 Xiaolei Lan
Xiaolei Lan Hai Zhao
Hai Zhao- Department of Neurosurgery, The Affiliated Hospital of Qingdao University, Qingdao, Shandong, China
Cancer-associated fibroblasts (CAFs) are integral components of the tumor microenvironment playing key roles in tumor progression, metastasis, and therapeutic resistance. However, challenges persist in understanding their heterogeneity, origin, and functional diversity. One major obstacle is the lack of standardized naming conventions for CAF subpopulations, with current systems failing to capture their full complexity. Additionally, the identification of CAFs is hindered by the absence of specific biomarkers, limiting the precision of diagnostic and therapeutic strategies. In vitro culture conditions often fail to maintain the in vivo characteristics of CAFs, which complicates their study and the translation of findings to clinical practice. Although current detection methods, such as antibodies, mRNA probes, and single-cell transcriptomics, offer insights into CAF biology, they lack standardization and fail to provide reliable quantitative measures. Furthermore, the dynamic interactions between CAFs, tumor cells, and immune cells within the TME remain insufficiently understood, and the role of CAFs in immune evasion and therapy resistance is an area of ongoing research. Understanding how CAFs influence drug resistance and the immune response is essential for developing more effective cancer therapies. This review aims to provide an in-depth analysis of the challenges in CAF research, propose future research directions, and emphasize the need for improved CAF-targeted therapeutic strategies. By addressing these gaps, it seeks to highlight the potential of CAFs as targets for overcoming therapeutic resistance and enhancing the efficacy of cancer treatments.
Introduction
Cancer-associated fibroblasts (CAFs) are a critical component of the tumor microenvironment (TME) and play pivotal roles in cancer progression, metastasis, and therapeutic resistance (1–3). As the most abundant stromal cells within tumors, CAFs are involved in various processes that facilitate tumor growth, including remodeling of the extracellular matrix (ECM), promoting angiogenesis, and influencing immune responses. Additionally, CAFs contribute to the metabolic reprogramming of the tumor, establishing a pro-tumorigenic microenvironment (4). Recent research has highlighted the complex and often dualistic nature of CAFs, as they can exhibit both tumor-promoting and tumor-suppressing activities depending on their activation state, subpopulations, and interactions within the TME (2).
Despite their central role in cancer biology, significant challenges remain in understanding CAFs due to their heterogeneity, dynamic plasticity, and the lack of specific markers for accurate identification (5). The classification of CAF subpopulations is further complicated by the absence of standardized naming conventions, making it difficult to compare findings across studies (2). Moreover, CAFs exhibit a high degree of functional diversity that varies between different tumor types and stages, complicating efforts to develop universal therapeutic strategies targeting CAFs (6). Their ability to modulate immune responses, promote resistance to conventional therapies, and contribute to immune evasion has made them a significant target in cancer treatment (7). For instance, CAFs can inhibit the infiltration of immune cells into the tumor, enhance the immunosuppressive microenvironment, and reduce the efficacy of both chemotherapy and immunotherapy.
The potential for CAF-targeted therapies is considerable, but several obstacles remain, including the difficulty in isolating CAFs and maintaining their characteristics in vivo and in vitro (4). Furthermore, understanding the molecular mechanisms underlying CAF interactions with tumor cells, immune cells, and the ECM is essential for the development of effective CAF-targeted treatments. This review aims to provide a comprehensive analysis of the current challenges in CAF research, exploring their roles in tumor progression, therapeutic resistance, and immune modulation, while also discussing promising future directions for targeted therapies that aim to overcome the barriers CAFs present in cancer treatment.
Fibroblasts and CAFs
Despite ongoing discussions about the precise definition of fibroblasts, they were originally identified about 150 years ago as spindle-shaped cells that produce collagen in connective tissues (8) (Figure 1). Current evidence suggests that fibroblasts in healthy tissues are dormant mesenchymal cells located within the extracellular matrix’s interstitial fibers. These cells can become activated in response to specific conditions such as wound healing, tissue inflammation, and organ fibrosis. In the context of cancer, analogous processes include cancer development (referred to as the ‘cancer wound’), inflammation that promotes tumor growth, and tumor-associated fibrosis (9–11). Activated fibroblasts in cancer settings are variously known as CAFs, tumor-associated fibroblasts, peritumoral fibroblasts, myofibroblasts, or reactive stromal fibroblasts (12, 13). These CAFs adapt alongside cancer cells, adopting a pro-tumor phenotype that allows them to thrive and expand within the complex tumor microenvironment, thereby facilitating tumor progression (14).
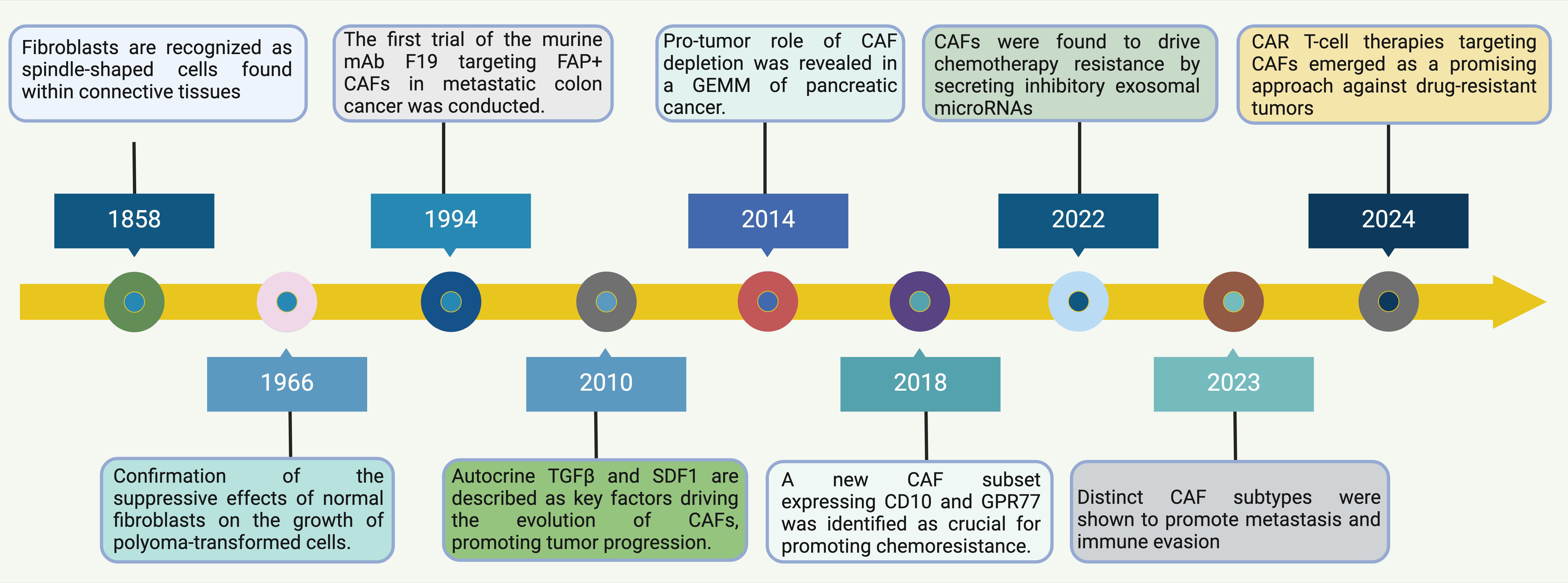
Figure 1. Timeline of Discoveries in Normal Fibroblasts and CAFs. This timeline illustrates significant milestones and the evolving focus of research on fibroblasts, especially in relation to their role in cancer. Starting from the historical identification of fibroblasts as spindle-shaped cells in connective tissues in 1858, the understanding of CAFs has evolved significantly (15). By 1966, research confirmed the suppressive effects of normal fibroblasts on the growth of polyoma-transformed cells, highlighting a functional aspect of fibroblasts in modulating cellular transformation (16). Progressing through the timeline, by 1994, the first trial targeting FAP+ CAFs using murine mAb F19 in metastatic colon cancer was conducted, illustrating a move towards targeted CAF therapies (17). In 2010, autocrine factors like TGFβ and SDF1 were identified as key drivers in the evolution of CAFs, promoting tumor progression, showcasing the increasing understanding of the molecular mechanisms by which CAFs influence cancer dynamics (18). Further advances in 2014 highlighted the role of CAFs in the depletion processes in a genetically engineered mouse model of pancreatic cancer, signifying the impact of CAFs beyond the tumor cells themselves (19). By 2018, the identification of a new CAF subset expressing CD10 and GPR77 was crucial for understanding chemoresistance, pointing towards the heterogeneity within CAF populations and their varied roles in cancer progression (20). Recent discoveries in 2022 and 2023 have established that CAFs drive chemotherapy resistance by secreting inhibitory exosomal microRNAs and promote metastasis and immune evasion through distinct CAF subtypes, respectively (21). These findings underscore the multifaceted roles of CAFs in facilitating tumor aggression and resisting therapeutic interventions. Finally, CAR T-cell therapies targeting CAFs have emerged as a promising approach against drug-resistant tumors, marking a pivotal shift towards leveraging immune system modifications to combat the complex interactions within the TME driven by CAFs (22). This shift not only represents a novel therapeutic strategy but also highlights the ongoing evolution of understanding in the field of tumor immunology and the critical role CAFs play in shaping cancer outcomes. Abbreviations: CAF, Cancer-Associated Fibroblast; FAP, Fibroblast Activation Protein; GEMM, Genetically Engineered Mouse Model; CAR, Chimeric Antigen Receptor; TGF, Transforming Growth Factor; SDF1, Stromal Cell-Derived Factor 1; GPR77, G Protein-Coupled Receptor 77.
Fibroblasts are spindle-shaped, non-epithelial, non-immune cells embedded within the extracellular matrix (ECM) that are easily propagated in adherent cell culture (23, 24). They play a key role in the stroma of gastrointestinal organs, where they are well-organized, much like in other tissues. Throughout the gastrointestinal tract, a reticular network of stromal cells is closely associated with the epithelial basement membrane (25). The subepithelial plexus, consisting of reticular stromal cells, completely surrounds the glandular axis from the stomach to the rectum (26). This compartment is dynamic, with a radial axis of proliferation and differentiation similar to that of the epithelium, originating from gremlin 1-expressing intestinal reticular stem cells (27). These stem cells give rise to intestinal reticular cells, likely overlapping with FOXL1+ subepithelial telocytes and GLI1+ mesenchymal cells, which together form an essential mesenchymal niche supporting intestinal stem cells (26–28). Beneath this compartment is a loosely organized network of fibroblasts within the lamina propria that interact with one another as well as with deeper stromal elements, including smooth muscle, blood vessels, nerves, and inflammatory cells (25, 29, 30). Functionally, fibroblasts are crucial in regulating ECM synthesis and facilitating paracrine and juxtacrine signaling to adjacent epithelial cells, thereby influencing their growth and differentiation (23, 31). They are also poised to respond to tissue damage, whether due to injury or tumorigenesis.
Cancer-associated fibroblasts (CAFs) are broadly recognized as the fibroblasts located within and surrounding tumors (14). This group comprises native, normal fibroblasts, as well as activated, proliferating (Ki67+) or recruited fibroblasts in response to cancer-derived stimuli. These newly formed CAFs may arise through several mechanisms, which will be discussed later. Despite the advancements in immune cell immunophenotyping and subtyping, a definitive marker for CAFs remains elusive (14, 32, 33). This gap has led to the identification of overlapping, incomplete, or distinct CAF populations in different studies, with markers that label both CAFs and other cell types. These challenges have complicated the interpretation of many studies, which will be addressed further.
The origin of CAFs
Due to the absence of specific biomarkers, identifying the origin of CAFs remains challenging. Current evidence primarily supports that fibroblasts originate from primitive mesenchymal cells, while CAFs arise from activated fibroblasts within local tissues (34, 35). Studies have shown that normal fibroblasts can proliferate, become activated, and express CAF markers by internalizing exosomes released from bladder cancer cells. Further research indicates that bladder cancer cells transform normal fibroblasts into CAFs through exosome-mediated transmission of transforming growth factor-beta (TGF-β) and SMAD signaling pathways (35, 36). Figure 2 illustrates the intricate and multifactorial processes involved in the transformation from normal fibroblasts (NFs) to CAFs. The recent research highlights the roles of key molecules and pathways, including transforming growth factor-beta 1 (TGF-β1), osteopontin, and interleukin-1β (IL-1β) (37–39). These elements initiate the transformation by engaging their respective receptors on NFs, subsequently activating downstream signaling cascades such as TGF-β/Smads and NF-κB, pivotal for modulating gene expression linked to the CAF phenotype (40). Exosomes from cancer cells, carrying miRNAs and lncRNAs, significantly contribute to the transformation of NFs into CAFs, mediated by pathways including TGF-β/Smads, JAK/STAT, and MAPK (41–43). These exosomes facilitate a feedback loop that enhances the conversion process. Additionally, the diagram delineates how alterations in glucose metabolism, driven by the hypoxia-inducible factor-1α (HIF-1α) pathway, are crucial for metabolic reprogramming essential for the survival and function of CAFs (44). HIF-1α are stabilized through both hypoxia-dependent mechanisms, where low oxygen levels inhibit their degradation, and hypoxia-independent pathways, such as activation by growth factors that affect the proteasome pathway (45). Key target genes involved in the transition from NFs to CAFs include VEGF for angiogenesis, GLUT1 for glucose metabolism, LOX for extracellular matrix remodeling, and CA9 for pH regulation (42, 46, 47). These genes contribute to the CAF phenotype, enhancing their ability to support tumor progression. Moreover, the transition to a CAF phenotype is driven by changes in cellular homeostasis, regulated by the activation of cytoskeletal proteins and the secreted phenotype, primarily through JAK/STAT and p53 signaling pathways. This comprehensive portrayal underscores the dynamic network of interactions that define CAF biology (Figure 2).
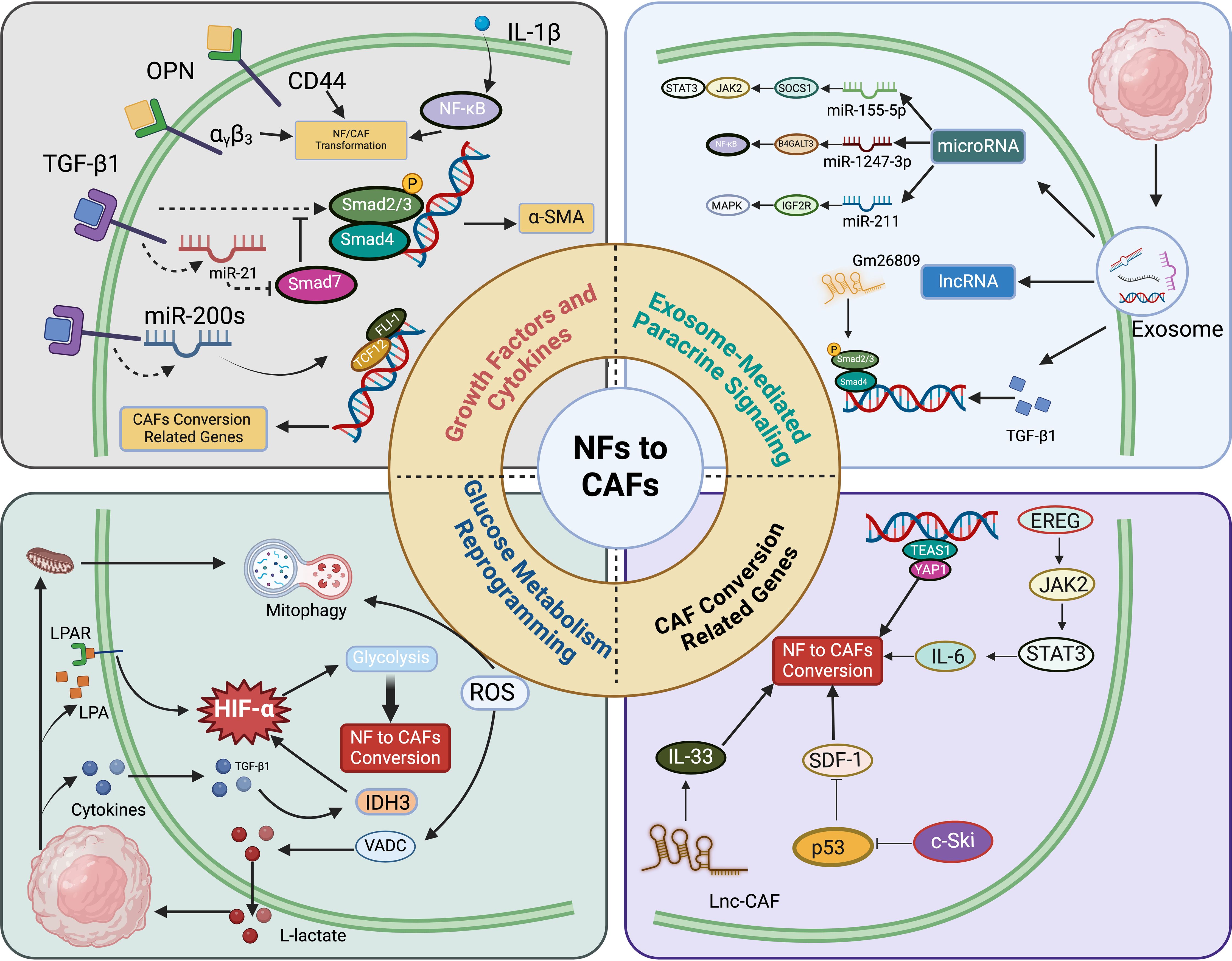
Figure 2. Conversion from normal fibroblasts (NFs) to cancer-associated fibroblasts (CAFs) involves multiple molecular mechanisms. a) Growth factors and cytokines like transforming growth factor-beta 1 (TGF-β1), osteopontin (OPN), and IL-1β interact with their respective receptors in NFs, subsequently activating downstream effectors such as miRNAs and CD44 (61–63). These molecules modulate the expression of genes associated with the CAF phenotype through the TGF-β/Smads and NF-κB signaling pathways (64, 65). b) Exosomes derived from cancer cells, carrying entities such as miRNAs and lncRNAs, induce the transformation of NFs into CAFs (65). This conversion is facilitated by signaling pathways including TGF-β/Smads, JAK/STAT, NF-κB, and MAPK cascades. c) The shift from NF to CAF is also driven by alterations in glucose metabolism, with the HIF-1α signaling pathway playing a crucial role in this metabolic reprogramming (66). d) Variations in cellular homeostasis prompt a self-driven transition to CAFs by regulating the activation of cytoskeletal proteins and the secreted phenotype, primarily through the JAK/STAT and p53 signaling pathways (67).
Additionally, a novel microfluidic model has been developed to regulate the three-dimensional tumor microenvironment (TME) in vitro, revealing that exosomes derived from melanoma can drive the differentiation of endothelial cells into CAFs through endothelial-mesenchymal transition (EndMT). Moreover, exosomes derived from mesenchymal stem cells (MSCs) have been shown to inhibit EndMT and induce CAFs to undergo reverse differentiation back into endothelial cells (48). This suggests that exosomes with the ability to reverse CAF differentiation could serve as effective carriers for anti-tumor drugs. Furthermore, epithelial cells in the TME can differentiate into CAFs via epithelial-mesenchymal transition (EMT) (49). Subsequently, CAFs secrete cytokines that promote EMT in tumor cells, ultimately facilitating tumor invasion and metastasis (50). Other studies have demonstrated that TGFβ1 can induce the differentiation of bone marrow-derived MSCs into CAFs by activating the JAK/STAT3 signaling pathway, promoting the migration and invasion of colorectal cancer cells (51). Various studies also support that PDGFα-CAF cells originate from MSCs (52). Additionally, several other sources of CAFs have been identified, including hematopoietic stem cells (HSCs), cancer stem cells (CSCs), adipocytes, pericytes, and stellate cells (47, 53–60). However, there is limited evidence supporting these origins, and their relevance to different tumor types remains uncertain.
In summary, CAFs originate from fibroblasts, epithelial cells, endothelial cells, bone marrow-derived MSCs, HSCs, CSCs, adipocytes, pericytes, and stellate cells (47, 53–60). This diversity in origin contributes to the heterogeneity of CAFs, with different origins being regulated by distinct signals or factors, resulting in varied differentiation pathways. Further research into CAF origins may provide insights into the discovery of biomarkers, therapeutic targets, signaling pathways, and activation mechanisms, all of which hold significant clinical potential. However, regardless of their cellular origin, the state, phenotype, and function of CAFs dynamically evolve throughout tumor progression, varying across different pathological stages (Figure 3).
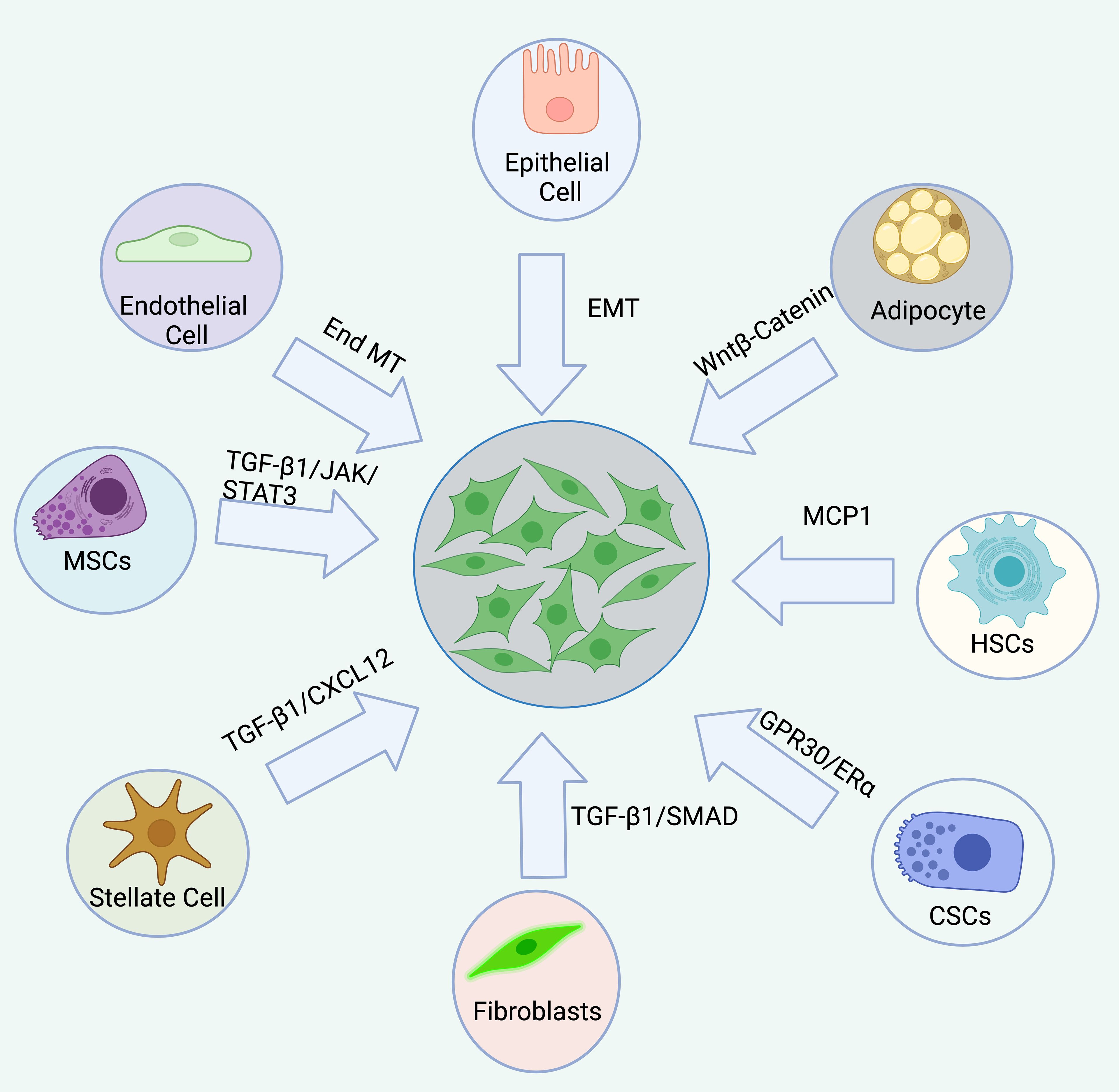
Figure 3. The origin of CAFs are complex, diverse, and heterogeneous. CAFs originate from a variety of sources, including resident fibroblasts, epithelial cells, endothelial cells, bone marrow-derived mesenchymal stem cells (MSCs), hematopoietic stem cells (HSCs), cancer stem cells (CSCs), adipocytes, pericytes, and stellate cells. Each origin is associated with distinct signaling pathways.
CAFs originate from a variety of sources, including resident fibroblasts, epithelial cells, endothelial cells, bone marrow-derived mesenchymal stem cells (MSCs), hematopoietic stem cells (HSCs), cancer stem cells (CSCs), adipocytes, pericytes, and stellate cells. Each origin is associated with distinct signaling pathways.Common CAF markers and gene signatures
In general, normal fibroblasts exhibit significant heterogeneity, which is reflected in differences in their morphology, behavior, and gene expression (as reviewed in (68)). In addition to the intrinsic heterogeneity within fibroblasts, other stromal cells share similarities with fibroblasts, particularly in the expression of so-called ‘specific’ markers. As a result, the complexity of the stromal compartment, along with the existence of multiple fibroblast subpopulations, makes it challenging to accurately isolate and define CAFs based on specific marker expression. Despite these challenges, numerous CAF markers are well-documented in the literature. Among the downregulated markers are caveolin-1 (69) and laminin (70), while frequently reported upregulated CAF markers include alpha-smooth muscle actin (aSMA) (71), vimentin (72), and fibroblast-specific protein 1 (FSP1) (32). However, Sugimoto et al., using a cancer mouse model, compared six markers from various mesenchymal cell groups residing in the stroma. They found that aSMA, platelet-derived growth factor receptor beta (PDGFRβ), and chondroitin sulfate proteoglycan 4 were expressed by fibroblasts, myofibroblasts, and vascular-associated cells, while vimentin and type I collagen also lacked specificity for fibroblasts (32). These findings raise questions about the specificity of these commonly accepted CAF markers. Moreover, aSMA was found to be downregulated, rather than upregulated, in the stromal compartment of prostate cancer (73). This highlights the heterogeneity within the stromal compartment and suggests that no definitive marker exists to clearly distinguish between mesenchymal subpopulations. Despite the fact that aSMA is often elevated in a cancer-type-specific manner and is considered a general marker for mesenchymal cells, it is incorrectly regarded as a universal marker for all CAFs (12, 74).
Several studies have identified a ‘CAF gene expression profile’ (75–77) and even ‘CAF signatures’ that can predict tumor outcomes (78, 79). A closer analysis of these studies reveals that, at the individual gene level, the differences between these profiles outweigh the similarities. However, when viewed from a broader perspective, examining gene families rather than individual genes, different cancer types and stages reveal similar groups of genes involved in processes such as cell adhesion, immune response, and extracellular matrix (ECM) modulation (75, 80, 81). This pattern aligns with expectations, as various cell types exposed to similar conditions tend to converge in performing the same functional tasks, albeit through different sets of genes.
Function of CAFs
The functions of CAFs has employed a range of methodologies from basic cell culture experiments and animal models to correlational studies within extensive patient groups. These methodologies have disclosed a broad spectrum of CAF functions. The relatively straightforward process of culturing CAFs and corresponding normal fibroblasts from patient samples has significantly advanced the understanding of CAF mechanisms. Notably, CAFs are extremely efficient at depositing and remodeling the extracellular matrix (ECM) in the tumor microenvironment. This capability is regulated by RHO and RAB GTPase-mediated mechanisms controlling integrin-linked adhesions and the actomyosin cytoskeleton, which is associated with the suppression of the CD36 transmembrane receptor (82–85). Furthermore, CAFs synthesize enzymes that form matrix crosslinks and engage in force-driven ECM remodeling, contributing to the increased rigidity of tumor tissues. Although these chemical crosslinks are stable, the secretion of matrix proteases by CAFs facilitates dynamic remodeling of the tumor matrix, creating pathways that enable cancer cell migration. Eph–ephrin signaling mediated by direct cell contact also affects cancer cell motility (86). Beyond fostering local invasion, CAFs enhance metastasis in experimental settings, a process linked to their ECM remodeling activities (87–90). At secondary metastatic sites, newly activated fibroblasts support the development of extensive metastases through various mechanisms, including the production of matrix molecules like tenascin and periostin, which fortify cancer cell signaling via WNT pathways (91–93). Recent findings suggest that alterations in ECM structure can also impact the migration of immune cells, with significant implications for tumor immune surveillance (90, 94, 95).
Alterations in matrix production and tumor mechanics driven largely by CAF activities have multifaceted implications for cancer. Enhanced tissue stiffness activates signaling pathways in cancer cells that promote survival and proliferation (96). Additionally, increased mechanical stress can compress blood vessels, inducing hypoxia, which fosters more aggressive cancer phenotypes and hampers drug delivery (97–99). Changes in tissue mechanics might also play a role in the onset of cancer and pre-malignant conditions, as evidenced by the relationship between mammographic density and breast cancer risk (85). Strategies aimed at disrupting the interactions between CAFs and the mechanical properties of tumors for therapeutic benefit are currently under investigation.
CAFs are also significant producers of growth factors, cytokines, and exosomes that encourage tumor growth and influence responses to therapy (42, 100, 101). They secrete TGF-β, leukemia inhibitory factor (LIF), growth arrest-specific protein 6 (GAS6), fibroblast growth factor 5 (FGF5), growth differentiation factor 15 (GDF15), and hepatocyte growth factor (HGF), which drive the invasive and proliferative behaviors in cancer cells (102–107). HGF, in particular, is noted for promoting resistance to BRAF-targeted therapies by activating an alternative BRAF-independent pathway for ERK–MAPK signaling (108, 109).
The secretome of CAFs additionally affects other components of the tumor microenvironment (110–114). VEGF from stromal cells can initiate angiogenesis, while a variety of cytokines and chemokines from CAFs act on different leukocytes, including CD8+ T cells, regulatory T cells, and macrophages, yielding both suppressive and promotional immune responses (114). However, the overarching impact of CAFs is immunosuppressive, mediated by factors like IL-6, CXCL9, and TGFβ, which notably reduce T cell activity (115). Recent observations of antigen cross-presentation by CAFs have highlighted their role in modulating CD4+ and CD8+ T cell responses (116, 117). Clinical studies further corroborate a negative correlation between CAF presence and CD8+ T cell levels (118). Moreover, IL-6 may also foster systemic immuno-suppression through metabolic effects (119). Disrupting CXCL12 from CAFs enhances T cell-mediated tumor control, and targeting focal adhesion kinase (FAK) in cancer cells simultaneously reduces stromal fibroblast activation and the formation of an immunosuppressive milieu (120). Nonetheless, the role of tumor necrosis factor (TNF) produced by CAFs is complex; while TNF can activate fibroblasts, its tumor-promoting, immunosuppressive effects are tied to the suppression of TNF signaling (121, 122).
Lastly, the metabolic exchange between cancer cells and CAFs provides another layer of interaction, where autophagy in stromal fibroblasts produces alanine used by pancreatic ductal adenocarcinoma cells to energize the tricarboxylic acid cycle (123–125). Additionally, metabolic irregularities in CAFs may link to altered immune regulation, potentially through IL-6 production or the depletion of immunomodulatory amino acids (126).
CAFs in cancer
CAFs play a multifaceted role in cancer progression and metastasis by fostering a conducive tumor microenvironment through several mechanisms (103, 127, 128). Firstly, CAFs contribute to cancer cell stemness and metastatic capabilities by engaging in paracrine signaling with cancer stem cells, enhancing their self-renewal and propagation abilities which are crucial for tumor aggressiveness and metastasis (42, 129). Secondly, they promote tumor angiogenesis by secreting pro-angiogenic factors like VEGFA and PDGF and altering the extracellular matrix to enhance vascular formation, facilitating tumor growth and the dissemination of cancer cells (130–132). Thirdly, CAFs are instrumental in mediating immunosuppression within the tumor microenvironment by secreting factors such as TGF-β and IL-6, which modulate immune cell function and contribute to immune evasion by the tumor (133–135). Lastly, they are involved in metabolic reprogramming known as the “Reverse Warburg Effect,” where they supply cancer cells with metabolic intermediates necessary for energy production, thus supporting the energetic demands of rapidly proliferating tumor cells (136, 137). Through these interactions, CAFs are key players in enabling cancer progression and the establishment of metastatic sites, making them significant targets for therapeutic intervention.
CAFs contribute to cancer stemness
CAFs are integral to the tumor microenvironment (TME) and play a pivotal role in the maintenance and enhancement of cancer cell stemness, characterized by self-renewal and the ability to propagate, which are key traits of cancer stem cells (CSCs) (138–140). These cells are known to drive tumor aggression, contribute to resistance against therapies, and facilitate metastasis. CSCs are identified through various markers such as CD44, CD24, CD133, LGR5, SOX2, AQP5, ESA, PAF1, and CXCR4, though these markers lack high specificity (141). CAFs interact with CSCs predominantly through paracrine signaling, supporting a conducive niche for tumor growth (142). Research has highlighted that certain CAF subsets secrete molecules that directly enhance CSC properties. For example, a specific subset of CAFs expressing CD10 and GPR77, activated by NF-kB, has been shown to enrich CSCs in breast and lung cancers by releasing IL-6 and IL-8 (20). Similarly, in bladder cancer, CAFs stimulated by interferon from cancer cells can enhance stemness through the WNT5a/β-catenin signaling pathway (143). In the context of hepatocellular carcinoma, CAFs boost cancer cell stemness via the ERK1/2-FRA1-HEY1 pathway by secreting hepatocyte growth factor (144). Additionally, CAFs are known to produce exosomes that perpetuate stemness across various cancer types (145, 146). They also indirectly facilitate the recruitment and stemness of myeloid-derived suppressor cells through FAP-dependent mechanisms (146). Given the significant role of these paracrine interactions, targeting such pathways might offer new therapeutic avenues, particularly through manipulation of WNT signaling which is crucial in mediating interactions between CAFs and CSCs, affecting both the active and dormant CSCs (147).
CAFs promote angiogenesis
Angiogenesis, the formation of new blood vessels, is essential for tumors to secure a greater supply of oxygen and nutrients. This process is predominantly driven by hypoxia within the tumor environment. Under these low-oxygen conditions, cancer cells release vascular endothelial growth factor A (VEGFA), which targets VEGF receptor 2 (VEGFR2) on adjacent endothelial cells (ECs) or on circulating endothelial progenitor cells derived from the bone marrow, thus initiating angiogenesis (148). This angiogenic cascade includes degradation of the basal lamina and extracellular matrix, EC proliferation, the development of vascular sprouts, and eventual vessel stabilization. Additionally, molecules like delta-like ligand 4 (DLL4) and angiopoietin 2 (ANGPT2) are vital for angiogenesis (149).
CAFs secrete a range of pro-angiogenic growth factors such as VEGFA, CXC-chemokine ligand 12 (CXCL12), fibroblast growth factor 2 (FGF2), and platelet-derived growth factor (PDGF) (150). CXCL12, also recognized as stromal cell-derived factor 1 (SDF-1), promotes tumor proliferation and angiogenesis via the CXCL12/CXCR4 pathway (151). This interaction triggers diverse signaling pathways, including the G-protein coupled/PI3K/AKT/NF-κB axis and the Ras-MEK1/2-Erk1/2 axis, leading to angiogenic responses (152). FGF2, part of the heparin-binding growth factor family, engages FGF receptors to stimulate multiple angiogenic activities and interacts with the VEGF pathway (153). Moreover, the PDGF/PDGFR signaling pathway is crucial in the development of connective tissue and wound healing, with studies indicating that PDGF-C upregulation in CAFs can promote angiogenesis even in the absence of VEGF activity (154, 155).
Beyond direct activation through paracrine signaling, CAFs also indirectly enhance angiogenesis via biomechanical properties of the tumor microenvironment, such as matrix stiffness (156). CAFs produce enzymes like lysyl oxidase (LOX) and lysyl hydroxylase 2 (LH2), which increase collagen and elastin cross-linking, thus raising matrix stiffness (157, 158). Research has shown that higher matrix stiffness correlates with enhanced VEGF binding by endothelial cells, which is influenced by β1 integrins (159). This relationship extends to complex pathways involving Ca2+ influx and HIF-1α ubiquitination in hepatocellular carcinoma angiogenesis, highlighting the multifaceted role of the ECM in angiogenic regulation (160). However, conflicting findings in studies like those by Bao et al., which suggest that increased stiffness may suppress VEGF secretion in certain cancer types, underscore the complexity of these interactions and the need for more nuanced research (161).
CAFs dedicate in metabolic changes in cancer
Despite residing in a nutrient-limited tumor microenvironment (TME), cancer cells demonstrate a remarkable capability for continuous proliferation, aided by metabolic adaptations within the TME. Notably, Warburg et al., about a century ago, documented that cancer cells preferentially convert glucose to lactate to generate ATP—even in the presence of sufficient oxygen—a process now known as the “Warburg Effect” (162). Warburg attributed this metabolic peculiarity to mitochondrial dysfunction in cancer cells. However, subsequent studies in cancer metabolism have revealed that not all tumor cells are wholly reliant on this pathway; some retain mitochondrial functionality and can engage in oxidative phosphorylation (OXPHOS), illustrating the Warburg Effect’s variability across different tumor environments (163).
This reverse Warburg Effect is primarily propelled by oxidative stress induced by cancer cells. The release of reactive oxygen species (ROS) by cancer cells heightens oxidative stress in stromal components, causing autophagosomes to merge with lysosomes, which leads to mitochondrial degradation in CAFs. This process also results in the breakdown of caveolin-1 (Cav-1) via the HIF-1α/NF-κB pathway (136). The subsequent reduction of Cav-1 in CAFs further increases ROS in cancer cells, fostering a feedback loop that amplifies oxidative stress and disrupts the NF-kB pathway (164). Additionally, TGF-β, a key regulator in cancer metabolism, influences ROS levels by modulating the expression of α-SMA and NOX4 in fibroblasts, thereby promoting oxidative stress (165–167).
Through the reverse Warburg Effect, CAFs under oxidative stress due to cancer-derived ROS undergo aerobic glycolysis, producing lactate and pyruvate. These metabolites are then utilized by neighboring oxidative cancer cells for further metabolic activities. While the transmission of ROS in this process has been documented, the detailed mechanisms by which cancer cells and CAFs initiate and adapt to these metabolic changes remain less explored. Nevertheless, targeting the Reverse Warburg Effect presents a theoretical possibility to disrupt cancer metabolism, potentially offering new avenues for therapeutic intervention aimed at curbing tumor growth by altering the metabolic interplay between cancer cells and their stromal environment.
CAFs mediate immunosuppression
Chronic inflammation, immune cell infiltration, and the ability of cancer cells to evade immune surveillance are recognized as key hallmarks of cancer progression (168). Research has demonstrated the dual role of the immune system in both suppressing and promoting tumor development, a phenomenon termed “cancer immunoediting.” This process involves three distinct stages: elimination, equilibrium, and escape (169).
During the elimination phase, innate and adaptive immune responses work in tandem to identify and destroy dysplastic cells before they progress to clinically detectable tumors. However, certain cancer cells may acquire immune-evasive or poorly immunogenic traits, allowing them to survive immune attacks and transition into the equilibrium phase. In this stage, the growth of neoplastic cells is restricted, and their immunogenicity is shaped under the selective pressure exerted by adaptive immunity, predominantly involving T cells and associated cytokines. Persistent immune selection pressure can lead these cancer cells to develop immunosuppressive or immune-evasive phenotypes, ultimately facilitating their progression to the immune escape phase. At this stage, cancer cells evade immune control entirely, resulting in unchecked growth, the formation of clinically evident tumors, and potentially metastasis (170). Despite significant progress, the complex mechanisms underlying cancer immunoediting remain incompletely understood, posing a barrier to developing effective immunotherapy strategies.
CAFs, as major components of the TME, play a pivotal role in promoting immune evasion. One of their key immunosuppressive mediators is TGF-β, which modulates the immune microenvironment by affecting T cell differentiation and proliferation through the inhibition of transcription factor activation triggered by Ca2+ influx (171). In ovarian tumors with T cell exclusion, elevated TGF-β expression and stromal activation are critical drivers of T cell exclusion. TGF-β reduces MHC-I expression on ovarian cancer cells in vitro and activates fibroblasts to promote extracellular matrix production, forming a physical barrier that impedes T cell infiltration (172). Additionally, TGF-β suppresses dendritic cell function, inhibits the development of cytotoxic natural killer (NK) cells and their secretion of IFN-γ, and polarizes macrophages towards an M2 phenotype with immunosuppressive, anti-inflammatory, and pro-angiogenic properties (173, 174). Beyond TGF-β, CAF-derived CXCL12 is a potent chemokine that modulates immune suppression. It restricts the migration of CD8+ T cells, sequestering them away from the tumor stroma, and inhibits NK cell proliferation, keeping them in a quiescent state (175, 176). Another significant CAF-secreted molecule in the immune microenvironment is IL-6, which is highly expressed in the inflammatory CAF (iCAF) subtype (176). IL-6 contributes to the accumulation of tumor-infiltrating lymphocytes and regulates neutrophil survival, activation, and function via the IL-6/STAT3/PD-L1 signaling axis (177, 178). Furthermore, CAFs secrete inhibitory immune checkpoints (iICPs) such as PD-1 and LAG3, further enhancing immune suppression within the TME (179).
These findings underscore the critical role of CAFs in facilitating tumor immune escape by creating an immunosuppressive microenvironment, thereby supporting cancer progression and resistance to immune-mediated therapies.
Non-follicular adaptive immune stimulating CAFs and Antigen-presenting CAF
In the context of cancer, non-follicular adaptive immune-stimulating CAFs and antigen-presenting CAFs (apCAFs) play distinct roles in modulating the immune response (180, 181). Non-follicular adaptive immune-stimulating CAFs, which predominantly reside outside organized lymphoid structures like cancer-associated tertiary lymphoid organs (CaTLOs), demonstrate a capacity to interact with the adaptive immune system beyond traditional immuno-suppressive roles (180). These CAFs are primarily characterized by their ability to activate and recruit adaptive immune cells rather than just innate immune interactions, marking a significant shift from the traditional view of CAFs as solely tumor-promoting entities (182).
Antigen-presenting CAFs (apCAFs), on the other hand, have a specialized role in direct immune modulation by presenting antigens to T cells via MHC class II molecules (116). CAFs expressing MHC class II, initially identified by Tuveson and colleagues in pancreatic cancer as apCAFs, have been subsequently confirmed by other research groups to be present in pancreatic, breast, and lung cancers (116, 183–187). This subset of CAFs is effective in initiating T cell responses, including the activation of effector CD4+ T cells (183–186). By presenting cancer antigens, apCAFs can influence T cell phenotypes and contribute to the immunological architecture of the tumor microenvironment (114). This function is critical for orchestrating localized immune responses against tumors and for supporting ongoing immune surveillance and anti-tumor activity.
Together, these roles underscore a complex interplay where CAFs can both suppress and stimulate immune responses, highlighting their dual potential as targets for cancer therapy (114, 188, 189). This emerging understanding challenges the traditional paradigm of CAFs and opens new avenues for therapeutic strategies aimed at modulating the tumor microenvironment to enhance anti-cancer immunity.
CAFs facilitate cancer metastasis
Cancer metastasis is a multifaceted process encompassing various stages. It initiates with the migration and invasion of tumor cells into adjacent tissues, proceeds through intravasation into the bloodstream, follows by circulation and extravasation, and culminates in the colonization of new sites (190). CAFs significantly facilitate this process via paracrine signaling and direct physical interactions. The mobility of tumor cells, crucial for their migration and invasion, is often enhanced by the epithelial-mesenchymal transition (EMT), characterized by the loss of cell polarity and adhesion, which imparts a mesenchymal phenotype conducive to migration and invasion (191, 192). The necessity of EMT in all metastatic events remains debated.
CAFs are known to boost the migratory and invasive capabilities of cancer cells by secreting chemokines and exosomes. For instance, in gastric cancer, CAFs stimulated by TGF-β1/Smad2/3 signaling significantly upregulate hyaluronan and proteoglycan link protein 1 (HAPLN1), enhancing tumor migration and invasion (193, 194). In esophageal squamous cell carcinoma, CAF-like cells produce plasminogen activator inhibitor-1 (PAI-1), which augments migration and invasion through the PAI-1/low-density lipoprotein receptor-related protein 1 (LRP1) axis via Akt-Erk1/2 pathways. Furthermore, exosomes secreted by CAFs containing miR-18b and miR-382-5p have been documented to promote cancer cell migration and invasion through EMT induction (194). Additionally, CAFs facilitate EMT by modulating matrix stiffness and signaling pathways, such as the TWIST1/G3BP2 and EPHA2/LYN/TWIST1 pathways (195).
Apart from inducing EMT, CAFs also directly drive cancer cell migration through exerted physical forces. Labernadie and colleagues discovered a mechanism wherein CAFs apply physical force to cancer cells via heterophilic adhesion involving N-cadherin on CAFs and E-cadherin on cancer cells, mediated by β-catenin and α-catenin/vinculin interactions (196). Erdogan and team demonstrated that CAFs create and align a fibronectin-rich matrix, facilitating CAF-cancer cell association and directional migration through the nonmuscle myosin II/PDGFRα/α5β1-integrin/fibronectin pathway (197). Moreover, CAFs express membrane-anchored metalloproteinases (MT1-MMPs) that degrade collagen, easing tumor cell penetration and movement within the ECM (198).
Intravasation, a critical phase preceding circulation, involves tumor cells penetrating leaky, immature blood vessels formed during angiogenesis, often characterized by inadequate endothelial cell junctions and abnormal pericyte coverage (199). Various factors, including TGF-β, VEGF, and SOX2, have been identified as regulators of both intravasation and extravasation processes in metastasis (200, 201). CAFs enhance both hematogenous and lymphatic metastasis, with mechanisms involving signaling pathways such as periostin/integrin/FAK/Src/VE-cadherin, VEGFC/VEGFR3, and IL-6/IL-6R (202–204).
CAFs are dynamic participants in the metastatic process, not merely passive entities. For instance, circulating CAFs (cCAFs) found in the blood of patients with metastatic breast cancer correlate with clinical metastasis (205). These cCAFs, along with other cell types, form heterotypic clusters that influence survival and proliferation of circulating tumor cells (CTCs), potentially via soluble factors (206).
The target site microenvironment, often hostile to CTCs, is pre-emptively modified by the primary tumor, creating pre-metastatic niches (PMNs) (207). PMNs are shaped by cytokines and exosomes from the tumor and TME, with CAFs playing a dual role in their activation. CAF-derived factors, such as the non-coding RNA LncSNHG5 and extracellular vesicles, modify distant fibroblasts to enhance their tumor-supportive capabilities (208). Additionally, CAFs themselves undergo activation during PMN formation, further emphasizing their central role in cancer metastasis and providing potential therapeutic targets to hinder tumor growth and spread (209).
CAF-targeting therapy
Recent years have witnessed significant advancements in therapies targeting cancer-associated fibroblasts (CAFs), focusing on three main objectives (1): directly or indirectly depleting CAFs, (2) mitigating or abolishing their tumor-promoting and immunosuppressive activities, or (3) reprogramming or normalizing CAFs towards a more dormant state. These strategies are outlined below.
Chemotherapy targeting CAFs
Initially identified by Tuveson and colleagues, CAFs in various tumors express fibroblast activation protein (FAP), a unique membrane-bound serine postprolyl peptidase known for its additional endopeptidase activity (210). Val-boroPro (Talabostat), a competitive inhibitor of prolyl peptidase and an orally administered drug, demonstrated some control over tumor growth by degrading the extracellular matrix (ECM) in mouse models (211). However, it failed to show therapeutic efficacy in human clinical trials for metastatic colorectal cancers (212). Sibrotuzumab, a humanized anti-FAP monoclonal antibody, inhibited the dipeptidyl peptidase activity of FAP but did not show efficacy in suppressing pancreatic cancer growth in patients, despite the radiolabeled version of the antibody accumulating in tumors as visualized by SPECT (213, 214).
Utilizing FAP’s enzymatic activity, anti-CAF prodrugs or protoxins that couple cytotoxic agents with a dipeptide containing a FAP cleavage site have been developed (210, 215). These prodrugs, which remain inactive until cleaved by FAP upon systemic delivery, have induced tumor lysis and growth inhibition when injected intratumorally in human breast and prostate cancer xenografts (215). Similarly, immunotoxins like Anti-FAP-PE39 have suppressed tumor growth and enhanced recruitment of tumor-infiltrating lymphocytes (216). Other novel approaches include monoclonal antibodies conjugated with cytotoxic agents or bispecific antibodies that target both FAP on CAFs and death receptor 5 on tumor cells, showing potent antitumor effects (216).
Immunotherapy targeting CAFs
Numerous approaches have been developed to bolster immunity against FAP-expressing cells, notably CAFs, and to curb cancer proliferation (2, 21, 180, 189, 217). Immunization using dendritic cells transfected with FAP mRNA has effectively curtailed the growth of both implanted and intravenously introduced tumors (218). This effect was amplified when the vaccine targeted both FAP and a tumor-associated antigen simultaneously. These dendritic cell vaccines, when used in conjunction with an anti-fibrotic agent, have effectively activated both innate and adaptive immune responses, leading to enhanced NK cell function, boosted anti-tumor humoral responses, and potentiated cytotoxic CD8+ T cell activity across diverse tumor models (218). Additionally, adenoviral vaccines targeting FAP have selectively eliminated CAFs by activating a CD8+ T cell response, thereby reducing tumor growth and metastasis in various murine cancer models (114, 189, 219). A significant study employing a transgenic mouse model engineered to express the diphtheria toxin receptor under the FAP promoter demonstrated that depletion of FAP+ CAFs via diphtheria toxin enhanced the efficacy of anti-cancer vaccines (220). Oral administration of an anti-FAP DNA vaccine markedly reduced new blood vessel formation, tumor growth, and metastasis in orthotopically injected breast carcinoma models (221), and the co-administration of doxorubicin significantly improved the drug’s intratumoral absorption and extended the survival of the treated mice (221).
Adoptive transfer of chimeric antigen receptor (CAR)-T cells specifically designed to target FAP-expressing cells has shown promise in depleting FAP+ populations, including CAFs, thus limiting tumor stroma formation and enhancing the effectiveness of chemotherapeutic agents (222–224). However, this strategy has been linked with severe adverse effects such as profound bone marrow toxicity and cachexia, underscoring the need for more selective targeting in CAF-based therapies, an area that continues to be vigorously researched (225). Furthermore, near-infrared photoimmunotherapy (NIR-PIT) represents a novel technique for selectively depleting FAP-positive cells within the tumor microenvironment, showing efficacy in inhibiting tumor growth in a co-culture xenograft model of human esophageal squamous cell carcinoma without negative side effects (226, 227). Combining anti-FAP+ CAF therapy with 5-fluorouracil (5-FU) has proven to surpass the effectiveness of 5-FU alone in overcoming chemoresistance (228).
Functional modification/reprogramming targeting CAFs
Reverting activated CAFs to a quiescent state involves the use of agents like all-trans-retinoic acid (ATRA), minnelide (which disrupts the TGF-β signaling pathway), and calcipotriol (228–232). The angiotensin receptor II antagonist losartan has reduced TGF-β-mediated CAF activation, enhancing drug delivery and the efficacy of immunotherapy, and is being studied in clinical trials for pancreatic cancer treatment (98, 233–235). Efforts to block immunosuppressive ligands of key CAF signaling pathways, including IL-6 (185, 186), LIF (187), and TGF-β (124, 126) aim to suppress or eliminate cancer cells (236–240).
The CXCL12/CXCR4 axis, crucial in cancer progression and immunosuppression, involves CXCL12 from CAFs recruiting CXCR4-expressing cells that support angiogenesis and tumor growth (241–243). Inhibiting this pathway using the CXCR4 antagonist plerixafor has significantly reduced fibrosis and improved immune cell infiltration and checkpoint inhibitor efficacy (244). Other strategies that inhibit CAF functions include TGF-β blockade, NFkB inhibitors to overcome chemotherapy resistance, and Smoothened hedgehog pathway inhibitors (IPI-926) (102, 245–247) (Figure 4).
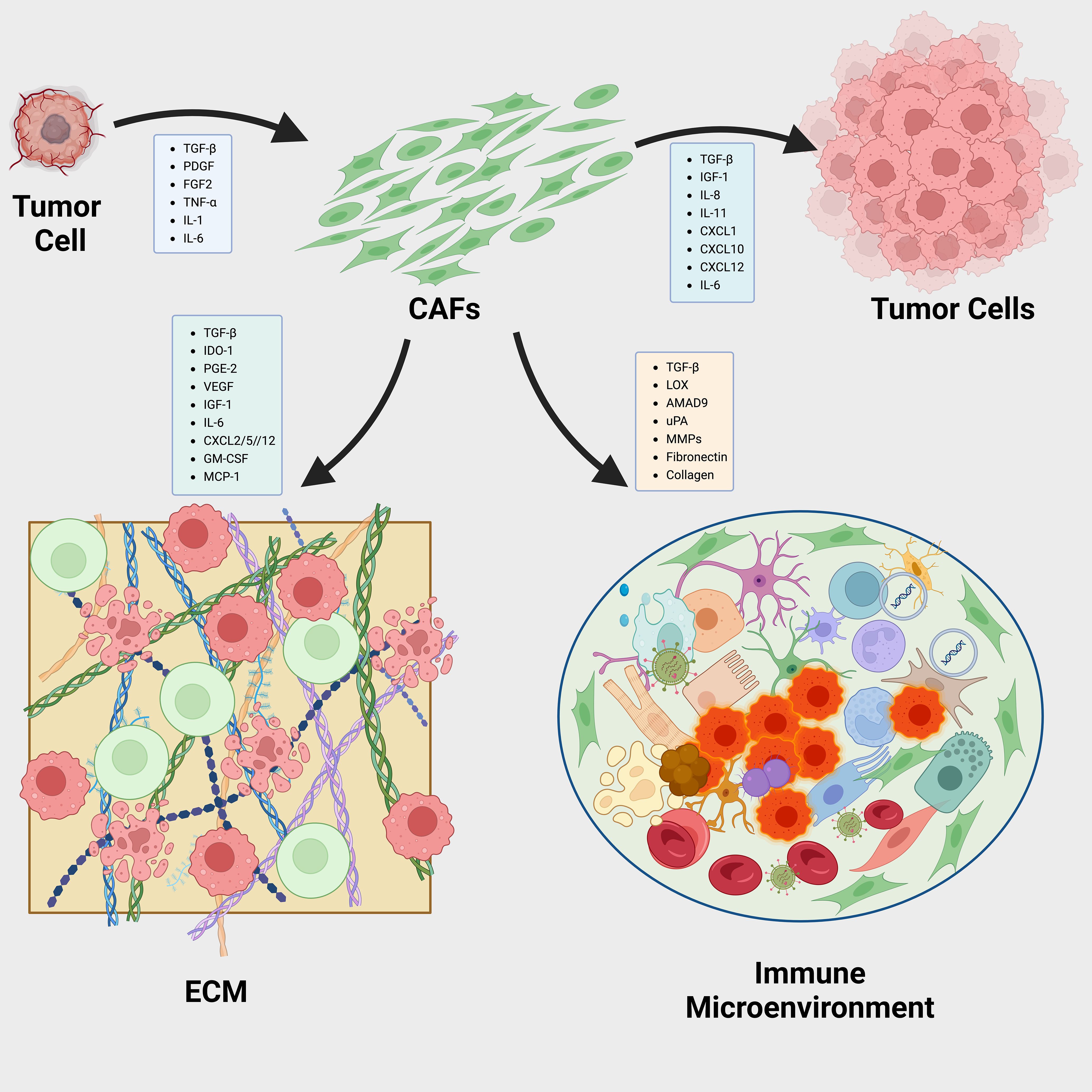
Figure 4. Dynamic Interactions Between CAFs and the Tumor Microenvironment in Cancer Progression. Reciprocal interactions between cancer-associated fibroblasts (CAFs) and the tumor microenvironment (TME) play pivotal roles in cancer progression via three primary mechanisms: a).Tumor-derived cytokines influence the behavior of CAFs, prompting them to release pro-tumorigenic factors that further drive the cancerous process. b).CAFs are instrumental in organizing and cross-linking the extracellular matrix (ECM) components, thus modifying the ECM’s structure and functionality to facilitate tumor growth and metastasis. c).Factors secreted by CAFs also modulate immune cells within the TME, including tumor-associated macrophages (TAMs), tumor-associated neutrophils (TANs), and dendritic cells (DCs). This modulation helps establish a tumor immune microenvironment (TIME) that promotes tumor development.
Targeting CAF control cancer cell dormancy
Initial research has established cancer cell dormancy as a critical factor contributing to drug resistance and recurrence, yet dormant cancer cells (DCC) continue to be challenging to detect clinically, highlighting a significant obstacle in overcoming drug resistance (248). The process of dormancy entry and escape involves complex interactions between tumor cells and the TME, with cytokines and chemokines secreted by CAFs playing a vital role (249). Consequently, a promising strategy to prevent cancer recurrence involves targeting these CAF-secreted factors. This approach focuses on drugs that specifically target these factors, potentially inhibiting dormancy-associated mechanisms.
Inhibiting microenvironment interactions to prevent cancer cell reawakening
Maintaining tumor cells in a dormant state is critical to preventing metastasis and recurrence. It is imperative to develop therapeutic strategies that inhibit the communication between CAF-driven signaling and the supportive TME involved in dormancy escape (250). An effective target could be uPAR, which maintains the dormant state of cancer cells and limits metastasis (251, 252). For instance, ATN-292 reduces migration in human pancreatic cancer cells by blocking the uPA to uPAR binding (253), and a novel anti-uPAR monoclonal antibody has shown antitumor effects in gastric cancer by disrupting this interaction (254). The small molecule uPA inhibitor, WX-671, combined with gemcitabine, although well tolerated, did not improve survival outcomes compared to gemcitabine alone in a phase II trial (255).
High levels of TGF-β1 in the TME trigger dormancy escape, with CAFs being a primary source of TGF-β. Inhibitors targeting TGF-β1 interactions or receptor kinase activities are strategies to keep tumor cells dormant. Agents like SRK-181, which binds to the pro-segment of TGF-β1 preventing its activation, and LY2157299, a small molecule TGF-βRI kinase inhibitor known as Galunisertib, have shown promise in clinical trials (256–259). Another TGF-βRI inhibitor, Ki26894, has demonstrated efficacy in reducing invasiveness and bone metastasis in gastric cancer (260). Moreover, anti-inflammatory therapies targeting pro-inflammatory cytokines secreted by CAFs have been studied. NSAIDs such as sulindac and celecoxib, which inhibit COX-2 activity, have shown efficacy in gastrointestinal cancers in both preclinical and clinical settings. Sulindac is under investigation in a phase III trial for its potential to reduce adenomas and secondary cancers (261, 262), and celecoxib is being studied to enhance response rates in advanced colorectal cancer treatment (263, 264).
CAF-mediated ECM remodeling significantly contributes to dormancy escape. Targeting ECM molecules like collagen and FN, which regulate integrin roles in dormancy to proliferation transitions, is a potential strategy. Anti-integrin therapies like Volociximab have shown positive results in clinical trials (265, 266).
Lastly, inhibiting enzymes like LOX or LOXL2, which are implicated in chemoresistance and metastasis growth, could prevent dormant cancer cell awakening. Agents like Simtuzumab and EGCG have been evaluated for their efficacy in reducing LOXL2 activity and TGF-β1 signaling, showing potential in clinical trials (267, 268).
In conclusion, reinforcing the dormant state and inhibiting the pathways facilitating dormancy escape through targeted therapies offers a promising avenue for managing cancer progression and recurrence.
Activate dormant cancer cells for improved treatment response
Understanding cancer dormancy has led to strategies aimed at preventing cells from becoming dormant or awakening dormant cancer cells (DCCs) to increase their sensitivity to treatment. One approach involves targeting dormancy-inducing factors influenced by CAFs. For instance, TGF-β2, which promotes dormancy via the TGF-βRIII pathway, is targeted by AP 12009 (Trabedersen), an antisense oligonucleotide (ASO) that has shown safety in phase I/II studies for pancreatic and colorectal cancer (269, 270).
Another CAF-secreted factor, DKK-1, helps maintain cancer cell dormancy (271). DKN-01, a humanized monoclonal antibody that inhibits DKK-1, is currently being evaluated in clinical trials for gastrointestinal (GI) cancers (272, 273). Notably, a phase II trial is investigating DKN-01 in combination with Tislelizumab and possibly chemotherapy for metastatic gastric cancer or gastroesophageal junction adenocarcinoma (274). Additionally, DKN-01 is being tested with pembrolizumab in advanced esophageal cancer in a phase Ib trial and in combination with bevacizumab and chemotherapy for advanced colorectal cancer in another ongoing phase II study (NCT05480306) (272, 275).
Other factors like GDF-10 and BMP4, also secreted by CAFs, have been implicated in promoting dormancy, although targeted therapies for these factors in GI cancers have yet to be explored (276–278). By inhibiting these CAF-derived factors, it is possible to either prevent entry into or trigger exit from dormancy. Implementing such strategies early in treatment may prevent tumor cells from developing robust malignancy. A summary of targeted factors by therapeutic agents in GI cancers emphasizes potential approaches to manage cancer dormancy at various stages (Figure 5).
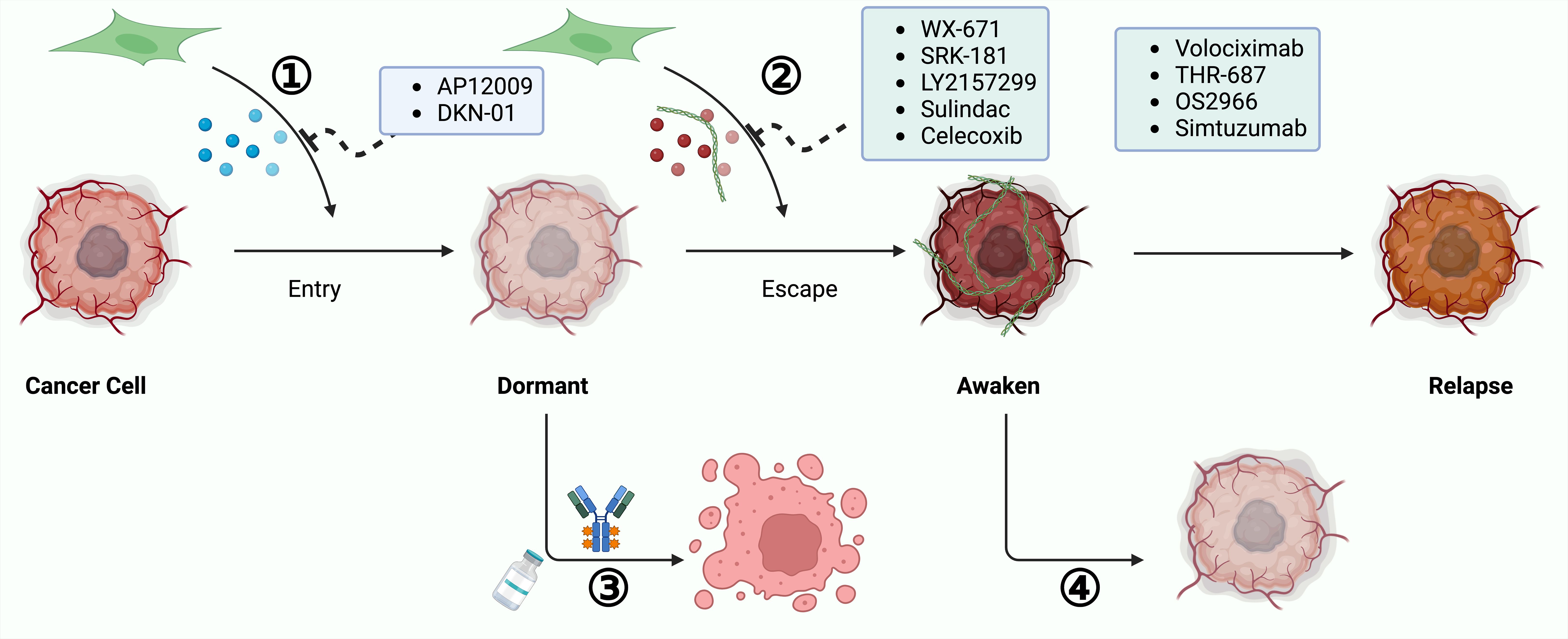
Figure 5. Approaches to Combat Cancer Recurrence by Targeting Dormant Cancer Cells. Targeting dormant cancer cells presents a viable strategy for preventing cancer recurrence. 1). Reactivating Dormant Cancer Cells for Therapeutic Sensitivity: This involves awakening dormant cancer cells to increase their susceptibility to treatments. Strategies include obstructing the secretion of dormancy-inducing factors by CAFs or reactivating cellular proliferation signals. 2).Sustaining Permanent Dormancy of Cancer Cells: This strategy aims to prevent cancer cell reactivation and subsequent growth by blocking pathways facilitated by CAFs that enable dormancy escape. By focusing on these dormant cancer cells, it is possible to avert cancer recurrence and enhance patient outcomes.
CAFs and therapeutic resistance
Resistance to cancer therapy often results in poor patient outcomes, underpinned by complex and dynamic mechanisms. Konieczkowski et al. introduced a convergence-based framework to understand cancer drug resistance, identifying major causes such as pathway reactivation, pathway bypass, and pathway indifference (279). Beyond genomic alterations in tumor cells, the role of CAFs in therapeutic resistance has been well-established, with their influence extending across multiple facets. CAFs affect the mechanical properties of the tumor microenvironment (TME), enhancing matrix stiffness which can impede the penetration of chemotherapeutic drugs. For example, gastric CAFs that express calponin 1 activate the ROCK1/MLC pathway, increasing matrix stiffness and contributing to resistance against 5-fluorouracil (5-FU) in cancer cells by activating the YAP signaling pathway (280). CAF-derived exosomes are also pivotal in mediating resistance within the TME (281). Annexin A6 in CAF-derived extracellular vesicles (EVs) activates the integrin β1-FAK-YAP signaling pathway, promoting the formation of a tubular network in the ECM that reinforces chemotherapeutic resistance (282). In breast cancer, CAF-derived circulating EVs containing the full mitochondrial genome enhance estrogen receptor (ER)-independent oxidative phosphorylation (OXPHOS), which induces therapy-resistant dormant cancer stem-like cells, leading to resistance to endocrine therapy (283). Targeting the YAP signaling pathway may be effective in overcoming the mechanical resistance encountered in targeted therapy. Regarding immunotherapy, CAFs activated by the IL-17/Act1/HIF1α pathway can lead to collagen deposition, enhancing PD-L1 resistance and reducing cytotoxic T cell infiltration (284). Another subtype of CAF, ecm-myCAF, has been shown to elevate PD-1 and CTLA4 protein levels in Tregs, boosting TGFβ-myCAF cellular content and mediating primary resistance to immunotherapy. Combining tumor-targeted therapy with CAF-targeted strategies, such as the FAP5-DM1 monoclonal antibody conjugated to maytansinoid, has demonstrated prolonged inhibition of tumor growth and complete regressions in xenograft models of multiple cancers (222). Additionally, CAFs contribute to radiotherapy resistance; upon irradiation, CAFs polarize towards the iCAF subtype via IL-1a, leading to oxidative DNA damage and p53-mediated therapy-induced senescence in iCAFs, which in turn facilitates chemoradiotherapy resistance and disease progression (285) (Figure 6).
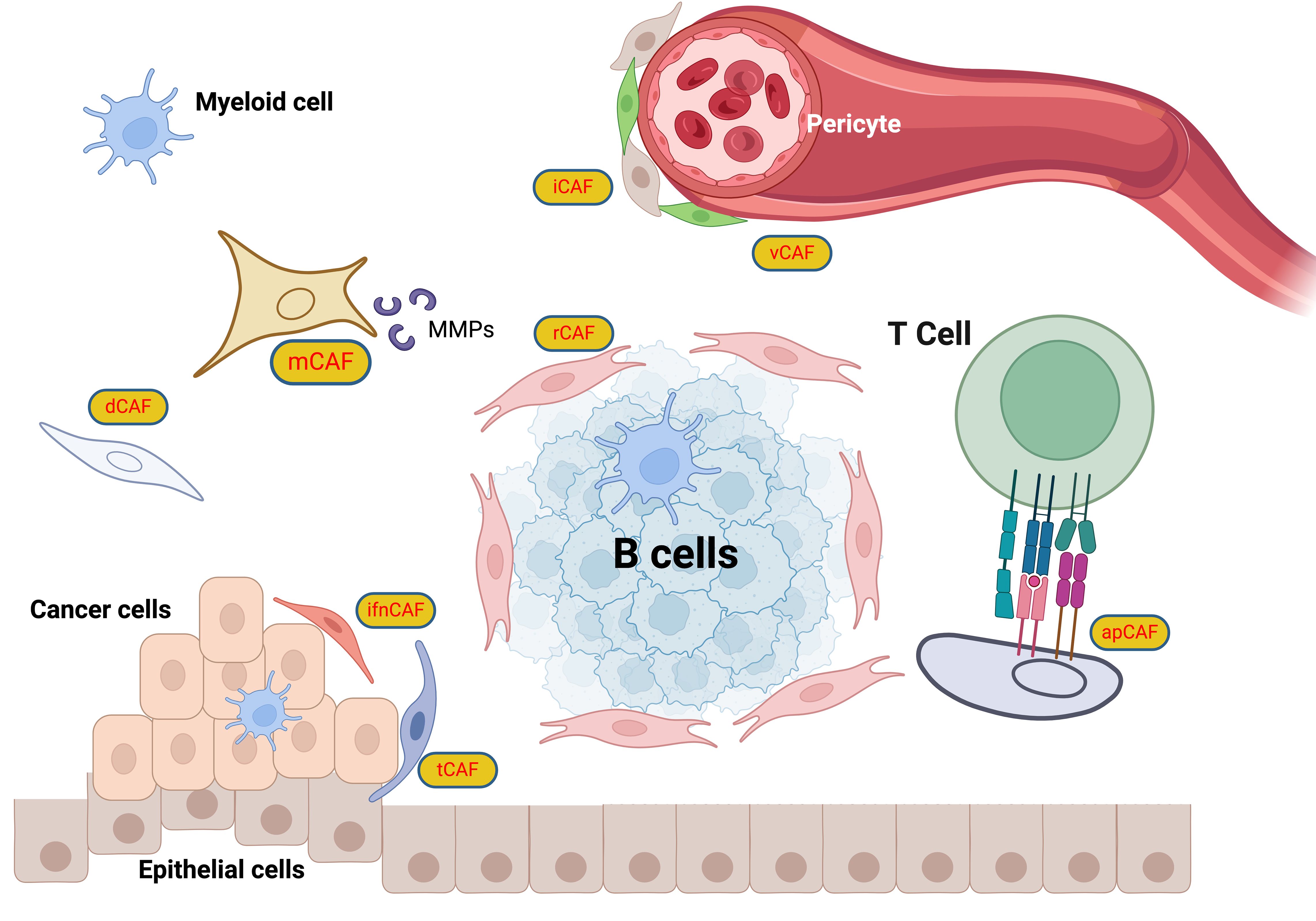
Figure 6. CAF classification scheme. The CAFs are categorized into specific roles: iCAFs (inflammatory CAFs) that modulate inflammation within the TME (286); rCAFs (regulatory CAFs), which might play roles in tumor regulation (2); dCAFs (developmental CAFs) associated with developmental processes (103); tCAFs (TGF-β producing CAFs), known for TGF-beta production influencing tumor growth and immune evasion (287); vCAFs (vascular CAFs) involved in vascular dynamics (288); ifnCAFs (interferon-producing CAFs), which might interact with immune pathways through interferon production (289); and apCAFs (antigen-presenting CAFs) that potentially present antigens to T cells, facilitating immune system interactions (290). This classification highlights the multifunctional nature of CAFs, underlining their importance in tumor progression, immune modulation, and the structural integrity of tumors, thus providing crucial insights for targeting these cells in cancer therapy strategies.
Challenges and directions
The challenges surrounding CAFs are multifaceted, ranging from classification issues to their dynamic roles within the TME. One major challenge lies in the lack of a uniform and comprehensive naming standard for CAF subgroups. An ideal naming convention should consider factors such as cell lineage, functional roles, biomarkers, clinical correlations, immune regulation, immune response, and metabolic status, integrating all these aspects to advance our understanding of CAFs. Another obstacle is the difficulty in identifying the origin of CAFs, which is compounded by the absence of specific biomarkers. A promising approach to this issue would involve the combined use of multiple biomarkers and the quantitative assessment of their variations to enhance specificity, particularly by focusing on distinct CAF subpopulations. Moreover, in vitro culture of CAFs presents significant challenges, as most of their in vivo characteristics tend to change due to alterations in culture conditions and passage (291, 292). I t is crucial to establish culture environments that closely mimic the TME and to track phenotypic changes during cultivation to preserve CAF traits. Despite the use of various methods for detecting CAFs, including antibodies, mRNA probes, and single-cell transcriptome analysis, there is still a lack of standardized, accurate, and universally applicable quantitative methods for their detection. While single-cell transcriptomics is already shedding light on CAF heterogeneity, further application of this technology is essential for deepening our understanding of CAF subpopulations (293). Another critical gap is the lack of longitudinal studies examining CAFs across different experimental stages, such as primary tumor growth, early isolation, and long-term passage, as well as across varying clinical stages (294). Such studies are necessary to improve our knowledge of CAF origins, subpopulations, heterogeneity, and plasticity in relation to tumor progression. Additionally, there is a need for horizontal studies that compare CAF subpopulations between different types of tumors and correlate these populations with clinical features to better understand the impact of CAFs on disease progression and treatment responses (2).
The regulation of CAFs within the TME is also not fully understood, and a deeper exploration of the dynamic interplay between CAFs, tumor cells, and other elements of the TME from biochemical, metabolic, immunological, and physical perspectives is necessary. Understanding how CAFs evolve in response to tumor progression and TME changes is equally crucial. Furthermore, while CAFs are known to influence immune responses within the TME, their crosstalk with immune cells remains poorly defined, highlighting the need for more research into how CAFs contribute to immune evasion and therapy resistance.
Discussion
CAFs represent a crucial, yet complex, component of the TME, influencing various aspects of cancer progression, metastasis, and therapeutic resistance. This review has highlighted the multifaceted roles of CAFs in shaping the TME, including their involvement in ECM remodeling, immune modulation, angiogenesis, and metabolic reprogramming. The dynamic and heterogeneous nature of CAFs, however, complicates their classification and therapeutic targeting. Current research on CAFs has emphasized the need for a more standardized system to categorize the various CAF subpopulations based on their lineage, function, biomarkers, and interactions within the TME. Understanding the underlying mechanisms of CAF activation and their dualistic roles—either promoting or suppressing tumor growth depending on the context—remains a critical challenge for therapeutic strategies.
To effectively address the highlighted lack of specific CAF markers, concerted efforts are needed to identify and validate reliable markers that enhance specificity. This can be achieved by employing advanced genomic and proteomic technologies to analyze diverse cancer types, facilitating the discovery of unique CAF profiles. Additionally, the lack of specific biomarkers for CAF identification remains a significant hurdle, limiting their effective targeting in clinical practice. Although several biomarkers have been proposed, the lack of specificity for CAFs means that they cannot be universally applied in clinical settings. The use of advanced techniques, such as single-cell transcriptomics, holds promise for resolving the complexity of CAF subpopulations and identifying precise markers for their targeting. Another critical issue need to be discussed is the challenge of maintaining CAF phenotypes in vitro, as their characteristics often change when cultured outside the TME. Optimizing culture conditions to better preserve the in vivo-like properties of CAFs is crucial for advancing CAF-based research and therapeutic development.
Single-cell technologies have profoundly refined our understanding of CAFs by revealing their cellular heterogeneity within the tumor microenvironment. Techniques like single-cell RNA sequencing (scRNA-seq) have identified distinct CAF subtypes with unique gene expressions and roles, enhancing our insight into their contributions to tumor progression and potential as therapeutic targets. Future research should integrate single-cell data with spatial transcriptomics to explore the dynamic interactions of CAFs with the TME across tumor development and therapy response, aiming to develop targeted treatments that disrupt crucial CAF-driven pathways.
The resistance mechanisms mediated by CAFs, particularly in the context of chemotherapy, immunotherapy, and radiotherapy, further complicate treatment efficacy. CAFs influence drug resistance through several mechanisms, including the promotion of ECM stiffness, secretion of pro-inflammatory cytokines, and modification of immune responses within the TME. Notably, CAFs enhance immune evasion by modulating the activity of immune cells, such as Tregs and NK cells, which significantly impacts the success of immunotherapies. Therefore, strategies that combine CAF-targeted therapies with conventional treatments, such as chemotherapy or immune checkpoint inhibitors, may offer a promising approach to overcome therapeutic resistance.
In addressing the current challenges and gaps identified in CAF research, we propose several specific experimental approaches and therapeutic strategies to advance this field. Firstly, the development of innovative CAF-specific markers through high-throughput screening techniques could greatly refine the targeting of these cells in diverse cancer types. Additionally, leveraging cutting-edge technologies such as CRISPR-Cas9 for gene editing within CAFs offers a promising avenue to dissect their functional roles in tumor progression and resistance mechanisms (295). Therapeutically, exploring bi-specific antibodies that target both CAFs and tumor cells could provide a dual approach to disrupt the supportive tumor microenvironment. Furthermore, employing organoid models incorporating CAFs from patient-derived samples would enhance our understanding of their interaction with tumor cells in a controlled, yet biologically relevant system. These approaches not only aim to fill the existing gaps but also pave the way for novel interventions that could be translated into clinical applications.
However, further research is still essential to gain a deeper understanding of the molecular mechanisms that regulate CAF behavior, particularly their interactions with tumor cells, immune cells, and the extracellular matrix. Investigating the dynamic roles of CAFs in different stages of tumor progression, as well as their involvement in the establishment of pre-metastatic niches, will be crucial for developing more effective therapeutic strategies. Additionally, refining methods to accurately identify and classify CAF subpopulations, along with developing therapies that can specifically modulate CAF function, holds great promise in advancing cancer treatment. Combining CAF-targeted therapies with current immunotherapies and other treatment modalities could significantly improve clinical outcomes and provide more effective treatment options for cancer patients.
Author contributions
XL: Methodology, Writing – original draft. WL: Validation, Writing – original draft. KZ: Formal Analysis, Writing – original draft. JW: Visualization, Writing – original draft. SL: Methodology, Writing – original draft. HZ: Conceptualization, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the Shandong Province Natural Science Foundation grants (grant no. ZR2022QH372).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Zhou S, Zhao Z, Wang Z, Xu H, Li Y, Xu K, et al. Cancer-associated fibroblasts in carcinogenesis. J Transl Med. (2025) 23:50. doi: 10.1186/s12967-025-06071-8
2. Cords L, de Souza N, Bodenmiller B. Classifying cancer-associated fibroblasts—The good, the bad, and the target. Cancer Cell. (2024) 42:1480–5. doi: 10.1016/j.ccell.2024.08.011
3. Chen Y, McAndrews KM, Kalluri R. Clinical and therapeutic relevance of cancer-associated fibroblasts. Nat Rev Clin Oncol. (2021) 18:792–804. doi: 10.1038/s41571-021-00546-5
4. Yamazaki M, Ishimoto T. Targeting cancer-associated fibroblasts: eliminate or reprogram? Cancer Sci. (2025) 16:613–21. doi: 10.1111/cas.v116.3
5. Lv K, He T. Cancer-associated fibroblasts: heterogeneity, tumorigenicity and therapeutic targets. Mol BioMed. (2024) 5:1–22. doi: 10.1186/s43556-024-00233-8
6. Kazakova AN, Lukina MM, Anufrieva KS, Bekbaeva IV, Ivanova OM, Shnaider PV, et al. Exploring the diversity of cancer-associated fibroblasts: insights into mechanisms of drug resistance. Front Cell Dev Biol. (2024) 12:1403122. doi: 10.3389/fcell.2024.1403122
7. Cao Z, Quazi S, Arora S, Osellame LD, Burvenich IJ, Janes PW, et al. Cancer-associated fibroblasts as therapeutic targets for cancer: advances, challenges, and future prospects. J BioMed Sci. (2025) 32:7. doi: 10.1186/s12929-024-01099-2
8. Virchow R. Die Cellularpathologie in ihrer Begründung auf physiologische und pathologische Gewebelehre. Good Press. (2020) 147:2236–44.
9. Flier JS, Underhill LH, Dvorak HF. Tumors: wounds that do not heal. N Engl J Med. (1986) 315:1650–9. doi: 10.1056/NEJM198612253152606
10. Hanahan D, Weinberg RA. Hallmarks of cancer: The next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
11. Wynn TA, Ramalingam TR. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med. (2012) 18:1028–40. doi: 10.1038/nm.2807
12. Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. (2006) 6:392–401. doi: 10.1038/nrc1877
13. Mueller MM, Fusenig NE. Friends or foes - Bipolar effects of the tumour stroma in cancer. Nat Rev Cancer. (2004) 4:839–49. doi: 10.1038/nrc1477
14. Öhlund D, Elyada E, Tuveson D. Fibroblast heterogeneity in the cancer wound. J Exp Med. (2014) 211:1503–23. doi: 10.1084/jem.20140692
15. Virchow R. Die Cellularpathologie in ihrer Begründung auf physiologische und pathologische Gewebelehre: 20 Vorlesungen, gehalten während d. Monate Febr., März u. April 1858 im Patholog. Inst zu Berlin. Hirschwald. (1858).
16. Stoker MGP, Shearer M, O’neill C. Growth inhibition of polyoma transformed cells by contact with static normal fibroblasts. J Cell Sci. (1966) 1:297–310. doi: 10.1242/jcs.1.3.297
17. Welt S, Divgi CR, Scott AM, Garin-Chesa P, Finn RD, Graham M, et al. Antibody targeting in metastatic colon cancer: a phase I study of monoclonal antibody F19 against a cell-surface protein of reactive tumor stromal fibroblasts. J Clin Oncol. (1994) 12:1193–203. doi: 10.1200/JCO.1994.12.6.1193
18. Kojima Y, Acar A, Eaton EN, Mellody KT, Scheel C, Ben-Porath I, et al. Autocrine TGF-β and stromal cell-derived factor-1 (SDF-1) signaling drives the evolution of tumor-promoting mammary stromal myofibroblasts. Proc Natl Acad Sci. (2010) 107:20009–14. doi: 10.1073/pnas.1013805107
19. Özdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu C-C, Simpson TR, et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell. (2014) 25:719–34. doi: 10.1016/j.ccr.2014.04.005
20. Su S, Chen J, Yao H, Liu J, Yu S, Lao L, et al. CD10+GPR77+ Cancer-associated fibroblasts promote cancer formation and chemoresistance by sustaining cancer stemness. Cell. (2018) 172:841–56.e16. doi: 10.1016/j.cell.2018.01.009
21. Saw PE, Chen J, Song E. Targeting CAFs to overcome anticancer therapeutic resistance. Trends Cancer. (2022) 8:527–55. doi: 10.1016/j.trecan.2022.03.001
22. Meng S, Hara T, Miura Y, Ishii H. Fibroblast activation protein constitutes a novel target of chimeric antigen receptor T-cell therapy in solid tumors. Cancer Sci. (2024) 115:3532–42. doi: 10.1111/cas.v115.11
23. Kalluri R. The biology and function of fibroblasts in cancer. Nat Rev Cancer. (2016). doi: 10.1038/nrc.2016.73
25. Furuya S, Furuya K. Subepithelial fibroblasts in intestinal villi: roles in intercellular communication. Int Rev Cytol. (2007). doi: 10.1016/S0074-7696(07)64004-2
26. Shoshkes-Carmel M, Wang YJ, Wangensteen KJ, Tóth B, Kondo A, Massassa EE, et al. Subepithelial telocytes are an important source of Wnts that supports intestinal crypts. Nature. (2018) 557:242–46. doi: 10.1038/s41586-018-0084-4
27. Worthley DL, Churchill M, Compton JT, Tailor Y, Rao M, Si Y, et al. Gremlin 1 identifies a skeletal stem cell with bone, cartilage, and reticular stromal potential. Cell. (2015) 160:269–84. doi: 10.1016/j.cell.2014.11.042
28. Degirmenci B, Valenta T, Dimitrieva S, Hausmann G, Basler K. GLI1-expressing mesenchymal cells form the essential Wnt-secreting niche for colon stem cells. Nature. (2018). doi: 10.1038/s41586-018-0190-3
29. Powell DW, Pinchuk IV, Saada JI, Chen X, Mifflin RC. Mesenchymal cells of the intestinal lamina propria. Annu Rev Physiol. (2011) 73:213–37. doi: 10.1146/annurev.physiol.70.113006.100646
30. Koliaraki V, Pallangyo CK, Greten FR, Kollias G. Mesenchymal cells in colon cancer. Gastroenterology. (2017) 152:964–79. doi: 10.1053/j.gastro.2016.11.049
31. Biswas S, Davis H, Irshad S, Sandberg T, Worthley D, Leedham S. Microenvironmental control of stem cell fate in intestinal homeostasis and disease. J Pathol. (2015) 237:135–45. doi: 10.1002/path.4563
32. Sugimoto H, Mundel TM, Kieran MW, Kalluri R. Identification of fibroblast heterogeneity in the tumor microenvironment. Cancer Biol Ther. (2006) 5:1640–6. doi: 10.4161/cbt.5.12.3354
33. Madar S, Goldstein I, Rotter V. Cancer associated fibroblasts” - more than meets the eye. Trends Mol Med. (2013) 19:447–53. doi: 10.1016/j.molmed.2013.05.004
34. Bu L, Baba H, Yoshida N, Miyake K, Yasuda T, Uchihara T, et al. Biological heterogeneity and versatility of cancer-associated fibroblasts in the tumor microenvironment. Oncogene. (2019) 38:4887–901. doi: 10.1038/s41388-019-0765-y
35. LeBleu VS, Kalluri R. A peek into cancer-associated fibroblasts: Origins, functions and translational impact. DMM Dis Model Mech. (2018) 11. doi: 10.1242/dmm.029447
36. Goulet CR, Bernard G, Tremblay S, Chabaud S, Bolduc S, Pouliot F. Exosomes induce fibroblast differentiation into cancer-associated fibroblasts through TGFb signaling. Mol Cancer Res. (2018) 16:1196–204. doi: 10.1158/1541-7786.MCR-17-0784
37. Ping Q, Wang C, Cheng X, Zhong Y, Yan R, Yang M, et al. TGF-β1 dominates stromal fibroblast-mediated EMT via the FAP/VCAN axis in bladder cancer cells. J Transl Med. (2023) 21:475. doi: 10.1186/s12967-023-04303-3
38. Nallasamy P, Nimmakayala RK, Karmakar S, Leon F, Seshacharyulu P, Lakshmanan I, et al. Pancreatic tumor microenvironment factor promotes cancer stemness via SPP1–CD44 axis. Gastroenterology. (2021) 161:1998–2013. doi: 10.1053/j.gastro.2021.08.023
39. IL-1β derived from mixed-polarized macrophages activates fibroblasts and synergistically forms a cancer-promoting microenvironment. (2024)
40. Mao Y, Xie Z, Zhang X, Fu Y, Yu X, Deng L, et al. Ergothioneine ameliorates liver fibrosis by inhibiting glycerophospholipids metabolism and TGF-β/smads signaling pathway: based on metabonomics and network pharmacology. J Appl Toxicol. (2025) 45:514–30. doi: 10.1002/jat.v45.3
41. Nedaeinia R, Najafgholian S, Salehi R, Goli M, Ranjbar M, Nickho H, et al. The role of cancer-associated fibroblasts and exosomal miRNAs-mediated intercellular communication in the tumor microenvironment and the biology of carcinogenesis: a systematic review. Cell Death Discovery. (2024) 10:380. doi: 10.1038/s41420-024-02146-5
42. Li X, González-Maroto C, Tavassoli M. Crosstalk between CAFs and tumour cells in head and neck cancer. Cell Death Discovery. (2024) 10:303. doi: 10.1038/s41420-024-02053-9
43. Lei Y, Shu D, Xia J, Zhang T, Wei H. Extracellular nicotinamide phosphoribosyltransferase visfatin activates JAK2-STAT3 pathway in cancer-associated fibroblasts to promote colorectal cancer metastasis. Genes Genomics. (2024), 1–10. doi: 10.1007/s13258-024-01596-6
44. Zhi S, Chen C, Huang H, Zhang Z, Zeng F, Zhang S. Hypoxia-inducible factor in breast cancer: role and target for breast cancer treatment. Front Immunol. (2024) 15:1370800. doi: 10.3389/fimmu.2024.1370800
45. Yang Z, Su W, Wei X, Pan Y, Xing M, Niu L, et al. Hypoxia inducible factor-1α drives cancer resistance to cuproptosis. Cancer Cell. (2025). doi: 10.1016/j.ccell.2025.02.015
46. He R, Hu C, Yuan Y, Li T, Tian Q, Huang T, et al. Glycolysis reprogramming in CAFs promotes oxaliplatin resistance in pancreatic cancer through circABCC4 mediated PKM2 nuclear translocation. Cell Death Dis. (2025) 16:126. doi: 10.1038/s41419-025-07431-4
47. Kou Z, Liu C, Zhang W, Sun C, Liu L, Zhang Q. Heterogeneity of primary and metastatic CAFs: From differential treatment outcomes to treatment opportunities. Int J Oncol. (2024) 64:54. doi: 10.3892/ijo.2024.5642
48. Yeon JH, Jeong HE, Seo H, Cho S, Kim K, Na D, et al. Cancer-derived exosomes trigger endothelial to mesenchymal transition followed by the induction of cancer-associated fibroblasts. Acta Biomater. (2018) 76:146–53. doi: 10.1016/j.actbio.2018.07.001
49. Krzysiek-Maczka G, Targosz A, Szczyrk U, Strzalka M, Brzozowski T, Ptak-Belowska A. Involvement of epithelial-mesenchymal transition-inducing transcription factors in the mechanism of helicobacter pylori-induced fibroblasts activation. J Physiol Pharmacol. (2019) 70. doi: 10.26402/jpp.2019.5.08
50. Sun Q, Zhang B, Hu Q, Qin Y, Xu W, Liu W, et al. The impact of cancer-associated fibroblasts on major hallmarks of pancreatic cancer. Theranostics. (2018) 8:5072–87. doi: 10.7150/thno.26546
51. Tan HX, Bin CZ, TT H, Huang T, CL X, Liu Y. TGFβ1 is essential for MSCs-CAFs differentiation and promotes HCT116 cells migration and invasion via JAK/STAT3 signaling. Onco Targets Ther. (2019) 12:5323–34. doi: 10.2147/OTT.S178618
52. Raz Y, Cohen N, Shani O, Bell RE, Novitskiy SV, Abramovitz L, et al. Bone marrow-derived fibroblasts are a functionally distinct stromal cell population in breast cancer. J Exp Med. (2018) 215:3075–93. doi: 10.1084/jem.20180818
53. Borzone FR, Giorello MB, Sanmartin MC, Yannarelli G, Martinez LM, Chasseing NA. Mesenchymal stem cells and cancer-associated fibroblasts as a therapeutic strategy for breast cancer. Br J Pharmacol. (2024) 181:238–56. doi: 10.1111/bph.15861
54. Nair N, Calle AS, Zahra MH, Prieto-Vila M, Oo AKK, Hurley L, et al. A cancer stem cell model as the point of origin of cancer-associated fibroblasts in tumor microenvironment. Sci Rep. (2017) 7:6838. doi: 10.1038/s41598-017-07144-5
55. Fang Y, Xiao X, Wang J, Dasari S, Pepin D, Nephew KP, et al. Cancer associated fibroblasts serve as an ovarian cancer stem cell niche through noncanonical Wnt5a signaling. NPJ Precis Oncol. (2024) 8:7. doi: 10.1038/s41698-023-00495-5
56. Sasaki R, Devhare P, Ray RB, Ray R. Hepatitis C virus–induced tumor-initiating cancer stem–like cells activate stromal fibroblasts in a xenograft tumor model. Hepatology. (2017) 66:1766–78. doi: 10.1002/hep.29346
57. Jia B, Gao Y, Li M, Shi J, Peng Y, Du X, et al. GPR30 promotes prostate stromal cell activation via suppression of erα expression and its downstream signaling pathway. Endocrinology. (2016) 157:3023–35. doi: 10.1210/en.2016-1035
58. Bochet L, Lehuédé C, Dauvillier S, Wang YY, Dirat B, Laurent V, et al. Adipocyte-derived fibroblasts promote tumor progression and contribute to the desmoplastic reaction in breast cancer. Cancer Res. (2013) 73:5657–68. doi: 10.1158/0008-5472.CAN-13-0530
59. Ning X, Zhang H, Wang C, Song X. Exosomes released by gastric cancer cells induce transition of pericytes into cancer- associated fibroblasts. Med Sci Monit. (2018) 24:2350–59. doi: 10.12659/MSM.906641
60. Tan HX, Gong WZ, Zhou K, Xiao ZG, Hou FT, Huang T, et al. CXCR4/TGF-β1 mediated hepatic stellate cells differentiation into carcinoma-associated fibroblasts and promoted liver metastasis of colon cancer. Cancer Biol Ther. (2020) 21:258–68. doi: 10.1080/15384047.2019.1685157
61. Zhang X, Zhang M, Sun H, Wang X, Wang X, Sheng W, et al. The role of transcription factors in the crosstalk between cancer-associated fibroblasts and tumor cells. J Adv Res. (2025) 67:121–32. doi: 10.1016/j.jare.2024.01.033
62. Kothari A, Arffa M, Chang V, Blackwell R, Syn W-K, Zhang J, et al. Osteopontin—A master regulator of epithelial-mesenchymal transition. J Clin Med. (2016) 5:39. doi: 10.3390/jcm5040039
63. Rao G, Wang H, Li B, Huang L, Xue D, Wang X, et al. Reciprocal interactions between tumor-associated macrophages and CD44-positive cancer cells via osteopontin/CD44 promote tumorigenicity in colorectal cancer. Clin Cancer Res. (2013). doi: 10.1158/1078-0432.CCR-12-2788
64. Sharon Y, Raz Y, Cohen N, Ben-Shmuel A, Schwartz H, Geiger T, et al. Tumor-derived osteopontin reprograms normal mammary fibroblasts to promote inflammation and tumor growth in breast cancer. Cancer Res. (2015) 75:963–73. doi: 10.1158/0008-5472.CAN-14-1990
65. Barcellos-de-Souza P, Comito G, Pons-Segura C, Taddei ML, Gori V, Becherucci V, et al. Mesenchymal stem cells are recruited and activated into carcinoma-associated fibroblasts by prostate cancer microenvironment-derived TGF-β1. Stem Cells. (2016) 34:2536–47. doi: 10.1002/stem.2412
66. Becker LM, O’Connell JT, Vo AP, Cain MP, Tampe D, Bizarro L, et al. Epigenetic reprogramming of cancer-associated fibroblasts deregulates glucose metabolism and facilitates progression of breast cancer. Cell Rep. (2020) 31. doi: 10.1016/j.celrep.2020.107701
67. Procopio M-G, Laszlo C, Al Labban D, Kim DE, Bordignon P, Jo S-H, et al. Combined CSL and p53 downregulation promotes cancer-associated fibroblast activation. Nat Cell Biol. (2015) 17:1193–204. doi: 10.1038/ncb3228
68. Baglole CJ, Smith TJ, Foster D, Sime PJ, Feldon S, Phipps RP. “Functional assessment of fibroblast heterogeneity by the cell-surface glycoprotein thy-1,” In Tissue Repair Contraction Myofibroblast. Boston, MA: Springer US (2007) p. 32–39. doi: 10.1007/0-387-33650-8_4
69. Mercier I, Casimiro MC, Wang C, Rosenberg AL, Quong J, Minkeu A, et al. Human breast cancer-associated fibroblasts (CAFs) show caveolin-1 downregulation and RB tumor suppressor functional inactivation: Implications for the response to hormonal therapy. Cancer Biol Ther. (2008) 7:1212–25. doi: 10.4161/cbt.7.8.6220
70. Tlsty TD. Stromal cells can contribute oncogenic signals. Semin Cancer Biol. (2001) 11:97–104. doi: 10.1006/scbi.2000.0361
71. Huang M, Li Y, Zhang H, Nan F. Breast cancer stromal fibroblasts promote the generation of CD44 +CD24- cells through SDF-1/CXCR4 interaction. J Exp Clin Cancer Res. (2010) 29:80. doi: 10.1186/1756-9966-29-80
72. Liu Y, Hu T, Shen J, Li SF, Lin JW, Zheng XH, et al. Separation, cultivation and biological characteristics of oral carcinoma-associated fibroblasts. Oral Dis. (2006) 12:375–80. doi: 10.1111/j.1601-0825.2005.01207.x
73. Ayala G, Tuxhorn JA, Wheeler TM, Frolov A, Scardino PT, Ohori M, et al. Reactive stroma as a predictor of biochemical-free recurrence in prostate cancer. Clin Cancer Res. (2003) 9:4792–801.
74. Gonda TA, Varro A, Wang TC, Tycko B. Molecular biology of cancer-associated fibroblasts: Can these cells be targeted in anti-cancer therapy? Semin Cell Dev Biol. (2010) 21:2–10. doi: 10.1016/j.semcdb.2009.10.001
75. Casey T, Bond J, Tighe S, Hunter T, Lintault L, Patel O, et al. Molecular signatures suggest a major role for stromal cells in development of invasive breast cancer. Breast Cancer Res Treat. (2009) 114:47–62. doi: 10.1007/s10549-008-9982-8
76. Ma XJ, Dahiya S, Richardson E, Erlander M, Sgroi DC. Gene expression profiling of the tumor microenvironment during breast cancer progression. Breast Cancer Res. (2009) 11:R7. doi: 10.1186/bcr2222
77. Saadi A, Shannon NB, Lao-Sirieix P, O’Donovan M, Walker E, Clemons NJ, et al. Stromal genes discriminate preinvasive from invasivedisease, predict outcome, and highlight inflammatory pathways in digestive cancers. Proc Natl Acad Sci U.S.A. (2010) 107:2177–82. doi: 10.1073/pnas.0909797107
78. Chang HY, Nuyten DSA, Sneddon JB, Hastie T, Tibshirani R, Sørlie T, et al. Robustness, scalability, and integration of a wound-response gene expression signature in predicting breast cancer survival. Proc Natl Acad Sci U.S.A. (2005) 102:3738–43. doi: 10.1073/pnas.0409462102
79. Finak G, Bertos N, Pepin F, Sadekova S, Souleimanova M, Zhao H, et al. Stromal gene expression predicts clinical outcome in breast cancer. Nat Med. (2008) 14:518–27. doi: 10.1038/nm1764
80. Dulauroy S, Di Carlo SE, Langa F, Eberl G, Peduto L. Lineage tracing and genetic ablation of ADAM12 + perivascular cells identify a major source of profibrotic cells during acute tissue injury. Nat Med. (2012) 18:1262–70. doi: 10.1038/nm.2848
81. Jotzu C, Alt E, Welte G, Li J, Hennessy BT, Devarajan E, et al. Adipose tissue derived stem cells differentiate into carcinoma-associated fibroblast-like cells under the influence of tumor derived factors. Cell Oncol. (2011). doi: 10.1007/s13402-011-0012-1
82. Gaggioli C, Hooper S, Hidalgo-Carcedo C, Grosse R, Marshall JF, Harrington K, et al. Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nat Cell Biol. (2007) 9:1392–400. doi: 10.1038/ncb1658
83. Goetz JG, Minguet S, Navarro-Lérida I, Lazcano JJ, Samaniego R, Calvo E, et al. Biomechanical remodeling of the microenvironment by stromal caveolin-1 favors tumor invasion and metastasis. Cell. (2011) 146:148–63. doi: 10.1016/j.cell.2011.05.040
84. Hooper S, Gaggioli C, Sahai E. A chemical biology screen reveals a role for Rab21-mediated control of actomyosin contractility in fibroblast-driven cancer invasion. Br J Cancer. (2010) 102:392–402. doi: 10.1038/sj.bjc.6605469
85. DeFilippis RA, Chang H, Dumont N, Rabban JT, Chen YY, Fontenay GV, et al. CD36 repression activates a multicellular stromal program shared by high mammographic density and tumor tissues. Cancer Discovery. (2012) 2:826–39. doi: 10.1158/2159-8290.CD-12-0107
86. Pasquale EB. Eph receptors and ephrins in cancer progression. Nat Rev Cancer. (2024) 24:5–27. doi: 10.1038/s41568-023-00634-x
87. Kechagia JZ, Ivaska J, Roca-Cusachs P. Integrins as biomechanical sensors of the microenvironment. Nat Rev Mol Cell Biol. (2019). doi: 10.1038/s41580-019-0134-2
88. Zeltz C, Primac I, Erusappan P, Alam J, Noel A, Gullberg D. Cancer-associated fibroblasts in desmoplastic tumors: emerging role of integrins. Semin Cancer Biol. (2020) 62:166–81. doi: 10.1016/j.semcancer.2019.08.004
89. Nguyen EV, Pereira BA, Lawrence MG, Ma X, Rebello RJ, Chan H, et al. Proteomic profiling of human prostate cancer-associated fibroblasts (CAF) reveals LOXL2-dependent regulation of the tumor microenvironment. Mol Cell Proteomics. (2019) 18:1410–27. doi: 10.1074/mcp.RA119.001496
90. Tang X, Hou Y, Yang G, Wang X, Tang S, Du YE, et al. Stromal miR-200s contribute to breast cancer cell invasion through CAF activation and ECM remodeling. Cell Death Differ. (2016) 23:132–45. doi: 10.1038/cdd.2015.78
91. Arpinati L, Carradori G, Scherz-Shouval R. CAF-induced physical constraints controlling T cell state and localization in solid tumours. Nat Rev Cancer. (2024). doi: 10.1038/s41568-024-00740-4
92. Zhao Z, Zhang Y, Guo E, Zhang Y, Wang Y. Periostin secreted from podoplanin-positive cancer-associated fibroblasts promotes metastasis of gastric cancer by regulating cancer stem cells via AKT and YAP signaling pathway. Mol Carcinog. (2023) 62:685–99. doi: 10.1002/mc.23517
93. Aizawa T, Karasawa H, Funayama R, Shirota M, Suzuki T, Maeda S, et al. Cancer-associated fibroblasts secrete Wnt2 to promote cancer progression in colorectal cancer. Cancer Med. (2019) 8:6370–82. doi: 10.1002/cam4.2523
94. Karampoga A, Tzaferi K, Koutsakis C, Kyriakopoulou K, Karamanos NK. Exosomes and the extracellular matrix: a dynamic interplay in cancer progression. Int J Dev Biol. (2021) 66:97–102. doi: 10.1387/ijdb.210120nk
95. Prakash J, Shaked Y. The interplay between extracellular matrix remodeling and cancer therapeutics. Cancer Discovery. (2024) 14:1375–88. doi: 10.1158/2159-8290.CD-24-0002
96. Paszek MJ, Zahir N, Johnson KR, Lakins JN, Rozenberg GI, Gefen A, et al. Tensional homeostasis and the Malignant phenotype. Cancer Cell. (2005) 8:241–54. doi: 10.1016/j.ccr.2005.08.010
97. Barbazan J, Pérez-González C, Gómez-González M, Dedenon M, Richon S, Latorre E, et al. Cancer-associated fibroblasts actively compress cancer cells and modulate mechanotransduction. Nat Commun. (2023) 14. doi: 10.1038/s41467-023-42382-4
98. Diop-Frimpong B, Chauhan VP, Krane S, Boucher Y, Jain RK. Losartan inhibits collagen I synthesis and improves the distribution and efficacy of nanotherapeutics in tumors. Proc Natl Acad Sci U.S.A. (2011) 108:2909–14. doi: 10.1073/pnas.1018892108
99. Dufort CC, DelGiorno KE, Carlson MA, Osgood RJ, Zhao C, Huang Z, et al. Interstitial pressure in pancreatic ductal adenocarcinoma is dominated by a gel-fluid phase. Biophys J. (2016) 110:2106–19. doi: 10.1016/j.bpj.2016.03.040
100. Bhome R, Goh RW, Bullock MD, Pillar N, Thirdborough SM, Mellone M, et al. Exosomal microRNAs derived from colorectal cancer-associated fibroblasts: Role in driving cancer progression. Aging (Albany NY). (2017) 9:2666–94. doi: 10.18632/aging.101355
101. Luga V, Zhang L, Viloria-Petit AM, Ogunjimi AA, Inanlou MR, Chiu E, et al. Exosomes mediate stromal mobilization of autocrine Wnt-PCP signaling in breast cancer cell migration. Cell. (2012) 151:1542–56. doi: 10.1016/j.cell.2012.11.024
102. Wang Y, Ding W, Hao W, Gong L, Peng Y, Zhang J, et al. CXCL3/TGF-β-mediated crosstalk between CAFs and tumor cells augments RCC progression and sunitinib resistance. iScience. (2024). doi: 10.1016/j.isci.2024.110224
103. Ye J, Baer JM, Faget DV, Morikis VA, Ren Q, Melam A, et al. Senescent CAFs mediate immunosuppression and drive breast cancer progression. Cancer Discovery. (2024) 14:1302–23. doi: 10.1158/2159-8290.CD-23-0426
104. Maker AV, Karakousis G. Educating cancer-associated fibroblasts (CAFs): are they the student or the teacher? Ann Surg Oncol. (2024) 31:2181–82. doi: 10.1245/s10434-023-14812-6
105. Solek J, Braun M, Sadej R, Romanska HM. FGFR-related phenotypic and functional profile of CAFs in prognostication of breast cancer. Int J Oncol. (2024) 65:94. doi: 10.3892/ijo.2024.5682
106. Isaacson A, Barki D, Scherz-Shouval R. Unlocking the role of age-related changes to fibroblasts in pancreatic cancer. Cancer Res. (2024) 84:1185–7. doi: 10.1158/0008-5472.CAN-24-0439
107. Li P, Zhang H, Chen T, Zhou Y, Yang J, Zhou J. Cancer-associated fibroblasts promote proliferation, angiogenesis, metastasis and immunosuppression in gastric cancer. Matrix Biol. (2024). doi: 10.1016/j.matbio.2024.06.004
108. Knauf JA, Luckett KA, Chen KY, Voza F, Socci ND, Ghossein R, et al. Hgf/Met activation mediates resistance to BRAF inhibition in murine anaplastic thyroid cancers. J Clin Invest. (2018) 128:4086–97. doi: 10.1172/JCI120966
109. Straussman R, Morikawa T, Shee K. Tumor microenvironment induces innate RAF-inhibitor resistance through HGF secretion. Nature. (2012) 487:500–4. doi: 10.1038/nature11183
110. Che S, Yan Z, Feng Y, Zhao H. Unveiling the intratumoral microbiota within cancer landscapes. Iscience. (2024). doi: 10.1016/j.isci.2024.109893
111. Linares J, Marín-Jiménez JA, Badia-Ramentol J, Calon A. Determinants and functions of CAFs secretome during cancer progression and therapy. Front Cell Dev Biol. (2021) 8. doi: 10.3389/fcell.2020.621070
112. Mishra D, Banerjee D. Secretome of stromal cancer-associated fibroblasts (CAFs): relevance in cancer. Cells. (2023) 12. doi: 10.3390/cells12040628
113. Varveri A, Papadopoulou M, Papadovasilakis Z, Compeer EB, Legaki A-I, Delis A, et al. Immunological synapse formation between T regulatory cells and cancer-associated fibroblasts promotes tumour development. Nat Commun. (2024) 15:4988.
114. Chen M, Chen F, Gao Z, Li X, Hu L, Yang S, et al. CAFs and T cells interplay: The emergence of a new arena in cancer combat. BioMed Pharmacother. (2024) 177:117045.
115. Fearon DT. The carcinoma-associated fibroblast expressing fibroblast activation protein and escape from immune surveillance. Cancer Immunol Res. (2014) 2:187–93. doi: 10.1158/2326-6066.CIR-14-0002
116. Elyada E, Bolisetty M, Laise P, Flynn WF, Courtois ET, Burkhart RA, et al. Cross-species single-cell analysis of pancreatic ductal adenocarcinoma reveals antigen-presenting cancer-associated fibroblasts. Cancer Discovery. (2019) 9:1102–23. doi: 10.1158/2159-8290.CD-19-0094
117. Lakins MA, Ghorani E, Munir H, Martins CP, Shields JD. Cancer-associated fibroblasts induce antigen-specific deletion of CD8 + T Cells to protect tumour cells. Nat Commun. (2018) 9. doi: 10.1038/s41467-018-03347-0
118. Costa A, Kieffer Y, Scholer-Dahirel A, Pelon F, Bourachot B, Cardon M, et al. Fibroblast heterogeneity and immunosuppressive environment in human breast cancer. Cancer Cell. (2018) 33:463–79.e10. doi: 10.1016/j.ccell.2018.01.011
119. Flint TR, Janowitz T, Connell CM, Roberts EW, Denton AE, Coll AP, et al. Tumor-induced IL-6 reprograms host metabolism to suppress anti-tumor immunity. Cell Metab. (2016) 24:672–84. doi: 10.1016/j.cmet.2016.10.010
120. Jiang H, Hegde S, Knolhoff BL, Zhu Y, Herndon JM, Meyer MA, et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat Med. (2016) 22:851–60. doi: 10.1038/nm.4123
121. Chaudhry SI, Hooper S, Nye E, Williamson P, Harrington K, Sahai E. Autocrine IL-1β-TRAF6 signalling promotes squamous cell carcinoma invasion through paracrine TNFα signalling to carcinoma-associated fibroblasts. Oncogene. (2013) 32:747–58.
122. Kennel KB, Bozlar M, De Valk AF, Greten FR. Cancer-associated fibroblasts in inflammation and antitumor immunity. Clin Cancer Res. (2023) 29:1009–16.
123. Sanford-Crane H, Abrego J, Sherman MH. Fibroblasts as modulators of local and systemic cancer metabolism. Cancers (Basel). (2019) 11:619. doi: 10.3390/cancers11050619
124. Bertero T, Oldham WM, Grasset EM, Bourget I, Boulter E, Pisano S, et al. Tumor-stroma mechanics coordinate amino acid availability to sustain tumor growth and Malignancy. Cell Metab. (2019) 29:124–40.e10. doi: 10.1016/j.cmet.2018.09.012
125. Martinez-Outschoorn UE, Lisanti MP, Sotgia F. Catabolic cancer-associated fibroblasts transfer energy and biomass to anabolic cancer cells, fueling tumor growth. Semin Cancer Biol. (2014) 25:47–60. doi: 10.1016/j.semcancer.2014.01.005
126. Chang CH, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell. (2015) 162:1229–41. doi: 10.1016/j.cell.2015.08.016
127. Joshi RS, Kanugula SS, Sudhir S, Pereira MP, Jain S, Aghi MK. The role of cancer-associated fibroblasts in tumor progression. Cancers (Basel). (2021) 13:1–27. doi: 10.3390/cancers13061399
128. Zhang C, Fei Y, Wang H, Hu S, Liu C, Hu R, et al. CAFs orchestrates tumor immune microenvironment—A new target in cancer therapy? Front Pharmacol. (2023) 14:1113378.
129. Guo Q, Zhou Y, Xie T, Yuan Y, Li H, Shi W, et al. Tumor microenvironment of cancer stem cells: Perspectives on cancer stem cell targeting. Genes Dis. (2024) 11. doi: 10.1016/j.gendis.2023.05.024
130. Yin Y, Liu Y, Wang Y, Li J, Liang S, Zhang W, et al. DZIP1 expressed in fibroblasts and tumor cells may affect immunosuppression and metastatic potential in gastric cancer. Int Immunopharmacol. (2023) 117:109886. doi: 10.1016/j.intimp.2023.109886
131. Ma J, Chen X, Chen Y, Tao N, Qin Z. Ligustilide inhibits tumor angiogenesis by downregulating VEGFA secretion from cancer-associated fibroblasts in prostate cancer via TLR4. Cancers (Basel). (2022) 14. doi: 10.3390/cancers14102406
132. Wang H, Chen J, Chen X, Liu Y, Wang J, Meng Q, et al. Cancer-associated fibroblasts expressing sulfatase 1 facilitate VEGFA-dependent microenvironmental remodeling to support colorectal cancer. Cancer Res. (2024). doi: 10.1158/0008-5472.c.7494054
133. Tsoumakidou M. The advent of immune stimulating CAFs in cancer. Nat Rev Cancer. (2023) 23:258–69. doi: 10.1038/s41568-023-00549-7
134. Luo Q, Hu Z, Zhao H, Fan Y, Tu X, Wang Y, et al. The role of TGF-β in the tumor microenvironment of pancreatic cancer. Genes Dis. (2023) 10:1513–24. doi: 10.1016/j.gendis.2022.10.019
135. Sun X, Mao Y, Wang J, Zu L, Hao M, Cheng G, et al. IL-6 secreted by cancer-associated fibroblasts induces tamoxifen resistance in luminal breast cancer. Oncogene. (2014). doi: 10.1038/onc.2014.158
136. Jaworska M, Szczudło J, Pietrzyk A, Shah J, Trojan SE, Ostrowska B, et al. The Warburg effect: a score for many instruments in the concert of cancer and cancer niche cells. Pharmacol Rep. (2023). doi: 10.1007/s43440-023-00504-1
137. Akter R, Awais M, Boopathi V, Ahn JC, Yang DC, Kang SC, et al. Inversion of the warburg effect: unraveling the metabolic nexus between obesity and cancer. ACS Pharmacol Transl Sci. (2024) 7:560–69. doi: 10.1021/acsptsci.3c00301
138. Nassar D, Blanpain C. Cancer stem cells: basic concepts and therapeutic implications. Annu Rev Pathol Mech Dis. (2016) 11:47–76. doi: 10.1146/annurev-pathol-012615-044438
139. Islam F, Lam AK. eds. Cancer stem cells: basic concept and therapeutic implications. Singapore: Springer Nature Singapore (2023). doi: 10.1007/978-981-99-3185-9.
140. Zhao K, Wu C, Li X, Niu M, Wu D. From mechanism to therapy: the journey of CD24 in cancer. doi: 10.3389/fimmu.2024.1401528.
141. Lan L, Behrens A. Are there specific cancer stem cell markers? Cancer Res. (2023) 83:170–2. doi: 10.1158/0008-5472.CAN-22-2053
142. Liu Q, Guo Z, Li G, Zhang Y, Liu X, Li B, et al. Cancer stem cells and their niche in cancer progression and therapy. Cancer Cell Int. (2023) 23. doi: 10.1186/s12935-023-03130-2
143. Ma Z, Li X, Mao Y, Wei C, Huang Z, Li G, et al. Interferon-dependent SLC14A1+ cancer-associated fibroblasts promote cancer stemness via WNT5A in bladder cancer. Cancer Cell. (2022) 40:1550–65.e7. doi: 10.1016/j.ccell.2022.11.005
144. Peng H, Zhu E, Zhang Y. Advances of cancer-associated fibroblasts in liver cancer. Biomark Res. (2022) 10. doi: 10.1186/s40364-022-00406-z
145. Zhuang J, Shen L, Li M, Sun J, Hao J, Li J, et al. Cancer-associated fibroblast-derived miR-146a-5p generates a niche that promotes bladder cancer stemness and chemoresistance. Cancer Res. (2023) 83:1611–27. doi: 10.1158/0008-5472.CAN-22-2213
146. Hu JL, Wang W, Lan XL, Zeng ZC, Liang YS, Yan YR, et al. CAFs secreted exosomes promote metastasis and chemotherapy resistance by enhancing cell stemness and epithelial-mesenchymal transition in colorectal cancer. Mol Cancer. (2019) 18:91. doi: 10.1186/s12943-019-1019-x
147. Katoh M, Katoh M. WNT signaling and cancer stemness. Essays Biochem. (2022) 66:319–31. doi: 10.1042/EBC20220016
148. Chen L, Endler A, Shibasaki F. Hypoxia and angiogenesis: Regulation of hypoxia-inducible factors via novel binding factors. Exp Mol Med. (2009) 41:849–57. doi: 10.3858/emm.2009.41.12.103
149. Baeriswyl V, Christofori G. The angiogenic switch in carcinogenesis. Semin Cancer Biol. (2009) 19:329–37. doi: 10.1016/j.semcancer.2009.05.003
150. De Palma M, Biziato D, Petrova TV. Microenvironmental regulation of tumour angiogenesis. Nat Rev Cancer. (2017). doi: 10.1038/nrc.2017.51
151. Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. (2005) 121:335–48. doi: 10.1016/j.cell.2005.02.034
152. Teicher BA, Fricker SP. CXCL12 (SDF-1)/CXCR4 pathway in cancer. Clin Cancer Res. (2010) 16:2927–31. doi: 10.1158/1078-0432.CCR-09-2329
153. Presta M, Dell’Era P, Mitola S, Moroni E, Ronca R, Rusnati M. Fibroblast growth factor/fibroblast growth factor receptor system in angiogenesis. Cytokine Growth Factor Rev. (2005) 16:159–78. doi: 10.1016/j.cytogfr.2005.01.004
154. Papadopoulos N, Lennartsson J. The PDGF/PDGFR pathway as a drug target. Mol Aspects Med. (2018) 62:75–88. doi: 10.1016/j.mam.2017.11.007
155. Crawford Y, Kasman I, Yu L, Zhong C, Wu X, Modrusan Z, et al. PDGF-C mediates the angiogenic and tumorigenic properties of fibroblasts associated with tumors refractory to anti-VEGF treatment. Cancer Cell. (2009) 15:21–34. doi: 10.1016/j.ccr.2008.12.004
156. Zhang JY, Zhu WW, Wang MY, Zhai RD, Wang Q, Shen WL, et al. Cancer-associated fibroblasts promote oral squamous cell carcinoma progression through LOX-mediated matrix stiffness. J Transl Med. (2021) 19. doi: 10.1186/s12967-021-03181-x
157. Zhang W, Zhang S, Zhang W, Yue Y, Qian W, Wang Z. Matrix stiffness and its influence on pancreatic diseases. Biochim Biophys Acta - Rev Cancer. (2021) 1876. doi: 10.1016/j.bbcan.2021.188583
158. Jiang Y, Zhang H, Wang J, Liu Y, Luo T, Hua H. Targeting extracellular matrix stiffness and mechanotransducers to improve cancer therapy. J Hematol Oncol. (2022) 15. doi: 10.1186/s13045-022-01252-0
159. Sack KD, Teran M, Nugent MA. Extracellular matrix stiffness controls VEGF signaling and processing in endothelial cells. J Cell Physiol. (2016) 231:2026–39. doi: 10.1002/jcp.25312
160. Li M, Zhang X, Wang M, Wang Y, Qian J, Xing X, et al. Activation of Piezo1 contributes to matrix stiffness-induced angiogenesis in hepatocellular carcinoma. Cancer Commun. (2022) 42:1162–84. doi: 10.1002/cac2.12364
161. Bao M, Chen Y, Liu JT, Bao H, Bin WW, Qi YX, et al. Extracellular matrix stiffness controls VEGF165 secretion and neuroblastoma angiogenesis via the YAP/RUNX2/SRSF1 axis. Angiogenesis. (2022) 25:71–86. doi: 10.1007/s10456-021-09804-7
162. Warburg O, Wind F, Negelein E. The metabolism of tumors in the body. J Gen Physiol. (1927) 8:519–30. doi: 10.1085/jgp.8.6.519
163. Liang L, Li W, Li X, Jin X, Liao Q, Li Y, et al. Reverse Warburg effect” of cancer-associated fibroblasts (Review). Int J Oncol. (2022) 60. doi: 10.3892/ijo.2022.5357
164. Mao X, Wong SYS, Tse EYT, Ko FCF, Tey SK, Yeung YS, et al. Mechanisms through which hypoxia-induced caveolin-1 drives tumorigenesis and metastasis in hepatocellular carcinoma. Cancer Res. (2016) 76:7242–53. doi: 10.1158/0008-5472.CAN-16-1031
165. Cruz-Bermúdez A, Laza-Briviesca R, Vicente-Blanco RJ, García-Grande A, Coronado MJ, Laine-Menéndez S, et al. Cancer-associated fibroblasts modify lung cancer metabolism involving ROS and TGF-β signaling. Free Radic Biol Med. (2019) 130:163–73. doi: 10.1016/j.freeradbiomed.2018.10.450
166. Zhao H, Liao X, Kang Y. Tregs: Where we are and what comes next? Front Immunol. (2017) 8:1578. doi: 10.3389/fimmu.2017.01578
167. Zhao H, Feng R, Peng A, Li G, Zhou L. The expanding family of noncanonical regulatory cell subsets. J Leukoc Biol. (2019) 106:369–83. doi: 10.1002/JLB.6RU0918-353RRRR
168. Rimal R, Desai P, Daware R, Hosseinnejad A, Prakash J, Lammers T, et al. Cancer-associated fibroblasts: Origin, function, imaging, and therapeutic targeting. Adv Drug Delivery Rev. (2022) 189:114504. doi: 10.1016/j.addr.2022.114504
169. Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science. (2011) 80-331:1565–70. doi: 10.1126/science.1203486
170. O’Donnell JS, Teng MWL, Smyth MJ. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat Rev Clin Oncol. (2019) 16:151–67. doi: 10.1038/s41571-018-0142-8
171. Chen C-H, Seguin-Devaux C, Burke NA, Oriss TB, Watkins SC, Clipstone N, et al. Transforming growth factor beta blocks Tec kinase phosphorylation, Ca2+ influx, and NFATc translocation causing inhibition of T cell differentiation. J Exp Med. (2003) 197:1689–99. doi: 10.1084/jem.20021170
172. Desbois M, Udyavar AR, Ryner L, Kozlowski C, Guan Y, Dürrbaum M, et al. Integrated digital pathology and transcriptome analysis identifies molecular mediators of T-cell exclusion in ovarian cancer. Nat Commun. (2020) 11:5583. doi: 10.1038/s41467-020-19408-2
173. Batlle E, Massagué J. Transforming growth factor-β Signaling in immunity and cancer. Immunity. (2019) 50:924–40. doi: 10.1016/j.immuni.2019.03.024
174. Ghahremanifard P, Chanda A, Bonni S, Bose P. TGF-β mediated immune evasion in cancer—spotlight on cancer-associated fibroblasts. Cancers (Basel). (2020) 12:1–11. doi: 10.3390/cancers12123650
175. Correia AL, Guimaraes JC, Auf der Maur P, De Silva D, Trefny MP, Okamoto R, et al. Hepatic stellate cells suppress NK cell-sustained breast cancer dormancy. Nature. (2021) 594:566–71. doi: 10.1038/s41586-021-03614-z
176. Ene-Obong A, Clear AJ, Watt J, Wang J, Fatah R, Riches JC, et al. Activated pancreatic stellate cells sequester CD8+ T cells to reduce their infiltration of the juxtatumoral compartment of pancreatic ductal adenocarcinoma. Gastroenterology. (2013) 145:1121–32. doi: 10.1053/j.gastro.2013.07.025
177. Cheng Y, Li H, Deng Y, Tai Y, Zeng K, Zhang Y, et al. Cancer-associated fibroblasts induce PDL1+ neutrophils through the IL6-STAT3 pathway that foster immune suppression in hepatocellular carcinoma. Cell Death Dis. (2018) 9:422. doi: 10.1038/s41419-018-0458-4
178. Kato T, Noma K, Ohara T, Kashima H, Katsura Y, Sato H, et al. Cancer-associated fibroblasts affect intratumoral CD8+ and foxP3+ T cells via IL6 in the tumor microenvironment. Clin Cancer Res. (2018) 24:4820–33. doi: 10.1158/1078-0432.CCR-18-0205
179. Eskandari-Malayeri F, Rezaei M. Immune checkpoint inhibitors as mediators for immunosuppression by cancer-associated fibroblasts: A comprehensive review. Front Immunol. (2022) 13. doi: 10.3389/fimmu.2022.996145
180. Tsoumakidou M. The advent of immune stimulating CAFs in cancer. Nat Rev Cancer. (2023) 23:258–69. doi: 10.1038/s41568-023-00549-7
181. Fukui Y, Kasashima H, Wang Z, Yonemitsu K, Kitayama K, Miki Y, et al. Analysis for the role of antigen-presenting cancer-associated fibroblasts in tumor microenvironment of colorectal cancer. Cancer Res. (2024) 84:287. doi: 10.1158/1538-7445.AM2024-287
182. Barrett RL, Pure E. Cancer-associated fibroblasts and their influence on tumor immunity and immunotherapy. Elife. (2020) 9:1–20. doi: 10.7554/ELIFE.57243
183. Friedman G, Levi-Galibov O, David E, Bornstein C, Giladi A, Dadiani M, et al. Cancer-associated fibroblast compositions change with breast cancer progression linking the ratio of S100A4+ and PDPN+ CAFs to clinical outcome. Nat Cancer. (2020) 1:692–708. doi: 10.1038/s43018-020-0082-y
184. Grauel AL, Nguyen B, Ruddy D, Laszewski T, Schwartz S, Chang J, et al. TGFβ-blockade uncovers stromal plasticity in tumors by revealing the existence of a subset of interferon-licensed fibroblasts. Nat Commun. (2020) 11:6315. doi: 10.1038/s41467-020-19920-5
185. Hutton C, Heider F, Blanco-Gomez A, Banyard A, Kononov A, Zhang X, et al. Single-cell analysis defines a pancreatic fibroblast lineage that supports anti-tumor immunity. Cancer Cell. (2021) 39:1227–44.e20. doi: 10.1016/j.ccell.2021.06.017
186. Kieffer Y, Hocine HR, Gentric G, Pelon F, Bernard C, Bourachot B, et al. Single-cell analysis reveals fibroblast clusters linked to immunotherapy resistance in cancer. Cancer Discovery. (2020) 10:1330–51. doi: 10.1158/2159-8290.CD-19-1384
187. Dominguez CX, Müller S, Keerthivasan S, Koeppen H, Hung J, Gierke S, et al. Single-cell RNA sequencing reveals stromal evolution into LRRC15+ Myofibroblasts as a determinant of patient response to cancer immunotherapy. Cancer Discovery. (2020) 10:232–53. doi: 10.1158/2159-8290.CD-19-0644
188. Chua ZMX, Tajebe F, Abuwarwar M, Fletcher AL. Differential induction of T-cell tolerance by tumour fibroblast subsets. Curr Opin Immunol. (2024) 86:102410. doi: 10.1016/j.coi.2023.102410
189. Arpinati L, Carradori G, Scherz-Shouval R. CAF-induced physical constraints controlling T cell state and localization in solid tumours. Nat Rev Cancer. (2024) 24:676–93. doi: 10.1038/s41568-024-00740-4
190. Chen X, Song E. Turning foes to friends: targeting cancer-associated fibroblasts. Nat Rev Drug Discovery. (2019) 18:99–115. doi: 10.1038/s41573-018-0004-1
191. Aiello NM, Brabletz T, Kang Y, Nieto MA, Weinberg RA, Stanger BZ. Upholding a role for EMT in pancreatic cancer metastasis. Nature. (2017) 547:E7–8. doi: 10.1038/nature22963
192. Hao Y, Baker D, Ten DP. TGF-β-mediated epithelial-mesenchymal transition and cancer metastasis. Int J Mol Sci. (2019) 20. doi: 10.3390/ijms20112767
193. Zhang T, Li X, He Y, Wang Y, Shen J, Wang S, et al. Cancer-associated fibroblasts-derived HAPLN1 promotes tumour invasion through extracellular matrix remodeling in gastric cancer. Gastric Cancer. (2022) 25:346–59. doi: 10.1007/s10120-021-01259-5
194. Sakamoto H, ichiro KY, Higashino N, Kodama T, Tanigawa K, Shimizu M, et al. PAI-1 derived from cancer-associated fibroblasts in esophageal squamous cell carcinoma promotes the invasion of cancer cells and the migration of macrophages. Lab Investig. (2021) 101:353–68. doi: 10.1038/s41374-020-00512-2
195. Fattet L, Jung H-Y, Matsumoto MW, Aubol BE, Kumar A, Adams JA, et al. Matrix rigidity controls epithelial-mesenchymal plasticity and tumor metastasis via a mechanoresponsive EPHA2/LYN complex. Dev Cell. (2020) 54:302–316.e7. doi: 10.1016/j.devcel.2020.05.031
196. Labernadie A, Kato T, Brugués A, Serra-Picamal X, Derzsi S, Arwert E, et al. A mechanically active heterotypic E-cadherin/N-cadherin adhesion enables fibroblasts to drive cancer cell invasion. Nat Cell Biol. (2017) 19:224–37. doi: 10.1038/ncb3478
197. Erdogan B, Ao M, White LM, Means AL, Brewer BM, Yang L, et al. Cancer-associated fibroblasts promote directional cancer cell migration by aligning fibronectin. J Cell Biol. (2017) 216:3799–816. doi: 10.1083/jcb.201704053
198. Sabeh F, Ota I, Holmbeck K, Birkedal-Hansen H, Soloway P, Balbin M, et al. Tumor cell traffic through the extracellular matrix is controlled by the membrane-anchored collagenase MT1-MMP. J Cell Biol. (2004) 167:769–81. doi: 10.1083/jcb.200408028
199. Leone P, Malerba E, Susca N, Favoino E, Perosa F, Brunori G, et al. Endothelial cells in tumor microenvironment: insights and perspectives. Front Immunol. (2024) 15:1367875. doi: 10.3389/fimmu.2024.1367875
200. Reymond N, D’Água BB, Ridley AJ. Crossing the endothelial barrier during metastasis. Nat Rev Cancer. (2013) 13:858–70. doi: 10.1038/nrc3628
201. Wu D, Deng S, Li L, Liu T, Zhang T, Li J, et al. TGF-β1-mediated exosomal lnc-MMP2-2 increases blood–brain barrier permeability via the miRNA-1207-5p/EPB41L5 axis to promote non-small cell lung cancer brain metastasis. Cell Death Dis. (2021). doi: 10.1038/s41419-021-04004-z
202. Gong MM, Lugo-Cintron KM, White BR, Kerr SC, Harari PM, Beebe DJ. Human organotypic lymphatic vessel model elucidates microenvironment-dependent signaling and barrier function. Biomaterials. (2019) 214:119225. doi: 10.1016/j.biomaterials.2019.119225
203. Wei W-F, Chen X-J, Liang L-J, Yu L, Wu X-G, Zhou C-F, et al. Periostin+ cancer-associated fibroblasts promote lymph node metastasis by impairing the lymphatic endothelial barriers in cervical squamous cell carcinoma. Mol Oncol. (2021) 15:210–27. doi: 10.1002/1878-0261.12837
204. Tacconi C, Correale C, Gandelli A, Spinelli A, Dejana E, D’Alessio S, et al. Vascular endothelial growth factor C disrupts the endothelial lymphatic barrier to promote colorectal cancer invasion. Gastroenterology. (2015) 148:1438–51.e8. doi: 10.1053/j.gastro.2015.03.005
205. Ao Z, Shah SH, Machlin LM, Parajuli R, Miller PC, Rawal S, et al. Identification of cancer-associated fibroblasts in circulating blood from patients with metastatic breast cancer. Cancer Res. (2015) 75:4681–7. doi: 10.1158/0008-5472.CAN-15-1633
206. Hurtado P, Martínez-Pena I, Yepes-Rodríguez S, Bascoy-Otero M, Abuín C, Fernández-Santiago C, et al. Modelling metastasis in zebrafish unveils regulatory interactions of cancer-associated fibroblasts with circulating tumour cells. Front Cell Dev Biol. (2023) 11:1076432. doi: 10.3389/fcell.2023.1076432
207. Peinado H, Zhang H, Matei IR, Costa-Silva B, Hoshino A, Rodrigues G, et al. Pre-metastatic niches: Organ-specific homes for metastases. Nat Rev Cancer. (2017) 17:302–17. doi: 10.1038/nrc.2017.6
208. Zeng H, Hou Y, Zhou X, Lang L, Luo H, Sun Y, et al. Cancer-associated fibroblasts facilitate premetastatic niche formation through lncRNA SNHG5-mediated angiogenesis and vascular permeability in breast cancer. Theranostics. (2022) 12:7351–70. doi: 10.7150/thno.74753
209. Dong G, Chen P, Xu Y, Liu T, Yin R. Cancer-associated fibroblasts: Key criminals of tumor pre-metastatic niche. Cancer Lett. (2023) 566:216234. doi: 10.1016/j.canlet.2023.216234
210. LeBeau AM, Brennen WN, Aggarwal S, Denmeade SR. Targeting the cancer stroma with a fibroblast activation protein-activated promelittin protoxin. Mol Cancer Ther. (2009) 8:1378–86. doi: 10.1158/1535-7163.MCT-08-1170
211. Adams S, Miller GT, Jesson MI, Watanabe T, Jones B, Wallner BP. PT-100, a small molecule dipeptidyl peptidase inhibitor, has potent antitumor effects and augments antibody-mediated cytotoxicity via a novel immune mechanism. Cancer Res. (2004) 64:5471–80. doi: 10.1158/0008-5472.CAN-04-0447
212. Narra K, Mullins SR, Lee H-O, Strzemkowski-Brun B, Magalong K, Christiansen VJ, et al. Phase II trial of single agent Val-boroPro (talabostat) inhibiting fibroblast activation protein in patients with metastatic colorectal cancer. Cancer Biol Ther. (2007) 6:1691–9. doi: 10.4161/cbt.6.11.4874
213. Hofheinz R-D, Al-Batran S-E, Hartmann F, Hartung G, Jäger D, Renner C, et al. Stromal antigen targeting by a humanised monoclonal antibody: an early phase II trial of sibrotuzumab in patients with metastatic colorectal cancer. Onkologie. (2003) 26:44–8. doi: 10.1159/000069863
214. Scott AM, Wiseman G, Welt S, Adjei A, Lee F-T, Hopkins W, et al. A Phase I dose-escalation study of sibrotuzumab in patients with advanced or metastatic fibroblast activation protein-positive cancer. Clin Cancer Res. (2003) 9:1639–47.
215. Huang S, Fang R, Xu J, Qiu S, Zhang H, Du J, et al. Evaluation of the tumor targeting of a FAPα-based doxorubicin prodrug. J Drug Target. (2011) 19:487–96. doi: 10.3109/1061186X.2010.511225
216. Peng X, Zheng J, Liu T, Zhou Z, Song C, Geng Y, et al. Tumor microenvironment heterogeneity, potential therapeutic avenues, and emerging therapies. Curr Cancer Drug Targets. (2024) 24:288–307. doi: 10.2174/1568009623666230712095021
217. Tian H, Wang W, Liang S, Ding J, Hua D. From darkness to light: Targeting CAFs as a new potential strategy for cancer treatment. Int Immunopharmacol. (2024) 143:113482. doi: 10.1016/j.intimp.2024.113482
218. Lee J, Fassnacht M, Nair S, Boczkowski D, Gilboa E. Tumor immunotherapy targeting fibroblast activation protein, a product expressed in tumor-associated fibroblasts. Cancer Res. (2005) 65:11156–63. doi: 10.1158/0008-5472.CAN-05-2805
219. Assouline B, Kahn R, Hodali L, Condiotti R, Engel Y, Elyada E, et al. Senescent cancer-associated fibroblasts in pancreatic adenocarcinoma restrict CD8+ T cell activation and limit responsiveness to immunotherapy in mice. Nat Commun. (2024) 15:6162. doi: 10.1038/s41467-024-50441-7
220. Herrera M, Mezheyeuski A, Villabona L, Corvigno S, Strell C, Klein C, et al. Prognostic interactions between FAP+ Fibroblasts and CD8a+ T cells in colon cancer. Cancers (Basel). (2020) 12:3238. doi: 10.3390/cancers12113238
221. Su J, Desmarais J, Chu C-Q, Zhu J. Potential therapeutic targets of fibrosis in inflammatory rheumatic diseases. Best Pract Res Clin Rheumatol. (2024), 101945. doi: 10.1016/j.berh.2024.101945
222. Ostermann E, Garin-Chesa P, Heider KH, Kalat M, Lamche H, Puri C, et al. Effective immunoconjugate therapy in cancer models targeting a serine protease of tumor fibroblasts. Clin Cancer Res. (2008) 14:4584–92. doi: 10.1158/1078-0432.CCR-07-5211
223. Freedman JD, Duffy MR, Lei-Rossmann J, Muntzer A, Scott EM, Hagel J, et al. An oncolytic virus expressing a T-cell engager simultaneously targets cancer and immunosuppressive stromal cells. Cancer Res. (2018) 78:6852–65. doi: 10.1158/0008-5472.CAN-18-1750
224. Wang L-CS, Lo A, Scholler J, Sun J, Majumdar RS, Kapoor V, et al. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol Res. (2014) 2:154–66. doi: 10.1158/2326-6066.CIR-13-0027
225. Mao Y, Yao C, Zhang S, Zeng Q, Wang J, Sheng C, et al. Targeting fibroblast activation protein with chimeric antigen receptor macrophages. Biochem Pharmacol. (2024) 230:116604. doi: 10.1016/j.bcp.2024.116604
226. Allen D, Szoo MJ, van Bergen TD, Seppelin A, Oh J, Saad MA. Near-infrared Photoimmunotherapy: mechanisms, applications, and future perspectives in cancer research. Antib Ther. (2025), tbaf001. doi: 10.1093/abt/tbaf001
227. Nakajima K, Ogawa M. Near-infrared photoimmunotherapy and anti-cancer immunity. Int Immunol. (2024) 36:57–64. doi: 10.1093/intimm/dxad042
228. Wen Y-F, Huang W-J, Chen X-L, Cai H-T, Zhang Y-B, Song X-L, et al. Predictive value of CXCL1+ _FAP+ phenotype in CAFs for distant metastasis and its correlation with PD-L1 expression in locoregionally advanced nasopharyngeal carcinoma patients. Oral Oncol. (2024) 157:106963. doi: 10.1016/j.oraloncology.2024.106963
229. Łabędź N, Anisiewicz A, Stachowicz-Suhs M, Banach J, Kłopotowska D, Maciejczyk A, et al. Dual effect of vitamin D3 on breast cancer-associated fibroblasts. BMC Cancer. (2024) 24:209. doi: 10.1186/s12885-024-11961-z
230. Owaki T, Iida T, Miyai Y, Kato K, Hase T, Ishii M, et al. Synthetic retinoid-mediated preconditioning of cancer-associated fibroblasts and macrophages improves cancer response to immune checkpoint blockade. Br J Cancer. (2024), 1–15. doi: 10.1038/s41416-024-02734-3
231. Choi B, Zhou Y, Kim S, Mumenthaler SM. Abstract C031: Targeting colorectal cancer-associated fibroblasts with all-trans retinoic acid to reprogram the tumor microenvironment. Cancer Res. (2024) 84:C031–1. doi: 10.1158/1538-7445.TUMBODY-C031
232. Yamashita K, Kumamoto Y. CAFs-associated genes (CAFGs) in pancreatic ductal adenocarcinoma (PDAC) and novel therapeutic strategy. Int J Mol Sci. (2024) 25:6003. doi: 10.3390/ijms25116003
233. Kluz N, Kraj L, Chmiel P, Przybyłkowski AM, Wyrwicz L, Stec R, et al. Correlation between antihypertensive drugs and survival among patients with pancreatic ductal adenocarcinoma. Cancers (Basel). (2024) 16:3945. doi: 10.3390/cancers16233945
234. Jain RK. Abstract SY33-01: Improving immunotherapy of cancer by normalizing tumor vessels. Cancer Res. (2019) 79:SY33–01-SY33-01. doi: 10.1158/1538-7445.AM2019-SY33-01
235. Chauhan VP, Martin JD, Liu H, Lacorre DA, Jain SR, Kozin SV, et al. Angiotensin inhibition enhances drug delivery and potentiates chemotherapy by decompressing tumour blood vessels. Nat Commun. (2013) 4:2516. doi: 10.1038/ncomms3516
236. Sheybani ND, Price RJ. Perspectives on recent progress in focused ultrasound immunotherapy. Theranostics. (2019) 9:7749–58. doi: 10.7150/thno.37131
237. van den Bijgaart RJE, Eikelenboom DC, Hoogenboom M, Fütterer JJ, den Brok MH, Adema GJ. Thermal and mechanical high-intensity focused ultrasound: perspectives on tumor ablation, immune effects and combination strategies. Cancer Immunol Immunother. (2017) 66:247–58. doi: 10.1007/s00262-016-1891-9
238. Morishita K, Karasuno H, Yokoi Y, Morozumi K, Ogihara H, Ito T, et al. Effects of therapeutic ultrasound on intramuscular blood circulation and oxygen dynamics. . J Japanese Phys Ther Assoc. (2014) 17:1–7. doi: 10.1298/jjpta.Vol17_001
239. Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. (2018) 554:544–8. doi: 10.1038/nature25501
240. Tauriello DVF, Palomo-Ponce S, Stork D, Berenguer-Llergo A, Badia-Ramentol J, Iglesias M, et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature. (2018) 554:538–43. doi: 10.1038/nature25492
241. Lian S-L, Lu Y-T, Lu Y-J, Yao Y-L, Wang X-L, Jiang R-Q. Tumor-associated macrophages promoting PD-L1 expression in infiltrating B cells through the CXCL12/CXCR4 axis in human hepatocellular carcinoma. Am J Cancer Res. (2024) 14:832. doi: 10.62347/ZIAX8828
242. Garg P, Jallepalli VR, Verma S. Unravelling the CXCL12/CXCR4 Axis in breast cancer: Insights into metastasis, microenvironment interactions, and therapeutic opportunities. Hum Gene. (2024), 201272. doi: 10.1016/j.humgen.2024.201272
243. Tang L, Chen X, Hou J, Wei X. CXCL14 in prostate cancer: complex interactions in the tumor microenvironment and future prospects. J Transl Med. (2025) 23:9. doi: 10.1186/s12967-024-06022-9
244. Yao Z, Qi C, Zhang F, Yao H, Wang C, Cao X, et al. Hollow Cu2MoS4 nanoparticles loaded with immune checkpoint inhibitors reshape the tumor microenvironment to enhance immunotherapy for pancreatic cancer. Acta Biomater. (2024) 173:365–77. doi: 10.1016/j.actbio.2023.10.024
245. Mancarella S, Gigante I, Pizzuto E, Serino G, Terzi A, Dituri F, et al. Targeting cancer-associated fibroblasts/tumor cells cross-talk inhibits intrahepatic cholangiocarcinoma progression via cell-cycle arrest. J Exp Clin Cancer Res. (2024) 43:286. doi: 10.1186/s13046-024-03210-9
246. Fang X, Chen D, Yang X, Cao X, Cheng Q, Liu K, et al. Cancer associated fibroblasts-derived SULF1 promotes gastric cancer metastasis and CDDP resistance through the TGFBR3-mediated TGF-β signaling pathway. Cell Death Discovery. (2024) 10:111. doi: 10.1038/s41420-024-01882-y
247. Pang N, Yang Z, Zhang W, Du Y, Zhang L, Li X, et al. Cancer-associated fibroblasts barrier breaking via TGF-β blockade paved way for docetaxel micelles delivery to treat pancreatic cancer. Int J Pharm. (2024) 665:124706. doi: 10.1016/j.ijpharm.2024.124706
248. Agudo J, Aguirre-Ghiso JA, Bhatia M, Chodosh LA, Correia AL, Klein CA. Targeting cancer cell dormancy. Nat Rev Cancer. (2024) 24:97–104. doi: 10.1038/s41568-023-00642-x
249. Liu Y, Zhang X, Gu W, Su H, Wang X, Wang X, et al. Unlocking the crucial role of cancer-associated fibroblasts in tumor metastasis: mechanisms and therapeutic prospects. J Adv Res. (2024). doi: 10.1016/j.jare.2024.05.031
250. Ghajar CM. Metastasis prevention by targeting the dormant niche. Nat Rev Cancer. (2015) 15:238–47. doi: 10.1038/nrc3910
251. Ma Y-Y, Tao H-Q. Role of urokinase plasminogen activator receptor in gastric cancer: A potential therapeutic target. Cancer Biother Radiopharm. (2012) 27:285–90. doi: 10.1089/cbr.2012.1232
252. Zhai B-T, Tian H, Sun J, Zou J-B, Zhang X-F, Cheng J-X, et al. Urokinase-type plasminogen activator receptor (uPAR) as a therapeutic target in cancer. J Transl Med. (2022) 20:135. doi: 10.1186/s12967-022-03329-3
253. Bauer TW, Liu W, Fan F, Camp ER, Yang A, Somcio RJ, et al. Targeting of urokinase plasminogen activator receptor in human pancreatic carcinoma cells inhibits c-Met- and insulin-like growth factor-I receptor-mediated migration and invasion and orthotopic tumor growth in mice. Cancer Res. (2005). doi: 10.1158/0008-5472.CAN-05-0946
254. Qin L, Wang L, Zhang J, Zhou H, Yang Z, Wang Y, et al. Therapeutic strategies targeting uPAR potentiate anti–PD-1 efficacy in diffuse-type gastric cancer. Sci Adv. (2022) 8:eabn3774. doi: 10.1126/sciadv.abn3774
255. Heinemann V, Ebert MP, Laubender RP, Bevan P, Mala C, Boeck S. Phase II randomised proof-of-concept study of the urokinase inhibitor upamostat (WX-671) in combination with gemcitabine compared with gemcitabine alone in patients with non-resectabl e, locally advanced pancreatic cancer. Br J Cancer. (2013) 108:766–70. doi: 10.1038/bjc.2013.62
256. Sabbadini F, Bertolini M, De Matteis S, Mangiameli D, Contarelli S, Pietrobono S, et al. The multifaceted role of TGF-β in gastrointestinal tumors. Cancers (Basel). (2021) 13:3960. doi: 10.3390/cancers13163960
257. Martin CJ, Datta A, Littlefield C, Kalra A, Chapron C, Wawersik S, et al. Selective inhibition of TGFβ1 activation overcomes primary resistance to checkpoint blockade therapy by altering tumor immune landscape. Sci Transl Med. (2020) 12. doi: 10.1126/scitranslmed.aay8456
258. Yin L. Combination therapy of bevacizumab and galunisertib extends TVN time window. Mol Ther Oncol. (2024) 32. doi: 10.1016/j.omton.2024.200888
259. Yu J, Du X, Zhang S, Long J, Wu P, Li Z, et al. Galunisertib promotes bevacizumab-induced vascular normalization in nasopharyngeal carcinoma: Multi-parameter MRI evaluation. Mol Ther Oncol. (2024) 32. doi: 10.1016/j.omton.2024.200858
260. Sadri S, Aghajani A, Soleimani H, Ghorbani Kalkhajeh S, Nazari H, Brouki Milan P, et al. Exploring the role of the TGF-β Signaling pathway in colorectal precancerous polyps biochemical genetics. Biochem Genet. (2024), 1–33.
261. Lemmens G, Brouwers J, Snoeys J, Augustijns P, Vanuytsel T. Insight into the colonic disposition of sulindac in humans. J Pharm Sci. (2021) 110:259–67. doi: 10.1016/j.xphs.2020.09.034
262. Bowen CM, Walter L, Borras E, Wu W, Ozcan Z, Chang K, et al. Combination of sulindac and bexarotene for prevention of intestinal carcinogenesis in familial adenomatous polyposis. Cancer Prev Res. (2021) 14:851–62. doi: 10.1158/1940-6207.CAPR-20-0496
263. Fadlallah H, El Masri J, Fakhereddine H, Youssef J, Chemaly C, Doughan S, et al. Colorectal cancer: Recent advances in management and treatment. World J Clin Oncol. (2024) 15:1136. doi: 10.5306/wjco.v15.i9.1136
264. Lev-Ari S, Strier L, Kazanov D, Madar-Shapiro L, Dvory-Sobol H, Pinchuk I, et al. Celecoxib and curcumin synergistically inhibit the growth of colorectal cancer cells. Clin Cancer Res. (2005) 11:6738–44. doi: 10.1158/1078-0432.CCR-05-0171
265. Sleeboom JJF, van Tienderen GS, Schenke-Layland K, van der Laan LJW, Khalil AA, Verstegen MMA. The extracellular matrix as hallmark of cancer and metastasis: From biomechanics to therapeutic targets. Sci Transl Med. (2024) 16. doi: 10.1126/scitranslmed.adg3840
266. Almokadem S, Belani CP. Volociximab in cancer. Expert Opin Biol Ther. (2012) 12:251–7. doi: 10.1517/14712598.2012.646985
267. Liu M, Wang J, Liu M. Lysyl oxidase inhibitors in colorectal cancer progression. Transl Oncol. (2025) 52:102233. doi: 10.1016/j.tranon.2024.102233
268. Pericàs JM, Anstee QM, Augustin S, Bataller R, Berzigotti A, Ciudin A, et al. A roadmap for clinical trials in MASH-related compensated cirrhosis. Nat Rev Gastroenterol Hepatol. (2024) 21:809–23. doi: 10.1038/s41575-024-00955-8
269. Kreatsoulas D, Damante M, Cua S, Lonser RR. Adjuvant convection-enhanced delivery for the treatment of brain tumors. J Neurooncol. (2024) 166:243–55. doi: 10.1007/s11060-023-04552-8
270. Aghajani M, Jalilzadeh N, Aghebati-Maleki A, Yari A, Tabnak P, Mardi A, et al. Current approaches in glioblastoma multiforme immunotherapy. Clin Transl Oncol. (2024) 26:1584–1612. doi: 10.1007/s12094-024-03395-7
271. Dunbar KJ, Efe G, Cunningham K, Esquea E, Navaridas R, Rustgi AK. Regulation of metastatic organotropism. Trends Cancer. (2025) 11:216–31. doi: 10.1016/j.trecan.2024.11.012
272. Klempner SJ, Bendell JC, Villaflor VM, Tenner LL, Stein SM, Rottman JB, et al. Safety, efficacy, and biomarker results from a phase Ib study of the anti-DKK1 antibody DKN-01 in combination with pembrolizumab in advanced esophagogastric cancers. Mol Cancer Ther. (2021) 20:2240–9. doi: 10.1158/1535-7163.MCT-21-0273
273. Wall JA, Klempner SJ, Arend RC. The anti-DKK1 antibody DKN-01 as an immunomodulatory combination partner for the treatment of cancer. Expert Opin Investig Drugs. (2020) 29:639–44. doi: 10.1080/13543784.2020.1769065
274. Klempner SJ, Chao J, Uronis HE, Sirard CA, Kagey M, Baum J, et al. DKN-01 and tislelizumab ± chemotherapy as a first-line (1L) and second-line (2L) investigational therapy in advanced gastroesophageal adenocarcinoma (GEA): DisTinGuish Trial. J Clin Oncol. (2022) 40:292–2. doi: 10.1200/JCO.2022.40.4_suppl.292
275. Chen J, Duan Y, Che J, Zhu J. Dysfunction of dendritic cells in tumor microenvironment and immunotherapy. Cancer Commun. (2024) 44:1047–70. doi: 10.1002/cac2.12596
276. Gao H, Chakraborty G, Lee-Lim AP, Mo Q, Decker M, Vonica A, et al. The BMP inhibitor coco reactivates breast cancer cells at lung metastatic sites. Cell. (2012) 150:764–79. doi: 10.1016/j.cell.2012.06.035
277. Senft D, Ronai ZA. Immunogenic, cellular, and angiogenic drivers of tumor dormancy-a melanoma view. Pigment Cell Melanoma Res. (2016) 29:27–42. doi: 10.1111/pcmr.12432
278. Huang D, Li Q, Wang Y, Liu Z, Wang Z, Li H, et al. Brain-specific NRSF deficiency aggravates dopaminergic neurodegeneration and impairs neurogenesis in the MPTP mouse model of Parkinson’s disease. Aging (Albany NY). (2019) 11:3280–97. doi: 10.18632/aging.101979
279. Konieczkowski DJ, Johannessen CM, Garraway LA. A convergence-based framework for cancer drug resistance. Cancer Cell. (2018) 33:801–15. doi: 10.1016/j.ccell.2018.03.025
280. Lu Y, Jin Z, Hou J, Wu X, Yu Z, Yao L, et al. Calponin 1 increases cancer-associated fibroblasts-mediated matrix stiffness to promote chemoresistance in gastric cancer. Matrix Biol. (2023) 115:1–15. doi: 10.1016/j.matbio.2022.11.005
281. Li I, Nabet BY. Exosomes in the tumor microenvironment as mediators of cancer therapy resistance. Mol Cancer. (2019) 18:32. doi: 10.1186/s12943-019-0975-5
282. Uchihara T, Miyake K, Yonemura A, Komohara Y, Itoyama R, Koiwa M, et al. Extracellular vesicles from cancer-associated fibroblasts containing annexin A6 induces FAK-YAP activation by stabilizing b1 integrin, enhancing drug resistance. Cancer Res. (2020) 80:3222–35. doi: 10.1158/0008-5472.CAN-19-3803
283. Sansone P, Savini C, Kurelac I, Chang Q, Amato LB, Strillacci A, et al. Packaging and transfer of mitochondrial DNA via exosomes regulate escape from dormancy in hormonal therapy-resistant breast cancer. Proc Natl Acad Sci U.S.A. (2017) 114:E9066–75. doi: 10.1073/pnas.1704862114
284. Chen X, Zhao J, Herjan T, Hong L, Liao Y, Liu C, et al. IL-17–induced HIF1α drives resistance to anti–PD-L1 via fibroblast-mediated immune exclusion. J Exp Med. (2022) 219. doi: 10.1084/jem.20210693
285. Nicolas AM, Pesic M, Engel E, Ziegler PK, Diefenhardt M, Kennel KB, et al. Inflammatory fibroblasts mediate resistance to neoadjuvant therapy in rectal cancer. Cancer Cell. (2022) 40:168–184.e13. doi: 10.1016/j.ccell.2022.01.004
286. Lee CM, Hwang Y, Jeong JW, Kim M, Lee J, Bae SJ, et al. BRCA1 mutation promotes sprouting angiogenesis in inflammatory cancer-associated fibroblast of triple-negative breast cancer. Cell Death Discovery. (2024) 10:5. doi: 10.1038/s41420-023-01768-5
287. Yoon H, Tang C-M, Banerjee S, Delgado AL, Yebra M, Davis J, et al. TGF-β1-mediated transition of resident fibroblasts to cancer-associated fibroblasts promotes cancer metastasis in gastrointestinal stromal tumor. Oncogenesis. (2021) 10:13. doi: 10.1038/s41389-021-00302-5
288. Coursier D, Calvo F. CAFs vs. TECs: when blood feuds fuel cancer progression, dissemination and therapeutic resistance. Cell Oncol. (2024) 47:1091–112. doi: 10.1007/s13402-024-00931-z
289. Hisano K, Mizuuchi Y, Ohuchida K, Kawata J, Torata N, Zhang J, et al. Microenvironmental changes in familial adenomatous polyposis during colorectal cancer carcinogenesis. Cancer Lett. (2024) 589:216822. doi: 10.1016/j.canlet.2024.216822
290. Song J, Wei R, Liu C, Zhao Z, Liu X, Wang Y, et al. Antigen-presenting cancer associated fibroblasts enhance antitumor immunity and predict immunotherapy response. Nat Commun. (2025) 16:2175. doi: 10.1038/s41467-025-57465-7
291. Strating E, Verhagen MP, Wensink E, Dünnebach E, Wijler L, Aranguren I, et al. Co-cultures of colon cancer cells and cancer-associated fibroblasts recapitulate the aggressive features of mesenchymal-like colon cancer. Front Immunol. (2023) 14:1053920. doi: 10.3389/fimmu.2023.1053920
292. Rizzolio S, Giordano S, Corso S. The importance of being CAFs (in cancer resistance to targeted therapies). J Exp Clin Cancer Res. (2022) 41:319. doi: 10.1186/s13046-022-02524-w
293. Wang Z, Yan N, Sheng H, Xiao Y, Sun J, Cao C. Single-cell transcriptomic analysis reveals an immunosuppressive network between POSTN CAFs and ACKR1 ECs in TKI-resistant lung cancer. Cancer Genomics Proteomics. (2024) 21:65–78. doi: 10.21873/cgp.20430
294. Li J, Bai L, Cao J, Zhi W, Cheng Q. Longitudinal photoacoustic monitoring of collagen evolution modulated by cancer-associated fibroblasts: simulation and experiment studies. In: 2024 IEEE ultrasonics, ferroelectrics, and frequency control joint symposium (UFFC-JS). IEEE (2024). p. 1–4.
Keywords: cancer-associated fibroblasts, tumor microenvironment, therapeutic resistance, biomarkers, immune modulation, targeted therapies
Citation: Lan X, Li W, Zhao K, Wang J, Li S and Zhao H (2025) Revisiting the role of cancer-associated fibroblasts in tumor microenvironment. Front. Immunol. 16:1582532. doi: 10.3389/fimmu.2025.1582532
Received: 24 February 2025; Accepted: 31 March 2025;
Published: 17 April 2025.
Edited by:
Mohanraj Sadasivam, The University of Iowa, United StatesReviewed by:
Mrinmoy Sarkar, Texas A&M University College Station, United StatesGopinath Prakasam, University of Texas Southwestern Medical Center, United States
Copyright © 2025 Lan, Li, Zhao, Wang, Li and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hai Zhao, eWlkYW9AcWR1LmVkdS5jbg==