 Yichen Zhou
Yichen Zhou Fanxing Shi
Fanxing Shi Junyu Zhu
Junyu Zhu Yi Yuan
Yi Yuan- 1School of Elderly Care Services and Management, Nanjing University of Chinese Medicine, Nanjing, Jiangsu, China
- 2School of Medicine, Nanjing University of Chinese Medicine, Nanjing, Jiangsu, China
- 3Nanjing University of Chinese Medicine, Nanjing, Jiangsu, China
Glioblastoma multiforme (GBM) is a common primary malignant brain tumor in adults, characterized by a high rate of recurrence and mortality. The median overall survival is less than 2 years with the current standard therapy. As immunotherapy has begun to show promising results in solid tumors such as non-small cell lung cancer and melanoma in recent years, immnunotherapy for patients with glioblastoma is also in full swing, which is mainly consisted of immune checkpoint inhibitors, cancer vaccines, chimeric antigen receptor T-cell and oncolytic viral therapy. However, the application of immunotherapy in glioblastoma is severely hampered by cognitive impairment of intracerebral lymphatic system, the existence of blood-brain barrier, highly immunosuppressive tumor microenvironment and GBM’s intrinsic features, including low tumor mutation burden and high heterogeneity. This review systematically evaluates recently published clinical trial outcomes of GBM immunotherapy, critically analyses both the progress and limitations of these trials, thoroughly examines current barriers to effective immunotherapy, and highlights promising preclinical studies that may guide future therapeutic development.
1 Introduction
Glioblastoma represents the most prevalent primary malignant brain tumor in adults (1), comprising 50.1% of all primary malignant central nervous system (CNS) neoplasms (2). With an overall incidence rate of approximately 3.22 cases per 100,000 individuals, its occurrence demonstrates both age-dependent progression and male predominance (3). GBM has consistently been characterized by its rapid disease progression, elevated mortality rates, unfavorable clinical outcomes, and frequent tumor recurrence (4). The 2021 World Health Organization (WHO) classification of CNS tumors categorizes glioblastoma as a grade IV, IDH wild-type adult diffuse glioma, reinforcing its recognition as an exceptionally aggressive malignancy with dismal prognosis (5).
Currently, the standard treatment for newly diagnosed glioblastoma (ndGBM) involves maximal safe surgical resection followed by concurrent chemoradiotherapy (CCRT), with subsequent adjuvant temozolomide (TMZ) and tumor treating fields (TTF). However, no consensus treatment exists for recurrent GBM (rGBM) (3). Alternative options include DNA damage response (DDR) inhibitors and targeted molecular therapies. Despite these treatments, the median overall survival (mOS) remains limited to approximately 20.9 months, with nearly universal tumor recurrence (6).
The high recurrence rate stems from multiple factors: the infiltrative growth pattern of GBM prevents complete surgical removal (7); the blood-brain barrier restricts delivery of large molecules and poorly lipid-soluble drugs (8) and the prevalent emergence of resistance to radiotherapy develops through various mechanisms, including the presence of quiescent glioma stem cells (GSCs), protective peritumoral brain (PTB) tissue (9) and intact DNA repair systems in O6-methylguanine-DNA methyltransferase(MGMT) unmethylated gliomas (10). The low tumor mutation burden and high degree of tumor heterogeneity present significant challenges for targeted molecular therapies. Furthermore, the profoundly immunosuppressive tumor microenvironment (TME) contributes to diminished T-cell infiltration and function through multiple mechanisms, including upregulation of inhibitory immune checkpoints (11), HIF-1α-mediated lactate accumulation (12, 13) and elevated TGF-β expression (14), collectively rendering GBM an immunologically "cold" tumor. These limitations underscore the critical need for novel therapeutic approaches, with immunotherapy emerging as one of the most promising strategies for GBM patients.
The concept of immunotherapy dates back to 1890 when Dr. William Coley pioneered the use of bacterial injections for treating inoperable malignancies. However, its application in glioblastoma faced considerable delays owing to the prevailing belief in the brain's "immune-privileged" status (15, 16). This scientific paradigm remained unchallenged until the 20th century when the discovery of the brain's dural lymphatic system fundamentally altered our understanding of CNS immunity (17). Moreover, clinical observations of immune-mediated neurological pathology in patients with autoimmune inflammatory diseases (18) and neurodevelopmental disorders (19, 20) provided definitive evidence for the bidirectional communication between central nervous system immunity and peripheral immune function. These seminal findings revolutionized neuroimmunological concepts and established the scientific foundation for developing immunotherapies against glioblastoma.
Immunotherapy for glioblastoma has evolved to encompass both established and emerging modalities. The classical approaches, including immune checkpoint inhibitors and cancer vaccines, function indirectly by stimulating the host immune system to identify and destroy tumor cells (21). Recent biotechnological advances have introduced more direct therapeutic strategies such as T cell therapies and oncolytic viruses. Currently, multiple immunotherapies for GBM have advanced to Phase II/III clinical trials, which can be systematically classified into four categories according to their mechanisms: immune checkpoint inhibitors, cancer vaccines, chimeric antigen receptor T-cell (CAR-T), and oncolytic viral therapy.
This review offers a thorough evaluation of recent clinical progress across these four principal immunotherapeutic strategies for glioblastoma. We perform a critical assessment of existing limitations in immunotherapy development and provide insightful perspectives on future therapeutic directions for GBM management.
2 Immune checkpoint inhibitors
For decades, it has been well-established that tumor cells evade anti-tumor immune responses by overexpressing inhibitory immune checkpoints (e.g., PD-1, PD-L1). These checkpoints interact with their respective ligands on T lymphocytes, effectively suppressing T-cell activation and enabling immune escape (22). To counteract this immunosuppressive mechanism, several immune checkpoint inhibitors targeting PD-1, PD-L1, CTLA-4 and other ones have been developed and implemented in clinical practice (Table 1), such as Nivolumab, Pembrolizumab, and Cemiplimab (23–26).
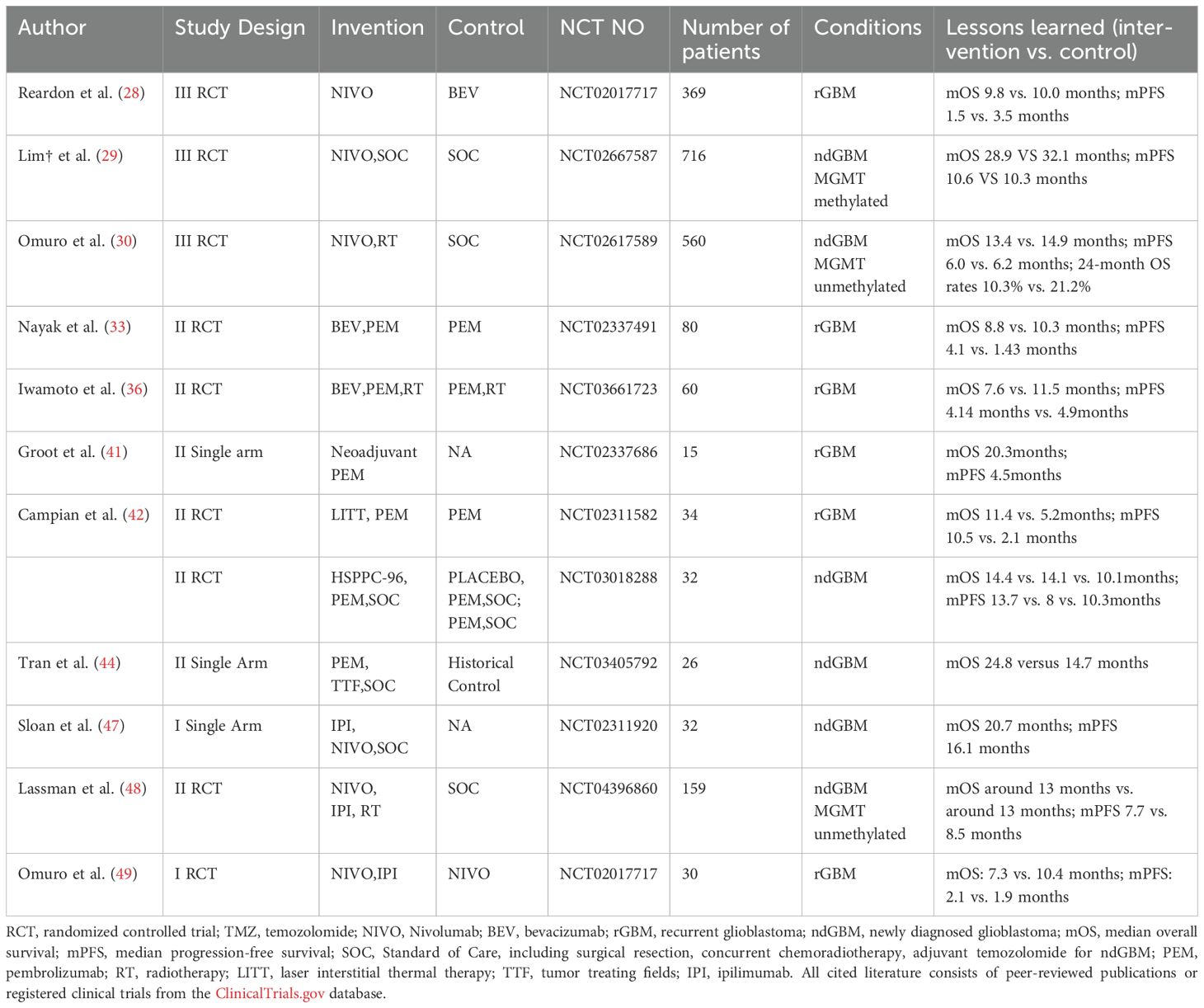
Table 1. Summary of Immune checkpoint inhibitors clinical trials in glioblastoma.
2.1 Nivolumab
Nivolumab, a common PD-1 immune checkpoint inhibitor, exerts its antitumor effect by binding to PD-L1 on tumor cells, thereby blocking the PD-1-mediated tyrosine phosphorylation signal that inhibits T-cell activation (27).
CheckMate 143 demonstrated that nivolumab monotherapy failed to show a statistically significant overall survival benefit compared to bevacizumab (Bev) in the overall rGBM population (28). Nevertheless, subgroup analysis revealed a potential survival advantage for nivolumab in patients with MGMT methylation.
For ndGBM patients, neither nivolizumab combined with standard of care (SOC) in MGMT-methylated cases (29), nor nivolizumab combined with radiotherapy alone in MGMT-unmethylated cases (30) demonstrated significant improvement in median overall survival compared to SOC, which was defined as maximal safe resection followed by CCRT and adjuvant TMZ according to 2016 WHO Classification of Tumors of the Central Nervous System (31).
This limited efficacy may be attributed to chemotherapy or RT induced T-cell depletion (32) and suppression of T cell function as a consequence of suppressive immune microenvironment and low immunogenicity of GBM. Supporting evidence comes from a phase II clinical study conducted by Schalper et al (32), where immunohistochemical analysis revealed that while nivolumab treatment increased intratumoral immune cell infiltration likely mediated by elevated chemokine levels, it failed to significantly enhance either T-cell cytotoxic activity or proliferative capacity.
To maximize the clinical benefit of nivolumab, a multifaceted optimization strategy is required. First, comprehensive pharmacodynamic studies are needed to validate the effects of intravenous nivolumab on tumor-infiltrating T-cell activity in vivo. Second, treatment combinations should be carefully considered with chemoradiotherapy. Third, precision patient selection through refined enrollment criteria is crucial, as evidenced by differential treatment responses between nivolumab and bevacizumab in patient subgroups stratified by PD-L1 expression levels (28). Notably, nivolumab demonstrates particular efficacy in the B7-H4-high glioma subgroup - a population intrinsically characterized by profoundly depleted tumor-infiltrating lymphocytes (TILs) (33).
2.2 Pembrolizumab
Originally developed for unresectable or metastatic melanoma (34), the PD-1 antagonist pembrolizumab has shown modest activity in recurrent glioblastoma, with phase II randomized controlled trials reporting a median overall survival of 10.3 months for monotherapy (35) and 11.5 months when combined with radiotherapy (36). However, combination strategies with bevacizumab—including both Pem+Bev and Pem+RT+Bev regimens—paradoxically compromised survival outcomes (35, 36). This observed reduction in therapeutic efficacy may stem from increased treatment-related adverse events due to Bev-associated toxicity (35, 37, 38) and Bev-mediated exacerbation of tumor hypoxia, which may further potentiate the immunosuppressive tumor microenvironment (39, 40).
Surprisingly, neoadjuvant pembrolizumab has demonstrated efficacy in recurrent glioblastoma, with a phase II trial showing prolonged survival through preoperative tumor reduction (41). This promising outcome has prompted an ongoing phase IV confirmatory study (NCT05235737, completion May 2026).
Further enhancing this approach, a separate phase II randomized controlled trial (RCT) revealed that laser interstitial thermal therapy (LITT) synergizes with Pem, significantly improving median survival (42) via blood-brain barrier disruption and pro-apoptotic effects (43). Building on these findings, an active phase II trial is currently evaluating a multimodal regimen combining tumor-treating fields, Pem and LITT (NCT06558214).
For newly diagnosed glioblastoma, phase II RCT data indicate a median survival of 10.1 months with pembrolizumab plus SOC (NCT03018288), though this may attribute to the limited 26-month follow-up. More promisingly, Pem combined with maintenance TMZ and tumor-treating fields after SOC demonstrated significantly improved median survival (25.2 months) versus historical controls (44). These results have propelled an ongoing phase III trial evaluating this triple combination (NCT06556563), with completion anticipated by April 2029.
2.3 Ipilimumab
CTLA-4 is a negative co-stimulatory molecule expressed on activated T cells. Due to its high homology with the extracellular domain of the T cell co-stimulatory molecule CD28, and its higher affinity for CD80/CD86, CTLA-4 often inhibits T cell activation by competitively binding to CD80/CD86 (45). Additionally, it can reduce the ex-pression of CD80/CD86 on the surface of antigen-presenting cells through trans-endocytosis (46). Ipilimumab, the most clinically established CTLA-4 inhibitor, counteracts these immunosuppressive effects.
For newly diagnosed GBM, a phase I trial demonstrated promising mOS of 20.7 months with ipilimumab, nivolumab on the basis of SOC, though requiring phase II validation (47). In MGMT-unmethylated ndGBM, the combination of ipilimumab, nivolumab and RT showed comparable mOS but inferior median progression-free survival (mPFS) versus SOC at 13.7-month follow-up (48).
In recurrent GBM, combination of ipilimumab and nivolumab yielded modest mOS of 7.3 months (49), showing no significant advantage over nivolumab monotherapy, warranting cautious evaluation of this combinatorial approach (Nivo, Ipi).
In summary, the limited survival benefit of immune checkpoint inhibitor monotherapy primarily stems from the profoundly immunosuppressive tumor microenvironment, low tumor mutational burden and inherent immunogenicity of GBM which consequently restrict both quantity and functionality of tumor-infiltrating T cells. Remarkably, the aforementioned TTF and LITT modalities may offer a safer alternative to conventional chemoradiation by promoting tumor apoptosis and tumor-associated antigen (TAA) release, though their precise mechanisms of action require further investigation.
Beyond classical targets, novel immune checkpoints have also entered the phase I / II clinical trial stage,including TIM-3 checkpoint inhibitors (NCT03961971), LAG-3 checkpoint inhibitors relatlimab (NCT02658981 and NCT03493932), TIGIT checkpoint inhibitors ( NCT04656535 ), CD137 checkpoint inhibitors ( NCT02658981) and IDO checkpoint inhibitors ( NCT04047706, NCT02052648).
3 Tumor vaccines
Tumor vaccines for glioblastoma can be broadly classified into four main categories: cellular vaccines, protein/synthetic peptide vaccines, nucleic acid vaccines, and viral vector vaccines (Table 2). These therapeutic approaches utilize various forms of tumor-specific antigens (TSAs), TAAs or antigen-encoding genes, which are either administered directly or delivered through artificial carriers such as dendritic cells or viral vectors. Following administration, these vaccines aim to fully activate the patient's anti-tumor immune responses, thereby inhibiting tumor growth and progression.
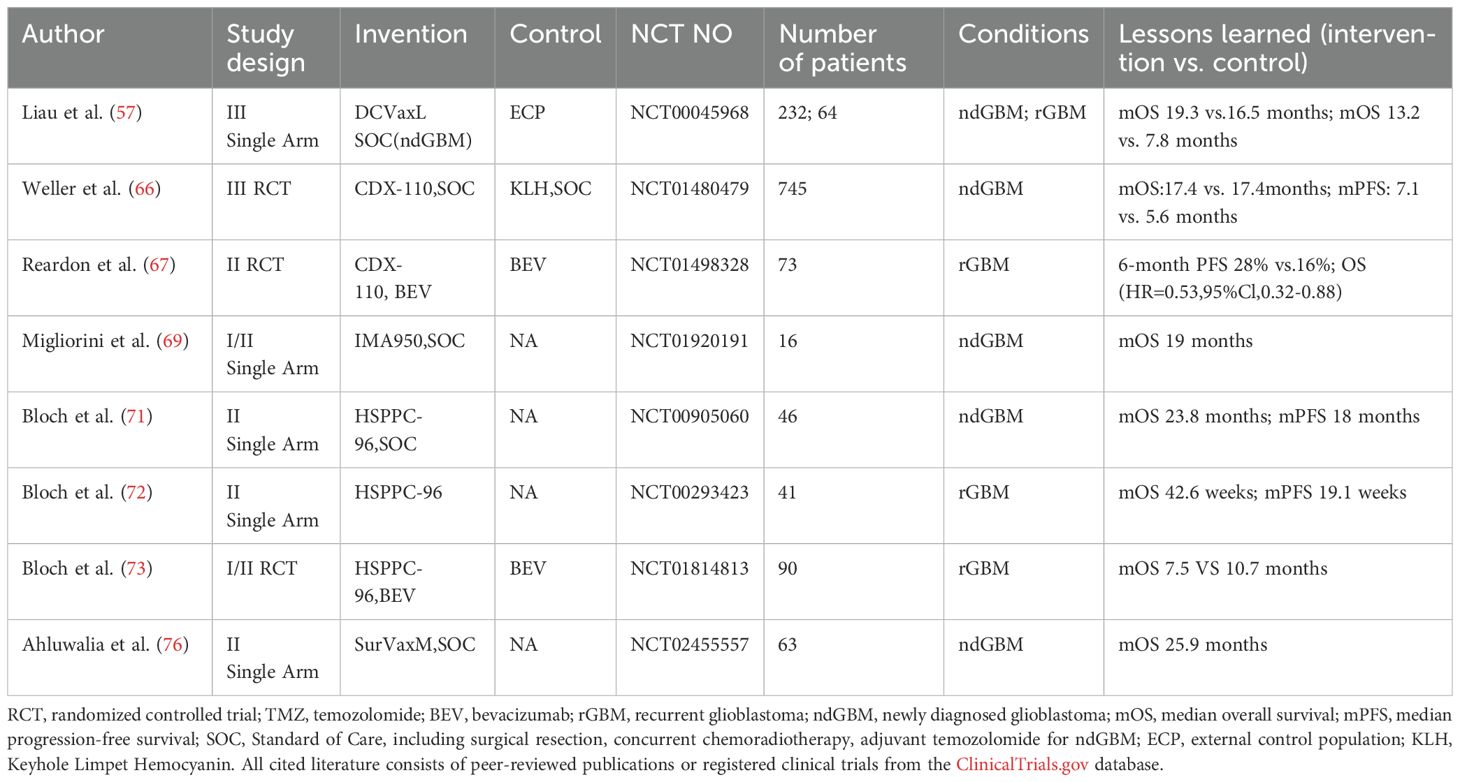
Table 2. Summary of tumor vaccine clinical trials in glioblastoma.
3.1 DC vaccine
Dendritic cells (DCs), as the body's primary antigen-presenting cells, play a crucial role in antitumor immunity by capturing antigens through phagocytosis, pinocytosis, or receptor-mediated endocytosis, processing them, and presenting antigenic peptides via MHC class I and II pathways to activate CD8+ and CD4+ T cells, respectively (50–52).
In glioblastoma immunotherapy, several DC vaccine platforms have been developed, including DCVax-L (loaded with tumor lysates) (53), GSC-DCV (pulsed with glioma stem cell antigens) (54), ICT-107 (loaded with synthetic peptides targeting glioma stem cell-associated antigens) (55), and CMV-DC (electroporated with CMV pp65 mRNA) (56). While all four approaches begin by isolating and differentiating patient-derived monocytes into immature DCs, they differ in their antigen-loading strategies.
DCVax-L has demonstrated significant clinical benefits in glioblastoma treatment, with phase III trial data showing mOS of 19.3 months for ndGBM patients overall, 30.2 months for MGMT-methylated ndGBM patients, and 13.2 months for rGBM patients (53, 57). The vaccine's personalized manufacturing approach, involving co-culture with patient-derived tumor lysates, enables broad targeting of tumor antigens and effectively addresses GBM's spatial heterogeneity. Although these results showed marked improvement over external controls, the original randomized controlled trial design was complicated by a late-stage crossover, necessitating external control comparisons for analysis. Current investigations include an ongoing phase III randomized trial evaluating DCVax-L combined with radiotherapy specifically in MGMT-methylated glioblastoma patients to further validate these promising findings (NCT03548571).
Additionally, clinical trial data demonstrate that the efficacy of glioma stem cell-targeted DC vaccines is strongly influenced by molecular biomarkers: the phase II single-center RCT of GSC-DCV revealed superior survival benefits in patients with IDH1 wild-type, TERT mutation, and low B7-H4 expression profiles (54), while the phase II multicenter trial of ICT-107 showed significantly improved median progression-free survival in newly diagnosed GBM patients, particularly those with HLA- A1 methylated tumors (55).
3.2 Peptide vaccine
Peptide vaccines are the most classic form of cancer vaccines. They consist of TSAs or TAAs that are typically 8 to 30 amino acids in length. These vaccines stimulate an adaptive immune response tar-geting the specific antigen to exert anti-tumor effects (58, 59), including CDX-110,HSPPC-96,SurVaxM and so on.
3.2.1 CDX-110
EGFRvIII, the most common EGFR mutation in glioblastoma with 25-30% prevalence (60), drives tumorigenesis through constitutive activation (61) and promotes radioresistance by enhancing DNA repair capacity (62).
While the EGFRvIII-targeted peptide vaccine rindopepimut (CDX-110) showed promising survival benefits in three phase II trials for ndGBM patients when combined with or without adjuvant TMZ on the basis of CCRT (63–65), these results were not replicated in a subsequent multicenter phase III RCT, potentially due to the unreliability of the results from the previous single-arm clinical trial (66).
Interestingly, in recurrent GBM, rindopepimut combined with bevacizumab demonstrated improved 6-month PFS and OS versus Bev alone (67), suggesting TMZ may compromise vaccine efficacy while Bev's modulation of the immunosuppressive microenvironment (68) could be synergistic.
Anyway, CDX-110, as a single-target peptide vaccine, ultimately demonstrates limited clinical efficacy due to the intrinsic heterogeneity of glioblastoma tumors. This aligns with emerging data on multi-target vaccines like IMA950 (targeting 11 TAAs), which achieved 19-month median survival in ndGBM (69), highlighting the therapeutic advantage of broad antigen targeting over single-epitope approaches.
3.2.2 HSPPC-96
HSPPC-96, a heat shock protein-based vaccine, enhances dendritic cell (DC)-mediated uptake and presentation of tumor antigens by binding to both tumor-derived peptides and CD91 on DCs (70).
In ndGBM, a phase II single-arm trial reported a median survival of 23.8 months with HSPPC-96 following SOC (71). However, a subsequent phase II RCT found no significant survival benefit when combining HSPPC-96 with Pem after SOC (NCT03018288). For rGBM, phase II trials indicate that HSPPC-96—either alone or with bevacizumab—fails to substantially improve long-term survival compared to Bev alone (72, 73). This limited efficacy may stem from GBM’s immunosuppressive microenvironment, which impairs T-cell function, and its inherently low tumor mutational burden.
3.2.3 SurVaxM
Integrin, an anti-apoptotic protein highly expressed on glioma stem cells (GSCs), critically regulates GSCs self-renewal, proliferation, migration, and invasion (9, 74, 75). The survivin-targeting vaccine SurVaxM recently completed a phase II trial in ndGBM patients, demonstrating a median survival of 25.9 months when combined with SOC (76). A phase II RCT (NCT05163080) is now evaluating this regimen.
In summary, GBM's temporal and spatial heterogeneity poses a fundamental challenge for tumor vaccines. Nonetheless, the DCaxL vaccine offers a promising strategy to address spatial heterogeneity. Further exploration of its safety and preparation protocols will be essential if considering its long-term cyclical administration to overcome temporal heterogeneity.
4 CAR-T
T cells are central to adaptive anti-tumor immunity, but tumors often evade detection by downregulating MHC expression, suppressing T cell activation (77, 78). Therefore, we engineered chimeric antigen receptors (CARs) on autologous or allogeneic T cells to enable MHC-independent tumor recognition (79, 80). Upon reinfusion into patients, these genetically modified T cells achieve dual therapeutic effects-precise tumor-targeted cytotoxicity and synergistic restoration of tumor-suppressed host immunity (81). For glioblastoma, CAR-T therapies targeting IL13Rα2, EGFRvIII, and HER2 remain in early-phase (I/II) trials (Table 3).
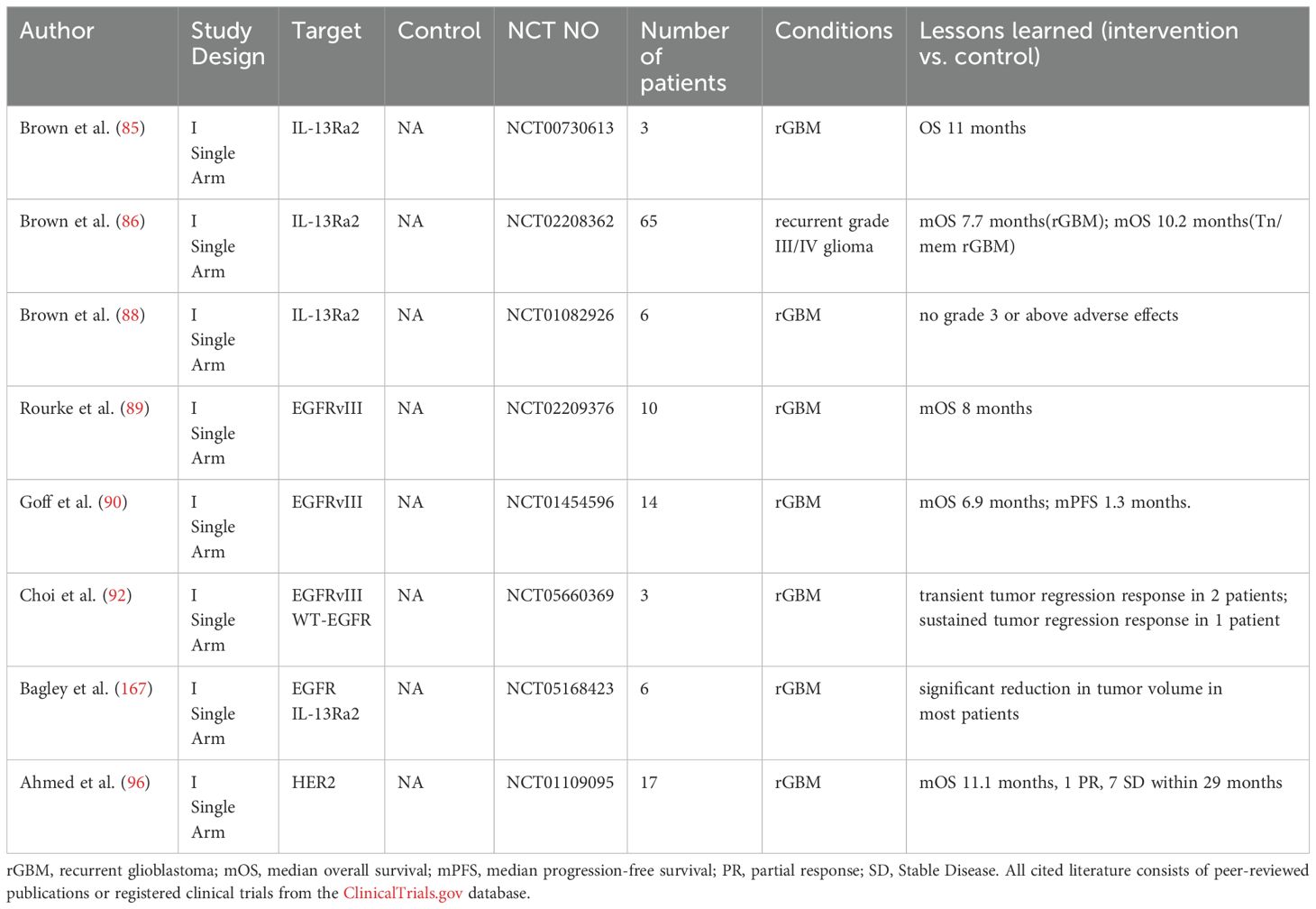
Table 3. Summary of CAR-T cell clinical trials in high grade glioma.
4.1 IL13Rα2
IL13Rα2 expressed in over 50% of GBM patients (82, 83), which has emerged as one of the most promising CAR-T targets for GBM, promotes tumor progression by inhibiting the STAT6 signaling pathway (84). Clinical development has progressed through three generations.
First-generation autologous IL13Rα2 CAR-T achieved 11-month OS in rGBM patients (n=3) via intratumoral delivery (85). However, the clinical significance of these findings remains limited by the small cohort size.
The second-generation IL-13Rα2 CAR-T builds upon the first-generation construct by incorporating the BBζ costimulatory domain and employing a novel dual intratumoral and ventricular delivery approaches combined with the Tn/mem production platform, which enhances undifferentiated memory T-cell populations with favorable marker profiles (increased memory markers, decreased senescence markers. Clinical results demonstrated a median overall survival of 10.2 months in rGBM patients with Tn/mem platform, outperforming Tcm comparator groups (86), effectively verifying the feasibility of the Tn/mem production platform for CAR-T preparation in vivo trials.
Although the mOS of second-generation IL-13Rα2 CAR-T cells remained comparable to first-generation therapy from a survival standpoint, imaging assessments revealed more significant benefits.Following 10 weeks of ventricular infusion, one of the patients achieved complete resolution of all detectable intracranial and spinal lesions on both PET and MRI, although disease recurrence occurred at four distinct new locations (87). These observations suggest that the unique tumor-reducing capability of this combined infusion approach in multifocal GBM patients may have important implications for designing future clinical trials.
To address limitations of autologous CAR-T therapy including prolonged manufacturing time and restricted eligibility due to poor T-cell quality, an allogeneic IL-13Rα2 CAR-T product has completed phase I testing (88). The therapy demonstrated a favorable safety profile with no grade ≥3 adverse events or graft-versus-host disease observed, though efficacy evaluation requires further clinical investigation.
4.2 EGFRvIII
While EGFRvIII-targeted approaches are well-established in GBM treatment (pioneered by rindopepimut), EGFRvIII CAR-T therapy represents an alternative modality. Phase I trials of intravenously administered EGFRvIII-CAR-T demonstrated limited efficacy in rGBM, with monotherapy achieving median overall survival of 8 months (89). While in another phase I single-arm trial evaluating intravenous infusion of EGFRvIII-CAR-T combined with IL-2, rGBM patients demonstrated not only a limited median survival of just 6.9 months, but also experienced dose-limiting toxicity (90). Most notably, one patient developed pulmonary edema and succumbed to the condition despite resuscitation attempts approximately 4 hours following infusion of 6×10¹° cells.
The observed toxicity profile primarily stems from intravenous CAR-T administration inducing pulmonary vascular toxicity, while the limited clinical efficacy relates to EGFRvIII's special biological behavior whereby the targeted variant co-amplifies wild-type EGFR through paracrine signaling (91) - a mechanistic explanation for both the development of treatment resistance and the frequent detection of wild-type EGFR overexpression in recurrent tumors following EGFRvIII-CAR-T therapy (60, 90).
To address these challenges, Bryan D. Choi's team developed CARv3-TEAM-E T-cells, a dual-targeting approach against both EGFRvIII and wild-type EGFR, delivered via ventricular infusion to circumvent vascular toxicity (92). Initial clinical results demonstrated a manageable safety profile with promising efficacy, including durable tumor regression persisting >150 days after single infusion. However, the preliminary nature of these findings constrained by small sample size and trial design limitations (Single Arm). Thus, larger-scale studies are required to comprehensively evaluate overall and progression-free survival outcomes.
4.3 HER2
The HER2 proto-oncogene (17q21) encodes ErbB2, a key EGFR-family tyrosine kinase receptor that is overexpressed in 80% of GBM cases (93). HER2/ErbB2 overexpression drives gliomagenesis through ErbB2 heterodimerization-induced tyrosine autophosphorylation and constitutive activation of proliferative signaling pathways, ultimately promoting uncontrolled cellular proliferation and malignant transformation in glioblastoma (94, 95).
Results from a phase I single-arm trial demonstrated that intravenous HER2-CAR VSTs in rGBM patients achieved a median overall survival of 11.1 months, suggesting potential clinical benefit (96); However, this finding requires validation in larger phase II/III trials.
The therapeutic efficacy of CAR-T cells in GBM remains constrained by late-adaptive resistance mechanisms arising from tumor heterogeneity and the correlation between CAR-T persistence and clinical response continues to be debated. Insights can be drawn from the NY-ESO-1 CAR-T clinical trial for synovial sarcoma and melanoma: the correlation between patients' objective clinical responses and CAR-T persistence was only validated short-term (≤28 days) (97), with stronger association observed for peak peripheral blood CAR-T cell counts (98).
5 Oncolytic viral therapy
Oncolytic viruses (OVs) are genetically modified, weakly pathogenic viruses that selectively infect and lyse tumor cells while inducing immunogenic cell death (ICD) in both cancer and stromal cells (99, 100). Their therapeutic potential is mediated through the release of TAAs, damage-associated molecular patterns (DAMPs) from lysed cells, OV-derived pathogen-associated molecular patterns (PAMPs), which collectively mobilize potent antitumor immunity (101–103).
Several engineered viral platforms have been evaluated in clinical trials for glioblastoma, including oncolytic herpes simplex virus, oncolytic adenovirus, poliovirus, retrovirus, Newcastle disease virus (NDV)-based vectors and so on (Table 4).
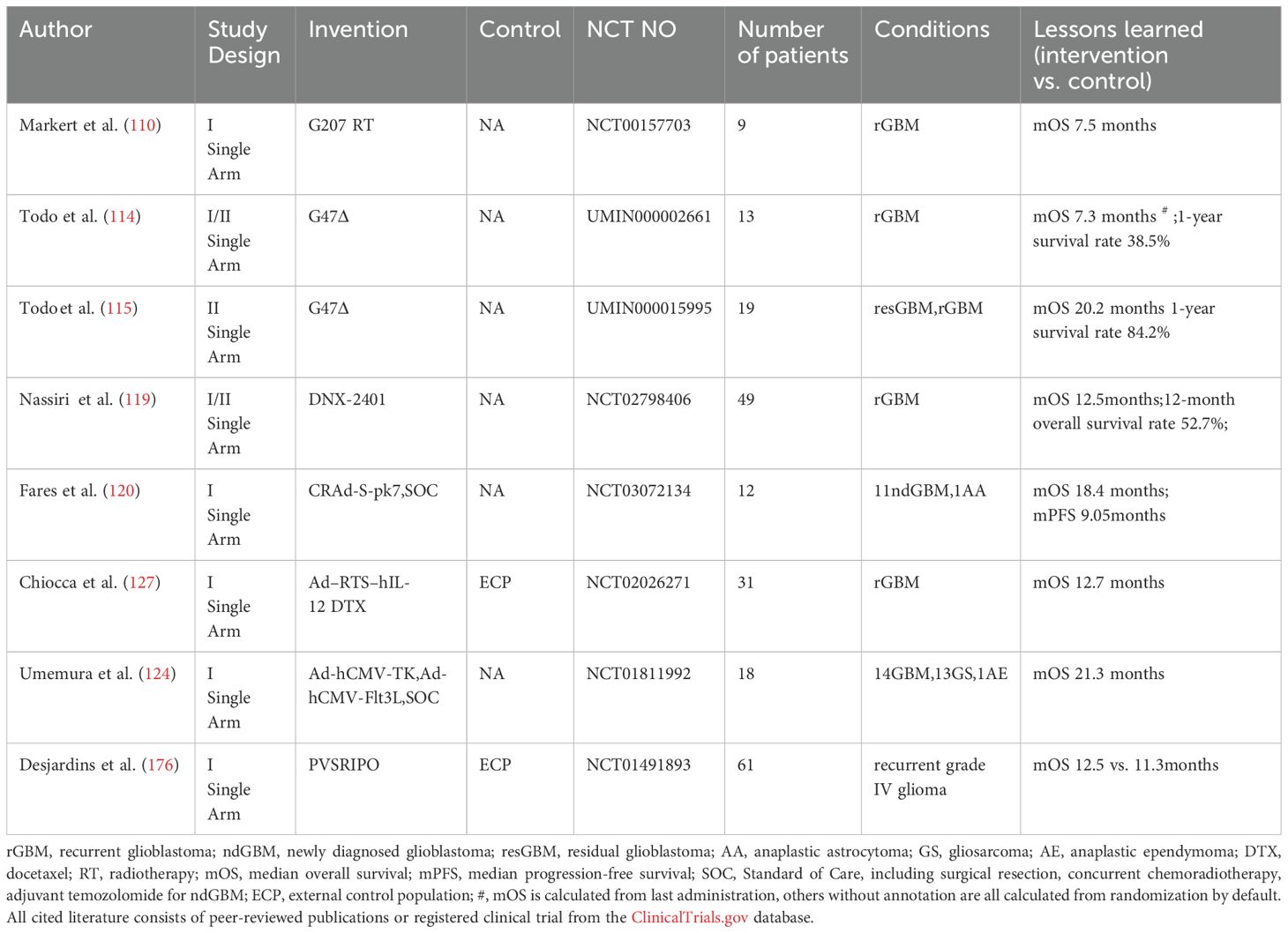
Table 4. Summary of oncolytic virus clinical trials in high grade glioma.
5.1 Oncolytic herpes simplex virus
Herpes simplex viruses (HSV) are neurotropic, double-stranded DNA viruses with large genomes (~150 kb) (104). Key oncolytic variants include HSV1716, HSV-G207, and G47Δ.
HSV1716, a first-generation oncolytic HSV, achieves tumor-selective replication through deletion of the RL1 gene (encoding ICP34.5), restricting viral propagation to actively dividing cells and thereby enhancing safety (105, 106). However, due to rapid tumor recurrence observed in Phase I trials following intracavitary infusion (107), HSV-G207 rapidly superseded HSV1716 in clinical development.
HSV-G207, a second-generation oncolytic herpes virus derived from HSV1716 through lacZ insertion-mediated inactivation of the ICP6 gene (UL39), exhibits restricted replication in normal cells by inhibiting late viral protein synthesis (108, 109). In a Phase I clinical trial for recurrent glioblastoma (rGBM),stereotactically guided intratumoral injection of HSV-G207 combined with a single 5 Gy radiotherapy fraction yielded a median overall survival of 7.5 months (110). The suboptimal therapeutic outcome likely reflects protocol limitations including use of a subtherapeutic radiation dose (5 Gy) and minimal therapeutic exposure (single viral injection plus single radiotherapy session), which collectively may have been insufficient to achieve adequate tumor cell destruction or elicit a robust antitumor immune response.
G47Δ, the third-generation oncolytic HSV-1, is engineered from the second-generation backbone by additionally inhibiting expression of the α47 gene in the G207 genome. This modification prevents downregulation of MHC class I molecules mediated through binding of the transporter associated with antigen presentation (TAP) in host cells (111, 112). Concurrently, it partially restores the function of the deleted γ34.5 gene, enhancing viral replication capability in tumor cells (113).
In the earliest phase I/II single-arm clinical trial conducted by Tomoki Todo's team, stereotactically guided intratumoral injection of G47Δ in 13 rGBM patients yielded a median survival of 7.3 months (114). However, subsequent analysis accounting for treatment parameters,including limited administration frequency (only 2 intratumoral infusions) and dose (3 patients received low-dose virus), revealed significantly improved outcomes in the latest phase II trial. The optimized protocol demonstrated a median survival of 20.2 months and 1-year survival rate of 84.2% in patients with rGBM or residual GBM (115).
5.2 Oncolytic adenovirus
Adenoviruses are non-enveloped, double-stranded DNA viruses that have been genetically modified for tumor-selective replication (116). Among the clinically evaluated oncolytic adenovirus platforms for glioblastoma treatment are DNX-2401, CRAd-S-pk7, and Onyx-015.
DNX-2401 in particular incorporates two key genetic modifications: a 24-base pair deletion in the E1A gene that restricts viral replication to cancer cells with aberrant retinoblastoma (Rb) pathway activity and insertion of an RGD-4C peptide motif in the fiber protein's HI loop to enhance tumor cell targeting through integrin binding (117, 118).
Phase I clinical trial results demonstrate that stereotactic intratumoral administration of DNX-2401 in combination with intravenous pembrolizumab yielded a median overall survival of 12.5 months in patients with rGBM (119), which represents a clinically meaningful improvement over historical monotherapy controls, suggesting synergistic therapeutic benefits from the combined viral immunotherapy and immune checkpoint inhibition approach.
CRAd-S-pk7 represents an innovative oncolytic adenovirus platform that utilizes neural stem cells (NSCs) as delivery vectors to penetrate the blood-brain barrier, while its tumor-specific survivin promoter restricts viral replication to survivin-overexpressing glioma cells. Phase I clinical trial data demonstrated promising efficacy in ndGBM, with the overall cohort achieving a median overall survival of 18.4 months in the treatment of the combination of intracavitary infusion of CRAd-S-pk7 and SOC, representing a 3.8-month improvement over SOC monotherapy, while the non-MGMT methylated subgroup showed particularly significant benefit with 18.0-month median survival (120).
5.3 PVSRIPO
PVSRIPO is an engineered oncolytic virus generated by replacing the poliovirus internal ribosomal entry site (IRES) with that of human rhinovirus type 2. This modification attenuates neurovirulence while preserving the natural CD155 tropism of poliovirus, enabling selective targeting of CD155-overexpressing tumor cells (121). Phase II clinical trial results demonstrated that convection-enhanced intratumoral delivery of PVSRIPO in recurrent glioblastoma (rGBM) patients yielded a median overall survival of 12.5 months - a significant improvement over historical controls (120). Based on these promising outcomes, an ongoing phase II trial is currently evaluating PVSRIPO in combination with pembrolizumab (NCT04479241).
5.4 Viral vectors
Viral vectors are engineered viral systems composed of modified structural elements from parent viruses (e.g. adenovirus, retrovirus) (122, 123) and therapeutic transgenes, such as immunomodulatory cytokines (IL-12, Flt3L), pro-apoptotic factors (Fas-L) and suicide genes (HSV-thymidine kinase) (124, 125), which are designed to safely deliver into target cells while maintaining efficient transduction capabilities and minimized pathogenicity.
IL-12 is a potent cytokine that enhances T-cell and natural killer (NK) cell activation/proliferation while exhibiting anti-angiogenic effects (126). The adenoviral vector Ad-RTS-hIL-12 carries an IL-12 expression cassette, enabling pharmacologically inducible IL-12 upregulation upon oral administration of veledimex (VDX). Phase I single-arm trial data demonstrated that intracavitary Ad-RTS-hIL-12 administration combined with oral veledimex (VDX) achieved a median overall survival of 12.7 months in patients with recurrent high-grade glioma (127).
The HSV-tk gene encodes thymidine kinase, which converts prodrugs (e.g., valacyclovir) into DNA synthesis-terminating metabolites (128). Flt3L stimulates dendritic cell development by binding Flt3 receptors on hematopoietic progenitors (129). In a Phase I trial for primary high-grade glioma, intracavitary delivery of adenoviral vectors (Ad-hCMV-TK + Ad-hCMV-Flt3L) combined with oral valacyclovir and SOC achieved a median overall survival of 21.3 months, warranting further validation (124).
In conclusion, as previously described, three delivery approaches are commonly utilized in oncolytic virus administration, including post-operative intracavitary infusion, stereotactically guided intratumoral injection for non-resectable lesions, and convection-enhanced delivery (CED) to achieve wider intraparenchymal distribution. The impact of pre-existing antiviral immunity remains a subject of active investigation,while low serum neutralizing antibody titers may theoretically permit enhanced viral spread due to reduced neutralization (130), they could alternatively indicate compromised host immune function that might limit therapeutic efficacy (130, 131). Besides, Radiographic assessment of treatment response presents unique challenges, as conventional criteria (e.g., RANO) may not fully capture virus-specific phenomena. For instance, G47Δ-treated tumors frequently demonstrate characteristic "exploded crater" morphology on MRI (114). These distinct imaging signatures underscore the need for revised response criteria specific to viral immunotherapies.
6 Challenges and countermeasures of GBM immunotherapy
Clinical trials of immunotherapies for glioblastomas are currently in full swing, but according to the above statement, most of the immunotherapies tend to be efficacious in early trials or single-arm studies, with few trials being able to demonstrate significant efficacy in phase III RCTs as they all face 4 main problems mentioned below-immunosuppressive tumor microenvironment, GBM intrinsic properties, drug infusion disorder and cognitive impairment of the lymphatic drainage pathways in the brain (17) (Figure 1).
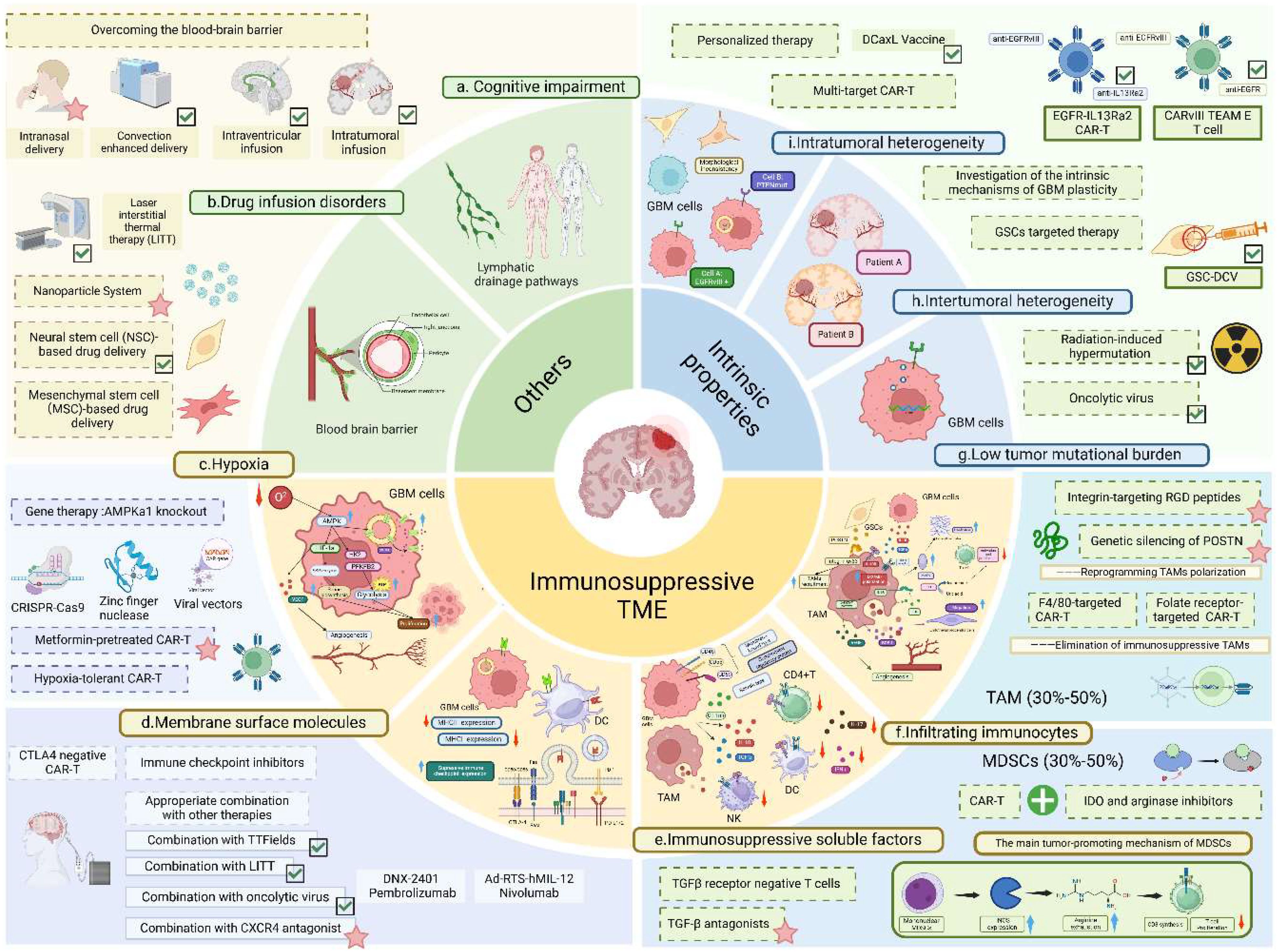
Figure 1. Challenges and countermeasures of glioblastoma immunotherapy. This figure provides a detailed illustration of the challenges in immunotherapy and corresponding countermeasures. GBM, glioblastoma multiforme; CAR-T, chimeric antigen receptor T-cell; DC, dendritic cell; NK, natural killer cell; TAM, tumor-associated macrophage; GSC, glioma stem cell; VEGF, vascular endothelial growth factor; SDF-1, stromal cell-derived factor-1;POSTN, periostin; IDO, indoleamine 2,3-dioxygenase; MMP9, matrix metalloproteinase-9; IL, interleukin; GSC-DCV glioma stem cell-derived dendritic cell vaccine; iNOS, inducible nitric oxide synthase. Detailed challenges: (A) Cognitive impairment of intracranial lymphatic drainage pathways; (B) Drug delivery obstacles due to the existence of blood-brain barrier; (C) Hypoxia.The hypoxic microenvironment upregulates intratumoral AMPK activity and mediates HIF-1α activation, thereby promoting glycolysis and glucose-derived de novo serine biosynthesis to facilitate tumor proliferation; (D) Membrane surface molecules. GBM cells evade immune surveillance by overexpressing inhibitory immune checkpoint ligands (e.g., PD-L1, Fas-L) and downregulating MHC expression; (E) Immunosuppressive soluble cytokines. Glioma cells express membrane-bound regulatory proteins (e.g., CD46, CD55, CD59) and secrete cytokines (e.g., IL-10, TGF-β) to suppress host immunity; (F) Infiltrating immune cells- GSCs recruit and polarize TAMs toward the M2 phenotype via secretion of POSTN and TGF-β. TAMs enhance tumor invasion by producing MMP9 to degrade the extracellular matrix, inhibit T-cell function via IDO1 expression, and promote angiogenesis by inducing VEGF secretion through the JAK-STAT pathway. Monocytic MDSCs suppress T-cell proliferation via iNOS expression; (G) Low tumor mutation burden; (H). Intertumoral heterogeneity; (I) Intratumoral heterogeneity. Detailed countermeasures: Approaches including convection-enhanced delivery, intraventricular and intratumoral infusion, LITT, and neural stem cell-based delivery to overcome the blood-brain barrier; DCaxL and multi-targeted CAR-T to address tumor heterogeneity; radiotherapy, chemotherapy, and combined use of oncolytic viruses to tackle low tumor mutational burden; and the combination of immune checkpoint inhibitors with TTF and LITT to counteract T-cell suppression have been preliminarily validated in clinical trials (marked with green ticks). Meanwhile, methods including intranasal exosome delivery, AMPK kinase-preconditioned T cells, CXCR4 antagonists, and RGD peptides have shown promising results in preclinical GBM mouse studies (marked with pink stars). Additionally, strategies like mesenchymal stem cell-based drug delivery, low-oxygen-demand CAR-T, gene therapy targeting AMPKα1 knockout in tumor cells, CTLA4-negative CAR-T cells, F4/80-targeted CAR-T, and folate receptor-targeted CAR-T have demonstrated efficacy in preclinical studies of other cancers (e.g., ovarian cancer, lung cancer), offering valuable insights for the future development of GBM immunotherapy. The figure is created in BioRender.com.
6.1 immunosuppressive tumor microenvironment
The immunosuppressive TME comprises four key interrelated components: profound hypoxia, aberrantly expressed membrane surface molecules, immunosuppressive soluble factors and infiltrating immunocytes with regulatory functions.
6.1.1 membrane surface molecules
Similar to other malignancies, glioblastoma (GBM) achieves immune escape through multiple pathways, comprising elevated expression of immune checkpoint ligands (e.g., PD-L1, Fas-L), reduced major histocompatibility complex (MHC) presentation and diminished co-stimulatory molecule expression (132–134). Beyond conventional immune checkpoint inhibitors, emerging strategies show promise: CTLA4-negative CAR-T cells have exhibited enhanced proliferative capacity and antitumor activity in leukemia patients, suggesting potential applicability to GBM (135). Checkpoint inhibitors combined with LITT/TTF has shown improved efficacy (42, 44) with more immunotherapy combinations mentioned below.
6.1.2 Hypoxia
Hypoxic tumor microenvironment triggers AMPK activation in cancer cells, which stabilizes HIF-1α and subsequently induces the transcriptional upregulation of key metabolic regulators including glucose transporters (GLUT1, GLUT3 and GLUT4), glycolytic enzymes (HK2 and PFKFB2), and serine biosynthesis pathway (SSP) enzymes. These HIF-1α-mediated metabolic reprogramming events collectively enhance glycolytic flux and glucose-derived de novo serine biosynthesis, thereby promoting tumor cell proliferation (136–138). Paradoxically, this metabolic shift generates an immunosuppressive milieu through HIF-1α-mediated lactate accumulation, which impairs NK and T cell function (139). Preclinical studies demonstrate that metformin-pretreated CAR-T cells exhibit enhanced tumor infiltration and significantly prolong survival in glioma-bearing mouse models (140).Engineered hypoxia-tolerant CAR-T cells demonstrate potent antitumor activity with minimal toxicity in ovarian cancer models (141) and CRISPR/Cas9-mediated AMPKα1 knockout in lung cancer cells has been shown to generate stable cell lines with markedly reduced proliferation rates (142), both of which suggest potential translational relevance for GBM.
6.1.3 Immunosuppressive soluble factors
Gliomas employ effectively shield themselves from complement-mediated destruction by expressing membrane-bound complement regulators like CD46, CD55, and CD59 (143). Simultaneously, their secretion of potent immunosuppressive cytokines such as IL-10 and TGF-β potently inhibits various immune populations including CD4+ T cells, NK cells, and dendritic cells leading to suppression of both antigen-specific and innate immune functions in patients (144, 145).
Recent preclinical advances highlight TGF-β inhibition as a particularly promising therapeutic avenue. When combined with radiotherapy, pharmacological TGF-β antagonists have been shown to reduce tumor invasiveness and reverse mesenchymal transition in GBM mice models, significantly extending survival (146). Equally encouraging, TGFBR2-knockout T cells demonstrate markedly enhanced tumor-killing capacity in both melanoma and lung adenocarcinoma models (147), providing valuable insights for its application in GBM treatment in the future.
6.1.4 Infiltrating immunocytes with regulatory functions
By secreting periostin, GSCs recruit Tumor-Associated Macrophages (TAMs) into the tumor mass, where periostin-integrin αvβ3 binding initiates their pro-tumoral activation (148). Once recruited, these TAMs undergo functional reprogramming through sustained exposure to GSC-derived factors (TGF-β, IL-4, IL-10), adopting an immunosuppressive phenotype that actively supports tumor progression (149). The activated TAMs then facilitate tumor invasion through MMP9-mediated extracellular matrix degradation (150). Concurrently, they establish an immunosuppressive niche via IDO1-driven tryptophan metabolism, which inhibits effector T-cell function (151, 152). Furthermore, TAMs coordinate angiogenic processes through dual mechanisms: activating the IL-6/JAK-STAT/VEGF axis and secreting SDF-1 to recruit CXCR4+ endothelial progenitor cells (153, 154).
As is validated in preclinical trials, the CXCR4 antagonist plerixafor combined with anti-PD-1 (pembrolizumab) significantly improved survival of GBM mice (155); Treatment with integrin-targeting RGD peptides significantly suppressed POSTN-dependent TAM infiltration, resulting in reduction of tumor burden (148); Genetic silencing of POSTN in GSC-derived GBM models significantly prolonged overall survival (148); Both F4/80-targeted and folate receptor-directed CAR-T cells demonstrated significant tumor growth delay in ovarian cancer models (156, 157), which may be potentially applicable to GBM.
Clinically, combining CAR-T therapy with IDO/arginase inhibitors enhanced interferon production in solid tumors like colon cancer by blocking bone marrow-derived MDSC migration to tumor sites - a critical mechanism that prevents iNOS-mediated arginine depletion and subsequent impairment of CD3-TCR synthesis and T-cell proliferation (158, 159), which could be relevant for GBM therapy.
6.2 GBM intrinsic properties
The inherently low tumor mutational burden in glioblastoma creates a dual therapeutic challenge: it provides insufficient immune activation to overcome immune escape (160) while simultaneously restricting the pool of targetable neoantigens for immunotherapy development. Emerging strategies to enhance tumor immunogenicity include leveraging radiation-induced hypermutation (161, 162) or employing oncolytic viruses to stimulate TAAs release (163). Early clinical validation of these approaches is already emerging, with combination regimens such as DNX-2401 plus pembrolizumab (119) and Ad-RTS-hIL-12 with nivolumab (127) demonstrating preliminary efficacy in GBM patients.
Glioblastoma exhibits profound intratumoral and intertumoral heterogeneity, manifested through multiple dimensions. Molecular analyses reveal significant differences between the tumor core (TC) and peripheral brain tissue (PTB) regions (164). This variability is further compounded by treatment-induced antigen downregulation and phenotypic transformation in recurrent GBM (165). While molecular profiling initially classified GBM into three major subgroups (TCGA-PN, TCGA-CL, and TCGA-MES), most tumors demonstrate dynamic co-existence of these subtypes in varying proportions over time, underscoring the extensive spatial and temporal heterogeneity (3). Furthermore, GSCs serve as key drivers in establishing and maintaining this heterogeneous landscape (166).
Therapeutically, while multi-targeted approaches such as CART-EGFR-IL13Rα2 cells have achieved measurable tumor reduction in clinical trials (167), and DC vaccines combined with anti-PD-1 therapy (nivolumab) show survival benefits in rGBM (NCT02529072) (168), these strategies face inherent limitations due to tumor plasticity. Ultimately, targeting the fundamental mechanisms governing GSC plasticity may be required to achieve durable therapeutic responses.
6.3 Drug infusion disorder
While the blood-tumor barrier (BTB) represents a modified version of the blood-brain barrier (BBB) with altered integrity in tumor-affected regions (169, 170), neuroimaging studies reveal critical spatial heterogeneity in its disruption. MRI analyses demonstrate that in both glioblastoma (GBM) and medulloblastoma patients, BBB breakdown primarily localizes to the tumor core, while peritumoral areas often retain partial or even complete barrier functionality (171, 172). This persisting barrier function in infiltrative tumor regions necessitates targeted strategies to overcome residual drug delivery obstacles.
Emerging preclinical evidence highlights the promise of intranasal delivery platforms for treating central nervous system pathologies. In glioma models, curcumin-encapsulated exosomes administered intranasally significantly enhanced tumor cell apoptosis and improved survival in GL26-bearing mice (173), while engineered microglial exosomes expressing anti-LAG3 antibodies synergized with laser irradiation to boost glioblastoma responses to immune checkpoint inhibitors without developing resistance (174). In addition, mesenchymal stem cell (MSC)-based drug delivery systems have demonstrated efficacy in modulating neuroinflammatory processes in Alzheimer's disease models (175), suggesting their potential utility in transporting anti-tumor drugs.
7 Discussion
Glioblastoma (GBM) remains the most aggressive primary brain tumor despite multimodal therapeutic approaches including radiotherapy, TMZ chemotherapy, DNA damage response (DDR) inhibitors, and targeted molecular therapies. When it comes to immunotherapy, we must acknowledge the fact that while numerous previous immunotherapeutic agents have demonstrated remarkable survival benefits in early-phase clinical trials and single-arm studies, very few have shown efficacy in Phase III randomized controlled trials. Nevertheless, through optimization of drug design (such as continuously advanced oncolytic viruses and upgraded CAR-T cells), appropriate implementation of combination strategies and innovative treatment paradigms ( such as the appearance of neoadjuvant therapy) continue to make immunotherapy a promising approach for GBM patients, especially recurrent ones (Tables 5, 6).
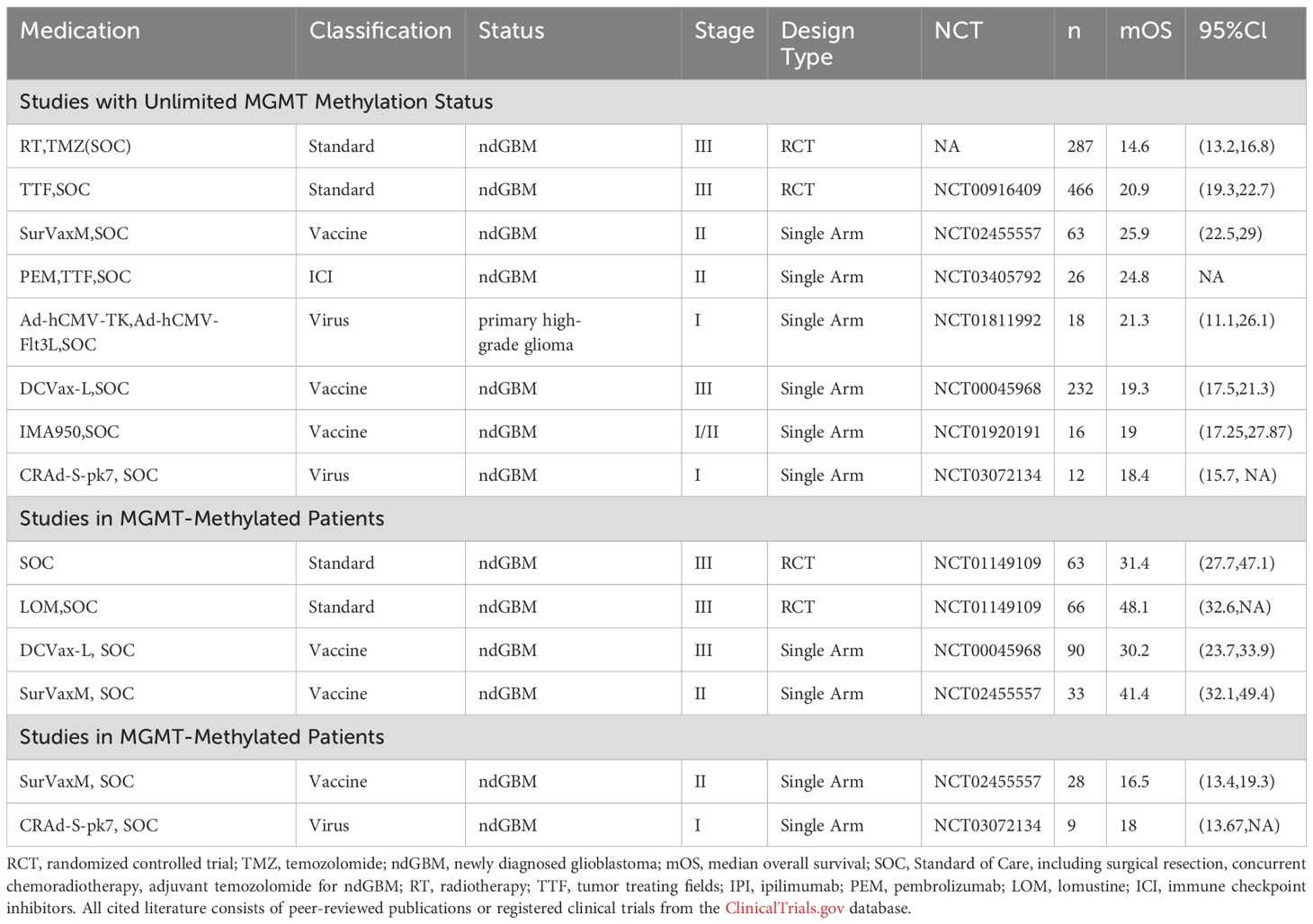
Table 5. Summary of promising drugs of immunotherapy for newly diagnosed patients.
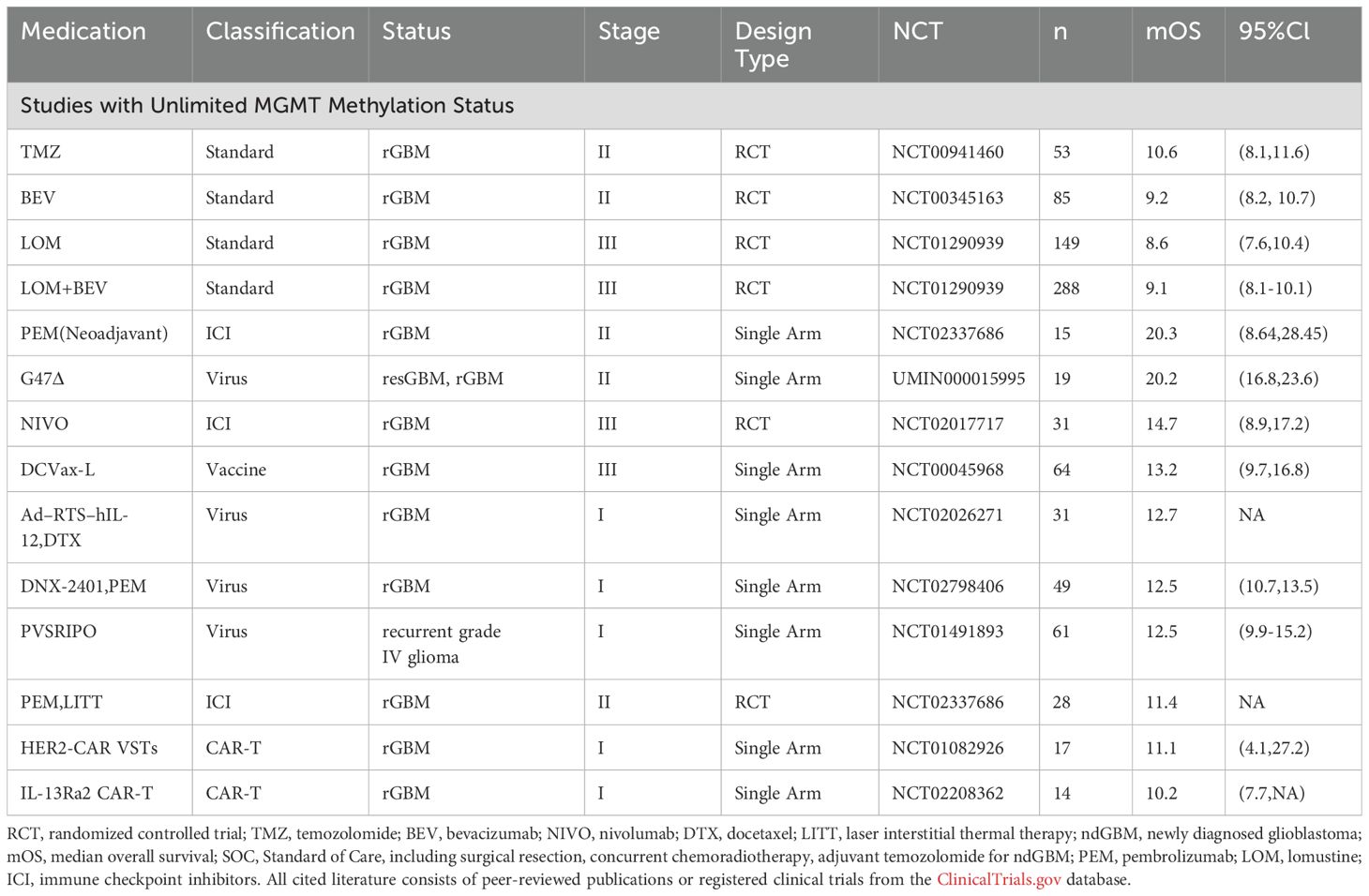
Table 6. Summary of promising drugs of immunotherapy for recurrent patients.
Notably, as diagnostic criteria evolution has introduced prognostic complexities: the 2021 WHO classification now defines GBM specifically as IDH-wildtype (IDHwt) tumors (5), earlier clinical trials, however, included IDH-mutant cases, potentially yielding more optimistic survival outcomes than those expected in trials conducted under current diagnostic standards. This historical discrepancy underscores the importance of molecular stratification in interpreting therapeutic results across different eras of GBM research. Furthermore, since tumor-treating fields were only formally incorporated into the SOC for glioblastoma in the 2021 WHO Classification of Central Nervous System Tumors (5), the control arms across cited in clinical trials consistently employed conventional SOC regimens consisting of concurrent chemoradiotherapy followed by adjuvant TMZ therapy.
In summary, the effectiveness of immunotherapy in glioblastoma is limited by several key factors, including the tumor's low mutational burden, its highly heterogeneous nature, the strongly immunosuppressive microenvironment, drug delivery obstacles, and the brain's unique lymphatic anatomy.
To counteract the hypoxic tumor microenvironment, potential approaches include using gene therapy to knock down AMPKα1 in tumor cells or pretreating T cells with AMPK kinase activators, developing CAR-T cells with low oxygen requirements. For GBM’s escape immunity by downregulating TGFβ receptors while overexpressing TGFβ, solutions involve creating TGFβ receptor-negative T cells or administering TGFβ antagonists. The recruitment of tumor-associated macrophages by POSTN might be blocked using integrin-inhibiting RGD peptides, while CAR-T cells targeting F4/80 and folate receptors could directly eliminate TAMs.
To improve drug delivery, potential strategies include intranasal administration, convection-enhanced delivery, direct tumor or ventricular infusion, and laser interstitial thermal therapy (LITT). Alternative approaches using neural stem cells or nanoparticles (including exosomes) may help drugs cross the blood-brain barrier.
Addressing glioblastoma's fundamental characteristics requires more than just increasing mutational load through radiotherapy or oncolytic viruses. The core challenge lies in overcoming the tumor's subclonal diversity and poor immunogenicity, which are critical for generating effective anti-tumor immune responses.
Beyond the inherent biological and technical hurdles in treating glioblastoma (GBM), the clinical implementation of immunotherapies faces critical logistical and financial barriers that demand urgent attention. Logistically, the cold-chain dependence and time-sensitive nature of many immunotherapeutic agents (e.g., CAR-T cells) pose significant supply-chain challenges. Potential solutions include accelerating the development of universal CAR-T products to reduce manufacturing delays and advancing lyophilized formulations (e.g., BioNTech's room-temperature-stable RNA vaccine technology) to minimize cold storage requirements.
Financially, the prohibitive costs of GBM immunotherapy remain a major obstacle, driven by both the expensive personalized production of immunotherapies (e.g., tumor-specific peptide vaccines) and limited insurance coverage for most emerging treatments. To address affordability, broader adoption of risk-sharing agreements (e.g., the UK NHS's "pay-for-performance" model) and global funding initiatives (e.g., WHO's immunotherapy accessibility programs) could help alleviate the economic burden on patients and healthcare systems.
Author contributions
YZ: Conceptualization, Writing – original draft, Writing – review & editing, Supervision, Funding acquisition, Visualization. FS: Writing – review & editing, Formal analysis, Validation, Project administration. JZ: Writing – review & editing, Software. YY: Writing – review & editing, Data curation, Investigation.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Tan AC, Ashley DM, López GY, Malinzak M, Friedman HS, Khasraw M. Management of glioblastoma: State of the art and future directions. CA Cancer J Clin. (2020) 70:299–312. doi: 10.3322/caac.21613
2. Ostrom QT, Price M, Neff C, Cioffi G, Waite KA, Kruchko C, et al. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2015-2019. Neuro Oncol. (2022) 24:v1–v95. doi: 10.1093/neuonc/noac202
3. Wen PY, Weller M, Lee EQ, Alexander BM, Barnholtz-Sloan JS, Barthel FP, et al. Glioblastoma in adults: a Society for Neuro-Oncology (SNO) and European Society of Neuro-Oncology (EANO) consensus review on current management and future directions. Neuro Oncol. (2020) 22:1073–113. doi: 10.1093/neuonc/noaa106
4. Tykocki T, Eltayeb M. Ten-year survival in glioblastoma. A systematic review. J Clin Neurosci. (2018) 54:7–13. doi: 10.1016/j.jocn.2018.05.002
5. Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, et al. The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro Oncol. (2021) 23:1231–51. doi: 10.1093/neuonc/noab106
6. Stupp R, Taillibert S, Kanner A, Read W, Steinberg D, Lhermitte B, et al. Effect of tumor-treating fields plus maintenance temozolomide vs maintenance temozolomide alone on survival in patients with glioblastoma: A randomized clinical trial. Jama. (2017) 318:2306–16. doi: 10.1001/jama.2017.18718
7. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. (2005) 352:987–96. doi: 10.1056/NEJMoa043330
9. Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. (2006) 444:756–60. doi: 10.1038/nature05236
10. Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. (2005) 352:997–1003. doi: 10.1056/NEJMoa043331
11. Woroniecka K, Chongsathidkiet P, Rhodin K, Kemeny H, Dechant C, Farber SH, et al. T-cell exhaustion signatures vary with tumor type and are severe in glioblastoma. Clin Cancer Res. (2018) 24:4175–86. doi: 10.1158/1078-0432.CCR-17-1846
12. Engel I, Seumois G, Chavez L, Samaniego-Castruita D, White B, Chawla A, et al. Innate-like functions of natural killer T cell subsets result from highly divergent gene programs. Nat Immunol. (2016) 17:728–39. doi: 10.1038/ni.3437
13. Feng Q, Liu Z, Yu X, Huang T, Chen J, Wang J, et al. Lactate increases stemness of CD8 + T cells to augment anti-tumor immunity. Nat Comms. (2022) 13:4981. doi: 10.1038/s41467-022-32521-8
14. Miller TE, Liau BB, Wallace LC, Morton AR, Xie Q, Dixit D, et al. Transcription elongation factors represent in vivo cancer dependencies in glioblastoma. Nature. (2017) 547:355–9. doi: 10.1038/nature23000
15. Niederkorn JY. See no evil, hear no evil, do no evil: the lessons of immune privilege. Nat Immunol. (2006) 7:354–9. doi: 10.1038/ni1328
16. Romagnani S. Immunological tolerance and autoimmunity. Intern Emerg Med. (2006) 1:187–96. doi: 10.1007/BF02934736
17. Castellani G, Croese T, Peralta Ramos JM, Schwartz M. Transforming the understanding of brain immunity. Science. (2023) 380:eabo7649. doi: 10.1126/science.abo7649
18. Sørensen TL, Tani M, Jensen J, Pierce V, Lucchinetti C, Folcik VA, et al. Expression of specific chemokines and chemokine receptors in the central nervous system of multiple sclerosis patients. J Clin Invest. (1999) 103:807–15. doi: 10.1172/JCI5150
19. Deverman BE, Patterson PH. Cytokines and CNS development. Neuron. (2009) 64:61–78. doi: 10.1016/j.neuron.2009.09.002
20. Hickey WF, Hsu BL, Kimura H. T-lymphocyte entry into the central nervous system. J Neurosci Res. (1991) 28:254–60. doi: 10.1002/jnr.490280213
21. Liu Z, Liu X, Liang J, Liu Y, Hou X, Zhang M, et al. Immunotherapy for hepatocellular carcinoma: current status and future prospects. Front Immunol. (2021) 12:765101. doi: 10.3389/fimmu.2021.765101
22. Puleo J, Polyak K. A Darwinian perspective on tumor immune evasion. Biochim Biophys Acta Rev Cancer. (2022) 1877:188671. doi: 10.1016/j.bbcan.2021.188671
23. Gunturi A, McDermott DF. Nivolumab for the treatment of cancer. Expert Opin Invest Drugs. (2015) 24:253–60. doi: 10.1517/13543784.2015.991819
24. Twomey JD, Zhang B. Cancer immunotherapy update: FDA-approved checkpoint inhibitors and companion diagnostics. Aaps j. (2021) 23:39. doi: 10.1208/s12248-021-00574-0
25. Balar AV, Galsky MD, Rosenberg JE, Powles T, Petrylak DP, Bellmunt J, et al. Atezolizumab as first-line treatment in cisplatin-ineligible patients with locally advanced and metastatic urothelial carcinoma: a single-arm, multicentre, phase 2 trial. Lancet. (2017) 389:67–76. doi: 10.1016/S0140-6736(16)32455-2
26. Patel RP, Parikh R, Gunturu KS, Tariq RZ, Dani SS, Ganatra S, et al. Cardiotoxicity of immune checkpoint inhibitors. Curr Oncol Rep. (2021) 23:79. doi: 10.1007/s11912-021-01070-6
27. Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci U S A. (2002) 99:12293–7. doi: 10.1073/pnas.192461099
28. Reardon DA, Brandes AA, Omuro A, Mulholland P, Lim M, Wick A, et al. Effect of nivolumab vs bevacizumab in patients with recurrent glioblastoma: the checkMate 143 phase 3 randomized clinical trial. JAMA Oncol. (2020) 6:1003–10. doi: 10.1001/jamaoncol.2020.1024
29. Lim M, Weller M, Idbaih A, Steinbach J, Finocchiaro G, Raval RR, et al. Phase III trial of chemoradiotherapy with temozolomide plus nivolumab or placebo for newly diagnosed glioblastoma with methylated MGMT promoter. Neuro Oncol. (2022) 24:1935–49. doi: 10.1093/neuonc/noac116
30. Omuro A, Brandes AA, Carpentier AF, Idbaih A, Reardon DA, Cloughesy T, et al. Radiotherapy combined with nivolumab or temozolomide for newly diagnosed glioblastoma with unmethylated MGMT promoter: An international randomized phase III trial. Neuro Oncol. (2023) 25:123–34. doi: 10.1093/neuonc/noac099
31. Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, et al. The 2016 world health organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. (2016) 131:803–20. doi: 10.1007/s00401-016-1545-1
32. Scaringi C, De Sanctis V, Minniti G, Enrici RM. Temozolomide-related hematologic toxicity. Onkologie. (2013) 36:444–9. doi: 10.1159/000353755
33. Schalper KA, Rodriguez-Ruiz ME, Diez-Valle R, López-Janeiro A, Porciuncula A, Idoate MA, et al. Neoadjuvant nivolumab modifies the tumor immune microenvironment in resectable glioblastoma. Nat Med. (2019) 25:470–6. doi: 10.1038/s41591-018-0339-5
34. Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, et al. Pembrolizumab versues ipilimumab in advanced melanoma. N Engl J Med. (2015) 372:2521–32. doi: 10.1056/NEJMoa1503093
35. Nayak L, Molinaro AM, Peters K, Clarke JL, Jordan JT, de Groot J, et al. Randomized Phase II and Biomarker Study of Pembrolizumab plus Bevacizumab versus Pembrolizumab Alone for Patients with Recurrent Glioblastoma. Clin Cancer Res. (2021) 27:1048–57. doi: 10.1158/1078-0432.CCR-20-2500
36. Iwamoto F, Tanguturi S, Desai A, Nayak L, Uhlmann E, Wang T, et al. Ctim-18. Phase 2 study of pd-1 blockade with pembrolizumab plus re-irradiation for recurrent glioblastoma (Rgbm) (Nct03661723). Neuro-Oncology. (2022) 24:vii63–4. doi: 10.1093/neuonc/noac209.233
37. Tol J, Koopman M, Cats A, Rodenburg CJ, Creemers GJ, Schrama JG, et al. Chemotherapy, bevacizumab, and cetuximab in metastatic colorectal cancer. N Engl J Med. (2009) 360:563–72. doi: 10.1056/NEJMoa0808268
38. Hapani S, Sher A, Chu D, Wu S. Increased risk of serious hemorrhage with bevacizumab in cancer patients: a meta-analysis. Oncology. (2010) 79:27–38. doi: 10.1159/000314980
39. Palazón A, Aragonés J, Morales-Kastresana A, de Landázuri MO, Melero I. Molecular pathways: hypoxia response in immune cells fighting or promoting cancer. Clin Cancer Res. (2012) 18:1207–13. doi: 10.1158/1078-0432.CCR-11-1591
40. Grantz KH, Meredith HR, Cummings DAT, Metcalf CJE, Grenfell BT, Giles JR, et al. The use of mobile phone data to inform analysis of COVID-19 pandemic epidemiology. Nat Comms. (2020) 11:4961. doi: 10.1038/s41467-020-18190-5
41. de Groot J, Penas-Prado M, Alfaro-Munoz K, Hunter K, Pei BL, O’Brien B, et al. Window-of-opportunity clinical trial of pembrolizumab in patients with recurrent glioblastoma reveals predominance of immune-suppressive macrophages. Neuro Oncol. (2020) 22:539–49. doi: 10.1093/neuonc/noz185
42. Campian J, Butt O, Ghinaseddin A, Rahman M, Chheda M, Johanns T, et al. Ctim-26. Phase I/ii study of the combination of pembrolizumab (Mk-3475) and laser interstitial thermal therapy (Litt) in recurrent glioblastoma. Neuro-Oncology. (2021) 23:vi56–6. doi: 10.1093/neuonc/noab196.220
43. Hawasli AH, Kim AH, Dunn GP, Tran DD, Leuthardt EC. Stereotactic laser ablation of high-grade gliomas. Neurosurg Focus. (2014) 37:E1. doi: 10.3171/2014.9.FOCUS14471
44. Tran D, Ghiaseddin A, Chen D, Le S, Rahman M. Ctim-05. Final results of 2-the-top: A pilot phase 2 study of ttfields (Optune) plus pembrolizumab plus maintenance temozolomide (Tmz) in patients with newly diagnosed glioblastoma (Ndgbm). Neuro-Oncology. (2022) 24:vii60–0. doi: 10.1093/neuonc/noac209.220
45. Edner NM, Carlesso G, Rush JS, Walker LSK. Targeting co-stimulatory molecules in autoimmune disease. Nat Rev Drug Discov. (2020) 19:860–83. doi: 10.1038/s41573-020-0081-9
46. Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM, et al. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science. (2011) 332:600–3. doi: 10.1126/science.1202947
47. Sloan AE, Winter K, Gilbert MR, Aldape K, Choi S, Wen PY, et al. NRG-BN002: Phase I study of ipilimumab, nivolumab, and the combination in patients with newly diagnosed glioblastoma. Neuro Oncol. (2024) 26:1628–37. doi: 10.1093/neuonc/noae058
48. Lassman A, Polly M-Y, Iwamoto F, Sloan A, Wang T, Aldape K, et al. Ctim-18. Nrg oncology study bn007: randomized phase ii/iii trial of ipilimiumab (Ipi) plus nivolumab (Nivo) vs. Temozolomide (Tmz) in mgmt-unmethylated (Umgmt) newly diagnosed glioblastoma (Ngbm). Neuro-Oncology. (2023) 25:v65–5. doi: 10.1093/neuonc/noad179.0024
49. Omuro A, Vlahovic G, Lim M, Sahebjam S, Baehring J, Cloughesy T, et al. Nivolumab with or without ipilimumab in patients with recurrent glioblastoma: results from exploratory phase I cohorts of CheckMate 143. Neuro Oncol. (2018) 20:674–86. doi: 10.1093/neuonc/nox208
50. Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. (1998) 392:245–52. doi: 10.1038/32588
51. Heath WR, Carbone FR. Cross-presentation, dendritic cells, tolerance and immunity. Annu Rev Immunol. (2001) 19:47–64. doi: 10.1146/annurev.immunol.19.1.47
52. Villadangos JA, Schnorrer P. Intrinsic and cooperative antigen-presenting functions of dendritic-cell subsets in vivo. Nat Rev Immunol. (2007) 7:543–55. doi: 10.1038/nri2103
53. Liau LM, Ashkan K, Tran DD, Campian JL, Trusheim JE, Cobbs CS, et al. First results on survival from a large Phase 3 clinical trial of an autologous dendritic cell vaccine in newly diagnosed glioblastoma. J Transl Med. (2018) 16:142. doi: 10.1186/s12967-018-1507-6
54. Yao Y, Luo F, Tang C, Chen D, Qin Z, Hua W, et al. Molecular subgroups and B7-H4 expression levels predict responses to dendritic cell vaccines in glioblastoma: an exploratory randomized phase II clinical trial. Cancer Immunol Immunother. (2018) 67:1777–88. doi: 10.1007/s00262-018-2232-y
55. Wen PY, Reardon DA, Armstrong TS, Phuphanich S, Aiken RD, Landolfi JC, et al. A randomized double-blind placebo-controlled phase II trial of dendritic cell vaccine ICT-107 in newly diagnosed patients with glioblastoma. Clin Cancer Res. (2019) 25:5799–807. doi: 10.1158/1078-0432.CCR-19-0261
56. Batich KA, Reap EA, Archer GE, Sanchez-Perez L, Nair SK, Schmittling RJ, et al. Long-term survival in glioblastoma with cytomegalovirus pp65-targeted vaccination. Clin Cancer Res. (2017) 23:1898–909. doi: 10.1158/1078-0432.CCR-16-2057
57. Liau LM, Ashkan K, Brem S, Campian JL, Trusheim JE, Iwamoto FM, et al. Association of autologous tumor lysate-loaded dendritic cell vaccination with extension of survival among patients with newly diagnosed and recurrent glioblastoma: A phase 3 prospective externally controlled cohort trial. JAMA Oncol. (2023) 9:112–21. doi: 10.1001/jamaoncol.2022.5370
58. Melief CJ, van der Burg SH. Immunotherapy of established (pre)malignant disease by synthetic long peptide vaccines. Nat Rev Cancer. (2008) 8:351–60. doi: 10.1038/nrc2373
59. Slingluff CL Jr. The present and future of peptide vaccines for cancer: single or multiple, long or short, alone or in combination? Cancer J. (2011) 17:343–50. doi: 10.1097/PPO.0b013e318233e5b2
60. Furnari FB, Cloughesy TF, Cavenee WK, Mischel PS. Heterogeneity of epidermal growth factor receptor signalling networks in glioblastoma. Nat Rev Cancer. (2015) 15:302–10. doi: 10.1038/nrc3918
61. Nishikawa R, Ji XD, Harmon RC, Lazar CS, Gill GN, Cavenee WK, et al. A mutant epidermal growth factor receptor common in human glioma confers enhanced tumorigenicity. Proc Natl Acad Sci U S A. (1994) 91:7727–31. doi: 10.1073/pnas.91.16.7727
62. Mukherjee B, McEllin B, Camacho CV, Tomimatsu N, Sirasanagandala S, Nannepaga S, et al. EGFRvIII and DNA double-strand break repair: a molecular mechanism for radioresistance in glioblastoma. Cancer Res. (2009) 69:4252–9. doi: 10.1158/0008-5472.CAN-08-4853
63. Sampson JH, Heimberger AB, Archer GE, Aldape KD, Friedman AH, Friedman HS, et al. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. J Clin Oncol. (2010) 28:4722–9. doi: 10.1200/JCO.2010.28.6963
64. Sampson JH, Aldape KD, Archer GE, Coan A, Desjardins A, Friedman AH, et al. Greater chemotherapy-induced lymphopenia enhances tumor-specific immune responses that eliminate EGFRvIII-expressing tumor cells in patients with glioblastoma. Neuro Oncol. (2011) 13:324–33. doi: 10.1093/neuonc/noq157
65. Schuster J, Lai RK, Recht LD, Reardon DA, Paleologos NA, Groves MD, et al. A phase II, multicenter trial of rindopepimut (CDX-110) in newly diagnosed glioblastoma: the ACT III study. Neuro Oncol. (2015) 17:854–61. doi: 10.1093/neuonc/nou348
66. Weller M, Butowski N, Tran DD, Recht LD, Lim M, Hirte H, et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): a randomised, double-blind, international phase 3 trial. Lancet Oncol. (2017) 18:1373–85. doi: 10.1016/S1470-2045(17)30517-X
67. Reardon DA, Desjardins A, Vredenburgh JJ, O’Rourke DM, Tran DD, Fink KL, et al. Rindopepimut with bevacizumab for patients with relapsed EGFRvIII-expressing glioblastoma (ReACT): results of a double-blind randomized phase II trial. Clin Cancer Res. (2020) 26:1586–94. doi: 10.1158/1078-0432.CCR-18-1140
68. Liu Z, Zhao Q, Zheng Z, Liu S, Meng L, Dong L, et al. Vascular normalization in immunotherapy: A promising mechanisms combined with radiotherapy. Biomed Pharmacother. (2021) 139:111607. doi: 10.1016/j.biopha.2021.111607
69. Migliorini D, Dutoit V, Allard M, Grandjean Hallez N, Marinari E, Widmer V, et al. Phase I/II trial testing safety and immunogenicity of the multipeptide IMA950/poly-ICLC vaccine in newly diagnosed adult Malignant astrocytoma patients. Neuro Oncol. (2019) 21:923–33. doi: 10.1093/neuonc/noz040
70. Binder RJ, Srivastava PK. Essential role of CD91 in re-presentation of gp96-chaperoned peptides. Proc Natl Acad Sci U S A. (2004) 101:6128–33. doi: 10.1073/pnas.0308180101
71. Bloch O, Lim M, Sughrue ME, Komotar RJ, Abrahams JM, O’Rourke DM, et al. Autologous heat shock protein peptide vaccination for newly diagnosed glioblastoma: impact of peripheral PD-L1 expression on response to therapy. Clin Cancer Res. (2017) 23:3575–84. doi: 10.1158/1078-0432.CCR-16-1369
72. Bloch O, Crane CA, Fuks Y, Kaur R, Aghi MK, Berger MS, et al. Heat-shock protein peptide complex-96 vaccination for recurrent glioblastoma: a phase II, single-arm trial. Neuro Oncol. (2014) 16:274–9. doi: 10.1093/neuonc/not203
73. Bloch O, Shi Q, Anderson SK, Knopp M, Raizer J, Clarke J, et al. Parney I. Atim-14. Alliance A071101: A phase ii randomized trial comparing the efficacy of heat shock protein peptide complex-96 (Hsppc-96) vaccine given with bevacizumab versus bevacizumab alone in the treatment of surgically resectable recurrent glioblastoma. Neuro-Oncology. (2017) 19:vi29–9. doi: 10.1093/neuonc/nox168.118
74. Lathia JD, Gallagher J, Heddleston JM, Wang J, Eyler CE, Macswords J, et al. Integrin alpha 6 regulates glioblastoma stem cells. Cell Stem Cell. (2010) 6:421–32. doi: 10.1016/j.stem.2010.02.018
75. Haas TL, Sciuto MR, Brunetto L, Valvo C, Signore M, Fiori ME, et al. Integrin α7 is a functional marker and potential therapeutic target in glioblastoma. Cell Stem Cell. (2017) 21:35–50.e39. doi: 10.1016/j.stem.2017.04.009
76. Ahluwalia MS, Reardon DA, Abad AP, Curry WT, Wong ET, Figel SA, et al. Phase IIa study of surVaxM plus adjuvant temozolomide for newly diagnosed glioblastoma. J Clin Oncol. (2023) 41:1453–65. doi: 10.1200/JCO.22.00996
77. Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. (2002) 3:991–8. doi: 10.1038/ni1102-991
78. Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q, et al. Loss of IFN-γ Pathway genes in tumor cells as a mechanism of resistance to anti-CTLA-4 therapy. Cell. (2016) 167:397–404.e399. doi: 10.1016/j.cell.2016.08.069
79. Sadelain M, Brentjens R, Rivière I. The basic principles of chimeric antigen receptor design. Cancer Discov. (2013) 3:388–98. doi: 10.1158/2159-8290.CD-12-0548
80. Sun Y, Florio TJ, Gupta S, Young MC, Marshall QF, Garfinkle SE, et al. Structural principles of peptide-centric chimeric antigen receptor recognition guide therapeutic expansion. Sci Immunol. (2023) 8:eadj5792. doi: 10.1126/sciimmunol.adj5792
81. Cherkassky L, Morello A, Villena-Vargas J, Feng Y, Dimitrov DS, Jones DR, et al. Adusumilli PS. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J Clin Invest. (2016) 126:3130–44. doi: 10.1172/JCI83092
82. Brown CE, Warden CD, Starr R, Deng X, Badie B, Yuan YC, et al. Glioma IL13Rα2 is associated with mesenchymal signature gene expression and poor patient prognosis. PloS One. (2013) 8:e77769. doi: 10.1371/journal.pone.0077769
83. Thaci B, Brown CE, Binello E, Werbaneth K, Sampath P, Sengupta S. Significance of interleukin-13 receptor alpha 2-targeted glioblastoma therapy. Neuro Oncol. (2014) 16:1304–12. doi: 10.1093/neuonc/nou045
84. McCormick SM, Heller NM. Commentary: IL-4 and IL-13 receptors and signaling. Cytokine. (2015) 75:38–50. doi: 10.1016/j.cyto.2015.05.023
85. Brown CE, Badie B, Barish ME, Weng L, Ostberg JR, Chang WC, et al. Bioactivity and safety of IL13Rα2-redirected chimeric antigen receptor CD8+ T cells in patients with recurrent glioblastoma. Clin Cancer Res. (2015) 21:4062–72. doi: 10.1158/1078-0432.CCR-15-0428
86. Brown CE, Hibbard JC, Alizadeh D, Blanchard MS, Natri HM, Wang D, et al. Locoregional delivery of IL-13Rα2-targeting CAR-T cells in recurrent high-grade glioma: a phase 1 trial. Nat Med. (2024) 30:1001–12. doi: 10.1038/s41591-024-02875-1
87. Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N Engl J Med. (2016) 375:2561–9. doi: 10.1056/NEJMoa1610497
88. Brown CE, Rodriguez A, Palmer J, Ostberg JR, Naranjo A, Wagner JR, et al. Off-the-shelf, steroid-resistant, IL13Rα2-specific CAR T cells for treatment of glioblastoma. Neuro Oncol. (2022) 24:1318–30. doi: 10.1093/neuonc/noac024
89. O’Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med. (2017) 9:eaaj0981. doi: 10.1126/scitranslmed.aaa0984
90. Goff SL, Morgan RA, Yang JC, Sherry RM, Robbins PF, Restifo NP, et al. Pilot trial of adoptive transfer of chimeric antigen receptor-transduced T cells targeting EGFRvIII in patients with glioblastoma. J Immunother. (2019) 42:126–35. doi: 10.1097/CJI.0000000000000260
91. Inda MM, Bonavia R, Mukasa A, Narita Y, Sah DW, Vandenberg S, et al. Tumor heterogeneity is an active process maintained by a mutant EGFR-induced cytokine circuit in glioblastoma. Genes Dev. (2010) 24:1731–45. doi: 10.1101/gad.1890510
92. Choi BD, Gerstner ER, Frigault MJ, Leick MB, Mount CW, Balaj L, et al. Intraventricular CARv3-TEAM-E T cells in recurrent glioblastoma. N Engl J Med. (2024) 390:1290–8. doi: 10.1056/NEJMoa2314390
93. Schlegel J, Stumm G, Brändle K, Merdes A, Mechtersheimer G, Hynes NE, et al. Amplification and differential expression of members of the erbB-gene family in human glioblastoma. J Neurooncol. (1994) 22:201–7. doi: 10.1007/BF01052920
94. Merkhofer EC, Cogswell P, Baldwin AS. Her2 activates NF-kappaB and induces invasion through the canonical pathway involving IKKalpha. Oncogene. (2010) 29:1238–48. doi: 10.1038/onc.2009.410
95. Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, et al. The somatic genomic landscape of glioblastoma. Cell. (2013) 155:462–77. doi: 10.1016/j.cell.2013.09.034
96. Ahmed N, Brawley V, Hegde M, Bielamowicz K, Kalra M, Landi D, et al. HER2-specific chimeric antigen receptor-modified virus-specific T cells for progressive glioblastoma: A phase 1 dose-escalation trial. JAMA Oncol. (2017) 3:1094–101. doi: 10.1001/jamaoncol.2017.0184
97. King AC, Orozco JS. Axicabtagene ciloleucel: the first FDA-approved CAR T-cell therapy for relapsed/refractory large B-cell lymphoma. J Adv Pract Oncol. (2019) 10:878–82. doi: 10.6004/jadpro.2019.10.8.7
98. Kochenderfer JN, Somerville RPT, Lu T, Shi V, Bot A, Rossi J, et al. Lymphoma remissions caused by anti-CD19 chimeric antigen receptor T cells are associated with high serum interleukin-15 levels. J Clin Oncol. (2017) 35:1803–13. doi: 10.1200/JCO.2016.71.3024
99. Shao X, Wang X, Guo X, Jiang K, Ye T, Chen J, et al. STAT3 contributes to oncolytic newcastle disease virus-induced immunogenic cell death in melanoma cells. Front Oncol. (2019) 9:436. doi: 10.3389/fonc.2019.00436
100. Diaconu I, Cerullo V, Hirvinen ML, Escutenaire S, Ugolini M, Pesonen SK, et al. Immune response is an important aspect of the antitumor effect produced by a CD40L-encoding oncolytic adenovirus. Cancer Res. (2012) 72:2327–38. doi: 10.1158/0008-5472.CAN-11-2975
101. Bartlett DL, Liu Z, Sathaiah M, Ravindranathan R, Guo Z, He Y, et al. Oncolytic viruses as therapeutic cancer vaccines. Mol Cancer. (2013) 12:103. doi: 10.1186/1476-4598-12-103
102. Jhawar SR, Thandoni A, Bommareddy PK, Hassan S, Kohlhapp FJ, Goyal S, et al. Oncolytic viruses-natural and genetically engineered cancer immunotherapies. Front Oncol. (2017) 7:202. doi: 10.3389/fonc.2017.00202
103. Inoue H, Tani K. Multimodal immunogenic cancer cell death as a consequence of anticancer cytotoxic treatments. Cell Death Differ. (2014) 21:39–49. doi: 10.1038/cdd.2013.84
104. Weller SK, Coen DM. Herpes simplex viruses: mechanisms of DNA replication. Cold Spring Harb Perspect Biol. (2012) 4:a013011. doi: 10.1101/cshperspect.a013011
105. Chou J, Kern ER, Whitley RJ, Roizman B. Mapping of herpes simplex virus-1 neurovirulence to gamma 134.5, a gene nonessential for growth in culture. Science. (1990) 250:1262–6. doi: 10.1126/science.2173860
106. Rampling R, Cruickshank G, Papanastassiou V, Nicoll J, Hadley D, Brennan D, et al. Toxicity evaluation of replication-competent herpes simplex viru (ICP 34.5 null mutant 1716) in patients with recurrent Malignant glioma. Gene Ther. (2000) 7:859–66. doi: 10.1038/sj.gt.3301184
107. Harrow S, Papanastassiou V, Harland J, Mabbs R, Petty R, Fraser M, et al. HSV1716 injection into the brain adjacent to tumour following surgical resection of high-grade glioma: safety data and long-term survival. Gene Ther. (2004) 11:1648–58. doi: 10.1038/sj.gt.3302289
108. Mineta T, Rabkin SD, Yazaki T, Hunter WD, Martuza RL. Attenuated multi-mutated herpes simplex virus-1 for the treatment of Malignant gliomas. Nat Med. (1995) 1:938–43. doi: 10.1038/nm0995-938
109. Goldstein DJ, Weller SK. Herpes simplex virus type 1-induced ribonucleotide reductase activity is dispensable for virus growth and DNA synthesis: isolation and characterization of an ICP6 lacZ insertion mutant. J Virol. (1988) 62:196–205. doi: 10.1128/jvi.62.1.196-205.1988
110. Markert JM, Razdan SN, Kuo HC, Cantor A, Knoll A, Karrasch M, et al. A phase 1 trial of oncolytic HSV-1, G207, given in combination with radiation for recurrent GBM demonstrates safety and radiographic responses. Mol Ther. (2014) 22:1048–55. doi: 10.1038/mt.2014.22
111. York IA, Roop C, Andrews DW, Riddell SR, Graham FL, Johnson DC. A cytosolic herpes simplex virus protein inhibits antigen presentation to CD8+ T lymphocytes. Cell. (1994) 77:525–35. doi: 10.1016/0092-8674(94)90215-1
112. Todo T. Oncolytic virus therapy using genetically engineered herpes simplex viruses. Front Biosci. (2008) 13:2060–4. doi: 10.2741/2823
113. He B, Chou J, Brandimarti R, Mohr I, Gluzman Y, Roizman B. Suppression of the phenotype of gamma(1)34.5- herpes simplex virus 1: failure of activated RNA-dependent protein kinase to shut off protein synthesis is associated with a deletion in the domain of the alpha47 gene. J Virol. (1997) 71:6049–54. doi: 10.1128/jvi.71.8.6049-6054.1997
114. Todo T, Ino Y, Ohtsu H, Shibahara J, Tanaka M. A phase I/II study of triple-mutated oncolytic herpes virus G47Δ in patients with progressive glioblastoma. Nat Commun. (2022) 13:4119. doi: 10.1038/s41467-022-31262-y
115. Todo T, Ito H, Ino Y, Ohtsu H, Ota Y, Shibahara J, et al. Intratumoral oncolytic herpes virus G47Δ for residual or recurrent glioblastoma: a phase 2 trial. Nat Med. (2022) 28:1630–9. doi: 10.1038/s41591-022-01897-x
116. Cervera-Carrascon V, Havunen R, Hemminki A. Oncolytic adenoviruses: a game changer approach in the battle between cancer and the immune system. Expert Opin Biol Ther. (2019) 19:443–55. doi: 10.1080/14712598.2019.1595582
117. Fueyo J, Gomez-Manzano C, Alemany R, Lee PS, McDonnell TJ, Mitlianga P, et al. A mutant oncolytic adenovirus targeting the Rb pathway produces anti-glioma effect in vivo. Oncogene. (2000) 19:2–12. doi: 10.1038/sj.onc.1203251
118. Dmitriev I, Krasnykh V, Miller CR, Wang M, Kashentseva E, Mikheeva G, et al. An adenovirus vector with genetically modified fibers demonstrates expanded tropism via utilization of a coxsackievirus and adenovirus receptor-independent cell entry mechanism. J Virol. (1998) 72:9706–13. doi: 10.1128/JVI.72.12.9706-9713.1998
119. Nassiri F, Patil V, Yefet LS, Singh O, Liu J, Dang RMA, et al. Oncolytic DNX-2401 virotherapy plus pembrolizumab in recurrent glioblastoma: a phase 1/2 trial. Nat Med. (2023) 29:1370–8. doi: 10.1038/s41591-023-02347-y
120. Fares J, Ahmed AU, Ulasov IV, Sonabend AM, Miska J, Lee-Chang C, et al. Neural stem cell delivery of an oncolytic adenovirus in newly diagnosed Malignant glioma: a first-in-human, phase 1, dose-escalation trial. Lancet Oncol. (2021) 22:1103–14. doi: 10.1016/S1470-2045(21)00245-X
121. Gromeier M, Lachmann S, Rosenfeld MR, Gutin PH, Wimmer E. Intergeneric poliovirus recombinants for the treatment of Malignant glioma. Proc Natl Acad Sci U S A. (2000) 97:6803–8. doi: 10.1073/pnas.97.12.6803
122. Perez OD, Logg CR, Hiraoka K, Diago O, Burnett R, Inagaki A, et al. Design and selection of Toca 511 for clinical use: modified retroviral replicating vector with improved stability and gene expression. Mol Ther. (2012) 20:1689–98. doi: 10.1038/mt.2012.83
123. Immonen A, Vapalahti M, Tyynelä K, Hurskainen H, Sandmair A, Vanninen R, et al. AdvHSV-tk gene therapy with intravenous ganciclovir improves survival in human Malignant glioma: a randomised, controlled study. Mol Ther. (2004) 10:967–72. doi: 10.1016/j.ymthe.2004.08.002
124. Umemura Y, Orringer D, Junck L, Varela ML, West MEJ, Faisal SM, et al. Combined cytotoxic and immune-stimulatory gene therapy for primary adult high-grade glioma: a phase 1, first-in-human trial. Lancet Oncol. (2023) 24:1042–52. doi: 10.1016/S1470-2045(23)00347-9
125. Brenner AJ, Peters KB, Vredenburgh J, Bokstein F, Blumenthal DT, Yust-Katz S, et al. Safety and efficacy of VB-111, an anticancer gene therapy, in patients with recurrent glioblastoma: results of a phase I/II study. Neuro Oncol. (2020) 22:694–704. doi: 10.1093/neuonc/noz231
126. Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol. (2003) 3:133–46. doi: 10.1038/nri1001
127. Chiocca EA, Yu JS, Lukas RV, Solomon IH, Ligon KL, Nakashima H, et al. Regulatable interleukin-12 gene therapy in patients with recurrent high-grade glioma: Results of a phase 1 trial. Sci Transl Med. (2019) 11:eaaw5680. doi: 10.1126/scitranslmed.aaw5680
128. Moolten FL. Tumor chemosensitivity conferred by inserted herpes thymidine kinase genes: paradigm for a prospective cancer control strategy. Cancer Res. (1986) 46:5276–81. doi: 10.1158/0008-5472.CAN-46-10-5276
129. Shurin MR, Pandharipande PP, Zorina TD, Haluszczak C, Subbotin VM, Hunter O, et al. FLT3 ligand induces the generation of functionally active dendritic cells in mice. Cell Immunol. (1997) 179:174–84. doi: 10.1006/cimm.1997.1152
130. Todo T, Feigenbaum F, Rabkin SD, Lakeman F, Newsome JT, Johnson PA, et al. Viral shedding and biodistribution of G207, a multimutated, conditionally replicating herpes simplex virus type 1, after intracerebral inoculation in aotus. Mol Ther. (2000) 2:588–95. doi: 10.1006/mthe.2000.0200
131. Markert JM, Liechty PG, Wang W, Gaston S, Braz E, Karrasch M, et al. Phase Ib trial of mutant herpes simplex virus G207 inoculated pre-and post-tumor resection for recurrent GBM. Mol Ther. (2009) 17:199–207. doi: 10.1038/mt.2008.228
132. Hao C, Chen G, Zhao H, Li Y, Chen J, Zhang H, et al. PD-L1 expression in glioblastoma, the clinical and prognostic significance: A systematic literature review and meta-analysis. Front Oncol. (2020) 10:1015. doi: 10.3389/fonc.2020.01015
133. Burster T, Gärtner F, Bulach C, Zhanapiya A, Gihring A, Knippschild U. Regulation of MHC I molecules in glioblastoma cells and the sensitizing of NK cells. Pharmaceuticals (Basel). (2021) 14:236. doi: 10.3390/ph14030236
134. Gratas C, Tohma Y, Van Meir EG, Klein M, Tenan M, Ishii N, et al. Fas ligand expression in glioblastoma cell lines and primary astrocytic brain tumors. Brain Pathol. (1997) 7:863–9. doi: 10.1111/j.1750-3639.1997.tb00889.x
135. Agarwal S, Aznar MA, Rech AJ, Good CR, Kuramitsu S, Da T, et al. Deletion of the inhibitory co-receptor CTLA-4 enhances and invigorates chimeric antigen receptor T cells. Immunity. (2023) 56:2388–2407.e2389. doi: 10.1016/j.immuni.2023.09.001
136. Hu YL, DeLay M, Jahangiri A, Molinaro AM, Rose SD, Carbonell WS, et al. Hypoxia-induced autophagy promotes tumor cell survival and adaptation to antiangiogenic treatment in glioblastoma. Cancer Res. (2012) 72:1773–83. doi: 10.1158/0008-5472.CAN-11-3831
137. Yun HJ, Li M, Guo D, Jeon SM, Park SH, Lim JS, et al. AMPK-HIF-1α signaling enhances glucose-derived de novo serine biosynthesis to promote glioblastoma growth. J Exp Clin Cancer Res. (2023) 42:340. doi: 10.1186/s13046-023-02927-3
138. Keerthana CK, Rayginia TP, Shifana SC, Anto NP, Kalimuthu K, Isakov N, et al. The role of AMPK in cancer metabolism and its impact on the immunomodulation of the tumor microenvironment. Front Immunol. (2023) 14:1114582. doi: 10.3389/fimmu.2023.1114582
139. . Brand A, Singer K, Koehl GE, Kolitzus M, Schoenhammer G, Thiel A, et al. LDHA-associated lactic acid production blunts tumor immunosurveillance by T and NK cells. Cell Metab. (2016) 24:657–71. doi: 10.1016/j.cmet.2016.08.011
140. Hatae R, Kyewalabye K, Yamamichi A, Chen T, Phyu S, Chuntova P, et al. Enhancing CAR-T cell metabolism to overcome hypoxic conditions in the brain tumor microenvironment. bioRxiv. (2023) 9(7):e177141. doi: 10.1101/2023.11.13.566775
141. Kosti P, Opzoomer JW, Larios-Martinez KI, Henley-Smith R, Scudamore CL, Okesola M, et al. Hypoxia-sensing CAR T cells provide safety and efficacy in treating solid tumors. Cell Rep Med. (2021) 2:100227. doi: 10.1016/j.xcrm.2021.100227
142. Huang X, Tu M, Yu H, Jia Y, Yan X, Tang X, et al. Application of CRISPR/Cas 9 System to Construct Lung Cancer Cell Lines to Obtain Stable Cell Lines with AMPK α1 Knockdown. J Modern Lab Med. (2023) 38(1):107–11. doi: 10.3969/j.issn.1671-7414.2023.01.020
143. Gorter A, Meri S. Immune evasion of tumor cells using membrane-bound complement regulatory proteins. Immunol Today. (1999) 20:576–82. doi: 10.1016/S0167-5699(99)01537-6
144. Han J, Alvarez-Breckenridge CA, Wang QE, Yu J. TGF-β signaling and its targeting for glioma treatment. Am J Cancer Res. (2015) 5:945–55. doi: 10.7150/ijbs.5.945
145. Ravi VM, Neidert N, Will P, Joseph K, Maier JP, KüDckelhaus J, et al. T-cell dysfunction in the glioblastoma microenvironment is mediated by myeloid cells releasing interleukin-10. Nat Commun. (2022) 13:925. doi: 10.1038/s41467-022-28523-1
146. Zhang M, Kleber S, Röhrich M, Timke C, Han N, Tuettenberg J, et al. Blockade of TGF-β signaling by the TGFβR-I kinase inhibitor LY2109761 enhances radiation response and prolongs survival in glioblastoma. Cancer Res. (2011) 71:7155–67. doi: 10.1158/0008-5472.CAN-11-1212
147. Silk JD, Abbott RJM, Adams KJ, Bennett AD, Brett S, Cornforth TV, et al. Engineering cancer antigen-specific T cells to overcome the immunosuppressive effects of TGF-β. J Immunol. (2022) 208:169–80. doi: 10.4049/jimmunol.2001357
148. Zhou W, Ke SQ, Huang Z, Flavahan W, Fang X, Paul J, et al. Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes Malignant growth. Nat Cell Biol. (2015) 17:170–82. doi: 10.1038/ncb3090
149. Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med. (2013) 19:1264–72. doi: 10.1038/nm.3337
150. Markovic DS, Vinnakota K, Chirasani S, Synowitz M, Raguet H, Stock K, et al. Gliomas induce and exploit microglial MT1-MMP expression for tumor expansion. Proc Natl Acad Sci U S A. (2009) 106:12530–5. doi: 10.1073/pnas.0804273106
151. Zhai L, Ladomersky E, Lauing KL, Wu M, Genet M, Gritsina G, et al. Infiltrating T cells increase IDO1 expression in glioblastoma and contribute to decreased patient survival. Clin Cancer Res. (2017) 23:6650–60. doi: 10.1158/1078-0432.CCR-17-0120
152. Stergiopoulos GM, Concilio SC, Galanin E. An update on the clinical status, challenges, and future directions of oncolytic virotherapy for Malignant gliomas. Curr Treat Opt Oncol. (2024) 25:952–91. doi: 10.1007/s11864-024-01211-6
153. Zagzag D, Lukyanov Y, Lan L, Ali MA, Esencay M, Mendez O, et al. Hypoxia-inducible factor 1 and VEGF upregulate CXCR4 in glioblastoma: implications for angiogenesis and glioma cell invasion. Lab Invest. (2006) 86:1221–32. doi: 10.1038/labinvest.3700482
154. Zhang L, Xu Y, Sun J, Chen W, Zhao L, Ma C, et al. M2-like tumor-associated macrophages drive vasculogenic mimicry through amplification of IL-6 expression in glioma cells.. Oncotarget. (2017) 8:819–32. doi: 10.18632/oncotarget.13661
155. Wu A, Maxwell R, Xia Y, Cardarelli P, Oyasu M, Belcaid Z, et al. Combination anti-CXCR4 and anti-PD-1 immunotherapy provides survival benefit in glioblastoma through immune cell modulation of tumor microenvironment. J Neurooncol. (2019) 143:241–9. doi: 10.1007/s11060-019-03172-5
156. Rodriguez-Garcia A, Lynn RC, Poussin M, Eiva MA, Shaw LC, O’Connor RS, et al. CAR-T cell-mediated depletion of immunosuppressive tumor-associated macrophages promotes endogenous antitumor immunity and augments adoptive immunotherapy. Nat Commun. (2021) 12:877. doi: 10.1038/s41467-021-20893-2
157. Sánchez-Paulete AR, Mateus-Tique J, Mollaoglu G, Nielsen SR, Marks A, Lakshmi A, et al. Targeting macrophages with CAR T cells delays solid tumor progression and enhances antitumor immunity. Cancer Immunol Res. (2022) 10:1354–69. doi: 10.1158/2326-6066.CIR-21-1075
158. Nayak-Kapoor A, Hao Z, Sadek R, Dobbins R, Marshall L, Vahanian NN, et al. Phase Ia study of the indoleamine 2,3-dioxygenase 1 (IDO1) inhibitor navoximod (GDC-0919) in patients with recurrent advanced solid tumors. J Immunother Cancer. (2018) 6:61. doi: 10.1186/s40425-018-0351-9
159. Umansky V, Adema GJ, Baran J, Brandau S, Van Ginderachter JA, Hu X, et al. Interactions among myeloid regulatory cells in cancer. Cancer Immunol Immunother. (2019) 68:645–60. doi: 10.1007/s00262-018-2200-6
160. Yadav M, Jhunjhunwala S, Phung QT, Lupardus P, Tanguay J, Bumbaca S, et al. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature. (2014) 515:572–6. doi: 10.1038/nature14001
161. Draaisma K, Chatzipli A, Taphoorn M, Kerkhof M, Weyerbrock A, Sanson M, et al. Molecular evolution of IDH wild-type glioblastomas treated with standard of care affects survival and design of precision medicine trials: A report from the EORTC 1542 study. J Clin Oncol. (2020) 38:81–99. doi: 10.1200/JCO.19.00367
162. Wang J, Cazzato E, Ladewig E, Frattini V, Rosenbloom DI, Zairis S, et al. Clonal evolution of glioblastoma under therapy. Nat Genet. (2016) 48:768–76. doi: 10.1038/ng.3590
163. Chiocca EA, Rabkin SD. Oncolytic viruses and their application to cancer immunotherapy. Cancer Immunol Res. (2014) 2:295–300. doi: 10.1158/2326-6066.CIR-14-0015
164. Wang X, Sun Q, Liu T, Lu H, Lin X, Wang W, et al. Single-cell multi-omics sequencing uncovers region-specific plasticity of glioblastoma for complementary therapeutic targeting. Sci Adv. (2024) 10:eadn4306. doi: 10.1126/sciadv.adn4306
165. Larsson I, Dalmo E, Elgendy R, Niklasson M, Doroszko M, Segerman A, et al. Modeling glioblastoma heterogeneity as a dynamic network of cell states. Mol Syst Biol. (2021) 17:e10105. doi: 10.15252/msb.202010105
166. Liu L, Liu Z, Liu Q, Wu W, Lin P, Liu X, et al. LncRNA INHEG promotes glioma stem cell maintenance and tumorigenicity through regulating rRNA 2’-O-methylation. Nat Commun. (2023) 14:7526. doi: 10.1038/s41467-023-43113-5
167. Bagley SJ, Logun M, Fraietta JA, Wang X, Desai AS, Bagley LJ, et al. Intrathecal bivalent CAR T cells targeting EGFR and IL13Rα2 in recurrent glioblastoma: phase 1 trial interim results. Nat Med. (2024) 30:1320–9. doi: 10.1038/s41591-024-02893-z
168. Das S, Dash BS, Premji TP, Chen JP. Immunotherapeutic approaches for the treatment of glioblastoma multiforme: mechanism and clinical applications. Int J Mol Sci. (2023) 24:11146. doi: 10.3390/ijms241310546
169. Rathi S, Griffith JI, Zhang W, Zhang W, Oh JH, Talele S, et al. The influence of the blood-brain barrier in the treatment of brain tumours. J Intern Med. (2022) 292(1):3–30. doi: 10.1111/joim.13438
170. Banks WA. From blood–brain barrier to blood–brain interface: new opportunities for CNS drug delivery. Nat Rev Drug Discov. (2016) 15:275–92. doi: 10.1038/nrd.2015.21
171. Sarkaria JN, Hu LS, Parney IF, Pafundi DH, Brinkmann DH, Laack NN, et al. Is the blood-brain barrier really disrupted in all glioblastomas? A critical assessment of existing clinical data. Neuro Oncol. (2018) 20:184–91. doi: 10.1093/neuonc/nox175
172. Varlet P, le teuff G, le deley MC, giangaspero F, haberler C, Jacques TS, et al. WHO grade has no prognostic value in the pediatric high-grade glioma included in the HERBY trial. Neuro Oncol. (2020) 22:116–27. doi: 10.1093/neuonc/noz142
173. Zhuang X, Xiang X, Grizzle W, Sun D, Zhang S, Axtell RC, et al. Treatment of brain inflammatory diseases by delivering exosome encapsulated anti-inflammatory drugs from the nasal region to the brain. Mol Ther. (2011) 19:1769–79. doi: 10.1038/mt.2011.164
174. Lin X, Sun Z, Huang S, Liu C, Peng J, Li Y, et al. Engineered microglia-exosomes coated highly twisting AIE photothermal agents to efficiently cross blood-brain-barrier for mild photothermal-immune checkpoint blockade therapy in glioblastoma. Adv Funct Matls. (2024) 34:2407527. doi: 10.1002/adfm.202407527
175. Jin Y, Tang Z, Shang S, Chen Y, Han G, Song M, et al. A nanodisc-paved biobridge facilitates stem cell membrane fusogenicity for intracerebral shuttling and bystander effects. Adv Matls. (2023) 35:2302367. doi: 10.1002/adma.202302367
Keywords: glioblastoma, immune checkpoint inhibitors, CAR-T, tumor vaccine, oncolytic virus therapy, PD-1 inhibitors, CTLA-4 inhibitors, DC vaccines
Citation: Zhou Y, Shi F, Zhu J and Yuan Y (2025) An update on the clinical trial research of immunotherapy for glioblastoma. Front. Immunol. 16:1582296. doi: 10.3389/fimmu.2025.1582296
Received: 24 February 2025; Accepted: 07 April 2025;
Published: 02 May 2025.
Edited by:
Vinay Kumar, The Pennsylvania State University, United StatesReviewed by:
Hina Sultana, University of North Carolina System, United StatesTara Chand Yadav, University of North Carolina at Chapel Hill, United States
Anuradha Tyagi, Institute of Nuclear Medicine & Allied Sciences (DRDO), India
Copyright © 2025 Zhou, Shi, Zhu and Yuan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yichen Zhou, eWljaGVuX3pob3VfbWRzQDE2My5jb20=