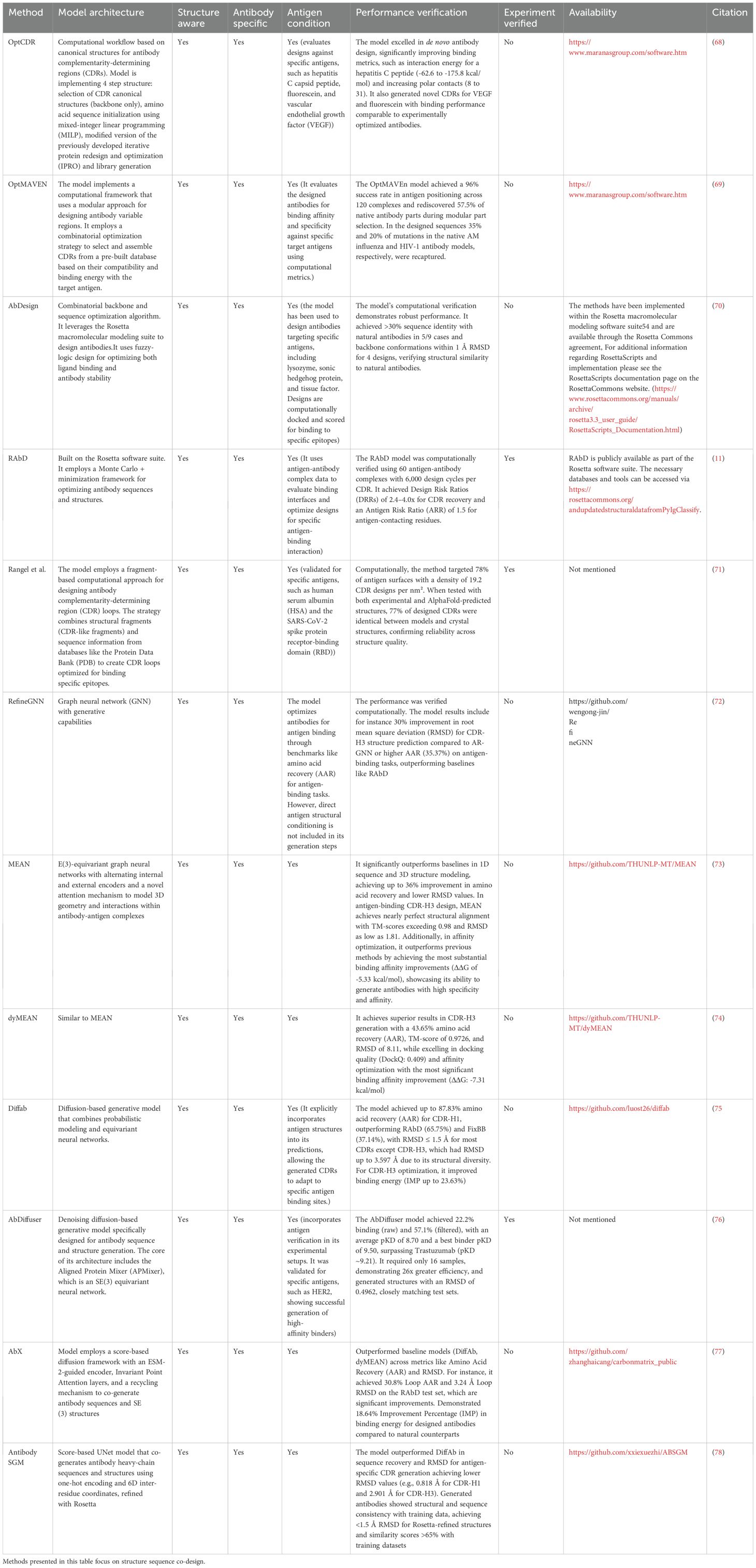
Table 5. Methods in antibody design.
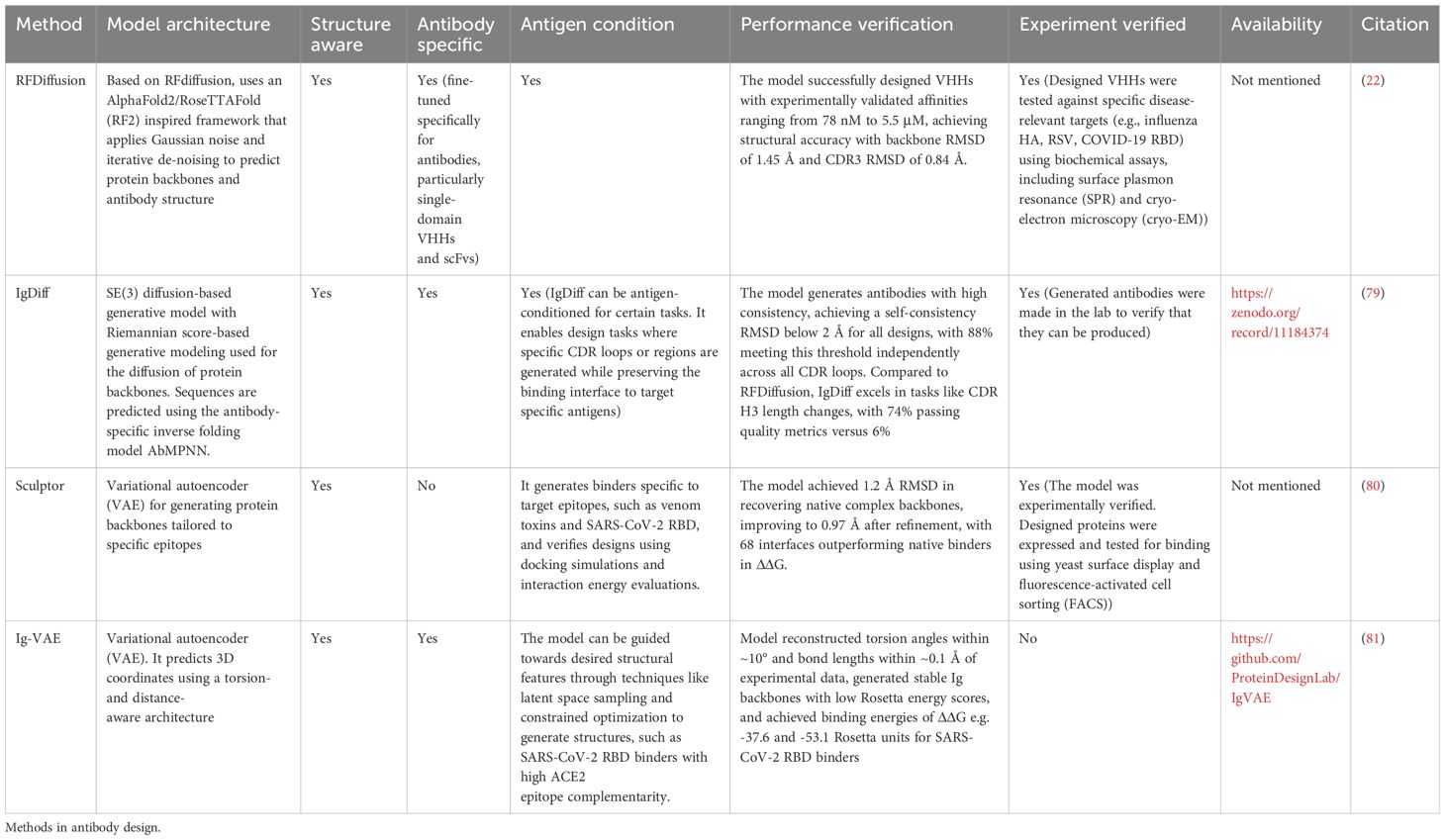
Table 6. Methods presented in this table focus on scaffold generation.

Table 7. Methods presented in this table focus on sequence design with structure, inverse folding.
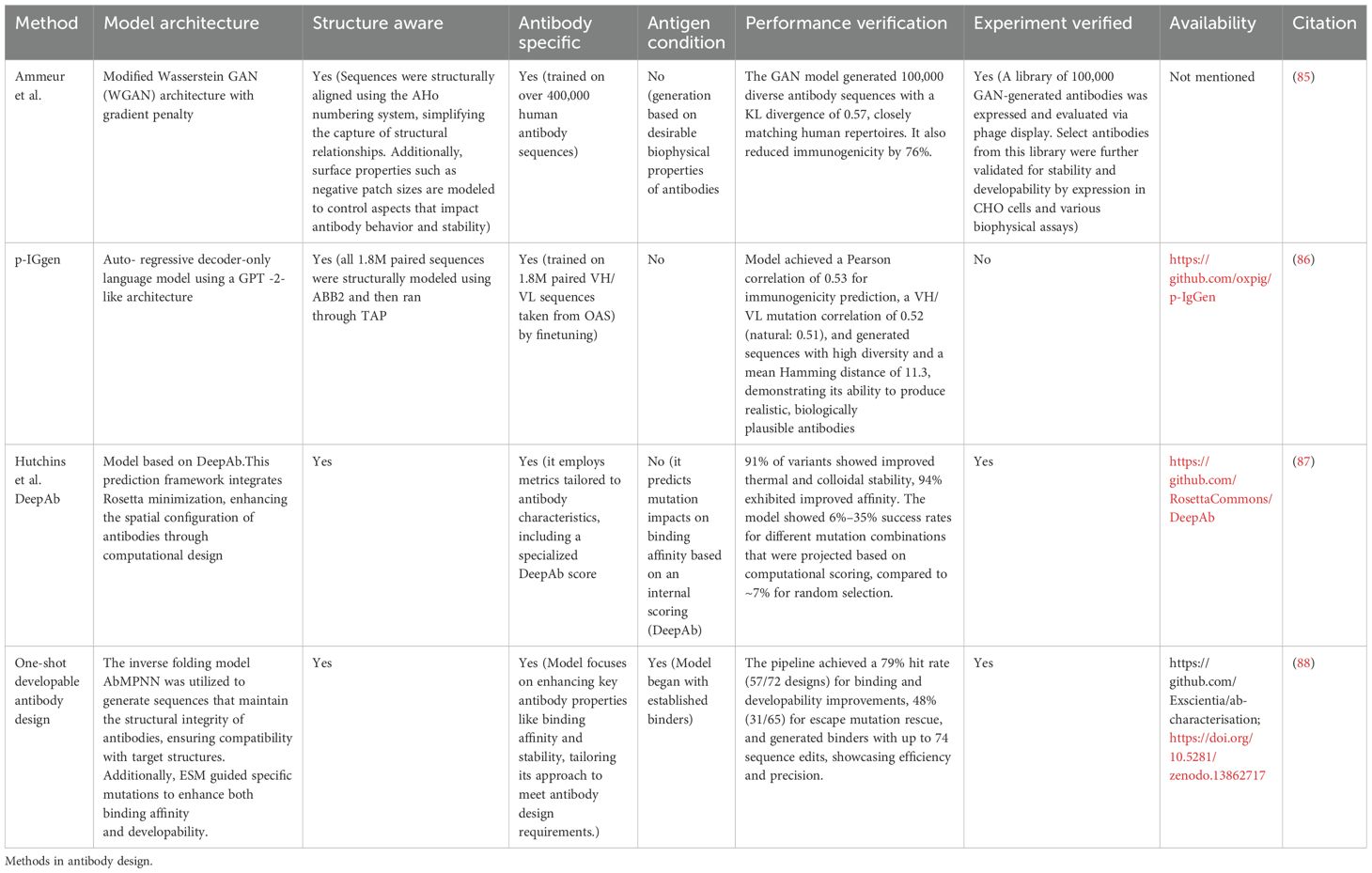
Table 8. Methods presented in this table focus on developability.
Central to any of such efforts would be to have a leveling metric, as is possible with structure prediction in the form of RMSD (89) or docking in the form of the DockQ score (90). Central to antibody design would be to quantify how many of the novel sequences bind the antigen and with what affinity. In most cases the methods report that for different antigens, making comparisons difficult. Furthermore, there is hardly any reliable control for such isolated experiments, as the randomly generated sequences or libraries are not produced. For this reason, most of the performance and experimental validation portions are given. The methods vary in the way they accept input (structure or sequence) and what output they produce (ready-to-use antibody or just a scaffold). This is not to say that some methods are incomplete; rather they have different applications within antibody design, as laid out in the following sections.
Maintaining binding function of the parent antibody - efficient exploration of the binder space without knowledge of the antigen in zero-shot fashion
If we already have an antibody that binds to a target, we can employ it to generate more binders of antibodies (Table 1). Different methods exist depending on the availability of associated experimental data - i.e. structure-enabled or not. For purely sequence-based tasks, we can use the starting antibody sequence to explore evolutionarily plausible mutations. This approach is cognate to general protein language models, where one learns to explore the fitness landscape of sequence-function relationships (91). For instance, models such as ProGEN or ESM3, can be used for generating novel sequences that maintain certain functions (92, 93) (e.g. fluorescence). Maintaining binding in antibodies is not a trivial task as introducing single mutations or combining favorable mutations is not guaranteed to maintain binding (12). Therefore, exploring the space of ‘favorable’ mutations is desirable for binder development.
The approach was pioneered by Hie et al. (92) where the authors used the ESM-1b language model and the ESM-1v ensemble of five models (six models in total) to guide the evolution of seven antibodies targeting viral antigens of SARS-CoV-2, Ebola, and Influenza A. Mutations were introduced based on the evolutionary likelihood of single-residue substitutions in the antibody variable regions (VH and VL), with substitutions that had higher evolutionary likelihood than the wild-type selected. A consensus of the six models was used to identify the most plausible substitutions. In the first round of evolution, variants with single-residue substitutions were experimentally tested for improved binding, and in the second round, combinations of beneficial substitutions were introduced to further enhance antibody affinity. Interestingly, most of the mutations recommended by the models occurred in the framework regions rather than the complementarity-determining regions (CDRs), with around half of the affinity-enhancing mutations located in these typically less mutated regions.
The aforementioned model had no notion of three dimensional structure - this was addressed in ProseLM (51), where structural adapter layers were introduced to include three dimensional information. ProseLM, builds upon the Progen family of models, incorporating structural information to improve the design of therapeutic antibodies. This structural information is integrated through structural adapter layers added after the language model layers, encoding backbone details and associated functional annotations. Models with more parameters show significant improvements in perplexity, with further gains observed when incorporating additional context information such as ligands. An antibody-specific version of ProseLM was trained exclusively on the SAbDab (94) dataset, achieving superior sequence recovery performance compared to larger models. The model was used to propose mutations for therapeutic antibodies Nivolumab and Secukinumab, targeting both the CDRs and framework regions, with designs based on structures from the PDB. Experimental results revealed that redesigning frameworks led to a much higher success rate in maintaining binding (92%), while redesigning CDRs resulted in a lower success rate (25% for Nivolumab).
A notable study that performed large-scale validation is by Shanehsazzadeh et al. (52). The authors employed their AI model to generate CDR-h3s and all-CDR variants of trastuzumab. Care was taken to remove all close-homologs of trastuzumab. The model focused on generating heavy chain complementarity-determining regions (HCDRs) in a zero-shot fashion - without prior exposure of the model to the target antigen. The study generated a library of about 400,000 HCDR variants and validated binders using high-throughput surface plasmon resonance (SPR) experiments. The results identified 421 diverse binders, with 71 showing low nanomolar affinity to HER2, and some antibodies performing on par or better than the therapeutic antibody trastuzumab. The top-performing generative models significantly outperformed biological baselines such as OAS and SAbDab databases, achieving a 10.6% binding rate for HCDR3 designs and a 1.8% rate for full HCDR123 designs.
The tool of choice for the few shot design is a language model that was trained using autoregressive or masking procedure. Although there is already a fair number of language models and their antibody-specific varieties (95–97), there were not multiple zero-shot/few-shot exercises like the ones above. Though such approaches appear to be well suited to explore the space around a specific binder, they do not offer a way to radically deviate from it. Therefore it is also desirable to have a more precise set of binders against a given target, which can be achieved by a combination of data-generation and supervised learning.
Oracles - large-scale binder generation and subsequent machine learning model training
One of the main tasks of an antibody design exercise is to develop a binder towards an antigen. Machine learning methods are notoriously data-intensive so an approach that has been explored by some groups was to generate prediction-first machine learning datasets of binders and non-binders and train the ‘oracle’ models on these (Table 2, Figure 3A).
Recent advancements in computational antibody design have leveraged high-throughput experimental data to train machine learning (ML) models, achieving remarkable success in predicting antigen specificity and binding affinity. Mason et al. (38) pioneered this approach by deep-sequencing libraries of trastuzumab variants and training a convolutional neural network (CNN) to predict HER2 specificity, achieving an area under the ROC curve (AUC) of 0.91. Similarly, Lim et al. (53) (Lim, Adler, and Johnson 2022) generated datasets for antibodies targeting CTLA-4 and PD-1 by sorting yeast-displayed libraries, with their CNN achieving AUC values of 0.90 and 0.94, respectively. Building on this foundation, Chinery et al. (39) expanded the scope, creating a dataset of over 524,000 trastuzumab variants classified by binding affinity to HER2 and benchmarking multiple ML models, including CNNs, Fast Library for Automated Machine Learning (FLAML), and Equivariant Graph Neural Networks (EGNN). Notably, the CNN excelled in low-data scenarios, while FLAML performed better on larger datasets. This work also integrated computational methods like AbLang and ProteinMPNN to enrich high-affinity variants, underscoring the potential of ML in optimizing antibody libraries with efficiencies comparable to traditional experimental methods.
The above-mentioned models were trained on datasets generated for this purpose. However, it is believed that fine-tuning models offers much improvement (98). Here, fine-tuning is understood as taking a feature-representation model, such as a language model, trained on many unrelated antibody/protein sequences, and focusing it on a library of antibody-specific ones.
For instance, Engelhart et al. (99) generated the AlphASeq SARS-CoV-2 dataset of 104,972 antibody sequences with quantitative binding data, enabling Deutschmann et al. (100) to fine-tune and benchmark domain-agnostic and domain-specific models. The ESM2 model outperformed AbLang in predicting binding affinities, demonstrating the power of generalist models when trained on large datasets. Similarly, (74) (101) fine-tuned ProGen with 60 CD40-targeting antibodies to bias sequence generation toward improved affinity. Barton et al. (102) introduced FAbCon, a generative antibody-specific language model fine-tuned on datasets like AlphASeq and others, achieving state-of-the-art predictive performance (e.g., AUROC of 0.815 for SARS-CoV-2 binding) and generating low-immunogenicity antibodies validated through computational developability assessments. Finally, AlphaBind (55) utilized pre-training on 7.5 million affinity measurements and fine-tuning on experimental data, incorporating sequence embeddings from ESM-2nv to optimize antibodies for binding affinity and developability. These studies highlight how fine-tuning enhances ML models’ ability to predict and generate optimized antibodies for diverse therapeutic targets.
Altogether the DMS-based methods demonstrate that it is possible to train machine learning models if enough data is available (according to Lim et al. in the order of hundreds of binders/non binders is enough). Such approaches require generating a large experimental dataset to then train a neural network. The overhead is justified by the subsequent ability to computationally scan a much larger space of binders, in search of antibodies with better developability or binding properties. Such approaches are paradoxically antigen-specific but do not require antigen at prediction time. The methods chiefly learn the distribution of the antibody-side, or just the CDR-H3 that recognizes the antigen. Therefore each is very constrained to the DMS antigen, lacking generalizability. The overarching task of antibody design remains to be able to generalize to any kind of antigen at the start. Such design is typically approached by structure-based and de novo methods.
Affinity maturation/structure optimization
Given the importance of enhancing antibody-antigen binding affinity, computational methods for affinity maturation have evolved significantly over the years. Early approaches, such as those by Lippow et al., utilized physics-based energy functions like CHARMM to systematically evaluate single-point mutations and their combinations (12). Such energy-based efforts were then combined with docking & early statistical methods (103–105). While these pioneering efforts laid the groundwork, novel strategies increasingly leverage machine learning and data-driven approaches to predict affinity or interaction energy and identify beneficial mutations (62). The input here is typically a co-crystal or co-folding structure of antibody-antigen with the model tasked in either predicting the energy of a set of mutations, or proposing a set of favorable ones (Table 3, Figure 3B). In broad terms, one can perform affinity maturation either in a supervised or unsupervised fashion.
Supervised methods for antibody affinity maturation rely on training models on structural affinity datasets such as SKEMPI and AB-BIND, often supplemented with synthetic data to address limited experimental availability. Notably the Antibody Random Forest Classifier (AbRFC) integrates structural and mutational data to predict affinity-enhancing mutations, successfully designing SARS-CoV-2 antibodies with up to 1000-fold binding improvements against Omicron variants. Graphinity (106), an equivariant graph neural network, learns atomic-resolution interaction patterns and achieves Pearson correlations nearing 0.9 on affinity datasets, demonstrating strong generalization through the use of both experimental and synthetic ΔΔG data. However, the study also highlighted the need for tens to hundreds of thousands of high-quality experimental data points for fully generalizable predictions, reflecting current limitations in dataset size and diversity.
By contrast, unsupervised methods for antibody affinity maturation focus on learning from structural data without requiring labeled binding affinities, offering data-efficient alternatives to supervised approaches. Models like FvHallucinator (56) use generative hallucination to design sequences by minimizing geometric loss between predicted and target structures, successfully recovering native-like CDR sequences and generating functional binders validated via Rosetta. GearBind (58), a geometric graph neural network, combines large-scale pretraining on protein structures with fine-tuning on datasets like SKEMPI to predict mutations that significantly enhance affinity, achieving up to 17-fold improvements experimentally. DSMBind (59) employs energy-based modeling with SE(3) denoising score matching, learning to reconstruct perturbed structures and generating nanobody designs validated by ELISA assays, showcasing its versatility across binding tasks. Similarly, RDE-PPI (61) leverages a flow-based generative model to estimate rotamer probability distributions, using entropy to predict binding free energy changes (ΔΔG). Trained on structural data, it outperformed traditional methods on the SKEMPI2 dataset and successfully ranked affinity-enhancing mutations in a SARS-CoV-2 antibody design. Together, these methods highlight the potential of unsupervised learning to generate and optimize antibodies with minimal reliance on labeled data.
While these computational strategies excel at affinity maturation, they generally rely on the availability of high-resolution antibody-antigen complex structures, such as those derived from X-ray crystallography. This dependency poses a challenge, as generating accurate models of antibody-antigen complexes remains non-trivial. Advances in structure prediction methods for co-folding are increasingly addressing this bottleneck, aiming to expand the applicability of affinity maturation techniques even in cases where experimental structures are unavailable.
Co-folding - structure-prediction-based design of binders for implicit flexible docking
Recent advances in structure prediction of monomers (15) have spurred an array of antibody variable region specific models (89). Such models now make it possible to provide predictions that are of higher quality than previous homology models, typically in high-throughput and with low memory requirements (107). For applications such as protein design, one would expect to model large numbers of variants, so the models have evolved to produce answers much faster than the pioneering AlphaFold-2 software, for which modeling even 1,000 antibodies would be cost and time prohibitive (108, 109; 110). High throughput modeling of single structures is desirable for scaffold design and inverse folding that are covered in later sections.
Modeling of individual structures has naturally evolved into tackling multimeric complex prediction or ‘co-folding’ - akin to classic global protein docking. In many ways, co-folding is an evolution of traditional docking methods exemplified by RosettaAntibody/SnugDock (13), ClusPro (111) or HADDOCK (112). Given two structures, these methods used a combination of pose sampling and re-scoring to obtain the final complex. One of the chief issues of such approaches was a limited way to deal with flexibility, which is crucial as models are not perfect, and rigid-only poses might prevent one from recreating the native pose because of atomic clashes. Co-folding addresses such issues by performing the folding and thus induced fit in one pass, which is also more computationally efficient.
The pioneer in co-folding was AlphaFold2-multimer that started a trend of using the scores (113) from the models to additionally assess the quality of binding between an antibody and the antigen (Table 4, Figure 3C). This is somewhat different from antibody-antigen docking, where one is interested in re-establishing the complex, but rather using the intermediary scores, such as iPTM+PTM to assess whether an arbitrary antibody (or protein) could bind to a given antigen as an oracle (114).
Recent studies have highlighted the application of advanced structural prediction methods in improving antibody-antigen docking and design. Yin and Pierce (64) evaluated AlphaFold2 (AF2) for refining docked antibody-antigen complexes by using stripped side-chain templates as input. AF2 improved docking performance, particularly in bound complexes, by retaining 50% of decoy contacts and refining interface structures with an average shift of 1.24Å, although its rescoring efficacy diminished with lower model quality. Wu et al. (63) introduced tfold-AB, a multi-task model leveraging AlphaFold2 and large language models for flexible docking and virtual screening, achieving DockQ scores of 0.217 in global and 0.416 in local docking scenarios. It showed potential for enriching antibody hits against targets like PD1 and SARS-CoV-2 antigens. Bang et al. (44) developed GaluxDesign, which achieved near-atomic accuracy (1.4Å RMSD) in challenging CDR-H3 loop predictions using inter-chain features and a novel G-pass scoring metric. The model outperformed AlphaFold and other tools in predicting HER2 binding, generating novel antibodies with high experimental success rates, including 13.2% for HER2-targeting designs.
Recently, diffusion-based improvements in AlphaFold3 were focused specifically on antibodies, improving the model performance on this modality upon AlphaFold2 (65). Nevertheless, antibodies appear to be a particularly problematic format that still eludes such state-of the art modeling attempts. Currently there are community efforts to reproduce the successful architecture of AlphaFold3. Such reproductions, however, appear to be running into the same issues, indicating that global antibody-antigen docking/co-folding is still out of reach, and to get at reasonable models one needs to provide some constraining epitope information to the model (66).
Collectively, these advances demonstrate significant progress in antibody docking, rescoring, and de novo design, but one that still needs to reach a level that can be translated into clinical applications. Antibody CDRs are consistently eluding attempts to predict them accurately (43). One confounding factor here might be CDR flexibility, as most of the methods treat 3D coordinates as static snapshots rather than means to an ensemble (115).
Given such shortcomings, predicting structures of an antibody-antigen complex can be seen as a proxy of assessing the viability of a given antibody sequence against an antigen. Exhaustive enumeration of such sequences is possible, but most of the structural methods above would make it computationally prohibitive to score. For this reason designing an antibody given an antigen, the so-called ‘de novo’ design has always been of great interest.
Sequence-structure co-design
The ultimate goal of structure-based antibody design is to develop a novel binder against a given epitope. Some of the early methods approached this without resorting to machine learning to assemble novel binding structures using fragments (Table 5; Figure 3D). Examples here include OptCDR, AbDesign, RosettaAntibodyDesign and the method by Rangel et al.
Early computational methods for antibody design leveraged structural data to generate and optimize complementarity-determining regions (CDRs) for antigen binding. OptCDR (68) focused on canonical CDR structures, utilizing energy minimization and mutational libraries to design diverse CDRs, although its designs were not experimentally validated. AbDesign (70) used structural and sequence data from the Protein Data Bank (PDB) combined with Rosetta-based docking to optimize binding affinity and stability, successfully recapitulating natural backbone conformations in several benchmarks. RosettaAntibodyDesign (RAbD) (11) improved upon this by incorporating Monte Carlo minimization to graft and optimize CDRs, with experimental validation showing up to 12-fold improvements in binding affinity. More recently, fragment-based approaches like that of Rangel et al. (71) used structural fragments from PDB datasets to design single-domain antibodies with optimized stability and nanomolar affinities, validated against targets such as SARS-CoV-2. Together, these methods showcase the evolution of computational tools in antibody design, with increasing emphasis on experimental validation and real-world applicability.
Recent advances in antibody design have integrated machine learning with structural data, moving beyond traditional structure-based methods. RefineGNN (72) pioneered this approach by representing antibody sequences and structures as graphs, using message-passing networks to co-design complementarity-determining regions (CDRs) for improved binding affinity and neutralization. Trained on data from SAbDab and CoVAbDab, it showed strong performance in computational tasks like antigen-binding and SARS-CoV-2 neutralization but lacked experimental validation. Similarly, MEAN (Multi-channel Equivariant Attention Network) framed antibody design as a conditional graph translation problem, leveraging E(3)-equivariant message passing and attention mechanisms to predict CDR sequences and structures (73). It outperformed baseline methods in computational benchmarks, including CDR-H3 design and binding affinity optimization, yet also lacked direct experimental testing. Building on this, dyMEAN (73) introduced full-atom modeling and the shadow paratope concept to better capture antigen-antibody interactions, further enhancing computational performance in structure prediction and affinity optimization. While these methods demonstrate promising results in silico, their lack of in vitro validation remains a limitation.
Recent advancements in computational antibody design have utilized diffusion-based generative models, which iteratively refine antibody sequences and structures by reversing noise corruption processes. DiffAb (Luo, 75) was one of the first to apply this approach, using antibody-antigen complexes from SAbDab to co-generate CDR sequences and 3D structures. It demonstrated strong computational performance on targets such as SARS-CoV-2 and influenza but lacked experimental validation. Similarly, AbDiffuser (19, 76) combined sequence-structure relationships with physics-informed constraints, achieving successful in vitro validation, with 37.5% of HER2-specific antibodies showing tight binding affinities comparable to Trastuzumab. AbX (77) extended diffusion modeling by integrating evolutionary, physical, and geometric constraints, leveraging pre-trained protein language models and structural data to optimize antibody-antigen binding. Though computationally robust, it also lacked experimental validation. Antibody-SGM (78) focused on heavy-chain design, using a score-based diffusion process to generate full-atom structures, further refined by Rosetta, and confirmed stability via molecular dynamics simulations. Despite promising computational results, it too remains unvalidated in wet-lab settings. Together, these diffusion-based methods showcase significant potential for antibody design but highlight a recurring gap in experimental confirmation.
All the methods described were solving a problem of combining the structure and sequence optimization. As the last graph and diffusion-based methods exemplify, the focus is shifting more towards machine learning generative methods. Though the methods covered here approaches simultaneous sequence-structure optimization there is a set of modern methods that split the problem in firstly generating the backbone, followed by predicting a sequence that could fit it.
Scaffold design
Much of the previous work covered supervised learning, where we already have some sequence or structure template to work from. In such scenarios, as shown with Oracles even CNN networks can prove useful, especially in the low-n learning scenario (116, 117). It is known that multiple protein sequences can adopt similar structures. This paradigm is exploited in ‘scaffolding’ which aims to generate novel protein backbones able to interact with another protein of choice (Table 6, Figure 3E). Backbone generating methods are exemplified by methods such as IgDiff (79), Ig-VAE (81), and Sculptor (80).
IgDiff utilizes SE(3) diffusion to model antibody backbones and employs AbMPNN for sequence generation, trained on synthetic antibody structures from the Observed Antibody Space (OAS) and ABodyBuilder2 predictions (109). Experimentally, 28 IgDiff designs showed high expression yields, validating its practical applicability. Ig-VAE, designed specifically for antibodies, generates 3D atomic coordinates for immunoglobulin domains using a rotationally and translationally invariant VAE, trained on AbDb/abYbank datasets. It demonstrated strong computational performance, such as epitope-specific SARS-CoV-2 RBD design, but lacked experimental validation. Sculptor, an evolution of Ig-VAE, integrates molecular dynamics simulations and interaction-guided modeling to design binders for user-specified epitopes. Combining VAE-generated backbones, sequence optimization, and Rosetta refinement, Sculptor successfully designed a broadly neutralizing binder for snake venom toxins, experimentally validated for cross-reactivity with multiple toxins. These methods highlight the growing potential of generative models in antibody and binder design, however as in many other cases, experimental validation was limited.
To the best of our knowledge, the current state of the art in backbone generation, which includes full experimental validation, was the fine-tuning of RFDiffusion (18). It was trained on backbones with introduced noise, with a network tasked to recreate the original coordinates. In order to operate within the sphere of antibodies/nanobodies, RFDiffusion had to be fine-tuned on antibodies from the Protein Data Bank (PDB) (22). This approach was developed using a combination of nanobody and general protein structures from the Protein Data Bank to train the model. The algorithm works by using a noising and de-noising process to iteratively refine protein backbones, specifically focusing on generating diverse CDR loop conformations and nanobody-antigen binding orientations. After designing the structures, ProteinMPNN is used to optimize the sequences of the CDR loops. The method was benchmarked both computationally and experimentally: it was applied to design nanobodies targeting a range of disease-relevant antigens (including influenza hemagglutinin, RSV, and SARS-CoV-2), and the resulting designs were experimentally validated through surface plasmon resonance (SPR) and cryo-electron microscopy (cryo-EM). The cryo-EM results confirmed that one of the designed nanobodies closely matched its predicted structure, validating the method’s accuracy at atomic resolution. It should be noted that at the time of publication of this review, this promising method has not yet released any public tool associated with it.
For full protein/antibody design, the structural scaffolds generated need to be designed with sequences. This is the domain of inverse folding that models a sequence given a rigid structure. The scaffold generation and inverse folding algorithms are used currently in conjunction for a full protein/antibody design pipeline.
Sequence design in structural context - inverse folding
It is often desirable to improve upon a known protein sequence, and the task is made easier if its coordinates are known. Altogether the problem is known as ‘inverse folding’ (Table 7, Figure 3F) for its clear shift in the prediction objective to the protein folding problem. Though the problem has been known and tackled for a very long time (118), recent advancements in protein structure prediction and antibody sequence generation have resulted in a revival of such methods. Millions of predicted structures by AlphaFold2 can be used to train inverse folding algorithms. Large-scale generation of antibody sequences allows us to model these and develop antibody-specific antibody inverse folding methods.
Protein-generic inverse folding methods generally form the foundation for antibody-specific design approaches, with ProteinMPNN and ESM-IF being two prominent examples. ProteinMPNN (20) uses a message-passing neural network (MPNN) to predict sequences that fold into given protein structures by encoding features like atomic distances and frame orientations. It achieves high sequence recovery rates and structural fidelity, outperforming traditional methods such as Rosetta. ProteinMPNN has been validated both computationally and experimentally, with techniques like X-ray crystallography and cryoEM confirming its ability to accurately fold into target structures. ESM-IF (21, 23) leverages a GVP-Transformer model trained on a dataset of 16,000 experimental and 12 million AlphaFold2-predicted structures, achieving notable improvements in sequence recovery, particularly for buried residues. It demonstrated its utility in practical applications by introducing point mutations to anti-SARS-CoV-2 antibodies (23), which enhanced binding affinity in experimental validations.
ESM-IF and ProteinMPNN were trained on a large corpus of proteins, but arguably a very small sample of all allowed antibody structures. To the best of our knowledge, two antibody-specific inverse folding methods were developed, AbMPNN and AntiFold, fine-tuning ProteinMPNN and ESM-IF respectively on a modeled antibody corpus.
AbMPNN (83), trained on 3,500 antigen-binding fragments from SAbDab and 147,919 paired variable regions from OAS using ABodyBuilder2-derived structures, achieved 60% sequence recovery for CDR loops—outperforming ProteinMPNN’s 40%—and showed a 20% improvement in median RMSD for CDR-H3 loops, enhancing designability and stability. AntiFold (106), trained on 2,074 experimentally solved and 147,458 predicted antibody structures, excelled in amino acid recovery (60% for CDR-H3) and achieved a Spearman’s rank correlation of 0.418 for antibody-antigen binding affinity, surpassing AbMPNN and ESM-2. IgDesign (119) focused on designing complementarity-determining regions (CDRs) for eight therapeutic antigens, generating 1 million sequences per antigen and filtering them for in vitro testing. It achieved superior binding rates across antigens, with statistically significant improvements for 7 out of 8 HCDR3 targets, making it a standout for experimental validation. While IgDesign demonstrated experimental success, it is not freely available, unlike AbMPNN and AntiFold that are free, but did not demonstrate experimental validation.
Inverse folding methods represent a powerful alternative to simultaneous sequence-backbone design by assuming that the fold will not change. This is oftentimes desirable as structure is crucial to antibody-antigen recognition and only minute changes need to be introduced, maintaining the fold, but modulating the overall function of the antibody.
Generating antibodies with improved functions - developability optimization
The chief focus of this review, as much of antibody design, is focused on binder development. However developing a binder is arguably an experimentally solved problem with the bigger wet-lab hurdle being the optimization of subsequent developability properties (Table 8). The iterative process of fine-tuning the myriad biophysical properties is not linear and can account for much of the time and effort in the preclinical stage (48, 50, 120).
Antibody design methods that address developability issues are much more heterogeneous both in their approaches and goals. Arguably, binder development has one objective, which is generally a high affinity interaction. A single developability property on the other hand, such as self-association, can have several assays associated with it that might not be directly comparable to one another. There is also a great scarcity of data on developability points. To date, the Jain characterization of ca. 100 therapeutic antibodies, remains one of the most comprehensive characterizations of developable antibodies - however without negative data points (121).
Because of data scarcity, many methods, such as the Oracles (Table 2, Figure 3A), focus on close-to-exhaustive enumeration of binders, followed by computational filtering for developable antibodies using general tools such as the Therapeutic Antibody Profiler (122) or Camsol (123).
There also exists a plethora of work dedicated to the computational prediction of individual assay data and properties that can lead to more developable models and be incorporated in generative protein design models (48, 124). A full review of these models is beyond the scope of this review, but generally these include Hydrophobic Interaction Chromatography (HIC), expression (concentration, purity), stability, chemical modifications (D isomerization, N deamidation), enzymatic PTMs including phosphorylation and glycosylation, immunogenicity (T-cell epitope), Pk properties such as clearance, and viscosity. Many of the published methods for these properties are not commercially or academically available as they are developed using scarce proprietary data, however, with the advent and broad participation of consortia such as the FAITE consortium (https://faiteconsortium.org/), this could be shifting.
An alternative to enumeration followed by binder validation is biased generation of sequences through multi-property optimization (MPO). Amimeur et al. (85) pioneered this approach using a Generative Adversarial Network (GAN) trained on over 400,000 sequences from the Observed Antibody Space (OAS) to produce humanoid antibodies with human-like structural and functional diversity. The GAN, fine-tuned for therapeutic traits such as stability and low immunogenicity, was experimentally validated through assays like DSF, SEC, and SINS, achieving a high success rate in generating antibodies with desirable developability profiles. Building on this, Turnbull et al. (86) introduced p-IgGen, a GPT-2-based model fine-tuned on paired and developable antibody sequences, which excelled in immunogenicity prediction while maintaining computational efficiency. Complementing these generative approaches, Hutchins et al. (87) used the DeepAb model to design 200 anti-HEL antibody variants, optimizing thermostability and affinity through mutations informed by deep mutational scanning and achieving significant experimental success, with most variants showing increased stability and up to a 21-fold affinity improvement. Dreyer et al. (88) extended this pipeline for one-shot antibody discovery, using computational tools like AbMPNN and ESM to design SARS-CoV-2 RBD-binding antibodies with enhanced developability, validated experimentally through stability and aggregation assays, achieving a 54% success rate against escape mutations.
Altogether, though there appears to be progress in generating and designing antibodies with improved developability properties, most efforts are one off proofs of concept. To close the gap between experimentation and these methods being employed to develop novel drugs, data generation, method integration and benchmarking is necessary.
Outstanding challenges - data & experimental validation
The biggest issue within the protein design field remains not model development but data availability, both for training and benchmarking. Unlike in text or image generation fields, where it is fairly cheap to gather datasets of millions of data points, in biology it is not. Data generation is prohibitively expensive with large discrepancies between cheaper but less informative (sequences) and more expensive but more informative datasets (e.g. structures).
There are only ca. 15,000 non-redundant single chain protein structures in the protein data bank (28) - compared to several billion sequenced chains (125). On the antibody front, there are several billion sequences available (126–128), but only several thousand non-redundant structures (94). The number of non-redundant antibody-antigen complexes is smaller still, being in the order of around 1000 structures.
The structure datasets are crucial to the development of backbone generation protocols, such as RFDiffusion. Paucity of such data, especially on the antibody-antigen complex front is a blocker for development of better algorithms, as even the latest iteration of AlphaFold, falls short of providing an actionable solution to the antibody-antigen interaction problem. The structural antibody datasets such as SAbDab (94) and ABDB (129) have become proxies for benchmarks as they compile antibody-antigen information specific to antibodies. For the design tasks, the complexes are employed to train backbone generation algorithms. Much more specialized datasets that also gather affinity information are SKEMPI (130) and AB-BIND (131). Here, the structure of an antibody-antigen is accompanied by mutation and affinity measurement. Nevertheless, these datasets contain a small number of structures and measurements relative to the scale of the problem (several hundred data points each).
The copious sequence datasets are employed to learn meaningful representations of molecules, given the unavailability of the corresponding large-scale structure data. For structure-informed sequence design using inverse folding, the paucity of structural data is side-stepped by model generation. For instance ESM-IF was trained using 12m AlphaFold models. In the antibody space, the widely used resource is the Observed Antibody Space (96, 127), which curates repertoire data used chiefly for language model training. Similar to ESM-IF training, OAS data were modeled and used for training the ABMPNN and AntiFold, to extend the datasets beyond the several thousand available structures. Overall usage of such synthetic data is not only a way to train better models but also to estimate how much data would be needed to solve the problem altogether (106, 132). Though it is plausible to re-use such experimentally generated data, care needs to be exercised as it has been raised that such data might in fact be biasing the models in the undesirable direction (133). The fact remains that though there is a lot of sequence data, hardly any of it is associated with binding or developability data points.
The paucity of data highlights another large problem, which is experimental validation. Many design methods are proposed purely in silico, without follow-on experimental work. The ways in which in silico performance is measured typically focuses on ‘re-discovery’ of existing binders (e.g. amino acid recovery, DockQ score or RMSD to reference binder). In such a scenario truly novel binders cannot be discovered. Sometimes a handful of sequences are produced to test binding, however oftentimes without any control, so it is quite difficult to gauge what benefits the methods actually bring over purely experimental discovery.
A facet where computational antibody design/protein design can bring value is not only speed but explainability and safety. By producing antibodies in silico one would expect to shift the paradigm from discovery to informed design. However, for that, the performance of the novel binders would have to be measured not only via the prism of binding efficacy but also the myriad biophysical features a biologic should have.
Such benchmarking however is done on publicly available datasets - which have few data points for structures (~1000), affinity (100s) and even less for developability (10s, depending on which assay one focuses on (FLAb: Benchmarking Deep Learning Methods for Antibody Fitness Prediction, 134). Because of the paucity of the data, generative methods are often compared on pre-existing datasets (135). In an ideal scenario, the generated sequences would in each case be made in the lab and the structures solved, as is done in few cases.
Experimental validation, however, is very expensive and benchmarks on par with CASP or CACHE are only very recently coming into existence in the protein design world (136–138). In the antibody-world, such benchmarking needs to take into account not only whether one can develop a binder but also how developable such sequences are (FLAb: Benchmarking Deep Learning Methods for Antibody Fitness Prediction, 134). Arguably, developing a binder is an experimentally solved problem, with the biggest remaining issues being hitting the right epitope, specificity, and developability. A binding model achieving starling results on RMSD of the predicted complex, but that would not hit the right biophysical properties (e.g. off-target effects, CDR-H3 stability, glycosylation etc.) would not be as useful as a library-based method that does. Any kind of benchmarking needs to weigh the speed and cost of computation versus purely experimental discovery. For this reason, the antibody-specific benchmarking that comes into existence takes this into account through both developability challenges as well as carefully weighing the benefits of experimental versus purely computational approaches (139, 140).
Altogether, protein and antibody design fields are still in their infancy. Most methods correctly focus on binder development, as this problem needs to be solved to start tackling a wider multifactorial puzzle of developability (120), Fc-engineering (141) of pH-dependent effects (142). It will take time, both in development and adoption before computational methods become the driver of discovery and design. It is difficult to envisage how much of the non-linear biologics discovery workflow can be replaced. On the pre-clinical front there are many opportunities as many of the binding/developability experiments should be susceptible to modeling, with enough data generated. Effect on target discovery/validation and clinical trials that require much deeper understanding of biology are much more difficult to define.
Given the state of the field, we think that the change will be fueled chiefly by creating large complex datasets adorned with developability data together with conscientious benchmarking and collaboration across industry and academia.
Author contributions
WB: Data curation, Formal Analysis, Supervision, Validation, Writing – original draft, Writing – review & editing. IJ: Data curation, Formal Analysis, Supervision, Validation, Writing – original draft, Writing – review & editing. PD: Writing – original draft, Writing – review & editing. BJ: Writing – review & editing. DC: Writing – review & editing. SW: Writing – review & editing. VG: Writing – review & editing. RF: Writing – review & editing. JA-B: Writing – original draft, Writing – review & editing. KK: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
Authors WB, IJ, PD, BJ, DC, SW, and KK were employed by the company Natural Antibody. Author VG was employed by the company Imprint Labs, LLC. Authors RF and JA-B were employed by the company Janssen Pharmaceuticals.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Santuari L, Salvy MB, Xenarios I, and Arpat B. AI-accelerated therapeutic antibody development: practical insights. Front Drug Discov. (2024) 4:1447867. doi: 10.3389/fddsv.2024.1447867
2. Laustsen AH, Greiff V, Karatt-Vellatt A, Muyldermans S, and Jenkins TP. Animal immunization, in vitro display technologies, and machine learning for antibody discovery. Trends Biotechnol. (2021) 39:1263–73. doi: 10.1016/j.tibtech.2021.03.003
3. Köhler G and Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature. (1975) 256:495–97. doi: 10.1038/256495a0
4. Irvine EB and Reddy ST. Advancing antibody engineering through synthetic evolution and machine learning. J Immunol. (2024) 212:235–435. doi: 10.4049/jimmunol.2300492
5. Smith GP. Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science. (1985) 228:1315–17. doi: 10.1126/science.4001944
6. Vazquez-Lombardi R, Nevoltris D, Luthra A, Schofield P, Zimmermann C, and Christ D. Transient expression of human antibodies in mammalian cells. Nat Protoc. (2018) 13:99–1175. doi: 10.1038/nprot.2017.126
7. O’Donnell TJ, Kanduri C, Isacchini G, Limenitakis JP, Brachman RA, Alvarez RA, et al. Reading the repertoire: progress in adaptive immune receptor analysis using machine learning. Cell Syst. (2024) 15:1168–895. doi: 10.1016/j.cels.2024.11.006
8. Kuhlman B and Bradley P. Advances in protein structure prediction and design. Nat Rev Mol Cell Biol. (2019) 20:681–975. doi: 10.1038/s41580-019-0163-x
9. Martí-Renom MA, Stuart AC, Fiser A, Sánchez R, Melo F, and Sali A. Comparative protein structure modeling of genes and genomes. Annu Rev Biophysics Biomolecular Structure. (2000) 29:291–325. doi: 10.1146/annurev.biophys.29.1.291
10. Lindorff-Larsen K, Piana S, Dror RO, and Shaw DE. How fast-folding proteins fold. Science. (2011) 334:517–20. doi: 10.1126/science.1208351
11. Adolf-Bryfogle J, Kalyuzhniy O, Kubitz M, Weitzner BD, Hu X, Adachi Y, et al. RosettaAntibodyDesign (RAbD): A general framework for computational antibody design. PloS Comput Biol. (2018) 14:e10061125. doi: 10.1371/journal.pcbi.1006112
12. Lippow SM, Wittrup KD, and Tidor B. Computational design of antibody-affinity improvement beyond in vivo maturation. Nat Biotechnol. (2007) 25:1171–765. doi: 10.1038/nbt1336
13. Jeliazkov JR, Frick R, Zhou J, and Gray JJ. Robustification of rosettaAntibody and rosetta snugDock. PloS One. (2021) 16:e02342825. doi: 10.1371/journal.pone.0234282
14. Sircar A and Gray JJ. SnugDock: paratope structural optimization during antibody-Antigen docking compensates for errors in antibody homology models. PloS Comput Biol. (2010) 6:e10006445. doi: 10.1371/journal.pcbi.1000644
15. Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, et al. Highly accurate protein structure prediction with alphaFold. Nature. (2021) 596:583–89. doi: 10.1038/s41586-021-03819-2
16. Rives A, Meier J, Sercu T, Goyal S, Lin Z, Liu J, et al. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. Proc Natl Acad Sci United States America. (2021) 118. doi: 10.1073/pnas.2016239118
17. Varadi M, Anyango S, Deshpande M, Nair S, Natassia C, Yordanova G, et al. AlphaFold protein structure database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. (2022) 50:D439–44. doi: 10.1093/nar/gkab1061
18. Watson JL, Juergens D, Bennett NR, Trippe BL, Yim J, Eisenach HE, et al. De novo design of protein structure and function with RFdiffusion. Nature. (2023) 620:1089–100. doi: 10.1038/s41586-023-06415-8
19. Khakzad H, Igashov I, Schneuing A, Goverde C, Bronstein M, and Correia B. A new age in protein design empowered by deep learning. Cell Syst. (2023) 14:925–395. doi: 10.1016/j.cels.2023.10.006
20. Dauparas J, Anishchenko I, Bennett N, Bai H, Ragotte RJ, Milles LF, et al. Robust deep learning–based protein sequence design using proteinMPNN. Science. (2022) 378:49–56. doi: 10.1126/science.add2187
21. Hsu C, Verkuil R, Liu J, Lin Z, Hie B, Sercu T, et al. Learning inverse folding from millions of predicted structures. In: Chaudhuri K, Le S, Csaba S, Niu G, and Sivan S, editors. Proceedings of the 39 th International Conference on Machine Learning. Baltimore, Maryland, USA: PMLR (2022) 162 :8946–70.
22. Bennett NR, Watson JL, Ragotte RJ, Borst AJ, See D, Weidle C, et al. Atomically accurate de novo design of single-domain antibodies. bioRxiv: Preprint Server Biology March. (2024). doi: 10.1101/2024.03.14.585103
23. Shanker VR, Bruun TUJ, Hie BL, and Kim PS. Unsupervised evolution of protein and antibody complexes with a structure-informed language model. Science. (2024) 385:46–53. doi: 10.1126/science.adk8946
24. Hie BL, Shanker VR, Xu D, Theodora UJ, Weidenbacher PA, et al. Efficient evolution of human antibodies from general protein language models. Nat Biotechnol. (2024) 42:275–835. doi: 10.1038/s41587-023-01763-2
25. Listov D, Goverde CA, Correia BE, and Fleishman SJ. Opportunities and challenges in design and optimization of protein function. Nat Rev Mol Cell Biol. (2024) 25:639–53. doi: 10.1038/s41580-024-00718-y
26. Liu Y and Kuhlman B. RosettaDesign server for protein design. Nucleic Acids Res. (. 2006) 34:W235–38. doi: 10.1093/nar/gkl163
27. Leman JK, Weitzner BD, Lewis SM, Adolf-Bryfogle J, Alam N, Alford RF, et al. Macromolecular modeling and design in rosetta: recent methods and frameworks. Nat Methods. (2020) 17:665–80. doi: 10.1038/s41592-020-0848-2
28. Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, et al. The protein data bank. Nucleic Acids Res. (2000) 28:235–42. doi: 10.1093/nar/28.1.235
29. Evans R, O’Neill M, Pritzel A, Antropova N, Senior A, Green T, et al. Protein complex prediction with alphaFold-multimer. bioRxiv. (2022). doi: 10.1101/2021.10.04.463034
30. Goverde CA, Pacesa M, Goldbach N, Dornfeld LJ, Balbi PEM, Georgeon S, et al. Computational design of soluble and functional membrane protein analogues. Nature. (2024) 631:449–58. doi: 10.1038/s41586-024-07601-y
31. Woolfson DN. Understanding a protein fold: the physics, chemistry, and biology of α-helical coiled coils. J Biol Chem. (2023) 299:104579. doi: 10.1016/j.jbc.2023.104579
32. Kuhlman B, Dantas G, Gregory C, Varani IG, Stoddard BL, and Baker D. Design of a novel globular protein fold with atomic-level accuracy. Sci (New York N.Y.). (2003) 302:1364–68. doi: 10.1126/science.1089427
33. Pacesa M, Nickel L, Schellhaas C, Schmidt J, Pyatova E, Kissling L, et al. BindCraft: one-shot design of functional protein binders. bioRxiv. (2024). doi: 10.1101/2024.09.30.615802
34. Praljak N, Yeh H, Moore M, Socolich M, Ranganathan R, and Ferguson AL. Natural language prompts guide the design of novel functional protein sequences. Synthetic Biol bioRxiv. (2024). doi: 10.1101/2024.11.11.622734v1.full
35. NobelPrize.org. The nobel prize in chemistry 2024 (2025). Available online at: https://www.nobelprize.org/prizes/chemistry/2024/press-release/ (Accessed May 4, 2025).
36. Crescioli S, Kaplon H, Chenoweth A, Wang L, Visweswaraiah J, and Reichert JM. Antibodies to watch in 2024. mAbs. (2024) 16:22974505. doi: 10.1080/19420862.2023.2297450
37. Rees AR. Understanding the human antibody repertoire. mAbs. (2020) 12:1729683. doi: 10.1080/19420862.2020.1729683
38. Mason DM, Friedensohn S, Weber C, Jordi C, Wagner B, Meng SM, et al. Optimization of therapeutic antibodies by predicting antigen specificity from antibody sequence via deep learning. Nat Biomed Eng. (2021) 5:600–12. doi: 10.1038/s41551-021-00699-9
39. Chinery L, Hummer AM, Mehta BB, Akbar R, Rawat P, Slabodkin A, et al. Baselining the buzz trastuzumab-HER2 affinity, and beyond. bioRxiv. (2024). doi: 10.1101/2024.03.26.586756
40. Jeliazkov JR, Sljoka A, Kuroda D, Tsuchimura N, Katoh N, Tsumoto K, et al. Repertoire analysis of antibody CDR-H3 loops suggests affinity maturation does not typically result in rigidification. Front Immunol. (2018) 9:413. doi: 10.3389/fimmu.2018.00413
41. Regep C, Georges G, Shi J, Popovic B, and Deane CM. The H3 loop of antibodies shows unique structural characteristics: CDR H3 of antibodies shows unique structural characteristics. Proteins. (2017) 85:1311–185. doi: 10.1002/prot.25291
42. Fernández-Quintero ML, Kraml J, Georges G, and Liedl KR. CDR-H3 loop ensemble in solution - conformational selection upon antibody binding. mAbs. (2019) 11:1077–885. doi: 10.1080/19420862.2019.1618676
43. Hitawala FN and Gray JJ. What has alphaFold3 learned about antibody and nanobody docking, and what remains unsolved? Bioengineering. bioRxiv. (2024). doi: 10.1101/2024.09.21.614257v1
44. Bang Y, Choi Y-A, Gu J, Kwon S, Lee D, Lee MS, et al. Accurate antibody loop structure prediction enables zero-shot design of target-specific antibodies. bioRxiv. (2024). doi: 10.1101/2024.08.23.609114
45. Sela-Culang I, Kunik V, and Ofran Y. The structural basis of antibody-antigen recognition. Front Immunol. (2013) 4:302. doi: 10.3389/fimmu.2013.00302
46. Prihoda D, Maamary J, Waight A, and Juan V. BioPhi: A platform for antibody design, humanization and humanness evaluation based on natural antibody repertoires and deep learning. mAbs. (2022) 14(1):2020203. doi: 10.1080/19420862.2021.2020203
47. Tennenhouse A, Khmelnitsky L, Khalaila R, Yeshaya N, Noronha A, Lindzen M, et al. Computational optimization of antibody humanness and stability by systematic energy-based ranking. Nat Biomed Eng. (2024) 8(1):30–44. doi: 10.1038/s41551-023-01079-1
48. Khetan R, Curtis R, Deane CM, Hadsund JT, Kar U, Krawczyk K, et al. Current advances in biopharmaceutical informatics: guidelines, impact and challenges in the computational developability assessment of antibody therapeutics. mAbs. (2022) 14:2020082. doi: 10.1080/19420862.2021.2020082
49. Akbar R, Bashour H, Rawat P, Robert PA, Smorodina E, Cotet T-S, et al. Progress and challenges for the machine learning-based design of fit-for-purpose monoclonal antibodies. mAbs. (2022) 14:2008790. doi: 10.1080/19420862.2021.2008790
50. Bashour H, Smorodina E, Pariset M, Zhong J, Akbar R, Chernigovskaya M, et al. Biophysical cartography of the native and human-engineered antibody landscapes quantifies the plasticity of antibody developability. Commun Biol. (2024) 7:922. doi: 10.1038/s42003-024-06561-3
51. Ruffolo JA, Bhatnagar A, Beazer J, Nayfach S, Russ J, Hill E, et al. Adapting protein language models for structure-conditioned design. bioRxiv. (2024) https. doi: 10.1101/2024.08.03.606485
52. Shanehsazzadeh A, Bachas S, Kasun G, Sutton JM, Steiger AK, Shuai R, et al. Unlocking de novo antibody design with generative artificial intelligence. bioRxiv. (2023). doi: 10.1101/2023.01.08.523187
53. Lim YW, Adler AS, and Johnson DS. Predicting antibody binders and generating synthetic antibodies using deep learning. mAbs. (2022) 14:20690755. doi: 10.1080/19420862.2022.2069075
54. Ursu E, Minnegalieva A, Rawat P, Chernigovskaya M, Tacutu R, Sandve GK, et al. Training data composition determines machine learning generalization and biological rule discovery. Bioinf bioRxiv. (2024). doi: 10.1101/2024.06.17.599333v1.full.pdf
55. Agarwal, Aditya A, Harrang J, Noble D, McGowan KL, Lange AW, et al. AlphaBind, a domain-specific model to predict and optimize antibody-antigen binding affinity. Synthetic Biol bioRxiv. (2024). doi: 10.1101/2024.11.11.622872v1.full
56. Mahajan SP, Ruffolo JA, Frick R, and Gray JJ. Hallucinating structure-conditioned antibody libraries for target-specific binders. Front Immunol. (2022) 13:999034. doi: 10.3389/fimmu.2022.999034
57. Clark T, Subramanian V, Jayaraman A, Fitzpatrick E, Gopal R, Pentakota N, et al. Enhancing antibody affinity through experimental sampling of non-deleterious CDR mutations predicted by machine learning. Commun Chem. (2023) 6:244. doi: 10.1038/s42004-023-01037-7
58. Cai H, Zhang Z, Wang M, Zhong B, Li Q, Zhong Y, et al. Pretrainable geometric graph neural network for antibody affinity maturation. Nat Commun. (2024) 15:77855. doi: 10.1038/s41467-024-51563-8
59. Jin W, Chen X, Vetticaden A, Sarzikova S, Raychowdhury R, Uhler C, et al. DSMBind: SE(3) denoising score matching for unsupervised binding energy prediction and nanobody design. Bioinf bioRxiv. (2023). doi: 10.1101/2023.12.10.570461v1.full.pdf
60. Li M, Shi Y, Hu S, Hu S, Guo P, Wan W, et al. MVSF-AB: accurate antibody-antigen binding affinity prediction via multi-view sequence feature learning. Bioinf (Oxford England). (2024), btae579. doi: 10.1093/bioinformatics/btae579
61. Luo S, Su Y, Wu Z, Su C, Peng J, and Ma J. Rotamer density estimator is an unsupervised learner of the effect of mutations on protein-protein interaction (2022). Available online at: https://openreview.net/forum?id=_X9Yl1K2mD (Accession May 4, 2025).
62. Myung Y, Pires DEV, and Ascher DB. CSM-AB: graph-based antibody-antigen binding affinity prediction and docking scoring function. Bioinf (Oxford England). (2022) 38:1141–435. doi: 10.1093/bioinformatics/btab762
63. Wu F, Zhao Y, Wu J, Jiang B, He B, Huang L, et al. Fast and accurate modeling and design of antibody-antigen complex using tFold. Bioinf bioRxiv. (2024). doi: 10.1101/2024.02.05.578892v1
64. Yin R and Pierce BG. Evaluation of alphaFold antibody-antigen modeling with implications for improving predictive accuracy. Protein Science: A Publ Protein Soc. (2024) 33:e48655. doi: 10.1002/pro.v33.1
65. Abramson J, Adler J, Dunger J, Evans R, Green T, Pritzel A, et al. Accurate structure prediction of biomolecular interactions with alphaFold 3. Nature. (2024) 630:493–500. doi: 10.1038/s41586-024-07487-w
66. Boitreaud J, Dent J, McPartlon M, Meier J, Reis V, Rogozhnikov A, et al. Chai-1: decoding the molecular interactions of life. Synthetic Biol bioRxiv. (2024). doi: 10.1101/2024.10.10.615955v1
67. Wohlwend J, Corso G, Passaro S, Reveiz M, Leidal K, Swiderski W, et al. Boltz-1 democratizing biomolecular interaction modeling. Biophysics. bioRxiv. (2024). doi: 10.1101/2024.11.19.624167v1.full
68. Pantazes RJ and Maranas CD. OptCDR: A general computational method for the design of antibody complementarity determining regions for targeted epitope binding. Protein Engineering Design Selection: PEDS. (2010) 23:849–58. doi: 10.1093/protein/gzq061
69. Li T, Pantazes RJ, and Maranas CD. OptMAVEn–a new framework for the de novo design of antibody variable region models targeting specific antigen epitopes. PloS One. (2014) 9:e1059545. doi: 10.1371/journal.pone.0105954
70. Lapidoth GD, Baran D, Pszolla GM, Norn C, Alon A, Tyka MD, et al. AbDesign: an algorithm for combinatorial backbone design guided by natural conformations and sequences: combinatorial backbone design in antibodies. Proteins. (2015) 83:1385–14065. doi: 10.1002/prot.24779
71. Rangel MA, Bedwell A, Costanzi E, Taylor RJ, Russo R, Bernardes G, et al. Fragment-based computational design of antibodies targeting structured epitopes. . Sci Adv. (2022) 8:eabp9540. doi: 10.1126/sciadv.abp9540
72. Jin W, Wohlwend J, Barzilay R, and Jaakkola T. Iterative refinement graph neural network for antibody sequence-structure co-design. arXiv [q-bio.BM]. arXiv. (2021). doi: 10.04624
73. Kong X, Huang W, and Liu Y. Conditional antibody design as 3D equivariant graph translation. arXiv[q-bio.BM].arXiv. (2022). http://arxiv.org/abs/2208.06073.
74. Kong X, Huang W, and Liu Y. End-to-end full-atom antibody design. arXiv. (2023). doi: 10.48550/arXiv.2302.00203
75. Luo S. Diffab: Antigen-specific antibody design and optimization with diffusion-based generative models for protein structures (NeurIPS 2022). Github (2024). Available at: https://github.com/luost26/diffab (Accessed May 4, 2025).
76. Martinkus K, Ludwiczak J, Cho K, Liang W-C, LaFrance-Vanasse J, Hotzel I, et al. AbDiffuser: full-atom generation of in-vitro functioning antibodies. arXiv [q-bio.BM]. arXiv. (2023). Available at: http://arxiv.org/abs/2308.05027.
77. Zhu T, Ren M, and Zhang H. Antibody design using a score-based diffusion model guided by evolutionary, physical and geometric constraints (2024). Available online at: https://openreview.net/forum?id=1YsQI04KaN&referrer=%5Bthe%20profile%20of%20Milong%20Ren%5D(%2Fprofile%3Fid%3D~Milong_Ren2 (Accessed May 4, 2025).
78. Xie X, Valiente PA, Lee JS, Kim J, and Kim PM. Antibody-SGM, a score-based generative model for antibody heavy-chain design. J Chem Inf Modeling. (2024) 64:6745–575. doi: 10.1021/acs.jcim.4c00711
79. Cutting D, Dreyer F, Errington D, Schneider C, and Deane CM. De novo antibody design with SE(3) diffusion. arXiv [q-bio.BM]. arXiv. (2024). Available at: http://arxiv.org/abs/2405.07622.
80. Eguchi RR, Choe CA, and Huang P-S. Ig-VAE: generative modeling of protein structure by direct 3D coordinate generation. PloS Comput Biol. (2022) 18:e10102715. doi: 10.1371/journal.pcbi.1010271
81. Eguchi RR, Choe CA, Parekh U, Irene S, Ward MD, et al. Deep generative design of epitope-specific binding proteins by latent conformation optimization. Biochem bioRxiv. (2022). doi: 10.1101/2022.12.22.521698v1.full.pdf
82. Shanehsazzadeh A, Alverio J, Kasun G, Levine S, Calman I, Khan JA, et al. IgDesign: in vitro validated antibody design against multiple therapeutic antigens using inverse folding. Synthetic Biol bioRxiv. (2023). doi: 10.1101/2023.12.08.570889v2.full.pdf
83. Dreyer F, Cutting D, Schneider C, Kenlay H, and Deane CM. Inverse folding for antibody sequence design using deep learning. arXiv [q-bio.BM]. arXiv. (2023). doi: 10.19513
84. Hummer AM, Høie MH, Olsen TH, Nielsen M, and Deane CM. AntiFold improved antibody structure design using inverse folding. In: NeurIPS machine learning for structural biology workshop. Machine Learning for Structural Biology Workshop, NeurIPS (2023). Available at: https://www.mlsb.io/papers_2023/AntiFold_Improved_antibody_structure_design_using_inverse_folding.pdf (Accessed May 4, 2025).
85. Amimeur T, Shaver JM, Ketchem RR, Taylor JA, Clark RH, Smith J, et al. Designing feature-controlled humanoid antibody discovery libraries using generative adversarial networks. bioRxiv. (2020). doi: 10.1101/2020.04.12.024844
86. Turnbull OM, Oglic D, Croasdale-Wood R, and Deane CM. P-igGen: A paired antibody generative language model. Bioinformatics (2024) 40:btae659. doi: 10.1093/bioinformatics/btae659
87. Hutchinson M, Ruffolo JA, Haskins N, Iannotti M, Vozza G, Pham T, et al. Toward enhancement of antibody thermostability and affinity by computational design in the absence of antigen. mAbs. (2024) 16:2362775. doi: 10.1080/19420862.2024.2362775
88. Dreyer F, Schneider C, Kovaltsuk A, Cutting D, Byrne MJ, Nissley DA, et al. Computational design of developable therapeutic antibodies: efficient traversal of binder landscapes and rescue of escape mutations. Bioinf bioRxiv. (2024). doi: 10.1101/2024.10.03.616038v1.full.pdf
89. Jaszczyszyn I, Bielska W, Gawlowski T, Dudzic P, Satława T, Kończak J, et al. Structural modeling of antibody variable regions using deep learning—progress and perspectives on drug discovery. Front Mol Biosci. (2023) 10:1214424. doi: 10.3389/fmolb.2023.1214424
90. Basu S and Wallner B. DockQ: A quality measure for protein-protein docking models. PloS One. (2016) 11:e01618795. doi: 10.1371/journal.pone.0161879
91. Yang KK, Wu Z, and Arnold FH. Machine-learning-guided directed evolution for protein engineering. Nat Methods. (2019) 16:687–945. doi: 10.1038/s41592-019-0496-6
92. Hayes T, Rao R, Akin H, Sofroniew NJ, Oktay D, Lin Z, et al. Simulating 500 million years of evolution with a language model. Synthetic Biol bioRxiv. (2024). doi: 10.1101/2024.07.01.600583v1
93. Madani A, Krause B, Greene ER, Subramanian S, Mohr BP, Holton JM, et al. Large language models generate functional protein sequences across diverse families. Nat Biotechnol. (2023) 41:1099–106. doi: 10.1038/s41587-022-01618-2
94. Dunbar J, Krawczyk K, Leem J, Baker T, Fuchs A, Georges G, et al. SAbDab: the structural antibody database. Nucleic Acids Res. (2014) 42:D1140–46. doi: 10.1093/nar/gkt1043
95. Ruffolo JA, Gray JJ, and Sulam J. Deciphering antibody affinity maturation with language models and weakly supervised learning. arXiv [q-bio.BM]. arXiv. (2021). Available at: http://arxiv.org/abs/2112.07782.
96. Olsen TH, Boyles F, and Deane CM. Observed antibody space: A diverse database of cleaned, annotated, and translated unpaired and paired antibody sequences. Protein Science: A Publ Protein Soc. (2022) 31:141–465. doi: 10.1002/pro.v31.1
97. Leem J, Mitchell LS, Farmery JHR, Barton J, and Galson JD. Deciphering the language of antibodies using self-supervised learning. Patterns (New York N.Y.). (2022) 3:1005135. doi: 10.1016/j.patter.2022.100513
98. Barton J, Gaspariunas A, Galson JD, and Leem J. Building representation learning models for antibody comprehension. Cold Spring Harbor Perspect Biol. (2023) 16(3):a041462. doi: 10.1101/cshperspect.a041462
99. Engelhart E, Emerson R, Shing L, Lennartz C, Guion D, Kelley M, et al. A dataset comprised of binding interactions for 104,972 antibodies against a SARS-coV-2 peptide. Sci Data. (2022) 9:6535. doi: 10.1038/s41597-022-01779-4
100. Deutschmann N, Pelissier A, Weber A, Gao S, Bogojeska J, and Martínez M. Do domain-specific protein language models outperform general models on immunology-related tasks? ImmunoInformatics. (2024) 14:100036. doi: 10.1016/j.immuno.2024.100036
101. Krause B, Subramanian S, Yuan T, Yang M, Sato A, and Naik N. Improving antibody affinity using laboratory data with language model guided design. Synthetic Biol bioRxiv. (2023). doi: 10.1101/2023.09.13.557505v1.full
102. Barton J, Gaspariunas A, Yadin DA, Dias J, Nice FL, Minns DH, et al. A generative foundation model for antibody sequence understanding. bioRxiv. (2024). doi: 10.1101/2024.05.22.594943
103. Kurumida Y, Saito Y, and Kameda T. Predicting antibody affinity changes upon mutations by combining multiple predictors. Sci Rep. (2020) 10:195335. doi: 10.1038/s41598-020-76369-8
104. Sevy AM, Wu NC, Gilchuk IM, Parrish EH, Burger S, Yousif D, et al. Multistate design of influenza antibodies improves affinity and breadth against seasonal viruses. Proc Natl Acad Sci United States America. (2019) 116:1597–602. doi: 10.1073/pnas.1806004116
105. Poosarla VG, Li T, Goh BC, Schulten K, Wood TK, and Maranas CD. Computational de novo design of antibodies binding to a peptide with high affinity. Biotechnol Bioengineering. (2017) 114:1331–425. doi: 10.1002/bit.v114.6
106. Hummer AM, Schneider C, Chinery L, and Deane CM. Investigating the volume and diversity of data needed for generalizable antibody-antigen ΔΔG prediction. bioRxiv. (2023). doi: 10.1101/2023.05.17.541222
107. Almagro JC, Teplyakov A, Luo J, Sweet RW, Kodangattil S, Hernandez-Guzman F, et al. Second antibody modeling assessment (AMA-II). Proteins. (2014) 82:1553–625. doi: 10.1002/prot.24567
108. Kończak J, Janusz B, Młokosiewicz J, Satława T, Wróbel S, Dudzic P, et al. Structural pre-training improves physical accuracy of antibody structure prediction using deep learning. ImmunoInformatics. (2023) 11:100028. doi: 10.1016/j.immuno.2023.100028
109. Abanades B, Wong WK, Boyles F, Georges G, Bujotzek A, and Deane CM. ImmuneBuilder: deep-learning models for predicting the structures of immune proteins. Commun Biol. (2023) 6:5755. doi: 10.1038/s42003-023-04927-7
110. Ruffolo JA, Chu L-S, Mahajan SP, and Gray JJ. Fast, accurate antibody structure prediction from deep learning on massive set of natural antibodies. Nat Commun. (2023) 14:23895. doi: 10.1038/s41467-023-38063-x
111. Desta IT, Kotelnikov S, Jones G, Ghani U, Abyzov M, Kholodov Y, et al. The clusPro abEMap web server for the prediction of antibody epitopes. Nat Protoc. (2023) 18:1814–405. doi: 10.1038/s41596-023-00826-7
112. Ambrosetti F, Jiménez-García B, Roel-Touris J, and Bonvin AMJJ. Modeling antibody-antigen complexes by information-driven docking. Structure. (2020) 28:119–295.e2. doi: 10.1016/j.str.2019.10.011
113. Akbar R, Robert PA, Pavlović M, Jeliazko R, Snapkov JI, Slabodkin A, et al. A compact vocabulary of paratope-epitope interactions enables predictability of antibody-antigen binding. Cell Rep. (2021) 34:108856. doi: 10.1016/j.celrep.2021.108856
114. Roney JP and Ovchinnikov S. State-of-the-art estimation of protein model accuracy using alphaFold. Phys Rev Lett. (2022) 129:2381015. doi: 10.1103/PhysRevLett.129.238101
115. Wong WK, Leem J, and Deane CM. Comparative analysis of the CDR loops of antigen receptors. Front Immunol. (2019) 10:2454. doi: 10.3389/fimmu.2019.02454
116. Erlach L, Friedensohn S, Neumeier D, Mason DM, and Reddy ST. Antibody affinity engineering using antibody repertoire data and machine learning. Bioengineering. bioRxiv. (2025). doi: 10.1101/2025.01.10.632313v1.abstract
117. Lin JY-Y, Hofmann JL, Leaver-Fay A, Liang W-C, Vasilaki S, Lee E, et al. DyAb: sequence-based antibody design and property prediction in a low-data regime. Bioengineering. bioRxiv. (2025). doi: 10.1101/2025.01.28.635353v1.full.pdf
118. Hu WP, Godzik A, and Skolnick J. Sequence-structure specificity–how does an inverse folding approach work? Protein Eng. (1997) 10:317–31. doi: 10.1093/protein/10.4.317
119. Shanehsazzadeh A, Alverio J, Kasun G, Levine S, Khan JA, Chung C, et al. In vitro validated antibody design against multiple therapeutic antigens using generative inverse folding. bioRxiv. (2023). doi: 10.1101/2023.12.08.570889
120. Svilenov HL, Arosio P, Menzen T, Tessier P, and Sormanni. P. Approaches to expand the conventional toolbox for discovery and selection of antibodies with drug-like physicochemical properties. mAbs. (2023) 15. doi: 10.1080/19420862.2022.2164459
121. Jain T, Sun T, Durand S, Hall A, Houston NR, Nett JH, et al. Biophysical properties of the clinical-stage antibody landscape. Proc Natl Acad Sci United States America. (2017) 114:944–49. doi: 10.1073/pnas.1616408114
122. Raybould MIJ, Marks C, Krawczyk K, Taddese B, Nowak J, Lewis AP, et al. Five computational developability guidelines for therapeutic antibody profiling. Proc Natl Acad Sci United States America. (2019) 116:4025–305. doi: 10.1073/pnas.1810576116
123. Sormanni P, Aprile FA, and Vendruscolo M. The camSol method of rational design of protein mutants with enhanced solubility. J Mol Biol. (2015) 427:478–905. doi: 10.1016/j.jmb.2014.09.026
124. Sormanni P, Aprile FA, and Vendruscolo M. Third generation antibody discovery methods: in silico rational design. Chem Soc Rev. (2018) 47:9137–57. doi: 10.1039/C8CS00523K
125. UniProt Consortium. UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res. (2021) 49:D480–89. doi: 10.1093/nar/gkaa1100
126. Dudzic P, Chomicz D, Kończak J, Satława T, Janusz B, Wrobel S, et al. Large-scale data mining of four billion human antibody variable regions reveals convergence between therapeutic and natural antibodies that constrains search space for biologics drug discovery. mAbs. (2024) 16:2361928. doi: 10.1080/19420862.2024.2361928
127. Kovaltsuk A, Leem J, Kelm S, Snowden J, Deane CM, and Krawczyk K. Observed antibody space: A resource for data mining next-generation sequencing of antibody repertoires. J Immunol. (2018) 201:2502–95. doi: 10.4049/jimmunol.1800708
128. Olsen TH, Moal IH, and Deane CM. AbLang: an antibody language model for completing antibody sequences. Bioinf Adv. (2022) 2:vbac046. doi: 10.1093/bioadv/vbac046
129. Ferdous S and Martin ACR. AbDb: antibody structure database-a database of PDB-derived antibody structures. atabase: J Biol Databases Curation. (2018) 2018. doi: 10.1093/database/bay040
130. Jankauskaite J, Jiménez-García B, Dapkunas J, Fernández-Recio J, and Moal IH. SKEMPI 2.0: an updated benchmark of changes in protein-protein binding energy, kinetics and thermodynamics upon mutation. Bioinformatics. (2019) 35:462–695. doi: 10.1093/bioinformatics/bty635
131. Sirin S, Apgar JR, Bennett EM, and Keating AE. AB-bind: antibody binding mutational database for computational affinity predictions. Protein Science: A Publ Protein Soc. (2016) 25:393–4095. doi: 10.1002/pro.v25.2
132. Robert PA, Akbar R, Frank R, Pavlović M, Widrich M, Snapkov I, et al. Unconstrained generation of synthetic antibody-antigen structures to guide machine learning methodology for antibody specificity prediction. Nat Comput Sci. (2022) 2:845–65. doi: 10.1038/s43588-022-00372-4
133. Shumailov I, Shumaylov Z, Zhao Y, Papernot N, Anderson R, and Gal Y. AI models collapse when trained on recursively generated data. Nature. (2024) 631:755–59. doi: 10.1038/s41586-024-07566-y
134. FLAb: benchmarking deep learning methods for antibody fitness prediction (2023). Available online at: https://neurips.cc/virtual/2023/77449 (Accessed May 4, 2025).
135. Uçar T, Malherbe C, and Gonzalez F. Exploring log-likelihood scores for ranking antibody sequence designs. Bioinf bioRxiv. (2024). doi: 10.1101/2024.10.07.617023v1
136. Armer C, Kane H, Cortade DL, Redestig H, Estell DA, Yusuf A, et al. Results of the protein engineering tournament: an open science benchmark for protein modeling and design. Bioengineering. bioRxiv. (2024). doi: 10.1101/2024.08.12.606135v1
137. Notin P, Kollasch AW, Ritter D, van Niekerk L, Paul S, Spinner H, et al. ProteinGym: large-scale benchmarks for protein design and fitness prediction. bioRxiv.org: Preprint Server Biol. (2023). doi: 10.1101/2023.12.07.570727
138. Ackloo S, Al-Awar R, Amaro RE, Arrowsmith CH, Azevedo H, Batey RA, et al. CACHE (Critical assessment of computational hit-finding experiments): A public-private partnership benchmarking initiative to enable the development of computational methods for hit-finding. Nat Rev Chem. (2022) 6:287–95. doi: 10.1038/s41570-022-00363-z
139. Erasmus MF, Spector L, Ferrara F, DiNiro R, Pohl TJ, Perea-Schmittle K, et al. AIntibody: an experimentally validated in silico antibody discovery design challenge. Nat Biotechnology. (2024) 42(11):1637–42. doi: 10.1038/s41587-024-02469-9
140. Wossnig L, Furtmann N, Buchanan A, kumar S, and greiff v. Best practices for machine learning in antibody discovery and development. Drug Discov Today. (2024) 29:1040255. doi: 10.1016/j.drudis.2024.104025
141. Spreter Von Kreudenstein T, Lario PI, and Dixit SB. Protein engineering and the use of molecular modeling and simulation: the case of heterodimeric fc engineering. Methods (San Diego Calif.). (2014) 65:77–945. doi: 10.1016/j.ymeth.2013.10.016
Keywords: antibody discovery, AlphaFold 2, machine learning, drug discovery, therapeutic antibodies, protein design
Citation: Bielska W, Jaszczyszyn I, Dudzic P, Janusz B, Chomicz D, Wrobel S, Greiff V, Feehan R, Adolf-Bryfogle J and Krawczyk K (2025) Applying computational protein design to therapeutic antibody discovery - current state and perspectives. Front. Immunol. 16:1571371. doi: 10.3389/fimmu.2025.1571371
Received: 05 February 2025; Accepted: 24 April 2025;
Published: 22 May 2025.
Edited by:
Traian Sulea, National Research Council Canada (NRC), CanadaReviewed by:
Frederic Cadet, INSERM Dynamique des Structures et Interactions des Macromolécules Biologiques (DSIMB), FranceChristopher Corbeil, National Research Council Canada (NRC), Canada
Copyright © 2025 Bielska, Jaszczyszyn, Dudzic, Janusz, Chomicz, Wrobel, Greiff, Feehan, Adolf-Bryfogle and Krawczyk. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Konrad Krawczyk, a29ucmFkQG5hdHVyYWxhbnRpYm9keS5jb20=
†These authors have contributed equally to this work and share first authorship