 Noha M. Elemam
Noha M. Elemam Iman M. Talaat
Iman M. Talaat Omar A. El Meligy
Omar A. El Meligy- 1Clinical Sciences Department, College of Medicine, University of Sharjah, Sharjah, United Arab Emirates
- 2Research Institute of Medical & Health Sciences, University of Sharjah, Sharjah, United Arab Emirates
- 3Pathology Department, Faculty of Medicine, Alexandria University, Alexandria, Egypt
- 4Pediatric Dentistry and Dental Public Health Department, Faculty of Dentistry, Alexandria University, Alexandria, Egypt
Systemic Lupus Erythematosus (SLE) is a multifaceted autoimmune disorder characterized by widespread inflammation and immune dysregulation, impacting various organ systems and generating autoantibodies. Oral lesions are a common and distressing manifestation of SLE, significantly affecting patients’ quality of life. Cytokines, key mediators of immune responses, play a crucial role in the pathogenesis of both systemic and oral manifestations of SLE. This review sheds the light on current research on the involvement of various cytokines, including interleukins different interferon types, and growth factors in SLE. The intricate interplay between pro-inflammatory and anti-inflammatory cytokines contributes to the disease’s initiation, progression, and diverse clinical presentations. Elevated levels of pro-inflammatory cytokines exacerbate inflammation, promote apoptosis, and drive autoantibody production. Understanding the specific roles of these cytokines offers potential therapeutic targets for managing SLE and improving patient outcomes.
1 Introduction
Systemic Lupus Erythematosus (SLE) is a complex autoimmune disease that affects multiple organ systems, including the skin, kidneys, and joints. The systemic nature of SLE arises from widespread immune system dysfunction, leading to autoimmunity against nucleic acids and their associated proteins, along with tissue-damaging inflammation. Among its numerous manifestations, oral lesions are a significant concern for patients, often resulting in pain, discomfort, and diminished quality of life. The pathogenesis of these oral manifestations is multifactorial, with cytokines playing a pivotal role. Recent literature has increasingly highlighted the importance of cytokines in the development and progression of oral lesions in SLE (1). This review aims to synthesize current findings on the role of cytokines in SLE’s systemic and oral manifestations and explore their therapeutic implications. Although genetic susceptibility and environmental factors, such as microbial infections, create a predisposition for SLE, specific triggers of immune activation are necessary to initiate the production of type 1 interferons and the generation of autoantibodies targeting self-antigens (2).
In SLE patients, elevated levels of various cytokines were detected throughout the disease course, appearing in circulation, saliva, urine, and affected tissues such as the skin, kidneys, and synovia (3). While most of these cytokines exhibit pro-inflammatory properties, some play immunomodulatory or anti-inflammatory roles (4).
Notably, multiple immunological abnormalities play a role in the breakdown of self-tolerance and the persistence of autoimmune responses in SLE. In particular, defects in apoptotic cell clearance—such as impaired phagocytosis and complement activation—lead to the accumulation of self-antigens. Dysregulated innate immune responses further drive adaptive immune activation, intensifying the inflammatory cascade (5). For instance, plasmacytoid dendritic cells (pDCs), myeloid dendritic cells (mDCs), monocytes and macrophages are key contributors to the inflammatory cascade in SLE through their enhanced antigen-presenting capacity and increased production of proinflammatory cytokines such as type I interferons and TNF-α. These cells exhibit an activated phenotype in SLE patients, promoting T cell activation and perpetuating autoimmunity (6–10). Additionally, abnormalities in B cell development, activation, and differentiation promote the production of autoreactive antibodies, which target self-antigens and form immune complexes that contribute to tissue damage and inflammation. Consistently, disturbances in T cell compartments, dysfunctional regulatory T cells (Tregs), and altered cytokine production further exacerbate tissue damage in this disease. B cells are central to SLE pathogenesis as the overexpression of B lymphocyte stimulator (BLyS) significantly contributes to disease progression (Figure 1). BLyS enhances B-cell survival by inhibiting apoptosis and promoting proliferation and differentiation. This process ultimately leads to increased autoantibody production, a hallmark of SLE (11).
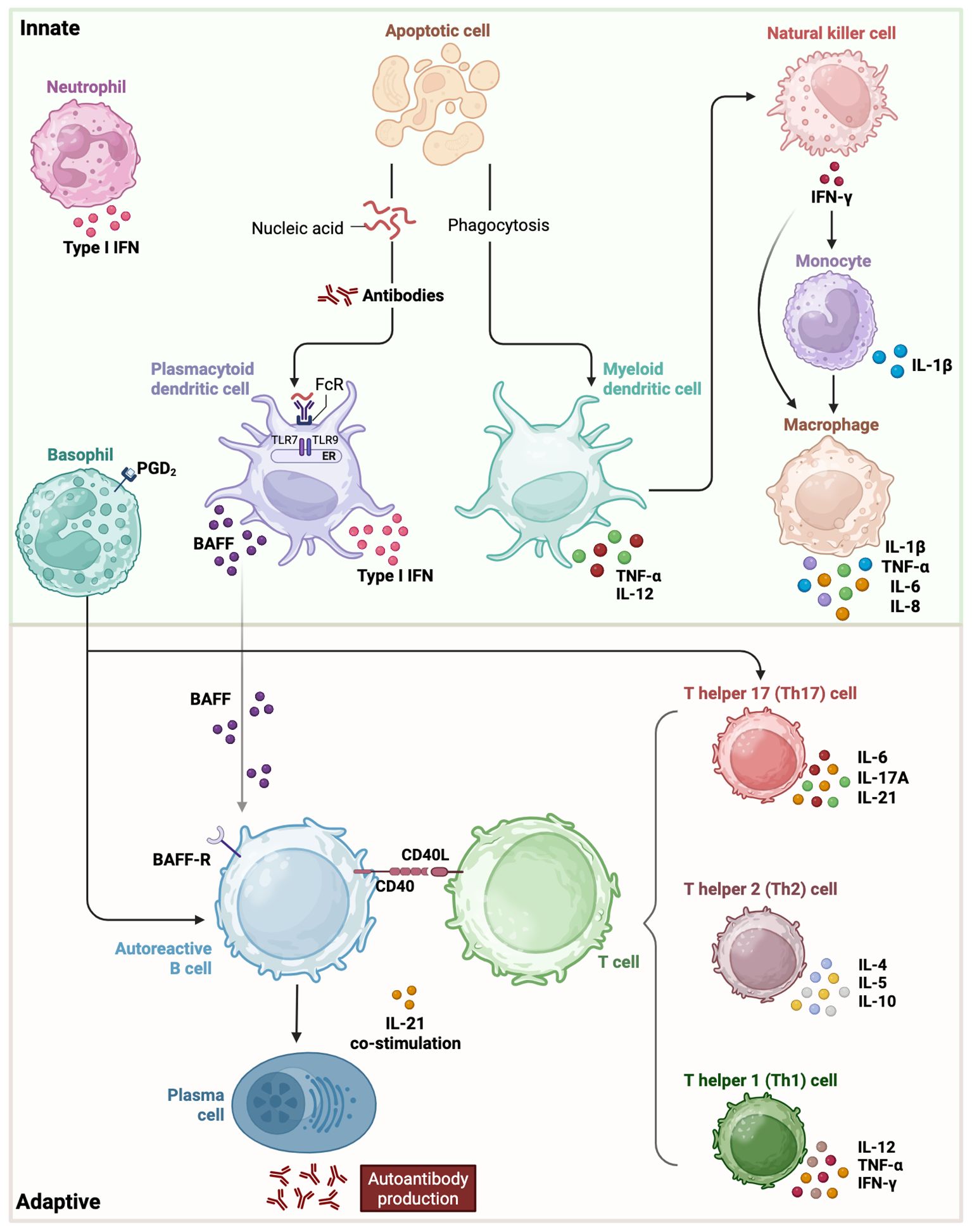
Figure 1. Innate and adaptive immune players with their respective cytokine involvement in SLE pathogenesis. In the innate phase, type I interferons, cell apoptosis and other early signaling pathways initiate activation of basophils, neutrophils, monocytes, macrophages, myeloid and plasmacytoid dendritic cells. The adaptive phase involves an imbalance among T helper cell subsets, their interaction with B cells, perpetuating autoantibody production and tissue damage. BAFF, B-cell-activating factor of the tumor-necrosis-factor family; IFN, interferon; IL, interleukin; PGD2, prostaglandin D2; TNF, tumor necrosis factor.
2 Cytokines involvement in the pathophysiology of lupus erythematosus
SLE is characterized by an aberrant immune system that generates autoantibodies and induces widespread inflammation, affecting various tissues. As shown in Figure 1, this immune dysregulation involves the activation of both innate and adaptive immune systems including B and T cells, leading to the formation of immune complexes, and the production of pro-inflammatory cytokines (12, 13). Excessive or insufficient cytokine production can contribute to the pathogenesis of several diseases (14). An imbalance between pro-inflammatory and anti-inflammatory cytokines is a well-recognized feature of SLE. Numerous studies suggest that cytokine levels play a role in SLE, with an elevation of pro-inflammatory cytokines, potentially exacerbating inflammation, promoting apoptosis, and driving autoantibody production, thus contributing to disease initiation and progression (15, 16). These cytokines contribute to the diverse clinical manifestations of lupus, including oral lesions and systemic features. Interleukins, including IL-1, IL-6, IL-10, IL-17, along with interferon-alpha (IFN-α) and tumor necrosis factor-alpha (TNF-α), are key cytokines that serve as biomarkers for assessing disease activity and severity. Therefore, modulating these cytokines could be a therapeutic approach towards the management of SLE (17, 18).
2.1 Interferons
2.1.1 Type I interferons
Type I IFNs, particularly IFN-α, are the predominant cytokines of this category and have been extensively studied in the context of SLE (19, 20). Also, IFN-κ signaling via the type I interferon receptor modulates the expression and release of cytokines from monocytes (21, 22). All type I IFNs transmit signals through the same receptor complex, composed of IFNAR1 and IFNAR2 (23). Elevated IFN activity has been associated with greater disease severity in SLE patients (24–26). Circulating IFN-α levels have been shown to correlate with disease activity, as measured by the SLEDAI score, as well as with anti-dsDNA antibody titers and complement activity. This suggests that IFN-α quantification could serve as a potential biomarker for disease monitoring (4). Specific autoantibody profiles, including anti-dsDNA, anti-RNP, anti-Sm, and anti-Ro, have been linked to heightened IFN activity (26–28). Multiple studies have demonstrated that between 50% and 75% of adults and up to 90% of children with SLE exhibit heightened expression of type I IFN-regulated genes, a phenomenon known as the IFN signature (29–32). Notably, younger patients display more pronounced IFN activity compared to older individuals, and SLE disease activity has been found to correlate with IFN-α levels and the intensity of the IFN signature (30, 33–35).
In patients with SLE, pDCs are reduced in circulation but can be identified in inflamed tissues such as the skin and kidneys, where they appear to be activated (36–38). These cells play a central role in sustaining IFN production in SLE. Hence, targeting pDCs in SLE patients was found to reduce the expression of IFN response genes in the blood, decrease immune cell infiltration in the skin, and ameliorate cutaneous lesions (39). This approach was done using the monoclonal antibody targeting blood DC antigen 2 (BDCA2), BIIB059, a known pDC-specific receptor that inhibits the production of type I IFNs (39). However, recent research indicated that BDCA2 is not exclusive to pDCs and is also expressed on differentiated monocytes. Consequently, BIIB059 may exert effects on broader myeloid populations, which could have an impact on the safety and expanded therapeutic applications in SLE (40, 41).
On the other hand, other studies demonstrated that pDCs in both preclinical autoimmunity and established SLE are functionally impaired, showing signs of stress, senescence, and reduced cytokine and T cell activation capacity. Instead, non-hematopoietic cells, particularly keratinocytes producing interferon-κ, could drive early interferon responses in the skin, implicating them as key sources of IFN prior to clinical disease onset (42). Also, another study on discoid lupus and cutaneous lupus erythematosus (CLE) identified that pDCs are not major producers of type I interferons, as they exhibit markedly reduced IFN-α production upon toll like receptor 7 (TLR7) stimulation (43).
Neutrophils also possess the capacity to produce type I IFN, and bone marrow-derived neutrophils from SLE patients have been shown to generate IFN-α (44, 45). This IFN production appears to drive alterations in B-cell development, leading to a reduction in pro/pre-B cells while expanding transitional B-cell populations. Such changes may represent early events in the disruption of immune tolerance and the initiation of autoimmunity, culminating in autoantibody production. A study by Klein B et al. identified Z-DNA binding protein 1 (ZBP1) as a key mediator of UVB-induced inflammation in autoimmune photosensitivity by stabilizing cytosolic Z-DNA derived from oxidized mitochondrial DNA. Moreover, ZBP1 was found to be elevated in lupus keratinocytes, where it enhances type I IFN production via cGAS-STING signaling, highlighting its central role in priming cutaneous inflammation (46).
Besides, IFN-κ is constitutively and highly expressed in keratinocytes (21). In patients with SLE, keratinocytes contribute to skin injury pathogenesis by undergoing apoptosis or necrosis, leading to the release of autoantigens (47). IFN-κ was previously investigated in SLE where it was found to be elevated in the skin lesions of cutaneous lupus erythematosus (48). Further, IFN-κ was found to amplify and accelerate responsiveness of epithelia to IFN-α and increase keratinocyte sensitivity to UV irradiation (48). This indicated that IFN-κ could be a potential novel target for ultraviolet B prophylaxis and cutaneous lupus erythematosus-directed therapy (48). The IFN score was identified to be markedly enriched in the skin of SLE patients, even in the absence of clinical inflammation or in ANA positive individuals (42). Furthermore, IFN-κ was diffusely expressed not only in the epidermis of lesional skin from SLE patients, but also in non-lesional epidermis of ANA+ individuals with elevated systemic IFN activity (42). On another note, IFN-κ functions as an interferon-stimulated gene (ISG) that is regulated in an IFN-β–dependent manner. While IFN-β is produced rapidly and transiently in response to stimulation, IFN-κ expression is upregulated later and sustained, suggesting a potential role in maintaining more prolonged or chronic type I IFN responses (49). This could be the explanation to the elevated levels of IFN-κ in autoimmune diseases such as SLE. Furthermore, the study by Xu B. et al. suggests a mechanistic model in keratinocytes whereby IRF3-activating stimuli rapidly induce IFN-β leading to the subsequent upregulation of IFN-κ via IFN-β–mediated STAT1 activation. Further investigation into these pathways may uncover novel therapeutic targets for chronic IFN-driven inflammation in cutaneous lupus (49).
2.1.2 Type II interferons
In contrast, type II IFN consists of a single cytokine, IFN-γ, which is primarily secreted by CD4+ and CD8+ Th1 lymphocytes, as well as by NK cells, B cells, and professional antigen-presenting cells. IFN-γ signaling occurs through a receptor complex consisting of IFNGR1 and IFNGR2 subunits (20). Activated NK cells in SLE exhibit increased secretion of IFN-γ, while peripheral blood leukocytes from SLE patients have been found to express detectable levels of IFN-λ transcripts, although the precise cellular source remains uncertain (50). A crucial observation is that several activated immune cell types can further stimulate pDCs to enhance IFN production. Notably, NK cells, B cells, and T cells have all been shown to amplify IFN secretion when pDCs are exposed to nucleic acid–containing immune complexes (51, 52). Interleukin-12, alongside IFN-γ, drives T-cell differentiation toward a Th1 immune response. SLE patients exhibited an increase in the circulating levels of the IL-12p40 subunit, which correlates positively with disease activity and inversely with complement C3 levels (53, 54).
2.1.3 Type III interferons
In established SLE, there is a production of type III IFNs (IFN-λ) in the skin. This type of IFNs engages signaling pathways and elicit cellular effects similar to type I interferons (55). Notably, studies using the SLE animal model (MRL/lpr), reported that deletion of the IFN-λ receptor leads to reduced skin inflammation, highlighting its functional relevance (56). Additionally, loss of IFNB1 (gene for IFN-β1) in keratinocytes significantly diminished IFNL1 (gene for IFN-Λ1) expression, mirroring the pattern seen with IFN-κ. In contrast, IFNL3 remained strongly and rapidly induced even in the absence of IFNB. These findings underscore the complex interplay between type I and type III interferons in the epidermis, warranting further investigation.
2.2 Growth factors
Increasing evidence suggests that TNF-α plays a complex role in the pathogenesis of SLE (57). This cytokine appears to have dual and sometimes opposing functions, making its role in the disease a subject of ongoing debate. On one hand, TNF-α contributes to immune regulation by supporting the development, differentiation, and maintenance of immune cells, acting as a crucial component of immunosuppressive mechanisms. On the other hand, it can serve as a potent proinflammatory agent, being released in affected tissues during active disease and potentially exacerbating inflammation (58–60).
Also, studies have shown that individuals with active SLE tend to exhibit elevated serum levels of TNF-α and its soluble receptors (TNFR1 and THFR2) compared to those with inactive disease (57, 61–63). A study by Zhu et al. revealed that SLE patients exhibited significantly reduced expression of key TNF adaptor proteins including TNF receptor-associated death domain (TRADD), Fas-associated death domain (FADD), TRAF2, and receptor-interacting protein kinase 1 (RIPK1) in their peripheral blood mononuclear cells. Furthermore, these diminished protein levels were found to correlate inversely with disease activity, suggesting a potential link between impaired TNF signaling and SLE progression (64). However, conflicting findings suggest that TNF-α levels may be lower in certain SLE patients, particularly those experiencing severe disease manifestations. Interestingly, some research indicates that TNF-α concentrations are higher in individuals with inactive SLE than in both those with highly active disease and healthy controls (57, 58, 65). This paradoxical pattern has led to speculation that TNF-α may also play a protective role in certain contexts, potentially contributing to immune regulation in SLE.
Both the transmembrane and soluble form of TNF interact with the two known receptors of TNF, TNF receptor 1 (TNFR1), and TNFR2 (66). They play distinct yet complementary roles in immune regulation and inflammation (67). TNFR1 (p55), is ubiquitously expressed on most cell types and mediates the majority of pro-inflammatory and apoptotic effects of TNF-α through activation of NF-κB, MAPK, and caspase pathways. In contrast, TNFR2 (p75) is primarily expressed on immune cells, including Tregs, endothelial cells, and certain neuronal populations, and is associated with tissue regeneration, immune modulation, and cell survival via alternative NF-κB and PI3K/Akt signaling pathways (68, 69). Overactivation of TNFR1 contributes to heightened inflammation, apoptosis of key immune and tissue cells, and amplification of the autoimmune response. Meanwhile, dysregulation or insufficient activation of TNFR2 may impair Treg function and tissue repair mechanisms, exacerbating disease progression in SLE (70). Emerging studies suggest that selective modulation of TNFR2 could restore immune tolerance without triggering widespread inflammation, offering a promising therapeutic strategy for SLE and other autoimmune diseases (71).
CD40, a transmembrane receptor of the TNF family, is broadly expressed on immune and non-immune cells and plays a pivotal role in regulating both humoral and cellular immunity through its interaction with CD40 ligand (CD40L) (72–74). This pathway is essential for dendritic cell/T-cell and T-cell/B-cell communication, promoting B cell proliferation, differentiation, and immunoglobulin production (73, 75). In SLE, CD40 signaling contributes to disease pathology by as its activation upregulates CD40L, which in turn further engages CD40 on B cells (76). Additionally, CD40-induced telomerase activity supports long-lived B cell memory, perpetuating chronic inflammation and autoantibody generation (77).
2.3 Interleukins
2.3.1 Interleukins belonging to IL-1 family
The cytokine interleukin-1 superfamily comprises IL-1α, IL-1β, IL-18, IL-33, and IL-38, all of which contribute to innate immunity and are regulated by inflammasome activation as an early pathogen response. Some studies reported that peripheral IL-1α levels in SLE patients were comparable to those of controls, while other studies indicated elevated IL-1α levels in individuals with renal and joint manifestations in SLE (78). Additionally, increased tissue IL-1β levels have been observed in skin lesions triggered by photoprovocation in CLE, accompanied by heightened TNF-α expression (79). In SLE, opsonized red blood cells retaining mitochondria (Mito+ RBCs) stimulate monocytes to co-produce type I interferons and mature IL-1β. This response is driven by cyclic GMP-AMP synthase (cGAS) and RIG-I-like receptors detecting mitochondrial DNA and RNA from Mito+ RBCs, activating both interferons signaling and inflammasome pathways. Notably, IL-1β secretion occurs through the activation of an IFN-inducible myxovirus-resistant protein 1 (MxA) and trans-Golgi network dependent mechanisms that are independent of gasdermin D or pyroptosis. This sheds the light on a subset of monocytes that express IFN-stimulated genes (ISGs) and released IL-1β and are enriched in patients with active SLE (8).
IL-18 is secreted by macrophages and plays a crucial role in promoting IFN-γ production by Th1 cells and splenocytes, often acting synergistically with IL-12 (80). Elevated serum IL-18 levels are commonly observed in SLE patients, particularly in those with active renal disease who exhibit an increased risk of developing kidney damage over time (81). Additionally, high IL-18 expression has been detected in cutaneous lupus erythematosus (CLE) lesions (82). IL-18 is identified as a potential biomarker for active SLE, whereas blocking IL-18 was found to delay the onset of SLE-like autoimmunity (80, 83).
2.3.2 Interleukins belonging to Type I cytokine family
Elevated plasma IL-6 levels have been detected in patients with renal impairment and are a key mediator in lupus nephritis (78, 84, 85). Urinary IL-6 levels have been proposed as a potential biomarker for the condition (86, 87). Additionally, SLE has been linked to impaired IL-2 production, with IL-2 deficiency associated with renal dysfunction (88). Notably, some SLE patients develop anti-IL-2 autoantibodies, which have been correlated with disease activity (89). IL-21 is an autocrine cytokine primarily produced by follicular helper T (Tfh) cells and Th17 cells, playing a key role in their development. It also facilitates B-cell differentiation into plasma cells through various signaling pathways, including JAK/STAT (90). IL-21 may contribute to the expansion of autoreactive B cells, further driving disease pathology (91). Genetic variations in IL-21 and its receptor (IL-21R) have been linked to increased susceptibility to SLE. There are controversial reports regarding the expression of IL-21 where some studies found elevated levels of IL-21-producing cells in the circulation of SLE patients (92), while others have found reduced circulating IL-21 levels in affected individuals (93).
2.3.3 Interleukins belonging to Type II cytokine family
Despite its anti-inflammatory functions, IL-10 can have pro-inflammatory effects, potentially influenced by type I IFNs (94). The genetic variations in the IL-10 gene are linked to increased SLE susceptibility (95, 96). Plasma IL-10 concentrations were elevated in SLE patients compared to healthy controls, and there was an association between IL-10 levels, disease activity, and anti-dsDNA antibody titers (78).
2.3.4 Interleukins belonging to IL-17 family
Another proinflammatory cytokine is IL-17 which plays a key role in the pathogenesis of autoimmune rheumatic diseases, including SLE (18). Its involvement in SLE development and disease activity is supported by findings that SLE patients exhibit significantly higher circulating IL-17 levels compared to healthy individuals, with its levels correlating positively with disease severity (97). Beyond driving inflammation, IL-17 also contributes to the progression of SLE-related comorbidities (98). The differentiation of naïve T cells into Th17 cells is driven by the combined effects of TGF-β and IL-6, while IL-23 is essential for the maturation and activation of pathogenic Th17 cells. IL-6 plays a crucial role in the differentiation of Th17 cells, and its inhibition may help mitigate SLE activity by indirectly suppressing IL-17-driven inflammation (99). IL-17, either alone or in conjunction with B-cell activating factor (BAFF), regulates B-cell survival, proliferation, and differentiation into immunoglobulin-secreting cells (97). By promoting B-cell expansion, IL-17 enhances autoantibody synthesis, which in turn facilitates immune complex formation, complement activation, and subsequent tissue damage in target organs. Additionally, these immune complexes activate plasmacytoid dendritic cells (pDCs), further fueling inflammation (100). The IL-23/IL-17 axis plays a central role in inflammation, coordinating Th17 cell differentiation and function, thereby perpetuating immune dysregulation in SLE (11). IL-17 interacts with other cytokines such as IL-23, IL-17F, and IL-21 to form a complex proinflammatory network that drives tissue damage and inflammation in SLE. It also amplifies the inflammatory cascade by inducing the release of other cytokines, such as IL-1 and IL-6. IL-21 plays a crucial role in maintaining the balance between Th17 and Treg cells, while IL-10, a member of the IL-2 cytokine family, is linked to chronic inflammation (97, 100).
Higher plasma IL-17 levels and an increased presence of circulating Th17 cells were reported in SLE patients (101–104). Multiple investigations have shown that serum IL-17A levels are markedly higher in patients with active SLE than in healthy individuals (105–107). Furthermore, IL-17 concentrations have been positively correlated with SLE disease activity index (SLEDAI) scores, indicating a direct link between IL-17 expression and disease severity (101, 108, 109). In newly diagnosed SLE patients, IL-17A levels were also found to be strongly associated with RORγt mRNA expression, erythrocyte sedimentation rate, and immunoglobulin levels (IgG and IgA), further emphasizing the crucial role of the Th17–IL-17 axis in SLE pathogenesis (110).
3 Oral manifestations of lupus erythematosus
Oral manifestations of SLE are common and range from non-specific findings such as xerostomia (dry mouth) and ulcers to more specific lesions like oral lichen planus and candidiasis (111). Oral manifestations of SLE can provide valuable clues for diagnosis and management. These manifestations can include oral ulcers, lupus cheilitis, discoid lupus erythematosus (DLE), dry mouth (xerostomia), gingival inflammation, periodontal disease, pale mucosa, oral candidiasis, telangiectasia (dilated blood vessels), and altered taste (dysgeusia) (112).
Aphthous-like ulcers are painful, round, or oval lesions often found on the hard palate, buccal mucosa, and tongue. These are similar to typical aphthous ulcers but can occur in a variety of sizes. Painless or painful ulcerations may appear as shallow, necrotic, and have a raised border (112).
Lupus cheilitis is characterized by red, scaly, or fissured lips. The lower lip is often more affected. This may resemble a form of cheilitis, with dry, cracked, and inflamed lips. DLE may present as localized lesions on the face, scalp, or mucous membranes, including the oral cavity. White, lacy lesions can be seen on the buccal mucosa and palate, and they may be accompanied by erythema or ulceration. These lesions are often referred to as lichen planus-like lesions.
Due to autoimmune damage to salivary glands, SLE can result in decreased saliva production, leading to a sensation of dryness in the mouth, difficulty swallowing, and increased risk of dental caries and oral infections. Gingivitis may be present, characterized by redness, swelling, and bleeding of the gums, often exacerbated by the systemic inflammation associated with SLE. Moreover, SLE patients may be at an increased risk for periodontal disease due to both autoimmune factors and dry mouth (xerostomia), which can lead to a higher incidence of plaque accumulation and bacterial growth. In addition, immunosuppression due to SLE or its treatment (such as corticosteroids) can increase the risk of opportunistic infections like oral thrush (candidiasis). Occasionally, visible dilated blood vessels may appear on the hard and soft palate. Some patients may experience changes in their sense of taste, which can be related to the disease or its treatment. Further, mucosal tissues can appear pale due to anemia, a common complication of SLE, reflecting the decreased red blood cell count.
3.1 Role of cytokines in oral manifestations of SLE
The relationship between cytokines and oral lesions in lupus suggests that these cytokines may not only reflect disease activity but also contribute to the inflammation and tissue damage observed in the oral mucosa (113).
Type I interferons, particularly IFN-α and IFN-β, play a crucial role in the immunopathogenesis of lupus and its related oral lesions. These cytokines enhance antigen presentation and promote the activation of autoreactive immune cells, contributing to ongoing inflammation. Elevated levels of IFN-α have been observed in lupus patients with oral manifestations, correlating with increased mucosal damage and immune dysregulation (114). Additionally, type I interferons stimulate the production of other inflammatory mediators, reinforcing the chronic immune activation observed in the oral mucosa (115).
TNF-α is a significant cytokine involved in lupus oral pathology. It promotes the apoptosis of keratinocytes, resulting in ulcer formation and destruction of mucosal tissue. Studies have shown significantly elevated levels of TNF-α in saliva and serum samples from lupus patients with oral lesions, indicating its role in amplifying inflammatory responses in the oral mucosa (116). Targeting TNF-α with biologic therapies has been investigated as a potential strategy for alleviating both oral and systemic lupus symptoms (117).
IL-6 is a critical cytokine involved in the chronic inflammation associated with lupus oral lesions. It drives B-cell hyperactivity and promotes autoantibody production, exacerbating immune-mediated tissue damage. Salivary and tissue samples from lupus patients show elevated IL-6 levels, further highlighting its contribution to disease progression (118). This cytokine also interacts with type I interferons, creating an inflammatory loop that sustains mucosal lesions (119). IL-8, a potent chemoattractant for neutrophils, plays a crucial role in the chronic inflammatory process associated with lupus oral lesions. Its overexpression in oral mucosal tissues has been linked to excessive infiltration of immune cells and prolonged persistence of lesions (117). IL-8 recruits neutrophils and monocytes to the affected tissues, exacerbating tissue damage and further sustaining the inflammatory environment (120).
IL-1 is a key mediator of inflammation and has been implicated in the pathogenesis of oral lesions in lupus. It exists in two primary forms, IL-1α and IL-1β, both of which enhance immune cell activation and amplify the release of inflammatory cytokines. IL-1 has been shown to potentiate the effects of TNF-α and IL-6, worsening mucosal tissue damage in lupus patients (121). The elevated levels of IL-1 in saliva and oral biopsies support its role in promoting ulceration and chronic inflammation (122). IL-18 is recognized as a crucial cytokine that influences the severity of oral lesions in lupus. IL-18 promotes the production of IFN-γ, leading to increased immune activation and progressive tissue destruction (123). Elevated IL-18 levels in lupus patients correlate with the severity of oral lesions, underscoring its potential as a biomarker for disease monitoring (124).
IFN-γ serves as a key regulator of Th1-mediated immune responses in lupus. It promotes macrophage activation and elevates the expression of adhesion molecules that facilitate immune cell infiltration into oral tissues. The increased levels of IFN-γ in lupus oral lesions indicate its role in sustaining chronic inflammation and immune dysfunction (125). Additionally, IFN-γ works in synergy with type I interferons, establishing a self-sustaining inflammatory cascade that contributes to the persistence of oral lesions (126). Furthermore, IL-17 contributes to the recruitment of neutrophils and macrophages, sustaining chronic inflammation and tissue destruction in the oral mucosa (127). Elevated levels of IL-17 in lupus patients have been linked to disease severity and increased lesion persistence, making it an emerging therapeutic target (128).
Macrophage inflammatory protein-1 alpha (MIP-1α), also known as CCL3, plays a significant role in the recruitment of immune cells and the inflammation associated with lupus oral lesions. Its elevated expression correlates with increased leukocyte infiltration and persistent mucosal inflammation (117). By promoting the migration of immune cells to inflamed oral tissues, MIP-1α aids in the maintenance of chronic immune activation (129). These cytokines work together to contribute to the complex immunopathology of oral lupus lesions by sustaining chronic inflammation, disrupting mucosal homeostasis, and driving disease progression. Understanding their roles offers insight into potential therapeutic strategies to alleviate oral manifestations in lupus patients.
4 Systemic manifestations of lupus erythematosus
SLE is characterized by widespread organ involvement, primarily driven by cytokine dysregulation. Key cytokines, including TNF-α, IL-6, IL-17, and type I interferons (particularly IFN-α), contribute to immune complex deposition, chronic inflammation, and tissue injury across multiple systems. In musculoskeletal tissues, these cytokines promote synovitis, joint inflammation, and cartilage degradation (130, 131). Similarly, renal involvement is mediated by glomerular immune complex deposition and cytokine-driven mesangial proliferation, ultimately contributing to lupus nephritis (132, 133). In parallel, within the skin, cytokines induce keratinocyte apoptosis and sustain cutaneous inflammation (134–136). Moreover, cardiovascular complications arise from cytokine-mediated endothelial dysfunction, which promotes the development of atherosclerosis and thrombosis (137, 138). Likewise, pulmonary manifestations reflect cytokine-induced interstitial inflammation, fibrosis, and vascular remodeling (139, 140). In addition, gastrointestinal features include mesenteric vasculitis and mucosal inflammation (141, 142), whereas hematologic abnormalities result from bone marrow suppression and an increased thrombotic risk (143, 144). Finally, Neuropsychiatric symptoms are associated with blood-brain barrier disruption, neuroinflammation, and cerebrovascular damage, which are linked to elevated cytokine levels (145, 146). Altogether, understanding these cytokine-mediated mechanisms provides the basis for targeted therapeutic interventions across the diverse systemic manifestations of SLE.
5 Therapeutic targeting of cytokines in SLE
The significant role of cytokines in the pathogenesis of SLE’s systemic and oral manifestations opens up potential therapeutic strategies targeting specific cytokines. Their inhibition has been investigated in preclinical models and targeted biologic therapies have been developed and assessed in clinical trials. While current treatments for lupus, such as hydroxychloroquine and immunosuppressants, aim to reduce systemic inflammation, biologic therapies that target cytokines have shown promise in treating both systemic and oral manifestations (3). Table 1 summarizes the key cytokine-targeted biologics in SLE with their mechanisms and clinical applications. Cytokine-targeted therapies, particularly IL-1 blockers (e.g., anakinra), and TNF-α inhibitors, represent a promising approach for reducing the severity of oral lesions and improving the oral health of lupus patients (147). A major challenge in SLE clinical trials has been the inclusion of heterogeneous patient populations, which may obscure treatment effects. To address this, it has been suggested that patients be stratified based on clinical and genetic phenotypes or cytokine profiles before trial enrollment (24, 148).
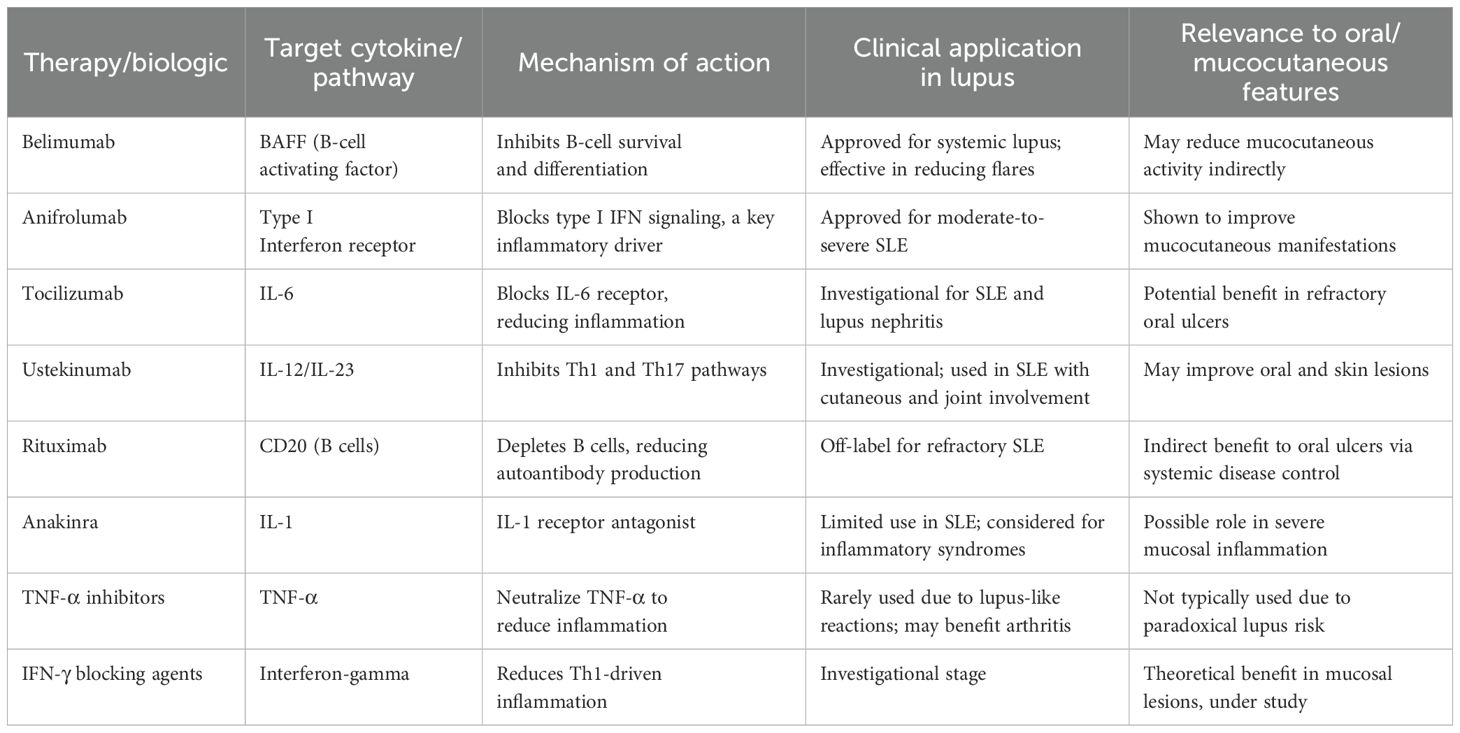
Table 1. Cytokine-targeted biologics in lupus erythematosus: mechanisms and clinical applications.
Guselkumab is a monoclonal antibody that binds with high affinity to human IL-23, preventing its interaction with the cell surface receptor. By blocking IL-23-mediated signaling, guselkumab inhibits the activation and cytokine production associated with this pathway. This is being explored in a phase 2 clinical trial (NCT04376827) by evaluating the safety and efficacy of guselkumab in combination with standard-of-care therapy, compared to a placebo plus standard-of-care (149). Ustekinumab, which targets the p40 subunit shared by IL-12 and IL-23, was evaluated in a phase 2 trial as an add-on therapy to standard SLE treatment. The study demonstrated positive effects on both clinical and laboratory markers, particularly in improving cutaneous and articular manifestations, while maintaining a favorable safety profile. Responders showed a decline in IFN-γ levels (150). Furthermore, ustekinumab suppresses both the Th1 and Th17 pathways (11). Emerging evidence supports the potential of IL-17A as a therapeutic target for SLE, particularly in patients whose disease is primarily driven by IL-17 (99). This was tried directly in the SELUNE study (NCT04181762), a randomized, double-blind trial designed to assess the efficacy and safety of secukinumab (Cosentyx), an anti-IL-17A monoclonal antibody, in combination with standard-of-care therapy for patients with active lupus nephritis (151). Another approach to indirectly target IL-17 involves inhibiting the synthesis of Th17 cells. Specifically, blocking cytokine pathways such as IL-6, IL-1, or IL-23 can interfere with Th17 cell development, thereby reducing IL-17 levels (99).
Tocilizumab, a monoclonal antibody that inhibits IL-6 signaling, was first evaluated for its efficacy in SLE patients in 2010 (152, 153). While it may be beneficial for certain patient subgroups with high inflammatory activity, caution is necessary, as higher doses can lead to immunosuppression (153). Targeting IL-6 with agents such as tocilizumab may help alleviate chronic oral inflammation and promote mucosal healing. Sirukumab is another human monoclonal antibody designed to selectively and effectively target IL-6. By inhibiting STAT-3 phosphorylation, it neutralizes IL-6 activity and mitigates its biological effects (154). To date, no IL-6-targeting therapies have received approval for SLE treatment. On the other hand, clinical trials investigating an anti-IL-10 monoclonal antibody showed a reduction in disease activity among SLE patients. However, the development of anti-drug antibodies raises concerns, and further studies are needed to assess long-term treatment feasibility (155).
The potential of anti-TNF-α monoclonal antibodies to suppress immune mechanisms initially suggested they could be beneficial for SLE treatment. While some studies yielded less promising results, early expectations remained optimistic (59). Studies have demonstrated the effectiveness of anti-TNF-α therapies in treating connective tissue diseases (CTD) (156), particularly SLE and cutaneous lupus erythematosus (CLE) (157). Among the most extensively studied TNF-α inhibitors are infliximab and etanercept (59). Infliximab, due to its chimeric nature, has the highest immunogenicity among anti-TNF-α agents. However, research indicates that infliximab maintains a favorable safety and tolerability profile in SLE patients. Notably, short-term induction therapy with infliximab, in combination with azathioprine or methotrexate, led to sustained improvement in lupus nephritis (57, 158, 159). Moreover, drugs such as etanercept, which target TNF-α, have been shown to reduce oral ulcers and improve overall oral health in lupus patients (160).
Etanercept has also been evaluated in several clinical trials for lupus nephritis treatment (NCT00447265) and discoid lupus erythematosus (DLE) (NCT02656082 and NCT00797784). An observational study found long-term etanercept therapy to be relatively safe and effective for refractory lupus arthritis (57). A separate study investigating SLE patients treated with adalimumab and etanercept revealed a significant reduction in the median prednisone dose, from 15 mg/day to 5 mg/day, during the observation period (59, 161). These findings support the notion that anti-TNF-α therapies may play a role in managing refractory lupus arthritis (59).
Etanercept has also shown efficacy in treating rhupus, a condition that exhibits features of both rheumatoid arthritis (RA) and SLE (162). Additionally, a combination of etanercept, plasmapheresis, and high-dose intravenous gamma globulin has been successfully used to manage severe diffuse proliferative nephritis in pregnant SLE patients (57, 163). Furthermore, a case report highlighted that etanercept could alleviate clinical symptoms and enhance overall quality of life in individuals with subacute cutaneous lupus erythematosus (SCLE) (164).
TNF-α inhibitors have been linked to the development of ANA and anti-dsDNA antibodies, potentially leading to clinical manifestations resembling idiopathic lupus. When this occurs, the condition is referred to as anti-TNF-α-induced lupus (ATIL) (59). This condition, also known as anti-TNF-α-induced lupus erythematosus (ATIL), is typically diagnosed based on a distinct temporal association between symptom onset and the initiation or dosage escalation of anti-TNF-α therapy (57, 157). Picardo et al. conducted a study to evaluate the incidence and clinical-serological features of ATIL in patients undergoing anti-TNF-α treatment. Their findings indicated a higher prevalence of ATIL among patients treated with infliximab compared to those receiving adalimumab (165). Current TNF-α blockers contribute to these side effects by blocking TNF-α interaction with both its regulatory receptor (TNFR2) and its pro-inflammatory/pro-apoptotic receptor (TNFR1) (57).
Rontalizumab and anifrolumab, which block type I interferons, have shown potential in reducing both systemic and oral manifestations of lupus (2).
Clinical trials targeting IFN-γ have begun to yield promising results in SLE treatment. AMG 811, a fully human IgG1 monoclonal antibody against IFN-γ, has exhibited good tolerability in patients with mild to moderate SLE. A single dose of AMG 811 has been shown to normalize IFN-regulated gene expression and induce a dose-dependent decrease in serum CXCL10 levels (166, 167). While AMG 811 effectively modulated IFN-γ-associated biomarkers and maintained a favorable safety profile, it did not produce significant clinical benefits for patients with DLE (168). However, encouraging results from phase Ib trials suggest that inhibiting the IFN-γ pathway may hold therapeutic potential for extrarenal manifestations of lupus (169). Given IFN-γ’s critical role in LN, further exploration of its inhibition in this context is warranted, especially considering the tolerability of its targeted blockade. Two completed studies registered as NCT00818948 and NCT02291588, have evaluated the safety of AMG 811 in SLE treatment.
It is worth mentioning that many other targeted therapies have been used or investigated in SLE treatment. These include targeting CD20, BAFF and cytokine downstream signaling molecules that play a role in the pathogenesis of SLE (149).
6 Conclusion
In summary, cytokines exert a profound influence on the pathogenesis of SLE, affecting both systemic and oral manifestations (Table 2, Figure 2). The imbalance between pro-inflammatory and anti-inflammatory cytokines, particularly the elevation of IFN-α, IL-6, IL-17, and TNF-α, contributes significantly to the disease’s progression and severity. Further research into the precise mechanisms of cytokine involvement is crucial for developing targeted therapeutic strategies. Modulating cytokine activity holds promise for improving the management of SLE, alleviating symptoms, and ultimately enhancing the quality of life for individuals affected by this complex autoimmune disease.
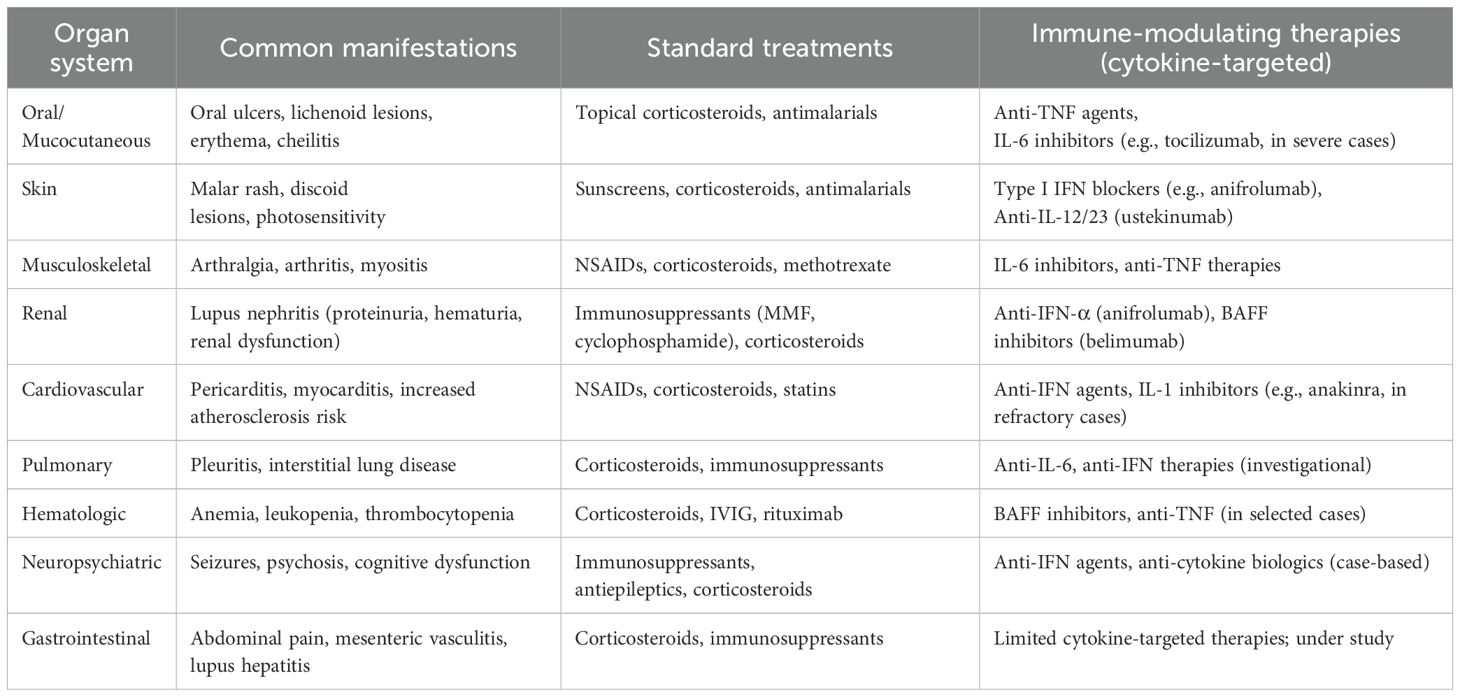
Table 2. Organ system manifestations and treatments in lupus erythematosus with immunological focus.
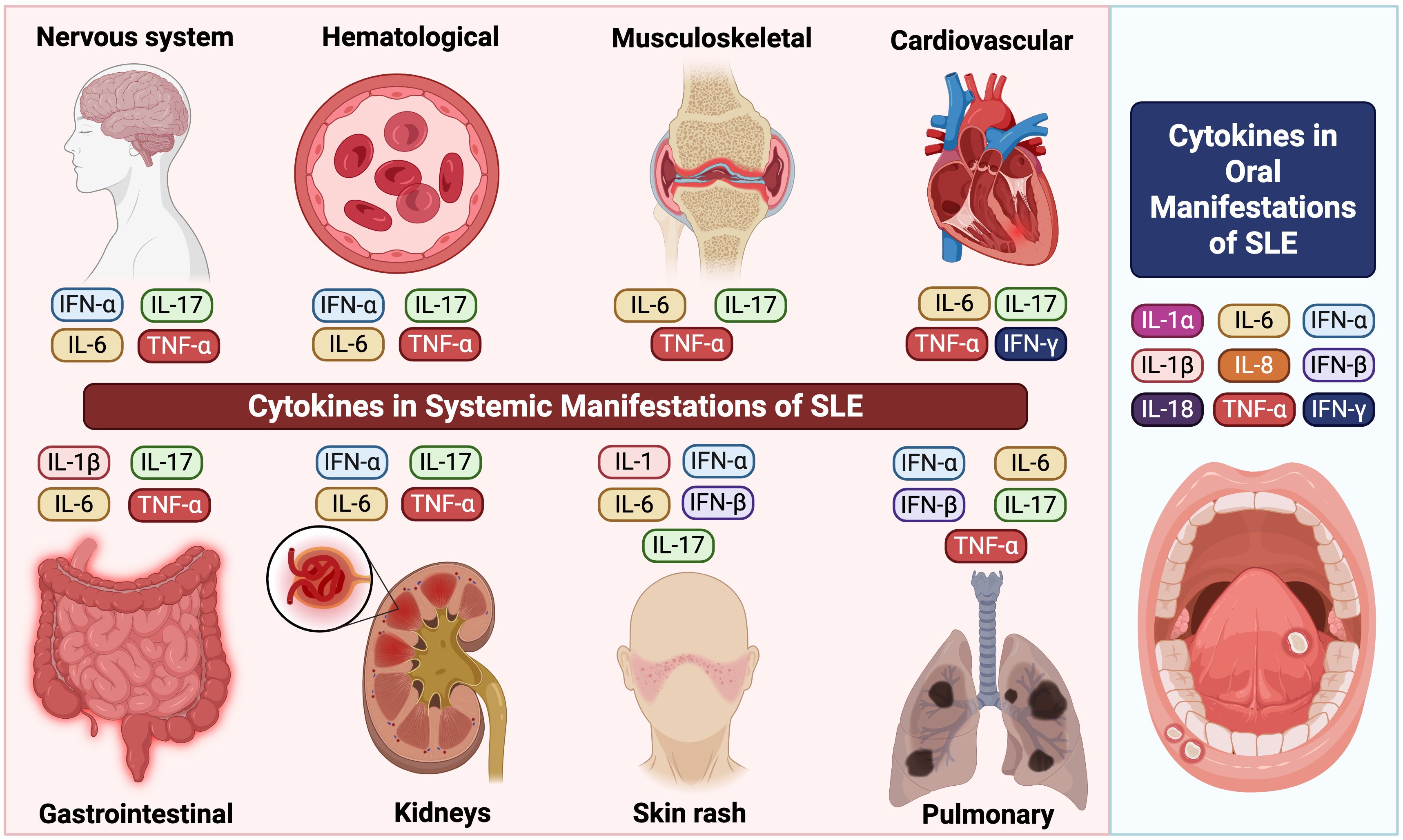
Figure 2. Cytokine-mediated immune mechanisms in SLE manifestations including oral and systemic compartments.
Author contributions
NE: Writing – original draft, Writing – review & editing. IT: Writing – original draft, Writing – review & editing. OE: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Wang R, Zhang X, and Wang S. Differential genotypes of TNF-α and IL-10 for immunological diagnosis in discoid lupus erythematosus and oral lichen planus: A narrative review. Front Immunol. (2022) 13:967281. doi: 10.3389/fimmu.2022.967281
2. Crow MK. Pathogenesis of systemic lupus erythematosus: risks, mechanisms and therapeutic targets. Ann Rheumatic Dis. (2023) 82:999. doi: 10.1136/ard-2022-223741
3. La Cava A. Anticytokine therapies in systemic lupus erythematosus. Immunotherapy. (2010) 2:575–82. doi: 10.2217/imt.10.29
4. Idborg H and Oke V. Cytokines as biomarkers in systemic lupus erythematosus: value for diagnosis and drug therapy. Int J Mol Sci. (2021) 22:11327. doi: 10.3390/ijms222111327
5. Mathian A, Felten R, Alarcon-Riquelme ME, Psarras A, Mertz P, Chasset F, et al. Type 1 interferons: A target for immune-mediated inflammatory diseases (IMIDs). Joint Bone Spine. (2024) 91:105627. doi: 10.1016/j.jbspin.2023.105627
6. Banchereau J, Pascual V, and Palucka AK. Autoimmunity through cytokine-induced dendritic cell activation. Immunity. (2004) 20:539–50. doi: 10.1016/s1074-7613(04)00108-6
7. Caielli S, Cardenas J, de Jesus AA, Baisch J, Walters L, Blanck JP, et al. Erythroid mitochondrial retention triggers myeloid-dependent type I interferon in human SLE. Cell. (2021) 184:4464–79.e19. doi: 10.1016/j.cell.2021.07.021
8. Caielli S, Balasubramanian P, Rodriguez-Alcazar J, Balaji U, Robinson L, Wan Z, et al. Type I IFN drives unconventional IL-1β secretion in lupus monocytes. Immunity. (2024) 57:2497–513.e12. doi: 10.1016/j.immuni.2024.09.004
9. Hänsel A, Günther C, Baran W, Bidier M, Lorenz H-M, Schmitz M, et al. Human 6-sulfo LacNAc (slan) dendritic cells have molecular and functional features of an important pro-inflammatory cell type in lupus erythematosus. J Autoimmun. (2013) 40:1–8. doi: 10.1016/j.jaut.2012.07.005
10. Billi AC, Ma F, Plazyo O, Gharaee-Kermani M, Wasikowski R, Hile GA, et al. Nonlesional lupus skin contributes to inflammatory education of myeloid cells and primes for cutaneous inflammation. Sci Transl Med. (2022) 14:eabn2263. doi: 10.1126/scitranslmed.abn2263
11. Farah Izati A, Wong KK, and Che Maraina CH. IL-23/IL-17 axis in the pathogenesis and treatment of systemic lupus erythematosus and rheumatoid arthritis. Malays J Pathol. (2020) 42:333–47.
12. Davis LS, Hutcheson J, and Mohan C. The role of cytokines in the pathogenesis and treatment of systemic lupus erythematosus. J Interferon Cytokine Res. (2011) 31:781–9. doi: 10.1089/jir.2011.0047
13. Munroe ME, Young KA, Guthridge JM, Kamen DL, Gilkeson GS, Weisman MH, et al. Pre-clinical autoimmunity in lupus relatives: self-reported questionnaires and immune dysregulation distinguish relatives who develop incomplete or classified lupus from clinically unaffected relatives and unaffected, unrelated individuals. Front Immunol. (2022) 13:866181. doi: 10.3389/fimmu.2022.866181
14. Quan W, An J, Li G, Qian G, Jin M, Feng C, et al. Th cytokine profile in childhood-onset systemic lupus erythematosus. BMC Pediatr. (2021) 21:187. doi: 10.1186/s12887-021-02659-3
15. Guimarães P, Scavuzzi B, Stadtlober N, Silva L, Lozovoy M, Iriyoda T, et al. Cytokines in systemic lupus erythematosus: Far beyond Th1/Th2 dualism. Immunol Cell Biol. (2017) 95:824–31. doi: 10.1038/icb.2017.53
16. Lu R, Munroe ME, Guthridge JM, Bean KM, Fife DA, Chen H, et al. Dysregulation of innate and adaptive serum mediators precedes systemic lupus erythematosus classification and improves prognostic accuracy of autoantibodies. J Autoimmun. (2016) 74:182–93. doi: 10.1016/j.jaut.2016.06.001
17. Postal M and Appenzeller S. The role of Tumor Necrosis Factor-alpha (TNF-α) in the pathogenesis of systemic lupus erythematosus. Cytokine. (2011) 56:537–43. doi: 10.1016/j.cyto.2011.08.026
18. Richter P, Macovei LA, Mihai IR, Cardoneanu A, Burlui MA, and Rezus E. Cytokines in systemic lupus erythematosus-focus on TNF-α and IL-17. Int J Mol Sci. (2023) 24:14413. doi: 10.3390/ijms241914413
19. Chasset F and Arnaud L. Targeting interferons and their pathways in systemic lupus erythematosus. Autoimmun Rev. (2018) 17:44–52. doi: 10.1016/j.autrev.2017.11.009
20. Schroder K, Hertzog PJ, Ravasi T, and Hume DA. Interferon-γ: an overview of signals, mechanisms and functions. J Leukocyte Biol. (2004) 75:163–89. doi: 10.1189/jlb.0603252
21. LaFleur DW, Nardelli B, Tsareva T, Mather D, Feng P, Semenuk M, et al. Interferon-κ, a novel type I interferon expressed in human keratinocytes. J Biol Chem. (2001) 276:39765–71. doi: 10.1074/jbc.M102502200
22. Nardelli B, Zaritskaya L, Semenuk M, Cho YH, LaFleur DW, Shah D, et al. Regulatory effect of IFN-κ, A novel type I IFN, on cytokine production by cells of the innate immune system. J Immunol. (2002) 169:4822–30. doi: 10.4049/jimmunol.169.9.4822
23. Rönnblom L and Leonard D. Interferon pathway in SLE: one key to unlocking the mystery of the disease. Lupus Sci Med. (2019) 6:e000270. doi: 10.1136/lupus-2018-000270
24. Oke V, Gunnarsson I, Dorschner J, Eketjäll S, Zickert A, Niewold TB, et al. High levels of circulating interferons type I, type II and type III associate with distinct clinical features of active systemic lupus erythematosus. Arthritis Res Ther. (2019) 21:107. doi: 10.1186/s13075-019-1878-y
25. Bengtsson A, Sturfelt G, Truedsson L, Blomberg J, Alm G, Vallin H, et al. Activation of type I interferon system in systemic lupus erythematosus correlates with disease activity but not with antiretroviral antibodies. Lupus. (2000) 9:664–71. doi: 10.1191/096120300674499064
26. Weckerle CE, Franek BS, Kelly JA, Kumabe M, Mikolaitis RA, Green SL, et al. Network analysis of associations between serum interferon-α activity, autoantibodies, and clinical features in systemic lupus erythematosus. Arthritis Rheumatism. (2011) 63:1044–53. doi: 10.1002/art.30187
27. Paradowska-Gorycka A, Wajda A, Stypinska B, Walczuk E, Rzeszotarska E, Walczyk M, et al. Variety of endosomal TLRs and Interferons (IFN-α, IFN-β, IFN-γ) expression profiles in patients with SLE, SSc and MCTD. Clin Exp Immunol. (2021) 204:49–63. doi: 10.1111/cei.13566
28. Furie R, Khamashta M, Merrill JT, Werth VP, Kalunian K, Brohawn P, et al. Anifrolumab, an anti–interferon-α receptor monoclonal antibody, in moderate-to-severe systemic lupus erythematosus. Arthritis Rheumatol. (2017) 69:376–86. doi: 10.1002/art.39962
29. Bennett L, Palucka AK, Arce E, Cantrell V, Borvak J, Banchereau J, et al. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med. (2003) 197:711–23. doi: 10.1084/jem.20021553
30. Baechler EC, Batliwalla FM, Karypis G, Gaffney PM, Ortmann WA, Espe KJ, et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci. (2003) 100:2610–5. doi: 10.1073/pnas.0337679100
31. Han GM, Chen SL, Shen N, Ye S, Bao CD, and Gu YY. Analysis of gene expression profiles in human systemic lupus erythematosus using oligonucleotide microarray. Genes Immun. (2003) 4:177–86. doi: 10.1038/sj.gene.6363966
32. Crow MK, Kirou KA, and Wohlgemuth J. Microarray analysis of interferon-regulated genes in SLE. Autoimmunity. (2003) 36:481–90. doi: 10.1080/08916930310001625952
33. Niewold TB, Adler JE, Glenn SB, Lehman TJA, Harley JB, and Crow MK. Age- and sex-related patterns of serum interferon-α activity in lupus families. Arthritis Rheumatism. (2008) 58:2113–9. doi: 10.1002/art.23619
34. Bengtsson AA, Sturfelt G, Truedsson L, Blomberg J, Alm G, Vallin H, et al. Activation of type I interferon system in systemic lupus erythematosus correlates with disease activity but not with antiretroviral antibodies. Lupus. (2000) 9:664–71. doi: 10.1191/096120300674499064
35. Feng X, Wu H, Grossman JM, Hanvivadhanakul P, FitzGerald JD, Park GS, et al. Association of increased interferon-inducible gene expression with disease activity and lupus nephritis in patients with systemic lupus erythematosus. Arthritis Rheumatism. (2006) 54:2951–62. doi: 10.1002/art.22044
36. Tucci M, Quatraro C, Lombardi L, Pellegrino C, Dammacco F, and Silvestris F. Glomerular accumulation of plasmacytoid dendritic cells in active lupus nephritis: role of interleukin-18. Arthritis Rheum. (2008) 58:251–62. doi: 10.1002/art.23186
37. Farkas L, Beiske K, Lund-Johansen F, Brandtzaeg P, and Jahnsen FL. Plasmacytoid dendritic cells (natural interferon- alpha/beta-producing cells) accumulate in cutaneous lupus erythematosus lesions. Am J Pathol. (2001) 159:237–43. doi: 10.1016/s0002-9440(10)61689-6
38. Blomberg S, Eloranta ML, Cederblad B, Nordlin K, Alm GV, and Ronnblom L. Presence of cutaneous interferon-alpha producing cells in patients with systemic lupus erythematosus. Lupus. (2001) 10:484–90. doi: 10.1191/096120301678416042
39. Furie R, Werth VP, Merola JF, Stevenson L, Reynolds TL, Naik H, et al. Monoclonal antibody targeting BDCA2 ameliorates skin lesions in systemic lupus erythematosus. J Clin Invest. (2019) 129:1359–71. doi: 10.1172/JCI124466
40. Antonios P, Zoe W, Ade A, and Edward MV. P24 Differentiated monocytes express the pDC markers BDCA-2 and CD123. Lupus Sci Med. (2024) 11:824–31. doi: 10.1136/lupus-2024-el.78
41. Psarras A, Wigston Z, and Vital E. AB0115 CD14+ MYELOID CELLS EXPRESS THE PDC MARKERS BDCA-2, BDCA-4, CD123 UPON DIFFERENTIATION IN BOTH HEALTHY INDIVIDUALS AND SLE PATIENTS. Ann Rheumatic Dis. (2023) 82:1236. doi: 10.1136/annrheumdis-2023-eular.2330
42. Psarras A, Alase A, Antanaviciute A, Carr IM, Md Yusof MY, Wittmann M, et al. Functionally impaired plasmacytoid dendritic cells and non-haematopoietic sources of type I interferon characterize human autoimmunity. Nat Commun. (2020) 11:6149. doi: 10.1038/s41467-020-19918-z
43. Vazquez T, Patel J, Kodali N, Diaz D, Bashir MM, Chin F, et al. Plasmacytoid dendritic cells are not major producers of type 1 IFN in cutaneous lupus: an in-depth immunoprofile of subacute and discoid lupus. J Invest Dermatol. (2024) 144:1262–72.e7. doi: 10.1016/j.jid.2023.10.039
44. Palanichamy A, Bauer JW, Yalavarthi S, Meednu N, Barnard J, Owen T, et al. Neutrophil-mediated IFN activation in the bone marrow alters B cell development in human and murine systemic lupus erythematosus. J Immunol. (2014) 192:906–18. doi: 10.4049/jimmunol.1302112
45. Denny MF, Yalavarthi S, Zhao W, Thacker SG, Anderson M, Sandy AR, et al. A distinct subset of proinflammatory neutrophils isolated from patients with systemic lupus erythematosus induces vascular damage and synthesizes type I IFNs. J Immunol. (2010) 184:3284–97. doi: 10.4049/jimmunol.0902199
46. Klein B, Reynolds MB, Xu B, Gharaee-Kermani M, Gao Y, Berthier CC, et al. Epidermal ZBP1 stabilizes mitochondrial Z-DNA to drive UV-induced IFN signaling in autoimmune photosensitivity. Sci Immunol. (2025) 10:eado1710. doi: 10.1126/sciimmunol.ado1710
47. Deng GM and Tsokos GC. Pathogenesis and targeted treatment of skin injury in SLE. Nat Rev Rheumatol. (2015) 11:663–9. doi: 10.1038/nrrheum.2015.106
48. Sarkar MK, Hile GA, Tsoi LC, Xing X, Liu J, Liang Y, et al. Photosensitivity and type I IFN responses in cutaneous lupus are driven by epidermal-derived interferon kappa. Ann Rheum Dis. (2018) 77:1653–64. doi: 10.1136/annrheumdis-2018-213197
49. Xu B, Musai J, Tan YS, Hile GA, Swindell WR, Klein B, et al. A critical role for IFN-β signaling for IFN-κ induction in keratinocytes. Front Lupus. (2024) 2:1359714. doi: 10.3389/flupu.2024.1359714
50. Lin S-C, Kuo C-C, Tsao J-T, and Lin L-J. Profiling the expression of interleukin (IL)-28 and IL-28 receptor α in systemic lupus erythematosus patients. Eur J Clin Invest. (2012) 42:61–9. doi: 10.1111/j.1365-2362.2011.02557.x
51. Leonard D, Eloranta M-L, Hagberg N, Berggren O, Tandre K, Alm G, et al. Activated T cells enhance interferon-&x3b1; production by plasmacytoid dendritic cells stimulated with RNA-containing immune complexes. Ann Rheumatic Dis. (2016) 75:1728–34. doi: 10.1136/annrheumdis-2015-208055
52. Hagberg N, Berggren O, Leonard D, Weber G, Bryceson YT, Alm GV, et al. IFN-α Production by plasmacytoid dendritic cells stimulated with RNA-containing immune complexes is promoted by NK cells via MIP-1β and LFA-1. J Immunol. (2011) 186:5085–94. doi: 10.4049/jimmunol.1003349
53. Klavdianou K, Lazarini A, and Fanouriakis A. Targeted biologic therapy for systemic lupus erythematosus: emerging pathways and drug pipeline. BioDrugs. (2020) 34:133–47. doi: 10.1007/s40259-020-00405-2
54. Aragón CC, Tafúr R-A, Suárez-Avellaneda A, Martínez MT, de Las Salas A, and Tobón GJ. Urinary biomarkers in lupus nephritis. J Trans Autoimmun. (2020) 3:100042. doi: 10.1016/j.jtauto.2020.100042
55. Alase AA, El-Sherbiny YM, Vital EM, Tobin DJ, Turner NA, and Wittmann M. IFNλ Stimulates mxA production in human dermal fibroblasts via a MAPK-dependent STAT1-independent mechanism. J Invest Dermatol. (2015) 135:2935–43. doi: 10.1038/jid.2015.317
56. Goel RR, Wang X, O’Neil LJ, Nakabo S, Hasneen K, Gupta S, et al. Interferon lambda promotes immune dysregulation and tissue inflammation in TLR7-induced lupus. Proc Natl Acad Sci. (2020) 117:5409–19. doi: 10.1073/pnas.1916897117
57. Ghorbaninezhad F, Leone P, Alemohammad H, Najafzadeh B, Nourbakhsh NS, Prete M, et al. Tumor necrosis factor−α in systemic lupus erythematosus: Structure, function and therapeutic implications (Review). Int J Mol Med. (2022) 49:43. doi: 10.3892/ijmm.2022.5098
58. Gomez D, Correa PA, Gomez LM, Cadena J, Molina JF, and Anaya JM. Th1/Th2 cytokines in patients with systemic lupus erythematosus: is tumor necrosis factor alpha protective? Semin Arthritis Rheum. (2004) 33:404–13. doi: 10.1016/j.semarthrit.2003.11.002
59. Lorenzo-Vizcaya A and Isenberg DA. The use of anti-TNF-alpha therapies for patients with systemic lupus erythematosus. Where are we now? Expert Opin Biol Ther. (2021) 21:639–47. doi: 10.1080/14712598.2021.1853096
60. Mageed RA and Isenberg DA. Tumour necrosis factor alpha in systemic lupus erythematosus and anti-DNA autoantibody production. Lupus. (2002) 11:850–5. doi: 10.1191/0961203302lu306rr
61. McCarthy EM, Smith S, Lee RZ, Cunnane G, Doran MF, Donnelly S, et al. The association of cytokines with disease activity and damage scores in systemic lupus erythematosus patients. Rheumatol (Oxford). (2014) 53:1586–94. doi: 10.1093/rheumatology/ket428
62. Davas EM, Tsirogianni A, Kappou I, Karamitsos D, Economidou I, and Dantis PC. Serum IL-6, TNFalpha, p55 srTNFalpha, p75srTNFalpha, srIL-2alpha levels and disease activity in systemic lupus erythematosus. Clin Rheumatol. (1999) 18:17–22. doi: 10.1007/s100670050045
63. Aringer M and Smolen JS. The role of tumor necrosis factor-alpha in systemic lupus erythematosus. Arthritis Res Ther. (2008) 10:202. doi: 10.1186/ar2341
64. Zhu L, Yang X, Chen W, Li X, Ji Y, Mao H, et al. Decreased expressions of the TNF-alpha signaling adapters in peripheral blood mononuclear cells (PBMCs) are correlated with disease activity in patients with systemic lupus erythematosus. Clin Rheumatol. (2007) 26:1481–9. doi: 10.1007/s10067-006-0531-8
65. Studnicka-Benke A, Steiner G, Petera P, and Smolen JS. Tumour necrosis factor alpha and its soluble receptors parallel clinical disease and autoimmune activity in systemic lupus erythematosus. Br J Rheumatol. (1996) 35:1067–74. doi: 10.1093/rheumatology/35.11.1067
66. Wajant H and Siegmund D. TNFR1 and TNFR2 in the control of the life and death balance of macrophages. Front Cell Dev Biol. (2019) 7:91. doi: 10.3389/fcell.2019.00091
67. Watts TH, Yeung KKM, Yu T, Lee S, and Eshraghisamani R. TNF/TNFR superfamily members in costimulation of T cell responses-revisited. Annu Rev Immunol. (2025) 43:113–42. doi: 10.1146/annurev-immunol-082423-040557
68. Faustman D and Davis M. TNF receptor 2 pathway: drug target for autoimmune diseases. Nat Rev Drug Discov. (2010) 9:482–93. doi: 10.1038/nrd3030
69. Chen G and Goeddel DV. TNF-R1 signaling: a beautiful pathway. Science. (2002) 296:1634–5. doi: 10.1126/science.1071924
70. Liu Z and Davidson A. Taming lupus-a new understanding of pathogenesis is leading to clinical advances. Nat Med. (2012) 18:871–82. doi: 10.1038/nm.2752
71. Chen Y, Jiang M, and Chen X. Therapeutic potential of TNFR2 agonists: a mechanistic perspective. Front Immunol. (2023) 14:1209188. doi: 10.3389/fimmu.2023.1209188
72. Peters AL, Stunz LL, and Bishop GA. CD40 and autoimmunity: The dark side of a great activator. Semin Immunol. (2009) 21:293–300. doi: 10.1016/j.smim.2009.05.012
73. Schönbeck U and Libby P. The CD40/CD154 receptor/ligand dyad. Cell Mol Life Sci. (2001) 58:4–43. doi: 10.1007/pl00000776
74. Ramanujam M, Steffgen J, Visvanathan S, Mohan C, Fine JS, and Putterman C. Phoenix from the flames: Rediscovering the role of the CD40–CD40L pathway in systemic lupus erythematosus and lupus nephritis. Autoimmun Rev. (2020) 19:102668. doi: 10.1016/j.autrev.2020.102668
75. Bishop GA and Hostager BS. The CD40-CD154 interaction in B cell-T cell liaisons. Cytokine Growth Factor Rev. (2003) 14:297–309. doi: 10.1016/s1359-6101(03)00024-8
76. Menard LC, Habte S, Gonsiorek W, Lee D, Banas D, Holloway DA, et al. B cells from African American lupus patients exhibit an activated phenotype. JCI Insight. (2016) 1:e87310. doi: 10.1172/jci.insight.87310
77. Hiepe F, Dörner T, Hauser AE, Hoyer BF, Mei H, and Radbruch A. Long-lived autoreactive plasma cells drive persistent autoimmune inflammation. Nat Rev Rheumatol. (2011) 7:170–8. doi: 10.1038/nrrheum.2011.1
78. Idborg H, Eketjäll S, Pettersson S, Gustafsson JT, Zickert A, Kvarnström M, et al. TNF-α and plasma albumin as biomarkers of disease activity in systemic lupus erythematosus. Lupus Sci Med. (2018) 5:e000260. doi: 10.1136/lupus-2018-000260
79. Barkauskaite V, Ek M, Popovic K, Harris H, Wahren-Herlenius M, and Nyberg F. Translocation of the novel cytokine HMGB1 to the cytoplasm and extracellular space coincides with the peak of clinical activity in experimentally UV-induced lesions of cutaneous lupus erythematosus. Lupus. (2007) 16:794–802. doi: 10.1177/0961203307081895
80. Kinoshita K, Yamagata T, Nozaki Y, Sugiyama M, Ikoma S, Funauchi M, et al. Blockade of IL-18 receptor signaling delays the onset of autoimmune disease in MRL-Faslpr mice. J Immunol. (2004) 173:5312–8. doi: 10.4049/jimmunol.173.8.5312
81. Mende R, Vincent FB, Kandane-Rathnayake R, Koelmeyer R, Lin E, Chang J, et al. Analysis of serum interleukin (IL)-1β and IL-18 in systemic lupus erythematosus. Front Immunol. (2018) 9:1250. doi: 10.3389/fimmu.2018.01250
82. Wang D, Drenker M, Eiz-Vesper B, Werfel T, and Wittmann M. Evidence for a pathogenetic role of interleukin-18 in cutaneous lupus erythematosus. Arthritis Rheumatism: Off J Am Coll Rheumatol. (2008) 58:3205–15. doi: 10.1002/art.23868
83. Xiang M, Feng Y, Wang Y, Wang J, Zhang Z, Liang J, et al. Correlation between circulating interleukin-18 level and systemic lupus erythematosus: A meta-analysis. Sci Rep. (2021) 11:4707. doi: 10.1038/s41598-021-84170-4
84. Cigni A, Pileri PV, Faedda R, Gallo P, Sini A, Satta AE, et al. Interleukin 1, interleukin 6, interleukin 10, and tumor necrosis factor α in active and quiescent systemic lupus erythematosus. J Invest Med. (2014) 62:825–9. doi: 10.2310/JIM.0000000000000085
85. Su H, Lei CT, and Zhang C. Interleukin-6 signaling pathway and its role in kidney disease: an update. Front Immunol. (2017) 8:405. doi: 10.3389/fimmu.2017.00405
86. Navarra SV, Guzmán RM, Gallacher AE, Hall S, Levy RA, Jimenez RE, et al. Efficacy and safety of belimumab in patients with active systemic lupus erythematosus: a randomised, placebo-controlled, phase 3 trial. Lancet. (2011) 377:721–31. doi: 10.1016/S0140-6736(10)61354-2
87. Furie R, Petri M, Zamani O, Cervera R, Wallace DJ, Tegzová D, et al. randomized, placebo-controlled study of belimumab, a monoclonal antibody that inhibits B lymphocyte stimulator, in patients with systemic lupus erythematosus. Arthritis Rheumatism. (2011) 63:3918–30. doi: 10.1002/art.30613
88. Shao M, He J, Zhang R, Zhang X, Yang Y, Li C, et al. Interleukin-2 deficiency associated with renal impairment in systemic lupus erythematosus. J Interferon Cytokine Res. (2019) 39:117–24. doi: 10.1089/jir.2018.0016
89. Shao M, Sun X-L, Sun H, He J, Zhang R-J, Zhang X, et al. Clinical relevance of autoantibodies against interleukin-2 in patients with systemic lupus erythematosus. Chin Med J. (2018) 131:1520–6. doi: 10.4103/0366-6999.235114
90. Long D, Chen Y, Wu H, Zhao M, and Lu Q. Clinical significance and immunobiology of IL-21 in autoimmunity. J Autoimmun. (2019) 99:1–14. doi: 10.1016/j.jaut.2019.01.013
91. Wang S, Wang J, Kumar V, Karnell JL, Naiman B, Gross PS, et al. IL-21 drives expansion and plasma cell differentiation of autoreactive CD11chiT-bet+ B cells in SLE. Nat Commun. (2018) 9:1758. doi: 10.1038/s41467-018-03750-7
92. Nakou M, Papadimitraki ED, Fanouriakis A, Bertsias GK, Choulaki C, Goulidaki N, et al. Interleukin-21 is increased in active systemic lupus erythematosus patients and contributes to the generation of plasma B cells. Clin Exp Rheumatol. (2013) 31:172–9.
93. Pan HF, Wu GC, Fan YG, Leng RX, Peng H, Zhou M, et al. Decreased serum level of IL-21 in new-onset systemic lupus erythematosus patients. Rheumatol Int. (2013) 33:2337–42. doi: 10.1007/s00296-013-2724-1
94. Sharif MN, Tassiulas I, Hu Y, Mecklenbräuker I, Tarakhovsky A, and Ivashkiv LB. IFN-alpha priming results in a gain of proinflammatory function by IL-10: implications for systemic lupus erythematosus pathogenesis. J Immunol. (2004) 172:6476–81. doi: 10.4049/jimmunol.172.10.6476
95. Peng H, Wang W, Zhou M, Li R, Pan H-F, and Ye D-Q. Role of interleukin-10 and interleukin-10 receptor in systemic lupus erythematosus. Clin Rheumatol. (2013) 32:1255–66. doi: 10.1007/s10067-013-2294-3
96. Rönnblom L and Elkon KB. Cytokines as therapeutic targets in SLE. Nat Rev Rheumatol. (2010) 6:339–47. doi: 10.1038/nrrheum.2010.64
97. Yin R, Xu R, Ding L, Sui W, Niu M, Wang M, et al. Circulating IL-17 level is positively associated with disease activity in patients with systemic lupus erythematosus: A systematic review and meta-analysis. BioMed Res Int. (2021) 2021:9952463. doi: 10.1155/2021/9952463
98. Qian W and La Cava A. IL-17 in systemic lupus erythematosus. Clin Invest. (2012) 2:417–21. doi: 10.4155/cli.12.21
99. Robert M and Miossec P. Interleukin-17 and lupus: enough to be a target? For which patients? Lupus. (2020) 29:6–14. doi: 10.1177/0961203319891243
100. Tabarkiewicz J, Pogoda K, Karczmarczyk A, Pozarowski P, and Giannopoulos K. The role of IL-17 and th17 lymphocytes in autoimmune diseases. Arch Immunol Ther Exp (Warsz). (2015) 63:435–49. doi: 10.1007/s00005-015-0344-z
101. Wong CK, Lit LC, Tam LS, Li EK, Wong PT, and Lam CW. Hyperproduction of IL-23 and IL-17 in patients with systemic lupus erythematosus: implications for Th17-mediated inflammation in auto-immunity. Clin Immunol. (2008) 127:385–93. doi: 10.1016/j.clim.2008.01.019
102. Lopez P, Rodriguez-Carrio J, Caminal-Montero L, Mozo L, and Suarez A. A pathogenic IFNalpha, BLyS and IL-17 axis in Systemic Lupus Erythematosus patients. Sci Rep. (2016) 6:20651. doi: 10.1038/srep20651
103. Henriques A, Ines L, Couto M, Pedreiro S, Santos C, Magalhaes M, et al. Frequency and functional activity of Th17, Tc17 and other T-cell subsets in Systemic Lupus Erythematosus. Cell Immunol. (2010) 264:97–103. doi: 10.1016/j.cellimm.2010.05.004
104. Rafael-Vidal C, Pérez N, Altabás I, Garcia S, and Pego-Reigosa JM. Blocking IL-17: A promising strategy in the treatment of systemic rheumatic diseases. Int J Mol Sci. (2020) 21:7100. doi: 10.3390/ijms21197100
105. Yang J, Chu Y, Yang X, Gao D, Zhu L, Yang X, et al. Th17 and natural Treg cell population dynamics in systemic lupus erythematosus. Arthritis Rheum. (2009) 60:1472–83. doi: 10.1002/art.24499
106. Wong CK, Ho CY, Li EK, and Lam CW. Elevation of proinflammatory cytokine (IL-18, IL-17, IL-12) and Th2 cytokine (IL-4) concentrations in patients with systemic lupus erythematosus. Lupus. (2000) 9:589–93. doi: 10.1191/096120300678828703
107. Mok MY, Wu HJ, Lo Y, and Lau CS. The relation of interleukin 17 (IL-17) and IL-23 to Th1/Th2 cytokines and disease activity in systemic lupus erythematosus. J Rheumatol. (2010) 37:2046–52. doi: 10.3899/jrheum.100293
108. Tang Y, Tao H, Gong Y, Chen F, Li C, and Yang X. Changes of serum IL-6, IL-17, and complements in systemic lupus erythematosus patients. J Interferon Cytokine Res. (2019) 39:410–5. doi: 10.1089/jir.2018.0169
109. Abdel Galil SM, Ezzeldin N, and El-Boshy ME. The role of serum IL-17 and IL-6 as biomarkers of disease activity and predictors of remission in patients with lupus nephritis. Cytokine. (2015) 76:280–7. doi: 10.1016/j.cyto.2015.05.007
110. Chen XQ, Yu YC, Deng HH, Sun JZ, Dai Z, Wu YW, et al. Plasma IL-17A is increased in new-onset SLE patients and associated with disease activity. J Clin Immunol. (2010) 30:221–5. doi: 10.1007/s10875-009-9365-x
111. Benli M, Batool F, Stutz C, Petit C, Jung S, and Huck O. Orofacial manifestations and dental management of systemic lupus erythematosus: A review. Oral Dis. (2021) 27:151–67. doi: 10.1111/odi.13271
112. García-Ríos P, Pecci-Lloret MP, and Oñate-Sánchez RE. Oral manifestations of systemic lupus erythematosus: A systematic review. Int J Environ Res Public Health. (2022) 19:11910. doi: 10.3390/ijerph191911910
113. Marques ERMC, Lourenço SV, Lima DM, and Nico MMS. Oral lesions in lupus erythematosus–cytokines profiles of inflammatory infiltrate. J Cutaneous Pathol. (2010) 37:439–45. doi: 10.1111/j.1600-0560.2009.01424.x
114. Arriens C, Van Vollenhoven R, Gottlieb A, Hobar C, Pomponi S, Koti R, et al. OP0048 CUTANEOUS LUPUS ERYTHEMATOSUS DISEASE AREA AND SEVERITY INDEX (CLASI) ACHIEVEMENT AND SUSTAINED RESPONSE WITH DEUCRAVACITINIB, AN ORAL, SELECTIVE, ALLOSTERIC TYROSINE KINASE 2 INHIBITOR, IN A PHASE 2 TRIAL IN SYSTEMIC LUPUS ERYTHEMATOSUS. Ann Rheumatic Dis. (2024) 83:138–9. doi: 10.1136/annrheumdis-2024-eular.584
115. Leber A, Hontecillas R, Tubau-Juni N, and Bassaganya-Riera J. Safety, tolerability, and pharmacokinetics of NIM-1324 an oral LANCL2 agonist in a randomized, double-blind, placebo-controlled phase I clinical trial. Clin Trans Sci. (2025) 18:e70129. doi: 10.1111/cts.70129
116. Morand E, Arriens C, Geraldino-Pardilla L, Clarke A, Pomponi S, Hobar C, et al. POS0527 DEUCRAVACITINIB, A FIRST-IN-CLASS, ORAL, SELECTIVE, ALLOSTERIC TYROSINE KINASE 2 (TYK2) INHIBITOR, IN SYSTEMIC LUPUS ERYTHEMATOSUS (SLE): EFFICACY BY BASELINE DEMOGRAPHICS AND DISEASE CHARACTERISTICS IN THE PHASE 2 PAISLEY TRIAL. Ann Rheumatic Dis. (2024) 83:945. doi: 10.1136/annrheumdis-2024-eular.250
117. Baribaud F, Saini J, Ignatenko S, Chadwick K, Zhu L, Bach HYT, et al. POS0551 PHARMACODYNAMIC EFFECTS AND EXPLORATORY EFFICACY OF AFIMETORAN, A TLR7/8 INHIBITOR, IN PATIENTS WITH CUTANEOUS LUPUS ERYTHEMATOSUS. Ann Rheumatic Dis. (2024) 83:1110–1. doi: 10.1136/annrheumdis-2024-eular.1540
118. Hobayan CGP, Korman A, and Lin J. Flesh-colored pinpoint papules with fine white spicules on the upper body. Cutis. (2024) 113:E11–E3. doi: 10.12788/cutis
119. AlDomyati RM, Showlag RA, Alghamdi GA, Al Ibrahim ZA, Alhemaidi GS, and Aljumaydi AT. Pediatric autoimmune disorders and their impact on dental development. J Oral Health Sci. (2024) 4:876–83. doi: 10.52533/JOHS.2024.41230
120. Tyagi S, Sihag Y, and Singh A. Bullous aplasia cutis congenita: A rare entity in the paradigm of congenital skin defect disorder. CosmoDerma (2024) 4:101. doi: 10.25259/CSDM_109_2024
121. Kailashiya V, Singh U, and Kailashiya J. Associations between serum autoantibodies, cytokines, complements, and clinical manifestations in SLE patients. Avicenna J Med Biochem. (2024) 12:106–13. doi: 10.34172/ajmb.2538
122. Schulz S. Periodontitis: current status and the future. Switzerland: MDPI-Multidisciplinary Digital Publishing Institute (2024).
123. Nahar N, Badhan RE, Ahmed S, Khan RR, and Setu SK. Association of interleukin-6 and interleukin-10 gene polymorphisms with susceptibility and clinical features of systemic lupus erythematosus in Bangladeshi population. Fortune J Health Sci. (2024) 7:676–85. doi: 10.26502/fjhs.237
124. Shaily A, Ryan M, and Gilbert M. Mere coincidence or an association? Case of juvenile idiopathic arthritis in a patient with Klinefelter syndrome. Pediatr Rheumatol. (2024) 22:1–2. doi: 10.1186/s12969-024-01042-7
125. Hanai S, Kobayashi Y, Watanabe M, Ikeda K, Kubota S, and Nakagomi D. Improvement of proteinuria by upadacitinib in a patient with refractory lupus membranous nephropathy. Rheumatology. (2025) 64:1548–9. doi: 10.1093/rheumatology/keae552
126. Ochi D, Cheema KS, Raychaudhuri SK, and Raychaudhuri SP. JAK-STAT targeted therapy for autoimmune diseases. In: Critical thinking in contemporary dermatology: cognitive essays. Singapore: Springer (2024). p. 251–64.
127. Hennebicq S, Silberberg D, and Dubois P. Libman-sacks endocarditis. Acta Cardiologica. (2024), 1–2. doi: 10.1080/00015385.2024.2445344
128. Gall S and Benson R. P02 Haemophagocytic lymphohistiocytosis in mixed connective tissue disease and infection. Rheumatol Adv Pract. (2024) 8:rkae117. 033. doi: 10.1093/rap/rkae117.033
129. Huang JJ, Singh N, Shadman M, Basom RS, Kalus A, Portuguese AJ, et al. Autoimmune outcomes in patients with concurrent autoimmune disease receiving CD19 CAR T-cell therapy for lymphoma. Blood. (2024) 144:7132. doi: 10.1182/blood-2024-201809
130. Şahin N, Uçak K, Atamyıldız Uçar S, Sönmez HE, and Sözeri B. Impact of perception of illness on quality of life in juvenile systemic lupus erythematosus. Lupus. (2024) 33:1476–82. doi: 10.1177/09612033241285622
131. Craig E and Cappelli LC. Gastrointestinal and hepatic disease in rheumatoid arthritis. Rheum Dis Clin North Am. (2018) 44:89–111. doi: 10.1016/j.rdc.2017.09.005
132. Zhang X, Liu L, Lin S, Duan X, Luo H, Wang Y, et al. The conditions that patients with systemic lupus erythematosus should fulfill before pregnancy to optimize outcomes: a large-scale multicenter cohort study from China. Arthritis Res Ther. (2025) 27:31. doi: 10.1186/s13075-025-03497-9
133. Kadoh Y, Yoshino J, Oka T, Itoga K, Hanada M, Niino D, et al. A case of posterior and reversible encephalopathy syndrome in a patient previously undiagnosed with lupus nephritis. CEN Case Rep. (2025) 14:328–34. doi: 10.1007/s13730-025-00973-8
134. Santacruz JC, Mantilla MJ, Pulido S, Bocanegra-Oyola N, Londoño JD, Agudelo C, et al. Superficial skin ulcers as an initial manifestation of systemic lupus erythematosus. Cureus. (2025) 17:e77701. doi: 10.7759/cureus.77701
135. Alharbe A, Almohnna H, Alqahtani A, Alshihry H, and AlShihry H. An uncommon presentation of tumid lupus erythematosus manifesting as annular, non-scarring alopecia on the scalp. Cureus. (2025) 17:e77085. doi: 10.7759/cureus.77085
136. Pazhyanur S, Lamberg O, Hauptman M, Cristiu J, Khan N, Billi AC, et al. Characterization of clinicopathological features and autoantibody profiles in patients with cutaneous lupus erythematous: A single-center retrospective study. Am J Clin Dermatol. (2025) 26:265–73. doi: 10.1007/s40257-024-00916-6
137. Huang C, Ding Y, Chen Z, Wu L, Wei W, Zhao C, et al. Future atherosclerotic cardiovascular disease in systemic lupus erythematosus based on CSTAR (XXVIII): the effect of different antiphospholipid antibodies isotypes. BMC Med. (2025) 23:8. doi: 10.1186/s12916-024-03843-9
138. Wang C, Chen B, Yu X, and Guan X. Macrophages unmasked: their pivotal role in driving atherosclerosis in systemic lupus erythematosus. Clin Rev Allergy Immunol. (2025) 68:1–17. doi: 10.1007/s12016-025-09025-6
139. Hasan MA, Alismail MA, Bokhari DR, Alghamdi RF, Alhalal ZE, AlQatari SG, et al. Pleuropulmonary involvement in patients with systemic lupus erythematosus as detected by high-resolution CT scans: clinical and immunological association. Medicina. (2025) 61:181. doi: 10.3390/medicina61020181
140. Guo A, Chen Y, Liu H, Gao S, Huang X, Liu D, et al. Predicting and validating the risk of interstitial lung disease in systemic lupus erythematosus. Int J Med Inf. (2025) 197:105839. doi: 10.1016/j.ijmedinf.2025.105839
141. Jain S, Khormi A, Nel L, Sangle S, and D’Cruz D. P49 Belimumab as a potential therapy for lupus enteritis–a case report. Rheumatol Adv Pract. (2024) 8:rkae117.080. doi: 10.1093/rap/rkae117.080
142. Odah AB, Awashra A, Sawaftah Z, Sawafta A, Sawafta O, Hamdan D, et al. Mixed connective tissue disease: A case of aggressive progression and multisystem involvement. Radiol Case Rep. (2025) 20:488–91. doi: 10.1016/j.radcr.2024.09.148
143. Feroz MF, Qazi ZA, and Bukhari SY. An investigation into blood-related disorders in individuals diagnosed with systemic lupus erythematosus (SLE). Int. J. Innov. Res. Technol. (2025) 11:1783–9.
144. Bathina A, Chintada DC, Yellu NKR, Vijayashree J, and Unnikrishnan P. Clinical and hematological manifestations of systemic lupus erythematosus at initial presentation in a tertiary healthcare center. Cureus. (2024) 16:e75956. doi: 10.7759/cureus.75956
145. Wang JJ, Lin MW, Suan D, Beroukas D, Gordon TP, and Lee AY. Clinical correlations of serum anti-dsDNA immunoglobulin subfamilies in patients with systemic lupus erythematosus (SLE). Autoimmunity. (2025) 58:2441992. doi: 10.1080/08916934.2024.2441992
146. Tang Z and Huang L. Systemic lupus erythematosus is associated with changes in brain function and structure: A multimodal meta-analysis of neuroimaging studies. (2025) 4:101.
147. Fortuna G and Brennan MT. Systemic lupus erythematosus: epidemiology, pathophysiology, manifestations, and management. Dent Clin North Am. (2013) 57:631–55. doi: 10.1016/j.cden.2013.06.003
148. Touma Z, Sayani A, Pineau CA, Fortin I, Matsos M, Ecker GA, et al. Belimumab use, clinical outcomes and glucocorticoid reduction in patients with systemic lupus erythematosus receiving belimumab in clinical practice settings: results from the OBSErve Canada Study. Rheumatol Int. (2017) 37:865–73. doi: 10.1007/s00296-017-3682-9
149. Su X, Yu H, Lei Q, Chen X, Tong Y, Zhang Z, et al. Systemic lupus erythematosus: pathogenesis and targeted therapy. Mol Biomedicine. (2024) 5:54. doi: 10.1186/s43556-024-00217-8
150. van Vollenhoven RF, Hahn BH, Tsokos GC, Wagner CL, Lipsky P, Touma Z, et al. Efficacy and safety of ustekinumab, an IL-12 and IL-23 inhibitor, in patients with active systemic lupus erythematosus: results of a multicentre, double-blind, phase 2, randomised, controlled study. Lancet. (2018) 392:1330–9. doi: 10.1016/S0140-6736(18)32167-6
151. A Two-year, Phase III Randomized, Double-blind, Parallel-group, Placebo-controlled Trial to Evaluate the Safety, Efficacy, and Tolerability of 300 mg s.c. Secukinumab Versus Placebo, in Combination With SoC Therapy, in Patients With Active Lupus Nephritis. Clinicaltrials.gov Identifier NCT04181762 (2019).
152. Illei GG, Shirota Y, Yarboro CH, Daruwalla J, Tackey E, Takada K, et al. Tocilizumab in systemic lupus erythematosus: data on safety, preliminary efficacy, and impact on circulating plasma cells from an open-label phase I dosage-escalation study. Arthritis Rheumatism: Off J Am Coll Rheumatol. (2010) 62:542–52. doi: 10.1002/art.27221
153. Wallace DJ, Strand V, Merrill JT, Popa S, Spindler AJ, Eimon A, et al. Efficacy and safety of an interleukin 6 monoclonal antibody for the treatment of systemic lupus erythematosus: a phase II dose-ranging randomised controlled trial. Ann Rheum Dis. (2017) 76:534–42. doi: 10.1136/annrheumdis-2016-209668
154. Xu Z, Bouman-Thio E, Comisar C, Frederick B, Van Hartingsveldt B, Marini JC, et al. Pharmacokinetics, pharmacodynamics and safety of a human anti-IL-6 monoclonal antibody (sirukumab) in healthy subjects in a first-in-human study. Br J Clin Pharmacol. (2011) 72:270–81. doi: 10.1111/j.1365-2125.2011.03964.x
155. Llorente L, Richaud-Patin Y, García-Padilla C, Claret E, Jakez-Ocampo J, Cardiel MH, et al. Clinical and biologic effects of anti–interleukin-10 monoclonal antibody administration in systemic lupus erythematosus. Arthritis Rheumatism: Off J Am Coll Rheumatol. (2000) 43:1790–800. doi: 10.1002/1529-0131(200008)43:8<1790::AID-ANR15>3.0.CO;2-2
156. Horiuchi T, Mitoma H, Harashima S, Tsukamoto H, and Shimoda T. Transmembrane TNF-alpha: structure, function and interaction with anti-TNF agents. Rheumatol (Oxford). (2010) 49:1215–28. doi: 10.1093/rheumatology/keq031
157. Said JT, Elman SA, and Merola JF. Evaluating safety and compatibility of anti-tumor necrosis factor therapy in patients with connective tissue disorders. Ann Transl Med. (2021) 9:430. doi: 10.21037/atm-20-5552
158. Aringer M, Houssiau F, Gordon C, Graninger WB, Voll RE, Rath E, et al. Adverse events and efficacy of TNF-alpha blockade with infliximab in patients with systemic lupus erythematosus: long-term follow-up of 13 patients. Rheumatol (Oxford). (2009) 48:1451–4. doi: 10.1093/rheumatology/kep270
159. Aringer M, Graninger WB, Steiner G, and Smolen JS. Safety and efficacy of tumor necrosis factor alpha blockade in systemic lupus erythematosus: an open-label study. Arthritis Rheum. (2004) 50:3161–9. doi: 10.1002/art.20576
160. Jacob N and Stohl W. Cytokine disturbances in systemic lupus erythematosus. Arthritis Res Ther. (2011) 13:228. doi: 10.1186/ar3349
161. Danion F, Sparsa L, Arnaud L, Alsaleh G, Lefebvre F, Gies V, et al. Long-term efficacy and safety of antitumour necrosis factor alpha treatment in rhupus: an open-label study of 15 patients. RMD Open. (2017) 3:e000555. doi: 10.1136/rmdopen-2017-000555
162. Yang BB, Xiao H, Li XJ, and Zheng M. Safety and efficacy of etanercept-methotrexate combination therapy in patients with rhupus: an observational study of non-glucocorticoid treatment for rheumatic diseases. Discov Med. (2018) 25:14–20.
163. Micheloud D, Nuño L, Rodríguez-Mahou M, Sánchez-Ramón S, Ortega MC, Aguarón A, et al. Efficacy and safety of Etanercept, high-dose intravenous gammaglobulin and plasmapheresis combined therapy for lupus diffuse proliferative nephritis complicating pregnancy. Lupus. (2006) 15:881–5. doi: 10.1177/0961203306070970
164. Norman R, Greenberg RG, and Jackson JM. Case reports of etanercept in inflammatory dermatoses. J Am Acad Dermatol. (2006) 54:S139–42. doi: 10.1016/j.jaad.2005.11.1090
165. Picardo S, So K, and Venugopal K. Anti-TNF-induced lupus in patients with inflammatory bowel disease. JGH Open. (2020) 4:507–10. doi: 10.1002/jgh3.12291
166. Chen P, Vu T, Narayanan A, Sohn W, Wang J, Boedigheimer M, et al. Pharmacokinetic and pharmacodynamic relationship of AMG 811, an anti-IFN-gamma IgG1 monoclonal antibody, in patients with systemic lupus erythematosus. Pharm Res. (2015) 32:640–53. doi: 10.1007/s11095-014-1492-2
167. Welcher AA, Boedigheimer M, Kivitz AJ, Amoura Z, Buyon J, Rudinskaya A, et al. Blockade of interferon-gamma normalizes interferon-regulated gene expression and serum CXCL10 levels in patients with systemic lupus erythematosus. Arthritis Rheumatol. (2015) 67:2713–22. doi: 10.1002/art.39248
168. Werth VP, Fiorentino D, Sullivan BA, Boedigheimer MJ, Chiu K, Wang C, et al. Brief report: pharmacodynamics, safety, and clinical efficacy of AMG 811, a human anti-interferon-γ Antibody, in patients with discoid lupus erythematosus. Arthritis Rheumatol. (2017) 69:1028–34. doi: 10.1002/art.40052
169. Boedigheimer MJ, Martin DA, Amoura Z, Sánchez-Guerrero J, Romero-Diaz J, Kivitz A, et al. Safety, pharmacokinetics and pharmacodynamics of AMG 811, an anti-interferon-γ monoclonal antibody, in SLE subjects without or with lupus nephritis. Lupus Sci Med. (2017) 4:e000226. doi: 10.1136/lupus-2017-000226
Keywords: SLE, cytokines, systemic features, oral features, therapeutic targets
Citation: Elemam NM, Talaat IM and El Meligy OA (2025) Cytokines unveiled: their impact on oral and multisystem features of lupus erythematosus. Front. Immunol. 16:1585280. doi: 10.3389/fimmu.2025.1585280
Received: 28 February 2025; Accepted: 30 June 2025;
Published: 28 July 2025.
Edited by:
Yasuhiro Shimojima, Shinshu University, JapanReviewed by:
Melissa E. Munroe, Oklahoma Medical Research Foundation, United StatesBenjamin Klein, University of Michigan, United States
Copyright © 2025 Elemam, Talaat and El Meligy. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Iman M. Talaat, aXRhbGFhdEBzaGFyamFoLmFjLmFl