 Alexander F. vom Stein
Alexander F. vom Stein Phuong-Hien Nguyen
Phuong-Hien Nguyen Elisa ten Hacken
Elisa ten Hacken- 1Faculty of Medicine and University Hospital Cologne, Department I of Internal Medicine, Center for Integrated Oncology Aachen Bonn Cologne Duesseldorf, University of Cologne, Cologne, Germany
- 2Center for Molecular Medicine Cologne, University of Cologne, Cologne, Germany
- 3Center of Excellence on Cellular Stress Responses in Aging-Associated Diseases (CECAD), University of Cologne, Cologne, Germany
- 4Department of Medicine, Division of Hematology and Oncology, Weill Cornell Medicine, New York City, NY, United States
Immunotherapy has revolutionized the treatment landscape for many cancers, including some B- cell lymphomas. Immune checkpoint blockade, CAR-T cells and bispecific antibodies have shown promise for the treatment of Richter Transformation (RT) but have displayed reduced activity in chronic lymphocytic leukemia (CLL). These observations suggest that, besides the intrinsic differences between CLL cells and transformed RT cells, there are also marked differences in tumor immune microenvironmental (TiME) composition and tumor-immune cell interactions between these two entities, which remain to be fully characterized. In this perspective, we highlight recent studies describing the TiME in CLL and RT, utilizing both patient-derived tissues and novel mouse models. We then provide a brief overview of current clinical trials employing immunotherapy in CLL and RT and offer a perspective on current challenges and future research efforts in the field.
1 Introduction
Chronic lymphocytic leukemia (CLL) remains the most common adult leukemia in Western countries, characterized by substantial variability in patient characteristics and clinical outcomes (1). Treatment of CLL has witnessed remarkable success with targeted therapies, including the use of BTK and BCL2 inhibitors (1). However, a subset of patients develop resistance to these treatments, resulting in poor clinical outcomes without established therapeutic options (2). Another challenge in the clinical management of CLL is the emergence of Richter Transformation (RT), an aggressive B-cell lymphoma occurring in up to 10% of patients, which is associated with dismal clinical outcomes (~12 months median survival) and displays refractoriness to most existing therapies (3).
Despite significant progress in immunotherapies thanks to the introduction of immune checkpoint inhibition, CAR-T cells, and bispecific antibodies (BsAbs), clinical responses to these new agents in CLL have stayed behind those observed in other B-cell malignancies. Importantly, some of these approaches have shown more promise in RT, implying that the tumor immune microenvironment (TiME) in CLL and RT is fundamentally distinct, with the RT-TiME enabling enhanced response to immunotherapy. To better understand these biological and therapeutic disparities, refined model systems and a deeper exploration of the TiME in both preclinical and clinical settings are necessary. Here, we discuss emerging transgenic mouse models for these entities, summarize the current landscape of targeted immunotherapies in CLL and RT, and offer a perspective on how insights from these systems can inform future clinical trials.
2 Advances in modeling CLL and RT in vivo
2.1 Immuno-competent mouse models of CLL and RT
Mouse models of CLL have been extensively developed and studied, yielding major advancements in our understanding of CLL biology (Figure 1). A variety of models capturing the spectrum of disease progression from early-stage monoclonal B-cell lymphocytosis (MBL) to advanced, aggressive disease have provided critical insights into disease pathogenesis and preclinical therapeutic response. Among them, the Eµ-TCL1 transgenic mouse remains the most commonly employed, owing to its high disease penetrance (100%) and well-characterized disease characteristics, which are recognized to recapitulate an aggressive variant of CLL-like disease (4). Conditional knock-in/out strategies have been used to model CLL genetic drivers including del(13q) (5), Sf3b1/Atm co-mutation (6), Ikzf3 (7) and Rps15 (8), all of which lead to incomplete disease penetrance but faithful disease characteristics to indolent CLL-like disease (as previously reviewed) (9).
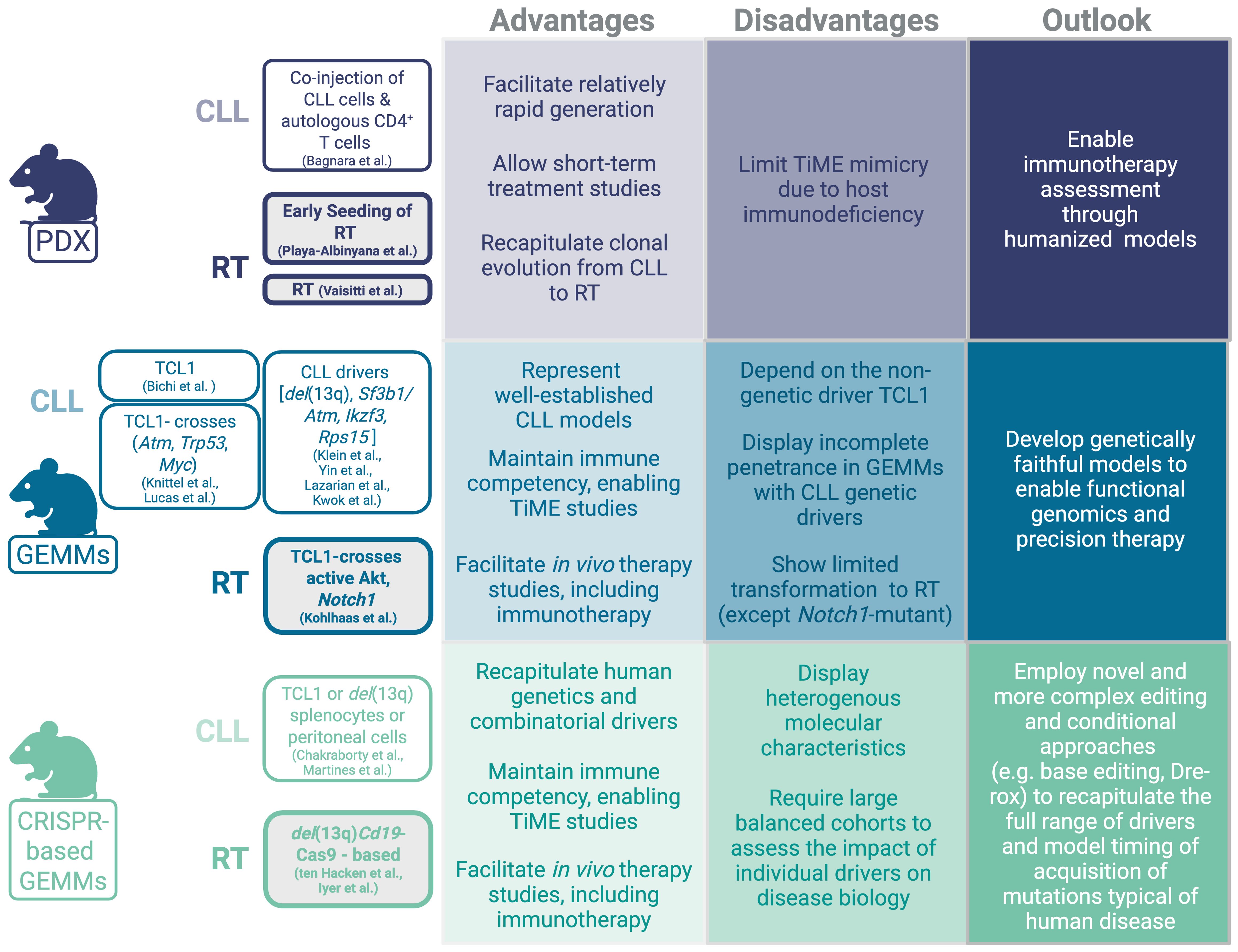
Figure 1. Patient-derived xenograft and genetically-engineered models for CLL and RT.
Novel immuno-competent mouse models that recapitulate CLL transformation into RT are now also available. Some of the initial studies have relied on intercrosses based on the Eµ-TCL1 background and provided functional insight into the relevance of selected genetic drivers and/or signaling pathways in RT, including loss or Trp53 or Atm (10), MYC overexpression (11), and aberrant expression of the protein arginine methyltransferase (PRMT5) (12). However, in these early models, RT was observed only in less than 20% of mice lacking Trp53, and none in mice lacking Atm, potentially due to the small size of the analyzed cohorts (10), while MYC overexpression resulted in a clonally-unrelated lymphoma co-developing alongside CLL rather than arising through a clonally-related transformation process (11). More recently, B-cell specific activation of Akt or Notch1 in the Eµ-TCL1 transgenic background resulted in 100% RT and significantly shortened survival compared to Eµ-TCL1 mice, thus showing the highest reported transformation rate among CLL mouse models (13). Notably, NOTCH signaling hyper-activation mediated by AKT constitutive activation on the Eµ-TCL1 background was associated with increased interaction of the transforming clone with CD4+ T-cells overexpressing the NOTCH ligand DLL1, thus providing a functional link between presence of selected molecular changes and their impact on tumor microenvironmental reprogramming.
Several studies have now implemented the CRISPR/Cas9 technology to model complex co-occurring alterations typical of RT via either stem-cell engineering of del(13q)-Cd19Cas9 donor mice [i.e., animals expressing Cas9-GFP in a B-cell restricted fashion together with the leukemogenic del(13q)- background] (14, 15) or of splenocytes or peritoneal cells derived from either Eµ-TCL1 or del(13q) mice (16, 17). Modeling combinatorial molecular drivers directly in stem cells or B-cells allowed faithful recapitulation of the molecular events leading to clonally-related RT arising from antecedent indolent CLL. Collectively, these models provided important knowledge on the functional role of recurrent genetic drivers of RT discovered through large-scale next generation sequencing studies (18, 19), including the MYC signaling regulator Mga, recently demonstrated to contribute to aberrant mitochondrial oxidative phosphorylation and glycerolipid metabolism via upregulation of the MYC target Nme (14, 20). Recent studies have also demonstrated the relevance of cooperative events in transformation biology, including co-occurrence of mutation in the ribosomal protein Rps15 and in the Trp53 tumor suppressor which jointly alter DNA damage response pathways (8), and the presence of autoreactive (i.e. ‘stereotyped’) B-cell receptors in models carrying concomitant alterations in Trp53 and the cell cycle regulators Cdkn2a/b (21), implying chronic auto-antigenic stimulation as a predisposing factor for transformation. The notion that B-cell receptor (BCR) stimulation may be implicated in transformation is further supported by the evidence that loss of the anergy regulator NFAT2 led to transformation into RT in vivo (22). Importantly, all these newly developed mouse models are fully immuno-competent and transplantable into syngeneic recipients, thus providing a valuable platform amenable to short-term (1-2 months) treatment studies, including of novel immunotherapies.
2.2 Patient-derived xenograft models of CLL and RT: recent advances and current challenges
In contrast to the wide range of transgenic mouse models, developing patient-derived xenograft (PDX) models for CLL and RT has proven to be significantly more challenging (Figure 1). CLL-PDX models could be established when neoplastic CLL cells were co-injected with autologous pre-activated CD4+ T-cells in NSG mice (23). CD4+ T-cell co-injection supported the engraftment of CLL cells in the perivascular area of spleens and highlighted the functional contribution of CD4+ T-cells to CLL progression. Although these PDX models successfully recapitulated the activation of NF-ĸB and BCR signaling in malignant B-cells, mimicking what is typically observed in the CLL lymph node microenvironment, the incomplete homology of murine and human signaling receptors and cytokines/chemokines limited the faithful recapitulation of key microenvironmental interactions. Although CLL engraftment in these PDX models could not persist long-term, these models have proven to be useful to investigate efficacy of targeted therapies, including BTK inhibitors (24).
The relatively rare occurrence of RT (~2-10% CLL patients) and the general difficulties in tissue procurement have thus far limited the generation of stable cell lines for in vitro functional analyses –with the sole exception of the U-RT1 cell line – (25) and of PDX models. Nonetheless, some RT-PDX models were thus far generated and have allowed the assessment of novel therapeutic modalities, including the combination of PI3K inhibition via duvelisib with BCL2 inhibition by venetoclax (26), anti-ROR1 monoclonal antibodies (27), anti-CD37 immunotoxins (28), or BET-PROTACs alone or combined with the BTK inhibitor ibrutinib or the BCL2 inhibitor venetoclax (29). More recently, PDX models were used to validate the existence of an ‘early seeding’ process in RT, defined as presence of subclonal ‘RT-like’ populations already present in CLL samples up to ~20 years prior to clinical and histological diagnosis of RT in patients (19). By engraftment of cells at the CLL stage into NSG recipients, Playa-Albinyana et al. observed transformation into clonally-related RT in vivo, further demonstrating clonal-relatedness of the two malignancies and providing proof-of-principle for the transforming potential of RT seeds (30). While PDX models represent valuable tools for the pre-clinical evaluation of therapeutics, particularly those targeting human antigens (e.g., anti-CD37 immunotoxins), the lack of functional T-, B- and NK-cells characterizing the NSG strain limits the ability to interrogate immune-related changes underlying response and resistance to immunotherapy. It is tempting to speculate that the TiME might contribute to the suppression of early RT seeds, thus the lack of immune surveillance after transplantation into NSG mice might facilitate this outgrowth process. To this end, novel humanized mouse models generated via engraftment of human stem cells and subsequent repopulation of the recipient mouse with a functional human immune system are underway (31) and will allow the evaluation of efficacy of novel immunotherapeutic strategies on both tumor-intrinsic and tumor-extrinsic pathways at greater depth.
3 The distinct TiME of RT in comparison to CLL
The TiME has long been understood to play a crucial role in CLL initiation and progression (Figure 2). Bi-directional interactions with different cell types in the tissue microenvironment, in particular macrophages and stromal cells, promote CLL survival and resistance to therapy by activation of BCR, NF-ĸB and Toll-like receptor (TLR) signaling, as well as upregulation of antiapoptotic proteins in leukemic cells. In addition, CD4+ T-cells significantly support CLL-cell proliferation and disease progression, while the cytotoxic capacity of CD8+ T-cells and NK-cells is limited, thus allowing leukemic cells to evade killing (32, 33). T-cell exhaustion with upregulation of immune checkpoint molecules such as PD-1, TIGIT or TIM-3 on T-cells (34, 35), TiME-interaction-induced PD-L1 and CTLA-4 expression on CLL cells (36), and proliferative signaling (37) contribute substantially to immune evasion. Abundance of precursor exhausted CD8+ T cells is a characteristic feature of the CLL lymph node TiME, and upregulation of Galectin 9 (the ligand for TIM-3) on CLL tumors is associated with inferior CLL patient survival (38). Interestingly, immunohistochemical (IHC) studies detect only minimal PD-L1 expression on the malignant cells, instead PD-L1 is predominantly expressed on non-malignant bystander cells, such as macrophages (39, 40). This observation is even more pronounced in RT than in CLL samples (41) and indicates further contribution of other TiME cells to immune suppression in both entities. Recent data demonstrates that RT preserves a core CLL-specific gene expression profile, which includes upregulated genes involved in the BCR signaling and downregulated genes related to immune response, TP53 signaling, and the JAK-STAT pathway (42). Kohlhas et al. have recently shown that macrophage interactions can activate the JAK-STAT pathway in CLL cells (43), highlighting the critical role of macrophages in enhancing CLL viability via inflammatory signaling.

Figure 2. The human CLL and RT TiME and immunotherapeutic strategies.
Key insights into TiME composition of RT have primarily been gathered via IHC analysis of lymph node tissues, including increased presence of regulatory T-cells and CD163+ tumor-associated macrophages (41), together with enhanced PD-1 staining on clonally-related malignant B-cells. PD-1 expression on malignant B-cells appears to be a unique characteristic of human (and murine) RT, that is generally not observed in CLL or de novo DLBCL (44, 45). Diminished T-cell receptor (TCR) polyclonality has also been observed in RT compared to CLL (41), which suggests presence of common antigenic determinants underlying evolution from antecedent CLL, that –together with BCR polyreactivity– further supports the relevance of antigenic drive in transformation biology. More recently, single-cell RNA sequencing studies of bone marrow from RT patients showing differential response to checkpoint blockade therapy allowed the identification of a population of CD8+ effector/effector memory T-cells, which were marked by the transcription factor ZNF683, and which displayed preserved cytotoxicity and intermediate exhaustion, in association with response to therapy. Similarly, baseline peripheral T-cells in responding patients overexpress ZNF683 and PD-1, whereas non-responding patients demonstrated upregulation of NK-/T-cell-related genes (46). In separate studies, RT patients responding to checkpoint inhibition show increased abundance of circulating CD8+ T-cells with reduced exhaustion markers and an interferon-gamma (IFN-γ) signature (47), which is consistent with in vitro observations of increased IFN-γ secretion by activated T-cells upon PD-1/PD-L1 blockade (34). A recent study cross-compared TiME characteristics of murine RT splenocytes and human RT lymph nodes identifying abundant (and shared) presence of pro-inflammatory CXCL9+ tumor-associated macrophages and CD8+ PD-1+ polyfunctional T-cells in patient and murine samples showing favorable response to checkpoint blockade therapy with anti-PD-1 (48). In line with the known relevance of monocyte/macrophages in mediating CLL progression in vivo in mouse models (49), TLR inhibition via IRAK4 blocking reduces macrophages and delays RT cell engraftment in vivo (17). Despite the increased infiltration of M2-skewed CD163+ macrophages in RT compared to CLL (41), the role of macrophages in RT seems less important for direct tumor cell support in patient samples. Although the functional significance of these macrophages in RT patients is not yet well-defined, their high PD-L1 expression (41) suggests a stronger contribution to immune suppression rather than direct tumor survival support. A thorough phenotypic and functional analysis of RT-associated macrophages would be invaluable to clarify their precise contributions to RT.
4 Clinical insights into immunotherapeutic approaches in CLL and RT
Restoring immune surveillance by rewiring the immune-suppressive TiME is a major goal for treating CLL and RT. Since CLL cells strongly promote an exhausted T-cell phenotype, therapy with BCL2 and BTK inhibitors (BCL2i/BTKi) that efficiently reduce leukemic burden generally help to restore T-cell functionality (50–52). The pronounced beneficial effects of BTKi and other kinase inhibitors on the T-cell compartment —partially mediated by off-target effects on T-cell specific kinases— make them attractive combination partners to enhance immunotherapy efficacy (2), as currently tested in clinical trials for both CLL and RT (Figure 2).
4.1 Immune checkpoint inhibition
The biological differences in the TiME are particularly reflected in different clinical responses to immune checkpoint inhibitors (ICIs). Single-agent pembrolizumab showed no efficacy in relapsed CLL patients (40), but achieved modest responses with limited durability in RT patients (objective response rate [ORR] ~10–44%) (40, 53). Notably, in cases of concurrent CLL and RT, ICIs selectively improved the RT phase while sparing the CLL phase, emphasizing differences in ICI susceptibility of these two entities, even when they co-exist within the same patient (40). Evaluating a BTKi plus ICI combination (Ibrutinib plus Nivolumab) in high-risk CLL showed comparable responses to BTKi monotherapy, corroborating the inefficiency of ICI in CLL (54). Subsequent combination therapies have demonstrated greater promise in RT. Trials combining ICIs with BTKi or PI3K inhibitors have yielded response rates of 42–65%, with longer progression-free survival compared to ICI or kinase inhibitor monotherapy (47, 54–56). The mechanisms by which BTKi enhance the efficacy of ICI in RT remains unclear and is somewhat surprising. This is particularly notable given that: (i) BTKi monotherapy demonstrated moderate activity in RT (ORR 40% for acalabrutinib and 50% for pirtobrutinib) but responses were only short-lived with median durations of 6-8 months (57, 58), (ii) preclinical studies have indicated reduced dependence of RT cells on BCR signaling (19), and (iii) most RT patients had prior exposure to BTKi as part of CLL treatment (e.g., 66% in the RT1 trial, 25% in the MOLTO trial), although the relevance of BTKi resistance mutations for RT transformation compared to those occurring in the CLL phase is incompletely understood (59). Biomarker analyses from trials of BTKi-ICI combinations revealed only a weak correlation between baseline PD-1/PD-L1 expression and clinical responses (39, 40, 54). Additional combinatorial strategies are being explored, including the potential of triple combinations. The MOLTO trial demonstrated the efficiency of combining BCL2i (Venetoclax) with ICI (anti-PD-L1 antibody Atezolizumab) plus a CD20 Antibody (Obinutuzumab), resulting in similar ORR and lasting responses as BTKi plus ICI (39). Consequently, ongoing trials are investigating triple combinations involving ICI, BTKi and BCL2i (NCT04271956 and NCT05388006).
Besides the PD-1/PD-L1 axis, other immune checkpoints such as LAG-3, TIGIT and CTLA-4 are overexpressed on CLL cells, contribute to immune evasion and are potential targets for immunotherapy. CTLA-4 expression on leukemic cells is regulated by microenvironmental interactions (36); contact with activated CD4+ T cells upregulates, whereas contact with stroma cells and the lymph node TiME reduces its expression. Inhibition of CTLA-4 reduces tumor burden in a murine model and augmented cytotoxicity of bispecific antibody treatment in vitro (36, 60, 61). Similarly, LAG-3 inhibition in combination with PD-1 inhibition reduces tumor burden in vivo (62). Targeting Galectin 9, the ligand for TIM-3, has also shown promise in preclinical studies in the Eμ-TCL1 mouse model (38). To our knowledge, no clinical trial has tested corresponding ICI in CLL or RT so far, but they represent promising therapeutic targets in upcoming combination therapies.
4.2 Chimeric antigen receptor T-cells
Overcoming immune evasion by CAR-T-cells, has been extensively investigated in CLL and RT. Based on the pivotal phase 1-2 TRANSCEND CLL 004 trial (63), lisocabtagene maraleucel (liso-cel) was approved for treatment of relapsed CLL after prior BTKi and BCL2i therapy (double-exposed) by the FDA in March 2024. In this trial, 43% of double-exposed CLL patients responded to CAR-T therapy, 18% achieving a complete response (CR), with a median duration of response of 35 months. Although response rates for CLL are much lower compared to other B-cell lymphomas, responses in CLL can be very lasting. Most patients, who reach one year of progression-free survival (PFS) after infusion, remain progression-free for more than 5 years without further treatment (64). Characterization of such long-lasting responders identified an active, proliferative CD4+ CAR-T population with cytotoxic characteristics (65). Similarly, the infusion product in responding patients had significantly more CD4+ cells and less effector/memory-like CD8+ T-cells than in non-responding patients in a trial using a third generation academic CAR-T construct (66), indicating a role for cytotoxic CD4+ T-cells for lasting leukemia control. In addition, characterization of infusion products and immune composition at time of apheresis in responding patients shows increase of early-memory T-cell phenotypes, increased IL6/STAT3-signature, reduced T-cell exhaustion and effector differentiation, as well as abundance of a CD45RO- CD27+ CD8+ T-cell population (67). This demonstrates that the T-cell characteristics at time of apheresis are highly relevant for successful CAR-T cell therapy, and the generally strongly exhausted CLL T-cell compartment might underlie the low response rates for CAR-T in CLL compared to other B-cell lymphomas. In addition, a persisting residual lymphadenopathy upon CAR-T treatment, even in patients that achieve undetectable minimal residual disease (uMRD) in peripheral blood or bone marrow, indicates a role for an immune-suppressive TiME in limiting efficacy of CAR-T cells (63, 68). Macrophages, fibroblasts and other stromal cells are important components of the CLL microenvironment and have been associated with suppression of CAR-T cells in other entities (69, 70).To overcome these challenges, combination of CAR-T cells with BTKi is clinically feasible and safe (68), and data indicates a trend towards increased response rates (71, 72), and facilitated CAR-T-cell manufacturing after Ibrutinib pre-treatment (73). Moreover, CAR-T therapy in CLL is hampered by frequent side effects including cytokine release syndrome (CRS) and neurotoxicity and BTKi treatment might lower CRS rates (71, 72), although randomized trials are still needed to clearly confirm these effect.
Given the rarity of RT, little insight on CAR-T cell treatment has been collected in prospective trials and one prospective trial on the use of Brexucabtagene autoleucel (brexu-cel) in relapsed/refractory RT was stopped before full recruitment. In a large retrospective, multicenter analysis with different CAR-T cell products approved for DLBCL, treatment of RT demonstrated an ORR of 63% with 46% achieving CR. While median PFS was short (4.7 months), patients achieving a CR (46%) had a median duration of response of 27.6 months, indicating that CAR-T can induce comparatively longer responses in RT patients (74). However, compared to de novo DLBCL or transformed indolent non-Hodgkin lymphoma, RT shows reduced response rates and is a significant negative prognostic factor in multivariate analyses (75). Thus, current trials are aiming to improve response rates by combination of CAR-T cells with BTKi (NCT05873712), as well as BTKi and ICI (NCT05672173).
4.3 Bispecific antibodies
Emerging clinical trials in both CLL and RT are exploring the potential of BsAbs, which target CD3 on T-cells and CD19 or CD20 on B-cells thereby inducing T-cell-mediated cytotoxicity. Preclinical studies show that CD19/CD3 and CD20/CD3 BsAbs effectively induce CLL lysis in vitro and in PDX models, with their efficacy linked to effector-to-target ratios (76, 77). Clinically, the anti-CD20/CD3 BsAb Epcoritamab has shown promising activity in refractory, high-risk CLL patients, with an ORR of 61%, albeit with high CRS rates (78). Other BsAbs, such as the anti-CD20/CD3 Mosunetuzumab, are currently under evaluation (NCT05091424). BTKi pre-treatment can enhance BsAb-mediated cytotoxicity, demonstrating that interfering with CLL-associated immuno-suppression by combination approaches is a promising future treatment strategy (50, 76). BsAbs also synergize with CAR-T cells in murine CLL models, achieving complete leukemia ablation and prolonged survival compared to monotherapy (79), although the risk of exacerbated CRS or immune effector cell-associated neurotoxicity syndrome (ICANS) requires careful clinical validation. BsAbs are also effective in RT, demonstrating ORRs of up to 63% with the anti-CD20/CD3 Glofitamab in pre-treated patients (80–82). While early relapse is frequent in non-CR patients, patients achieving CR often sustain responses for over 20 months, potentially reflecting restored immunosurveillance. However, CRS rates remain a concern, reaching 80% with Epcoritamab (82). The anti-CD19/anti-CD3 bispecific T-cell engager Blinatumomab combined with R-CHOP has been shown to improve response depth in patients who did not initially achieve CR (80), highlighting a potential avenue for a new combination approach. Ongoing trials are investigating novel strategies, such as BsAbs with ICIs (NCT06043674) or BTKi (NCT06735664), to further improve efficacy.
5 Perspective
5.1 Deep multi-omic profiling of patient samples
A critical next step in advancing immunotherapy for CLL and RT lies in the comprehensive characterization of TiME characteristics in RT patients in direct comparison to CLL cohorts. The rapid development of high-throughput single-cell and spatial analytics has opened new possibilities for in-depth profiling of different immune cell subsets, offering unprecedented resolution in mapping cellular interactions and signaling networks. These analyses will be crucial for uncovering the fundamental differences in TiME between CLL and RT, ultimately guiding more precise TiME-focused therapeutic strategies. Genotype-aware multi-omics (e.g. Genotyping-of-Transcriptomes, Genotyping-of-Targeted loci with single-cell Chromatin Accessibility) (83, 84) will further allow to dissect the function of individual (or multiplexed) driver mutations when in complex admixtures, such as those occurring in CLL specimens evolving into RT. To this end, accurate biobanking of longitudinal samples and/or of lymph node material from CLL and paired RT will be essential, as RT specimens are currently mostly collected as paraffin-embedded tissue, while most multi-omic strategies require viable single-cell suspensions. Nodal single-cell suspensions could also be valuable to generate 3D cell culture models or organoids, a rapid tool to screen personalized therapeutics. These systems are just emerging for CLL (85, 86), as they have been notoriously challenged by the poor ex vivo viability of primary CLL patient-derived material.
5.2 Refinement of preclinical in vivo and ex vivo models
In parallel to the patient-focused studies, these comprehensive TiME analyses will also benefit substantially from the recently generated RT mouse models that faithfully recapitulate the transformation process, which offer a more controlled sampling setting along the longitudinal evolution of CLL into RT. Novel genetically-engineered mouse models also provide a fundamental tool to dissect the mechanistic relevance of genetic drivers in RT, which is still largely lacking. Incorporating novel CRISPR-based editing strategies such as cytosine base editing (87) will further allow a more comprehensive modeling of disease drivers, which is now limited to loss-of-function mutations achieved via conventional Cas9-based methods. Addition of inducible engineering strategies (e.g. Dre-rox) (88) would further facilitate the modeling of timing of acquisition of driver events typical of human disease. The development of well-annotated PDX models will further be advantageous to identify novel targets and personalized treatment strategies, enabling the rapid assessment of drug sensitivities and resistance patterns. Humanized mouse models, particularly those that enable reconstitution of the human myeloid compartment (89), represent a central tool for preclinical evaluation not only of T-cell-based immunotherapies but also of macrophage-reprogramming strategies. A key advantage of humanized mouse models is the faithful recapitulation of the human immune system, which can guide preclinical assessment of novel immunotherapies in a personalized manner and anticipate possible risks/side effects.
As patients with double-refractory CLL and RT have limited therapeutic options and dismal outcomes, immunotherapies hold promise for addressing this critical unmet need. Significant progress is being made to establish such therapies, yet a key challenge remains the relatively poor response of CLL. Preclinical studies will be essential to pave the way and improve these outcomes, and these can benefit from either faithful GEMMs or 3D/organoid models. Advanced culture systems for CLL have progressed from co-cultures with stromal- or nurse-like cells (43, 90, 91) to more sophisticated 3D multicellular spheroid assemblies that better recapitulate lymph node architecture and cellular interactions (86, 92). These spheroids incorporate multiple adaptive immune cells with leukemic cells and can provide a more physiologically relevant platform to study drug responses. The integration of 3D bioprinting, which supplements a close-to-native extracellular matrix, further offers significant advantages to faithfully recreating the complex structure of lymphoid tissues (92). Such culture systems for RT remain underdeveloped due to the scarcity of primary material. Adapting these advanced CLL culture models and employing 3D DLBCL spheroid models (69) will be valuable for dissecting RT-TiME interactions and evaluating therapeutic approaches preclinically.
5.3 Advanced diagnostic tools to guide novel combination therapies
Companion diagnostic tools derived from preclinical analyses, such as expression of ZNF683 in blood PBMCs (46), presence of IFN-γ signatures in the T-cell compartment (47), pro-inflammatory TiME signatures and PD-L1 expression on tumor cells or macrophages (48), could be incorporated in translational screening platforms to identify patients with a higher likelihood of exhibiting a favorable response to immunotherapy. Applying such pre-clinically defined diagnostic approaches along novel clinical trials, combined with thorough characterization of the TiME adaptations under treatment, holds promise not only for patient-specific prognostication of response to novel immunotherapies but also to identify novel combination treatments that promote induction of these TiME signatures. Such combination regimens, for example those integrating the use of kinase inhibitors, show potential to overcome resistance mechanisms mediated by the TiME and have already demonstrated to enhance immunotherapeutic efficacy in RT. Notably, combinations of novel BTK inhibitors with BsAbs or CAR-T cells —all of which exhibit single-agent activity in refractory disease— are now being planned in CLL. Moreover, innovative therapies must be increasingly tailored to specific targets identified through preclinical research. Next generation ICI intercepting CTLA-4, TIGIT, LAG-3 or TIM-3, are being developed and demonstrate preclinical potential in CLL and can be further tested in novel murine models and clinical trials. Another emerging concept involves combining multiple immunotherapeutic agents to augment anti-tumor responses. For instance, the combination of BsAbs with CAR-T cells, shown to be highly effective in preclinical studies (79), or integrating these modalities with ICIs, may further enhance immunotherapeutic potency as preclinically demonstrated. However, these novel approaches warrant careful safety monitoring given the risk of exacerbated CRS and ICANS. Here, novel immune-competent GEMMs and humanized murine models can further contribute to pre-clinical assessment of safety of combination strategies. Robust translational research must accompany ongoing clinical trials. This synergy between bench and bedside will provide real-time insights into the immunological shifts occurring in patients and foster the development of rational combination regimens designed to maximize efficacy and minimize toxicity.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Author contributions
AS: Writing – original draft. P-HN: Writing – original draft. ET: Writing – original draft.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Acknowledgments
We thank Dr. Barbara Eichhorst for critically reading the manuscript.
Conflict of interest
AS reports speaker honoraria from AstraZeneca.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that Generative AI was used in the creation of this manuscript. Generative AI was used to improve readability of the manuscript. The authors take full responsibility for the accuracy of the content.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Jain N, Wierda WG, and O’Brien S. Chronic lymphocytic leukaemia. Lancet. (2024) 404:694–706. doi: 10.1016/S0140-6736(24)00595-6
2. Lewis RI, Vom Stein AF, and Hallek M. Targeting the tumor microenvironment for treating double-refractory chronic lymphocytic leukemia. Blood. (2024) 144:601–14. doi: 10.1182/blood.2023022861
3. Parry EM, Ten Hacken E, and Wu CJ. Richter syndrome: novel insights into the biology of transformation. Blood. (2023) 142:11–22. doi: 10.1182/blood.2022016502
4. Bichi R, Shinton SA, Martin ES, Koval A, Calin GA, Cesari R, et al. Human chronic lymphocytic leukemia modeled in mouse by targeted TCL1 expression. Proc Natl Acad Sci U S A. (2002) 99:6955–60. doi: 10.1073/pnas.102181599
5. Klein U, Lia M, Crespo M, Siegel R, Shen Q, Mo T, et al. The DLEU2/miR-15a/16-1 cluster controls B cell proliferation and its deletion leads to chronic lymphocytic leukemia. Cancer Cell. (2010) 17:28–40. doi: 10.1016/j.ccr.2009.11.019
6. Yin S, Gambe RG, Sun J, Martinez AZ, Cartun ZJ, Regis FFD, et al. A murine model of chronic lymphocytic leukemia based on B cell-restricted expression of sf3b1 mutation and atm deletion. Cancer Cell. (2019) 35:283–296.e285. doi: 10.1016/j.ccell.2018.12.013
7. Lazarian G, Yin S, Ten Hacken E, Sewastianik T, Uduman M, Font-Tello A, et al. A hotspot mutation in transcription factor IKZF3 drives B cell neoplasia via transcriptional dysregulation. Cancer Cell. (2021) 39:380–393 e388. doi: 10.1016/j.ccell.2021.02.003
8. Kwok M, Gutierrez C, Ruthen N, Waddicor P, Ouspenskaia T, Curran C, et al. Impaired response to oxidative DNA damage underlies genomic instability in B-cell leukemia driven by RPS15 mutation. Blood. (2024) 144:758. doi: 10.1182/blood-2024-202115
9. Ten Hacken E and Wu CJ. Understanding CLL biology through mouse models of human genetics. Blood. (2021) 138:2621–31. doi: 10.1182/blood.2021011993
10. Knittel G, Rehkamper T, Korovkina D, Liedgens P, Fritz C, Torgovnick A, et al. Two mouse models reveal an actionable PARP1 dependence in aggressive chronic lymphocytic leukemia. Nat Commun. (2017) 8:153. doi: 10.1038/s41467-017-00210-6
11. Lucas F, Rogers KA, Harrington BK, Pan A, Yu L, Breitbach J, et al. Emu-TCL1xMyc: A novel mouse model for concurrent CLL and B-cell lymphoma. Clin Cancer Res. (2019) 25:6260–73. doi: 10.1158/1078-0432.CCR-19-0273
12. Hing ZA, Walker JS, Whipp EC, Brinton L, Cannon M, Zhang P, et al. Dysregulation of PRMT5 in chronic lymphocytic leukemia promotes progression with high risk of Richter’s transformation. Nat Commun. (2023) 14:97. doi: 10.1038/s41467-022-35778-1
13. Kohlhaas V, Blakemore SJ, Al-Maarri M, Nickel N, Pal M, Roth A, et al. Active Akt signaling triggers CLL toward Richter transformation via overactivation of Notch1. Blood. (2021) 137:646–60. doi: 10.1182/blood.2020005734
14. Iyer P, Zhang B, Liu T, Jin M, Hart K, Zhang J, et al. MGA deletion leads to Richter’s transformation by modulating mitochondrial OXPHOS. Sci Transl Med. (2024) 16:eadg7915. doi: 10.1126/scitranslmed.adg7915
15. Ten Hacken E, Sewastianik T, Yin S, Brunsting Hoffmann G, Gruber M, Clement K, et al. In vivo modeling of CLL transformation to Richter’s syndrome reveals convergent evolutionary paths and therapeutic vulnerabilities. Blood Cancer Discov. (2023) 4:150–69. doi: 10.1158/2643-3230.BCD-22-0082
16. Chakraborty S, Martines C, Porro F, Fortunati I, Bonato A, Dimishkovska M, et al. B-cell receptor signaling and genetic lesions in TP53 and CDKN2A/CDKN2B cooperate in Richter transformation. Blood. (2021) 138:1053–66. doi: 10.1182/blood.2020008276
17. Martines C, Chakraborty S, Vujovikj M, Gobessi S, Vaisitti T, Deaglio S, et al. Macrophage- and BCR-derived but not TLR-derived signals support the growth of CLL and Richter syndrome murine models in vivo. Blood. (2022) 140:2335–47. doi: 10.1182/blood.2022016272
18. Parry EM, Leshchiner I, Guieze R, Johnson C, Tausch E, Parikh SA, et al. Evolutionary history of transformation from chronic lymphocytic leukemia to Richter syndrome. Nat Med. (2023) 29:158–69. doi: 10.1038/s41591-022-02113-6
19. Nadeu F, Royo R, Massoni-Badosa R, Playa-Albinyana H, Garcia-Torre B, Duran-Ferrer M , et al. Detection of early seeding of Richter transformation in chronic lymphocytic leukemia. Nat Med. (2022) 28:1662–71. doi: 10.1038/s41591-022-01927-8
20. Iyer P, Jiang B, Venkataraman G, Song JY, Chung Chan W, Siddiqi T, et al. Integrating metabolomics and molecular pathways to uncover therapeutic vulnerabilities in richter’s transformatio. Blood. (2024) 144:760. doi: 10.1182/blood-2024-199843
21. Martines C, Chakraborty S, Felician G, Hofmann K, Blasutig S, Negara I, et al. Richter syndrome-associated genetic lesions in TP53 and the cell cycle inhibitors CDKN2A and CDKN2B induce transformation of a CD5+ B cell subset characterized by a restricted IGHV repertoire, autoreactivity and BCR dependence. Blood. (2024) 144:759. doi: 10.1182/blood-2024-207095
22. Marklin M, Heitmann JS, Fuchs AR, Truckenmuller FM, Gutknecht M, Bugl S, et al. NFAT2 is a critical regulator of the anergic phenotype in chronic lymphocytic leukaemia. Nat Commun. (2017) 8:755. doi: 10.1038/s41467-017-00830-y
23. Bagnara D, Kaufman MS, Calissano C, Marsilio S, Patten PE, Simone R, et al. A novel adoptive transfer model of chronic lymphocytic leukemia suggests a key role for T lymphocytes in the disease. Blood. (2011) 117:5463–72. doi: 10.1182/blood-2010-12-324210
24. Herman SE, Sun X, McAuley EM, Hsieh MM, Pittaluga S, Raffeld M, et al. Modeling tumor-host interactions of chronic lymphocytic leukemia in xenografted mice to study tumor biology and evaluate targeted therapy. Leukemia. (2013) 27:2311–21. doi: 10.1038/leu.2013.131
25. Schmid T, Maier J, Martin M, Tasdogan A, Tausch E, Barth TFE, et al. U-RT1 - A new model for Richter transformation. Neoplasia. (2021) 23:140–8. doi: 10.1016/j.neo.2020.11.010
26. Iannello A, Vitale N, Coma S, Arruga F, Chadburn A, Di Napoli A, et al. Synergistic efficacy of the dual PI3K-delta/gamma inhibitor duvelisib with the Bcl-2 inhibitor venetoclax in Richter syndrome PDX models. Blood. (2021) 137:3378–89. doi: 10.1182/blood.2020010187
27. Vaisitti T, Arruga F, Vitale N, Lee TT, Ko M, Chadburn A, et al. ROR1 targeting with the antibody-drug conjugate VLS-101 is effective in Richter syndrome patient-derived xenograft mouse models. Blood. (2021) 137:3365–77. doi: 10.1182/blood.2020008404
28. Vaisitti T, Vitale N, Micillo M, Brandimarte L, Iannello A, Papotti MG, et al. Anti-CD37 alpha-amanitin-conjugated antibodies as potential therapeutic weapons for Richter syndrome. Blood. (2022) 140:1565–9. doi: 10.1182/blood.2022016211
29. Sun B, Fiskus W, Qian Y, Rajapakshe K, Raina K, Coleman KG, et al. BET protein proteolysis targeting chimera (PROTAC) exerts potent lethal activity against mantle cell lymphoma cells. Leukemia. (2018) 32:343–52. doi: 10.1038/leu.2017.207
30. Playa-Albinyana H, Arenas F, Royo R, Giro A, Lopez-Oreja I, Aymerich M, et al. Chronic lymphocytic leukemia patient-derived xenografts recapitulate clonal evolution to Richter transformation. Leukemia. (2024) 38:557–69. doi: 10.1038/s41375-023-02095-5
31. Chuprin J, Buettner H, Seedhom MO, Greiner DL, Keck JG, Ishikawa F, et al. Humanized mouse models for immuno-oncology research. Nat Rev Clin Oncol. (2023) 20:192–206. doi: 10.1038/s41571-022-00721-2
32. Ramsay AG, Johnson AJ, Lee AM, Gorgun G, Le Dieu R, Blum W, et al. Chronic lymphocytic leukemia T cells show impaired immunological synapse formation that can be reversed with an immunomodulating drug. J Clin Invest. (2008) 118:2427–37. doi: 10.1172/JCI35017
33. Hofland T, Endstra S, Gomes CKP, de Boer R, de Weerdt I, Bobkov V, et al. Natural Killer Cell Hypo-responsiveness in Chronic Lymphocytic Leukemia can be Circumvented In Vitro by Adequate Activating Signaling. Hemasphere. (2019) 3:e308. doi: 10.1097/HS9.0000000000000308
34. Brusa D, Serra S, Coscia M, Rossi D, D'Arena G, Laurenti L, et al. The PD-1/PD-L1 axis contributes to T-cell dysfunction in chronic lymphocytic leukemia. Haematologica. (2013) 98:953–63. doi: 10.3324/haematol.2012.077537
35. Peters FS, Strefford JC, Eldering E, and Kater AP. T-cell dysfunction in chronic lymphocytic leukemia from an epigenetic perspective. Haematologica. (2021) 106:1234–43. doi: 10.3324/haematol.2020.267914
36. Do P, Beckwith KA, Cheney C, Tran M, Beaver L, Griffin BG, et al. Leukemic B cell CTLA-4 suppresses costimulation of T cells. J Immunol. (2019) 202:2806–16. doi: 10.4049/jimmunol.1801359
37. Bottcher M, Bruns H, Volkl S, Lu J, Chartomatsidou E, Papakonstantinou N, et al. Control of PD-L1 expression in CLL-cells by stromal triggering of the Notch-c-Myc-EZH2 oncogenic signaling axis. J Immunother Cancer. (2021) 9(4):e001889. doi: 10.1136/jitc-2020-001889
38. Cid L, Wong J, Fernandez Botana I, Paul Y, Wierz M, Florchinger A, et al. High-dimensional single-cell definition of CLL T cells identifies Galectin-9 as novel immunotherapy target. bioRxiv. (2023). doi: 10.1101/2022.12.15.519719
39. Tedeschi A, Frustaci AM, Condoluci A, Coscia M, Chiarle R, Zinzani PL, et al. Atezolizumab, venetoclax, and obinutuzumab combination in Richter transformation diffuse large B-cell lymphoma (MOLTO): a multicentre, single-arm, phase 2 trial. Lancet Oncol. (2024) 25:1298–309. doi: 10.1016/S1470-2045(24)00396-6
40. Ding W, LaPlant BR, Call TG, Parikh SA, Leis JF, He R, et al. Pembrolizumab in patients with CLL and Richter transformation or with relapsed CLL. Blood. (2017) 129:3419–27. doi: 10.1182/blood-2017-02-765685
41. Wang Y, Sinha S, Wellik LE, Secreto CR, Rech KL, Call TG, et al. Distinct immune signatures in chronic lymphocytic leukemia and Richter syndrome. Blood Cancer J. (2021) 11:86. doi: 10.1038/s41408-021-00477-5
42. Broseus J, Hergalant S, Vogt J, Tausch E, Kreuz M, Mottok A, et al. Molecular characterization of Richter syndrome identifies de novo diffuse large B-cell lymphomas with poor prognosis. Nat Commun. (2023) 14:309. doi: 10.1038/s41467-022-34642-6
43. Kohlhas V, Jestrabek H, Rebollido-Rios R, Truong TT, Zolzer R, Schreurs LD, et al. Comparative analysis of macrophage feeder systems reveals distinct behaviors and key transcriptional shifts in chronic lymphocytic leukemia cells via coculture. bioRxiv. (2025). doi: 10.1101/2025.1102.1113.638101
44. He R, Ding W, Viswanatha DS, Chen D, Shi M, Van Dyke D, et al. PD-1 expression in chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL) and large B-cell richter transformation (DLBCL-RT): A characteristic feature of DLBCL-RT and potential surrogate marker for clonal relatedness. Am J Surg Pathol. (2018) 42:843–54. doi: 10.1097/PAS.0000000000001077
45. Behdad A, Griffin B, Chen YH, Ma S, Kelemen K, Lu X, et al. PD-1 is highly expressed by neoplastic B-cells in Richter transformation. Br J Haematol. (2019) 185:370–3. doi: 10.1111/bjh.2019.185.issue-2
46. Parry EM, Lemvigh CK, Deng S, Dangle N, Ruthen N, Knisbacher BA, et al. ZNF683 marks a CD8(+) T cell population associated with anti-tumor immunity following anti-PD-1 therapy for Richter syndrome. Cancer Cell. (2023) 41:1803–1816 e1808. doi: 10.1016/j.ccell.2023.08.013
47. Shouse G, Popplewell L, Muir A, Siddiqi T, Zain J, Herrera AF, et al. Final update of safety, efficacy and T-cell predictive biomarkers from a phase I trial of copanlisib+Nivolumab in patients with richter’s transformation (RT) or transformed non-hodgkin lymphoma (tNHL). Blood. (2024) 144:4484. doi: 10.1182/blood-2024-209610
48. Ten Hacken E, Gustafsson J, Tomasoni C, Brunsting Hoffmann G, Cruz K, Lu W, et al. Polyfunctional effector PD-1+ CD8+ T cells and M1 macrophages promote immune checkpoint blockade response in richter syndrome mouse models. Blood. (2024) 144:761. doi: 10.1182/blood-2024-201006
49. Galletti G, Scielzo C, Barbaglio F, Rodriguez TV, Riba M, Lazarevic D, et al. Targeting macrophages sensitizes chronic lymphocytic leukemia to apoptosis and inhibits disease progression. Cell Rep. (2016) 14:1748–60. doi: 10.1016/j.celrep.2016.01.042
50. Papazoglou D, Wang XV, Shanafelt TD, Lesnick CE, Ioannou N, De Rossi G, et al. Ibrutinib-based therapy reinvigorates CD8+ T cells compared to chemoimmunotherapy: immune monitoring from the E1912 trial. Blood. (2024) 143:57–63. doi: 10.1182/blood.2023020554
51. Long M, Beckwith K, Do P, Mundy BL, Gordon A, Lehman AM, et al. Ibrutinib treatment improves T cell number and function in CLL patients. J Clin Invest. (2017) 127:3052–64. doi: 10.1172/JCI89756
52. Griggio V, Vitale C, Jones R, Mancin G, Bondielli G, Visentin , et al. Treatment with venetoclax positively modulates immunological parameters and T-cell features in patients with chronic lymphocytic leukemia. Blood. (2024) 144:1854. doi: 10.1182/blood-2024-203053
53. Armand P, Murawski N, Molin D, Zain J, Eichhorst B, Gulbas Z, et al. Pembrolizumab in relapsed or refractory Richter syndrome. Br J Haematol. (2020) 190:e117–20. doi: 10.1111/bjh.v190.2
54. Younes A, Brody J, Carpio C, Lopez-Guillermo A, Ben-Yehuda D, Ferhanoglu B, et al. Safety and activity of ibrutinib in combination with nivolumab in patients with relapsed non-Hodgkin lymphoma or chronic lymphocytic leukaemia: a phase 1/2a study. Lancet Haematol. (2019) 6:e67–78. doi: 10.1016/S2352-3026(18)30217-5
55. Jain N, Senapati J, Thakral B, Ferrajoli A, Thompson P, Burger J, et al. A phase 2 study of nivolumab combined with ibrutinib in patients with diffuse large B-cell Richter transformation of CLL. Blood Adv. (2023) 7:1958–66. doi: 10.1182/bloodadvances.2022008790
56. Al-Sawaf O, Ligtvoet R, Robrecht S, Stumpf J, Fink AM, Tausch E, et al. Tislelizumab plus zanubrutinib for Richter transformation: the phase 2 RT1 trial. Nat Med. (2024) 30:240–8. doi: 10.1038/s41591-023-02722-9
57. Eyre TA, Schuh A, Wierda WG, Brown JR, Ghia P, Pagel JM, et al. Acalabrutinib monotherapy for treatment of chronic lymphocytic leukaemia (ACE-CL-001): analysis of the Richter transformation cohort of an open-label, single-arm, phase 1-2 study. Lancet Haematol. (2021) 8:e912–21. doi: 10.1016/S2352-3026(21)00305-7
58. Wierda WG, Shah NN, Cheah CY, Lewis D, Hoffmann MS, Coombs CC, et al. Pirtobrutinib, a highly selective, non-covalent (reversible) BTK inhibitor in patients with B-cell Malignancies: analysis of the Richter transformation subgroup from the multicentre, open-label, phase 1/2 BRUIN study. Lancet Haematol. (2024) 11:e682–92. doi: 10.1016/S2352-3026(24)00172-8
59. Kanagal-Shamanna R, Jain P, Patel KP, Routbort M, Bueso-Ramos C, Alhalouli T, et al. Targeted multigene deep sequencing of Bruton tyrosine kinase inhibitor-resistant chronic lymphocytic leukemia with disease progression and Richter transformation. Cancer. (2019) 125:559–74. doi: 10.1002/cncr.31831
60. Yano M, Nunes J, Mo X, Rogers KA, Woyach JA, Byrd JC, et al. Differential regulation of CTLA4 expression through BTK-dependent and independent mechanisms in CLL. Blood Adv. (2022) 6:5440–8. doi: 10.1182/bloodadvances.2021005571
61. Mhibik M, Gaglione EM, Eik D, Kendall EK, Blackburn A, Keyvanfar K, et al. BTK inhibitors, irrespective of ITK inhibition, increase efficacy of a CD19/CD3-bispecific antibody in CLL. Blood. (2021) 138:1843–54. doi: 10.1182/blood.2020009686
62. Wierz M, Pierson S, Guyonnet L, Viry E, Lequeux A, Oudin A, et al. Dual PD1/LAG3 immune checkpoint blockade limits tumor development in a murine model of chronic lymphocytic leukemia. Blood. (2018) 131:1617–21. doi: 10.1182/blood-2017-06-792267
63. Siddiqi T, Maloney DG, Kenderian SS, Brander DM, Dorritie K, Soumerai J, et al. Lisocabtagene maraleucel in chronic lymphocytic leukaemia and small lymphocytic lymphoma (TRANSCEND CLL 004): a multicentre, open-label, single-arm, phase 1-2 study. Lancet. (2023) 402:641–54. doi: 10.1016/S0140-6736(23)01052-8
64. Frost BF, Frey N, Hexner E, Schuster SJ, Nasta SD, Loren AW, et al. Curing CLL: long-term outcomes of chronic lymphocytic leukemia patients with at least one year of response to CART-19 therapy. Blood. (2024) 144:588. doi: 10.1182/blood-2024-204197
65. Melenhorst JJ, Chen GM, Wang M, Porter DL, Chen C, Collins MA, et al. Decade-long leukaemia remissions with persistence of CD4(+) CAR T cells. Nature. (2022) 602:503–9. doi: 10.1038/s41586-021-04390-6
66. Derigs P, Schubert ML, Dreger P, Schmitt A, Yousefian S, Haas S, et al. Third-generation anti-CD19 CAR T cells for relapsed/refractory chronic lymphocytic leukemia: a phase 1/2 study. Leukemia. (2024) 38:2419–28. doi: 10.1038/s41375-024-02392-7
67. Fraietta JA, Lacey SF, Orlando EJ, Pruteanu-Malinici I, Gohil M, Lundh S, et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat Med. (2018) 24:563–71. doi: 10.1038/s41591-018-0010-1
68. Gill S, Vides V, Frey NV, Hexner EO, Metzger S, O'Brien M, et al. Anti-CD19 CAR T cells in combination with ibrutinib for the treatment of chronic lymphocytic leukemia. Blood Adv. (2022) 6:5774–85. doi: 10.1182/bloodadvances.2022007317
69. Apollonio B, Spada F, Petrov N, Cozzetto D, Papazoglou D, Jarvis P, et al. Tumor-activated lymph node fibroblasts suppress T cell function in diffuse large B cell lymphoma. J Clin Invest. (2023) 133(13):e166070. doi: 10.1172/JCI166070
70. Rodriguez-Garcia A, Lynn RC, Poussin M, Eiva MA, Shaw LC, O'Connor RS, et al. CAR-T cell-mediated depletion of immunosuppressive tumor-associated macrophages promotes endogenous antitumor immunity and augments adoptive immunotherapy. Nat Commun. (2021) 12:877. doi: 10.1038/s41467-021-20893-2
71. Gauthier J, Hirayama AV, Purushe J, Hay KA, Lymp J, Li DH, et al. Feasibility and efficacy of CD19-targeted CAR T cells with concurrent ibrutinib for CLL after ibrutinib failure. Blood. (2020) 135:1650–60. doi: 10.1182/blood.2019002936
72. Wierda W, Dorritie K, Gauthier J, Nath R, Kipps TJ, Riedell PA, et al. Lisocabtagene maraleucel (liso-cel) combined with ibrutinib (ibr) for patients (pts) with relapsed or refractory (R/R) chronic lymphocytic leukemia (CLL)/small lymphocytic lymphoma (SLL): primary results from the open-label, phase 1/2 transcend CLL 004 study. Blood. (2024) 144:887. doi: 10.1182/blood-2024-200339
73. Geyer MB, Riviere I, Senechal B, Wang X, Wang Y, Purdon TJ, et al. Safety and tolerability of conditioning chemotherapy followed by CD19-targeted CAR T cells for relapsed/refractory CLL. JCI Insight. (2019) 5(9):e122627. doi: 10.1172/jci.insight.122627
74. Kittai AS, Bond D, Huang Y, Bhat SA, Blyth E, Byrd JC, et al. Anti-CD19 chimeric antigen receptor T-cell therapy for richter transformation: an international, multicenter, retrospective study. J Clin Oncol. (2024) 42:2071–9. doi: 10.1200/JCO.24.00033
75. Benjamini O, Fried S, Shouval R, Flynn JR, Beyar-Katz O, Leslie LA, et al. Anti-CD19 chimeric antigen receptor T-cell therapy has less efficacy in Richter transformation than in de novo large B-cell lymphoma and transformed low-grade B-cell lymphoma. Haematologica. (2024) 109:3566–77. doi: 10.3324/haematol.2023.284664
76. Mhibik M, Gaglione EM, Eik D, Herrick J, Le J, Ahn IE, et al. Cytotoxicity of the CD3xCD20 bispecific antibody epcoritamab in CLL is increased by concurrent BTK or BCL-2 targeting. Blood Adv. (2023) 7:4089–101. doi: 10.1182/bloodadvances.2022009517
77. Robinson HR, Qi J, Cook EM, Nichols C, Dadashian EL, Underbayev C, et al. A CD19/CD3 bispecific antibody for effective immunotherapy of chronic lymphocytic leukemia in the ibrutinib era. Blood. (2018) 132:521–32. doi: 10.1182/blood-2018-02-830992
78. Danilov A, Fakhri B, Awan F, Bentzen H, Eradat H, Niemann C, et al. Epcoritamab monotherapy in patients (Pts) with relapsed or refractory (R/R) chronic lymphocytic leukemia (CLL): results from CLL expansion and optimization cohorts of epcore CLL-1. Blood. (2024) 144(Supplement 1):883. doi: 10.1182/blood-2024-199708
79. Brinkmann BJ, Floerchinger A, Schniederjohann C, Roider T, Coelho M, Mack N, et al. CD20-bispecific antibodies improve response to CD19-CAR T cells in lymphoma in vitro and CLL in vivo models. Blood. (2024) 144:784–9. doi: 10.1182/blood.2023022682
80. Thompson PA, Jiang X, Banerjee P, Basar R, Garg N, Chen K, et al. A phase two study of high dose blinatumomab in Richter’s syndrome. Leukemia. (2022) 36:2228–32. doi: 10.1038/s41375-022-01649-3
81. Cheah CY, Assouline S, Baker R, Bartlett NL, El-Sharkawi D, Giri P, et al. Mosunetuzumab monotherapy demonstrates activity and a manageable safety profile in patients with relapsed or refractory richter’s transformation. Blood. (2023) 142(Supplement 1):614. doi: 10.1182/blood-2023-173796
82. Kater AP, Janssens A, Eradat H, Offner F, Sandoval-Sus J, Shadman M, et al. Single-agent epcoritamab leads to deep responses in patients with Richter’s transformation: primary results from the Epcore CLL-1 Trial. EHA Conf abstracts. (2024). doi: 10.1016/S2152-2650(24)00580-9
83. Nam AS, Kim KT, Chaligne R, Izzo F, Ang C, Taylor J, et al. Somatic mutations and cell identity linked by Genotyping of Transcriptomes. Nature. (2019) 571:355–60. doi: 10.1038/s41586-019-1367-0
84. Izzo F, Myers RM, Ganesan S, Mekerishvili L, Kottapalli S, Prieto T, et al. Mapping genotypes to chromatin accessibility profiles in single cells. Nature. (2024) 629:1149–57. doi: 10.1038/s41586-024-07388-y
85. Barbaglio F, Belloni D, Scarfo L, Sbrana FV, Ponzoni M, Bongiovanni L, et al. Three-dimensional co-culture model of chronic lymphocytic leukemia bone marrow microenvironment predicts patient-specific response to mobilizing agents. Haematologica. (2021) 106:2334–44. doi: 10.3324/haematol.2020.248112
86. Haselager MV, van Driel BF, Perelaer E, de Rooij D, Lashgari D, Loos R, et al. In vitro 3D spheroid culture system displays sustained T cell-dependent CLL proliferation and survival. Hemasphere. (2023) 7:e938. doi: 10.1097/HS9.0000000000000938
87. Katti A, Vega-Perez A, Foronda M, Zimmerman J, Zafra MP, Granowsky E, et al. Generation of precision preclinical cancer models using regulated in vivo base editing. Nat Biotechnol. (2024) 42:437–47. doi: 10.1038/s41587-023-01900-x
88. Dunbar AJ, Bowman RL, Park YC, O'Connor K, Izzo F, Myers RM, et al. Jak2V617F reversible activation shows its essential requirement in myeloproliferative neoplasms. Cancer Discov. (2024) 14:737–51. doi: 10.1158/2159-8290.CD-22-0952
89. Rongvaux A, Willinger T, Martinek J, Strowig T, Gearty SV, Teichmann LL, et al. Development and function of human innate immune cells in a humanized mouse model. Nat Biotechnol. (2014) 32:364–72. doi: 10.1038/nbt.2858
90. Lagneaux L, Delforge A, Bron D, De Bruyn C, and Stryckmans P. Chronic lymphocytic leukemic B cells but not normal B cells are rescued from apoptosis by contact with normal bone marrow stromal cells. Blood. (1998) 91:2387–96. doi: 10.1182/blood.V91.7.2387
91. Kurtova AV, Balakrishnan K, Chen R, Ding W, Schnabl S, Quiroga MP, et al. Diverse marrow stromal cells protect CLL cells from spontaneous and drug-induced apoptosis: development of a reliable and reproducible system to assess stromal cell adhesion-mediated drug resistance. Blood. (2009) 114:4441–50. doi: 10.1182/blood-2009-07-233718
Keywords: microenvironment, chronic lymphocytic leukemia, Richter Transformation, immunotherapy, mouse models, preclinical studies, clinical trials
Citation: vom Stein AF, Nguyen P-H and ten Hacken E (2025) A question of TiME: how microenvironmental interactions shape response to immunotherapy in CLL and Richter Transformation. Front. Immunol. 16:1592574. doi: 10.3389/fimmu.2025.1592574
Received: 12 March 2025; Accepted: 05 May 2025;
Published: 29 May 2025.
Edited by:
Mandato Elisa, Dana–Farber Cancer Institute, United StatesReviewed by:
Rahul Shivahare, The Ohio State University, United StatesCopyright © 2025 vom Stein, Nguyen and ten Hacken. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elisa ten Hacken, ZWx0NDAxMEBtZWQuY29ybmVsbC5lZHU=