 Wei Shao
Wei Shao Zilong Wang
Zilong Wang Jian Wu
Jian Wu Tianting Guo
Tianting Guo Jianwen Mo2*
Jianwen Mo2*- 1The First Clinical College, Gannan Medical University, Ganzhou, China
- 2Department of Orthopedics, The First Affiliated Hospital of Gannan Medical University, Ganzhou, China
- 3Department of Orthopedics, Ganzhou Hospital of Guangdong Provincial People’s Hospital, Ganzhou Municipal Hospital, Ganzhou, China
Deep vein thrombosis (DVT) is a complex multifactorial vascular disease characterized by abnormal blood stasis and coagulation within the deep veins, primarily occurring in the lower limbs. This pathological condition not only causes local circulatory disruption but also has the potential to trigger life-threatening pulmonary embolism through thrombus detachment, thus posing a major threat to human health. Toll - like receptors (TLRs), essential components of the innate immune system, have been increasingly acknowledged as crucial determinants in the pathogenesis of DVT. TLRs possess the ability to recognize a diverse range of pathogen - associated molecular patterns (PAMPs) and endogenous danger - associated molecular patterns (DAMPs). Upon activation, they trigger a cascade of inflammatory responses that are intricately intertwined with the thrombotic process. This review comprehensively scrutinizes the extant knowledge pertaining to the role of TLRs in DVT. It systematically synthesizes the molecular mechanisms underpinning the participation of TLRs in DVT, spanning platelet activation, endothelial cell dysfunction, and leukocyte recruitment. Moreover, it delves profoundly into the potential of targeting TLRs as therapeutic strategies for DVT. This entails the exploration of the development and application of TLR inhibitors or antagonists. By elucidating these aspects, the objective is to proffer novel perspectives and insights for the prevention and treatment of DVT.
1 Introduction
DVT is a prevalent vascular condition that holds considerable clinical significance and exhibits a high occurrence rate, particularly in patients who have recently fracture, undergone surgery, individuals who remain immobile for extended periods, and those diagnosed with cancer (1). DVT not only causes lower limb swelling, pain, and functional impairment, but may also lead to serious complications such as pulmonary embolism, which can threaten patients’ lives (2). According to the latest epidemiological data, the incidence of DVT is on the rise globally, and its economic burden and social impact cannot be ignored (3). Data from a multinational cohort (2017–2023) involving over 1 million surgical patients revealed that DVT and pulmonary embolism (PE) accounted for 51% and 49% of venous thromboembolism (VTE) cases (4). Preoperative DVT prevalence in lower-extremity fractures reached 24.1%, with femoral fractures posing higher risks than tibiofibular injuries (5). In the pathophysiological process of DVT, Virchow’s triad—blood flow stasis, hypercoagulable state, and vascular endothelial damage—are considered the main pathogenic factors (6). However, research in recent years has shown that immune-inflammatory response plays a key role in the occurrence and development of DVT, especially the mediating effects of inflammatory cells and cytokines (7).
TLR, is widely present in various immune and non-immune cells. It plays a role in identifying PAMPs and DAMPs, which in turn triggers multiple signaling pathways and modulates immune responses (8). The activation of TLR signaling pathways is significant not only for anti-infection immunity but is also intricately connected to the development of various inflammatory diseases, such as atherosclerosis, rheumatoid arthritis, and DVT (9, 10). Recently, an increasing amount of research has concentrated on the particular mechanisms involved in TLR signaling pathways related to DVT, as well as their possible therapeutic targets. For example, the activation of TLR4 can lead to the activation of key transcription factors such as NF-κB and IRF3 through MyD88-dependent and MyD88-independent pathways, thereby inducing the expression of inflammatory factors and coagulation factors (11). In addition, TLR signaling pathways in endothelial cells have also been found to be closely related to vascular endothelial dysfunction, which in turn promotes thrombosis formation (12, 13). The results not only enhance our comprehension of how DVT develops but also offer a foundational theory for creating new treatment approaches.
In terms of treatment, the existing DVT treatment methods mainly include anticoagulant therapy and thrombolytic therapy. Although these methods have achieved certain effects in the prevention and treatment of thrombosis, they still have the risk of bleeding and therapeutic limitations (14–16). Therefore, the regulation of TLR signaling pathways has become a promising new therapeutic strategy. TLR antagonists and inhibitors have shown good antithrombotic effects in experimental models, indicating their potential for clinical application (17, 18). In addition, research on natural products has also shown that by regulating TLR signaling pathways, the inflammatory response and thrombosis formation of DVT can be effectively alleviated (19). This review aims to focus on the specific roles of TLR in the pathophysiological process of DVT, regulatory mechanisms, and therapeutic strategies based on TLR signaling pathways. By comprehensively analyzing the latest research progress, this article aims to provide new theoretical basis and research directions for the prevention and treatment of DVT.
2 The pathophysiological mechanisms of deep vein thrombosis
2.1 Virchow’s triad
DVT is a complex pathological process, the pathogenesis of which is mainly driven by Virchow’s triad, including blood flow stasis, hypercoagulable state, and vascular endothelial damage (6). Blood flow stasis is usually caused by venous valve dysfunction, long-term bed rest, or restricted activity after surgery, leading to a slowdown in venous blood flow and an increased risk of thrombosis (20, 21). Hypercoagulable state can be caused by hereditary coagulation factor abnormalities (such as prothrombin gene mutation, protein C/protein S deficiency) or acquired factors (such as cancer, pregnancy, oral contraceptives), which promote thrombosis by increasing the activity of coagulation factors or reducing the level of anticoagulant factors (22–24). Vascular endothelial damage promotes platelet adhesion and aggregation and initiates the coagulation cascade by exposing subendothelial matrix components (such as collagen) and procoagulant substances (such as tissue factor) (25–27).
2.2 The role of inflammatory response in DVT
In addition to Virchow’s triad, the inflammatory response is also significant in the pathophysiology of DVT (Figure 1). Inflammatory cells, such as neutrophils, monocytes, and macrophages, regulate the processes of thrombosis and dissolution by releasing cytokines, chemokines, and enzymes (28–31). Neutrophils release Neutrophil Extracellular Traps (NETs), which not only capture pathogens but also promote thrombus stabilization and formation. NETs are composed of DNA, histones, and antimicrobial proteins, which can enhance fibrin formation and promote platelet activation and aggregation, thus accelerating thrombus development (32). Monocytes and macrophages further enhance the coagulation response by expressing tissue factor(TF) and other procoagulant factors in DVT. Studies have shown that monocytes accumulate in the venous intima and promote thrombus formation and maintenance by secreting TF and inflammatory mediators (such as IL-6, TNF-α) (33–35). In addition, macrophages participate in thrombus dissolution and fibrin degradation by clearing red blood cells and fibrin in the thrombus, maintaining the dynamic balance of the thrombus (36, 37).
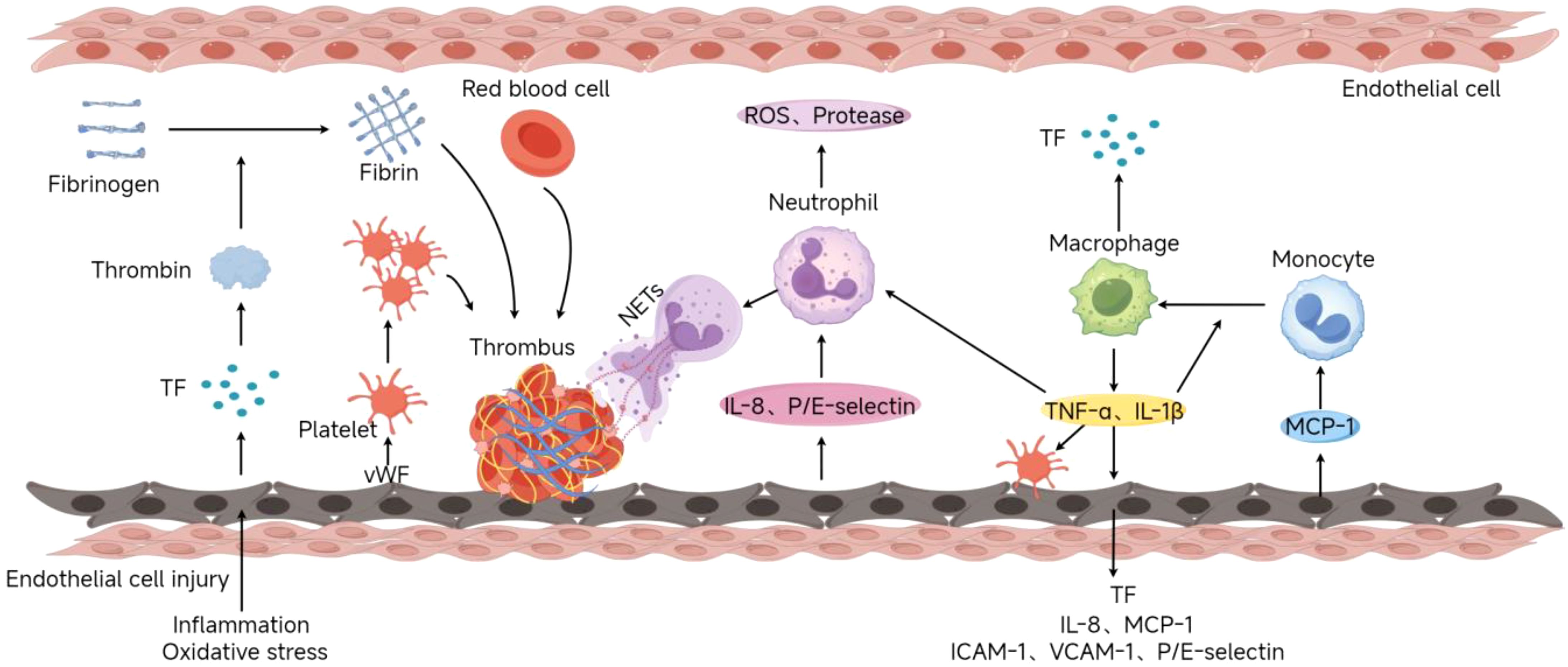
Figure 1. Deep Vein Thrombosis Formation Process(By Figdraw). Endothelial injury caused by inflammation or oxidative stress exposes the subendothelial matrix (e.g., collagen, vWF) and releases procoagulants like tissue factor (TF). Platelets bind to vWF via GPIb-IX-V, adhere to the site, and release mediators (ADP, TXA2) to recruit additional platelets, forming aggregates. TF activates the extrinsic coagulation pathway, generating thrombin to convert fibrinogen into fibrin mesh. Damaged endothelium releases chemokines (IL-8) and adhesion molecules (P/E-selectin), recruiting neutrophils that release NETs to stabilize thrombi. Monocytes recruited via MCP-1 differentiate into macrophages, secreting pro-inflammatory cytokines (TNF-α, IL-1β) and TF.
3 Overview of TLR signaling pathways
3.1 Types and structures of TLR
Pattern recognition receptors (PRRs) of the innate immune system, known as TLRs, play a crucial role. TLRs were first discovered in fruit flies, and their name originates from the description of their importance by German scientists (“Toll” means “amazing” in German) (38). In 1997, the first Toll-like receptor, TLR4, was identified in mammals, marking the beginning of a new era in TLR research (39). In humans, 10 TLRs (TLR1 to TLR10) have been identified, while in mice, there are 12 (TLR1 to TLR9 and TLR11 to TLR13) (40). These receptors are distributed on immune cells (such as macrophages, dendritic cells, neutrophils, etc.) and some non-immune cells (such as endothelial cells, epithelial cells, etc.). They trigger complex signaling cascades by recognizing specific exogenous or endogenous molecules (41, 42).
TLRs constitute a group of transmembrane proteins characterized by a distinct molecular architecture featuring three components: the extracellular region, the transmembrane segment, and the intracellular signaling region. The extracellular domain of TLRs consists of multiple leucine-rich repeats (LRRs), which are arranged in a horseshoe shape to form specific ligand-binding sites (43). The extracellular domain configuration of various TLRs defines the specificity of their ligands (Table 1). For example, TLR4 exerts anti-Gram-negative bacterial effects by binding to lipopolysaccharide (LPS) (44). TLR9 recognizes CpG DNA (DNA sequences rich in unmethylated CpG dinucleotides), which is a characteristic of bacterial and viral DNA (45). These recognition capabilities make TLRs play a key role in anti-infection immunity and inflammatory responses, and they are also involved in the pathogenesis of various autoimmune and inflammatory diseases. The transmembrane domain of TLRs consists of a hydrophobic α-helical structure embedded in the cell membrane or endosomal membrane, serving to stabilize the receptor and mediate signal transduction (46). The transmembrane region not only aids in the localization of TLRs within the membrane but is also involved in the dimerization of receptors, a crucial step in the activation of signaling. The intracellular part of TLRs is called the Toll/IL-1 receptor domain (TIR), which is the core region for signal transduction (47). After ligand binding, the TIR domain interacts with specific adaptor proteins (such as MyD88 or TRIF) to activate downstream signaling pathways (48). The diversity of the TIR domain also supports the specificity of signal transduction by different TLRs.
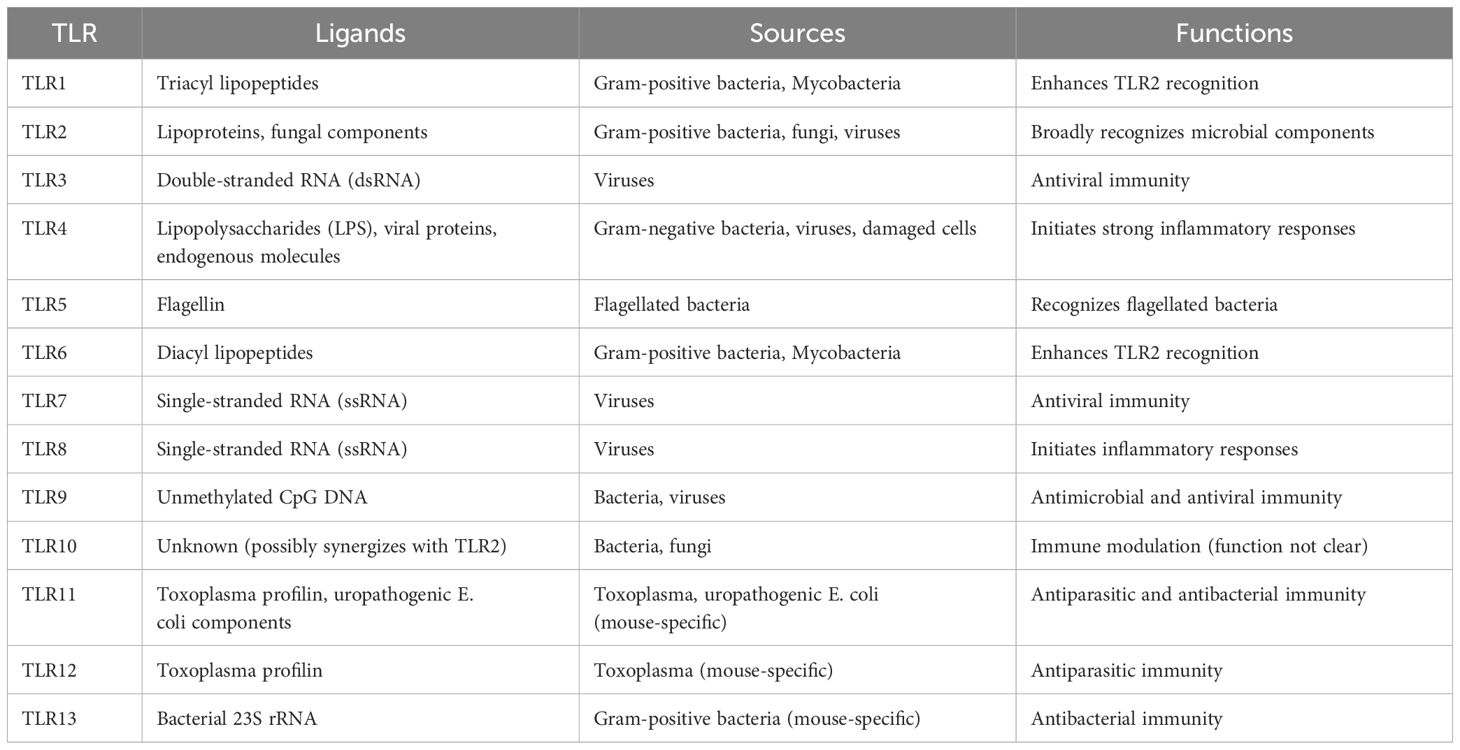
Table 1. TLR ligands, sources, and functions.
3.2 TLR signaling pathways
The activation of TLR signaling pathways mainly occurs through two different downstream pathways (Figure 2): the MyD88-dependent pathway and the MyD88-independent pathway (49). Most TLRs (except TLR3) activate downstream signaling molecules through the adaptor protein MyD88 (Myeloid differentiation primary response 88), this process activates the NF-κB (nuclear factor κB) and MAPK (mitogen-activated protein kinase) signaling pathways, leading to an increase in the production of pro-inflammatory cytokines, including TNF-α and IL-6 (50). TLR3 and TLR4, on the other hand, can use the adaptor protein TRIF (TIR-domain-containing adapter-inducing interferon-β) to activate IRF3 (interferon regulatory factor 3) and NF-κB, inducing the production of type I interferons (such as IFN-β) (51). Inside the cell, the activation process of TLR signaling pathways involves multiple key molecules and steps. First, after TLRs bind to their ligands, the receptors undergo dimerization or oligomerization, which then recruits adaptor proteins (such as MyD88, TRIF, TIRAP/MAL, TRAM) to the TIR domain of the receptor. These adaptor proteins further recruit downstream kinases (such as IRAK4, IRAK1) and E3 ubiquitin ligases (such as TRAF6), forming a signaling complex that promotes the activation of the IκB kinase (IKK) complex (52). The IKK complex adds phosphate groups to the IκB protein, resulting in its breakdown and the subsequent release of the NF-κB transcription factor into the nucleus, where it prompts the transcription of genes related to inflammation (53). Moreover, the stimulation of the MAPK signaling pathway increases the expression of inflammatory factors and other biological processes, such as cell growth and differentiation (54). In addition to the classical MyD88-dependent and -independent pathways, research in recent years has revealed the complexity and diversity of TLR signaling pathways. For instance, when LPS is recognized by TLR4, it not only activates NF-κB via the MyD88-dependent pathway but also triggers the expression of antiviral genes through the TRIF-dependent pathway, showcasing its diverse functions in combating infections (55). Moreover, the regulation of TLR signaling pathways is also influenced by various negative regulatory mechanisms, including the endocytosis of receptors, ubiquitination of adaptor proteins, and the involvement of negative regulatory proteins (such as A20, SARM). These mechanisms collectively maintain the balance of TLR signaling, preventing excessive inflammatory responses and the occurrence of autoimmune diseases (56, 57).
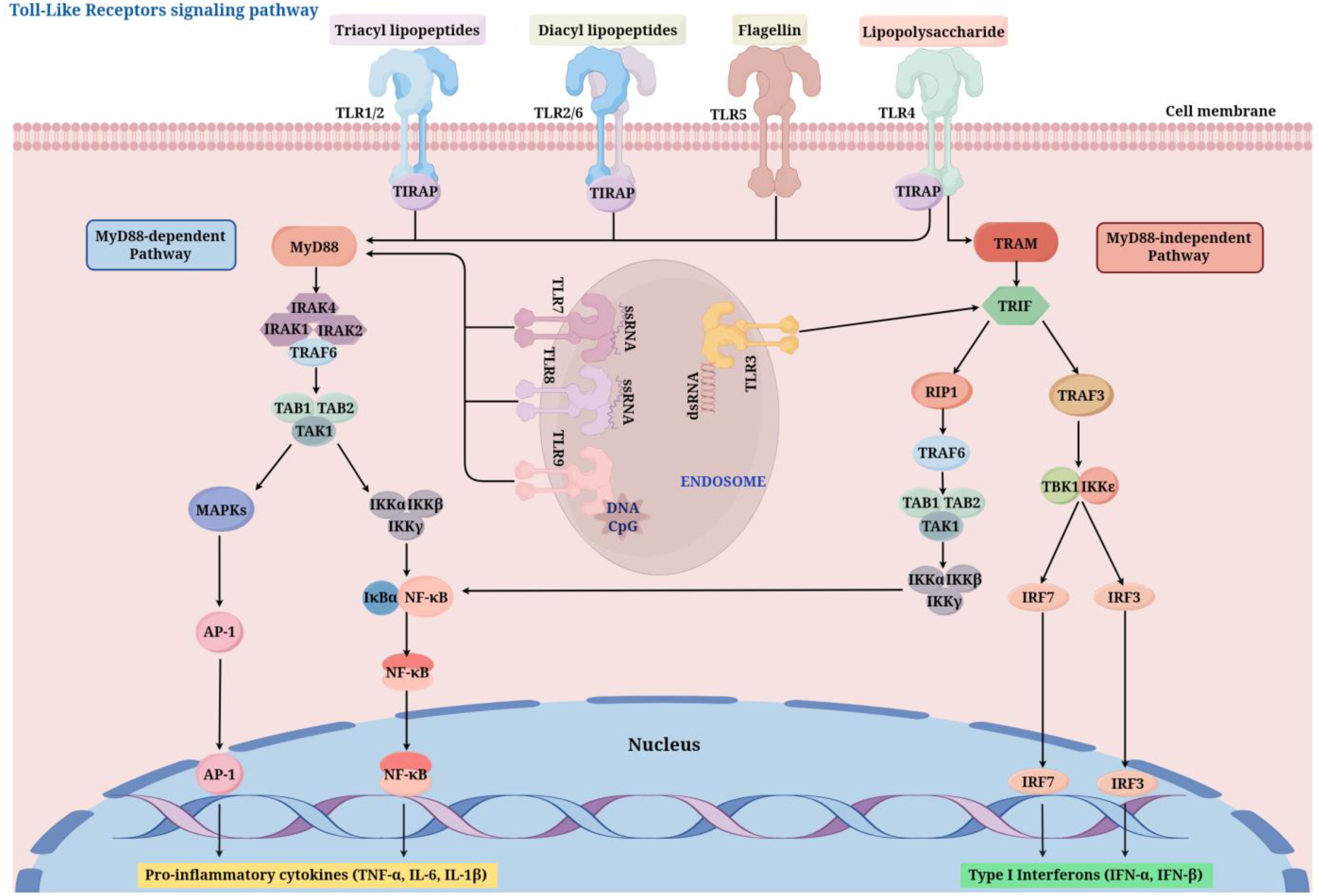
Figure 2. TLR signaling pathways (By Figdraw). After ligand binding (e.g., LPS for TLR4, dsRNA for TLR3), TLRs dimerize and recruit adaptor proteins such as MyD88 or TRIF through their TIR domains. The MyD88-dependent pathway activates IRAK and TRAF6, leading to NF-κB and MAPK activation, which induces pro-inflammatory cytokines. The MyD88-independent Pathway, utilized by TLR3 and TLR4, activates TBK1 and IRF3, promoting the production of type I interferons. TIRAP, TIR Domain-Containing Adaptor Protein; MyD88, myeloid differentiation factor 88; IRAK1/2/4, interleukin 1 Receptor Associated Kinase 1/2/4; TRAF, tumor necrosis factor receptor-associated factor ; TAB1/2, TGF-beta Activated Kinase 1 Binding Protein 1/2; TAK1, transforming growth factor activating kinase-1; NF-κB, nuclear factor kappa B; MAPK, mitogen-activated protein kinase; AP-1, Activator Protein 1; TRAM, TRIF-Related Adaptor Molecule; TRIF, toll receptor-associated interferon activator; RIP1, Receptor-Interacting Protein 1; IKKϵ, I-κB kinase ϵ; TBK1, TANK-binding kinase 1; IRF, interferon regulatory factor .
4 The role of TLR signaling pathways in DVT
4.1 TLR activation and thrombosis formation
TLR1 and TLR6 usually form dimers with TLR2, recognizing bacterial lipopeptides and other bacterial components, activating the MyD88-dependent signaling pathway, promoting the activation of NF-κB and MAPK, inducing the expression of inflammatory and coagulation factors, and thus promoting thrombosis formation (58). TLR3 mainly recognizes double-stranded RNA through the TRIF-dependent pathway, activating the IRF3 and NF-κB signaling pathways, inducing the production of type I interferons and pro-inflammatory cytokines, and enhancing the inflammatory environment for thrombosis formation (59).
One of the most thoroughly researched members of the TLR family is TLR4, which plays a crucial role in DVT and various other thrombotic conditions. This receptor is capable of identifying endogenous DAMPs like high-mobility group box 1 (HMGB1) as well as exogenous PAMPs such as bacterial LPS, activating downstream signaling through both MyD88-dependent and TRIF-dependent pathways, inducing the expression of inflammatory factors (e.g., TNF-α, IL-6) and coagulation factors (e.g., tissue factor), and promoting thrombus formation and stabilization (60, 61). Recent studies have shown that the activation of TLR4 also involves the interplay between the PI3K/Akt and MAPK signaling pathways, further enhancing inflammatory responses and thrombus stability (62–64).
TLR5 mainly recognizes bacterial flagellin, activating NF-κB and MAPK through the MyD88-dependent pathway, promoting the expression of inflammatory and coagulation factors, and thereby promoting thrombosis formation (65, 66). Despite receiving less attention, TLR7 and TLR8 have been demonstrated to contribute to the regulation of immune responses and coagulation processes in DVT. They mainly recognize single-stranded RNA, participate in inflammation associated with viral infections, and induce the production of type I interferons and pro-inflammatory cytokines through the IRF7 and NF-κB signaling pathways, promoting thrombosis formation (67, 68).
TLR9 recognizes CpG DNA, activating the IRF7 and NF-κB signaling pathways, inducing the production of type I interferons and pro-inflammatory cytokines, and further promoting thrombosis formation (69, 70). TLR10 is considered to have inhibitory functions in humans, negatively regulating the signaling pathways of TLR2 and TLR4 to limit excessive inflammatory responses and maintain vascular homeostasis (71–73). TLR11 to TLR13 in mice mainly participate in parasite infections and autoimmune regulation, and their specific roles in thrombosis formation require further research (74, 75).
4.2 Cellular sources and targets of TLRs in thrombosis
The role of TLRs in DVT depends not only on their expression levels but also on their distribution and functions in different cell types. The main TLR-expressing cells include endothelial cells, platelets, monocytes, macrophages and neutrophils.
4.2.1 Endothelial cells
Vascular endothelial cells are crucial for sustaining vascular balance and managing the process of blood clotting. The activation of TLR signaling pathways can lead to endothelial cell dysfunction, promoting thrombosis formation. Specifically, the activation of TLR4 in endothelial cells induces the expression of inflammatory factors (e.g., IL-6, TNF-α) and adhesion molecules (e.g., VCAM-1, ICAM-1), enhancing the adhesion and migration of white blood cells, and promoting the accumulation and activation of inflammatory cells in the vascular wall (76, 77). Further studies have revealed that TLR4 regulates the microenvironment of endothelial cells, inducing oxidative stress and apoptosis, disrupting the integrity of the vascular endothelium, increasing the permeability of the vascular wall, and promoting thrombus formation and expansion (78). Additionally, TLR4 regulates the metabolic pathways of endothelial cells, promoting lipid peroxidation and the generation of inflammatory mediators, further exacerbating endothelial cell dysfunction (79, 80). The mechanisms of TLR2 and TLR9 in endothelial cells have also been gradually revealed. TLR2 regulates the proliferation and migration of endothelial cells through the PI3K/Akt and NF-κB signaling pathways, promoting vascular remodeling and thrombosis formation (81). TLR9 affects the ability of endothelial cells to cope with oxidative stress by regulating their antioxidant defense system, thereby influencing thrombosis formation (82). Research indicates that the relationship between TLRs and vascular endothelial cells extends beyond inflammatory responses, encompassing mechanisms associated with metabolism, cell viability, and apoptosis, all of which play a role in the emergence and progression of DVT.
4.2.2 Platelets
Platelets are not only key participants in hemostasis and thrombosis formation but also play important roles in immune regulation and inflammatory responses. Recent research has shown that platelets regulate thrombo-inflammatory responses in various pathological conditions through TLR-mediated signaling pathways. TLR2 and TLR4 regulate platelet function through different mechanisms: the TLR2 agonist Pam3CSK4 directly induces platelet aggregation, while the TLR4 agonist LPS enhances platelet adhesion and glycoprotein Ib-dependent aggregation (83). In SARS-CoV-2 infection, TLR4 promotes platelet activation and thrombosis formation by binding to the viral spike protein, a mechanism of significant importance in COVID-19-related thrombotic complications (84). Furthermore, in individuals diagnosed with essential thrombocythemia (ET), there is a notable increase in the expression levels of TLR2 and TLR4, which contribute to the activation of platelets and the secretion of pro-inflammatory mediators through the activation of NF-κB and MAPK signaling pathways, thereby intensifying thrombo-inflammatory reactions (85). Calprotectin induces platelet apoptosis by binding to TLR4 on the platelet surface and activating the NLRP3 inflammasome, thereby reducing platelet survival (86). Tyrosyl-tRNA synthetase (YRSACT) induces the generation of “inflammatory” megakaryocytes and platelets by activating TLR and type I interferon signaling pathways, and these platelets exhibit enhanced agonist-induced activation and pro-coagulant activity (87). Low-grade endotoxemia (LGE) enhances platelet activation and thrombosis formation through the TLR4 signaling pathway, a mechanism of significant importance in cardiovascular diseases (88). Additionally, platelets are involved in the pathological mechanisms of hemolytic disorders by expressing TLR4 and the NLRP3 inflammasome. For instance, free hemin can stimulate the release of α-granules from platelets through the activation of the TLR4 signaling pathway, which in turn activates the production of mitochondrial reactive oxygen species (mtROS) (89). These results offer novel insights for advancing therapies aimed at anti-thrombosis and anti-inflammation that focus on the signaling pathways of platelet TLR.
4.2.3 Monocytes
LPS activates monocytes through TLR4, inducing the transient expression of TF, which triggers the coagulation cascade (90). Anti-phospholipid antibodies (aPL) stimulate monocytes and endothelial cells via TLR2, which activates the NF-κB signaling pathway and enhances the production of pro-inflammatory cytokines (such as TNF-α and IL-6). This process highlights the role of TLR2 in mediating inflammatory responses associated with antiphospholipid syndrome (APS) (91). Similarly, autoantibodies against β2-glycoprotein I (β2GPI) induce a pro-inflammatory phenotype in endothelial cells and monocytes by cross-reacting with TLR4, further elucidating the role of TLR4 in APS pathology (92). Furthermore, the exosomes found in body fluids activate monocytes through the TLR2 and TLR4 signaling pathways, which in turn promotes the secretion of inflammatory cytokines like IL-6 and TNF-α, underscoring the potential role of exosomes in inflammatory diseases (93). In addition to the TLR signaling pathway, the Notch signaling pathway significantly influences monocyte differentiation and inflammatory responses. The TLR7-Myd88 pathway and Notch2 together determine the fate of monocytes. When Notch2 signaling is absent, monocytes may aberrantly differentiate into dendritic cells and macrophages, resulting in an excessive elevation of systemic inflammation. This highlights the crucial role of Notch2 in the differentiation of monocytes mediated by TLR (94). Functional abnormalities of monocytes in pathological conditions can lead to severe clinical consequences.
4.2.4 Macrophages
The activation of TLR9 and TLR7/8 has been shown to induce the formation of multinucleated macrophages, indicating chronic inflammation and granuloma formation. This suggests that TLR signaling is central to macrophage activation and the progression of inflammatory responses (95). Additionally, TLR activation coordinates the mobilization of arachidonic acid in macrophages, a process regulated by group IVA and V phospholipases (96). This mechanism is important for initiating and amplifying inflammatory responses. The TRIM protein family also plays an important role in macrophage activation after TLR stimulation, especially after TLR3 and TLR4 activation, contributing to the regulation of immune responses (97). Moreover, interferon (IFN) has been shown to enhance the expression of TLR genes in macrophages during viral infections, thereby amplifying the innate immune response (98). In addition to pro-inflammatory responses, TLR7 and TLR9 ligands also shift macrophage function to a long-lived phagocytic state, characterized by M2-like polarization and reduced antigen-presenting capacity (99). Chronic signaling through TLR7 and TLR9 is also associated with driving the differentiation of inflammatory hemophagocytic cells, leading to anemia and thrombocytopenia observed in conditions such as macrophage activation syndrome (MAS) and malaria (100). These findings collectively emphasize the key role of TLR signaling in macrophage function, inflammation, and tissue-specific responses in various diseases.
4.2.5 Neutrophils
Neutrophils are the first responders in inflammatory reactions, promoting thrombus formation by releasing reactive oxygen species (ROS) and neutrophil extracellular traps (NETs), which disrupt the endothelial cell barrier. NETs are mesh-like structures composed of chromatin and antimicrobial proteins that can capture platelets and red blood cells, forming the core structure of thrombi (32, 101). TLR signaling pathways influence thrombus formation mechanisms by regulating the formation and stability of NETs. Activation of TLR4 prompts neutrophils to produce NETs, which contain histones and DNA that subsequently stimulate the TLR4 and TLR9 signaling pathways. This generates a positive feedback loop that amplifies inflammatory and coagulation responses, thereby facilitating thrombus formation (102, 103). Additionally, TLR2 promotes NET formation by recognizing bacterial lipopeptides, further increasing thrombus stability (104). Brill et al. (105) demonstrated that NETs play a key role in DVT in mice. They found that extracellular chromatin from neutrophils contributes to thrombus formation. The study further indicated that DNA and histones in NETs are involved in promoting DVT, providing new insights for DVT treatment strategies.
4.3 The role of TLR-mediated crosstalk between platelets and leukocytes in DVT
The pathological mechanisms of DVT involve complex biological processes including vascular endothelial injury, platelet activation, and inflammatory cell recruitment, where the cross-talk between platelets and leukocytes plays a pivotal role in disease development. The TLR-mediated interactions between platelets and leukocytes exert critical regulatory effects on DVT pathogenesis. Activated platelets express surface markers such as P-selectin and CD40L, enabling them to bind leukocytes via PSGL-1 and integrins, respectively (106–108). These interactions facilitate the formation of platelet-leukocyte aggregates, thereby enhancing leukocyte recruitment, tissue factor expression, and NET release—key drivers of venous thrombosis (7, 109, 110). In inflammatory microenvironments triggered by infection or tissue injury, DAMPs and PAMPs released from necrotic/apoptotic cells activate TLRs on leukocytes and platelets, establishing a vicious cycle of prothrombotic and proinflammatory responses (111, 112), particularly prominent in severe COVID-19, dengue virus infection, and other inflammatory crises (113, 114). Targeting the TLRs activation pathways may alleviate thrombotic events and hyperinflammatory states, thereby breaking the thrombo-inflammatory vicious cycle.
In sepsis and cardiovascular diseases, platelet TLRs promote leukocyte recruitment and create prothrombotic microenvironments through MyD88 signaling by inducing proinflammatory factors like IL-1β and CD40L (115). Human platelets recognize bacterial components (e.g., Pam3CSK4) via TLR2, activating the PI3-K/Akt pathway to induce platelet-neutrophil aggregation and oxidative stress, potentially contributing to thrombosis risk post-bacterial infection (116). TLR7 activation in platelets triggers p38 MAPK and Akt phosphorylation, leading to α-granule release (e.g., surface expression of P-selectin and CD40L) that promotes platelet-leukocyte aggregation (117). During viral infections, platelets activate TLR-MyD88-IRAK4 cascades through viral particle endocytosis, triggering complement C3 release and platelet-leukocyte aggregation (118). Influenza virus specifically induces C3 secretion and NET formation via platelet TLR7 (119), revealing molecular mechanisms of viral-associated thrombosis. Conversely, histones in NETs activate platelets through TLR2, TLR4 and NLRP3 inflammasomes, promoting P-selectin expression and phosphatidylserine (PtdSer) exposure to enhance coagulation factor binding (120–122). Additionally, extracellular vesicles (EVs) generated by platelet CLEC2 activation drive neutrophil NETosis via CLEC5A/TLR2 dual receptors and stimulate macrophages to secrete IL-6 and TNF-α (123). Platelet-derived HMGB1 facilitates monocyte migration and inhibits apoptosis through the RAGE/TLR4 axis (124), while disulfide HMGB1 from platelets mediates neutrophil recruitment and NET release via RAGE/TLR2 in experimental DVT, establishing a self-amplifying “thrombo-inflammatory loop” (125).
Notably, platelets exert homeostatic regulation on leukocyte functions. TLR2 agonists (e.g., Pam3CSK4 and FSL-1) suppress neutrophil CD66b expression and elastase release while enhancing phagocytosis (126). Platelets modulate cytokine secretion in peripheral blood mononuclear cells (PBMCs) via TLR4 and TLR2 signaling (e.g., reducing IL-6 but increasing IL-10) (127), suggesting TLRs balance pro- and anti-inflammatory responses through spatiotemporal signaling. This regulatory pattern may influence thrombus stability by dynamically modulating local inflammatory milieus in DVT. Furthermore, aberrant platelet-leukocyte interactions can induce sterile inflammation and tissue damage, particularly evident in immune-mediated vascular disorders (128). Therefore, elucidating TLR-mediated platelet-leukocyte cross-talk not only advances understanding of DVT pathogenesis but also provides insights for developing novel therapeutic strategies.
4.4 Interactions between TLRs and the coagulation system
The interaction between TLR signaling pathways and the coagulation system is particularly important in the pathophysiology of DVT. TLR activation promotes coagulation responses through various mechanisms, enhancing thrombus formation and stability.
The regulation of TF expression is one of the main mechanisms by which TLR signaling pathways promote coagulation responses. The activation of TLR4 and TLR2 directly induces TF expression, with TF being a key molecule initiating the coagulation cascade. By binding to coagulation factor VIIa, TF activates thrombin, promoting fibrin formation and thrombus development (120, 129). Xia et al. (130) found that TLR4 activation significantly increases TF expression through the NF-κB and AP-1 signaling pathways, further promoting coagulation responses.
The inhibition of anticoagulant factors is also an important pathway by which TLR signaling pathways promote a hypercoagulable state. Activation of TLR4 not only enhances the production of thrombin but also reduces the expression of anticoagulant proteins (such as protein C, protein S, and antithrombin III), resulting in a hypercoagulable environment that facilitates the formation and stabilization of thrombi (131). Biswas et al. (132)found that TLR2 senses oxidized phospholipids binding to CD36 on platelets, activating the MyD88 signaling pathway, thereby leading to a hypercoagulable state and accelerating thrombosis formation associated with hyperlipidemia. Haseeb et al. (133) discovered that TLR9 markedly boosts the generation of pro-inflammatory and coagulation factors through the activation of the MAPK and NF-κB signaling pathways, which in turn facilitates a hypercoagulable condition.
Additionally, TLR9 activation enhances thrombus stability by inducing the production of type I interferons and regulating thrombin generation and fibrin stability (134). These multiple mechanisms collectively indicate that TLR signaling pathways play an important role in the regulation of the coagulation system in DVT.
4.5 Downstream effects of TLR signaling pathways and their specific roles in DVT
The activation of TLR signaling pathways regulates the occurrence and development of DVT through multiple downstream effector pathways. The main downstream effector pathways include the NF-κB, MAPK, and IRF signaling pathways, which jointly regulate the expression of inflammatory factors and the enhancement of coagulation responses.
4.5.1 NF-κB signaling pathway
As an essential transcription factor that plays a significant role in TLR signaling pathways, NF-κB is vital for the development of DVT. In a rat model featuring femoral fractures, the activation of TLR2 significantly increased the concentrations of NF-κB, COX-2, along with inflammatory factors like IL-6 and P-selectin. Conversely, the inhibition of TLR2 diminished these effects, underscoring the critical role of the TLR2/NF-κB signaling pathway in the development of thrombotic disease following deep vein thrombosis (135). The multiple roles of the NF-κB signaling pathway in inflammation, immune regulation, and disease have been widely studied, with its interaction with TLR signaling being particularly prominent in thrombosis formation (136). In contrast, cotinine, a substance produced from nicotine metabolism, enhances the development of DVT and inflammatory responses through the TLR4/NF-κB signaling pathway, highlighting the essential role of NF-κB in thrombosis progression (137). At the molecular level, TLR signaling activates NF-κB through the myddosome complex, thereby regulating the expression of inflammatory genes, a mechanism of significant importance in thrombosis formation (138). To delve deeper into the function of NF-κB within TLR-related inflammatory responses, scientists have created a range of approaches, including the measurement of NF-κB’s movement into the nucleus using imaging techniques, which offer valuable resources for investigating its roles in conditions such as DVT (139).
4.5.2 MAPK signaling pathway
The MAPK signaling pathway serves as a crucial downstream effector of TLR signaling and is significant in the development of DVT. Studies have shown that the MAPK signaling pathway contributes to the development and advancement of DVT by affecting the expression of inflammatory mediators and various biological processes, such as cell growth and differentiation. For instance, the p38 MAPK and JNK components of the MAPK signaling pathway enhance the production of pro-inflammatory cytokines such as TNF-α and IL-6 by activating TLR signaling, which are crucial in the inflammatory responses associated with DVT (140). Moreover, the MAPK signaling pathway significantly contributes to inflammation triggered by endoplasmic reticulum stress and NOD2 activation, as its activation intensifies the secretion of inflammatory factors in macrophages (141). Within the MAPK signaling pathway, MAPK-activated protein kinases (MAPKAPKs) serve as downstream effectors and contribute to the progression of chronic inflammatory diseases, such as DVT, by influencing the expression of genes related to inflammation (142). In a mouse model of DVT, Fisetin markedly decreased the levels of pro-inflammatory cytokines and oxidative stress through the inhibition of the MAPK signaling pathway (such as p38 and JNK), thereby reinforcing the crucial function of the MAPK pathway in DVT (143). Additionally, UCHL1 plays a role in the inflammatory response of macrophages stimulated by LPS by modulating the MAPK and NF-κB pathways, highlighting the importance of MAPK signaling in inflammation linked to DVT (144). Finally, the MAPK signaling pathway’s independent regulation of TLR expression and cytokine production from NF-κB further emphasizes its central position in DVT inflammatory responses (145).
4.5.3 IRF signaling pathway
TLR signaling pathways play a key role in immune regulation and inflammatory responses by activating IRF transcription factors (e.g., IRF3 and IRF7). The triggering of type I interferon production (like IFN-β) occurs through TLR signaling, which activates IRF3 and IRF7, thereby regulating both antiviral and inflammatory responses (146). In particular, in sterile inflammatory diseases such as DVT, the TLR9-IRF1 signaling pathway affects thrombus formation and resolution by regulating inflammatory and coagulation responses (147). Additionally, the TLR3 signaling pathway induces the expression of type I interferons by activating IRF3, while certain antipsychotic drugs can inhibit TLR3-mediated inflammatory responses by suppressing the PI3K signaling pathway and inhibiting IRF3 activation (148). The irregular activation of the IRF signaling pathway is closely linked to inflammatory conditions, such as DVT. In this context, IRF3 and IRF7 play a crucial role in the regulation of thrombus formation and its resolution by modulating inflammatory responses and coagulation processes (149, 150).
4.6 Cross-regulation and feedback mechanisms of TLR signaling pathways
The involvement of TLR signaling pathways in DVT is evident not only through the activation of specific pathways but also through the interplay and feedback interactions among various signaling routes. Following the activation of TLR4, it triggers not only NF-κB and IRF3 via the MyD88 and TRIF pathways but also plays a role in the immunity and inflammation regulation by influencing additional signaling pathways, such as PI3K/Akt and JAK/STAT (151). These cross-regulation mechanisms enhance the regulatory effects of TLR signaling pathways in DVT and also provide multiple targets for DVT treatment.
4.6.1 PI3K/Akt signaling pathway
The role of the PI3K/Akt signaling pathway is crucial in the regulation of inflammation, cell survival, and metabolic activities, particularly within the framework of TLR signaling. TLR activation (including TLR2 and TLR4) of PI3K/Akt signaling contributes to the regulation of pro-inflammatory cytokines. The expression of TLR4 in macrophages is elevated by hypoxic stress via the PI3K/Akt signaling pathway, which intensifies inflammation associated with ischemic conditions (152). The interplay between the PI3K/Akt/mTOR and TLR/NF-κB signaling pathways is crucial for the response to streptococcal infections. This suggests that TLR2 and TLR4 initiate the activation of the PI3K/Akt/mTOR pathway to control inflammation within mammary epithelial cells (153). Furthermore, the TLR4/PI3K/Akt pathway regulates LPS-induced proliferation of vascular smooth muscle cells (VSMCs), linking this signaling pathway to diseases such as atherosclerosis (154). Collectively, these investigations emphasize the crucial involvement of PI3K/Akt in the modulation of inflammation and immune responses via TLR signaling, indicating possible therapeutic approaches for addressing inflammation across different diseases.
4.6.2 JAK/STAT signaling pathway
The JAK/STAT signaling pathway plays a key role in immune regulation, especially exhibiting important regulatory functions in its interaction with TLR signaling pathways. STAT1 plays a significant role in TLR signaling, particularly after TLR4 activation, where STAT1 enhances TLR-mediated inflammatory responses through interactions with TRAF6 (155). The study showed that TLR3 stimulation could “pre-activate” macrophages, thereby enhancing the immune response to subsequent TLR7 stimulation and promoting the synergistic production of cytokines through the JAK-STAT pathway (156). The crosstalk between JAK/STAT, TLR, and ITAM-dependent pathways in macrophages was also further explored, with the study showing that IFN-γ promotes TLR-mediated immune responses by modulating the interactions among these signaling pathways (157). At the same time, the signaling pathways of TLR work in conjunction with JAK/STAT signaling via cytokines like IFN-γ and IL-10, offering fresh perspectives on the intricate regulatory network of immune system signaling (158).
4.7 Negative regulatory mechanisms of TLR signaling pathways
To avoid excessive stimulation of TLR signaling pathways and mitigate uncontrolled inflammatory responses, the body employs various negative regulatory mechanisms. These mechanisms inhibit key nodes of signal transduction, limiting the intensity and duration of inflammatory responses to maintain immune balance.
4.7.1 A20 protein
A20 (TNFAIP3) functions as an important negative regulator, playing a significant role in modulating the NF-κB signaling pathway during immune responses. It regulates the intensity and duration of inflammatory responses by inhibiting ubiquitination modifications in TLR signaling pathways through its ubiquitin-editing functions. A20 directly affects the activation process of NF-κB through deubiquitination, ensuring the timely termination of immune responses (159). A20 interacts with E3 ligases such as TRAF6 to prevent the activation of these signaling molecules in TLR signaling. For example, Düwel et al. (160) discovered that A20 prevents the prolonged activation of NF-κB through the removal of K63-linked ubiquitin chains from Malt1, an action essential for the correct functioning of immune cells. Shembade et al. (161) further demonstrated that A20 inhibits the excessive activation of TLR signaling pathways by disrupting the E3 ligase activities of TRAF6, TRAF2, and cIAP1, thereby avoiding sustained inflammatory responses.
4.7.2 SOCS family proteins
Emerging evidence has identified Suppressors of Cytokine Signaling (SOCS) proteins as key regulators of TLR signaling networks. These cytokine-induced proteins act as molecular rheostats, maintaining immune homeostasis through negative feedback mechanisms activated by cytokine receptors and pattern recognition receptors, including TLRs themselves. Specific SOCS family members (SOCS1–3 and CIS) differentially regulate TLR signaling by selectively targeting interferon pathways while preserving NF-κB activation, enabling precise control of antiviral responses without compromising inflammatory signaling (162). SOCS proteins show functional specialization in different immune cells, with SOCS1 in myeloid cells like monocytes and macrophages specifically limiting IFN - dependent signaling (163). Recent advances highlight SOCS1 and SOCS3’s dual-layer regulation of the TLR7 pathway in human plasmacytoid dendritic cells, balancing antiviral responses and immune tolerance through IRF7 degradation (164). This complex regulatory paradigm highlights the context-dependent nature of SOCS protein functions, where the cellular microenvironment, receptor crosstalk, and temporal expression patterns collectively determine their immunomodulatory outcomes.
4.8 The role of TLRs in DVT resolution
Early murine model studies demonstrated that the TLR9 signaling pathway facilitates early thrombus resolution by regulating sterile inflammatory responses (including inflammatory cell infiltration, thrombus collagenization, and necrotic pathways), with TLR9 knockout mice exhibiting significantly increased thrombus volumes (134). Further investigations revealed that TLR9 deficiency impairs macrophage function, manifesting as reduced chemotactic capacity and downregulated fibrinolytic gene expression, while exogenous transfusion of wild-type macrophages effectively restored thrombus resolution capability (165). In neutrophils, TLR9 influences early venous thrombus formation through modulation of necrotic markers such as uric acid and NETs (166). Clinical studies showed that although TLR9 mRNA expression in DVT patients showed no significant correlation with thrombus remnants, it was markedly elevated in recurrent DVT cases, suggesting TLR9 may possess a dual regulatory role while also indicating potential mechanistic differences in TLR9 regulation between animal models and human clinical samples (167). Similar to TLR9’s mechanism, TLR4 deficiency was found to inhibit thrombus dissolution by reducing neutrophil/macrophage infiltration and downregulating MMP-9/MCP-1 expression, a process dependent on the NF-κB signaling pathway, thereby expanding understanding of TLR family roles in thrombus clearance (168). Current studies propose a two-phase immune model of thrombus resolution: an early phase where neutrophils stabilize thrombus structure through NETs, followed by a macrophage-dominated fibrotic and endothelialization phase (169). In summary, TLRs play multifaceted roles in resolving deep vein thrombosis by modulating inflammation, interacting with multiple signaling pathways, and influencing immune cell activities including macrophages. Understanding the mechanisms by which TLRs promote thrombus dissolution may facilitate development of novel therapeutic strategies aimed at enhancing DVT resolution while reducing thromboembolic complication risks (170).
5 The potential of TLR signaling pathways as therapeutic targets for DVT
Therapeutic strategies targeting TLR signaling pathways primarily focus on suppressing excessive TLR activation or blocking downstream signaling to mitigate acute-phase inflammatory and coagulation responses, thereby preventing early-stage inflammatory thrombus formation (Table 2).
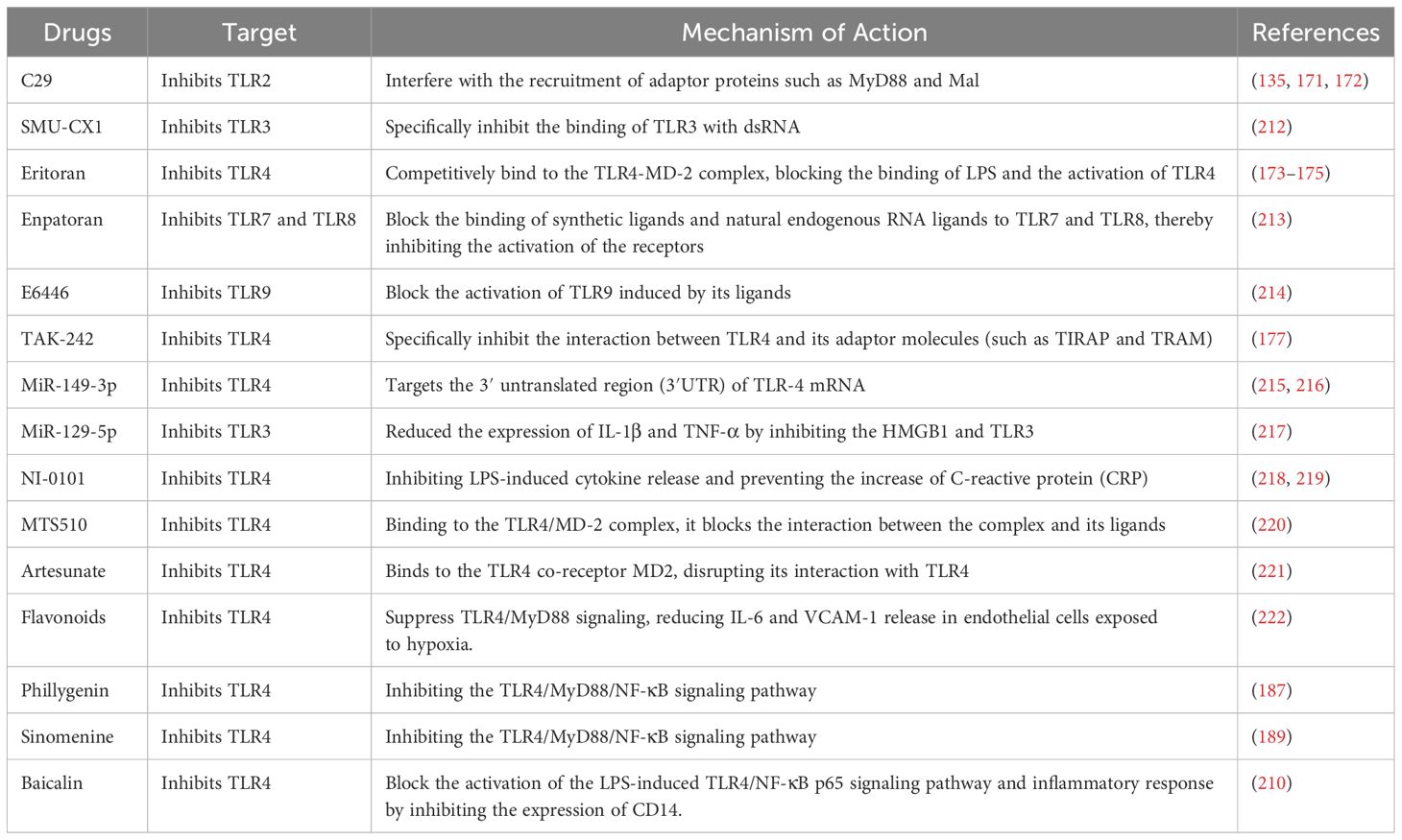
Table 2. Drugs associated with TLR signaling pathways.
5.1 TLR inhibitors/antagonists
TLR inhibitors/antagonists, by inhibiting TLR activation, may effectively alleviate the inflammatory response during thrombosis and reduce the incidence of DVT.
C29, which selectively inhibits TLR2, is able to obstruct the activation of downstream signaling pathways by preventing TLR2 from binding with its ligands. Research indicates that C29 interferes with the interaction between TLR2 and its adaptor protein MyD88 by binding to the BB loop pocket located in the TIR domain of TLR2, which prevents the activation of downstream signaling pathways involving MAPK and NF-κB (171). Subsequent investigations revealed that C29 effectively diminishes the generation of inflammatory substances by blocking NF-κB activation facilitated by TLR2/1. When combined with the analog MMG-11, it exhibits a synergistic effect that further amplifies the suppression of TLR2 signaling (172). Recent research has shown that C29 markedly decreases the expression of inflammatory markers, including IL-6 and TNF-α, through the inhibition of the TLR2/NF-κB/COX-2 signaling pathway, which helps relieve traumatic deep vein thrombosis (135). These studies collectively suggest that C29, as a TLR2 inhibitor, has broad application potential in various diseases involving inflammation and thrombosis.
Eritoran, which is an artificial antagonist of TLR4, has the ability to competitively attach to the TLR4-MD-2 complex, preventing LPS from binding and inhibiting TLR4 activation (173). However, despite showing significant TLR4 inhibitory effects in vitro, its clinical application has been limited. A multicenter randomized controlled trial showed that Eritoran did not significantly reduce the mortality rate of patients with severe sepsis, but it had good safety (174). On the other hand, in a mouse model of influenza virus infection, Eritoran significantly reduced pulmonary inflammatory responses and viral loads by inhibiting the TLR4 signaling pathway, indicating its potential application value in treating influenza-related acute lung injury (175). Recently, there has been considerable discourse surrounding Eritoran’s potential in addressing inflammation, infections, and cancer. Additionally, innovative approaches for regulating TLR4, utilizing disaccharide lipid A analogs, have been suggested, offering fresh avenues for enhancing and applying TLR4 antagonists in clinical settings (176).
TAK-242 (Resatorvid), a small molecule TLR4 inhibitor, selectively binds to the intracellular TIR domain of TLR4, blocking downstream signal transduction and thereby reducing the production of inflammatory factors. Initial research indicated that TAK-242 had the capability to selectively block the interaction of TLR4 with its adaptor proteins (including TIRAP and TRAM), leading to a marked decrease in the synthesis of inflammatory mediators triggered by LPS (like TNF-α and IL-6) and hindering the activation of NF-κB (177). Further research found that the specific inhibitory effect of TAK-242 on TLR4 was independent of other TLRs (such as TLR1/2, TLR3, etc.), indicating its high selectivity (178). Recent studies have shown that TAK-242 significantly reduces the production of inflammatory mediators, such as IL-1β, IL-6, and TNF-α, in synovial fibroblasts derived from patients with osteoarthritis. This effect is achieved by inhibiting the TLR4-NF-κB signaling pathway (179). These studies collectively suggest that TAK-242, as a TLR4 inhibitor, has broad application potential.
5.2 MicroRNAs
MicroRNAs (miRNAs) are small non-coding RNAs that serve important regulatory functions in a range of biological processes. In recent years, the role of microRNAs in TLR signaling pathways and DVT has gradually become a research hotspot. MiRNAs influence TLR signaling by targeting key components of the pathway. For instance, miR-146a and miR-155 are known to regulate the expression of proteins involved in TLR signaling, thereby modulating the immune response (180). A rat model demonstrated the expression patterns of miRNAs and mRNAs in DVT, identifying 22 miRNAs and 487 mRNAs that were significantly different between the DVT group and controls, primarily associated with endothelial cell activity. This suggests that miRNAs are crucial in the development of thrombosis through their regulatory influence on endothelial cells (181). Studies concerning human hemophilia and thrombosis have demonstrated that miRNAs significantly influence thrombosis development by regulating the expression of coagulation and inflammatory mediators, thereby providing a new perspective for the diagnosis and treatment of thrombotic conditions (182). Recent studies have discovered that miRNAs significantly influence the development of DVT by modulating processes like cell growth, differentiation, the release of inflammatory factors, and vascular remodeling. This offers a foundational understanding that supports the use of miRNAs in diagnosing and treating DVT (183). Understanding the roles of miRNAs in TLR signaling and DVT not only provides insights into the molecular mechanisms underlying these conditions but also offers potential targets for therapeutic interventions.
5.3 Monoclonal antibodies
Monoclonal antibodies targeting TLRs are also an effective inhibitory approach. Anti-TLR4 monoclonal antibodies, by specifically blocking the TLR4 signaling pathway, have shown significant therapeutic potential in various inflammatory and injury-related diseases. The Fab fragment derived from a humanized anti-TLR4 monoclonal antibody was discovered to effectively suppress inflammatory responses induced by LPS (184). This effect is achieved by preventing the interaction of TLR4 with its ligand, which leads to a marked decrease in the activation of both NF-κB and MAPK signaling pathways. Consequently, this action inhibits the release of cytokines, thereby offering a theoretical foundation for therapeutic strategies targeting TLR4 that utilize the Fab fragment. Additionally, anti-TLR4 monoclonal antibodies were found to significantly alleviate ventilator-induced lung injury (VILI) by inhibiting MyD88 and NF-κB-dependent signaling pathways, reducing pulmonary inflammatory cell infiltration and cytokine release, and improving pathological damage to lung tissue (185). In recent years, the development of a fully human anti-TLR4 IgG2 antibody has further expanded research in this area. This antibody demonstrated a high affinity for TLR4 in vitro, successfully reducing the levels of TNF-α, IFN-β, and IL-6 that were triggered by LPS, while simultaneously diminishing the phosphorylation of the NF-κB, MAPK, and IRF3 signaling cascades. Notably, in animal studies, the antibody greatly enhanced the survival rate of mice subjected to an LPS challenge (186). Collectively, these studies suggest that monoclonal antibodies targeting TLR4 hold significant potential for application across different disease models, offering novel insights for the clinical management of associated ailments.
5.4 Natural products and herbal
Natural products regulate TLR signaling pathways through various mechanisms and have good anti-inflammatory and anticoagulant effects. Natural products serve as significant potential drugs and offer a theoretical foundation for developing novel strategies to treat DVT.
Phillygenin, a lignan compound derived from the Forsythia suspensa fruit, exhibits notable anti-inflammatory, antioxidant, and neuroprotective properties. In recent years, its pharmacological activities in various disease models have attracted widespread attention, especially its potential therapeutic value in inflammation-related diseases and central nervous system diseases. Phillygenin can effectively inhibit LPS-induced activation and inflammatory response of LX2 cells by inhibiting the TLR4/MyD88/NF-κB signaling pathway, indicating its potential therapeutic value in hepatic fibrosis and related inflammatory diseases (187). In the field of neuroprotection, Phillygenin can reduce neuroinflammatory responses after spinal cord injury and promote functional recovery by inhibiting the TLR4/NF-κB signaling pathway, providing a scientific basis for its application in central nervous system diseases (188). These studies collectively reveal the multiple mechanisms of action of Phillygenin and Forsythia-related components in anti-inflammatory and neuroprotective effects, laying a theoretical foundation for their further development into clinical drugs.
Sinomenine (SIN), a alkaloid extracted from the Chinese medicine Sinomenium acutum, has significant anti-inflammatory, immunomodulatory, and neuroprotective effects. Recently, there has been significant interest in its potential use for treating inflammatory and autoimmune diseases. Studies show that Sinomenine exhibits significant anti-inflammatory and immunomodulatory effects across various disease models by influencing the TLR4/NF-κB signaling pathway (189). The investigation of rheumatoid arthritis (RA) revealed that Sinomenine markedly diminished the inflammatory reaction of fibroblast-like synoviocytes (FLS) while also decreasing the levels of MyD88 and TRAF6 by blocking the TLR4/MyD88/NF-κB signaling pathway, thereby offering molecular support for the treatment of RA (190). Recent research indicates that Sinomenine contributes to the equilibrium between the brain and gut through the modulation of the cholinergic anti-inflammatory pathway (CAP) and the inhibition of the TLR4/NF-κB signaling pathway in a mouse model of Alzheimer’s disease (AD) (191). This results in significant improvements in cognitive abilities and a decrease in inflammatory responses.
Baicalin, a flavonoid derived from the roots of Scutellaria baicalensis, exhibits notable anti-inflammatory, antioxidant, and immunomodulatory properties. In recent years, its role in regulating TLR signaling pathways and related inflammatory diseases has attracted much attention. Research indicates that baicalin may inhibit the activation of the TLR4/NF-κB p65 signaling pathway triggered by LPS, as well as the related inflammatory response, through the suppression of CD14 expression (192). Moreover, the findings indicated that baicalin has the potential to reduce joint inflammation and tissue damage in a rat model of collagen-induced arthritis (CIA) by modulating the TLR2/MYD88/NF-κB p65 signaling pathway, thus offering a novel therapeutic approach for rheumatoid arthritis management (193). Recent research has expanded the range of applications for baicalin, showcasing its significant anti-inflammatory and antioxidant effects in various inflammatory ailments such as hepatitis, rheumatoid arthritis, obesity, and type 2 diabetes by intervening in the TLR/NF-κB signaling pathway (194).
5.5 Combination therapy
In recent years, combination therapies have demonstrated remarkable potential in inflammatory regulation and organ protection, particularly showing significant advancements in synergistic suppression of immune signaling pathways (e.g., TLR) and multi-mechanism interventions. Multiple clinical studies have confirmed that the combination of antiplatelet therapy (Triflusal) with moderate-intensity anticoagulation (Acenocoumarol) reduces vascular event risk by 67% in moderate-to-high risk atrial fibrillation patients without significantly increasing major bleeding rates (195). This combined strategy shows greater efficacy in mechanical heart valve patients, achieving a 35% relative risk reduction in thrombotic events, though requires cautious selection in atrial fibrillation populations based on individual bleeding risks (196). Basic research reveals that the protease inhibitor Bortezomib (VELCADE) combined with tPA suppresses the TLR4/IRAK1 pathway in endothelial cells via miR-146a upregulation, reducing cerebral infarct volume by 46% in rat stroke models while reversing tPA-induced vascular inflammation (197). In oncology and surgical fields, the TLR4 inhibitor TAK-242 combined with estrogen antagonist Fulvestrant inhibits non-small cell lung cancer metastasis (198), while a compound formulation of TAK-242 with hyaluronic acid reduces postoperative abdominal adhesion incidence by 64% in mice through TLR4/ROS signaling inhibition (199). Notably, the combination of Pentoxifylline (PTX) and Dexamethasone demonstrates synergistic suppression of TLR4/7/8 pathways, showing 3.2-fold greater efficacy in neonates than adults through bidirectional regulation of TNF/IL-10 mRNA balance (200). Novel drug delivery systems like omeprazole nanoliposomes achieve targeted inhibition of endosomal TLR3/4/7/8 signaling in macrophages, validating the translational value of “nanocarrier + drug repurposing” strategies (201). These findings align with the critical role of dynamic TLR pathway modulation, exemplified by TAK-242 combined with G-CSF reducing mortality from 66% to 0% in ACLF mouse models (202), further highlighting the broad prospects of combination therapies.
6 Discussion
Although TLR signaling pathways have significant potential as therapeutic targets for DVT, their clinical application still faces many challenges. First, the key role of TLR signaling pathways in immune responses means that their inhibitors may suppress immune function and increase the risk of infection (9). Therefore, the development of TLR inhibitors with high selectivity and low toxicity is one of the current research priorities. Second, the complexity and diversity of TLR signaling pathways mean that the inhibition of a single target may not comprehensively suppress the enhanced inflammatory and coagulation responses, affecting therapeutic effects (203). Moreover, the polymorphism of TLR genes and differences in epigenetic regulation among individuals may also affect the therapeutic effects and safety of TLR inhibitors, requiring the support of personalized treatment strategies (204). Finally, regarding the dual role of TLR signaling: TLR activation can both exacerbate thrombosis (e.g., through pro-inflammatory factor release) (205, 206) and promote DVT resolution (e.g., by regulating macrophage polarization) (165, 207), with precise regulatory strategies remaining to be explored.
TLR signaling pathways play a key role in the inflammatory response of DVT. In-depth studies of their regulatory mechanisms provide important directions for the development of new therapeutic strategies. The development of more specific TLR inhibitors through high-throughput screening and structural optimization to precisely regulate inflammatory responses. For example, studies based on molecular dynamics simulations and molecular docking have found that the Arg241 residue of TLR4 and the Tyr102, Ser120, and Lys122 residues of MD2 are key sites for antagonist ligand binding, providing important information for structure-based TLR4 inhibitor design (208, 209). TLR signaling pathways interact with other signaling pathways such as PI3K/Akt and JAK/STAT, which are involved in the inflammatory response of DVT (210, 211). However, a thorough assessment of potential adverse effects (particularly hemorrhagic complications) is warranted to ensure clinical safety. Therefore, the development of multi-target drugs may more effectively treat DVT and reduce the occurrence of drug resistance. Finally, in-depth studies of the molecular mechanisms of TLR signaling pathways, especially the formation and dynamic changes of the myddosome, will help reveal their role in DVT. For example, recent studies have shown that the myddosome, as the core platform of TLR signal transduction, is crucial for the initiation and maintenance of inflammatory responses through its assembly and functional regulation (138).
7 Conclusion and outlook
In conclusion, the signaling pathways associated with TLR are crucial in the development of DVT. They facilitate the amplification of inflammatory and coagulation reactions via multiple mechanisms, thereby promoting thrombus formation and stabilization. Members of the TLR family, such as TLR4, TLR2, and TLR9, induce the expression of inflammatory and coagulation factors by activating downstream MyD88-dependent and independent signaling pathways. This process promotes the engagement and activation of immune cells, consequently enhancing the development and stability of blood clots. Moreover, TLR signaling pathways further promote thrombus formation and expansion through their interaction with vascular endothelial cells and the coagulation system. The sources of endogenous and exogenous TLR ligands in DVT provide an important driving force for the activation of TLR signaling pathways. Additionally, the negative regulatory mechanisms of TLR signaling pathways play a crucial role in maintaining immune balance and limiting inflammatory responses. The improper regulation of TLR signaling pathways may result in heightened activation, facilitating the onset and progression of DVT.
Based on these mechanisms, therapeutic strategies targeting TLR signaling pathways hold important clinical application potential. A variety of drugs, including TLR antagonists, inhibitors, as well as natural products, can effectively inhibit inflammatory and coagulation responses by regulating key molecules and steps in TLR signaling pathways, reducing thrombus formation and stability. Multi-target combined treatment strategies, by simultaneously regulating multiple key molecules and steps, further enhance therapeutic effects and reduce the occurrence of drug resistance, providing new ideas and methods for the treatment of DVT. However, the complexity and diversity of TLR signaling pathways, as well as their key role in immune responses, remain the main challenges for their clinical application. Future research needs to further explore the distinct regulatory mechanisms and interrelationships of TLR signaling pathways, develop highly selective and low-toxicity TLR inhibitors, formulate personalized treatment strategies, and verify their safety and effectiveness through clinical trials, in order to promote the application and translation of TLR signaling pathway-related therapeutic strategies in DVT.
Author contributions
WS: Investigation, Methodology, Writing – original draft, Writing – review & editing. ZW: Investigation, Supervision, Writing – review & editing. JW: Conceptualization, Investigation, Writing – review & editing. TG: Funding acquisition, Project administration, Resources, Writing – review & editing. JM: Conceptualization, Funding acquisition, Supervision, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This research was supported by the National Natural Science Foundation of China (NO. 82160375), Science and Technology Planning Project of Jiangxi Provincial Health Commission (NO. 202312146), Science and Technology Program of Jiangxi Provincial Administration of Traditional Chinese Medicine (Project NO. 2021A374) and Key research and development plan of Ganzhou (NO. 2023LNS26841).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Heit JA. Epidemiology of venous thromboembolism. Nat Rev Cardiol. (2015) 12:464–74. doi: 10.1038/nrcardio.2015.83
2. Stevens SM, Woller SC, Kreuziger LB, Bounameaux H, Doerschug K, Geersing G-J, et al. Antithrombotic therapy for VTE disease: second update of the CHEST guideline and expert panel report. Chest. (2021) 160:e545–608. doi: 10.1016/j.chest.2021.07.055
3. Martin SS, Aday AW, Allen NB, Almarzooq ZI, Anderson CAM, Arora P, et al. 2025 Heart disease and stroke statistics: A report of US and global data from the american heart association. Circulation. (2025) 151:e41–e660. doi: 10.1161/CIR.0000000000001303
4. Cowan S, Ghayyad K, Conlon MJ, Naik M, Zeini I, Hawks M, et al. Trends in the epidemiology of deep vein thrombosis and pulmonary embolism in patients undergoing surgery. Cureus. (2024) 16:e74925. doi: 10.7759/cureus.74925
5. Hu Y, Zhu L, Tian X, and Duan F. Prevalence of preoperative deep vein thrombosis in long bone fractures of lower limbs: a systematic review and meta-analysis. J Orthop Traumatol Off J Ital Soc Orthop Traumatol. (2023) 24:19. doi: 10.1186/s10195-023-00699-2
6. Mammen EF. Pathogenesis of venous thrombosis. Chest. (1992) 102:640S–4S. doi: 10.1378/chest.102.6_supplement.640s
7. von Brühl M-L, Stark K, Steinhart A, Chandraratne S, Konrad I, Lorenz M, et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice. vivo J Exp Med. (2012) 209:819–35. doi: 10.1084/jem.20112322
8. Akira S and Takeda K. Toll-like receptor signalling. Nat Rev Immunol. (2004) 4:499–511. doi: 10.1038/nri1391
9. Duan T, Du Y, Xing C, Wang HY, and Wang R-F. Toll-like receptor signaling and its role in cell-mediated immunity. Front Immunol. (2022) 13:812774. doi: 10.3389/fimmu.2022.812774
10. Wang K, Huang H, Zhan Q, Ding H, and Li Y. Toll-like receptors in health and disease. MedComm. (2024) 5:e549. doi: 10.1002/mco2.549
11. Fitzpatrick J-M, Minogue E, Curham L, Tyrrell H, Gavigan P, Hind W, et al. MyD88-dependent and -independent signalling via TLR3 and TLR4 are differentially modulated by Δ9-tetrahydrocannabinol and cannabidiol in human macrophages. J Neuroimmunol. (2020) 343:577217. doi: 10.1016/j.jneuroim.2020.577217
12. Ren M, Li R, Luo M, Chen N, Deng X, Yan K, et al. Endothelial cells but not platelets are the major source of Toll-like receptor 4 in the arterial thrombosis and tissue factor expression in mice. Am J Physiol Regul Integr Comp Physiol. (2014) 307:R901–907. doi: 10.1152/ajpregu.00324.2014
13. Mudaliar H, Pollock C, Ma J, Wu H, Chadban S, and Panchapakesan U. The role of TLR2 and 4-mediated inflammatory pathways in endothelial cells exposed to high glucose. PloS One. (2014) 9:e108844. doi: 10.1371/journal.pone.0108844
14. Janssen MCH, Wollersheim H, Schultze-Kool LJ, and Thien T. Local and systemic thrombolytic therapy for acute deep venous thrombosis. Neth J Med. (2005) 63:81–90.
15. Kahn SR. Review: Thrombolysis for DVT reduces postthrombotic syndrome but increases bleeding complications. Ann Intern Med. (2017) 166:JC30. doi: 10.7326/ACPJC-2017-166-6-030
16. Strijkers RHW, Cate-Hoek AJT, Bukkems SFFW, and Wittens CHA. Management of deep vein thrombosis and prevention of post-thrombotic syndrome. BMJ. (2011) 343:d5916. doi: 10.1136/bmj.d5916
17. Lin E, Freedman JE, and Beaulieu LM. Innate immunity and toll-like receptor antagonists: a potential role in the treatment of cardiovascular diseases. Cardiovasc Ther. (2009) 27:117–23. doi: 10.1111/j.1755-5922.2009.00077.x
18. Pawar RD, Ramanjaneyulu A, Kulkarni OP, Lech M, Segerer S, and Anders H-J. Inhibition of Toll-like receptor-7 (TLR-7) or TLR-7 plus TLR-9 attenuates glomerulonephritis and lung injury in experimental lupus. J Am Soc Nephrol JASN. (2007) 18:1721–31. doi: 10.1681/ASN.2006101162
19. Khan A, Khan SU, Khan A, Shal B, Rehman SU, Rehman SU, et al. Anti-inflammatory and anti-rheumatic potential of selective plant compounds by targeting TLR-4/AP-1 signaling: A comprehensive molecular docking and simulation approaches. Mol Basel Switz. (2022) 27:4319. doi: 10.3390/molecules27134319
20. Kaplan RE, Czyrny JJ, Fung TS, Unsworth JD, and Hirsh J. Electrical foot stimulation and implications for the prevention of venous thromboembolic disease. Thromb Haemost. (2002) 88:200–4.
21. Eberhardt RT and Raffetto JD. Chronic venous insufficiency. Circulation. (2014) 130:333–46. doi: 10.1161/CIRCULATIONAHA.113.006898
22. Thomas DP and Roberts HR. Hypercoagulability in venous and arterial thrombosis. Ann Intern Med. (1997) 126:638–44. doi: 10.7326/0003-4819-126-8-199704150-00009
23. Abramson N and Abramson S. Hypercoagulability: clinical assessment and treatment. South Med J. (2001) 94:1013–20. doi: 10.1097/00007611-200194100-00015
24. Douxfils J, Morimont L, and Bouvy C. Oral contraceptives and venous thromboembolism: focus on testing that may enable prediction and assessment of the risk. Semin Thromb Hemost. (2020) 46:872–86. doi: 10.1055/s-0040-1714140
25. Zwaginga JJ, Sixma JJ, and de Groot PG. Activation of endothelial cells induces platelet thrombus formation on their matrix. Studies of new in vitro thrombosis model with low molecular weight heparin as anticoagulant. Arterioscler Dallas Tex. (1990) 10:49–61. doi: 10.1161/01.atv.10.1.49
26. Varga-Szabo D, Pleines I, and Nieswandt B. Cell adhesion mechanisms in platelets. Arterioscler Thromb Vasc Biol. (2008) 28:403–12. doi: 10.1161/ATVBAHA.107.150474
27. Sang Y, Roest M, de Laat B, de Groot PG, and Huskens D. Interplay between platelets and coagulation. Blood Rev. (2021) 46:100733. doi: 10.1016/j.blre.2020.100733
28. Fanning NF, Kell MR, Shorten GD, Kirwan WO, Bouchier-Hayes D, Cotter TG, et al. Circulating granulocyte macrophage colony-stimulating factor in plasma of patients with the systemic inflammatory response syndrome delays neutrophil apoptosis through inhibition of spontaneous reactive oxygen species generation. Shock Augusta Ga. (1999) 11:167–74. doi: 10.1097/00024382-199903000-00003
29. Yago T, Liu Z, Ahamed J, and McEver RP. Cooperative PSGL-1 and CXCR2 signaling in neutrophils promotes deep vein thrombosis in mice. Blood. (2018) 132:1426–37. doi: 10.1182/blood-2018-05-850859
30. Mosevoll KA, Lindås R, Tvedt THA, Bruserud Ø, and Reikvam H. Altered plasma levels of cytokines, soluble adhesion molecules and matrix metalloproteases in venous thrombosis. Thromb Res. (2015) 136:30–9. doi: 10.1016/j.thromres.2015.04.002
31. Charo IF and Taubman MB. Chemokines in the pathogenesis of vascular disease. Circ Res. (2004) 95:858–66. doi: 10.1161/01.RES.0000146672.10582.17
32. Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD, et al. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci U S A. (2010) 107:15880–5. doi: 10.1073/pnas.1005743107
33. Napoleone E, Di Santo A, and Lorenzet R. Monocytes upregulate endothelial cell expression of tissue factor: a role for cell-cell contact and cross-talk. Blood. (1997) 89:541–9. doi: 10.1182/blood.V89.2.541
34. Laurance S, Bertin F-R, Ebrahimian T, Kassim Y, Rys RN, Lehoux S, et al. Gas6 promotes inflammatory (CCR2hiCX3CR1lo) monocyte recruitment in venous thrombosis. Arterioscler Thromb Vasc Biol. (2017) 37:1315–22. doi: 10.1161/ATVBAHA.116.308925
35. Burnand KG, Gaffney PJ, McGuinness CL, Humphries J, Quarmby JW, and Smith A. The role of the monocyte in the generation and dissolution of arterial and venous thrombi. Cardiovasc Surg Lond Engl. (1998) 6:119–25. doi: 10.1016/s0967-2109(97)00162-2
36. Lu M-J, Zhang J-Q, Nie Z-Y, Yan T-H, Cao Y-B, Zhang L-C, et al. Monocyte/macrophage-mediated venous thrombus resolution. Front Immunol. (2024) 15:1429523. doi: 10.3389/fimmu.2024.1429523
37. Loscalzo J. The macrophage and fibrinolysis. Semin Thromb Hemost. (1996) 22:503–6. doi: 10.1055/s-2007-999051
38. Nüsslein-Volhard C. The Toll gene in Drosophila pattern formation. Trends Genet TIG. (2022) 38:231–45. doi: 10.1016/j.tig.2021.09.006
39. Means TK, Golenbock DT, and Fenton MJ. The biology of Toll-like receptors. Cytokine Growth Factor Rev. (2000) 11:219–32. doi: 10.1016/s1359-6101(00)00006-x
40. Lim K-H and Staudt LM. Toll-like receptor signaling. Cold Spring Harb Perspect Biol. (2013) 5:a011247. doi: 10.1101/cshperspect.a011247
41. Li T-T, Ogino S, and Qian ZR. Toll-like receptor signaling in colorectal cancer: carcinogenesis to cancer therapy. World J Gastroenterol. (2014) 20:17699–708. doi: 10.3748/wjg.v20.i47.17699
42. Nagai Y, Garrett KP, Ohta S, Bahrun U, Kouro T, Akira S, et al. Toll-like receptors on hematopoietic progenitor cells stimulate innate immune system replenishment. Immunity. (2006) 24:801–12. doi: 10.1016/j.immuni.2006.04.008
43. Bell JK, Mullen GED, Leifer CA, Mazzoni A, Davies DR, and Segal DM. Leucine-rich repeats and pathogen recognition in Toll-like receptors. Trends Immunol. (2003) 24:528–33. doi: 10.1016/s1471-4906(03)00242-4
44. Park BS, Song DH, Kim HM, Choi B-S, Lee H, and Lee J-O. The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature. (2009) 458:1191–5. doi: 10.1038/nature07830
45. Latz E, Schoenemeyer A, Visintin A, Fitzgerald KA, Monks BG, Knetter CF, et al. TLR9 signals after translocating from the ER to CpG DNA in the lysosome. Nat Immunol. (2004) 5:190–8. doi: 10.1038/ni1028
46. Mineev KS, Goncharuk SA, Goncharuk MV, Volynsky PE, Novikova EV, and Aresinev AS. Spatial structure of TLR4 transmembrane domain in bicelles provides the insight into the receptor activation mechanism. Sci Rep. (2017) 7:6864. doi: 10.1038/s41598-017-07250-4
47. Xu Y, Tao X, Shen B, Horng T, Medzhitov R, Manley JL, et al. Structural basis for signal transduction by the Toll/interleukin-1 receptor domains. Nature. (2000) 408:111–5. doi: 10.1038/35040600
48. Watters TM, Kenny EF, and O’Neill LAJ. Structure, function and regulation of the Toll/IL-1 receptor adaptor proteins. Immunol Cell Biol. (2007) 85:411–9. doi: 10.1038/sj.icb.7100095
49. Takeda K and Akira S. TLR signaling pathways. Semin Immunol. (2004) 16:3–9. doi: 10.1016/j.smim.2003.10.003
50. Gohda J, Matsumura T, and Inoue J. Cutting edge: TNFR-associated factor (TRAF) 6 is essential for MyD88-dependent pathway but not toll/IL-1 receptor domain-containing adaptor-inducing IFN-beta (TRIF)-dependent pathway in TLR signaling. J Immunol Baltim Md 1950. (2004) 173:2913–7. doi: 10.4049/jimmunol.173.5.2913
51. Yamamoto M, Sato S, Mori K, Hoshino K, Takeuchi O, Takeda K, et al. Cutting edge: a novel Toll/IL-1 receptor domain-containing adapter that preferentially activates the IFN-beta promoter in the Toll-like receptor signaling. J Immunol Baltim Md 1950. (2002) 169:6668–72. doi: 10.4049/jimmunol.169.12.6668
52. Motshwene PG, Moncrieffe MC, Grossmann JG, Kao C, Ayaluru M, Sandercock AM, et al. An oligomeric signaling platform formed by the Toll-like receptor signal transducers MyD88 and IRAK-4. J Biol Chem. (2009) 284:25404–11. doi: 10.1074/jbc.M109.022392
53. Napetschnig J and Wu H. Molecular basis of NF-κB signaling. Annu Rev Biophys. (2013) 42:443–68. doi: 10.1146/annurev-biophys-083012-130338
54. Huang P, Han J, and Hui L. MAPK signaling in inflammation-associated cancer development. Protein Cell. (2010) 1:218–26. doi: 10.1007/s13238-010-0019-9
55. Kawai T, Takeuchi O, Fujita T, Inoue J, Mühlradt PF, Sato S, et al. Lipopolysaccharide stimulates the MyD88-independent pathway and results in activation of IFN-regulatory factor 3 and the expression of a subset of lipopolysaccharide-inducible genes. J Immunol Baltim Md. (2001) 1950:167. doi: 10.4049/jimmunol.167.10.5887
56. Guven-Maiorov E, Keskin O, Gursoy A, and Nussinov R. A structural view of negative regulation of the toll-like receptor-mediated inflammatory pathway. Biophys J. (2015) 109:1214–26. doi: 10.1016/j.bpj.2015.06.048
57. Coll RC and O’Neill LAJ. New insights into the regulation of signalling by toll-like receptors and nod-like receptors. J Innate Immun. (2010) 2:406–21. doi: 10.1159/000315469
58. Jin MS, Kim SE, Heo JY, Lee ME, Kim HM, Paik S-G, et al. Crystal structure of the TLR1-TLR2 heterodimer induced by binding of a tri-acylated lipopeptide. Cell. (2007) 130:1071–82. doi: 10.1016/j.cell.2007.09.008
59. Alexopoulou L, Holt AC, Medzhitov R, and Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. (2001) 413:732–8. doi: 10.1038/35099560
60. Stierschneider A and Wiesner C. Shedding light on the molecular and regulatory mechanisms of TLR4 signaling in endothelial cells under physiological and inflamed conditions. Front Immunol. (2023) 14:1264889. doi: 10.3389/fimmu.2023.1264889
61. Zhang C, Wang H, Wang H, Shi S, Zhao P, Su Y, et al. A microsatellite DNA-derived oligodeoxynucleotide attenuates lipopolysaccharide-induced acute lung injury in mice by inhibiting the HMGB1-TLR4-NF-κB signaling pathway. Front Microbiol. (2022) 13:964112. doi: 10.3389/fmicb.2022.964112
62. Atzeni F, Boiardi L, Vaglio A, Nicoli D, Farnetti E, Palmisano A, et al. TLR-4 and VEGF polymorphisms in chronic periaortitis. PloS One. (2013) 8:e62330. doi: 10.1371/journal.pone.0062330
63. Xu S, Wang J, Jiang J, Song J, Zhu W, Zhang F, et al. TLR4 promotes microglial pyroptosis via lncRNA-F630028O10Rik by activating PI3K/AKT pathway after spinal cord injury. Cell Death Dis. (2020) 11:693. doi: 10.1038/s41419-020-02824-z
64. Ahuja A, Kim E, Sung G-H, and Cho JY. STAT3 differentially regulates TLR4-mediated inflammatory responses in early or late phases. Int J Mol Sci. (2020) 21:7675. doi: 10.3390/ijms21207675
65. Yan J, Shen S, He Y, and Li Z. TLR5 silencing reduced hyperammonaemia-induced liver injury by inhibiting oxidative stress and inflammation responses via inactivating NF-κB and MAPK signals. Chem Biol Interact. (2019) 299:102–10. doi: 10.1016/j.cbi.2018.11.026
66. Hwang H-S, Kim J-W, Oh S-H, Song JH, Yang J-W, Zang Y, et al. TLR5 activation induces expression of the pro-inflammatory mediator Urokinase Plasminogen Activator via NF-κB and MAPK signalling pathways in human dental pulp cells. Int Endod J. (2019) 52:1479–88. doi: 10.1111/iej.13140
67. Eng H-L, Hsu Y-Y, and Lin T-M. Differences in TLR7/8 activation between monocytes and macrophages. Biochem Biophys Res Commun. (2018) 497:319–25. doi: 10.1016/j.bbrc.2018.02.079
68. Salvi V, Nguyen HO, Sozio F, Schioppa T, Gaudenzi C, Laffranchi M, et al. SARS-CoV-2-associated ssRNAs activate inflammation and immunity via TLR7/8. JCI Insight. (2021) 6:e150542. doi: 10.1172/jci.insight.150542
69. Aldawsari MF, Alalaiwe A, Khafagy E-S, Al Saqr A, Alshahrani SM, Alsulays BB, et al. Efficacy of SPG-ODN 1826 nanovehicles in inducing M1 phenotype through TLR-9 activation in murine alveolar J774A.1 cells: plausible nano-immunotherapy for lung carcinoma. Int J Mol Sci. (2021) 22:6833. doi: 10.3390/ijms22136833
70. Kulangara VK, Zhong Y, Yang X, Nagarkatti PS, and Nagarkatti M. TLR-9 agonist CpG activates macrophages and modulates expression of microRNAs involved in cancer drug resistance and various signaling pathways. J Immunol. (2022) 208:105.18. doi: 10.4049/jimmunol.208.Supp.105.18
71. Komai T, Inoue M, Okamura T, Morita K, Iwasaki Y, Sumitomo S, et al. Transforming growth factor-β and interleukin-10 synergistically regulate humoral immunity via modulating metabolic signals. Front Immunol. (2018) 9:1364. doi: 10.3389/fimmu.2018.01364
72. Oosting M, Cheng S-C, Bolscher JM, Vestering-Stenger R, Plantinga TS, Verschueren IC, et al. Human TLR10 is an anti-inflammatory pattern-recognition receptor. Proc Natl Acad Sci U S A. (2014) 111:E4478–4484. doi: 10.1073/pnas.1410293111
73. Deb P, Singh S, Kalyoussef E, Hess NJ, Tapping RI, and Fitzgerald-Bocarsly P. TLR10 (CD290) is a regulator of immune responses in human plasmacytoid dendritic cells. J Immunol Baltim Md 1950. (2024) 213:577–87. doi: 10.4049/jimmunol.2200468
74. López-Yglesias AH, Camanzo E, Martin AT, Araujo AM, and Yarovinsky F. TLR11-independent inflammasome activation is critical for CD4+ T cell-derived IFN-γ production and host resistance to Toxoplasma gondii. PloS Pathog. (2019) 15:e1007872. doi: 10.1371/journal.ppat.1007872
75. Rael VE, Yano JA, Huizar JP, Slayden LC, Weiss MA, Turcotte EA, et al. Large-scale mutational analysis identifies UNC93B1 variants that drive TLR-mediated autoimmunity in mice and humans. J Exp Med. (2024) 221:e20232005. doi: 10.1084/jem.20232005
76. Qu D, Wang L, Huo M, Song W, Lau C-W, Xu J, et al. Focal TLR4 activation mediates disturbed flow-induced endothelial inflammation. Cardiovasc Res. (2020) 116:226–36. doi: 10.1093/cvr/cvz046
77. Beckman JD, Abdullah F, Chen C, Kirchner R, Rivera-Rodriguez D, Kiser ZM, et al. Endothelial TLR4 expression mediates vaso-occlusive crisis in sickle cell disease. Front Immunol. (2020) 11:613278. doi: 10.3389/fimmu.2020.613278
78. Wang X, Yao W, Wang M, Zhu J, and Xia L. TLR4-SIRT3 mechanism modulates mitochondrial and redox homeostasis and promotes EPCs recruitment and survival. Oxid Med Cell Longev. (2022) 2022:1282362. doi: 10.1155/2022/1282362
79. Li Y, Huang L, Li J, Li S, Lv J, Zhong G, et al. Targeting TLR4 and regulating the Keap1/Nrf2 pathway with andrographolide to suppress inflammation and ferroptosis in LPS-induced acute lung injury. Chin J Nat Med. (2024) 22:914–28. doi: 10.1016/S1875-5364(24)60727-2
80. Ren G, Bai C, Yi S, Cong Q, and Zhu Y. Mechanisms and therapeutic strategies for MAFLD targeting TLR4 signaling pathways. J Innate Immun. (2024) 16:45–55. doi: 10.1159/000535524
81. Li X, Jiang S, and Tapping RI. Toll-like receptor signaling in cell proliferation and survival. Cytokine. (2010) 49:1–9. doi: 10.1016/j.cyto.2009.08.010
82. Loomis Z, Eigenberger P, Redinius K, Lisk C, Karoor V, Nozik-Grayck E, et al. Hemoglobin induced cell trauma indirectly influences endothelial TLR9 activity resulting in pulmonary vascular smooth muscle cell activation. PloS One. (2017) 12:e0171219. doi: 10.1371/journal.pone.0171219
83. Niklaus M, Klingler P, Weber K, Koessler A, Boeck M, Kobsar A, et al. The involvement of toll-like receptors 2 and 4 in human platelet signalling pathways. Cell Signal. (2020) 76:109817. doi: 10.1016/j.cellsig.2020.109817
84. Carnevale R, Cammisotto V, Bartimoccia S, Nocella C, Castellani V, Bufano M, et al. Toll-like receptor 4-dependent platelet-related thrombosis in SARS-coV-2 infection. Circ Res. (2023) 132:290–305. doi: 10.1161/CIRCRESAHA.122.321541
85. Marín Oyarzún CP, Glembotsky AC, Goette NP, Lev PR, De Luca G, Baroni Pietto MC, et al. Platelet toll-like receptors mediate thromboinflammatory responses in patients with essential thrombocythemia. Front Immunol. (2020) 11:705. doi: 10.3389/fimmu.2020.00705
86. Hoy CK, NaveenKumar SK, Navaz SA, Sugur K, Yalavarthi S, Sarosh C, et al. Calprotectin impairs platelet survival in patients with primary antiphospholipid syndrome. Arthritis Rheumatol Hoboken NJ. (2024) 76:928–35. doi: 10.1002/art.42801
87. Morodomi Y, Kanaji S, Sullivan BM, Zarpellon A, Orje JN, Won E, et al. Inflammatory platelet production stimulated by tyrosyl-tRNA synthetase mimicking viral infection. Proc Natl Acad Sci U S A. (2022) 119:e2212659119. doi: 10.1073/pnas.2212659119
88. Liberale L and Gorog DA. Low-grade endotoxaemia and platelets: a deadly aggregation. Eur Heart J. (2020) 41:3166–8. doi: 10.1093/eurheartj/ehz955
89. Annarapu GK, Nolfi-Donegan D, Reynolds M, Wang Y, Kohut L, Zuckerbraun B, et al. Heme stimulates platelet mitochondrial oxidant production to induce targeted granule secretion. Redox Biol. (2021) 48:102205. doi: 10.1016/j.redox.2021.102205
90. Sachetto ATA and Mackman N. Monocyte tissue factor expression: lipopolysaccharide induction and roles in pathological activation of coagulation. Thromb Haemost. (2023) 123:1017–33. doi: 10.1055/a-2091-7006
91. Satta N, Kruithof EKO, Fickentscher C, Dunoyer-Geindre S, Boehlen F, Reber G, et al. Toll-like receptor 2 mediates the activation of human monocytes and endothelial cells by antiphospholipid antibodies. Blood. (2011) 117:5523–31. doi: 10.1182/blood-2010-11-316158
92. Colasanti T, Alessandri C, Capozzi A, Sorice M, Delunardo F, Longo A, et al. Autoantibodies specific to a peptide of β2-glycoprotein I cross-react with TLR4, inducing a proinflammatory phenotype in endothelial cells and monocytes. Blood. (2012) 120:3360–70. doi: 10.1182/blood-2011-09-378851
93. Bretz NP, Ridinger J, Rupp A-K, Rimbach K, Keller S, Rupp C, et al. Body fluid exosomes promote secretion of inflammatory cytokines in monocytic cells via Toll-like receptor signaling. J Biol Chem. (2013) 288:36691–702. doi: 10.1074/jbc.M113.512806
94. Gamrekelashvili J, Kapanadze T, Sablotny S, Ratiu C, Dastagir K, Lochner M, et al. Notch and TLR signaling coordinate monocyte cell fate and inflammation. eLife. (2020) 9:e57007. doi: 10.7554/eLife.57007
95. Chinnery HR, Leong CM, Chen W, Forrester JV, and McMenamin PG. TLR9 and TLR7/8 activation induces formation of keratic precipitates and giant macrophages in the mouse cornea. J Leukoc Biol. (2015) 97:103–10. doi: 10.1189/jlb.3AB0414-216R
96. Ruipérez V, Astudillo AM, Balboa MA, and Balsinde J. Coordinate regulation of TLR-mediated arachidonic acid mobilization in macrophages by group IVA and group V phospholipase A2s. J Immunol Baltim Md 1950. (2009) 182:3877–83. doi: 10.4049/jimmunol.0804003
97. Jiang M-X, Hong X, Liao B-B, Shi S-Z, Lai X-F, Zheng H-Y, et al. Expression profiling of TRIM protein family in THP1-derived macrophages following TLR stimulation. Sci Rep. (2017) 7:42781. doi: 10.1038/srep42781
98. Miettinen M, Sareneva T, Julkunen I, and Matikainen S. IFNs activate toll-like receptor gene expression in viral infections. Genes Immun. (2001) 2:349–55. doi: 10.1038/sj.gene.6363791
99. Celhar T, Pereira-Lopes S, Thornhill SI, Lee HY, Dhillon MK, Poidinger M, et al. TLR7 and TLR9 ligands regulate antigen presentation by macrophages. Int Immunol. (2016) 28:223–32. doi: 10.1093/intimm/dxv066
100. Akilesh HM, Buechler MB, Duggan JM, Hahn WO, Matta B, Sun X, et al. Chronic TLR7 and TLR9 signaling drives anemia via differentiation of specialized hemophagocytes. Science. (2019) 363:eaao5213. doi: 10.1126/science.aao5213
101. Castanheira FVS and Kubes P. Neutrophils and NETs in modulating acute and chronic inflammation. Blood. (2019) 133:2178–85. doi: 10.1182/blood-2018-11-844530
102. Tsourouktsoglou T-D, Warnatsch A, Ioannou M, Hoving D, Wang Q, and Papayannopoulos V. Histones, DNA, and citrullination promote neutrophil extracellular trap inflammation by regulating the localization and activation of TLR4. Cell Rep. (2020) 31:107602. doi: 10.1016/j.celrep.2020.107602
103. Huang H, Tohme S, Al-Khafaji AB, Tai S, Loughran P, Chen L, et al. Damage-associated molecular pattern-activated neutrophil extracellular trap exacerbates sterile inflammatory liver injury. Hepatol Baltim Md. (2015) 62:600–14. doi: 10.1002/hep.27841
104. Tamarozzi F, Turner JD, Pionnier N, Midgley A, Guimaraes AF, Johnston KL, et al. Wolbachia endosymbionts induce neutrophil extracellular trap formation in human onchocerciasis. Sci Rep. (2016) 6:35559. doi: 10.1038/srep35559
105. Brill A, Fuchs TA, Savchenko AS, Thomas GM, Martinod K, De Meyer SF, et al. Neutrophil extracellular traps promote deep vein thrombosis in mice. J Thromb Haemost JTH. (2012) 10:136–44. doi: 10.1111/j.1538-7836.2011.04544.x
106. Schrottmaier WC, Mussbacher M, Salzmann M, and Assinger A. Platelet-leukocyte interplay during vascular disease. Atherosclerosis. (2020) 307:109–20. doi: 10.1016/j.atherosclerosis.2020.04.018
107. Nagata K, Tsuji T, Todoroki N, Katagiri Y, Tanoue K, Yamazaki H, et al. Activated platelets induce superoxide anion release by monocytes and neutrophils through P-selectin (CD62). J Immunol. (1993) 151:3267–73. doi: 10.4049/jimmunol.151.6.3267
108. Jin R, Yu S, Song Z, Zhu X, Wang C, Yan J, et al. Soluble CD40 ligand stimulates CD40-dependent activation of the β2 integrin mac-1 and protein kinase C zeda (PKCζ) in neutrophils: implications for neutrophil-platelet interactions and neutrophil oxidative burst. PloS One. (2013) 8:e64631. doi: 10.1371/journal.pone.0064631
109. Cerletti C, Tamburrelli C, Izzi B, Gianfagna F, and de Gaetano G. Platelet-leukocyte interactions in thrombosis. Thromb Res. (2012) 129:263–6. doi: 10.1016/j.thromres.2011.10.010
110. Ghasemzadeh M and Hosseini E. Platelet-leukocyte crosstalk: Linking proinflammatory responses to procoagulant state. Thromb Res. (2013) 131:191–7. doi: 10.1016/j.thromres.2012.11.028
111. D’ Atri LP and Schattner M. Platelet toll-like receptors in thromboinflammation. Front Biosci Landmark Ed. (2017) 22:1867–83. doi: 10.2741/4576
112. Swystun LL and Liaw PC. The role of leukocytes in thrombosis. Blood. (2016) 128:753–62. doi: 10.1182/blood-2016-05-718114
113. Ghasemzadeh M, Ahmadi J, and Hosseini E. Platelet-leukocyte crosstalk in COVID-19: How might the reciprocal links between thrombotic events and inflammatory state affect treatment strategies and disease prognosis? Thromb Res. (2022) 213:179–94. doi: 10.1016/j.thromres.2022.03.022
114. Amin A, Nikdoust F, Khorram S, Marashi SM, Ghanavati P, Ameri F, et al. Dengue virus infection: how platelet-leukocyte crosstalk shapes thrombotic events and inflammation. Mol Biol Rep. (2025) 52:119. doi: 10.1007/s11033-025-10222-x
115. Hally K, Fauteux-Daniel S, Hamzeh-Cognasse H, Larsen P, and Cognasse F. Revisiting platelets and toll-like receptors (TLRs): at the interface of vascular immunity and thrombosis. Int J Mol Sci. (2020) 21:6150. doi: 10.3390/ijms21176150
116. Blair P, Rex S, Vitseva O, Beaulieu L, Tanriverdi K, Chakrabarti S, et al. Stimulation of Toll-like receptor 2 in human platelets induces a thromboinflammatory response through activation of phosphoinositide 3-kinase. Circ Res. (2009) 104:346–54. doi: 10.1161/CIRCRESAHA.108.185785
117. Koupenova M, Vitseva O, MacKay CR, Beaulieu LM, Benjamin EJ, Mick E, et al. Platelet-TLR7 mediates host survival and platelet count during viral infection in the absence of platelet-dependent thrombosis. Blood. (2014) 124:791–802. doi: 10.1182/blood-2013-11-536003
118. Banerjee M, Huang Y, Joshi S, Popa GJ, Mendenhall MD, Wang QJ, et al. Platelets endocytose viral particles and are activated via TLR (Toll-like receptor) signaling. Arterioscler Thromb Vasc Biol. (2020) 40:1635–50. doi: 10.1161/ATVBAHA.120.314180
119. Koupenova M, Corkrey HA, Vitseva O, Manni G, Pang CJ, Clancy L, et al. The role of platelets in mediating a response to human influenza infection. Nat Commun. (2019) 10:1780. doi: 10.1038/s41467-019-09607-x
120. Semeraro F, Ammollo CT, Morrissey JH, Dale GL, Friese P, Esmon NL, et al. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: involvement of platelet TLR2 and TLR4. Blood. (2011) 118:1952–61. doi: 10.1182/blood-2011-03-343061
121. Elaskalani O, Abdol Razak NB, and Metharom P. Neutrophil extracellular traps induce aggregation of washed human platelets independently of extracellular DNA and histones. Cell Commun Signal CCS. (2018) 16:24. doi: 10.1186/s12964-018-0235-0
122. Campos J, Ponomaryov T, De Prendergast A, Whitworth K, Smith CW, Khan AO, et al. Neutrophil extracellular traps and inflammasomes cooperatively promote venous thrombosis in mice. Blood Adv. (2021) 5:2319–24. doi: 10.1182/bloodadvances.2020003377
123. Sung P-S, Huang T-F, and Hsieh S-L. Extracellular vesicles from CLEC2-activated platelets enhance dengue virus-induced lethality via CLEC5A/TLR2. Nat Commun. (2019) 10:2402. doi: 10.1038/s41467-019-10360-4
124. Vogel S, Rath D, Borst O, Mack A, Loughran P, Lotze MT, et al. Platelet-derived high-mobility group box 1 promotes recruitment and suppresses apoptosis of monocytes. Biochem Biophys Res Commun. (2016) 478:143–8. doi: 10.1016/j.bbrc.2016.07.078
125. Stark K, Philippi V, Stockhausen S, Busse J, Antonelli A, Miller M, et al. Disulfide HMGB1 derived from platelets coordinates venous thrombosis in mice. Blood. (2016) 128:2435–49. doi: 10.1182/blood-2016-04-710632
126. Hally KE, Bird GK, La Flamme AC, Harding SA, and Larsen PD. Platelets modulate multiple markers of neutrophil function in response to in vitro Toll-like receptor stimulation. PloS One. (2019) 14:e0223444. doi: 10.1371/journal.pone.0223444
127. Hally KE, La Flamme AC, Harding SA, and Larsen PD. Platelets regulate leucocyte responses to Toll-like receptor stimulation. Clin Transl Immunol. (2018) 7:e1036. doi: 10.1002/cti2.1036
128. Maugeri N, Baldini M, Ramirez GA, Rovere-Querini P, and Manfredi AA. Platelet-leukocyte deregulated interactions foster sterile inflammation and tissue damage in immune-mediated vessel diseases. Thromb Res. (2012) 129:267–73. doi: 10.1016/j.thromres.2011.12.001
129. Yang X, Li L, Liu J, Lv B, and Chen F. Extracellular histones induce tissue factor expression in vascular endothelial cells via TLR and activation of NF-κB and AP-1. Thromb Res. (2016) 137:211–8. doi: 10.1016/j.thromres.2015.10.012
130. Xia L, Zhou H, Hu L, Xie H, Wang T, Xu Y, et al. Both NF-κB and c-Jun/AP-1 involved in anti-β2GPI/β2GPI-induced tissue factor expression in monocytes. Thromb Haemost. (2013) 109:643–51. doi: 10.1160/TH12-09-0655
131. Lu Z, Li Y, Jin J, Zhang X, Lopes-Virella MF, and Huang Y. Toll-like receptor 4 activation in microvascular endothelial cells triggers a robust inflammatory response and cross talk with mononuclear cells via interleukin-6. Arterioscler Thromb Vasc Biol. (2012) 32:1696–706. doi: 10.1161/ATVBAHA.112.251181
132. Biswas S, Zimman A, Gao D, Byzova TV, and Podrez EA. TLR2 plays a key role in platelet hyperreactivity and accelerated thrombosis associated with hyperlipidemia. Circ Res. (2017) 121:951–62. doi: 10.1161/CIRCRESAHA.117.311069
133. Haseeb M, Choi YS, Patra MC, Jeong U, Lee WH, Qayyum N, et al. Discovery of novel small molecule dual inhibitor targeting toll-like receptors 7 and 9. J Chem Inf Model. (2024) 64:5090–107. doi: 10.1021/acs.jcim.4c00578
134. Henke PK, Mitsuya M, Luke CE, Elfline MA, Baldwin JF, Deatrick KB, et al. Toll-like receptor 9 signaling is critical for early experimental deep vein thrombosis resolution. Arterioscler Thromb Vasc Biol. (2011) 31:43–9. doi: 10.1161/ATVBAHA.110.216317
135. Guo T, Xiong L, Xie J, Zeng J, Huang Z, Yao M, et al. TLR2 promotes traumatic deep venous thrombosis of the lower extremity following femoral fracture by activating the NF−κB/COX−2 signaling pathway in rats. Exp Ther Med. (2024) 28:436. doi: 10.3892/etm.2024.12725
136. Guo Q, Jin Y, Chen X, Ye X, Shen X, Lin M, et al. NF-κB in biology and targeted therapy: new insights and translational implications. Signal Transduct Target Ther. (2024) 9:53. doi: 10.1038/s41392-024-01757-9
137. Cheng Z, Jia W, Tian X, Jiang P, Zhang Y, Li J, et al. Cotinine inhibits TLR4/NF-κB signaling pathway and improves deep vein thrombosis in rats. Biosci Rep. (2020) 40:BSR20201293. doi: 10.1042/BSR20201293
138. Fisch D, Zhang T, Sun H, Ma W, Tan Y, Gygi SP, et al. Molecular definition of the endogenous Toll-like receptor signalling pathways. Nature. (2024) 631:635–44. doi: 10.1038/s41586-024-07614-7
139. Ernst O, Vayttaden SJ, and Fraser IDC. Measurement of NF-κB activation in TLR-activated macrophages. Methods Mol Biol Clifton NJ. (2018) 1714:67–78. doi: 10.1007/978-1-4939-7519-8_5
140. Kaminska B. MAPK signalling pathways as molecular targets for anti-inflammatory therapy–from molecular mechanisms to therapeutic benefits. Biochim Biophys Acta. (2005) 1754:253–62. doi: 10.1016/j.bbapap.2005.08.017
141. Peng S, Zhao Y, Jiang W, Long Y, Hu T, Li M, et al. MAPK signaling mediated intestinal inflammation induced by endoplasmic reticulum stress and NOD2. Mol Cell Biochem. (2025) 480:3709–17. doi: 10.1007/s11010-025-05212-3
142. Moens U, Kostenko S, and Sveinbjørnsson B. The role of mitogen-activated protein kinase-activated protein kinases (MAPKAPKs) in inflammation. Genes. (2013) 4:101–33. doi: 10.3390/genes4020101
143. Liu H and Lu Q. Fisetin alleviates inflammation and oxidative stress in deep vein thrombosis via MAPK and NRF2 signaling pathway. Int J Mol Sci. (2024) 25:3724. doi: 10.3390/ijms25073724
144. Zhang Z, Liu N, Chen X, Zhang F, Kong T, Tang X, et al. UCHL1 regulates inflammation via MAPK and NF-κB pathways in LPS-activated macrophages. Cell Biol Int. (2021) 45:2107–17. doi: 10.1002/cbin.11662
145. Peroval MY, Boyd AC, Young JR, and Smith AL. A critical role for MAPK signalling pathways in the transcriptional regulation of toll like receptors. PloS One. (2013) 8:e51243. doi: 10.1371/journal.pone.0051243
146. Honda K and Taniguchi T. Toll-like receptor signaling and IRF transcription factors. IUBMB Life. (2006) 58:290–5. doi: 10.1080/15216540600702206
147. Colonna M. TLR pathways and IFN-regulatory factors: to each its own. Eur J Immunol. (2007) 37:306–9. doi: 10.1002/eji.200637009
148. Zhu J, Smith K, Hsieh PN, Mburu YK, Chattopadhyay S, Sen GC, et al. High-throughput screening for TLR3-IFN regulatory factor 3 signaling pathway modulators identifies several antipsychotic drugs as TLR inhibitors. J Immunol Baltim Md 1950. (2010) 184:5768–76. doi: 10.4049/jimmunol.0903559
149. Matta B, Song S, Li D, and Barnes BJ. Interferon regulatory factor signaling in autoimmune disease. Cytokine. (2017) 98:15–26. doi: 10.1016/j.cyto.2017.02.006
150. Moynagh PN. TLR signalling and activation of IRFs: revisiting old friends from the NF-kappaB pathway. Trends Immunol. (2005) 26:469–76. doi: 10.1016/j.it.2005.06.009
151. Liu Q and Ding JL. The molecular mechanisms of TLR-signaling cooperation in cytokine regulation. Immunol Cell Biol. (2016) 94:538–42. doi: 10.1038/icb.2016.18
152. Kim S, Jeong E, Joung S, and Lee J. PI3K/Akt contributes to increased expression of Toll-like receptor 4 in macrophages exposed to hypoxic stress. Biochem Biophys Res Commun. (2012) 419 3:466–71. doi: 10.1016/j.bbrc.2012.02.015
153. Li B, Xi P, Wang Z, Han X, Xu Y, Zhang Y, et al. PI3K/Akt/mTOR signaling pathway participates in Streptococcus uberis-induced inflammation in mammary epithelial cells in concert with the classical TLRs/NF-ĸB pathway. Vet Microbiol. (2018) 227:103–11. doi: 10.1016/j.vetmic.2018.10.031
154. Jiang D, Li D, Cao L, Wang L, Zhu S, Xu T, et al. Positive feedback regulation of proliferation in vascular smooth muscle cells stimulated by lipopolysaccharide is mediated through the TLR 4/Rac1/Akt pathway. PloS One. (2014) 9:e92398. doi: 10.1371/journal.pone.0092398
155. Luu K, Greenhill CJ, Majoros A, Decker T, Jenkins BJ, and Mansell A. STAT1 plays a role in TLR signal transduction and inflammatory responses. Immunol Cell Biol. (2014) 92:761–9. doi: 10.1038/icb.2014.51
156. Liu B, Liu Q, Yang L, Palaniappan SK, Bahar I, Thiagarajan P, et al. Innate immune memory and homeostasis may be conferred through crosstalk between the TLR3 and TLR7 pathways. Sci Signal. (2016) 9:70–0. doi: 10.1126/scisignal.aac9340
157. Hu X, Chen J, Wang L, and Ivashkiv LB. Crosstalk among Jak-STAT, Toll-like receptor, and ITAM-dependent pathways in macrophage activation. J Leukoc Biol. (2007) 82:237–43. doi: 10.1189/jlb.1206763
158. Ivashkiv L. “Crosstalk with the Jak-STAT Pathway in Inflammation.,” In: Decker T and Müller M, editors. Jak-Stat Signaling: From Basics to Disease. Vienna: Springer (2012). p. 353–70. doi: 10.1007/978-3-7091-0891-8_19
159. Dabbah-Krancher G, Ruchinskas A, and Snow A. A20 is a negative regulator of both NF-κB and JNK signaling following T cell receptor stimulation in humans. J Immunol. (2024) 212:0213_4454. doi: 10.4049/jimmunol.212.supp.0213.4454
160. Düwel M, Welteke V, Oeckinghaus A, Baens M, Kloo B, Ferch U, et al. A20 negatively regulates T cell receptor signaling to NF-κB by cleaving malt1 ubiquitin chains1. J Immunol. (2009) 182:7718–28. doi: 10.4049/jimmunol.0803313
161. Shembade N, Ma A, and Harhaj E. Inhibition of NF-κB signaling by A20 through disruption of ubiquitin enzyme complexes. Science. (2010) 327:1135–9. doi: 10.1126/science.1182364
162. Baetz A, Frey M, Heeg K, and Dalpke A. Suppressor of cytokine signaling (SOCS) proteins indirectly regulate toll-like receptor signaling in innate immune cells*♦. J Biol Chem. (2004) 279:54708–15. doi: 10.1074/JBC.M410992200
163. Prêle CM, Woodward EA, Bisley J, Keith-Magee A, Nicholson SE, and Hart PH. SOCS1 regulates the IFN but not NFkappaB pathway in TLR-stimulated human monocytes and macrophages. J Immunol Baltim Md 1950. (2008) 181:8018–26. doi: 10.4049/jimmunol.181.11.8018
164. Yu C-F, Peng W-M, Schlee M, Barchet W, Eis-Hübinger AM, Kolanus W, et al. SOCS1 and SOCS3 target IRF7 degradation to suppress TLR7-mediated type I IFN production of human plasmacytoid dendritic cells. J Immunol Baltim Md 1950. (2018) 200:4024–35. doi: 10.4049/jimmunol.1700510
165. Dewyer NA, El-Sayed OM, Luke CE, Elfline M, Kittan N, Allen R, et al. Divergent effects of Tlr9 deletion in experimental late venous thrombosis resolution and vein wall injury. Thromb Haemost. (2015) 114:1028–37. doi: 10.1160/TH14-12-1031
166. El-Sayed OM, Dewyer NA, Luke CE, Elfline M, Laser A, Hogaboam C, et al. Intact Toll-like receptor 9 signaling in neutrophils modulates normal thrombogenesis in mice. J Vasc Surg. (2016) 64:1450–1458.e1. doi: 10.1016/j.jvs.2015.08.070
167. Cheung YW, Bouman AC, Castoldi E, Wielders SJ, Spronk HMH, Ten Cate H, et al. Toll-like receptor 9 gene expression in the post-thrombotic syndrome, residual thrombosis and recurrent deep venous thrombosis: A case-control study. Thromb Res. (2016) 140:106–9. doi: 10.1016/j.thromres.2016.02.025
168. Yuan H, Huang X, and Ding J. Toll-like receptor 4 deficiency in mice impairs venous thrombus resolution. Front Mol Biosci. (2023) 10:1165589. doi: 10.3389/fmolb.2023.1165589
169. Nicklas JM, Gordon AE, and Henke PK. Resolution of deep venous thrombosis: proposed immune paradigms. Int J Mol Sci. (2020) 21:2080. doi: 10.3390/ijms21062080
170. Saha P, Humphries J, Modarai B, Mattock K, Waltham M, Evans CE, et al. Leukocytes and the natural history of deep vein thrombosis: current concepts and future directions. Arterioscler Thromb Vasc Biol. (2011) 31:506–12. doi: 10.1161/ATVBAHA.110.213405
171. Mistry P, Laird MHW, Schwarz RS, Greene S, Dyson T, Snyder GA, et al. Inhibition of TLR2 signaling by small molecule inhibitors targeting a pocket within the TLR2 TIR domain. Proc Natl Acad Sci U S A. (2015) 112:5455–60. doi: 10.1073/pnas.1422576112
172. Grabowski M, Murgueitio MS, Bermudez M, Wolber G, and Weindl G. The novel small-molecule antagonist MMG-11 preferentially inhibits TLR2/1 signaling. Biochem Pharmacol. (2020) 171:113687. doi: 10.1016/j.bcp.2019.113687
173. Kim HM, Park BS, Kim J-I, Kim SE, Lee J, Oh SC, et al. Crystal structure of the TLR4-MD-2 complex with bound endotoxin antagonist Eritoran. Cell. (2007) 130:906–17. doi: 10.1016/j.cell.2007.08.002
174. Opal SM, Laterre P-F, Francois B, LaRosa SP, Angus DC, Mira J-P, et al. Effect of eritoran, an antagonist of MD2-TLR4, on mortality in patients with severe sepsis: the ACCESS randomized trial. JAMA. (2013) 309:1154–62. doi: 10.1001/jama.2013.2194
175. Shirey KA, Lai W, Scott AJ, Lipsky M, Mistry P, Pletneva LM, et al. The TLR4 antagonist Eritoran protects mice from lethal influenza infection. Nature. (2013) 497:498–502. doi: 10.1038/nature12118
176. Heine H and Zamyatina A. Therapeutic targeting of TLR4 for inflammation, infection, and cancer: A perspective for disaccharide lipid A mimetics. Pharm Basel Switz. (2022) 16:23. doi: 10.3390/ph16010023
177. Kawamoto T, Ii M, Kitazaki T, Iizawa Y, and Kimura H. TAK-242 selectively suppresses Toll-like receptor 4-signaling mediated by the intracellular domain. Eur J Pharmacol. (2008) 584:40–8. doi: 10.1016/j.ejphar.2008.01.026
178. Matsunaga N, Tsuchimori N, Matsumoto T, and Ii M. TAK-242 (resatorvid), a small-molecule inhibitor of Toll-like receptor (TLR) 4 signaling, binds selectively to TLR4 and interferes with interactions between TLR4 and its adaptor molecules. Mol Pharmacol. (2011) 79:34–41. doi: 10.1124/mol.110.068064
179. Khomeijani-Farahani M, Karami J, Farhadi E, Soltani S, Delbandi A-A, Shekarabi M, et al. TAK-242 (Resatorvid) inhibits proinflammatory cytokine production through the inhibition of NF-κB signaling pathway in fibroblast-like synoviocytes in osteoarthritis patients. Adv Rheumatol Lond Engl. (2024) 64:46. doi: 10.1186/s42358-024-00385-9
180. Yang B, Yang R, Xu B, Fu J, Qu X, Li L, et al. miR-155 and miR-146a collectively regulate meningitic Escherichia coli infection-mediated neuroinflammatory responses. J Neuroinflammation. (2021) 18:114. doi: 10.1186/s12974-021-02165-4
181. Jin Q-Q, Sun J-H, Du Q-X, Lu X-J, Zhu X-Y, Fan H-L, et al. Integrating microRNA and messenger RNA expression profiles in a rat model of deep vein thrombosis. Int J Mol Med. (2017) 40:1019–28. doi: 10.3892/ijmm.2017.3105
182. Jankowska KI, Sauna ZE, and Atreya CD. Role of microRNAs in hemophilia and thrombosis in humans. Int J Mol Sci. (2020) 21:3598. doi: 10.3390/ijms21103598
183. Fang C, Huang F, Yao M, Wang Z, Ma J, Wu D, et al. Advances in microRNA regulation of deep vein thrombosis through venous vascular endothelial cells (Review). Mol Med Rep. (2024) 29:96. doi: 10.3892/mmr.2024.13220
184. Cai B, Wang M, Zhu X, Xu J, Zheng W, Zhang Y, et al. The fab fragment of a humanized anti-toll like receptor 4 (TLR4) monoclonal antibody reduces the lipopolysaccharide response via TLR4 in mouse macrophage. Int J Mol Sci. (2015) 16:25502–15. doi: 10.3390/ijms161025502
185. Huang C, Pan L, Lin F, Dai H, and Fu R. Monoclonal antibody against Toll-like receptor 4 attenuates ventilator-induced lung injury in rats by inhibiting MyD88- and NF-κB-dependent signaling. Int J Mol Med. (2017) 39:693–700. doi: 10.3892/ijmm.2017.2873
186. Wang Y, Gong D, Yao C, Zheng F, Zhou T, Cao Q, et al. Human monoclonal anti−TLR4 antibody negatively regulates lipopolysaccharide−induced inflammatory responses in mouse macrophages. Mol Med Rep. (2020) 22:4125–34. doi: 10.3892/mmr.2020.11500
187. Hu N, Wang C, Dai X, Zhou M, Gong L, Yu L, et al. Phillygenin inhibits LPS-induced activation and inflammation of LX2 cells by TLR4/MyD88/NF-κB signaling pathway. J Ethnopharmacol. (2020) 248:112361. doi: 10.1016/j.jep.2019.112361
188. Zhang Y, Xiao S, Dan F, Yao G, Hong S, Liu J, et al. Phillygenin inhibits neuroinflammation and promotes functional recovery after spinal cord injury via TLR4 inhibition of the NF-κB signaling pathway. J Orthop Transl. (2024) 48:133–45. doi: 10.1016/j.jot.2024.07.013
189. Jiang S, Li S, Pang S, Liu M, Sun H, Zhang N, et al. A systematic review: Sinomenine. Heliyon. (2024) 10:e29976. doi: 10.1016/j.heliyon.2024.e29976
190. Yao R-B, Zhao Z-M, Zhao L-J, and Cai H. Sinomenine inhibits the inflammatory responses of human fibroblast-like synoviocytes via the TLR4/MyD88/NF-κB signaling pathway in rheumatoid arthritis. Pharm. (2017) 72:355–60. doi: 10.1691/ph.2017.6946
191. Ni H, Liu M, Cao M, Zhang L, Zhao Y, Yi L, et al. Sinomenine regulates the cholinergic anti-inflammatory pathway to inhibit TLR4/NF-κB pathway and protect the homeostasis in brain and gut in scopolamine-induced Alzheimer’s disease mice. BioMed Pharmacother Biomedecine Pharmacother. (2024) 171:116190. doi: 10.1016/j.biopha.2024.116190
192. Fu Y-J, Xu B, Huang S-W, Luo X, Deng X-L, Luo S, et al. Baicalin prevents LPS-induced activation of TLR4/NF-κB p65 pathway and inflammation in mice via inhibiting the expression of CD14. Acta Pharmacol Sin. (2021) 42:88–96. doi: 10.1038/s41401-020-0411-9
193. Bai L, Bai Y, Yang Y, Zhang W, Huang L, Ma R, et al. Baicalin alleviates collagen−induced arthritis and suppresses TLR2/MYD88/NF−κB p65 signaling in rats and HFLS−RAs. Mol Med Rep. (2020) 22:2833–41. doi: 10.3892/mmr.2020.11369
194. Jiang M, Li Z, and Zhu G. Immunological regulatory effect of flavonoid baicalin on innate immune toll-like receptors. Pharmacol Res. (2020) 158:104890. doi: 10.1016/j.phrs.2020.104890
195. Pérez-Gómez F, Alegría E, Berjón J, Iriarte JA, Zumalde J, Salvador A, et al. Comparative effects of antiplatelet, anticoagulant, or combined therapy in patients with valvular and nonvalvular atrial fibrillation: A randomized multicenter study. J Am Coll Cardiol. (2004) 44:1557–66. doi: 10.1016/j.jacc.2004.05.084
196. Vedovati MC, Becattini C, and Agnelli G. Combined oral anticoagulants and antiplatelets: benefits and risks. Intern Emerg Med. (2010) 5:281–90. doi: 10.1007/s11739-010-0349-x
197. Zhang L, Chopp M, Liu X, Teng H, Tang T, Kassis H, et al. Combination therapy with VELCADE and tissue plasminogen activator is neuroprotective in aged rats after stroke and targets microRNA-146a and the toll-like receptor signaling pathway. Arterioscler Thromb Vasc Biol. (2012) 32:1856–64. doi: 10.1161/ATVBAHA.112.252619
198. Fan S, Liao Y, Qiu W, Li L, Li D, Cao X, et al. Targeting Toll-like receptor 4 with CLI-095 (TAK-242) enhances the antimetastatic effect of the estrogen receptor antagonist fulvestrant on non-small cell lung cancer. Clin Transl Oncol Off Publ Fed Span Oncol Soc Natl Cancer Inst Mex. (2020) 22:2074–86. doi: 10.1007/s12094-020-02353-3
199. Liu D, Tong H, Guo Y, Liu B, Ye C, Yang N, et al. The Toll-like receptor 4 antagonist TAK-242 in combination with sodium hyaluronate alleviates postoperative abdominal adhesion in a mouse model. BMC Med Genomics. (2024) 17:257. doi: 10.1186/s12920-024-02031-1
200. Speer EM, Dowling DJ, Xu J, Ozog LS, Mathew JA, Chander A, et al. Pentoxifylline, dexamethasone and azithromycin demonstrate distinct age-dependent and synergistic inhibition of TLR- and inflammasome-mediated cytokine production in human newborn and adult blood. vitro PloS One. (2018) 13:e0196352. doi: 10.1371/journal.pone.0196352
201. Sun L, Liu Y, Liu X, Wang R, Gong J, Saferali A, et al. Nano-enabled reposition of proton pump inhibitors for TLR inhibition: toward A new targeted nanotherapy for acute lung injury. Adv Sci Weinh Baden-Wurtt Ger. (2022) 9:e2104051. doi: 10.1002/advs.202104051
202. Engelmann C, Habtesion A, Hassan M, Kerbert AJ, Hammerich L, Novelli S, et al. Combination of G-CSF and a TLR4 inhibitor reduce inflammation and promote regeneration in a mouse model of ACLF. J Hepatol. (2022) 77:1325–38. doi: 10.1016/j.jhep.2022.07.006
203. Chen Y-H, Wu K-H, and Wu H-P. Unraveling the complexities of toll-like receptors: from molecular mechanisms to clinical applications. Int J Mol Sci. (2024) 25:5037. doi: 10.3390/ijms25095037
204. Foster SL, Hargreaves DC, and Medzhitov R. Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature. (2007) 447:972–8. doi: 10.1038/nature05836
205. Iba T, Levi M, and Levy JH. Intracellular communication and immunothrombosis in sepsis. J Thromb Haemost JTH. (2022) 20:2475–84. doi: 10.1111/jth.15852
206. Shafeghat M, Kazemian S, Aminorroaya A, Aryan Z, and Rezaei N. Toll-like receptor 7 regulates cardiovascular diseases. Int Immunopharmacol. (2022) 113:109390. doi: 10.1016/j.intimp.2022.109390
207. Watanabe S, Alexander M, Misharin AV, and Budinger GRS. The role of macrophages in the resolution of inflammation. J Clin Invest. (2019) 129:2619–28. doi: 10.1172/JCI124615
208. Yahaya M a. F, Bakar ARA, Stanslas J, Nordin N, Zainol M, and Mehat MZ. Insights from molecular docking and molecular dynamics on the potential of vitexin as an antagonist candidate against lipopolysaccharide (LPS) for microglial activation in neuroinflammation. BMC Biotechnol. (2021) 21:38. doi: 10.1186/s12896-021-00697-4
209. Zhang Y, Liang X, Bao X, Xiao W, and Chen G. Toll-like receptor 4 (TLR4) inhibitors: Current research and prospective. Eur J Med Chem. (2022) 235:114291. doi: 10.1016/j.ejmech.2022.114291
210. Guo L-T, Wang S-Q, Su J, Xu L-X, Ji Z-Y, Zhang R-Y, et al. Baicalin ameliorates neuroinflammation-induced depressive-like behavior through inhibition of toll-like receptor 4 expression via the PI3K/AKT/FoxO1 pathway. J Neuroinflammation. (2019) 16:95. doi: 10.1186/s12974-019-1474-8
211. Liu Y, Zhang M, Zeng L, Lai Y, Wu S, and Su X. Wogonin upregulates SOCS3 to alleviate the injury in Diabetic Nephropathy by inhibiting TLR4-mediated JAK/STAT/AIM2 signaling pathway. Mol Med Camb Mass. (2024) 30:78. doi: 10.1186/s10020-024-00845-4
212. Pan Y, Fu Q, Li Y, Yang J, and Cheng K. Discovery of an ellipticine derivative as TLR3 inhibitor against influenza A virus and SARS-CoV-2. Bioorg Med Chem Lett. (2024) 101:129672. doi: 10.1016/j.bmcl.2024.129672
213. Klopp-Schulze L, Gopalakrishnan S, Yalkinoglu Ö, Kuroki Y, Lu H, Goteti K, et al. Asia-inclusive global development of enpatoran: results of an ethno-bridging study, intrinsic/extrinsic factor assessments and disease trajectory modeling to inform design of a phase II multiregional clinical trial. Clin Pharmacol Ther. (2024) 115:1346–57. doi: 10.1002/cpt.3216
214. Yoshida K, Abe K, Ishikawa M, Saku K, Shinoda-Sakamoto M, Ishikawa T, et al. Inhibition of TLR9-NF-κB-mediated sterile inflammation improves pressure overload-induced right ventricular dysfunction in rats. Cardiovasc Res. (2019) 115:658–68. doi: 10.1093/cvr/cvy209
215. Shen W, Liu J, Zhao G, Fan M, Song G, Zhang Y, et al. Repression of Toll-like receptor-4 by microRNA-149-3p is associated with smoking-related COPD. Int J Chron Obstruct Pulmon Dis. (2017) 12:705–15. doi: 10.2147/COPD.S128031
216. Xue W, Tianrun W, Jiaqi Y, Xin L, Ruxue D, and Peng Z. Bta-miR-149-3p suppresses inflammatory response in bovine Sertoli cells exposed to microcystin-leucine arginine (MC-LR) through TLR4/NF-kB signaling pathway. Ecotoxicol Environ Saf. (2024) 281:116636. doi: 10.1016/j.ecoenv.2024.116636
217. Li X-Q, Chen F-S, Tan W-F, Fang B, Zhang Z-L, and Ma H. Elevated microRNA-129-5p level ameliorates neuroinflammation and blood-spinal cord barrier damage after ischemia-reperfusion by inhibiting HMGB1 and the TLR3-cytokine pathway. J Neuroinflammation. (2017) 14:205. doi: 10.1186/s12974-017-0977-4
218. Monnet E, Lapeyre G, Poelgeest EV, Jacqmin P, Graaf KD, Reijers J, et al. Evidence of NI-0101 pharmacological activity, an anti-TLR4 antibody, in a randomized phase I dose escalation study in healthy volunteers receiving LPS. Clin Pharmacol Ther. (2017) 101:200–8. doi: 10.1002/cpt.522
219. Monnet E, Choy EH, McInnes I, Kobakhidze T, de Graaf K, Jacqmin P, et al. Efficacy and safety of NI-0101, an anti-toll-like receptor 4 monoclonal antibody, in patients with rheumatoid arthritis after inadequate response to methotrexate: a phase II study. Ann Rheum Dis. (2020) 79:316–23. doi: 10.1136/annrheumdis-2019-216487
220. Andresen L, Theodorou K, Grünewald S, Czech-Zechmeister B, Könnecke B, Lühder F, et al. Evaluation of the therapeutic potential of anti-TLR4-antibody MTS510 in experimental stroke and significance of different routes of application. PloS One. (2016) 11:e0148428. doi: 10.1371/journal.pone.0148428
221. Koch L, Hoeft K, and Kramann R. Artesunate: attenuating TLR4/MD2 signaling to alleviate cardiac fibrosis. Signal Transduct Target Ther. (2025) 10:46. doi: 10.1038/s41392-025-02131-z
Keywords: deep vein thrombosis, toll-like receptors, inflammation, signal transduction, targeted therapy
Citation: Shao W, Wang Z, Wu J, Guo T and Mo J (2025) Targeting toll-like receptors: unveiling potential therapeutic strategies for deep vein thrombosis. Front. Immunol. 16:1579113. doi: 10.3389/fimmu.2025.1579113
Received: 18 February 2025; Accepted: 14 May 2025;
Published: 29 May 2025.
Edited by:
Eleni Gavriilaki, Aristotle University of Thessaloniki, GreeceReviewed by:
Anita Sahu, National Jewish Health (United States), United StatesMehran Ghasemzadeh, High Institute for Research and Education in Transfusion Medicine, Iran
Copyright © 2025 Shao, Wang, Wu, Guo and Mo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tianting Guo, Z3VvdGlhbnRpbmdAMTYzLmNvbQ==; Jianwen Mo, bWp3MTk5N0AxMjYuY29t