 Jiajia Li
Jiajia Li Zhangxue Hu
Zhangxue Hu- Department of Nephrology, West China Hospital, Sichuan University, Chengdu, China
Acute kidney injury (AKI) is a clinical syndrome characterized by a sudden dysfunction of the kidney, which is common worldwide, with a relatively high incidence and mortality rate. Damage to the proximal renal tubule is a pathological hallmark of AKI, and inflammation triggered by the overactivation of the immune system is a common cause of proximal renal tubular injury, which is an important contributing factor in AKI exacerbation. Damage-associated molecular patterns (DAMPs) are endogenous molecules released by cells in response to external stimuli that can trigger an inflammatory response by binding to specific pattern recognition receptors (PRRs). Numerous studies have indicated that when the kidney is exposed to external stress or chemical stimuli, injured cells actively secrete or passively release various DAMPs, which can exacerbate or attenuate kidney injury by stimulating or inhibiting the inflammatory response through binding to the appropriate receptor. Currently, there is a lack of early diagnostic biomarkers and specific therapeutic strategies for AKI in the clinic have been established, and given the important role of the release of DAMPs in the regulation of inflammatory response, they will highly likely become favorable candidate biomarkers and clinical therapeutic targets for AKI. Therefore, a deeper understanding of the types of DAMPs and the specific mechanisms of their actions will provide more possibilities for the specific AKI diagnosis and treatment.
1 Introduction
Acute kidney injury (AKI) is a clinical syndrome of rapid decline in renal function caused by various etiologies (1, 2). The decrease in the estimated glomerular filtration rate (eGFR) causes fluid retention and acid–base imbalance, leading to injuries of multiple systems such as the heart, brain, lung, and gastrointestinal (3–6). Severe AKI may cause chronic kidney disease (CKD) and even permanent loss of kidney function. Given its high incidence, mortality and risk for CKD, AKI has remained a global health problem (7–10). The incidence of AKI in patients during hospitalization was reported approximately 10%-15% (11). However, among patients in the intensive care unit (ICU), the incidence became as high as 50% (12). A Canadian study that included approximately 200,000 patients hospitalized for AKI revealed that the 1-year mortality rate for patients with AKI was approximately 28%. Among those who survived, approximately 14% were rehospitalized due to recurrence, with approximately 45% dying within 1 year of hospitalization (13). In China, the all-cause hospital mortality rate of patients with AKI reached 12.4%, and most of them died of multiple organ dysfunction and sepsis (14). Furthermore, some cases of AKI progress to CKD, even end-stage renal disease, with an unfavorable prognosis (5, 15). Apart from the above characteristics, the high hospitalization rate of patients with AKI and the large proportion of medical costs should not be ignored. According to a nationwide cross-sectional study conducted in China, approximately 1.4 million people with AKI were hospitalized in 2013, with hospitalization costs amounting to approximately $13 billion, accounting for 10% of China’s total healthcare expenditure (14). Despite the high proportion of AKI-related mortality and medical costs, no specific drugs have been approved for the treatment of patients with AKI. Current clinical management includes addressing the underlying causes, controlling blood glucose and blood pressure, avoiding nephrotoxic drugs, volume management, hemodynamic monitoring, and renal replacement therapy. Though these approaches partially improve the prognosis of patients with AKI, the CKD risk and death in patients with AKI remains dismal, and new therapeutic strategies need to be explored (4, 16–18).
Factors commonly associated with AKI development include ischemia–reperfusion injury (IRI), sepsis, hemodynamic alterations, systemic inflammation, and use of nephrotoxic drugs, which are often associated with sterile inflammation (3, 6, 10). In the early stage of AKI, various immune cells are recruited to the kidney, releasing proinflammatory mediators and exacerbating renal impairment. In the later AKI stage, the dynamic interaction of pro-inflammatory and anti-inflammatory mediators released by immune cells mediates AKI development, ultimately causing the inflammation to subside and renal tissue damage to be repaired or progressing to CKD (19–21). Therefore, an in-depth study of the mechanisms that regulate sterile renal inflammation will help in AKI prevention and treatment.
Sterile inflammation is an inflammatory response that occurs in the absence of pathogens and usually relies on the release of damage-associated molecular patterns (DAMPs) (22, 23). DAMPs are endogenous molecules released by cells in response to unfavorable stimuli from the internal or external environment. These endogenous molecules are normally sequestered in their respective intracellular compartments under physiological conditions (24, 25). However, when cells are exposed to deleterious stimuli, they are released into the extracellular space, activating the immune system and triggering a sterile inflammatory response (26–28). DAMPs are mainly released passively by dying cells, and the former includes various forms of death such as necrosis, necroptosis, and pyroptosis, which lead to DAMPs leakage through the destruction of the plasma membrane or formation of channel pores into the extracellular space (28, 29). In addition to passive release, some DAMPs can be actively secreted from living cells through vesicular transport by docking and fusion with the plasma membrane through the exosomal and lysosomal exocytosis pathways (30, 31). These released DAMPs bind to specific pattern recognition receptors (PRRs), such as toll-like receptors (TLRs) and receptor for advanced glycation end products (RAGE), inducing nuclear factor-kappa B (NF-κB) transcription and the activation of the NOD-like receptor family pyrin domain-containing protein 3 (NLRP3) inflammasome, which promotes the recruitment of various immune cells and the release of inflammatory factors, which subsequently trigger a sterile inflammatory response (22, 25, 28, 32).
The death of proximal renal tubular epithelial cells is a key factor that leads to AKI, and oxidative stress, necrosis, apoptosis, and inflammation are crucial in AKI development (20, 33–35). Given that inflammation is an important AKI driver and DAMPs are crucial in regulating inflammation, this review focuses on the role of the inflammatory response participated by DAMPs in AKI. When the kidney suffers from adverse stimuli such as major surgical trauma or nephrotoxic drugs, inflammatory DAMPs released by damaged cells participate in AKI development by binding to the PRRs, stimulating and amplifying inflammatory response signals (36–38). Interestingly, not all DAMPs exacerbate AKI, and some DAMPs have anti-inflammatory effects that promote the repair of damaged tissues and protect the kidney (39–41). Therefore, an in-depth understanding of certain roles played by different DAMPs during the immune response and damage–repair process involved in AKI may contribute to the early diagnosis and discovery of therapeutic targets for AKI. This review focuses on the types of DAMPs in AKI (Table 1) and describes the functions of DAMPs in AKI via binding to different PRRs (Figure 1).
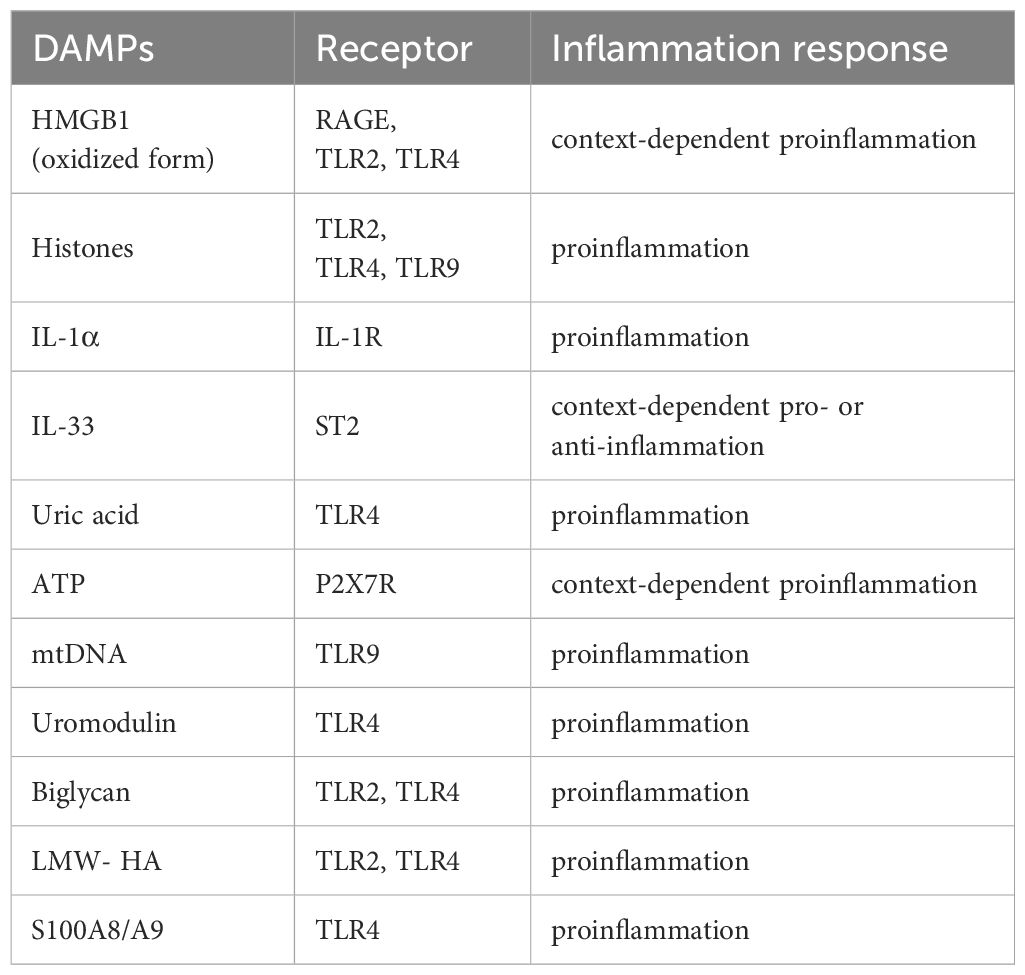
Table 1. Overview of the types of DAMPs in AKI.
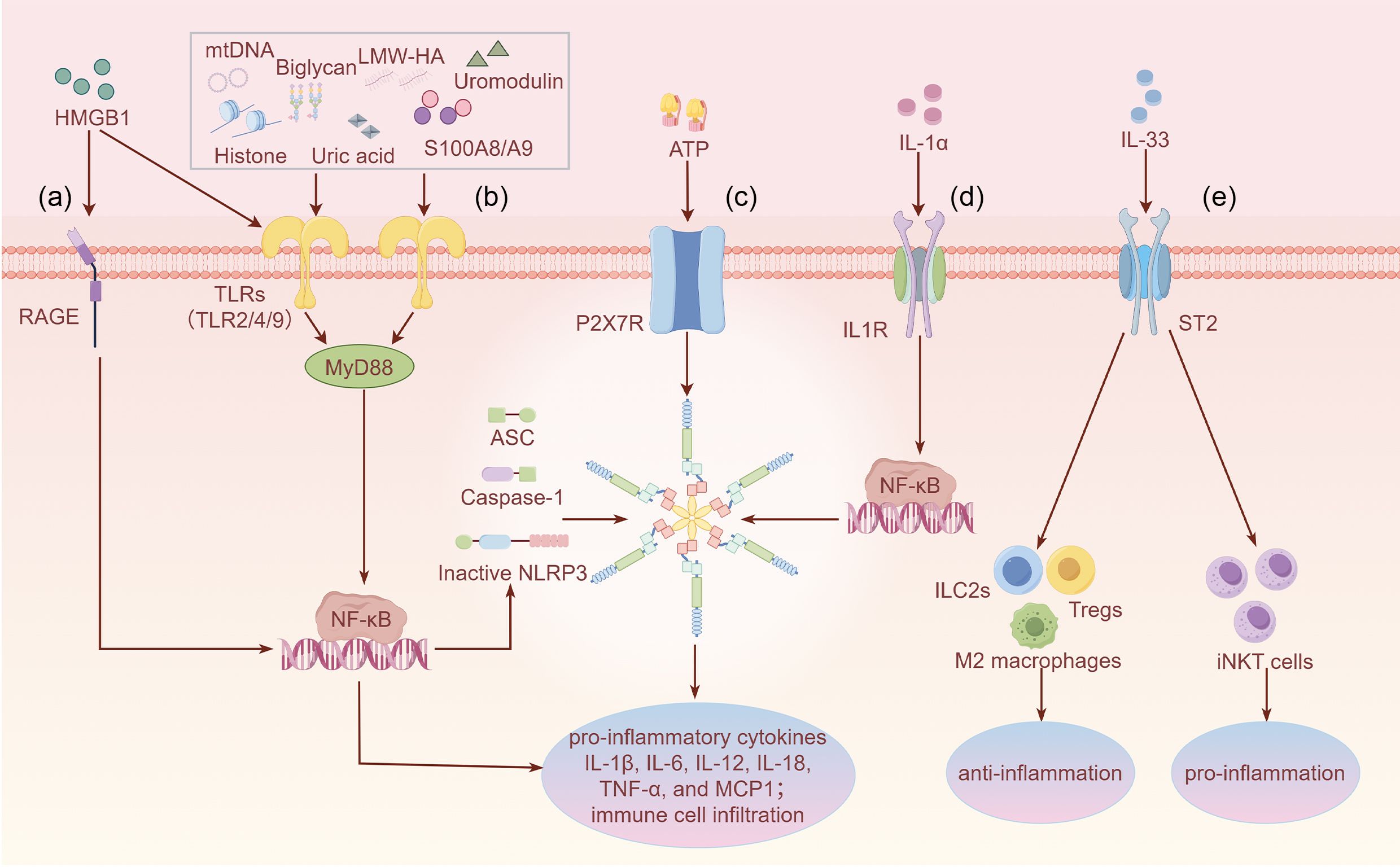
Figure 1. Overview of the specific functions of DAMPs in AKI through binding to different PRRs (a) HMGB1 activates RAGE, leading to downstream NF-κB signaling and inflammatory factor release. (b) Most DAMPs such as HMGB1, histones, uric acid, mtDNA, uromodulin, biglycan, LMW-HA, and S100A8/A9 activate TLR2/4/9, exacerbating the inflammatory signaling cascade. Only the downstream canonical MyD88/NF-κB signaling pathway is shown. (c) ATP regulates NLRP3 inflammasome signaling through the activation of P2X7R and subsequently promotes inflammatory responses. (d) IL-1α activates IL-1R, promoting NF-κB translocation to the nucleus and activating inflammatory signaling. (e) IL-33 activates ST2, which promotes polarization of M2 macrophages and induces the expansion of ILC2s and Tregs to fight inflammation. Meanwhile, IL-33 binds to ST2 on the surface of iNKT cells, amplifying inflammatory injury. IL-33 exhibits dual functions, which may depend on the specific immune microenvironment. Footnote: Figure 1 only shows the function of IL-1α and IL-33 as DAMPs rather their function as secreted cytokines.
2 Functions and release mechanisms of DAMPs
2.1 High mobility group box 1
HMGB1 is a non-histone DNA-binding protein that is widely expressed in the cellular environment and plays a crucial role in regulating DNA replication, DNA repair, and nucleosome stability, which are essential for cell growth and differentiation (42, 43). In response to adverse external stimuli, HMGB1 can be released passively by necrotic cells because of plasma membrane destruction (44, 45). Cells undergoing necroptosis, a regulated form of necrotic cell death, involving specific signaling molecules such as receptor-interacting protein kinase 1 and 3, and mixed-lineage kinase domain-like protein, also release HMGB1, which ultimately leads to membrane depolarization and cell rupture (46–49). In addition, HMGB1 can be actively secreted by immune cells, which involves multiple post-translational modifications (PTMs) of HMGB1, including oxidation, acetylation, lactation, and phosphorylation. Various PTMs promote interactions between HMGB1 and the nuclear transport receptors that mediate the translocation of HMGB1 from the nucleus to the cytoplasm and its subsequent release into the extracellular space through the exosomal and lysosomal exocytosis pathways (43, 50–52). Notably, the proinflammatory function of HMGB1 is determined by its redox status, and the oxidized form of HMGB1, but not its reduced form, determines the proinflammatory activity of HMGB1 once it is released (53). The oxidized form of extracellular HMGB1 interacts with TLR2, TLR4, and RAGE, expressed on various immune cells, including monocytes, macrophages, and dendritic cells, activating NF-κB transcription and inducing the expression of proinflammatory cytokines, such as interleukin-6 (IL-6), tumor necrosis factor α (TNF-α), and monocyte chemoattractant protein 1 (MCP1), leading to inflammation and worsening injury of the kidney (54–57). Several clinical studies have shown the high serum level of HMGB1 in patients suffering from AKI (58, 59). Studies have reported that HMGB1 inhibitors (60–62) and HMGB1-neutralizing antibodies (63, 64) are effective in improving kidney function and attenuating kidney damage in various AKI mouse models.
2.2 Histones
Histones are basic proteins with highly conserved sequences found in the nucleus and influence the stabilization chromatin structure and regulate gene expression under normal physiological conditions (65, 66). Histones can be released passively during apoptosis and necrosis, triggering an inflammatory response by binding to the corresponding PRRs (67–70). Recently, neutrophil extracellular traps (NETs) are a widely reported form of histone release, an extracellular DNA mesh-like structure composed of histones and anti-microbial protein, which are generally released by dying neutrophils (71). Adverse stimuli induce peptidylarginine deiminase 4 (PAD4) and neutrophil elastase (NE) to initiate chromatin decondensation in the nucleus and the breakdown of the nuclear membrane. Finally, with the breakdown of the plasma membrane, histones are released into the extracellular space (72, 73). As a proinflammatory DAMP, histones activate the downstream MyD88 and NF-κB signaling pathway by interacting with TLR2, TLR4, and TLR9, and produce inflammatory cytokines such as IL-6, TNF-α, and MCP1, exacerbating the renal inflammatory response (64, 74). NETs can also be actively secreted from living neutrophils through vesicular transport (71, 75).
2.3 Interleukin-1
Interleukin-1α (IL-1α) and interleukin-33 (IL-33) are members of the interleukin-1 (IL-1) family that are both cytokines and can act as DAMPs, which are passively released by necrotic cells into the extracellular space due to plasma membrane destruction (76–78). IL-1α can also be actively released by macrophages through gasdermin D (GSDMD) pore formation during pyroptosis, a form of regulated cell death (77, 79). IL-1α is present and biologically active in epithelial cells under physiological conditions (80). Upon external stimuli, IL-1α is released from damaged cells and binds to the IL-1 receptor (IL-1R) on nearby cells, activating the NF-κB signaling pathway, inducing TNF-α production, and mediating the recruitment of immune cells, which promotes kidney injury (34, 81–83). IL-33 is constitutively expressed in the nucleus of endothelial, epithelial, and other structural cells in the physiological state (84–86). IL-33 is a pleiotropic cytokine that mediates tissue inflammation and repair by binding to the specific receptor tumorigenicity 2 (ST2). ST2 is expressed on the surface of multiple immune cells, including nature killer (NK) cells, regulatory T cells (Tregs), neutrophils, macrophages, B cells, and natural killer T (NKT) cells (87). In mice model of IRI-induced AKI, IL-33 was passively released by dying endothelial cells and bound to ST2 as a ligand, which induced the conversion of proinflammatory M1 macrophages into anti-inflammatory M2 macrophages, reducing the release of proinflammatory factors, whereas IL-33 promotes the expansion of type II innate lymphoid cells (ILC2s) and Tregs, improving the immune microenvironment and reducing renal inflammation (88, 89). Interestingly, IL-33 released into the extracellular space can also act as a proinflammatory DAMP targeting invariant NKT (iNKT) cells, recruiting neutrophils to infiltrate the kidney and amplifying inflammatory injury in a mouse model of IRI-induced AKI (90–92). IL-33 exhibits dual functions in AKI, which may depend on the type of immune cells activated by IL-33 and the specific immune microenvironment, which will require more in-depth studies to elucidate the complex regulatory mechanisms of the IL-33/ST2 signaling pathway in AKI.
2.4 Uric acid
Uric acid is generated through the metabolic degradation of purines, approximately 70% of it is excreted via the kidney, and it has long been recognized as a significant risk factor for kidney-related diseases (93, 94). Uric acid is released passively from the damaged cells. Uric acid, particularly in its crystalline form, can act as a proinflammatory DAMP. Uric acid crystals bind to TLRs (e.g., TLR2 and TLR4), providing priming signals, and activate the NLRP3 inflammasome to induce the production of IL-1β, TNF-α, and MCP1, and induce the recruitment of immune cells, exacerbating renal inflammatory responses (28, 95–97). Many clinical approaches for the treatment of kidney-associated disorders, such as allopurinol and febuxostat administration, can effectively attenuate renal function partly by reducing circulating uric acid concentrations (98). This conclusion was validated in animal experiments, where allopurinol improved renal function by lowering the serum uric acid concentration in animal models of heat stress, rhabdomyolysis, and exercise-induced AKI (99, 100). Febuxostat can ameliorate contrast and IRI-induced AKI by the same mechanism (101–103). In-hospital mortality in patients with AKI appears to be associated with high serum uric acid levels, which is expected to be a prognostic marker for AKI (104).
2.5 Adenosine triphosphate
ATP serves as a vital energy source for cells, and mitochondrial oxidative phosphorylation regulates intracellular ATP levels (105, 106). ATP can be released into the extracellular space either passively or in a regulated manner. Passive release occurs from damaged or necrotic cells through plasma membrane rupture (22, 107). In contrast, regulated ATP release from living cells occurs via specific pathways such as pannexin or connexin hemichannels, as well as vesicular transport mechanisms (108, 109). Extracellular ATP is recognized as a proinflammatory DAMP that binds to the P2X7 purinergic receptor (P2X7R), causing potassium efflux, which activates NLRP3 inflammasome (110). In IRI and sepsis-induced AKI, ATP can activate NLRP3 inflammasome by binding to P2X7R, promoting the production of proinflammatory factors including IL-1β, IL-6, and MCP1 and impairing the kidney (111, 112). P2X7R knockdown ameliorated IRI and cisplatin-induced AKI and AKI progression to renal fibrosis by inhibiting the activation of NLRP3 inflammasome (112, 113). Similarly, studies have indicated that the use of P2X7R antagonists mediates the inactivation of NLRP3 inflammasome and treats IRI and sepsis-induced AKI (114, 115). This predicts an optimistic therapeutic prospect for P2X7R antagonists in AKI of multiple etiologies.
2.6 Mitochondrial DNA
mtDNA, a double-stranded circular DNA molecule, is involved in encoding the composition of the core subunits of the respiratory chain, which is important for the maintenance of mitochondrial function and cellular metabolism (116, 117). In recent years, mtDNA has also been recognized as a proinflammatory DAMP that plays a role in various diseases (118, 119). Depletion of mtDNA has been observed during AKI, which is closely related to the degree of renal damage (120). During cellular stress, mitochondrial homeostasis is disrupted, prompting the opening of the mitochondrial permeability transition pore, followed by the release of mtDNA into the cytoplasm, The leaking mtDNA acts as an endogenous ligand for the cyclic GMP–AMP synthase (cGAS), which triggers type I interferon responses via the stimulator of interferon gene (STING) signaling pathway, and also activates the classical NF-κB inflammatory response, which produces inflammatory cytokines such as TNF-α and IL-6 (118, 121). By binding to TLR9, mtDNA can also activate NF-κB transcription, leading to the release of proinflammatory factors and immune cell infiltration, amplifying the inflammatory response (122–124). In addition, mtDNA can directly activate NLRP3 inflammasome and enhances the release of the inflammatory cytokines IL-1β and IL-18 (125–127). Some studies have shown significant correlations among urinary mtDNA level and serum creatinine, eGFR, and the AKI marker neutrophil gelatinase-associated lipocalin (NGAL), suggesting the promising potential of urinary mtDNA as a biomarker for predicting AKI severity, which needs to be further validated by larger, multicenter-like cohort studies in the future to establish the role of urinary mtDNA in predicting AKI severity (128–130).
2.7 Uromodulin
Uromodulin, also known as Tamm-Horsfall protein (THP), is a kidney-specific glycoprotein produced mainly by the epithelial cells of the ascending limb of the Henle loop, which is excreted into the urine mainly by apical secretion, and is one of the most abundant urinary proteins (131). Physiologically, uromodulin is not immunologically active in the tubular lumen of the renal tubule but serves multiple protective functions, such as resisting urinary tract infections and preventing urinary stone formation (132–134). However, when renal tubular cells are damaged, uromodulin may be partially leaked into the renal interstitial compartment, and mislocalized uromodulin is thought to be a proinflammatory DAMP, which induces renal inflammation by binding to TLR4, activating NLRP3 inflammasome, and inducing the activated aggregation of immune cells and secretion of the proinflammatory factor IL-1β (135–137). The specific receptors and molecules involved in immune cells modulation by uromodulin leaking from the renal interstitial compartment require more in-depth studies.
2.8 Biglycan
Biglycan, a small leucine-rich proteoglycan expressed mainly in the renal mesenchyme and present in the extracellular matrix (ECM) in the physiological state, is a key ECM component that plays an important role in angiogenesis, inflammation, and autophagy (136, 138). Dying cells can release biglycan through protein hydrolysis. The released biglycan exists in its soluble form and is seen as a DAMP, which exerts proinflammatory function by binding to the corresponding receptors (19). Biglycan can activate the downstream MyD88 and NF-κB inflammatory signaling pathway by binding to TLR2 and TLR4, inducing the infiltration of various immune cells such as T-cells and neutrophils, polarization of M1 macrophages, and production of TNF-α, IL-1β, C-C motif chemokine ligand 2 (CCL2) and CCL5, thereby exacerbating the inflammatory injury in IRI-induced AKI (139–141).
2.9 Hyaluronic acid
HA is another important ECM component. It is primarily composed of repeating disaccharide chains of D-glucuronic acid and D-N-acetylglucosamine, which play important roles in maintaining tissue integrity and regulating cellular functions (142–144). The biological function of HA depends on its molecular weight, and in the physiological state, it exists mainly as high-molecular-weight HA (HMW-HA) (145, 146). HMW-HA binds to CD44, a main receptor of HA, to promote the polarization of M1 to M2 macrophages and inhibit NF-κB activation, reducing the expression of inflammatory factors including IL-6 and IL-8 and thus reducing systemic inflammation and maintaining immune balance (147–149). During cell injury, HMW-HA in the ECM is degraded by hyaluronidases to low-molecular-weight HA (LMW-HA), which is regarded as a DAMP. By binding to TLR2 and TLR4, LMW-HA activates dendritic cells and activates the NF-κB signaling pathway, increasing the release of the proinflammatory cytokines IL-1β, IL-6, TNF-α, and IL-12 (145, 150). LMW-HA also induces the expression of chemokines such as CCL3 and CCL4, recruiting immune cells and exacerbating inflammatory responses (150, 151). The inhibition of HMW-HA degradation alleviates renal inflammation and protects renal function in a mouse model of IRI-induced AKI (152).
2.10 S100A8/A9
S100A8 and S100A9 are calcium-binding proteins that belong to the S100 protein family and are usually present as heterodimers in a high-calcium environment. The S100A8/A9 heterodimer is predominantly expressed in the cytoplasm of myeloid cells and is involved in regulating the polymerization of microtubules (153). S100A8/A9 released into the extracellular space is regarded as a DAMP and helps in regulating immune responses (154, 155). S100A8/A9 can be actively secreted or passively released into the extracellular space. Passive release occurs through dying cells or GSDMD pore, or by neutrophils through the formation of NETs, whereas active secretion occurs mainly by macrophages via the lysosomal exocytosis pathway (153, 156, 157). In a mouse model of IRI-induced AKI, S100A8/A9 activates the NF-κB signaling pathway by binding to TLR4 and promotes the infiltration of the proinflammatory cytokines IL-6, IL-1β, and TNF-α and the aggregation of macrophages and neutrophils, thereby exacerbating renal inflammation (157–159). A study revealed that S100A8/A9 inhibitor attenuates the binding of S100A8/A9 to TLR4, alleviates the inflammatory response to sepsis-induced AKI, and protects the kidney (160).
3 Conclusions
Despite current advances in investigating the pathological mechanisms of AKI, the lack of diagnostic and prognostic biomarkers for AKI and specific AKI therapeutic strategies are major clinical challenges that need to be addressed. This review preliminarily explored the release mechanisms and functions of DAMPs in regulating immune responses, revealing that not all DAMPs are exacerbating factors for AKI; some DAMPs promote AKI repair and protect the kidney, which may depend in part on the specific immune microenvironment or molecular weight in which the DAMPs are located. In the mouse model of IRI-induced AKI, IL-33 binds to ST2 on the surface of macrophages, ILC2s, and Tregs, reducing renal inflammation; however, IL-33 also binds to ST2 on the surface of iNKT cells, recruiting neutrophils and amplifying inflammatory injury. The complex mechanisms by which the IL-33/ST2 signaling pathway expresses a dual function in AKI may be due to the binding of IL-33 to ST2 on the surface of different immune cells and the specific immune microenvironment in which it resides. The complex mechanisms through which the IL-33/ST2 signaling pathway expresses a dual function in AKI warrant more in-depth investigation. The dual function of HA depends on its molecular weight. In the physiological context, HA predominantly exists as HMW-HA, which can inhibit NF-κB activation, reduce the expression of inflammatory factors such as IL-6 and IL-8, and exhibit anti-inflammatory function. Conversely, during stress, HMW-HA is degraded by hyaluronidases to LMW-HA, which acts as a proinflammatory DAMP. Moreover, serum uric acid was highly correlated with in-hospital mortality in patients with AKI, and urinary mtDNA was significantly correlated with serum creatinine, eGFR, and NGAL, which all appear to indicate that some DAMPs have predictive value for AKI. In addition, antagonists or neutralizing antibodies targeting some of the DAMPs (HMGB1 and S100A8/A9) or PRR (P2X7R) could protect to a certain extent against multiple etiologically induced AKI, which in turn highlights the great potential of DAMPs in clinical therapy. However, further studies are needed, including the identification of more DAMPs and elucidation of the detailed pathway and relationship with clinical features, to help us identify AKI early and contribute to the development of drugs for the treatment of AKI.
Author contributions
JL: Conceptualization, Data curation, Investigation, Methodology, Software, Validation, Writing – original draft, Writing – review & editing. ZH: Conceptualization, Methodology, Resources, Supervision, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Acknowledgments
The authors are very grateful to West China Hospital for providing the research platform and resources to support the manuscript. Figure 1 was created by Figdraw (www.figdraw.com).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Li S, Zhou L, Huang Y, and Tang S. Emerging Frontiers in acute kidney injury: The role of extracellular vesicles. Bioactive materials. (2025) 48:149–70. doi: 10.1016/j.bioactmat.2025.02.018
2. Ostermann M, Lumlertgul N, Jeong R, See E, Joannidis M, and James M. Acute kidney injury. Lancet. (2025) 405:241–56. doi: 10.1016/S0140-6736(24)02385-7
3. Mehta RL, Cerda J, Burdmann EA, Tonelli M, Garcia-Garcia G, Jha V, et al. International Society of Nephrology’s 0by25 initiative for acute kidney injury (zero preventable deaths by 2025): a human rights case for nephrology. Lancet. (2015) 385:2616–43. doi: 10.1016/S0140-6736(15)60126-X
4. Kellum JA, Romagnani P, Ashuntantang G, Ronco C, Zarbock A, and Anders HJ. Acute kidney injury. Nat Rev Dis Primers. (2021) 7:52. doi: 10.1038/s41572-021-00284-z
5. Pickkers P, Darmon M, Hoste E, Joannidis M, Legrand M, Ostermann M, et al. Acute kidney injury in the critically ill: an updated review on pathophysiology and management. Intensive Care Med. (2021) 47:835–50. doi: 10.1007/s00134-021-06454-7
6. Takkavatakarn K, Oh W, Chan L, Hofer I, Shawwa K, Kraft M, et al. Machine learning derived serum creatinine trajectories in acute kidney injury in critically ill patients with sepsis. Crit Care. (2024) 28:156. doi: 10.1186/s13054-024-04935-x
7. Molema G, Zijlstra JG, van Meurs M, and Kamps J. Renal microvascular endothelial cell responses in sepsis-induced acute kidney injury. Nat Rev Nephrol. (2022) 18:95–112. doi: 10.1038/s41581-021-00489-1
8. Matsuura R, Doi K, and Rabb H. Acute kidney injury and distant organ dysfunction-network system analysis. Kidney Int. (2023) 103:1041–55. doi: 10.1016/j.kint.2023.03.025
9. Zarbock A, Forni LG, Ostermann M, Ronco C, Bagshaw SM, Mehta RL, et al. Designing acute kidney injury clinical trials. Nat Rev Nephrol. (2024) 20:137–46. doi: 10.1038/s41581-023-00758-1
10. Gong S, Zhang A, Yao M, Xin W, Guan X, Qin S, et al. REST contributes to AKI-to-CKD transition through inducing ferroptosis in renal tubular epithelial cells. JCI Insight. (2023) 8:e166001. doi: 10.1172/jci.insight.166001
11. Al-Jaghbeer M, Dealmeida D, Bilderback A, Ambrosino R, and Kellum JA. Clinical decision support for in-hospital AKI. J Am Soc Nephrol. (2018) 29:654–60. doi: 10.1681/ASN.2017070765
12. Hoste EA, Bagshaw SM, Bellomo R, Cely CM, Colman R, Cruz DN, et al. Epidemiology of acute kidney injury in critically ill patients: the multinational AKI-EPI study. Intensive Care Med. (2015) 41:1411–23. doi: 10.1007/s00134-015-3934-7
13. Silver SA, Harel Z, McArthur E, Nash DM, Acedillo R, Kitchlu A, et al. Causes of death after a hospitalization with AKI. J Am Soc Nephrol. (2018) 29:1001–10. doi: 10.1681/ASN.2017080882
14. Yang L, Xing G, Wang L, Wu Y, Li S, Xu G, et al. Acute kidney injury in China: a cross-sectional survey. Lancet. (2015) 386:1465–71. doi: 10.1016/S0140-6736(15)00344-X
15. James MT, Bhatt M, Pannu N, and Tonelli M. Long-term outcomes of acute kidney injury and strategies for improved care. Nat Rev Nephrol. (2020) 16:193–205. doi: 10.1038/s41581-019-0247-z
16. Levey AS and James MT. Acute kidney injury. Ann Intern Med. (2017) 167:ITC66–80. doi: 10.7326/AITC201711070
17. Ronco C, Bellomo R, and Kellum JA. Acute kidney injury. Lancet. (2019) 394:1949–64. doi: 10.1016/S0140-6736(19)32563-2
18. Landoni G, Monaco F, Ti LK, Baiardo Redaelli M, Bradic N, Comis M, et al. A randomized trial of intravenous amino acids for kidney protection. New Engl J Med. (2024) 391:687–98. doi: 10.1056/NEJMoa2403769
19. Meissner M, Viehmann SF, and Kurts C. DAMPening sterile inflammation of the kidney. Kidney Int. (2019) 95:489–91. doi: 10.1016/j.kint.2018.12.007
20. Li B, Xia Y, Mei S, Ye Z, Song B, Yan X, et al. Histone H3K27 methyltransferase EZH2 regulates apoptotic and inflammatory responses in sepsis-induced AKI. Theranostics. (2023) 13:1860–75. doi: 10.7150/thno.83353
21. Wu X, You D, Pan M, Weng M, Xie Q, Guan Y, et al. Knockout of the C3a receptor protects against renal ischemia reperfusion injury by reduction of NETs formation. Cell Mol Life sciences: CMLS. (2023) 80:322. doi: 10.1007/s00018-023-04967-6
22. Chen F, Tang H, Cai X, Lin J, Kang R, Tang D, et al. DAMPs in immunosenescence and cancer. Semin Cancer Biol. (2024) 106-107:123–42. doi: 10.1016/j.semcancer.2024.09.005
23. Sakai S and Shichita T. Role of alarmins in poststroke inflammation and neuronal repair. Semin immunopathology. (2023) 45:427–35. doi: 10.1007/s00281-022-00961-5
24. Kaur J, Singh H, and Naqvi S. Intracellular DAMPs in neurodegeneration and their role in clinical therapeutics. Mol neurobiology. (2023) 60:3600–16. doi: 10.1007/s12035-023-03289-9
25. Murao A, Aziz M, Wang H, Brenner M, and Wang P. Release mechanisms of major DAMPs. Apoptosis. (2021) 26:152–62. doi: 10.1007/s10495-021-01663-3
26. McQuitty CE, Williams R, Chokshi S, and Urbani L. Immunomodulatory role of the extracellular matrix within the liver disease microenvironment. Front Immunol. (2020) 11:574276. doi: 10.3389/fimmu.2020.574276
27. Sundaram B, Pandian N, Mall R, Wang Y, Sarkar R, Kim HJ, et al. NLRP12-PANoptosome activates PANoptosis and pathology in response to heme and PAMPs. Cell. (2023) 186:2783–801.e20. doi: 10.1016/j.cell.2023.05.005
28. Ma M, Jiang W, and Zhou R. DAMPs and DAMP-sensing receptors in inflammation and diseases. Immunity. (2024) 57:752–71. doi: 10.1016/j.immuni.2024.03.002
29. Deng T, Tang C, Zhang G, and Wan X. DAMPs released by pyroptotic cells as major contributors and therapeutic targets for CAR-T-related toxicities. Cell Death Dis. (2021) 12:129. doi: 10.1038/s41419-021-03428-x
30. Gong T, Liu YT, and Fan J. Exosomal mediators in sepsis and inflammatory organ injury: unraveling the role of exosomes in intercellular crosstalk and organ dysfunction. Military Med Res. (2024) 11:24. doi: 10.1186/s40779-024-00527-6
31. Yin K, Wang D, Zhang Y, Lu H, Hou L, Guo T, et al. Polystyrene microplastics promote liver inflammation by inducing the formation of macrophages extracellular traps. J hazardous materials. (2023) 452:131236. doi: 10.1016/j.jhazmat.2023.131236
32. Hu H, Ma J, Peng Y, Feng R, Luo C, Zhang M, et al. Thrombospondin-1 regulates trophoblast necroptosis via NEDD4-mediated ubiquitination of TAK1 in preeclampsia. Advanced Sci (Weinheim Baden-Wurttemberg Germany). (2024) 11:e2309002. doi: 10.1002/advs.202309002
33. Li J, Sun X, Yang N, Ni J, Xie H, Guo H, et al. Phosphoglycerate mutase 5 initiates inflammation in acute kidney injury by triggering mitochondrial DNA release by dephosphorylating the pro-apoptotic protein Bax. Kidney Int. (2023) 103:115–33. doi: 10.1016/j.kint.2022.08.022
34. Ren J, Liu K, Wu B, Lu X, Sun L, Privratsky J, et al. Divergent actions of renal tubular and endothelial type 1 IL-1 receptor signaling in toxin-induced AKI. J Am Soc Nephrology: JASN. (2023) 34:1629–46. doi: 10.1681/ASN.0000000000000191
35. Zhang J, Luan ZL, Huo XK, Zhang M, Morisseau C, Sun CP, et al. Direct targeting of sEH with alisol B alleviated the apoptosis, inflammation, and oxidative stress in cisplatin-induced acute kidney injury. Int J Biol Sci. (2023) 19:294–310. doi: 10.7150/ijbs.78097
36. Venereau E, Ceriotti C, and Bianchi ME. DAMPs from cell death to new life. Front Immunol. (2015) 6:422. doi: 10.3389/fimmu.2015.00422
37. Baatarjav C, Komada T, Karasawa T, Yamada N, Sampilvanjil A, Matsumura T, et al. dsDNA-induced AIM2 pyroptosis halts aberrant inflammation during rhabdomyolysis-induced acute kidney injury. Cell Death differentiation. (2022) 29:2487–502. doi: 10.1038/s41418-022-01033-9
38. Vallés PG, Gil Lorenzo AF, Garcia RD, Cacciamani V, Benardon ME, and Costantino VV. Toll-like receptor 4 in acute kidney injury. Int J Mol Sci. (2023) 24:1415. doi: 10.3390/ijms24021415
39. Jaroszynski A, Zaborowski T, Gluszek S, Zapolski T, Sadowski M, Zaluska W, et al. Heat shock protein 27 is an emerging predictor of contrast-induced acute kidney injury on patients subjected to percutaneous coronary interventions. Cells. (2021) 10(3):684. doi: 10.3390/cells10030684
40. Saad HM, Elekhnawy E, Shaldam MA, Alqahtani MJ, Altwaijry N, Attallah NGM, et al. Rosuvastatin and diosmetin inhibited the HSP70/TLR4/NF-κB p65/NLRP3 signaling pathways and switched macrophage to M2 phenotype in a rat model of acute kidney injury induced by cisplatin. Biomedicine pharmacotherapy = Biomedecine pharmacotherapie. (2024) 171:116151. doi: 10.1016/j.biopha.2024.116151
41. Huang Y, Jiang W, and Zhou R. DAMP sensing and sterile inflammation: intracellular, intercellular and inter-organ pathways. Nat Rev Immunol. (2024) 24:703–19. doi: 10.1038/s41577-024-01027-3
42. Zhao Z, Hu Z, Zeng R, and Yao Y. HMGB1 in kidney diseases. Life Sci. (2020) 259:118203. doi: 10.1016/j.lfs.2020.118203
43. Du S, Zhang X, Jia Y, Peng P, Kong Q, Jiang S, et al. Hepatocyte HSPA12A inhibits macrophage chemotaxis and activation to attenuate liver ischemia/reperfusion injury via suppressing glycolysis-mediated HMGB1 lactylation and secretion of hepatocytes. Theranostics. (2023) 13:3856–71. doi: 10.7150/thno.82607
44. Alaaeldin R, Bakkar SM, Mohyeldin RH, Ali FEM, Abdel-Maqsoud NMR, and Fathy M. Azilsartan modulates HMGB1/NF-κB/p38/ERK1/2/JNK and apoptosis pathways during renal ischemia reperfusion injury. Cells. (2023) 12(1):185. doi: 10.3390/cells12010185
45. Zhao ZB, Marschner JA, Iwakura T, Li C, Motrapu M, Kuang M, et al. Tubular epithelial cell HMGB1 promotes AKI-CKD transition by sensitizing cycling tubular cells to oxidative stress: A rationale for targeting HMGB1 during AKI recovery. J Am Soc Nephrol. (2023) 34:394–411. doi: 10.1681/ASN.0000000000000024
46. Wen S, Li X, Ling Y, Chen S, Deng Q, Yang L, et al. HMGB1-associated necroptosis and Kupffer cells M1 polarization underlies remote liver injury induced by intestinal ischemia/reperfusion in rats. FASEB journal: Off Publ Fed Am Societies Exp Biol. (2020) 34:4384–402. doi: 10.1096/fj.201900817R
47. Liu Y, Xu Q, Wang Y, Liang T, Li X, Wang D, et al. Necroptosis is active and contributes to intestinal injury in a piglet model with lipopolysaccharide challenge. Cell Death Dis. (2021) 12:62. doi: 10.1038/s41419-020-03365-1
48. Zhao G, Wang X, Lei H, Ruan N, Yuan B, Tang S, et al. Serum HMGB-1 released by ferroptosis and necroptosis as a novel potential biomarker for systemic lupus erythematosus. Int Immunopharmacol. (2024) 140:112886. doi: 10.1016/j.intimp.2024.112886
49. Linkermann A. Nonapoptotic cell death in acute kidney injury and transplantation. Kidney Int. (2016) 89:46–57. doi: 10.1016/j.kint.2015.10.008
50. Yang K, Fan M, Wang X, Xu J, Wang Y, Tu F, et al. Lactate promotes macrophage HMGB1 lactylation, acetylation, and exosomal release in polymicrobial sepsis. Cell Death differentiation. (2022) 29:133–46. doi: 10.1038/s41418-021-00841-9
51. Deng C, Zhao L, Yang Z, Shang JJ, Wang CY, Shen MZ, et al. Targeting HMGB1 for the treatment of sepsis and sepsis-induced organ injury. Acta pharmacologica Sinica. (2022) 43:520–8. doi: 10.1038/s41401-021-00676-7
52. Kwak MS, Kim HS, Lee B, Kim YH, Son M, and Shin JS. Immunological significance of HMGB1 post-translational modification and redox biology. Front Immunol. (2020) 11:1189. doi: 10.3389/fimmu.2020.01189
53. Ahmed A and Tait SWG. Targeting immunogenic cell death in cancer. Mol Oncol. (2020) 14:2994–3006. doi: 10.1002/1878-0261.12851
54. Xu C, Lu C, Wang Z, Hu X, Li S, Xie Y, et al. Liraglutide abrogates nephrotoxic effects of chemotherapies. Pharmacol Res. (2023) 189:106680. doi: 10.1016/j.phrs.2023.106680
55. Liu X, Qian N, Zhu L, Fan L, Fu G, Ma M, et al. Geniposide ameliorates acute kidney injury via enhancing the phagocytic ability of macrophages towards neutrophil extracellular traps. Eur J Pharmacol. (2023) 957:176018. doi: 10.1016/j.ejphar.2023.176018
56. Qiang X, Peng Y, Wang Z, Fu W, Li W, Zhao Q, et al. Synthesis of glycyrrhizin analogues as HMGB1 inhibitors and their activity against sepsis in acute kidney injury. Eur J medicinal Chem. (2023) 259:115696. doi: 10.1016/j.ejmech.2023.115696
57. Wu W, Wang W, Liang L, Chen J, Sun S, Wei B, et al. SARS-CoV-2 N protein induced acute kidney injury in diabetic db/db mice is associated with a Mincle-dependent M1 macrophage activation. Front Immunol. (2023) 14:1264447. doi: 10.3389/fimmu.2023.1264447
58. Liu Y, Yuan W, Fang M, Guo H, Zhang X, Mei X, et al. Determination of HMGB1 in hepatitis B virus-related acute-on-chronic liver failure patients with acute kidney injury: Early prediction and prognostic implications. Front Pharmacol. (2022) 13:1031790. doi: 10.3389/fphar.2022.1031790
59. Zang D, Li W, Cheng F, Zhang X, Rao T, Yu W, et al. Accuracy and sensitivity of high mobility group box 1 (HMGB1) in diagnosis of acute kidney injury caused by sepsis and relevance to prognosis. Clinica chimica acta; Int J Clin Chem. (2022) 535:61–7. doi: 10.1016/j.cca.2022.07.015
60. Oh H, Choi A, Seo N, Lim JS, You JS, and Chung YE. Protective effect of glycyrrhizin, a direct HMGB1 inhibitor, on post-contrast acute kidney injury. Sci Rep. (2021) 11:15625. doi: 10.1038/s41598-021-94928-5
61. Tang Y, Luo H, Xiao Q, Li L, Zhong X, Zhang J, et al. Isoliquiritigenin attenuates septic acute kidney injury by regulating ferritinophagy-mediated ferroptosis. Renal failure. (2021) 43:1551–60. doi: 10.1080/0886022X.2021.2003208
62. Jin X, He R, Lin Y, Liu J, Wang Y, Li Z, et al. Shenshuaifu granule attenuates acute kidney injury by inhibiting ferroptosis mediated by p53/SLC7A11/GPX4 pathway. Drug design Dev Ther. (2023) 17:3363–83. doi: 10.2147/DDDT.S433994
63. Ha ZL and Yu ZY. Downregulation of miR-29b-3p aggravates podocyte injury by targeting HDAC4 in LPS-induced acute kidney injury. Kaohsiung J Med Sci. (2021) 37:1069–76. doi: 10.1002/kjm2.12431
64. Nakazawa D, Kumar SV, Marschner J, Desai J, Holderied A, Rath L, et al. Histones and neutrophil extracellular traps enhance tubular necrosis and remote organ injury in ischemic AKI. J Am Soc Nephrol. (2017) 28:1753–68. doi: 10.1681/ASN.2016080925
65. Yu YS, Kim H, Kim KI, and Baek SH. Epigenetic regulation of autophagy by histone-modifying enzymes under nutrient stress. Cell Death differentiation. (2023) 30:1430–6. doi: 10.1038/s41418-023-01154-9
66. Wang H, Fan Z, Shliaha PV, Miele M, Hendrickson RC, Jiang X, et al. H3K4me3 regulates RNA polymerase II promoter-proximal pause-release. Nature. (2023) 615:339–48. doi: 10.1038/s41586-023-05780-8
67. Cheng Z, Abrams ST, Toh J, Wang SS, Wang Z, Yu Q, et al. The critical roles and mechanisms of immune cell death in sepsis. Front Immunol. (2020) 11:1918. doi: 10.3389/fimmu.2020.01918
68. Zhou M, Aziz M, and Wang P. Damage-associated molecular patterns as double-edged swords in sepsis. Antioxidants Redox Signaling. (2021) 35:1308–23. doi: 10.1089/ars.2021.0008
69. Li Y, Chen Y, Yang T, Chang K, Deng N, Zhao W, et al. Targeting circulating high mobility group box-1 and histones by extracorporeal blood purification as an immunomodulation strategy against critical illnesses. Crit Care. (2023) 27:77. doi: 10.1186/s13054-023-04382-0
70. de Vries F, Huckriede J, Wichapong K, Reutelingsperger C, and Nicolaes GAF. The role of extracellular histones in COVID-19. J Internal Med. (2023) 293:275–92. doi: 10.1111/joim.13585
71. Kim TS, Silva LM, Theofilou VI, Greenwell-Wild T, Li L, Williams DW, et al. Neutrophil extracellular traps and extracellular histones potentiate IL-17 inflammation in periodontitis. J Exp Med. (2023) 220(9):e20221751. doi: 10.1084/jem.20221751
72. Zhu YP, Speir M, Tan Z, Lee JC, Nowell CJ, Chen AA, et al. NET formation is a default epigenetic program controlled by PAD4 in apoptotic neutrophils. Sci Adv. (2023) 9:eadj1397. doi: 10.1126/sciadv.adj1397
73. Li X, Xiao S, Filipczak N, Yalamarty SSK, Shang H, Zhang J, et al. Role and therapeutic targeting strategies of neutrophil extracellular traps in inflammation. Int J nanomedicine. (2023) 18:5265–87. doi: 10.2147/IJN.S418259
74. Mohamed ME, Elmorsy MA, and Younis NS. Renal ischemia/reperfusion mitigation via geraniol: the role of nrf-2/HO-1/NQO-1 and TLR2,4/MYD88/NFκB pathway. Antioxidants (Basel Switzerland). (2022) 11(8):1568. doi: 10.3390/antiox11081568
75. Zhang R, Sun C, Han Y, Huang L, Sheng H, Wang J, et al. Neutrophil autophagy and NETosis in COVID-19: perspectives. Autophagy. (2023) 19:758–67. doi: 10.1080/15548627.2022.2099206
76. Castellanos-Molina A, Bretheau F, Boisvert A, Bélanger D, and Lacroix S. Constitutive DAMPs in CNS injury: From preclinical insights to clinical perspectives. Brain behavior immunity. (2024) 122:583–95. doi: 10.1016/j.bbi.2024.07.047
77. Leinardi R, Pochet A, Uwambayinema F, Yakoub Y, Quesniaux V, Ryffel B, et al. Gasdermin D membrane pores orchestrate IL-1α secretion from necrotic macrophages after NFS-rich silica exposure. Arch toxicology. (2023) 97:1001–15. doi: 10.1007/s00204-023-03463-x
78. Jiang W, Lian J, Yue Y, and Zhang Y. IL-33/ST2 as a potential target for tumor immunotherapy. Eur J Immunol. (2021) 51:1943–55. doi: 10.1002/eji.202149175
79. Tsuchiya K, Hosojima S, Hara H, Kushiyama H, Mahib MR, Kinoshita T, et al. Gasdermin D mediates the maturation and release of IL-1α downstream of inflammasomes. Cell Rep. (2021) 34:108887. doi: 10.1016/j.celrep.2021.108887
80. Cavalli G, ColaFrancesco S, Emmi G, Imazio M, Lopalco G, Maggio MC, et al. Interleukin 1α: a comprehensive review on the role of IL-1α in the pathogenesis and treatment of autoimmune and inflammatory diseases. Autoimmun Rev. (2021) 20:102763. doi: 10.1016/j.autrev.2021.102763
81. Privratsky JR, Zhang J, Lu X, Rudemiller N, Wei Q, Yu YR, et al. Interleukin 1 receptor (IL-1R1) activation exacerbates toxin-induced acute kidney injury. Am J Physiol Renal Physiol. (2018) 315:F682–F91. doi: 10.1152/ajprenal.00104.2018
82. Wang Y, Wang J, Zheng W, Zhang J, Wang J, Jin T, et al. Identification of an IL-1 receptor mutation driving autoinflammation directs IL-1-targeted drug design. Immunity. (2023) 56:1485–501.e7. doi: 10.1016/j.immuni.2023.05.014
83. Nakazawa D, Takeda Y, Kanda M, Tomaru U, Ogawa H, Kudo T, et al. Inhibition of Toll-like receptor 4 and Interleukin-1 receptor prevent SARS-CoV-2 mediated kidney injury. Cell Death discovery. (2023) 9:293. doi: 10.1038/s41420-023-01584-x
84. Donahue KL, Watkoske HR, Kadiyala P, Du W, Brown K, Scales MK, et al. Oncogenic KRAS-dependent stromal interleukin-33 directs the pancreatic microenvironment to promote tumor growth. Cancer discovery. (2024) 14:1964–89. doi: 10.1158/2159-8290.CD-24-0100
85. Chen L, Gao C, Yin X, Mo L, Cheng X, Chen H, et al. Partial reduction of interleukin-33 signaling improves senescence and renal injury in diabetic nephropathy. MedComm. (2024) 5:e742. doi: 10.1002/mco2.742
86. Gauvreau GM, Bergeron C, Boulet LP, Cockcroft DW, Côté A, Davis BE, et al. Sounding the alarmins-The role of alarmin cytokines in asthma. Allergy. (2023) 78:402–17. doi: 10.1111/all.15609
87. Xu H, Turnquist HR, Hoffman R, and Billiar TR. Role of the IL-33-ST2 axis in sepsis. Military Med Res. (2017) 4:3. doi: 10.1186/s40779-017-0115-8
88. Sabapathy V, Price A, Cheru NT, Venkatadri R, Dogan M, Costlow G, et al. ST2 + T-regulatory cells in renal inflammation and fibrosis after ischemic kidney injury. J Am Soc Nephrol. (2025) 36:73–86. doi: 10.1681/ASN.0000000000000471
89. Xu L, Xing Z, Yuan J, Han Y, Jiang Z, Han M, et al. Ultrasmall nanoparticles regulate immune microenvironment by activating IL-33/ST2 to alleviate renal ischemia-reperfusion injury. Advanced healthcare materials. (2024) 13:e2303276. doi: 10.1002/adhm.202303276
90. Liu L, Mao L, Wu X, Wu T, Liu W, Yang Y, et al. BRG1 regulates endothelial-derived IL-33 to promote ischemia-reperfusion induced renal injury and fibrosis in mice. Biochim Biophys Acta Mol Basis Dis. (2019) 1865:2551–61. doi: 10.1016/j.bbadis.2019.06.015
91. Ferhat M, Robin A, Giraud S, Sena S, Goujon JM, Touchard G, et al. Endogenous IL-33 contributes to kidney ischemia-reperfusion injury as an alarmin. J Am Soc Nephrol. (2018) 29:1272–88. doi: 10.1681/ASN.2017060650
92. Sehnine M, Ferhat M, Sena S, Gombert JM, Goujon JM, Thierry A, et al. IL-33 receptor ST2 deficiency attenuates renal ischemia-reperfusion injury in euglyaemic, but not streptozotocin-induced hyperglycemic mice. Diabetes Metab. (2018) 44:55–60. doi: 10.1016/j.diabet.2017.06.008
93. Tang Y, Du Y, Ye J, Deng L, and Cui W. Intestine-targeted explosive hydrogel microsphere promotes uric acid excretion for gout therapy. Advanced materials (Deerfield Beach Fla). (2024) 36:e2310492. doi: 10.1002/adma.202310492
94. Xu YX, Liu LD, Zhu JY, Zhu SS, Ye BQ, Yang JL, et al. Alistipes indistinctus-derived hippuric acid promotes intestinal urate excretion to alleviate hyperuricemia. Cell Host Microbe. (2024) 32:366–81.e9. doi: 10.1016/j.chom.2024.02.001
95. Milanesi S, Verzola D, Cappadona F, Bonino B, Murugavel A, Pontremoli R, et al. Uric acid and angiotensin II additively promote inflammation and oxidative stress in human proximal tubule cells by activation of toll-like receptor 4. J Cell Physiol. (2019) 234:10868–76. doi: 10.1002/jcp.27929
96. Li L, Cheng D, An X, Liao G, Zhong L, Liu J, et al. Mesenchymal stem cells transplantation attenuates hyperuricemic nephropathy in rats. Int Immunopharmacol. (2021) 99:108000. doi: 10.1016/j.intimp.2021.108000
97. Braga TT, Forni MF, Correa-Costa M, Ramos RN, Barbuto JA, Branco P, et al. Soluble uric acid activates the NLRP3 inflammasome. Sci Rep. (2017) 7:39884. doi: 10.1038/srep39884
98. Casanova AG, Morales AI, Vicente-Vicente L, and López-Hernández FJ. Effect of uric acid reduction on chronic kidney disease. Systematic review and meta-analysis. Front Pharmacol. (2024) 15:1373258. doi: 10.3389/fphar.2024.1373258
99. Sánchez-Lozada LG, García-Arroyo FE, Gonzaga G, Silverio O, Blas-Marron MG, Muñoz-Jimenez I, et al. Kidney injury from recurrent heat stress and rhabdomyolysis: protective role of allopurinol and sodium bicarbonate. Am J Nephrol. (2018) 48:339–48. doi: 10.1159/000494663
100. Hosoya T, Uchida S, Shibata S, Tomioka NH, Matsumoto K, and Hosoyamada M. Xanthine oxidoreductase inhibitors suppress the onset of exercise-induced AKI in high HPRT activity urat1-uox double knockout mice. J Am Soc Nephrol. (2022) 33:326–41. doi: 10.1681/ASN.2021050616
101. Yang KJ, Kim JH, Chang YK, Park CW, Kim SY, and Hong YA. Inhibition of xanthine oxidoreductase protects against contrast-induced renal tubular injury by activating adenosine monophosphate-activated protein kinase. Free Radical Biol Med. (2019) 145:209–20. doi: 10.1016/j.freeradbiomed.2019.09.027
102. El-Shoura EAM, Sharkawi SMZ, Abdelzaher LA, Abdel-Wahab BA, Ahmed YH, and Abdel-Sattar AR. Reno-protective effect of fenofibrate and febuxostat against vancomycin-induced acute renal injury in rats: Targeting PPARγ/NF-κB/COX-II and AMPK/Nrf2/HO-1 signaling pathways. Immunopharmacol immunotoxicology. (2024) 46:509–20. doi: 10.1080/08923973.2024.2373216
103. Zhou H, Li X, Li Y, Zhu X, Zhang L, and Li J. Synthesis and bioevaluation of 1-phenylimidazole-4-carboxylic acid derivatives as novel xanthine oxidoreductase inhibitors. Eur J medicinal Chem. (2020) 186:111883. doi: 10.1016/j.ejmech.2019.111883
104. Li X, Sun J, Bu Q, Zhou B, Li L, Man X, et al. Association between serum uric acid levels and clinical outcomes in patients with acute kidney injury. Renal failure. (2023) 45:2169617. doi: 10.1080/0886022X.2023.2169617
105. Xiao J, Zhou Y, Xie Y, Li T, Su X, He J, et al. ATP homeostasis and signaling in plants. Plant Commun. (2024) 5:100834. doi: 10.1016/j.xplc.2024.100834
106. Wang T, Sun F, Li C, Nan P, Song Y, Wan X, et al. MTA1, a novel ATP synthase complex modulator, enhances colon cancer liver metastasis by driving mitochondrial metabolism reprogramming. Advanced Sci (Weinheim Baden-Wurttemberg Germany). (2023) 10:e2300756. doi: 10.1002/advs.202300756
107. Chan NJ, Chen YY, Hsu CC, Lin YS, Zakeri M, Kim S, et al. Release of ATP in the lung evoked by inhalation of irritant gases in rats. J Appl Physiol (Bethesda Md: 1985). (2024) 137:581–90. doi: 10.1152/japplphysiol.00137.2024
108. van Megen WH, Canki E, Wagenaar VHA, van Waes C, Peters DJM, Van Asbeck-Van der Wijst J, et al. Fluid shear stress stimulates ATP release without regulating purinergic gene expression in the renal inner medullary collecting duct. FASEB journal: Off Publ Fed Am Societies Exp Biol. (2023) 37:e23232. doi: 10.1096/fj.202301434R
109. Dunne OM, Martin SL, Sergeant GP, McAuley DF, O’Kane CM, Button B, et al. TRPV2 modulates mechanically Induced ATP Release from Human bronchial epithelial cells. Respir Res. (2024) 25:188. doi: 10.1186/s12931-024-02807-0
110. Higashikuni Y, Liu W, Numata G, Tanaka K, Fukuda D, Tanaka Y, et al. NLRP3 inflammasome activation through heart-brain interaction initiates cardiac inflammation and hypertrophy during pressure overload. Circulation. (2023) 147:338–55. doi: 10.1161/CIRCULATIONAHA.122.060860
111. Rabadi M, Kim M, Li H, Han SJ, Choi Y, D’Agati V, et al. ATP induces PAD4 in renal proximal tubule cells via P2X7 receptor activation to exacerbate ischemic AKI. Am J Physiol Renal Physiol. (2018) 314:F293–305. doi: 10.1152/ajprenal.00364.2017
112. Qian Y, Qian C, Xie K, Fan Q, Yan Y, Lu R, et al. P2X7 receptor signaling promotes inflammation in renal parenchymal cells suffering from ischemia-reperfusion injury. Cell Death Dis. (2021) 12:132. doi: 10.1038/s41419-020-03384-y
113. Qian Y, Zhao N, Wang M, Zou Z, and Xie K. P2X7 receptor deficiency attenuates cisplatin-induced kidney injury via inhibiting NLRP3 inflammasome activation. Biochem Pharmacol. (2024) 226:116369. doi: 10.1016/j.bcp.2024.116369
114. Zhang R, Su K, Yang L, Tang M, Zhao M, Ye N, et al. Design, synthesis, and biological evaluation of novel P2X7 receptor antagonists for the treatment of septic acute kidney injury. J medicinal Chem. (2023) 66:11365–89. doi: 10.1021/acs.jmedchem.3c00837
115. El-Maadawy WH, Hassan M, Badawy MH, AbuSeada A, and Hafiz E. Probenecid induces the recovery of renal ischemia/reperfusion injury via the blockade of Pannexin 1/P2X7 receptor axis. Life Sci. (2022) 308:120933. doi: 10.1016/j.lfs.2022.120933
116. Li Y, Chen H, Yang Q, Wan L, Zhao J, Wu Y, et al. Increased Drp1 promotes autophagy and ESCC progression by mtDNA stress mediated cGAS-STING pathway. J Exp Clin Cancer research: CR. (2022) 41:76. doi: 10.1186/s13046-022-02262-z
117. Tábara LC, Burr SP, Frison M, Chowdhury SR, Paupe V, Nie Y, et al. MTFP1 controls mitochondrial fusion to regulate inner membrane quality control and maintain mtDNA levels. Cell. (2024) 187:3619–37.e27. doi: 10.1016/j.cell.2024.05.017
118. Yu CH, Davidson S, Harapas CR, Hilton JB, Mlodzianoski MJ, Laohamonthonkul P, et al. TDP-43 Triggers Mitochondrial DNA Release via mPTP to Activate cGAS/STING in ALS. Cell. (2020) 183:636–49 e18. doi: 10.1016/j.cell.2020.09.020
119. Newman LE, Weiser Novak S, Rojas GR, Tadepalle N, Schiavon CR, Grotjahn DA, et al. Mitochondrial DNA replication stress triggers a pro-inflammatory endosomal pathway of nucleoid disposal. Nat Cell Biol. (2024) 26:194–206. doi: 10.1038/s41556-023-01343-1
120. Zhao M, Wang Y, Li L, Liu S, Wang C, Yuan Y, et al. Mitochondrial ROS promote mitochondrial dysfunction and inflammation in ischemic acute kidney injury by disrupting TFAM-mediated mtDNA maintenance. Theranostics. (2021) 11:1845–63. doi: 10.7150/thno.50905
121. Liu J, Jia Z, and Gong W. Circulating mitochondrial DNA stimulates innate immune signaling pathways to mediate acute kidney injury. Front Immunol. (2021) 12:680648. doi: 10.3389/fimmu.2021.680648
122. Hu Y, Yang C, Amorim T, Maqbool M, Lin J, Li C, et al. Cisplatin-mediated upregulation of APE2 binding to MYH9 provokes mitochondrial fragmentation and acute kidney injury. Cancer Res. (2021) 81:713–23. doi: 10.1158/0008-5472.CAN-20-1010
123. Tsuji N, Tsuji T, Yamashita T, Hayase N, Hu X, Yuen PS, et al. BAM15 treats mouse sepsis and kidney injury, linking mortality, mitochondrial DNA, tubule damage, and neutrophils. J Clin Invest. (2023) 133(7):e152401. doi: 10.1172/JCI152401
124. Vringer E and Tait SWG. Mitochondria and cell death-associated inflammation. Cell Death differentiation. (2023) 30:304–12. doi: 10.1038/s41418-022-01094-w
125. Wang Z, He Z, Chang X, Xie L, Song Y, Wu H, et al. Mitochondrial damage-associated molecular patterns: New perspectives for mitochondria and inflammatory bowel diseases. Mucosal Immunol. (2025) 18:290–8. doi: 10.1016/j.mucimm.2025.01.013
126. West AP and Shadel GS. Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat Rev Immunol. (2017) 17:363–75. doi: 10.1038/nri.2017.21
127. Bu WJ, Li SS, Liu C, Wang YH, Lu JR, Dong CR, et al. Nepetin limits NLRP3 inflammasome activation and alleviates NLRP3-driven inflammatory diseases via PINK1-dependent mitophagy. Free Radical Biol Med. (2025) 227:420–33. doi: 10.1016/j.freeradbiomed.2024.12.027
128. Abayasekara K and Sullo N. The clinical use of urinary mitochondrial DNA in adult surgical critical care patients with acute kidney injury. Clin Exp Pharmacol Physiol. (2023) 50:277–86. doi: 10.1111/1440-1681.13746
129. Ho PW, Pang WF, Luk CC, Ng JK, Chow KM, Kwan BC, et al. Urinary mitochondrial DNA level as a biomarker of acute kidney injury severity. Kidney Dis (Basel). (2017) 3:78–83. doi: 10.1159/000475883
130. Hu Q, Ren J, Wu J, Li G, Wu X, Liu S, et al. Urinary mitochondrial DNA levels identify acute kidney injury in surgical critical illness patients. Shock (Augusta Ga). (2017) 48:11–7. doi: 10.1097/SHK.0000000000000830
131. Mo B, Scharf B, Gutheil C, Letzel MC, and Hensel A. Tamm-Horsfall protein in humane urine: sex-dependent differences in the excretion and N-glycosylation pattern. Sci Rep. (2023) 13:17815. doi: 10.1038/s41598-023-44650-1
132. Mercado-Evans V, Branthoover H, Chew C, Serchejian C, Saltzman AB, Mejia ME, et al. Tamm-Horsfall protein augments neutrophil NETosis during urinary tract infection. JCI Insight. (2025) 10(1):e180024. doi: 10.1172/jci.insight.180024
133. Franceschini N and Le TH. Urine uromodulin and genetics of its variation. J Am Soc Nephrol. (2022) 33:461–2. doi: 10.1681/ASN.2022010027
134. LaFavers KA, Gaddy AR, Micanovic R, Lingeman J, Williams JC Jr., Coe FL, et al. Water loading and uromodulin secretion in healthy individuals and idiopathic calcium stone formers. Clin J Am Soc Nephrol. (2023) 18:1059–67. doi: 10.2215/CJN.0000000000000202
135. Immler R, Lange-Sperandio B, Steffen T, Beck H, Rohwedder I, Roth J, et al. Extratubular polymerized uromodulin induces leukocyte recruitment and inflammation in vivo. Front Immunol. (2020) 11:588245. doi: 10.3389/fimmu.2020.588245
136. Ludes PO, de Roquetaillade C, Chousterman BG, Pottecher J, and Mebazaa A. Role of damage-associated molecular patterns in septic acute kidney injury, from injury to recovery. Front Immunol. (2021) 12:606622. doi: 10.3389/fimmu.2021.606622
137. Karagiannidis AG, Theodorakopoulou MP, Pella E, Sarafidis PA, and Ortiz A. Uromodulin biology. Nephrology dialysis transplantation: Off Publ Eur Dialysis Transplant Assoc - Eur Renal Assoc. (2024) 39:1073–87. doi: 10.1093/ndt/gfae008
138. Yu M, He X, Song X, Gao J, Pan J, Zhou T, et al. Biglycan promotes hepatic fibrosis through activating heat shock protein 47. Liver international: Off J Int Assoc Study Liver. (2023) 43:500–12. doi: 10.1111/liv.15477
139. Roedig H, Nastase MV, Frey H, Moreth K, Zeng-Brouwers J, Poluzzi C, et al. Biglycan is a new high-affinity ligand for CD14 in macrophages. Matrix Biol. (2019) 77:4–22. doi: 10.1016/j.matbio.2018.05.006
140. Li H, Ghorbani S, Ling CC, Yong VW, and Xue M. The extracellular matrix as modifier of neuroinflammation and recovery in ischemic stroke and intracerebral hemorrhage. Neurobiol disease. (2023) 186:106282. doi: 10.1016/j.nbd.2023.106282
141. Poluzzi C, Nastase MV, Zeng-Brouwers J, Roedig H, Hsieh LT, Michaelis JB, et al. Biglycan evokes autophagy in macrophages via a novel CD44/Toll-like receptor 4 signaling axis in ischemia/reperfusion injury. Kidney Int. (2019) 95(21):11917. doi: 10.1016/j.kint.2018.10.037
142. Klejch T, Buffa R, Šimek M, Nešporová K, Exnerová A, Bednařík J, et al. Enzymatically stable unsaturated hyaluronan-derived oligosaccharides with selective cytostatic properties. Carbohydr polymers. (2024) 336:122129. doi: 10.1016/j.carbpol.2024.122129
143. Kawai K, Ho MT, Ueno Y, Abdo D, Xue C, Nonaka H, et al. Hyaluronan improves photoreceptor differentiation and maturation in human retinal organoids. Acta biomaterialia. (2024) 181:117–32. doi: 10.1016/j.actbio.2024.05.001
144. Zhang Z, Tian X, Lu JY, Boit K, Ablaeva J, Zakusilo FT, et al. Increased hyaluronan by naked mole-rat Has2 improves healthspan in mice. Nature. (2023) 621:196–205. doi: 10.1038/s41586-023-06463-0
145. Ferreira NDR, Sanz CK, Raybolt A, Pereira CM, and DosSantos MF. Action of hyaluronic acid as a damage-associated molecular pattern molecule and its function on the treatment of temporomandibular disorders. Front Pain Res (Lausanne Switzerland). (2022) 3:852249. doi: 10.3389/fpain.2022.852249
146. Rabelink TJ, Wang G, van der Vlag J, and van den Berg BM. The roles of hyaluronan in kidney development, physiology and disease. Nat Rev Nephrol. (2024) 20:822–32. doi: 10.1038/s41581-024-00883-5
147. Lee CH, Chiang CF, Kuo FC, Su SC, Huang CL, Liu JS, et al. High-molecular-weight hyaluronic acid inhibits IL-1β-induced synovial inflammation and macrophage polarization through the GRP78-NF-κB signaling pathway. Int J Mol Sci. (2021) 22(21):11917. doi: 10.3390/ijms222111917
148. Lee JH, Liu A, Park JH, Kato H, Hao Q, Zhang X, et al. Therapeutic effects of hyaluronic acid in peritonitis-induced sepsis in mice. Shock (Augusta Ga). (2020) 54:488–97. doi: 10.1097/SHK.0000000000001512
149. Kang LJ, Yoon J, Rho JG, Han HS, Lee S, Oh YS, et al. Self-assembled hyaluronic acid nanoparticles for osteoarthritis treatment. Biomaterials. (2021) 275:120967. doi: 10.1016/j.biomaterials.2021.120967
150. Romo M, López-Vicario C, Pérez-Romero N, Casulleras M, Martínez-Puchol AI, Sánchez B, et al. Small fragments of hyaluronan are increased in individuals with obesity and contribute to low-grade inflammation through TLR-mediated activation of innate immune cells. Int J Obes (2005). (2022) 46:1960–9. doi: 10.1038/s41366-022-01187-z
151. Kim SM, Song GY, Shim A, Lee JH, Eom CB, Liu C, et al. Hyaluronan synthase 2, a target of miR-200c, promotes carbon tetrachloride-induced acute and chronic liver inflammation via regulation of CCL3 and CCL4. Exp Mol Med. (2022) 54:739–52. doi: 10.1038/s12276-022-00781-5
152. Zhang Y, Zhao H, and Zhang J. Hyaluronidase inhibitor sHA2.75 alleviates ischemia-reperfusion-induced acute kidney injury. Cell Cycle (Georgetown Tex). (2024) 23:248–61. doi: 10.1080/15384101.2024.2309019
153. Wang Q, Long G, Luo H, Zhu X, Han Y, Shang Y, et al. S100A8/A9: An emerging player in sepsis and sepsis-induced organ injury. Biomedicine pharmacotherapy = Biomedecine pharmacotherapie. (2023) 168:115674. doi: 10.1016/j.biopha.2023.115674
154. Wang Z, Wang Y, Yan Q, Cai C, Feng Y, Huang Q, et al. FPR1 signaling aberrantly regulates S100A8/A9 production by CD14(+)FCN1(hi) macrophages and aggravates pulmonary pathology in severe COVID-19. Commun Biol. (2024) 7:1321. doi: 10.1038/s42003-024-07025-4
155. von Wulffen M, Luehrmann V, Robeck S, Russo A, Fischer-Riepe L, van den Bosch M, et al. S100A8/A9-alarmin promotes local myeloid-derived suppressor cell activation restricting severe autoimmune arthritis. Cell Rep. (2023) 42:113006. doi: 10.1016/j.celrep.2023.113006
156. Jorch SK, McNally A, Berger P, Wolf J, Kaiser K, Chetrusca Covash A, et al. Complex regulation of alarmins S100A8/A9 and secretion via gasdermin D pores exacerbates autoinflammation in familial Mediterranean fever. J Allergy Clin Immunol. (2023) 152:230–43. doi: 10.1016/j.jaci.2023.01.037
157. Chen Y, Ouyang Y, Li Z, Wang X, and Ma J. S100A8 and S100A9 in cancer. Biochim Biophys Acta Rev cancer. (2023) 1878:188891. doi: 10.1016/j.bbcan.2023.188891
158. Huang J, Shi L, Xia Y, Zhu J, Zha H, Wu X, et al. S100-A8/A9 activated TLR4 in renal tubular cells to promote ischemia-reperfusion injury and fibrosis. Int Immunopharmacol. (2023) 118:110110. doi: 10.1016/j.intimp.2023.110110
159. Yao W, Chen Y, Li Z, Ji J, You A, Jin S, et al. Single cell RNA sequencing identifies a unique inflammatory macrophage subset as a druggable target for alleviating acute kidney injury. Advanced Sci (Weinheim Baden-Wurttemberg Germany). (2022) 9:e2103675. doi: 10.1002/advs.202103675
Keywords: AKI, DAMPs, PRRs, inflammation, immunity
Citation: Li J and Hu Z (2025) Research progress on damage-associated molecular patterns in acute kidney injury. Front. Immunol. 16:1590822. doi: 10.3389/fimmu.2025.1590822
Received: 10 March 2025; Accepted: 20 June 2025;
Published: 10 July 2025.
Edited by:
Alexandre M Carmo, Universidade do Porto, PortugalReviewed by:
Karsten Grote, University of Marburg, GermanySatoko Arai, The Institute for AIM Medicine, Japan
Copyright © 2025 Li and Hu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhangxue Hu, aHp4YXd5QHNjdS5lZHUuY24=