 Wei Chen1
Wei Chen1 Afang Zhou
Afang Zhou Yunfeng Zhou
Yunfeng Zhou- 1Department of Thoracic Surgery, West China School of Public Health and West China Fourth Hospital, Sichuan University, Chengdu, Sichuan, China
- 2State Key Laboratory of Biotherapy, Sichuan University, Chengdu, Sichuan, China
Lung cancer is a refractory malignancy. Although various therapeutic options, including targeted therapies, immune checkpoint inhibitors, and systemic chemotherapy, have significantly improved the prognosis of lung cancer patients, five-year survival rates are still low. Bispecific antibodies have attracted much attention because of their ability to bind different antigens or epitopes on the same antigen at once and because of their multiple novel functional mechanisms. Recently, three bispecific antibodies have been successively approved for lung cancer treatment, demonstrating the potential of bispecific drugs in lung cancer therapy. Various bispecific antibodies are currently under clinical trials to evaluate their safety and efficacy in lung cancer. In this review, we provide an overview of these antibodies’ structure and mechanism of action, summarize their clinical progress in lung cancer treatment, and discuss and analyze the challenges and future directions of bsAbs application in lung cancer.
1 Introduction
Lung cancer is one of the world’s most common cancers and the leading cause of cancer-related deaths, with an estimated 2.2 million new cases and 1.79 million deaths annually (1). In most parts of the world, the 5-year survival rate for lung cancer patients is only 10-20% (2). Non-small cell lung cancer (NSCLC) is one of the most common types of lung cancer, accounting for about 85% of lung cancers (1). The treatment landscape for NSCLC has changed dramatically over the past decade by introducing several new targeted and immunotherapeutic agents. Patients treated with protein kinase inhibitors [especially tyrosine kinase inhibitors (TKIs)] and monoclonal antibodies [e.g., immune checkpoint inhibitors (ICIs)] may have a relatively good prognosis. However, although the above treatment strategies significantly prolong the overall survival of patients, a common problem is drug resistance (3, 4). Small cell lung cancer (SCLC) is another type of lung cancer that accounts for 10-15% of all lung cancers (5–7). It is even a tumor type with an inferior prognosis and limited therapeutic options, with a median survival of 2 years for most patients with early-stage disease and 1 year for patients with metastatic disease (8). Therefore, there is still a significant unmet medical need in the field of lung cancer.
Bispecific antibodies (bsAbs) have been described as “next-generation antibodies” that overcome the limitation of natural monoclonal antibodies to bind only a single epitope (9). Amivantamab is the first bispecific antibody approved for the treatment of lung cancer. The drug was initially approved for treating adult patients with locally advanced or metastatic NSCLC harboring epidermal growth factor receptor (EGFR) Exon 20 insertion mutations whose disease has progressed on or after platinum-based chemotherapy (10). Several bsAbs with potential for lung cancer therapy are currently undergoing clinical trials, and many have produced exciting results. This review provides an overview of the bsAbs that have shown promise in treating lung cancer.
2 Bispecific antibody formats and mechanisms
One of the significant challenges of dual antibodies, which took about half a century to move from concept to the clinic, is that only 12.5% of the target molecules can be obtained by conventional production means, with the rest being mostly nonfunctional or monospecific molecules (11, 12). To address this challenge, researchers have developed various strategies based on natural antibodies such as IgG and heavy-chain antibodies (Figure 1A). These strategies aim to increase the proportion of the target molecule and facilitate its separation and purification, while enabling the modular combination of distinct antibody functional domains as required. Today, more than 100 types of bsAbs are known (9) and can be briefly classified into three categories: no IgG-like bsAbs (Figure 1B), asymmetric IgG-based bsAbs (Figure 1C), and symmetric IgG-based bsAbs (Figure 1D). No IgG-like bsAbs consist of combinations of partial structures of antibodies. Among them, bsAbs designed based on single-chain variable fragment (scFv) (e.g., BiTE, DART, etc.) are the simplest and the least difficult to generate. Moreover, due to their low molecular weight, they have better tissue permeability. However, the absence of fragment crystallizable (Fc) structure results in a short plasma half-life of these molecules and a lack of Fc-mediated effector function [e.g., antibody-dependent cell cytotoxicity (ADCC) or antibody-dependent cell phagocytosis (ADCP)]. To extend the half-life of such bsAbs, a common strategy is to fuse Fc fragments (including HEL-BiTE, Tetravalent DART Fc, VHH-Fc, etc.) or conjugate fragments of anti-human serum albumin (HSA) antibodies (e.g., TRACTr, TriTAC) with the bsAbs protein. IgG-based bsAbs retain Fc and have a longer half-life, and Fc function can be adjusted to enhance the therapeutic effect of the molecule according to specific needs. However, there will be more factors to consider in the molecular design of the protein. For example, asymmetric IgG-based antibodies are closer in form to natural IgG. However, as mentioned above, the target molecule content is only 12.5% when produced by conventional means, and it is not easy to purify the target molecule from the system to obtain a high-purity target molecule. Therefore, a series of molecular modifications are needed to promote the correct pairing of light and heavy chains to increase the target molecule content, and these strategies include knob-into-hole, DEKK mutation, common light chain, and the addition of alternative interchain disulfide. Symmetric IgG-based bsAbs are made by fusing another antigen-binding fragment to the conventional antibody (e.g., Tetrabody, IgG-VHH, FIT-Ig, etc.) or by mutating Fc to form a new antigen-binding site (e.g., mAb2) and thus do not need to consider the correct pairing of light and heavy chains. However, such modifications can change physicochemical properties such as antibody stability and solubility (13, 14). In addition, antigen-antibody binding is affected by the position of the fused fragment (15).
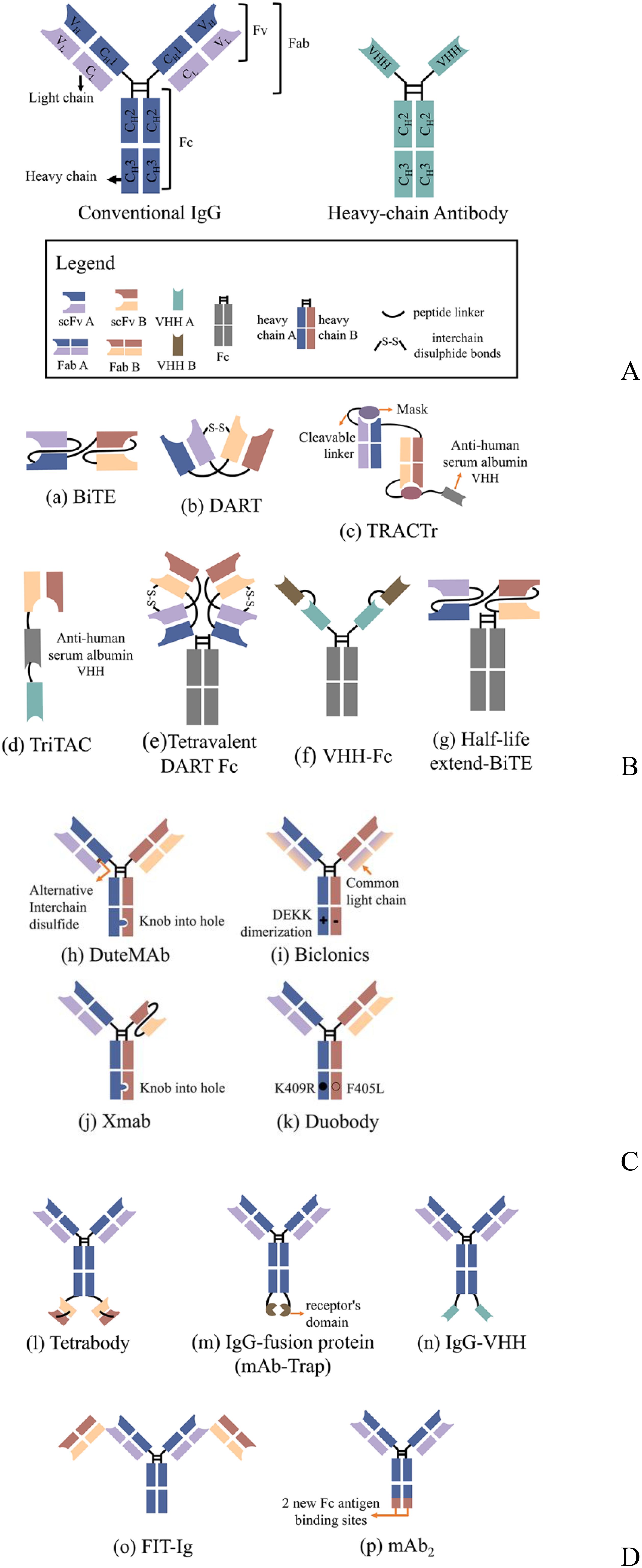
Figure 1. Schematic overview of the antibody structure and representations of several classical bsAbs formats; (A) Conventional IgG consists of 2 heavy chains and two light chains, while heavy-chain antibodies are only comprised of heavy chains; (B) No IgG-like bsAbs; those bsAb consist of antibody-based fragments, such as scFv, VHH, Fab, Fc; (C) Asymmetric IgG-based bsAbs; the molecules may contain mutations, knob-into-hole or DEKK for example, that affect chain pairing and other manufacturability parameters. (D) Symmetric IgG-based bsAbs; Symmetric bispecific antibodies are generated by a fusion of an additional binding site to the heavy/light chains or by making differential but overlapping use of the light and heavy complementarity determining regions as primary contacts for each antigen. scFv, single-chain variable fragment; Fab, antigen-binding fragments; VHH, variable heavy domain of heavy chain; Fc, fragment crystallizable.
3 The mechanisms of action of bispecific antibodies in lung cancer
Unlike the simple mixing of antibodies, bsAbs have become a primary focus of drug developers because they have new mechanisms of action (MOA) different from those of the parent antibody combination. Currently, bsAbs used in lung cancer therapy include three main mechanisms: dual inhibition (Figure 2A), engaging immune cells and tumor cells (Figure 2B), and immune cytokines (Figure 2C).
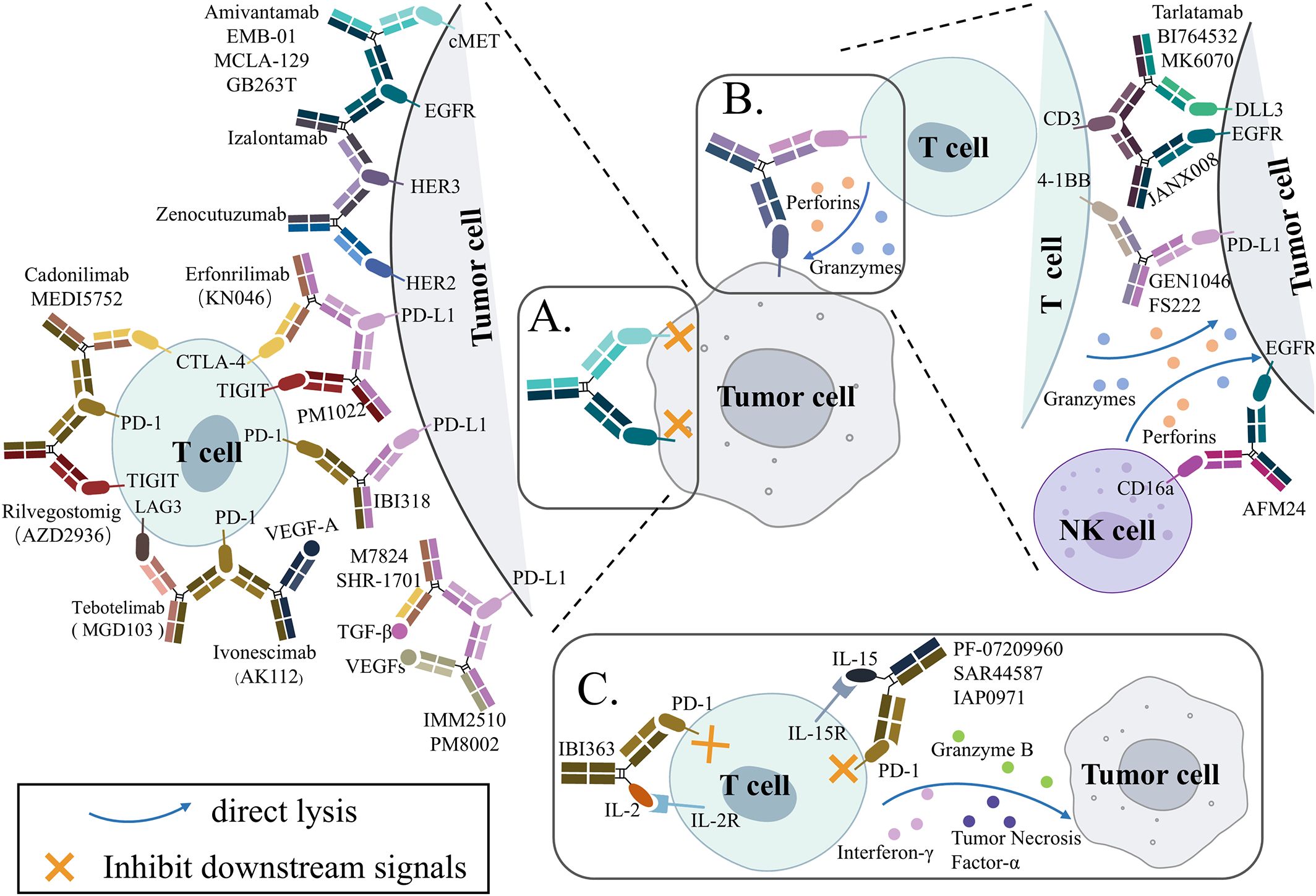
Figure 2. Simplified schematic overview of the proposed mechanisms of action for bispecific antibodies (bsAbs) in clinicals for lung cancer treatment. (A) Blocking signaling. Two targets are being disrupted by the bsAb. (B) Engagement of immune cells to the tumor cell. Immune cells can be engaged to tumor cells by bsAbs. (C) Immunocytokine. Increase cytokine accumulation within the tumor and block immune checkpoint. cMET, mesenchymal-epithelial transition; EGFR, epidermal growth factor receptor; HER2, human-epidermal growth factor receptor 2; HER3, human-epidermal growth factor receptor 3; PD-1, programmed cell death protein 1; PD-L1, programmed death-ligand 1; CTLA-4, cytotoxic T-lymphocyte-associated protein 4; TIGIT, T cell immunoglobulin and ITIM domain; LAG-3, lymphocyte activation gene 3; VEGF, vascular endothelial growth factor; TGF-β, transforming growth factor β; DLL3, Delta-like ligand 3; IL-2, interleukin-2.
By targeting two antigens at the same time, dual-inhibition bsAbs inhibited the pathways of two signals that are related to each other, exerting the effect of 1 + 1>2. More than half of the bsAbs currently applied in lung cancer treatment mainly exert anti-tumor effects by this mechanism of action. They can be further categorized into three types according to the difference in the signals they block: (i) simultaneous targeting of two surface receptors with specific signaling and functional overlap associated with tumorigenesis and progression, mainly ErbB family proteins; (ii) dual immune checkpoint molecule blockade; (iii) simultaneous inhibition of immune checkpoints and tumor microenvironmental pro-tumor growth factors.
Immune cell engagers (ICEs) redirect cytotoxic immune cells to disease-associated target cells that play a key role in the disease process to achieve direct killing of these cells by immune cells. Among them, T cell engagers (TCEs) are the typical application of bsAb, and about half of the bsAbs currently evaluated in clinical trials are TCEs (16). Recently, researchers have also been experimenting with Natural Killer cell engagers (NKCEs) based on the recruitment of cytotoxic NK cells (17, 18).
Immunocytokines are a type of antibody-cytokine fusion protein. Mechanistically, immunocytokines fuse a therapeutic cytokine to one end of an antibody. Through the specific targeting function of the antibody, the cytokine is specifically delivered near tumor cells, allowing it to bind to specific cytokine receptors on the surface of surrounding immune cells, thus significantly reducing non-specific toxicity (19, 20). Structurally, immunocytokines have a symmetric or asymmetric structure based on IgG (21). Therefore, in some literature and this article, immunocytokines are classified as a type of bispecific antibody with a special mechanism (22, 23).
The following is a further discussion and analysis of bispecific antibodies in lung cancer according to the different mechanisms of action described previously.
4 Dual inhibition bispecific antibodies
4.1 Dual receptors inhibition bispecific antibodies
The ErbB family of transmembrane receptor tyrosine kinases consist of four members: EGFR/ErbB1/HER1, ErbB2/Neu/HER2, ErbB3/HER3, ErbB4/HER4. ErbB receptors are activated upon homo-or heterodimerization, which activates many downstream signaling pathways, primarily the mitogen-activated protein kinase (MAPK), phosphatidylinositol 3-kinase (PI3K)/protein kinase B (PKB/AKT), and Janus kinase (JAK) and signal transducer/activator of transcription (STAT) signaling pathways (24). Moreover, all these pathways regulate cell metabolism, growth, and survival. Overexpression and overactivation of ErbB receptors are associated with poor prognosis, drug resistance, tumor metastasis, and lower survival in a variety of cancers, including lung cancer. There are two clinically important ErbB inhibitors: humanized antibodies targeting the extracellular structural domains of EGFR or HER2 and small-molecule TKIs that compete with adenosine triphosphate in the structural domain of the receptor tyrosine kinase. Eventually, however, a significant proportion of tumor cells develop resistance through the activation of another ErbB receptor signal or the activation of bypass pathways (25, 26). BsAbs inhibit tumor growth more effectively by simultaneously targeting two ErbB members or related bypass pathways, thereby blocking overlapping downstream signals.
4.1.1 EGFR × cMET
EGFR mutations exist in approximately 50% of Asian NSCLC patients and 11-16% of European NSCLC patients (27–29). Currently, the US Food and Drug Administration (FDA) has approved six EGFR TKIs, as well as a fully humanized monoclonal antibody targeting EGFR, as the standard of care for the first-line treatment of NSCLC patients with EGFR mutations (30). However, the selection pressure exerted by the above drugs inevitably leads to treatment resistance. Among cases of acquired resistance to EGFR TKIs, 5-10% are mesenchymal-epithelial transition factor (MET) amplified, which activates the EGFR-independent PI3K-AKT signaling pathway by driving ErbB3 dimerization and signal transduction (31, 32). Drug-resistant tumors are also able to activate the cMET pathway through increased cMET expression and/or increased cMET ligand expression, which provides an alternative mechanism for tumor cells to bypass the TKI blockade of EGFR and promote cancer cell survival (33–36). Due to the signaling crossover between EGFR and cMET, combined inhibition of both receptors may limit the activation of the compensatory pathway and improve overall efficacy.
Amivantamab (JNJ-61186372; Rybrevant™) is a fully humanized bsAb targeting EGFR and cMET and is the first approved therapeutic agent for NSCLC patients with EGFR exon 20 insertion (EGFR ex20ins) after failure of platinum-containing chemotherapy. In addition to its ability to block both EGFR- and cMET-mediated downstream signaling, the antibody exerts its anti-tumor effects through various Fc-mediated mechanisms, such as ADCC and ACDP (37, 38). The CHRYSALIS (NCT02609776) analyses the efficacy and safety of amivantamab in post-platinum NSCLC patients with EGFR Exon20ins. In the efficacy population, the reported overall response rate (ORR) was 40%, the median duration of response (mDOR) was 11.1 months, and the median overall survival (mOS) was 22.8 months, respectively (10, 39, 40). Besides, the drug has a favorable safety profile, with the most common side effects including rash (89%) and infusion-related events (67%). Based on the above data, the FDA approved the new drug application of amivantamab in 2021 (41). In a subsequent clinical trial called PAPILLON (NCT04538664), researchers analyzed the anti-tumor activity of amivantamab in combination with carboplatin-pemetrexed. Among treatment-naïve NSCLC patients with EGFR ex20ins, the progression-free survival (PFS) in the amivantamab plus chemotherapy group was significantly longer than that of the chemotherapy group (median, 11.4 months and 6.7 months, respectively) (42). Based on this clinical result, the FDA approved amivantamab plus chemotherapy as a first-line therapy for advanced NSCLC with EGFR ex20ins mutation (43).
The efficacy of amivantamab is not limited to EGFR ex20ins mutation. However, it has shown positive clinical benefits for the larger population of patients with other EGFR and MET mutations. For patients with locally advanced or metastatic NSCLC with EGFR mutations (Ex19del or L858R) (NCT06120140), compared to those receiving osimertinib, patients treated with amivantamab plus lazertinib had longer mPFS (23.7 months vs. 16.6 months, HR=0.7) and longer mDoR (25.8 months vs. 16.8 months) (44, 45). For primary METex14 patients with advanced NSCLC treated with amivantamab (NCT02609776), an ORR of 33% (56% in the treatment-naïve population) with a mDOR of 11.2 months was observed (46). In addition, a subcutaneous formulation of amivantamab, based on human hyaluronidase, was developed to improve patient tolerability and reduce administration time (47). It was shown (NCT05388669) that when administered subcutaneously, the drug was not only pharmacokinetically non-inferior to intravenous administration, but fewer patients in the subcutaneous group experienced infusion-related reactions (13% vs. 66%) and venous thromboembolism (9% vs. 14%). Concurrently subcutaneously administered patients had prolonged mPFS (6.1 months vs. 4.3 months, HR=0.84), prolonged mDOR (11.2 months vs. 8.3 months), and significantly prolonged OS (HR=0.62).
EMB-01 is an EGFR- and cMET-targeted bispecific antibody based on the FIT-Ig technology platform, which fuses the Fab of an anti-cMET antibody to the variable region of an anti-EGFR antibody to form a tetravalent bispecific antibody. EMB-01 induces endocytosis of EGFR and cMET receptors on the cell surface and their degradation. Preliminary clinical (NCT03797391) data suggest an ORR of 5.3% and a DCR of 42.1% in 38 evaluable patients with advanced NSCLC (48).
MCLA-129 is a 1 + 1 form of asymmetric Ig-G-like bispecific antibody designed based on the biclonics common light chain platform (49). It targets both EGFR and cMET and has a mechanism of action similar to amivantamab. Preliminary clinical data (NCT04930432) on fortnightly intravenous administration of 1,500 mg MCLA-129 in different NSCLC patients were recently published: for patients with METex14 mutation, the ORR was 43.5%, and the DCR was 95.7%; for EGFR20ins-mutated patients, the ORR was 28.6% and DCR of 84.1%; for patients with sensitized EGFR-mutated, ORR was 21.8% and DCR was 69.1% (50). In addition, this bispecific antibody in combination with osimertinib was observed in treatment-naïve patients with advanced EGFRmut NSCLC (NCT04868877) with an ORR of 75.0% and a DCR of 93.8%; in patients who progressed on osimertinib, the ORR was 35.3%, and the DCR was 73.5% (51).
4.1.2 HER2 × HER3
As a member of the ERBB receptor family, HER2 alterations are involved in the oncogenic process in a variety of solid tumors, mainly including HER2 mutation, HER2 amplification, and HER2 overexpression, with corresponding incidence rates of 1%-6.7%, 2%-22%, and 7.7%-23%, respectively, in NSCLC and all of them are associated with poor prognosis (52–55). HER3 is overexpressed in 83% of primary NSCLC tumors and is associated with advanced disease, shorter time to metastasis, and lower survival (56). HER2 is activated by dimerization or with other ERBB family members and further activates downstream signaling pathways. Of all possible EGFR family dimers, the HER2:HER3 heterodimer has the highest translational capacity (57–59). When HER3 binds to its ligands (neuregulin1-4, NRG1-4), its conformation is altered, exposing its dimerization sites with other proteins of the EGFR family, predominantly EGFR and HER2, which induces phosphorylation events downstream of the protein (60). Notably, NRG1 can form fusion proteins with various membrane proteins, which provide a transmembrane structural domain to anchor NRG1 to the membrane, thus enabling NRG1 to bind to HER3 in its own or neighboring cells (61). NRG1 fusions have been detected in a wide range of tumors, with the highest number of cases reported in NSCLC (62). Early reports observed relatively poor NRG1 fusion-positive (NRG1+) lung therapy outcomes. The ORR of those patients treated with platinum-doublet and taxane-based (post-platinum-doublet) chemotherapy was only 13% and 14%, with mPFS of 5.8 and 4.0 months, respectively (63).
Zenocutuzumab (MCLA-128) is an IgG-like asymmetric bsAb targeting HRE2 and HER3. The antibody preferentially binds to more abundant HER2 proteins on the cell surface via the higher affinity HER2-targeting arm, providing a high concentration of local antibody while at the same time positioning the HER3-targeting arm to block NRG1 or NRG1 fusion proteins binding to HER3 (64). Thereby, the formation of HER2:HER3 heterodimers is potently inhibited, preventing subsequent phosphorylation of the HER3 cytoplasmic structural domain and downstream oncogenic signaling. In addition, glycoengineering modifications enhanced the antibody’s ADCC activity (64). The FDA recently granted accelerated marketing approval for zenocutuzumab based on clinical results from a study called eNRGy (NCT02912949) (65). In 64 evaluable patients with NRG1+ advanced NSCLC, the confirmed ORR was 34% (22/64; [95% CI] 23-47), and the mDOR was 12.9 months, with responses ongoing in 11/22 (50%) patients (66). The drug’s safety profile was favorable, with <4% of patients experiencing grade ≥3 adverse events (66).
4.1.3 EGFR × HER3
In addition to HER3 forming dimers with HER2 to deliver proliferation and survival signals to the cells, another important dimerization partner is EGFR. It has been found that regardless of the type of EGFR-TKIs resistance mechanisms, such as EGFR T790M mutation, MET amplification, and HER2 amplification, HER3 amplification is observed in EGFR-mutated NSCLC tumors that progress after EGFR-TKI treatment (67). HER3 can also be activated independently of ligand binding through dysregulation of other tyrosine kinase receptors. For example, in NSCLC carrying activating EGFR mutations, EGFR can transactivate HER3 via heterodimers (68). In the presence of EGFR-TKIs, MET amplification activates HER3, thereby initiating a downstream PI3K/AKT survival mechanism (31).
Izalontamab (SI-B001) is a tetravalent symmetric bsAb targeting EGFR and HER3. The antibody is cetuximab-based and contains an anti-HER3 scFv at the end of the constant region of cetuximab (69). Since the antibody has a significantly lower affinity for the HER3 arm than the EGFR arm, the antibody can only bind to HER3 after definitive binding to the EGFR (69). As a result, it effectively inhibits tumor cells that express both EGFR and HER3, minimizing the effect on functioning HER3 in normal tissues. In a phase II clinical trial (NCT04603287), researchers enrolled 55 patients with locally advanced or metastatic EGFR/anaplastic lymphoma kinase (ALK) wild-type NSCLC who had failed first-line anti-PD-1/L1 therapy with or without platinum-based chemotherapy (PBC) to receive SI-B001 combined with docetaxel (70). Of the 48 evaluable patients, the ORR and the DCR were 31.3% and 77.1%, respectively (63). Among the patients in Cohort B who failed first-line anti-PD-1/L1 combined with PBC, 22 of the evaluable patients in this cohort were on a regimen of 16 + 9 mg/kg/week. These patients’ reported ORR and DCR were 45.5% and 68.2%. Among all, the most common≥grade 3 treatment-related adverse events (TRAEs) were myelosuppression (17%), decreased neutrophil count (15%), and decreased white blood cell count (12%).
4.2 Dual immune checkpoints inhibition bispecific antibodies
The immune system is an elaborate and complex network of multiple signals that activate immune cells to accurately recognize and eliminate pathogenic microorganisms and mutated cells in the body. At the same time, to avoid damage to normal tissues and organs by excessive immune response, the immune system has evolved a series of checkpoints to modulate the duration and amplitude of the immune response. However, tumor cells will hold these checkpoints hostage to escape immune surveillance (71). Reactivating or enhancing the immune system’s innate or adaptive immune response to strengthen the attack on cancer cells is an important strategy for cancer treatment.
4.2.1 PD-1/L1 × CTLA-4
Programmed cell death protein 1 (PD-1), programmed death-ligand 1(PD-L1), and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) are the relatively well-studied and most maturely applied immune checkpoints. Currently, immune checkpoint inhibitors are approved for treating various solid and hematological malignancies (72). Despite the success of anti-CTLA-4 and anti-PD-1/L1 therapies, only 10-25% of patients benefit from such treatments (73). Because of the similarities and differences between the CTLA-4 and PD-1 pathways and some complementarity between them (74), combination therapy with anti-PD-1/L1 and anti-CTLA-4 clinically improves overall survival. The combination of these two classes of antibodies has been approved for the treatment of various tumors, including non-small cell lung cancer. However, at the same time, the toxicities of the combination are more intense than those of the single agent (75–79). On the other hand, PD-1 and CTLA-4 are co-expressed in a high percentage of tumor-infiltrating lymphocytes (80). Therefore, it is possible to maximize clinical benefit and minimize additional toxicity by designing bispecific antibodies that can specifically target such tumor-infiltrating lymphocytes and avoid activation of immune cells in other normal tissues by the bsAbs.
Cadonilimab (AK104; 开坦尼®) is a humanized tetravalent symmetric bispecific antibody incorporating an anti-CTLA-4 scFv fragment at the C-terminus of each of the two heavy chains of the anti-PD-1 antibody (81). Also, to avoid T-cell depletion and adverse immune responses caused by Fc-mediated immune cell activation, the Fc segment was mutated to completely remove ADCC, ADCP, and CDC effects (81). Cadonilimab has a higher affinity at high PD-1 concentrations and a relatively low binding capacity at lower PD-1 concentrations (82). As a result, the antibody has higher activity in high PD-1-expressing tumor microenvironments and weaker activity in normal tissues. It received conditional approval for marketing in China in June 2022 for the treatment of patients with recurrent or metastatic cervical cancer who have failed prior treatment with platinum-containing chemotherapy (83). Several clinical trials of cadonilimab alone or combined with other drugs for treating NSCLC and SCLC are underway (83). A clinical trial called AK104-208 (NCT04646330) evaluated the efficacy of cadonilimab in combination with anilotinib, an anti-angiogenic drug, in treatment-naïve patients with NSCLC. In 69 evaluable patients, the overall ORR was 53.6% (95% CI, 41.2-65.7), the DCR was 92.8% (95% CI, 83.9-97.6), and grade 3+ TRAEs occurred in 49.3% of patients (84). Another study called AK104-IIT-018 (NCT05816499) included patients with histologically or cytologically confirmed stage IIIB/IIIC or IV NSCLC without sensitizing EGFR/ALK/ROS1 mutations, who must have progressed during or after a PD-1/L1 inhibitor and platinum-based chemotherapy and were treated with three-drug combination therapy (cadonilimab plus anilotinib plus docetaxel) (85). Among 33 evaluable patients, the overall ORR was 30.3% (95% CI:15.6-48.7%), the DCR was up to 94.0% (95% CI:79.8-99.3%), the mPFS was 6.5 months, and the proportion of ≥ grade 3 TRAEs was only 17.4% (85). Patients with advanced driver-negative NSCLC have limited therapeutic options after progression on first-line immune-combination chemotherapy (86). The standard of care recommended by the NCCN guidelines is chemotherapy monotherapy, such as docetaxel, gemcitabine, and albumin-conjugated paclitaxel. However, the efficacy was minimal, with ORR of 14%-17% and mPFS of 4.0-5.4 months (87–89). Therefore, combining cadonilimab with anlotinib and docetaxel offers a potentially attractive treatment option for this group of patients. The LungCadX study (NCT06424821)evaluated the efficacy and safety of cadonilimab in combination with chemotherapy as first-line treatment for patients with driver-negative, PD-L1-negative advanced NSCLC (90). Among 30 evaluable patients, the overall ORR was 66.7%, with an ORR of 93.3% in squamous cancer, 40% in non-squamous cancer, and a DCR of 100% (90). The drug safety was good, with an overall TRAE incidence of 61.4%, including 34.1% of ≥ grade 3 TRAEs (90). The above results indicate that the first-line treatment of PD-L1-negative advanced NSCLC with cadonilimab in combination with chemotherapy shows an auspicious therapeutic effect, especially in patients with squamous carcinoma.
MEDI5752 is a 1 + 1 form of an asymmetric bispecific antibody that preferentially binds CTLA-4 from PD-1+ T cells by reducing the affinity to the CTLA-4 (80). In a head-to-head comparative trial with Keytruda, patients treated with carboplatin plus pemetrexed plus MEDI5752 had better DOR, PFS, and OS than those treated with carboplatin plus pemetrexed plus Keytruda (mDOR: 20.5% vs. 9.9%, mPFS: 15.1 vs. 8.9 months, mDOR: NR vs. 16.5 months) (91). In addition, bsAbs based on PD-1 and CTLA-4 targeting include XmAb20717, SI-B003, and MGD019, some of which have published preliminary clinical data (Table 1). These bispecific antibodies, although not aligned with AK104 as well as MEDI5752 in terms of the specific protein sequences and format, are all designed to target both PD-1 and CTLA-4 and to bind PD-1/CTLA-4 double-positive cells to reduce CTLA-4 toxicity preferentially (124–126).
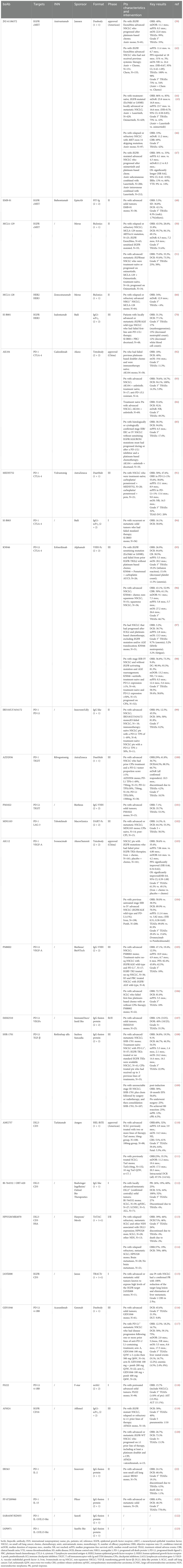
Table 1. Clinical results of bsAbs in lung cancer.
Erfonrilimab (KN046) is a tetravalent symmetric bsAb targeting PD-L1 and CTLA-4, consisting of two identical chains, each consisting of a PD-L1 single-domain antibody, a CTLA-4 single-domain antibody, and a Fc domain (87). By design, the antibody has a higher affinity for PD-L1 and, therefore, can preferentially target the tumor microenvironment with high PD-L1 expression to reduce toxicities (96). At the same time, the antibody retains the function of Fc to remove CTLA-4-expressing Treg in the tumor microenvironment (127). In the study named KN046-201 (NCT03838848), a total of 26 advanced NSCLC patients with EGFR sensitivity mutation who had failed EGFR-TKI(s) and without platinum-based chemotherapy were enrolled and were given KN046 in combination with pemetrexed and carboplatin as a second-line treatment (95). The reported ORR was 26.9%, mPFS was 5.5 months, and mOS was 20.2 months (95). And 57.7% of the patients experienced grade 3 or higher TRAEs (95). In another phase II study evaluating KN046 in combination with chemotherapy for the first-line treatment of patients with metastatic NSCLC (NCT04054531), the ORR was 46.0%, with a mPFS of 5.8 months and a mOS of 26.6 months (96). In addition, for patients with metastatic NSCLC who had failed previous immunotherapy and platinum-based chemotherapy, the mOS of patients given KN046 was up to 13.3 months (97). In a study evaluating KN046 in combination with axitinib in advanced NSCLC (NCT05420220), for previously untreated patients with PD-L1 TPS ≥1% and patients treated with CPIs, the ORR after receiving the combination therapy was 56.8% and 9.4% respectively, and the DCR was 90.9% and 81.3%, showing promising efficacy (98).
4.2.2 PD-1 × PD-L1
IBI318/LY3434172 is an IgG-like dual antibody targeting PD-L1 and PD-1, and preclinical data suggest that it has significant tumor-suppressive effects and is superior to equivalent doses of monoclonal antibodies, as well as the combination of PD-1 and PD-L1 monoclonal antibodies (128). Preliminary phase Ib clinical (NCT03875157) data suggests that this drug has significant efficacy in immunotherapy-naïve NSCLC patients who had failed or were intolerant to first-line chemotherapy. Its ORR in patients with PD-L1 scores of 1-49% and ≥50% was 12.5% (1/8) and 45.5% (5/11), respectively, and its DCR was 50% (4/8) and 81.8% (9/11), respectively (99).
4.2.3 PD-1/(L)1 × TIGIT
T cell immunoglobulin and ITIM domain (TIGIT) is an immune checkpoint that has recently attracted attention. TIGIT interacts with CD155 (poliovirus receptor, PVR, or NECL-5) on the surface of antigen-presenting cells or tumor cells and inhibits the anti-tumor response of T cells and NK cells (129). TIGIT is expressed in various lung cancers, including NSCLC, and overexpression of TIGIT/CD155 is an unfavorable prognostic factor in lung adenocarcinoma (130). TIGIT is generally co-expressed with immunosuppressive molecules, such as PD-1, on various T cells, and both inhibit CD8+ T cell activity through different mechanisms (131, 132). TIGIT blockade is a promising immunotherapy in terms of molecular mechanisms. However, vibostolimab and tiragolumab, two monoclonal antibody drugs targeting TITGI, are ineffective as monotherapy (133, 134). However, tiragolumab in combination with atezolizumab had an overall ORR of 37% and an ORR of 66% in the PD-L1 TPS>50% subgroup, which exceeded atezolizumab monotherapy (21% and 24%, respectively) (135). Therefore, developing bsAb targeting PD-1 and TIGIT is also interesting to researchers.
Rilvegostomig (AZD2936) is an asymmetric dual antibody targeting PD-1 and TIGIT. The initial efficacy of rilvegostomig in patients with advanced NSCLC treated with CPIs has been published (NCT04995523). Patients with PD-L1 TPS ≥50% treated with rilvegostomig 750mg had an ORR of 61.8% and a DCR of 88.3%, and the drug had a favorable safety profile, with a grade 3 or higher adverse events rate of 10.5% (100). The company developing the drug is currently conducting a head-to-head clinical trial with Keytruda, which shows the drug developer’s confidence in this drug.
PM1022 is a bispecific antibody targeting PD-L1 and TIGIT, with VHH targeting PD-L1 fused to the C-terminus of the anti-TIGIT antibody. The antibody is currently undergoing a dose extension clinical study (NCT05867771), and preliminary results show an ORR of 7.1%, a DCR of 35.7%, and an overall TRAE rate of 53.3% in treated patients (101). One of the NSCLC patients had received prior treatment, including chemotherapy and anti-PD-1 therapy, and had a 56.8% reduction in lesion size (101).
4.2.4 PD-1 × LAG-3
Lymphocyte activation gene 3 (LAG-3), the third-generation immune checkpoint receptor, is highly up-regulated on exhausted T cells in the tumor microenvironment (136). The binding of LAG-3 to its classical ligand, the major histocompatibility complex-II, leads to the down-regulation of T cell activation, proliferation, and cytokine production, ultimately causing T cell dysfunction (137). Immunohistochemistry analysis revealed that LAG-3 is expressed in various cancer tissues, with 90% of NSCLC and 52% of SCLC samples having detectable positive LAG-3 expression (102). In NCSLC, co-expression of PD-1 and LAG-3 was detectable in 59% of samples (102). Moreover, it was found that LAG-3 and PD-1 synergize on CD8+ T cells to drive T cell exhaustion (138). Currently, the FDA approved Opdualag (consisting of a fixed-dose combination of relatlimab and nivolumab) for treating unresectable or metastatic melanoma in 2022 (139). In addition, it was shown that compared to nivolumab combination chemotherapy, Opdualag combination chemotherapy improved ORR (53.2% vs. 40.8%), prolonged mPFS (9.8 vs. 6.1 months), and had an HR of 0.63 in this group of NSCLC patients with PDL1 ≥ 1%; and, for non-squamous NSCLC (non-sq-NSCLC) patients with PD-L1 TPS≥1%, the ORR improved to 58%, the mPFS increased to 11.6 months, and the HR was further reduced to 0.55 (140). The above study demonstrated that dual immune checkpoint therapy with LAG-3 and PD-1 has a better prospect in lung cancer treatment.
MGD103 (Tebotelimab) is a tetravalent DART-Fc (IgG4κ) fusion protein that blocks PD-1 and LAG-3. In vitro studies have shown that this bsAbs is significantly more potent than the combination of nivolumab and relatlimab in stimulating IFN-γ secretion (102). Tebotelimab had an ORR of 14.3% and a DCR of 64.3% in NSCLC patients who did not receive CPIs; however, no remission was observed in NSCLC patients who had previously received CPIs (NCT03219268) (102). Interestingly, there was a correlation between objective response to tebotelimab and LAG-3 expression (P < 0.05), but no statistical association between PD-1 and clinical response was observed (102).
In addition to the aforementioned immunomodulatory targets with more basic and clinical studies, some new immune checkpoint molecules (e.g., OX40, TIM3) have also attracted the attention of researchers in the past decade (141, 142). Moreover, there is much clinical or preclinical evidence that these new immune checkpoints synergize with anti-PD-1/L1 and/or anti-CTLA-4 monoclonal antibodies (143). Therefore, it is worth waiting for the final clinical outcome based on these newly discovered immune checkpoints combined with PD-1/L1 or anti-CTLA -4 to form a bsAbs.
4.3 Dual inhibition of tumor microenvironment and immune checkpoints
A tumor is a heterogeneous complex of tumor cells, various non-malignant cells (e.g., immune cells, stromal cells, endothelial cells, cancer-associated fibroblasts), and various non-cellular components (e.g., vascularised extracellular matrix, exosomes, cytokines) (144). The tumor microenvironment (TME) influences tumor growth, metastatic spread, and response to therapy. It has become common knowledge that TME-mediated immunosuppression impairs beneficial responses.
4.3.1 PD-1/L1 × VEGF
In NSCLC, high vascular endothelial growth factor (VEGFs) expression is associated with tumor recurrence, low survival, metastasis, and death in patients (145, 146). Overexpression of VEGF induces a reduction in the expression of endothelial cell adhesion molecules, which severely impairs T-cell homing and reduces the number of T lymphocytes entering the TME (147). It has been shown that VEGF-A also enhances the expression of PD-1 and other inhibitory checkpoints, such as CTLA-4, on the surface of T cells and inhibits the activity of CD8+ T cells, leading to a blockade of the effector function of T cells (148–150). Therefore, combining anti-angiogenic drugs with immunotherapy, which normalizes tumor vasculature through anti-angiogenic drugs and promotes the increase of tumor immune cells (e.g., tumor-infiltrating lymphocytes) in NSCLC, and utilizing immune checkpoint inhibitors which can unlock the functional inhibition of T cells by PD-1 and PD-L1, both act synergistically with each other thus showing better therapeutic effects within solid tumors (151–153). In the IMpower-150 trial, researchers tested atezolizumab plus bevacizumab plus chemotherapy as a first-line treatment for non-squamous NSCLC (non-sq-NSCLC), with an ORR and PFS of 63.5% and 8.3 months, respectively (154). Still, the incidence of adverse events in grades 3 or higher was as high as 58.5% (154). The FDA has approved atezolizumab, bevacizumab, and chemotherapy as first-line treatment for advanced non-squamous NSCLC (155).
AK112 (Ivonescimab) is a humanized anti-PD-1/VEGF-A bispecific monoclonal antibody in which an anti-PD-1 scFv fused to the end of each of the two heavy chains of the anti-VEGF-A antibody (bevacizumab) to form a tetravalent bis-antibody. This bispecific antibody can accumulate in the tumor microenvironment, effectively blocking both the PD-1 and VEGF pathways, inhibiting PD-1-mediated immunosuppression, and blocking tumor angiogenesis in the microenvironment (156, 157). In May 2024, China approved AK112 in combination with chemotherapy (pemetrexed + carboplatin) for patients with locally advanced or metastatic non-sq-NSCLC who are EGFR mutation-positive and have progressed after treatment with EGFR TKIs (158). The approval was based on a phase III clinical trial called HARMONi-A (NCT05184712), which showed that in patients resistant to EGFR TKIs, AK112 in combination with chemotherapy was sufficient to significantly prolong mPFS compared to placebo combination chemotherapy (7.06 months vs. 4.08 months; HR 0.46) and patients had a higher ORR (50.6% vs. 35.4%) (103). The primary results of HARMONi-2 (NCT05499390), a trial comparing the efficacy of ivonescimab with that of pembrolizumab, were also presented. It showed that in patients with previously untreated stage IIIB to IV advanced NSCLC who were EGFR/ALK wild-type and with PD-L1 TPS≥1%, AK112 significantly prolonged their mPFS compared to pembrolizumab (11.14 vs. 5.82 months; HR, 0.51; p<0.0001), increasing their ORR (50.0% vs. 38.5%) and DCR (89.9% vs. 70.5%) (104). Although the rate of serious adverse events in patients treated with AK112 was slightly higher than with pembrolizumab (Grade 3+ TRAEs: 29.4% vs. 15.6%), the results of HARMONi-2 are still encouraging (104). It is expected that AK112 will become a potential clinical option in the first-line treatment of lung cancer.
PM8002 is a humanized anti-PD-L1/VEGF-A bispecific antibody with the variable region of a humanized anti-PD-L1 nanobody fused at the end of the two heavy chains of the anti-VEGF-A antibody. Preliminary published data (NCT05918445) showed that in untreated EGFR/ALK wild-type and PD-L1+ patients who received PM8002, the ORR is 47.1%, the mPFS is 10.9 months, and the incidence of grade ≥3 TRAEs was 18% (105). In addition, another study (ChiCTR2200059911) demonstrated the efficiency of PM8002 combined with paclitaxel for second-line treatment of SCLC. It was reported that the ORR was 72.7% (16/22), the DCR was 81.8% (18/22), and the mPFS was 5.5 months (106). Notably, the recommended drug for second-line treatment of SCLC is topotecan, with an ORR of 22% and a mDoR of 7.6 months (159). Therefore, further observing the subsequent clinical efficacy of PM8002 in SCLC is worthwhile.
IMM2510 is a bispecific antibody targeting PD-L1 and VEGFs that incorporates a VEGF receptor 1 domain 2 (VEGFR1-D2) at the C-terminus of each of the two heavy chains of the anti-PD-L1 antibody, forming a VEGF trap capable of binding a wide range of VEGF receptor ligands in addition to VEGF-A (160). IMM2510 is currently in the preliminary phase I clinical studies (NCT05972460). As of 21 December 2023, 33 patients with advanced solid tumors were treated with IMM2510 at nine doses (0.007-20.0 mg/kg) (107). Among the 25 patients whose conditions were evaluable, 3 patients had achieved confirmed partial response, and 7 patients had stable disease (107). Among the PR patients, one patient with sq-NSCLC (onco-driver gene negative, previous IO treatment failure) who was treated with 3 mg/kg IMM2510 achieving tumor shrinkage of 46% and still on the treatment with treatment duration over 20 months; one sq-NSCLC treated with 10 mg/kg showed a tumor shrinkage of about 32% and a treatment duration of 9.4 months (107). TRAEs occurred in 32 pts (97.0%), and most were grade 1 or 2. Grade ≥3 TRAEs occurred in 11 pts (33.3%), and no DLT occurred (107).
4.3.2 PD-L1 × TGF-β
A central factor in tumor immune resistance is immunosuppressive cytokines in the tumor microenvironment, a major component of which is transforming growth factor β (TGF-β). It has been shown that TGF-β upregulates PD-L1 transcription in tumors (161). Besides, TGF-β-mediated T-cell rejection is one of the mechanisms by which tumors become resistant to anti-PD-L1 therapy (162). TGF-β also induces the release of PD-L1-containing exosomes by tumor cells, and PD-L1 exosomes in tumor regions hamper the effector activity of CD8+ T cells (163). In addition, there is a bidirectional interaction between TGF-β and tumor-area hypoxia, where hypoxia is considered a key inducer of TGF-β, and the activity of the latter further enhances tumor-area hypoxia (164). Hypoxia induces upregulation of PD-L1 expression on tumor cells and upregulation of PD-1 expression on immune cells (e.g., TAMs, DCs, Tregs) in the tumor microenvironment (165, 166). Aberrant TGF-β activity is associated with immunosuppressive TME, promoting progression and metastasis in NSCLC (167). Several companies have developed drug molecules with dual inhibitory effects on PD-1/L1 and TGF-β, among which M7824 and SHR1701 were developed earlier and have more preclinical and clinical data.
M7824 is a bifunctional fusion protein, a molecule that fuses a TGF-β receptor II (a TGF-β ‘trap’ which is capable of trapping all TGF-β) to the end of the constant region of avelumab via a flexible linker (168). M7824 has been noticed and given high expectations based on its phase I clinical (NCT02517398) results released in 2018. Among 40 patients with advanced NSCLC who relapsed after standard therapy treated with 1200 mg of M7824, the ORR was 40.7% for PD-L1-positive patients and 71.4% for patients with high PD-L1 expression (169). However, the subsequent publication of several clinical data has overshadowed M7824’s prospects. In the phase III clinical trial (NCT03631706) using either M7824 or pembrolizumab as first-line treatment for patients with advanced PD-L1 high-expression (TPS ≥80%) NSCLC, the results showed that first-line treatment with M7824 did not demonstrate superior efficacy compared to pembrolizumab and had a higher frequency of associated adverse events (170). Combined with unsatisfactory results from multiple other clinical trials, the outlook for M7824 is troubling, and clinical trials for the drug have now primarily been terminated.
SHR-1701 is also a bifunctional fusion protein that fuses an extracellular TGF-β receptor II structure to the C-terminus of a monoclonal antibody against PD-L1 (171). At the ESMO 2023 meeting, researchers preliminarily presented the results of SHR-1701 in 3 NSCLC clinical expansion cohorts (NCT03774979). For patients who had not received systemic chemotherapy and had a PD-L1 TPS ≥1%, ORR was 36.8%, DCR was 66.7%, and mOS was 24.2 months; for patients with an EGFR mutation who had failed prior treatment with standard EGFR TKIs or had no standard EGFR TKIs available, ORR was 19.5%, DCR was 46.3%, and mOS was 14.4 months; for patients who experienced disease progression after recent anti-PD-1/PD-L1 therapy and had received up to 3 lines of prior therapy ORR was 9.1%, DCR was 54.5%, mOS was 16.1 months (108). In this trial, 76.3% of patients treated with SHR-1701 experienced TRAEs, including 22.9% with grade ≥3 TRAEs (108). In addition, a trial (NCT04580498) was conducted to assess neoadjuvant SHR-1701 with or without chemotherapy, followed by surgery or radiotherapy, and then consolidation SHR-1701 in unresectable stage III NSCLC, and preliminary clinical results were recently published. Of the 97 evaluable patients treated with SHR-1701 plus chemotherapy, the postinduction ORR was 58%, and the 18-month event-free survival rate was 56.6% (109). Regarding safety, the rate of patients with grade ≥3 TRAEs was 75% in the SHR1701 plus chemotherapy arm (109). The drug is currently undergoing multiple multi-center phase III clinics, and there is much interest in the final outcome, especially as M7824 has been terminated from the clinic.
5 Immune cell engagement bispecific antibodies
5.1 T cell engagement
TCE is a typical application of bsAbs, with one targeting arm of most TCEs designed to bind specifically to selected tumor-associated antigen (TAA) on the surface of tumor cells and the other targeting arm designed to target the CD3ϵ chain in the TCR complex. Due to the signaling capacity of the CD3ϵ chain, TCEs can bypass the major histocompatibility complex restriction and, independently of the epitope specificity of the TCR, elicit T-cell activation and proliferation, as well as the subsequent release of transient inflammatory cytokines induced by the TCR and trigger tumor apoptosis via perforin and granzyme release (172). In addition, bsAbs targeting TAA and co-stimulatory receptors on T cells (e.g., 4-1BB, CD40, CD28, etc.) have a mechanism similar to that of a TCE.
5.1.1 DLL3 × CD3
Delta-like ligand 3 (DLL3) is a TAA highly expressed on the surface of tumor cells in patients with SCLC, with high expression detected in approximately 85%-94% of SCLC patients (173, 174). Under normal conditions, DLL3 is mainly localized in the Golgi apparatus and cytoplasmic vesicles and is hardly expressed on the surface of normal cells (175). In contrast, in cells of SCLC and other high-grade neuroendocrine tumors, DLL3 is highly up-regulated and aberrantly expressed on the cell surface, leading to abnormal growth of neuroendocrine tumor cells (176). Thus, DLL3 has become a potential target for treating SCLC.
Tarlatamab (AMG757) is a DLL3-based TCE. According to published data from the clinical trial called Delphi -301 (NCT05060016), patients with SCLC who had received two or more systemic therapies benefited significantly from fortnightly intravenous treatment with Tarlatamab (110, 177). This study demonstrated a more significant benefit in patients treated with 10 mg of Tarlatamab compared to 100 mg, with an ORR of 40.0%, mPFS of 4.9 months, and mOS of 14.3 months (110, 177). The most common adverse events were cytokine release syndrome (in 51% of the patients in the 10-mg group), decreased appetite (29%), pyrexia (35%), constipation (27%), and anemia (26%) (110, 177). Grade 3 or higher adverse events occurred in 59% of the patients in the 10-mg group (110, 177). Based on the clinical data published in Delphi-301, the FDA granted tarlatamab accelerated approval in May 2024 for the treatment of extensive-stage SCLC, where disease progression occurs during or after platinum-based chemotherapy (178). In addition, newly published long-term follow-up data from Delphi-300 (NCT03319940) showed that the overall ORR for patients treated with more than 10 mg of tarlatamab was 25%, with an mDOR of 11.2 months (111). Patients with brain metastases also showed significant benefits after treatment with tarlatamab, with intracranial disease control occurring in 87.5% (111). Safety data were consistent with Delphi -301, with no new safety signals (111). TCEs in solid tumors have long been considered challenging (172), and tarlatamab is the first TCE therapy to be approved for treating a solid tumor, marking a critical step forward.
BI764532 is an IgG-like TCE that induces strictly DLL3-dependent tumor killing (179). BI764532 is currently in phase I clinical trial (NCT04429087), and preliminary clinical data have been published. Of 71 evaluable patients treated with no less than 90ug/kg of BI764532, 25% achieved partial response, with a DCR of 52%, and tumor shrinkage was observed in all patients (112). A total of 86% of patients experienced adverse events of any grade, with 37% experiencing grade 3+ TRAEs and four non-Asian patients discontinuing due to TRAEs (112).
PN328/MK6070 is a TCE targeting DLL3 and CD3. In addition, to prolong the half-life of this TCE as well as to maintain its most substantial direct T-cell killing ability, an anti-HSA single-domain antibody was fused in the middle of the DLL3 and CD3 targeting arms (see Figure 1) (180). It is currently enrolling participants with SCLC, neuroendocrine prostate cancer, and other high-grade neuroendocrine neoplasms in a phase I/II clinical trial (NCT04471727). The data showed that in the dose-optimized cohort (1-mg priming dose and a 12- or 24-mg target dose), patients with SCLC had an ORR of 39% and a DCR of 71% (113). In addition, the data showed that SCLC patients with brain metastases also responded well to HPN328 (ORR of 37% and DCR of 78%) (114). In terms of the safety of the drug, the incidence of TRAEs at any grade was 93%, with grade 3+ TRAEs occurring in 26%, with four patients discontinuing the drug due to TRAEs and two patients dying (113).
5.1.2 EGFR × CD3
As mentioned earlier, EGFR is highly expressed in a variety of tumors. Meanwhile, EGFR regulates the development and homeostasis of regulated epithelial tissues in normal tissues and plays a key role in epithelial cell physiology (181). Therefore, a variety of drugs targeting EGFR have some skin toxicity (182, 183). In contrast, TCE-induced tumor cell killing is highly efficient, as TCE enables individual T cells to connect with multiple tumor cells, resulting in sequential killing (184). In addition, cytokines released from activated T cells can generate a cascade amplification effect to achieve a more excellent range of tumor cell killing (185). Therefore, TCEs targeting EGFR face more severe on-target off-tumor toxicity, resulting in a limited therapeutic window. However, suppose TCEs are engineered with a modified design to specifically recognize EGFR on tumor cells and bind no or less EGFR on normal tissues. In that case, they can significantly enhance the efficacy and expand the drug’s therapeutic window.
JANX008 is a prodrug form of TCE that targets EGFR and contains an EGFR-binding domain, a CD3-binding domain, and a HAS-binding domain that serves to extend the half-life of the molecule (see Figure 1c) (186). In addition, a mask is fused to each of the EGFR and CD3 binding domains through tumor protease cleavable linkers, and only when the molecule enters the tumor site and is recognized by the tumor protease can the peptide masks be released, ultimately generating the active molecules (186). The drug currently enrolls patients with advanced or metastatic cancers with high EGFR expression in a clinical phase Ia trial. It has been observed to achieve partial remission in one NSCLC subject, with a 100% reduction in targeted lung lesions and the elimination of liver metastases (115). Notably, JANX008 had a favorable safety profile, with grade 1 CRS observed in only 2 of 11 subjects at doses up to 1.25 mg (significantly higher than the expected maximum tolerated dose of the parental T-cell articulator) (115).
5.1.3 PDL-1 × 4-1BB
4-1BB [also known as CD137 or TNF receptor superfamily member 9 (TNFRSF9)] is an inducible co-stimulatory receptor expressed on activated T cells and NK cells (187, 188). The agonism of 4-1BB avoids tumor-infiltrating lymphocyte exhaustion and enhances the antitumor activity of ICIs (189, 190). Although preclinical results have shown excellent efficacy of anti-4-1BB antibodies in different tumors, the development of 4-1BB-based monoclonal antibodies has successively failed either because of fatal side effects (urelumab) or because of limited efficacy (utomilumab) (191). Therefore, preserving the efficacy of anti-4-1BB antibodies while reducing their toxicity is a priority for subsequent development. One such strategy is the use of bsAb, which minimizes off-target tumor toxicity by designing the antibody to preferentially bind specifically to tumor cells and be enriched in the tumor microenvironment, and then to bind to 4-1BB and achieve activation of 4-1BB signaling. In this strategy, the most studied is the PD-L1/4-1BB bispecific antibody. Because PD-L1 is not only expressed on the surface of a wide range of cancer cells, a variety of host cells in the TME and lymph nodes, including dendritic cells, macrophages, fibroblasts, and T cells, also express PD-L1 to reduce antitumor immunity. In addition, PD-L1 blockade combined with 4-1BB agonistic antibodies has shown enhanced antitumor responses in preclinical cancer models (189, 190, 192). Thus, dual targeting of PD-L1 and 41BB by bispecific antibodies may permit tumor cell-dependent 4-1BB activation of T cells and allow optimal antitumor immunity.
GEN1046 (acasunlimab) is currently the fastest advancing bsAb targeting PD-L1 and 4-1BB. It is an IgG-like bsAb, with one arm targeting PD-L1 to block PD-1/PD-L1 inhibitory signals and another targeting 4-1BB to activate co-stimulatory signals in immune cells (116). A clinical trial (NCT05117242) evaluating acasunlimab monotherapy and acasunlimab in combination with pembrolizumab in metastatic NSCL patients who were resistant to anti-PD-1/L1 antibodies is currently underway, and preliminary results have been published. The data showed that patients treated with GEN1046 100 mg Q6W in combination with pembrolizumab 400 mg Q6W had a favorable treatment outcome (117). This subset of patients had an ORR of 16.7%, a DCR of 75%, a mOS of 17.5 months, and a 12-month OS rate of 69% (117). The most common adverse events (all grades; grade ≥3) in patients treated with the combination therapy included liver-related events (18.7%; 13.3%), fatigue (14.7%; 0%), malaise (13.3%; 0%), and diarrhea (12%; 0%) (117).
FS222 is a 2 + 2 tetravalent bsAbs constructed on a platform called mAb2. The antibody is based on a PD-L1 antibody, engineered to make its Fc recognize 4-1BB (193). The antibody is, therefore, similar in size to a conventional monoclonal antibody. Additionally, the antibody undergoes Fc mutation to reduce associated effects such as ADCC and CDC. FIH (NCT04740424) is an ongoing phase I clinical trial investigating the efficacy of FS222 in advanced solid tumors. Currently, interim results from the Q4W cohorts have been reported. The cohort had a total of 90 patients enrolled, with a median of 2 (1–7) regimens previously treated. At the cut-off date (05 Dec 2023), 20 (22.2%) patients were still on treatment, and objective remissions (CR, PR) were observed in patients with melanoma, NSCLC, ovarian, triple-negative breast, liposarcoma, and colon cancer with an ORR of 15.7% (118). The most common TRAEs grade ≥3 (≥10% of pts) were increased AST (13.3%) and ALT (11.1%) (118).
5.2 NK cell engagement
NK cells are innate immune cells with cytotoxicity. NKCEs have one targeting arm targeting tumor cell surface-specific antigens and the other targeting arm targeting activating receptors on the surface of NK cells, such as CD16a, NNKG2D (atural Killer Group 2 Member D), NKp30 (Natural Killer cell p30), etc., resulting in the formation of antigen-specific immune synapses between the NK cells and the tumor cells that which in turn triggers NK cell-mediated killing of tumor cells (194). NKCEs are a new exploration, and several drugs of this type are undergoing preclinical or clinical evaluation.
5.2.1 EGFR × CD16a
AFM24 is a tetravalent bispecific antibody in which the anti-EGFR scFv is fused to the c-terminus of the anti-CD16a antibody via a connector, which mediates the killing of tumor cells by NK cells in an EGFR-dependent manner (195). Studies have demonstrated the preliminary efficacy of AMF24 in NSCLC patients with EGFR mutant NSCLC, relapsed or refractory to ≥1 prior lines of therapy. Of the 10 evaluable patients, 1 patient suffered a 45% reduction in tumor volume, and 4 had stable disease with a 50% tumor control rate (119). However, a high proportion of patients experienced TRAEs (13/14), with grade ≥3 TRAEs occurring in 4 patients and grade 5 pneumonitis occurring in 1 patient (13/14) (119). In NSCLC patients with EGFR-WT who relapsed after one or more first-line therapies, the combination of AFM24 and atezolizumab led to 1 complete response, 3 partial responses, and 7 cases of stable disease among the 15 evaluable patients (120). Moreover, this combination therapy was well tolerated. The main adverse events that occurred in the patients were infusion-related reactions (10/17) (120).
6 Immunocytokines
Cytokines are mediators and modulators of the innate and adaptive immune systems, and immunotherapy based on cytokines such as interferon and interleukin-2 (IL-2) has been used in the treatment of cancer as early as the end of the 20th century (196). However, the short half-life of cytokines results in the need for short periods of high-dose administration, which can lead to severe non-specific toxicity. In addition, the cytokine’s inhibitory or activating effect on immune cells is also related to the concentration and the environment of action. Therefore, engineering cytokines to preferentially target disease sites and to activate only specific lymphocyte types can increase the tolerability of cytokine therapy. The specific targeting of antibodies naturally makes them ideal ‘carriers’ for targeted delivery of therapeutic cytokines. In many mouse models, antibody-cytokine fusion proteins targeting tumor markers increase the selective accumulation of the corresponding cytokines at the tumor site (19, 20). Such antibody-cytokine fusion proteins, also known as immunecytokines, are another significant application of bispecific antibodies and have been called the next generation of cytokine products.
6.1 PD-1 × IL-2
High-dose recombinant IL-2 (Proleukin) was approved by the FDA for treating metastatic renal cell carcinoma in 1992, followed by metastatic melanoma in 1998 (197, 198). However, recombinant natural IL-2 is dose-limited (0.037 mg/kg admitted) and causes severe non-specific extravasation toxicity (197–199). In addition, IL-2 has two opposing functions: at low doses, IL-2 tends to stimulate Treg cells expressing high-affinity trimeric receptors (IL-2Rαβγ), resulting in immunosuppression (200, 201); at high doses, after saturation of the receptor on Treg cells, excess IL-2 also interacts with effector T cells via intermediate affinity receptors (IL-2Rβγ) binds to effector T and NK cells, promoting immune activation and anti-tumour responses (202). To improve the therapeutic index, researchers have engineered IL-2 through various strategies to promote a longer half-life of the modified drug and specific binding to IL2-Rβγ (197) without binding to IL2-Rα (203), expressed on Treg. However, these bias-modified drugs did not achieve better clinical results (204).
IBI363 is a PD-1/IL-2α-bias bispecific antibody fusion protein. Unlike the mainstream IL2-Rβγ-biased design, IBI363 fused an IL-2Rα-biased engineered modified IL-2 at the end of the anti-PD-1 antibody. This design was based on a preclinical correlative study, in which researchers found that newly activated tumor-specific CD8+ T cells expressed PD-1 while upregulating IL-2Rα and that the anti-tumor effect of anti-PD-1 was dependent on the activation of PD-1+ CD25+ CD8+ T cells via autocrine IL-2/IL2-Rα signaling (205). Thus, through specific guidance of PD-1, PD-1/IL-2α-bias can selectively stimulate and expand T cells expressing both PD-1 and IL-2Rα within the tumor, leading to more precise and effective targeting and activation of this T cell subpopulation. At WCLC 2024, researchers presented a phase I clinical study (NCT05460767) of IBI363 in advanced non-small cell lung cancer. The data showed an ORR of 24.1% and a DCR of 68.4% in 79 patients who received doses higher than 0.3 mg/kg and an ORR of 85.7% (6/7) in the 3 mg/kg dose group (22). The drug’s safety profile was good, with 87.6% of patients experiencing any grade of TRAEs and 19.1% of patients experiencing grade 3+ TRAEs (22). From the preliminary clinical data, it can be seen that IBI363 was administered at an unprecedented dose (3mg/kg) and demonstrated a favorable safety profile, breaking through the safety concerns of IL-2 therapy. Combined with its preliminary published efficacy data, the clinical results of subsequent treatments with IBI363 are promising.
6.2 PD-1 × IL-15
Interleukin 15 (IL-15) is an IL2-related cytokine. Both can bind IL-2Rβγ, but the high-affinity forms of both IL2 and IL15 receptors contain IL2-Rα (CD25) or IL-15Rα (CD215), respectively (206). IL-15 is produced by dendritic cells, macrophages, and stromal cells, and, like IL-2, IL-15 can stimulate T-cell proliferation, induce cytotoxic T lymphocyte production, and promote the maintenance of NK cells (207, 208). In addition, the unique role of IL-15 is to maintain NK cell and CD8+CD44h memory T cell function to provide a long-term immune response to pathogen (208). Like IL-2, IL-15 therapy was initially limited by the short molecular half-life and toxicity associated with systemic immune activation (209). Anktiva (N-803) is a recombinant IL-15 superagonist protein complex consisting of a high-affinity IL-15 mutant and IL-15Rα fused to Fc (and thus with an extended half-life). 2024 The FDA approved Anktiva in combination with Bacillus Calmette-Guérin (BCG) for treating adult BCG-naïve patients with non-muscle invasive bladder cancer with carcinoma in situ with or without papillary tumors (210). Animal studies have demonstrated that the combination of N-803 and anti-PD-L1 antibody can activate NK and CD8⁺ T cells and induce the production of immunostimulatory cytokines, demonstrating significant efficacy in various models that do not respond or respond poorly to monotherapy (211). Clinical studies have demonstrated that patients with ≥2nd line NSCLC who failed CPI therapy treated with Anktiva in combination with CPI have an mOS of up to 11.4 months (212).
PF-07209960 is a cytokine fusion protein fused with mutated IL-15 at the end of one of the heavy chains of the anti-PD-1 antibody (213). The mutation in IL-15 is designed so that PF-07209960 does not bind to IL-15Rα and has a significantly reduced affinity for IL-2/15Rβγ. The molecule, therefore, explicitly delivers IL-15 to CD8+ T cells with high PD1 expression in tumors without binding to IL-15Rα-expressing cells and PD1-negative IL-15Rβγ-positive NK cells (213). Preclinical studies demonstrated that PF-07209960 could increase the number of CD8⁺ TILs within tumors specifically, had excellent anti-tumor activity, and significantly reduced adverse effects (213). Researchers recently published preliminary clinical data on PF-07209960. Of the 29 evaluable patients, two patients, both with microsatellite-stable colorectal cancer, were confirmed to be in partial remission, with an ORR of 6.9% and a DCR of 48.3% (121). Regarding safety, 97.3% of patients experienced one or more adverse events, of which 78.4% experienced grade 3 or higher TRAEs, with a serious adverse events rate of 70.3% (121).
SAR44587, or KD050, consists of an Fc-silenced high-affinity human anti-PD-1 antibody (IgG1 subtype) fused to a mutated IL-15/IL-15Rα sushi domain (122). Through its anti-PD1 portion, SAR44587 binds to PD-1-expressing T cells and NK cells and may result in the specific expansion and activation of CD8+ T cells and NK cells expressing PD-1 and IL-2/15Rβγ (214). In preclinical models of PD-1 resistance, SAR445877 had a stronger tumor suppressive effect compared to pembrolizumab, increasing the ratio of CD8+/CD4+ T cells in tumors and significantly increasing the percentage of effector memory CD8+ T cells in tumors (215). The drug is currently undergoing a clinical phase I study (NCT05584670), which is planned to enroll 240 adult patients with advanced unresectable or metastatic solid tumors to confirm the safety, tolerability, pharmacokinetics, pharmacodynamics, and antitumor activity (214).
IAP0971 is an immunocytokine that binds specifically to PD-1 and fused IL-15/IL-15Rα complex (123). Mechanistically, IAP0971 can deregulate the immunosuppression of the PD-1/PD-L1 axis while increasing the targeting of IL-15 to the tumor microenvironment and avoiding systemic non-specific activation; furthermore, IAP0971 has an IgG4-based structure, resulting in a weak effect such as ADCC and ADCP, and a long half-life (123). In preclinical studies, IAP0971 can stimulate the proliferation of CD8+ T cells and NKT cells, activate NK cells to kill tumor cells, and significantly inhibit tumor growth in mice at as low as 0.1 mg/kg without affecting their body weights (123). IAP0971 is currently in Phase I/IIa clinical trial (NCT05396391) to evaluate its safety, tolerability, and preliminary efficacy in patients with locally advanced or metastatic malignancies (216). Indications include a variety of malignancies, including lung cancer, cervical cancer, squamous cell carcinoma of the head and neck, hepatocellular carcinoma, and lymphoma.
7 Challenges and limitations
BsAbs show great potential in lung cancer therapy, and several key clinical advances are exciting. However, as described below, the therapeutic use of bsAbs in lung cancer faces many challenges.
7.1 Production challenges
Compared with monoclonal antibodies, the challenges facing the development and industrialization of bsABs are enormous. Unlike the standardized preparation process of monoclonal antibodies, the expression titer of bsABs is usually lower than that of monoclonal antibodies if the traditional process is directly followed (217), and they are prone to the formation of by-products such as aggregation and mismatch products (218–220). Although it is possible to increase the content of target products through various engineering modification strategies, bsAb-specific by-products are generally present at low levels in the cell culture supernatant of bsAb. It is necessary to rely on multiple purification strategies (e.g., a combination of affinity chromatography, ion-exchange chromatography, and size-exclusion chromatography) in order to obtain high purity of the target products, which significantly prolongs the development cycle and increases the cost of production (221).
7.2 Immunogenicity risk
Immunogenicity is one of the key challenges in the development of bsAbs. While some bispecific antibodies, such as amivantamab, have a very low incidence of anti-drug antibodies (ADAs), other bispecific antibodies show a high incidence of ADAs and neutralizing antibody positivity (222). These antibodies, which, due to drug immunogenicity, will directly affect the pharmacokinetics、pharmacodynamics, and safety of the drug (223). How to reduce the ADAs of a drug is a relatively complex issue, which needs to start from the initial molecular design (e.g., optimization of humanized modified epitopes), early in vitro immunogenicity assessment (based on in silico algorithms, and in vitro T cell-based assays), manufacturing process improvement (e.g., reduction of drug aggregation tendency, reduction of impurity residues), and clinical intervention (e.g., adjustment of drug immunogenicity), production process improvement (e.g., reducing drug aggregation tendency, reducing impurity residues), and clinical interventions (e.g., adjusting drug dosage, dosing regimen, and route of administration) are multifaceted and synergistic, which is complex and time-consuming (224).
7.3 Side effects
The adverse effects of bsAbs are closely related to their mechanism of action. Among them, cytokine release syndrome (CRS) is a common adverse reaction of TCE. For example, 49% of patients receiving AMG757 developed CRS, and 26% of them had severe symptoms (110). BsAbs based on a dual blocking mechanism show adverse reactions that are superimposed on the two targets (although they can be significantly lower than the parent antibody combination). For example, adverse reactions such as rash, pruritus, and diarrhea associated with EGFR and hypoproteinemia and peripheral edema associated with cMET have been observed with amivantamab (42).
Researchers are currently attempting various strategies to reduce the adverse effects of bsAbs and expand the therapeutic window. For example, by lowering the affinity of CD3 to increase the tissue distribution of bispecific antibodies in tumors and significantly reduce the level of cytokine release in normal tissues (225–227); and by restricting the activity of bsAbs in normal tissues through shielding techniques (186, 228); or by using tumor poxviruses as vectors to deliver dual antibodies to tumor sites in a targeted manner (229). However, drugs based on these strategies are currently in the preclinical or early clinical stage, and more clinical data are needed to analyze their safety and efficacy more correctly. Another strategy is to change the mode of administration. Currently, bsAbs are administered mainly by intravenous infusion, but data have shown that subcutaneous administration improves patient compliance and reduces the incidence of adverse events. For example, subcutaneous administration significantly reduced the incidence of infusion-related reactions and venous thromboembolic events with amivantamab compared with intravenous administration (47). Besides, some TCEs have also been shown through clinical studies to have a lower incidence of CRS with subcutaneous administration (230). However, subcutaneous administration is currently not widely used in solid tumors, and adequate subsequent validation is needed before widespread use. Intervention by the clinical therapist is the final barrier against adverse reactions. Real-time monitoring, pretreatment, and symptomatic management can enhance the patient experience (172, 231). However, it then requires clinicians to individualize the assessment of the patient and adjust the treatment regimen, which undoubtedly adds to the widespread use of the drug. Finally, it is worth noting that pneumonia is a particular concern in the lung cancer population, as lung cancer patients often have poor lung reserve due to current or past smoking history (232, 233). Drug-induced pneumonitis can, therefore, severely compromise their already poor lung reserve, which, in some cases, can be fatal.
7.4 Tumour heterogeneity and microenvironmental limitations
The complexity of lung cancer itself also poses a challenge for drug development. Lung cancer, as a very heterogeneous type of solid tumor, has differences in response to the same drug in different patients (as seen in Table 1). Thus, screening the most appropriate target population for bsAbs will be a significant challenge. In addition, the TME plays a central role in the genesis and progression of primary lung cancer, where cancer cells can reprogram tumor-infiltrating stromal cells, thereby promoting carcinogenesis (234, 235). In lung cancer, tumors can reprogram the lung microenvironment, which in turn promotes inflammation, angiogenesis, immunosuppression, and unresponsiveness to therapy, ultimately leading to lung metastasis from both primary and extrapulmonary tumors (236). Moreover, the disturbed and inefficient vascular supply of the TME and the elevated interstitial fluid pressure due to lymphovascular dysfunction greatly limit tumor penetration and T-cell infiltration into the tumor by dual antibodies. Small molecular weight dual antibodies (e.g., BiTE) that have better penetration but short half-lives (e.g., blinatumomab has a half-life of 2-4 hours (172)) require repeated administration. Small molecular weight dual antibodies (e.g., BiTE) that have better penetration but short half-lives (e.g., blinatumomab has a half-life of 2-4 hours (167)) require repeated administration. Fusing the VHH fragment of anti-HSA is a strategy that prolongs the drug’s half-life while maintaining high drug penetration, and several such bispecific antibodies are currently under clinical investigation. Finally, the current bsAbs are mainly targeted at some mature targets (e.g., EGFR, PD1, etc.), and breakthroughs are urgently needed in the mining of new lung cancer-specific targets (e.g., c-MET, HER3) and TME regulation strategies (e.g., combining with anti-angiogenic drugs).
7.5 Pharmacoeconomic considerations
In addition to the technical challenges discussed above, pharmacoeconomic considerations are also a key obstacle to the widespread application of bsAbs in treating lung cancer. Compared with conventional antibody drugs, bispecific antibodies have higher production costs and greater clinical development risks, resulting in higher drug prices. Moreover, adverse reactions caused by the drugs require additional symptomatic medications, which further increase the treatment costs for patients. Therefore, balancing the cost of the drug and its corresponding therapeutic effect is a huge challenge. Some researchers have noted that although the treatment regimens of amivantamab in combination with chemotherapy or amivantamab in combination with lazertinib provide significant benefits to patients, when the economic costs are considered, they are higher than the cost-effectiveness thresholds given current US pricing (237, 238). Therefore, how to balance the cost and pricing to benefit more patients is an issue that needs to be addressed. In addition, the treatment cost can be reduced by further optimising the drug dosage. For example, compared with the recommended treatment regimen, using an optimised alternative dosing regimen can save 16% of the treatment cost of Amivantamab (239).
8 Conclusion
BsAbs have ushered in significant development, with more than 110 in clinical development and nearly 180 in preclinical development (240). There are three drugs approved for lung cancer treatment, of which the launch of AMG757 is a breakthrough in the field of small-cell lung cancer treatment for many years, and AK112 beat Keytruda in head-to-head clinical studies, both of which are of landmark significance. In the next phase of drug development, the therapeutic potential of bsAbs will be further unlocked through research to optimize the production of CMC, reduce the adverse effects, and research into the pathogenesis of lung cancer itself, enhancing the cost-effectiveness of the drugs, which is of great significance, and discovering new therapeutic targets. In conclusion, more new drugs will be developed for more lung cancer patients with unmet needs for quite some time to come.
Author contributions
WC: Conceptualization, Data curation, Writing – original draft, Writing – review & editing, Investigation. AZ: Conceptualization, Data curation, Writing – review & editing, Writing – original draft. YZ: Supervision, Writing – review & editing, Writing – original draft.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted without any commercial or financial relationships that could potentially create a conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Correction note
A correction has been made to this article. Details can be found at: 10.3389/fimmu.2025.1656082.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Thai AA, Solomon BJ, Sequist LV, Gainor JF, and Heist RS. Lung cancer. Lancet. (2021) 398:535–54. doi: 10.1016/s0140-6736(21)00312-3
2. Allemani C, Weir HK, Carreira H, Harewood R, Spika D, Wang X-S, et al. Global surveillance of cancer survival 1995–2009: analysis of individual data for 25 676 887 patients from 279 population-based registries in 67 countries (CONCORD-2). Lancet. (2015) 385:977–1010. doi: 10.1016/s0140-6736(14)62038-9
3. Rotow J and Bivona TG. Understanding and targeting resistance mechanisms in NSCLC. Nat Rev Cancer. (2017) 17:637–58. doi: 10.1038/nrc.2017.84
4. Horvath L, Thienpont B, Zhao L, Wolf D, and Pircher A. Overcoming immunotherapy resistance in non-small cell lung cancer (NSCLC) - novel approaches and future outlook. Mol Cancer. (2020) 19:141. doi: 10.1186/s12943-020-01260-z
5. Koinis F, Kotsakis A, and Georgoulias V. Small cell lung cancer (SCLC): no treatment advances in recent years. Trans Lung Cancer Res. (2016) 5(1):39–50. doi: 10.3978/j.issn.2218-6751.2016.01.03
6. Gazdar AF, Bunn PA, and Minna JD. Small-cell lung cancer: what we know, what we need to know and the path forward. Nat Rev Cancer. (2017) 17:725–37. doi: 10.1038/nrc.2017.87
7. Rudin CM, Giaccone G, and Ismaila N. Treatment of small-cell lung cancer: american society of clinical oncology endorsement of the american college of chest physicians guideline. J Oncol Pract. (2016) 12:83–6. doi: 10.1200/jop.2015.008201
8. Rudin CM, Brambilla E, Faivre-Finn C, and Sage J. Small-cell lung cancer. Nat Rev Dis Primers. (2021) 7(1):3. doi: 10.1038/s41572-020-00235-0
9. Brinkmann U and Kontermann RE. The making of bispecific antibodies. MAbs. (2017) 9:182–212. doi: 10.1080/19420862.2016.1268307
11. Krah S, Sellmann C, Rhiel L, Schröter C, Dickgiesser S, Beck J, et al. Engineering bispecific antibodies with defined chain pairing. N Biotechnol. (2017) 39:167–73. doi: 10.1016/j.nbt.2016.12.010
12. Krah S, Kolmar H, Becker S, and Zielonka S. Engineering igG-like bispecific antibodies-an overview. Antibodies (Basel). (2018) 7(3):28. doi: 10.3390/antib7030028
13. Chames P and Baty D. Bispecific antibodies for cancer therapy. Curr Opin Drug Discovery Devel. (2009) 12:276–83. doi: 10.4161/mabs.1.6.10015
14. Chan AC and Carter PJ. Therapeutic antibodies for autoimmunity and inflammation. Nat Rev Immunol. (2010) 10:301–16. doi: 10.1038/nri2761
15. Wu C, Ying H, Bose S, Miller R, Medina L, Santora L, et al. Molecular construction and optimization of anti-human IL-1alpha/beta dual variable domain immunoglobulin (DVD-Ig) molecules. MAbs. (2009) 1:339–47. doi: 10.4161/mabs.1.4.8755
16. Labrijn AF, Janmaat ML, Reichert JM, and Parren P. Bispecific antibodies: a mechanistic review of the pipeline. Nat Rev Drug Discovery. (2019) 18:585–608. doi: 10.1038/s41573-019-0028-1
17. Demaria O, Gauthier L, Debroas G, and Vivier E. Natural killer cell engagers in cancer immunotherapy: Next generation of immuno-oncology treatments. Eur J Immunol. (2021) 51:1934–42. doi: 10.1002/eji.202048953
18. Whalen KA, Rakhra K, Mehta NK, Steinle A, Michaelson JS, and Baeuerle PA. Engaging natural killer cells for cancer therapy via NKG2D, CD16A and other receptors. MAbs. (2023) 15:2208697. doi: 10.1080/19420862.2023.2208697
19. Carnemolla B, Borsi L, Balza E, Castellani P, Meazza R, Berndt A, et al. Enhancement of the antitumor properties of interleukin-2 by its targeted delivery to the tumor blood vessel extracellular matrix. Blood. (2002) 99:1659–65. doi: 10.1182/blood.v99.5.1659
20. Gillies SD, Lan Y, Williams S, Carr F, Forman S, Raubitschek A, et al. An anti-CD20-IL-2 immunocytokine is highly efficacious in a SCID mouse model of established human B lymphoma. Blood. (2005) 105:3972–8. doi: 10.1182/blood-2004-09-3533
21. Shi W, Liu N, and Lu H. Advancements and challenges in immunocytokines: A new arsenal against cancer. Acta Pharm Sin B. (2024) 14:4649–64. doi: 10.1016/j.apsb.2024.07.024
22. Zhou J, Xu N, Shan J, Wang Y, Fang W, Wang H, et al. MA11.04 first-in-class PD-1/IL-2 bispecific antibody IBI363 in patients with advanced non-small cell lung cancer in a phase I study. J Thorac Oncol. (2024) 19:S97. doi: 10.1016/j.jtho.2024.09.176
23. Polonskaya Z, Chang T-P, Martomo S, Luna X, Zhang Z, Ng S, et al. Abstract 5215: MOA study of KD050, an anti-PD-1/IL-15 bi-functional antibody selectively targeted PD-1 positive tumor-infiltrating lymphocytes resulted in robust anti-tumor activity and low systemic toxicity. Cancer Res. (2022) 82:5215–5. doi: 10.1158/1538-7445.Am2022-5215
24. Wang Z. ErbB receptors and cancer. Methods Mol Biol. (2017) 1652:3–35. doi: 10.1007/978-1-4939-7219-7_1
25. Wu SG and Shih JY. Management of acquired resistance to EGFR TKI-targeted therapy in advanced non-small cell lung cancer. Mol Cancer. (2018) 17:38. doi: 10.1186/s12943-018-0777-1
26. Lim SM, Syn NL, Cho BC, and Soo RA. Acquired resistance to EGFR targeted therapy in non-small cell lung cancer: Mechanisms and therapeutic strategies. Cancer Treat Rev. (2018) 65:1–10. doi: 10.1016/j.ctrv.2018.02.006
27. Shi Y, Au JS, Thongprasert S, Srinivasan S, Tsai CM, Khoa MT, et al. A prospective, molecular epidemiology study of EGFR mutations in Asian patients with advanced non-small-cell lung cancer of adenocarcinoma histology (PIONEER). J Thorac Oncol. (2014) 9:154–62. doi: 10.1097/jto.0000000000000033
28. Rosell R, Moran T, Queralt C, Porta R, Cardenal F, Camps C, et al. Screening for epidermal growth factor receptor mutations in lung cancer. N Engl J Med. (2009) 361:958–67. doi: 10.1056/NEJMoa0904554
29. Barlesi F, Mazieres J, Merlio JP, Debieuvre D, Mosser J, Lena H, et al. Routine molecular profiling of patients with advanced non-small-cell lung cancer: results of a 1-year nationwide programme of the French Cooperative Thoracic Intergroup (IFCT). Lancet. (2016) 387:1415–26. doi: 10.1016/s0140-6736(16)00004-0
30. Wu Q, Qian W, Sun X, and Jiang S. Small-molecule inhibitors, immune checkpoint inhibitors, and more: FDA-approved novel therapeutic drugs for solid tumors from 1991 to 2021. J Hematol Oncol. (2022) 15:143. doi: 10.1186/s13045-022-01362-9
31. Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. (2007) 316:1039–43. doi: 10.1126/science.1141478
32. Yu HA, Arcila ME, Rekhtman N, Sima CS, Zakowski MF, Pao W, et al. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin Cancer Res. (2013) 19:2240–7. doi: 10.1158/1078-0432.Ccr-12-2246
33. Sequist LV, Waltman BA, Dias-Santagata D, Digumarthy S, Turke AB, Fidias P, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. (2011) 3:75ra26. doi: 10.1126/scitranslmed.3002003
34. Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PloS Med. (2005) 2:e73. doi: 10.1371/journal.pmed.0020073
35. Yano S, Yamada T, Takeuchi S, Tachibana K, Minami Y, Yatabe Y, et al. Hepatocyte growth factor expression in EGFR mutant lung cancer with intrinsic and acquired resistance to tyrosine kinase inhibitors in a Japanese cohort. J Thorac Oncol. (2011) 6:2011–7. doi: 10.1097/JTO.0b013e31823ab0dd
36. Turke AB, Zejnullahu K, Wu YL, Song Y, Dias-Santagata D, Lifshits E, et al. Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer Cell. (2010) 17:77–88. doi: 10.1016/j.ccr.2009.11.022
37. Neijssen J, Cardoso RMF, Chevalier KM, Wiegman L, Valerius T, Anderson GM, et al. Discovery of amivantamab (JNJ-61186372), a bispecific antibody targeting EGFR and MET. J Biol Chem. (2021) 296:100641. doi: 10.1016/j.jbc.2021.100641
38. Vijayaraghavan S, Lipfert L, Chevalier K, Bushey BS, Henley B, Lenhart R, et al. Amivantamab (JNJ-61186372), an fc enhanced EGFR/cMet bispecific antibody, induces receptor downmodulation and antitumor activity by monocyte/macrophage trogocytosis. Mol Cancer Ther. (2020) 19:2044–56. doi: 10.1158/1535-7163.Mct-20-0071
39. Park K, Haura EB, Leighl NB, Mitchell P, Shu CA, Girard N, et al. Amivantamab in EGFR exon 20 insertion-mutated non-small-cell lung cancer progressing on platinum chemotherapy: initial results from the CHRYSALIS phase I study. J Clin Oncol. (2021) 39:3391–402. doi: 10.1200/jco.21.00662
40. Vyse S and Huang PH. Amivantamab for the treatment of EGFR exon 20 insertion mutant non-small cell lung cancer. Expert Rev Anticancer Ther. (2022) 22:3–16. doi: 10.1080/14737140.2022.2016397
41. US Food & Drug Administration. FDA grants accelerated approval to amivantamab-vmjw for metastatic non-small cell lung cancer [media release]. (2021). Available at: https://www.fda.gov/.
42. Zhou C, Tang KJ, Cho BC, Liu B, Paz-Ares L, Cheng S, et al. Amivantamab plus chemotherapy in NSCLC with EGFR exon 20 insertions. N Engl J Med. (2023) 389:2039–51. doi: 10.1056/NEJMoa2306441
43. US Food & Drug Administration. FDA approves amivantamab-vmjw for EGFR exon 20 insertion-mutated non-small cell lung cancer indications [media release]. (2024). Available at: https://www.fda.gov/.
44. Cho BC, Felip E, Spira AI, Girard N, Lee JS, Lee SH, et al. LBA14 Amivantamab plus lazertinib vs osimertinib as first-line treatment in patients with EGFR-mutated, advanced non-small cell lung cancer (NSCLC): Primary results from MARIPOSA, a phase III, global, randomized, controlled trial. Ann Oncol. (2023) 34:S1306. doi: 10.1016/j.annonc.2023.10.062
45. Cho BC, Lu S, Felip E, Spira AI, Girard N, Lee JS, et al. Amivantamab plus lazertinib in previously untreated EGFR-mutated advanced NSCLC. N Engl J Med. (2024). doi: 10.1056/NEJMoa2403614
46. Leighl N, Cho BC, Hiret S, Han JY, Lee KH, Llacer Perez C, et al. OA21.04 amivantamab in patients with advanced NSCLC and MET exon 14 skipping mutation: results from the CHRYSALIS study. J Thorac Oncol. (2023) 18:S93–4. doi: 10.1016/j.jtho.2023.09.105
47. Leighl NB, Akamatsu H, Lim SM, Cheng Y, Minchom AR, Marmarelis ME, et al. Subcutaneous versus intravenous amivantamab, both in combination with lazertinib, in refractory epidermal growth factor receptor-mutated non-small cell lung cancer: primary results from the phase III PALOMA-3 study. J Clin Oncol. (2024) 42:3593–605. doi: 10.1200/jco.24.01001
48. Qing Z, Gabrail N, Uprety D, Rotow J, Han B, Jänne PA, et al. 22P EMB-01: An EGFR-cMET bispecific antibody, in advanced/metastatic solid tumors phase I results. Ann Oncol. (2022) 33:S39–40. doi: 10.1016/j.annonc.2022.02.031
49. Geuijen C, d. Matteo M, Gallenne T, Cafarello ST, Nijhuis R, Visser T, et al. Abstract LB-C07: Preclinical evaluation of MCLA-129: A bispecific antibody targeting c-MET and EGFR. Mol Cancer Ther. (2019) 18:LB–C07-LB-C07. doi: 10.1158/1535-7163.Targ-19-lb-c07
50. Wang J, Zhong J, Wu L, Chen B, Wang Z-H, Yao Y, et al. Ding: Efficacy and safety of MCLA-129, an EGFR/c-MET bispecific antibody, in advanced non-small cell lung cancer (NSCLC). J Clin Oncol. (2024) 42:8604–4. doi: 10.1200/JCO.2024.42.16_suppl.8604
51. Cappuzzo F, Moreno Garcia V, Ou SHI, Brandão M, Sanmamed MF, Helissey C, et al. 516MO Efficacy and safety of MCLA-129, an anti-EGFR/c-MET bispecific antibody, combined with osimertinib, as first-line therapy or after progression on osimertinib in non-small cell lung cancer (NSCLC). Ann Oncol. (2023) 34:S1671. doi: 10.1016/j.annonc.2023.10.595
52. Pillai RN, Behera M, Berry LD, Rossi MR, Kris MG, Johnson BE, et al. HER2 mutations in lung adenocarcinomas: A report from the Lung Cancer Mutation Consortium. Cancer. (2017) 123:4099–105. doi: 10.1002/cncr.30869
53. Kim EK, Kim KA, Lee CY, and Shim HS. The frequency and clinical impact of HER2 alterations in lung adenocarcinoma. PloS One. (2017) 12:e0171280. doi: 10.1371/journal.pone.0171280
54. Tomizawa K, Suda K, Onozato R, Kosaka T, Endoh H, Sekido Y, et al. Prognostic and predictive implications of HER2/ERBB2/neu gene mutations in lung cancers. Lung Cancer. (2011) 74:139–44. doi: 10.1016/j.lungcan.2011.01.014
55. Li BT, Ross DS, Aisner DL, Chaft JE, Hsu M, Kako SL, et al. HER2 amplification and HER2 mutation are distinct molecular targets in lung cancers. J Thorac Oncol. (2016) 11:414–9. doi: 10.1016/j.jtho.2015.10.025
56. Trinder A, Ding K, and Zhang J. The therapeutic significance of HER3 in non-small cell lung cancer (NSCLC): A review study. Curr Med Chem. (2024). doi: 10.2174/0109298673269305231115102542
57. Lee-Hoeflich ST, Crocker L, Yao E, Pham T, Munroe X, Hoeflich KP, et al. A central role for HER3 in HER2-amplified breast cancer: implications for targeted therapy. Cancer Res. (2008) 68:5878–87. doi: 10.1158/0008-5472.Can-08-0380
58. Hsieh AC and Moasser MM. Targeting HER proteins in cancer therapy and the role of the non-target HER3. Br J Cancer. (2007) 97:453–7. doi: 10.1038/sj.bjc.6603910
59. Wallasch C, Weiss FU, Niederfellner G, Jallal B, Issing W, and Ullrich A. Heregulin-dependent regulation of HER2/neu oncogenic signaling by heterodimerization with HER3. EMBO J. (1995) 14:4267–75. doi: 10.1002/j.1460-2075.1995.tb00101.x
60. Haikala HM and Jänne PA. Thirty years of HER3: from basic biology to therapeutic interventions. Clin Cancer Res. (2021) 27:3528–39. doi: 10.1158/1078-0432.Ccr-20-4465
61. Dimou A and Camidge DR. Detection of NRG1 fusions in solid tumors: rare gold? Clin Cancer Res. (2019) 25:4865–7. doi: 10.1158/1078-0432.Ccr-19-1219
62. Jonna S, Feldman RA, Swensen J, Gatalica Z, Korn WM, Borghaei H, et al. Detection of NRG1 gene fusions in solid tumors. Clin Cancer Res. (2019) 25:4966–72. doi: 10.1158/1078-0432.Ccr-19-0160
63. Drilon A, Duruisseaux M, Han JY, Ito M, Falcon C, Yang SR, et al. Clinicopathologic features and response to therapy of NRG1 fusion-driven lung cancers: the eNRGy1 global multicenter registry. J Clin Oncol. (2021) 39:2791–802. doi: 10.1200/jco.20.03307
64. Geuijen CAW, De Nardis C, Maussang D, Rovers E, Gallenne T, Hendriks LJA, et al. Unbiased combinatorial screening identifies a bispecific igG1 that potently inhibits HER3 signaling via HER2-guided ligand blockade. Cancer Cell. (2018) 33:922–936.e10. doi: 10.1016/j.ccell.2018.04.003
65. US Food & Drug Administration. FDA grants accelerated approval to zenocutuzumab-zbco for non-small cell lung cancer and pancreatic adenocarcinoma [media release]. (2024). Available at: https://www.fda.gov/.
66. Schram A, Goto K, Kim DW, Hollebecque A, Rha SY, Nishino K, et al. 1315MO Durable efficacy of zenocutuzumab, a HER2 x HER3 bispecific antibody, in advanced NRG1 fusion-positive (NRG1+) non-small cell lung cancer (NSCLC). Ann Oncol. (2023) 34:S756–7. doi: 10.1016/j.annonc.2023.09.2349
67. Yonesaka K, Tanizaki J, Maenishi O, Haratani K, Kawakami H, Tanaka K, et al. HER3 augmentation via blockade of EGFR/AKT signaling enhances anticancer activity of HER3-targeting patritumab deruxtecan in EGFR-mutated non-small cell lung cancer. Clin Cancer Res. (2022) 28:390–403. doi: 10.1158/1078-0432.Ccr-21-3359
68. Engelman JA, Jänne PA, Mermel C, Pearlberg J, Mukohara T, Fleet C, et al. ErbB-3 mediates phosphoinositide 3-kinase activity in gefitinib-sensitive non-small cell lung cancer cell lines. Proc Natl Acad Sci U.S.A. (2005) 102:3788–93. doi: 10.1073/pnas.0409773102
69. Goulet DR, Tsai TI, Khalili J, Mak NSA, Zhu H, and Zhu Y. SPECIFICITY ENHANCED BISPECIFIC ANTIBODY (SEBA) WO 2022/061255 A1. (2022).
70. Zhao Y, Zhang L, Fang W, Yang Y, Huang Y, Zou W, et al. SI-B001 plus chemotherapy in patients with locally advanced or metastatic EGFR/ALK wild-type non-small cell lung cancer: A phase II, multicenter, open-label study. J Clin Oncol. (2023) 41:9025–5. doi: 10.1200/JCO.2023.41.16_suppl.9025
71. Kubli SP, Berger T, Araujo DV, Siu LL, and Mak TW. Beyond immune checkpoint blockade: emerging immunological strategies. Nat Rev Drug Discovery. (2021) 20:899–919. doi: 10.1038/s41573-021-00155-y
72. Darvin P, Toor SM, Sasidharan Nair V, and Elkord E. Immune checkpoint inhibitors: recent progress and potential biomarkers. Exp Mol Med. (2018) 50:1–11. doi: 10.1038/s12276-018-0191-1
73. Schoenfeld AJ and Hellmann MD. Acquired resistance to immune checkpoint inhibitors. Cancer Cell. (2020) 37:443–55. doi: 10.1016/j.ccell.2020.03.017
74. Buchbinder EI and Desai A. CTLA-4 and PD-1 pathways: similarities, differences, and implications of their inhibition. Am J Clin Oncol. (2016) 39:98–106. doi: 10.1097/coc.0000000000000239
75. Valsecchi ME. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. (2015) 373:1270. doi: 10.1056/NEJMc1509660
76. Hellmann MD, Paz-Ares L, Bernabe Caro R, Zurawski B, Kim SW, Carcereny Costa E, et al. Nivolumab plus ipilimumab in advanced non-small-cell lung cancer. N Engl J Med. (2019) 381:2020–31. doi: 10.1056/NEJMoa1910231
77. Motzer RJ, Tannir NM, McDermott DF, Arén Frontera O, Melichar B, Choueiri TK, et al. Nivolumab plus Ipilimumab versus Sunitinib in Advanced Renal-Cell Carcinoma. N Engl J Med. (2018) 378:1277–90. doi: 10.1056/NEJMoa1712126
78. Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Rutkowski P, Lao CD, et al. Five-year survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med. (2019) 381:1535–46. doi: 10.1056/NEJMoa1910836
79. Reck M, Borghaei H, and O’Byrne KJ. Nivolumab plus ipilimumab in non-small-cell lung cancer. Future Oncol. (2019) 15:2287–302. doi: 10.2217/fon-2019-0031
80. Dovedi SJ, Elder MJ, Yang C, Sitnikova SI, Irving L, Hansen A, et al. Design and efficacy of a monovalent bispecific PD-1/CTLA4 antibody that enhances CTLA4 blockade on PD-1(+) activated T cells. Cancer Discovery. (2021) 11:1100–17. doi: 10.1158/2159-8290.Cd-20-1445
81. Huang Z, Pang X, Zhong T, Chen N, He X, Xia D, et al. 289 Cadonilimab, an anti-PD1/CTLA4 bi-specific antibody with Fc effector null backbone. J ImmunoTherapy Cancer. (2021) 9:A313–4. doi: 10.1136/jitc-2021-SITC2021.289
82. Pang X, Huang Z, Zhong T, Zhang P, Wang ZM, Xia M, et al. Cadonilimab, a tetravalent PD-1/CTLA-4 bispecific antibody with trans-binding and enhanced target binding avidity. MAbs. (2023) 15:2180794. doi: 10.1080/19420862.2023.2180794
84. Chen B, Yao W, Li X, Lin G, Chu Q, Liu H, et al. A phase Ib/II study of cadonilimab (PD-1/CTLA-4 bispecific antibody) plus anlotinib as first-line treatment in patients with advanced non-small cell lung cancer. Br J Cancer. (2024) 130:450–6. doi: 10.1038/s41416-023-02519-0
85. Han B and Zhang Y. 647P A phase Ib/II study of second-line cadonilimab, anlotinib and docetaxel in patients with checkpoint inhibitor (CPI)-experienced advanced non-small cell lung cancer. Ann Oncol. (2024) 35:S1646. doi: 10.1016/j.annonc.2024.10.680
86. Insa A, Martín-Martorell P, Di Liello R, Fasano M, Martini G, Napolitano S, et al. Which treatment after first line therapy in NSCLC patients without genetic alterations in the era of immunotherapy? Crit Rev Oncol Hematol. (2022) 169:103538. doi: 10.1016/j.critrevonc.2021.103538
87. Neal J, Pavlakis N, Kim SW, Goto Y, Lim SM, Mountzios G, et al. CONTACT-01: A randomized phase III trial of atezolizumab + Cabozantinib versus docetaxel for metastatic non-small cell lung cancer after a checkpoint inhibitor and chemotherapy. J Clin Oncol. (2024) 42:2393–403. doi: 10.1200/jco.23.02166
88. Paz-Ares L, Goto Y, Wan-Teck Lim D, Halmos B, Chul Cho B, Cobo M, et al. Canakinumab in combination with docetaxel compared with docetaxel alone for the treatment of advanced non-small cell lung cancer following platinum-based doublet chemotherapy and immunotherapy (CANOPY-2): A multicenter, randomized, double-blind, phase 3 trial. Lung Cancer. (2024) 189:107451. doi: 10.1016/j.lungcan.2023.107451
89. Borghaei H, de Marinis F, Dumoulin D, Reynolds C, Theelen W, Percent I, et al. SAPPHIRE: phase III study of sitravatinib plus nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. Ann Oncol. (2024) 35:66–76. doi: 10.1016/j.annonc.2023.10.004
90. Wang L and Su C. EP.11D.01 A phase II study of cadonilimab plus chemotherapy as first-line treatment for PD-L1-negative advanced non-small cell lung cancer: lungCadX. J Thorac Oncol. (2024) 19:S614. doi: 10.1016/j.jtho.2024.09.1152
91. Ahn MJ, Kim SW, Costa EC, Rodríguez LM, Oliveira J, Insa Molla MA, et al. LBA56 MEDI5752 or pembrolizumab (P) plus carboplatin/pemetrexed (CP) in treatment-na&xef;ve (1L) non-small cell lung cancer (NSCLC): A phase Ib/II trial. Ann Oncol. (2022) 33:S1423. doi: 10.1016/j.annonc.2022.08.058
92. Zhao Y, Ma Y, Fan Y, Zhou J, Yang N, Yu Q, et al. A multicenter, open-label phase Ib/II study of cadonilimab (anti PD-1 and CTLA-4 bispecific antibody) monotherapy in previously treated advanced non-small-cell lung cancer (AK104-202 study). Lung Cancer. (2023) 184:107355. doi: 10.1016/j.lungcan.2023.107355
93. Wu L, Chen B, Yao W, Li X, Xiao Z, Liu H, et al. 1300P A phase Ib/II trial of AK104 (PD-1/CTLA-4 bispecific antibody) in combination with anlotinib in advanced NSCLC. Ann Oncol. (2021) 32:S1006. doi: 10.1016/j.annonc.2021.08.1902
94. Wang Y, Li Y, Liu J, S.-x. Luo Q, Zou W, Wang Z, et al. Shen: SI-B003 (PD-1/CTLA-4) in patients with advanced solid tumors: A phase I study. J Clin Oncol. (2023) 41:e14668–8. doi: 10.1200/JCO.2023.41.16_suppl.e14668
95. Zhou C, Xiong A, Fang J, Li X, Xie Q, Yu Q, et al. 1330P Updated results of the efficacy and safety of KN046 (a bispecific anti-PD-L1/CTLA-4) in patients with metastatic non-small cell lung cancer (NSCLC) who failed prior EGFR-TKI(s). Ann Oncol. (2023) 34:S767–8. doi: 10.1016/j.annonc.2023.09.2363
96. Zhao Y, Chen G, Li X, Wu J, Chang B, Hu S, et al. KN046, a bispecific antibody against PD-L1 and CTLA-4, plus chemotherapy as first-line treatment for metastatic NSCLC: A multicenter phase 2 trial. Cell Rep Medicine 5(3). (2024). doi: 10.1016/j.xcrm.2024.101470
97. Zhou C, Xiong A, Li X, Fan Y, Zhuang W, Yu Q, et al. 1459P Preliminary efficacy and safety of KN046 (a bispecific anti-PD-L1/CTLA-4) in patients with metastatic non-small cell lung cancer who previously treated with immune checkpoint inhibitor(s). Ann Oncol. (2023) 34:S829. doi: 10.1016/j.annonc.2023.09.2490
98. Zhang L, Zhao Y, Meng X, Huang Y, Fang W, Yang Y, et al. 133P The efficacy and safety of KN046 combined with axitinib for previously untreated and checkpoint inhibitor treated advanced non-small cell lung cancer: A single-arm, open-label, multicenter phase II clinical trial. Immuno-Oncology Technol. (2024) 24:100762. doi: 10.1016/j.iotech.2024.100762
99. Ruan D-Y, Wei X-L, Liu F-R, Hu X-C, Zhang J, Ji D-M, et al. The first-in-class bispecific antibody IBI318 (LY3434172) targeting PD-1 and PD-L1 in patients with advanced tumors: a phase Ia/Ib study. J Hematol Oncol. (2024) 17:118. doi: 10.1186/s13045-024-01644-4
100. Hiltermann TJN, Izumi H, Cho BC, Cunha S, Danchaivijitr P, Felip E, et al. OA11.03 efficacy and safety of rilvegostomig, an anti-PD-1/TIGIT bispecific, for CPI-naïve metastatic NSCLC with PD-L1 1-49% or ≥50%. J Thorac Oncol. (2024) 19:S33. doi: 10.1016/j.jtho.2024.09.061
101. Xue J, Zhang Y, Hu X, Zhao W, Sun Y, Li Q, et al. Phase I/II safety and preliminary efficacy of PM1022, a bispecific antibody targeting PD-L1 and TIGIT, in patients with advanced solid tumors. J Clin Oncol. (2024) 42:e14694–4. doi: 10.1200/JCO.2024.42.16_suppl.e14694
102. Luke JJ, Patel MR, Blumenschein GR, Hamilton E, Chmielowski B, Ulahannan SV, et al. The PD-1- and LAG-3-targeting bispecific molecule tebotelimab in solid tumors and hematologic cancers: a phase 1 trial. Nat Med. (2023) 29:2814–24. doi: 10.1038/s41591-023-02593-0
103. Zhang L, Fang W, Zhao Y, Luo Y, Yang R, Huang Y, et al. Ivonescimab combined with chemotherapy in patients with EGFR-mutant non-squamous non-small cell lung cancer who progressed on EGFR tyrosine-kinase inhibitor treatment (HARMONi-A): A randomized, double-blind, multi-center, phase 3 trial. J Clin Oncol. (2024) 42:8508–8. doi: 10.1200/JCO.2024.42.16_suppl.8508
104. Zhou JCC, Wu L, Wang L, Xiong A, Yao B, Zhong H, et al. Phase 3 Study of Ivonescimab (AK112) vs. Pembrolizumab as First-line Treatment for PD-L1 positive Advanced NSCLC: HARMONi-2. In WCLC. (2024). doi: 10.1016/j.jtho.2024.09.012
105. Wu C, Lv D, Cui J, Wang Z, Zhao H, Duan H, et al. A phase Ib/IIa trial to evaluate the safety and efficacy of PM8002, a bispecific antibody targeting PD-L1 and VEGF-A, as a monotherapy in patients with advanced NSCLC. J Clin Oncol. (2024) 42:8533–3. doi: 10.1200/JCO.2024.42.16_suppl.8533
106. Cheng Y, Qin Z, Meng X, Xu F, Wang Y, Yao Y, et al. 1992P A phase II safety and efficacy study of PM8002 (anti-PD-L1 x VEGF-A bispecific) combined with paclitaxel as a second-line therapy for small cell lung cancer (SCLC). Ann Oncol. (2023) 34:S1062. doi: 10.1016/j.annonc.2023.09.1223
107. Liu R, Hu X, Song Z, Zhang J, Jin J, Gao S, et al. IMM2510, an anti-PD-L1/VEGF bispecific antibody fusion protein, in patients with advanced solid tumors: A phase I dose-escalation study. J Clin Oncology 42(16_suppl) e14506-e14506. (2024). doi: 10.1200/JCO.2024.42.16_suppl.e14506
108. Feng J, Shi M, Chen J, Li K, Li X, Sun M, et al. 511MO A phase I study of SHR-1701, a bifunctional fusion protein targeting PD-L1 and TGF-&x3b2;, in patients with advanced non-small cell lung cancer (NSCLC). Ann Oncol. (2023) 34:S1667. doi: 10.1016/j.annonc.2023.10.590
109. Zhou Q, Pan Y, Yang X, Zhao Y, Han G, Pang Q, et al. Neoadjuvant SHR-1701 with or without chemotherapy in unresectable stage III non-small-cell lung cancer: A proof-of-concept, phase 2 trial. Cancer Cell. (2024) 42(7):1258–67.e2. doi: 10.1016/j.ccell.2024.05.024
110. Ahn MJ, Cho BC, Felip E, Korantzis I, Ohashi K, Majem M, et al. Tarlatamab for patients with previously treated small-cell lung cancer. N Engl J Med. (2023) 389:2063–75. doi: 10.1056/NEJMoa2307980
111. Dowlati A, Hummel H-D, Champiat S, Olmedo ME, Boyer M, He K, et al. Sustained clinical benefit and intracranial activity of tarlatamab in previously treated small cell lung cancer: deLLphi-300 trial update. J Clin Oncol. 42(29):3392–9. doi: 10.1200/jco.24.00553
112. Wermke M, Felip E, Kuboki Y, Morgensztern D, Sayehli C, Sanmamed MF, et al. First-in-human dose-escalation trial of BI 764532, a delta-like ligand 3 (DLL3)/CD3 IgG-like T-cell engager in patients (pts) with DLL3-positive (DLL3+) small-cell lung cancer (SCLC) and neuroendocrine carcinoma (NEC). J Clin Oncol. (2023) 41:8502–2. doi: 10.1200/JCO.2023.41.16_suppl.8502
113. Beltran H, Johnson ML, Jain P, Schenk EL, Sanborn RE, Thompson JR, et al. Updated results from a phase 1/2 study of HPN328, a tri-specific, half-life (T1/2) extended DLL3-targeting T-cell engager in patients (pts) with small cell lung cancer (SCLC) and other neuroendocrine cancers (NEC). J Clin Oncol. (2024) 42:8090–0. doi: 10.1200/JCO.2024.42.16_suppl.8090
114. Choudhury NJ, Beltran H, Johnson ML, Schenk EL, Sanborn RE, Thompson JR, et al. OA10.06 impact of brain metastases on safety and efficacy of MK-6070, a DLL3-targeting T cell engager, in small cell lung cancer. J Thorac Oncol. (2024) 19:S32–3. doi: 10.1016/j.jtho.2024.09.060
115. Yingling J. Interim clinical data for EGFR-TRACTr JANX008 in solid tumors as of February 12, 2024. In: Janux Ther News. (2024).
116. Muik A, Garralda E, Altintas I, Gieseke F, Geva R, Ben-Ami E, et al. Preclinical characterization and phase I trial results of a bispecific antibody targeting PD-L1 and 4-1BB (GEN1046) in patients with advanced refractory solid tumors. Cancer Discovery. (2022) 12:1248–65. doi: 10.1158/2159-8290.Cd-21-1345
117. Aerts J, Paz-Ares LG, Helissey C, Cappuzzo F, Quere G, Kowalski D, et al. Acasunlimab (DuoBody-PD-L1x4-1BB) alone or in combination with pembrolizumab (pembro) in patients (pts) with previously treated metastatic non-small cell lung cancer (mNSCLC): Initial results of a randomized, open-label, phase 2 trial. J Clin Oncol. (2024) 42:2533–3. doi: 10.1200/JCO.2024.42.16_suppl.2533
118. Garralda E, Oberoi A, de Velasco G, Victoria I, Pesantez D, Eguren Santamaría I, et al. First-in-human study (FIH) of FS222, a next-generation tetravalent PD-L1/CD137 bispecific antibody: Safety, pharmacodynamics (PD), and antitumor activity in patients (pts) with advanced solid tumors including PD-1 refractory melanoma. J Clin Oncol. (2024) 42:2505–5. doi: 10.1200/JCO.2024.42.16_suppl.2505
119. El-Khoueiry AB, Rivas D, Lee S-H, Thomas JS, Kim YJ, Cervantes A, et al. Leveraging innate immunity with AFM24, a novel CD16A and epidermal growth factor receptor (EGFR) bispecific innate cell engager: Interim results for the non-small cell lung cancer (NSCLC) cohort. J Clin Oncol. (2023) 41:2533–3. doi: 10.1200/JCO.2023.41.16_suppl.2533
120. Kim HR, Saavedra O, Cervantes A, Lugowska IA, Oberoi A, El-Khoueiry AB, et al. Preliminary results from the phase 2 study of AFM24 in combination with atezolizumab in patients with EGFR wild-type (EGFR-WT) non-small cell lung cancer (NSCLC). J Clin Oncol. (2024) 42:2522–2. doi: 10.1200/JCO.2024.42.16_suppl.2522
121. Naing A, McKean M, Rosen LS, Sommerhalder D, Shaik NM, Wang IM, et al. First-in-human phase I study to evaluate safety, tolerability, pharmacokinetics, pharmacodynamics, immunogenicity, and antitumor activity of PF-07209960 in patients with advanced or metastatic solid tumors. ESMO Open. (2025) 10:104291. doi: 10.1016/j.esmoop.2025.104291
122. Pratumchai I, Bernardo M, Marquardt KL, Zhu C, Menas F, Carrio R, et al. Abstract 4063: SAR445877, an anti-PD-1 antibody-IL-15 mutein fusion protein restores function to exhausted T cells. Cancer Res. (2024) 84:4063–3. doi: 10.1158/1538-7445.Am2024-4063
123. Chen J, Shen Z, Jiang X, Huang Z, Wu C, Jiang D, et al. Preclinical evaluation of IAP0971, a novel immunocytokine that binds specifically to PD1 and fuses IL15/IL15Rα complex. Antib Ther. (2023) 6:38–48. doi: 10.1093/abt/tbac031
124. Shum E, Reilley M, Najjar Y, Daud A, Thompson J, Baranda J, et al. 523 Preliminary clinical experience with XmAb20717, a PD-1 x CTLA-4 bispecific antibody, in patients with advanced solid tumors. J Immuno Therapy of Cancer. (2021) 8(3):A553–3. doi: 10.1136/jitc-2021-SITC2021.523%
125. Burton EM and Tawbi HA. Bispecific antibodies to PD-1 and CTLA4: doubling down on T cells to decouple efficacy from toxicity. Cancer Discovery. (2021) 11:1008–10. doi: 10.1158/2159-8290.CD-21-0257%
126. Berezhnoy A, Sumrow BJ, Stahl K, Shah K, Liu D, Li J, et al. Development and preliminary clinical activity of PD-1-guided CTLA-4 blocking bispecific DART molecule. Cell Rep Med. (2020) 1:100163. doi: 10.1016/j.xcrm.2020.100163
127. Coward J, Ganju V, Behzadigohar R, Kwong K, Xu J, Van H, et al. Preliminary safety, efficacy, and pharmacokinetics (PK) results of KN046 (bispecific anti-PD-L1/CTLA4) from a first-in-human study in subjects with advanced solid tumors. JCO. (2019) 37:2554–4. doi: 10.1200/JCO.2019.37.15_suppl.2554
128. Kotanides H, Li Y, Malabunga M, Carpenito C, Eastman SW, Shen Y, et al. Bispecific targeting of PD-1 and PD-L1 enhances T-cell activation and antitumor immunity. Cancer Immunol Res. (2020) 8:1300–10. doi: 10.1158/2326-6066.Cir-20-0304
129. Tang W, Chen J, Ji T, and Cong X. TIGIT, a novel immune checkpoint therapy for melanoma. Cell Death Dis. (2023) 14:466. doi: 10.1038/s41419-023-05961-3
130. Rousseau A, Parisi C, and Barlesi F. Anti-TIGIT therapies for solid tumors: a systematic review. ESMO Open. (2023) 8:101184. doi: 10.1016/j.esmoop.2023.101184
131. Chu X, Tian W, Wang Z, Zhang J, and Zhou R. Co-inhibition of TIGIT and PD-1/PD-L1 in cancer immunotherapy: mechanisms and clinical trials. Mol Cancer. (2023) 22:93. doi: 10.1186/s12943-023-01800-3
132. Banta KL, Xu X, Chitre AS, Au-Yeung A, Takahashi C, O’Gorman WE, et al. Mechanistic convergence of the TIGIT and PD-1 inhibitory pathways necessitates co-blockade to optimize anti-tumor CD8(+) T cell responses. Immunity. (2022) 55:512–526.e9. doi: 10.1016/j.immuni.2022.02.005
133. Niu J, Maurice-Dror C, Lee DH, Kim DW, Nagrial A, Voskoboynik M, et al. First-in-human phase 1 study of the anti-TIGIT antibody vibostolimab as monotherapy or with pembrolizumab for advanced solid tumors, including non-small-cell lung cancer(☆). Ann Oncol. (2022) 33:169–80. doi: 10.1016/j.annonc.2021.11.002
134. Bendell JC, Bedard P, Bang Y-J, LoRusso P, Hodi S, Gordon M, et al. Abstract CT302: Phase Ia/Ib dose-escalation study of the anti-TIGIT antibody tiragolumab as a single agent and in combination with atezolizumab in patients with advanced solid tumors. Cancer Res. (2020) 80:CT302–2. doi: 10.1158/1538-7445.Am2020-ct302
135. Cho BC, Abreu DR, Hussein M, Cobo M, Patel AJ, Secen N, et al. Tiragolumab plus atezolizumab versus placebo plus atezolizumab as a first-line treatment for PD-L1-selected non-small-cell lung cancer (CITYSCAPE): primary and follow-up analyses of a randomised, double-blind, phase 2 study. Lancet Oncol. (2022) 23:781–92. doi: 10.1016/s1470-2045(22)00226-1
136. Aggarwal V, Workman CJ, and Vignali DAA. LAG-3 as the third checkpoint inhibitor. Nat Immunol. (2023) 24:1415–22. doi: 10.1038/s41590-023-01569-z
137. Mariuzza RA, Shahid S, and Karade SS. The immune checkpoint receptor LAG3: Structure, function, and target for cancer immunotherapy. J Biol Chem. (2024) 300. doi: 10.1016/j.jbc.2024.107241
138. Andrews LP, Butler SC, Cui J, Cillo AR, Cardello C, Liu C, et al. LAG-3 and PD-1 synergize on CD8(+) T cells to drive T cell exhaustion and hinder autocrine IFN-γ-dependent anti-tumor immunity. Cell. (2024) 187:4355–4372.e22. doi: 10.1016/j.cell.2024.07.016
139. US Food & Drug Administration. FDA approves Opdualag for unresectable or metastatic melanoma [media release] (2022). Available at: https://www.fda.gov/.
140. Girard N, Burotto M, Paz-Ares LG, Reck M, Schenker M, Lingua A, et al. LBA53 Nivolumab (NIVO) plus relatlimab with platinum-doublet chemotherapy (PDCT) vs NIVO + PDCT as first-line (1L) treatment (tx) for stage IV or recurrent NSCLC: Results from the randomized phase II RELATIVITY-104 study. Ann Oncol. (2024) 35:S1243–4. doi: 10.1016/j.annonc.2024.08.2295
141. De Giglio A, Di Federico A, Nuvola G, Deiana C, and Gelsomino F. The landscape of immunotherapy in advanced NSCLC: driving beyond PD-1/PD-L1 inhibitors (CTLA-4, LAG3, IDO, OX40, TIGIT, vaccines). Curr Oncol Rep. (2021) 23:126. doi: 10.1007/s11912-021-01124-9
142. Sun Q, Hong Z, Zhang C, Wang L, Han Z, and Ma D. Immune checkpoint therapy for solid tumours: clinical dilemmas and future trends. Signal Transduct Target Ther. (2023) 8:320. doi: 10.1038/s41392-023-01522-4
143. Qin S, Xu L, Yi M, Yu S, Wu K, and Luo S. Novel immune checkpoint targets: moving beyond PD-1 and CTLA-4. Mol Cancer. (2019) 18:155. doi: 10.1186/s12943-019-1091-2
144. Anderson NM and Simon MC. The tumor microenvironment. Curr Biol 30(16) R921-r925. (2020). doi: 10.1016/j.cub.2020.06.081
145. Eguchi R and Wakabayashi I. HDGF enhances VEGF−dependent angiogenesis and FGF−2 is a VEGF−independent angiogenic factor in non−small cell lung cancer. Oncol Rep. (2020) 44:14–28. doi: 10.3892/or.2020.7580
146. Jung WY, Min KW, and Oh YH. Increased VEGF-A in solid type of lung adenocarcinoma reduces the patients’ survival. Sci Rep. (2021) 11:1321. doi: 10.1038/s41598-020-79907-6
147. Bellone M and Calcinotto A. Ways to enhance lymphocyte trafficking into tumors and fitness of tumor infiltrating lymphocytes. Front Oncol. (2013) 3:231. doi: 10.3389/fonc.2013.00231
148. Voron T, Colussi O, Marcheteau E, Pernot S, Nizard M, Pointet AL, et al. VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. J Exp Med. (2015) 212:139–48. doi: 10.1084/jem.20140559
149. Martino EC, Misso G, Pastina P, Costantini S, Vanni F, Gandolfo C, et al. Immune-modulating effects of bevacizumab in metastatic non-small-cell lung cancer patients. Cell Death Discov 2 16025. (2016). doi: 10.1038/cddiscovery.2016.25
150. de Almeida PE, Mak J, Hernandez G, Jesudason R, Herault A, Javinal V, et al. Anti-VEGF treatment enhances CD8(+) T-cell antitumor activity by amplifying hypoxia. Cancer Immunol Res. (2020) 8:806–18. doi: 10.1158/2326-6066.Cir-19-0360
151. Kudo M. Scientific rationale for combined immunotherapy with PD-1/PD-L1 antibodies and VEGF inhibitors in advanced hepatocellular carcinoma. Cancers (Basel). (2020) 12. doi: 10.3390/cancers12051089
152. Georganaki M, van Hooren L, and Dimberg A. Vascular targeting to increase the efficiency of immune checkpoint blockade in cancer. Front Immunol. (2018) 9:3081. doi: 10.3389/fimmu.2018.03081
153. Meder L, Schuldt P, Thelen M, Schmitt A, Dietlein F, Klein S, et al. Combined VEGF and PD-L1 blockade displays synergistic treatment effects in an autochthonous mouse model of small cell lung cancer. Cancer Res. (2018) 78:4270–81. doi: 10.1158/0008-5472.Can-17-2176
154. Socinski MA, Jotte RM, Cappuzzo F, Orlandi F, Stroyakovskiy D, Nogami N, et al. Atezolizumab for first-line treatment of metastatic nonsquamous NSCLC. N Engl J Med. (2018) 378:2288–301. doi: 10.1056/NEJMoa1716948
155. US Food & Drug Administration. FDA approves atezolizumabas adjuvant treatment for non-small cell lung cancer [media release]. (2021). Available at: https://www.fda.gov/.
156. Hu X, Song Z, Zhang J, Jin J, Gao S, Sun Y, et al. IMM2510, an anti-PD-L1/VEGF bispecific antibody fusion protein, in patients with advanced solid tumors: A phase I dose-escalation study. J Clin Oncol. (2024) 42:e14506–e14506. doi: 10.1200/JCO.2021.39.15_suppl.2515
157. Coward J, Lemech CR, Mislang ARA, Parakh S, Underhill C, Nagrial A, et al. A phase I study of AK112, a bispecific antibody that targets PD-1 and VEGF co-expressing T cells, in patients with advanced solid tumors. J Clin Oncol. (2020) 38:TPS3155–TPS3155. doi: 10.1200/JCO.2020.38.15_suppl.TPS3155
159. Ardizzoni A, Hansen H, Dombernowsky P, Gamucci T, Kaplan S, Postmus P, et al. Topotecan, a new active drug in the second-line treatment of small-cell lung cancer: a phase II study in patients with refractory and sensitive disease. The European Organization for Research and Treatment of Cancer Early Clinical Studies Group and New Drug Development Office, and the Lung Cancer Cooperative Group. J Clin Oncol. (1997) 15:2090–6. doi: 10.1200/jco.1997.15.5.2090
160. Cheng X, Song Z, Zhang J, Jin J, Gao S, Liu R, et al. Preliminary results of a phase I dose escalation study of IMM2510, a PD-L1 and VEGF bispecific fusion protein, in patients with advanced tumors. J Clin Oncol. (2023) 41:2535–5. doi: 10.1200/JCO.2023.41.16_suppl.2535
161. David JM, Dominguez C, McCampbell KK, Gulley JL, Schlom J, and Palena C. A novel bifunctional anti-PD-L1/TGF-β Trap fusion protein (M7824) efficiently reverts mesenchymalization of human lung cancer cells. Oncoimmunology. (2017) 6:e1349589. doi: 10.1080/2162402x.2017.1349589
162. Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, et al. Powles: TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. (2018) 554:544–8. doi: 10.1038/nature25501
163. Chatterjee S, Chatterjee A, Jana S, Dey S, Roy H, Das MK, et al. Transforming growth factor beta orchestrates PD-L1 enrichment in tumor-derived exosomes and mediates CD8 T-cell dysfunction regulating early phosphorylation of TCR signalome in breast cancer. Carcinogenesis. (2021) 42:38–47. doi: 10.1093/carcin/bgaa092
164. Mortezaee K. Normalization in tumor ecosystem: Opportunities and challenges. Cell Biol Int. (2021) 45:2017–30. doi: 10.1002/cbin.11655
165. Karami Z, Mortezaee K, and Majidpoor J. Dual anti-PD-(L)1/TGF-β inhibitors in cancer immunotherapy - Updated. Int Immunopharmacol. (2023) 122:110648. doi: 10.1016/j.intimp.2023.110648
166. Tschernia NP and Gulley JL. Tumor in the crossfire: inhibiting TGF-β to enhance cancer immunotherapy. BioDrugs. (2022) 36:153–80. doi: 10.1007/s40259-022-00521-1
167. Eser P and Jänne PA. TGFβ pathway inhibition in the treatment of non-small cell lung cancer. Pharmacol Ther. (2018) 184:112–30. doi: 10.1016/j.pharmthera.2017.11.004
168. Grenga I, Donahue RN, Gargulak ML, Lepone LM, Roselli M, Bilusic M, et al. Anti-PD-L1/TGFβR2 (M7824) fusion protein induces immunogenic modulation of human urothelial carcinoma cell lines, rendering them more susceptible to immune-mediated recognition and lysis. Urol Oncol. (2018) 36:93.e1–93.e11. doi: 10.1016/j.urolonc.2017.09.027
169. Paz-Ares LG, Kim TM, Baz DV, Felip E, Lee DH, Lee KH, et al. Results from a second-line (2L) NSCLC cohort treated with M7824 (MSB0011359C), a bifunctional fusion protein targeting TGF-β and PD-L1. J Clin Oncol. (2018) 36:9017–7. doi: 10.1200/JCO.2018.36.15_suppl.9017
170. Cho BC, Lee JS, Wu YL, Cicin I, Dols MC, Ahn MJ, et al. Bintrafusp alfa versus pembrolizumab in patients with treatment-naive, programmed death-ligand 1-high advanced NSCLC: A randomized, open-label, phase 3 trial. J Thorac Oncol. (2023) 18:1731–42. doi: 10.1016/j.jtho.2023.08.018
171. Liu D, Zhou J, Wang Y, Li M, Jiang H, Liu Y, et al. Bifunctional anti-PD-L1/TGF-βRII agent SHR-1701 in advanced solid tumors: a dose-escalation, dose-expansion, and clinical-expansion phase 1 trial. BMC Med. (2022) 20:408. doi: 10.1186/s12916-022-02605-9
172. van de Donk N and Zweegman S. T-cell-engaging bispecific antibodies in cancer. Lancet. (2023) 402:142–58. doi: 10.1016/s0140-6736(23)00521-4
173. Giffin MJ, Cooke K, Lobenhofer EK, Estrada J, Zhan J, Deegen P, et al. AMG 757, a half-life extended, DLL3-targeted bispecific T-cell engager, shows high potency and sensitivity in preclinical models of small-cell lung cancer. Clin Cancer Res. (2021) 27:1526–37. doi: 10.1158/1078-0432.Ccr-20-2845
174. Rojo F, Corassa M, Mavroudis D, Öz AB, Biesma B, Brcic L, et al. International real-world study of DLL3 expression in patients with small cell lung cancer. Lung Cancer. (2020) 147:237–43. doi: 10.1016/j.lungcan.2020.07.026
175. Chapman G, Sparrow DB, Kremmer E, and Dunwoodie SL. Notch inhibition by the ligand DELTA-LIKE 3 defines the mechanism of abnormal vertebral segmentation in spondylocostal dysostosis. Hum Mol Genet. (2011) 20:905–16. doi: 10.1093/hmg/ddq529
176. Saunders LR, Bankovich AJ, Anderson WC, Aujay MA, Bheddah S, Black K, et al. A DLL3-targeted antibody-drug conjugate eradicates high-grade pulmonary neuroendocrine tumor-initiating cells in vivo. Sci Transl Med. (2015) 7:302ra136. doi: 10.1126/scitranslmed.aac9459
177. Paz-Ares L, Ahn MJ, Felip E, Handzhiev S, Korantzis I, Izumi H, et al. LBA92 Tarlatamab for patients (pts) with previously treated small cell lung cancer (SCLC): Primary analysis of the phase II DeLLphi-301 study. Ann Oncol. (2023) 34:S1333–4. doi: 10.1016/j.annonc.2023.10.094
178. US Food & Drug Administration. FDA grants accelerated approval to tarlatamab-dlle for extensive stage small cell lung cancer [media release]. (2024). Available at: https://www.fda.gov/.
179. Hipp S, Voynov V, Drobits-Handl B, Giragossian C, Trapani F, Nixon AE, et al. A bispecific DLL3/CD3 igG-like T-cell engaging antibody induces antitumor responses in small cell lung cancer. Clin Cancer Res. (2020) 26:5258–68. doi: 10.1158/1078-0432.Ccr-20-0926
180. Molloy ME, Aaron WH, Barath M, Bush MC, Callihan EC, Carlin K, et al. HPN328, a trispecific T cell–activating protein construct targeting DLL3-expressing solid tumors. Mol Cancer Ther. (2024) 23:1294–304. doi: 10.1158/1535-7163.Mct-23-0524
181. Sigismund S, Avanzato D, and Lanzetti L. Emerging functions of the EGFR in cancer. Mol Oncol. (2018) 12:3–20. doi: 10.1002/1878-0261.12155
182. Soria JC, Ohe Y, Vansteenkiste J, Reungwetwattana T, Chewaskulyong B, Lee KH, et al. Osimertinib in untreated EGFR-mutated advanced non-small-cell lung cancer. N Engl J Med. (2018) 378:113–25. doi: 10.1056/NEJMoa1713137
183. Vermorken JB, Mesia R, Rivera F, Remenar E, Kawecki A, Rottey S, et al. Platinum-based chemotherapy plus cetuximab in head and neck cancer. N Engl J Med. (2008) 359:1116–27. doi: 10.1056/NEJMoa0802656
184. Hoffmann P, Hofmeister R, Brischwein K, Brandl C, Crommer S, Bargou R, et al. Serial killing of tumor cells by cytotoxic T cells redirected with a CD19-/CD3-bispecific single-chain antibody construct. Int J Cancer. (2005) 115:98–104. doi: 10.1002/ijc.20908
185. Arvedson T, Bailis JM, Britten CD, Klinger M, Nagorsen D, Coxon A, et al. Targeting solid tumors with bispecific T cell engager immune therapy. Annu Rev Cancer Biology 6(Volume 6. (2022) 2022):17–34. doi: 10.1146/annurev-cancerbio-070620-104325
186. DiRaimondo TR, Budimir N, Ma L, Shenhav S, Cicchini V, Wu H, et al. Abstract B04: Preclinical activity and safety profile or JANX008, a novel EGFR-targeting tumor-activated T cell engager for treatment of solid tumors. Cancer Immunol Res. (2022) 10:B04–4. doi: 10.1158/2326-6074.Tumimm22-b04
187. Kwon BS and Weissman SM. cDNA sequences of two inducible T-cell genes. Proc Natl Acad Sci U.S.A. (1989) 86:1963–7. doi: 10.1073/pnas.86.6.1963
188. Schwarz H, Valbracht J, Tuckwell J, von Kempis J, and Lotz M. ILA, the human 4-1BB homologue, is inducible in lymphoid and other cell lineages. Blood. (1995) 85:1043–52. doi: 10.1182/blood.V85.4.1043.bloodjournal8541043
189. Kim HD, Park S, Jeong S, Lee YJ, Lee H, Kim CG, et al. 4-1BB delineates distinct activation status of exhausted tumor-infiltrating CD8(+) T cells in hepatocellular carcinoma. Hepatology. (2020) 71:955–71. doi: 10.1002/hep.30881
190. Woroniecka KI, Rhodin KE, Dechant C, Cui X, Chongsathidkiet P, Wilkinson D, et al. 4-1BB agonism averts TIL exhaustion and licenses PD-1 blockade in glioblastoma and other intracranial cancers. Clin Cancer Res. (2020) 26:1349–58. doi: 10.1158/1078-0432.Ccr-19-1068
191. Claus C, Ferrara-Koller C, and Klein C. The emerging landscape of novel 4-1BB (CD137) agonistic drugs for cancer immunotherapy. MAbs. (2023) 15:2167189. doi: 10.1080/19420862.2023.2167189
192. Vezys V, Penaloza-MacMaster P, Barber DL, Ha SJ, Konieczny B, Freeman GJ, et al. 4-1BB signaling synergizes with programmed death ligand 1 blockade to augment CD8 T cell responses during chronic viral infection. J Immunol. (2011) 187:1634–42. doi: 10.4049/jimmunol.1100077
193. Lakins MA, Koers A, Giambalvo R, Munoz-Olaya J, Hughes R, Goodman E, et al. FS222, a CD137/PD-L1 tetravalent bispecific antibody, exhibits low toxicity and antitumor activity in colorectal cancer models. Clin Cancer Res. (2020) 26:4154–67. doi: 10.1158/1078-0432.Ccr-19-2958
194. Zhang M, Lam KP, and Xu S. Natural Killer Cell Engagers (NKCEs): a new frontier in cancer immunotherapy. Front Immunol. (2023) 14:1207276. doi: 10.3389/fimmu.2023.1207276
195. Wingert S, Reusch U, Knackmuss S, Kluge M, Damrat M, Pahl J, et al. Preclinical evaluation of AFM24, a novel CD16A-specific innate immune cell engager targeting EGFR-positive tumors. MAbs. (2021) 13:1950264. doi: 10.1080/19420862.2021.1950264
196. Kontermann RE. Antibody-cytokine fusion proteins. Arch Biochem Biophys. (2012) 526:194–205. doi: 10.1016/j.abb.2012.03.001
197. Fyfe G, Fisher RI, Rosenberg SA, Sznol M, Parkinson DR, and Louie AC. Results of treatment of 255 patients with metastatic renal cell carcinoma who received high-dose recombinant interleukin-2 therapy. J Clin Oncol. (1995) 13:688–96. doi: 10.1200/jco.1995.13.3.688
198. Atkins MB, Lotze MT, Dutcher JP, Fisher RI, Weiss G, Margolin K, et al. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J Clin Oncol. (1999) 17:2105–16. doi: 10.1200/jco.1999.17.7.2105
199. Rosenberg SA, Lotze MT, Muul LM, Chang AE, Avis FP, Leitman S, et al. A progress report on the treatment of 157 patients with advanced cancer using lymphokine-activated killer cells and interleukin-2 or high-dose interleukin-2 alone. N Engl J Med. (1987) 316:889–97. doi: 10.1056/nejm198704093161501
200. Rosenzwajg M, Lorenzon R, Cacoub P, Pham HP, Pitoiset F, El Soufi K, et al. Immunological and clinical effects of low-dose interleukin-2 across 11 autoimmune diseases in a single, open clinical trial. Ann Rheum Dis. (2019) 78:209–17. doi: 10.1136/annrheumdis-2018-214229
201. Hartemann A, Bensimon G, Payan CA, Jacqueminet S, Bourron O, Nicolas N, et al. Low-dose interleukin 2 in patients with type 1 diabetes: a phase 1/2 randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol. (2013) 1:295–305. doi: 10.1016/s2213-8587(13)70113-x
202. Rosenberg SA, Yang JC, Topalian SL, Schwartzentruber DJ, Weber JS, Parkinson DR, et al. Treatment of 283 consecutive patients with metastatic melanoma or renal cell cancer using high-dose bolus interleukin 2. Jama. (1994) 271:907–13. doi: 10.1001/jama.1994.03510360033032
203. Hernandez R, Põder J, LaPorte KM, and Malek TR. Engineering IL-2 for immunotherapy of autoimmunity and cancer. Nat Rev Immunol. (2022) 22:614–28. doi: 10.1038/s41577-022-00680-w
204. Bempeg failure unlikely to affect other IL2 drugs. Cancer Discovery. (2022) 12:1604–5. doi: 10.1158/2159-8290.Cd-nb2022-0036
205. Wu W, Chia T, Lu J, Li X, Guan J, Li Y, et al. IL-2Rα-biased agonist enhances antitumor immunity by invigorating tumor-infiltrating CD25(+)CD8(+) T cells. Nat Cancer. (2023) 4:1309–25. doi: 10.1038/s43018-023-00612-0
206. Ring AM, Lin JX, Feng D, Mitra S, Rickert M, Bowman GR, et al. Mechanistic and structural insight into the functional dichotomy between IL-2 and IL-15. Nat Immunol. (2012) 13:1187–95. doi: 10.1038/ni.2449
207. Cheever MA. Twelve immunotherapy drugs that could cure cancers. Immunol Rev. (2008) 222:357–68. doi: 10.1111/j.1600-065X.2008.00604.x
208. Waldmann TA. The shared and contrasting roles of IL2 and IL15 in the life and death of normal and neoplastic lymphocytes: implications for cancer therapy. Cancer Immunol Res. (2015) 3:219–27. doi: 10.1158/2326-6066.Cir-15-0009
209. Conlon KC, Lugli E, Welles HC, Rosenberg SA, Fojo AT, Morris JC, et al. Redistribution, hyperproliferation, activation of natural killer cells and CD8 T cells, and cytokine production during first-in-human clinical trial of recombinant human interleukin-15 in patients with cancer. J Clin Oncol. (2015) 33:74–82. doi: 10.1200/jco.2014.57.3329
210. Keam SJ. Nogapendekin alfa inbakicept: first approval. Drugs. (2024) 84:867–74. doi: 10.1007/s40265-024-02060-1
211. Knudson KM, Hicks KC, Alter S, Schlom J, and Gameiro SR. Mechanisms involved in IL-15 superagonist enhancement of anti-PD-L1 therapy. J Immunother Cancer. (2019) 7:82. doi: 10.1186/s40425-019-0551-y
212. Wrangle J, Reddy S, and Soon-shiong P. OA06.05 IL15 superagonist (N-803, anktiva) + Checkpoint inhibitor (CPI) prolongs OS in 2ndline or greater NSCLC patients failing CPI. J Thorac Oncol. (2024) 19:S21. doi: 10.1016/j.jtho.2024.09.043
213. Xu Y, Carrascosa LC, Yeung YA, Chu ML, Yang W, Djuretic I, et al. An engineered IL15 cytokine mutein fused to an anti-PD1 improves intratumoral T-cell function and antitumor immunity. Cancer Immunol Res. (2021) 9:1141–57. doi: 10.1158/2326-6066.Cir-21-0058
214. Gutierrez M, Garralda E, Calvo E, van Dongen M, Eskens FA, Finlay M, et al. 1074TiP A phase I/II, open label, first-in-human, dose escalation and expansion study of SAR445877 administered as monotherapy in adults with advanced solid tumors. Ann Oncol. (2023) 34:S646–7. doi: 10.1016/j.annonc.2023.09.2213
215. Bernardo M, Y.-a. Zhang D, Martomo S, Menas F, Zhu C, Perez R, et al. Li: Abstract 2972: Preclinical characterization of SAR445877, an anti-PD-1 antibody-IL-15 mutein fusion protein with robust anti-tumor efficacy as monotherapy and in combination with PD-L1 blockade. Cancer Res. (2023) 83:2972–2. doi: 10.1158/1538-7445.Am2023-2972
216. Yin L, Jiang X, Wu C, and Chen Z. Abstract 1889: IAP0971, A novel PD1/IL15 immunocytokine that binds specifically to PD1 and fuses IL15/IL15Rα complex. Cancer Res. (2023) 83:1889–9. doi: 10.1158/1538-7445.Am2023-1889
217. Peltret M, Vetsch P, Farvaque E, Mette R, Tsachaki M, Duarte L, et al. Development of a 10 g/L process for a difficult-to-express multispecific antibody format using a holistic process development approach. J Biotechnol. (2024) 389:30–42. doi: 10.1016/j.jbiotec.2024.04.017
218. Lu X, Domingo-Yenes B, Cohen N, and Grzincic E. Freezing-induced protein aggregation in a bispecific antibody: Characterization and mechanistic insights. J Pharm Sci. (2025), 103711. doi: 10.1016/j.xphs.2025.103711
219. Barzadd MM, Lundqvist M, Harris C, Malm M, Volk AL, Thalén N, et al. Autophagy and intracellular product degradation genes identified by systems biology analysis reduce aggregation of bispecific antibody in CHO cells. N Biotechnol. (2022) 68:68–76. doi: 10.1016/j.nbt.2022.01.010
220. Wang S, Zhang W, Yang B, Zhang X, Fang J, Rui H, et al. A case study of a bispecific antibody manufacturability assessment and optimization during discovery stage and its implications. Antib Ther. (2024) 7:189–98. doi: 10.1093/abt/tbae013
221. Chen SW and Zhang W. Current trends and challenges in the downstream purification of bispecific antibodies. Antib Ther. (2021) 4:73–88. doi: 10.1093/abt/tbab007
222. Lim K, Zhu XS, Zhou D, Ren S, and Phipps A. Clinical pharmacology strategies for bispecific antibody development: learnings from FDA-approved bispecific antibodies in oncology. Clin Pharmacol Ther. (2024) 116:315–27. doi: 10.1002/cpt.3308
223. Lim EA, Schweizer MT, Chi KN, Aggarwal R, Agarwal N, Gulley J, et al. Phase 1 study of safety and preliminary clinical activity of JNJ-63898081, a PSMA and CD3 bispecific antibody, for metastatic castration-resistant prostate cancer. Clin Genitourinary Cancer. (2023) 21:366–75. doi: 10.1016/j.clgc.2023.02.010
224. Zhou Y, Penny HL, Kroenke MA, Bautista B, Hainline K, Chea LS, et al. Immunogenicity assessment of bispecific antibody-based immunotherapy in oncology. J Immunother Cancer. (2022) 10. doi: 10.1136/jitc-2021-004225
225. Haber L, Olson K, Kelly MP, Crawford A, DiLillo DJ, Tavaré R, et al. Generation of T-cell-redirecting bispecific antibodies with differentiated profiles of cytokine release and biodistribution by CD3 affinity tuning. Sci Rep. (2021) 11:14397. doi: 10.1038/s41598-021-93842-0
226. Mandikian D, Takahashi N, Lo AA, Li J, Eastham-Anderson J, Slaga D, et al. Relative target affinities of T-cell-dependent bispecific antibodies determine biodistribution in a solid tumor mouse model. Mol Cancer Ther. (2018) 17:776–85. doi: 10.1158/1535-7163.Mct-17-0657
227. Valdes CR, Voorhees PM, D’Souza A, Chung A, Tuchman S, Safah H, et al. Efficacy, safety, and determination of RP2D of ABBV-383, a BCMA bispecific antibody, in patients with relapsed/refractory multiple myeloma (RRMM). J Clin Oncol. (2024) 42:7531–1. doi: 10.1200/JCO.2024.42.16_suppl.7531
228. To M, Yeung P, Fox M, Hammond M, Cattaruzza F, Nazeer A, et al. Abstract P193: AMX-818, a novel prodrug HER2-XPAT T-cell engager (TCE) with potent T cell activation, proteolytic cleavage and efficacy in xenograft tumors, and wide safety margins in NHP (Non Human Primate). Mol Cancer Ther. (2021) 20:P193–3. doi: 10.1158/1535-7163.Targ-21-p193
229. Wang Y and Cheng P. Arming oncolytic viruses with bispecific T cell engagers: The evolution and current status. Biochim Biophys Acta Mol Basis Dis. (2024) 1870:166962. doi: 10.1016/j.bbadis.2023.166962
230. Carlo-Stella C, Mazza R, Manier S, Facon T, Yoon S-S, Koh Y, et al. RG6234, a GPRC5DxCD3 T-cell engaging bispecific antibody, is highly active in patients (pts) with relapsed/refractory multiple myeloma (RRMM): updated intravenous (IV) and first subcutaneous (SC) results from a phase I dose-escalation study. Blood 140(Supplement. (2022) 1):397–9. doi: 10.1182/blood-2022-157988
231. Girard N, Li W, Spira A, Feldman J, Mak M, Sauder MB, et al. 10MO: Preventing moderate to severe dermatologic adverse events in first-line EGFR-mutant advanced NSCLC treated with amivantamab plus lazertinib: Early success of the COCOON trial. J Thorac Oncol. (2025) 20:S14–6. doi: 10.1016/S1556-0864(25)00205-9
232. Shepshelovich D, Barda N, Goldvaser H, Dagan N, Zer A, Diker-Cohen T, et al. Incidence of lung cancer following pneumonia in smokers: a population-based study. Qjm. (2022) 115:287–91. doi: 10.1093/qjmed/hcab030
233. Zhang Q, Tang L, Zhou Y, He W, and Li W. Immune checkpoint inhibitor-associated pneumonitis in non-small cell lung cancer: current understanding in characteristics, diagnosis, and management. Front Immunol. (2021) 12:663986. doi: 10.3389/fimmu.2021.663986
234. Chen Z, Fillmore CM, Hammerman PS, Kim CF, and Wong KK. Non-small-cell lung cancers: a heterogeneous set of diseases. Nat Rev Cancer. (2014) 14:535–46. doi: 10.1038/nrc3775
235. Hanahan D and Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. (2012) 21:309–22. doi: 10.1016/j.ccr.2012.02.022
236. Altorki NK, Markowitz GJ, Gao D, Port JL, Saxena A, Stiles B, et al. The lung microenvironment: an important regulator of tumour growth and metastasis. Nat Rev Cancer. (2019) 19:9–31. doi: 10.1038/s41568-018-0081-9
237. Yue P, Zhang M, Feng Y, Gao Y, Sun C, and Chen P. Cost-effectiveness analysis of amivantamab plus chemotherapy versus chemotherapy alone in NSCLC with EGFR Exon 20 insertions. Front Oncol. (2024) 14:1368804. doi: 10.3389/fonc.2024.1368804
238. Wu Q, Li Q, and Qin Y. A cost-effectiveness analysis of amivantamab plus lazertinib versus osimertinib in the treatment of US and Chinese patients with EGFR-mutated advanced non-small cell lung cancer. Lung Cancer. (2025) 203:108533. doi: 10.1016/j.lungcan.2025.108533
239. Ter Heine R, van den Heuvel MM, Piet B, Deenen MJ, van der Wekken AJ, Hendriks LEL, et al. A systematic evaluation of cost-saving dosing regimens for therapeutic antibodies and antibody-drug conjugates for the treatment of lung cancer. Target Oncol. (2023) 18:441–50. doi: 10.1007/s11523-023-00958-6
Keywords: bispecific antibodies, lung cancer, novel therapies, immunotherapy, targeted therapy
Citation: Chen W, Zhou A and Zhou Y (2025) Bispecific antibody for lung cancer: mechanisms and clinical insights. Front. Immunol. 16:1572802. doi: 10.3389/fimmu.2025.1572802
Received: 07 February 2025; Accepted: 09 May 2025;
Published: 29 May 2025; Corrected: 17 July 2025.
Edited by:
Renata Pacholczak-Madej, Maria Skłodowska-Curie National Institute of Oncology, PolandReviewed by:
Rosendo Luria-Perez, Children’s Hospital of Mexico Federico Gomez, MexicoMoshe Elkabets, Ben-Gurion University of the Negev, Israel
Copyright © 2025 Chen, Zhou and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yunfeng Zhou, eXVuZl96aG91X2h4c3lAMTYzLmNvbQ==