 Natália S. Vellozo
Natália S. Vellozo Thayane C. Matos-Silva
Thayane C. Matos-Silva Marcela F. Lopes
Marcela F. Lopes- Laboratório de Biologia Imunitária George DosReis, Instituto de Biofísica Carlos Chagas Filho, Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ, Brazil
Macrophage plasticity is remarkable, and recent studies have opened new prophylactic and therapeutic avenues for immunomodulation of macrophage phenotypes in inflammatory and infectious diseases. During infections caused by the pathogenic protozoans Leishmania spp. and Trypanosoma cruzi, susceptibility to disseminated or chronic infections and/or the development of inflammatory diseases depend on the balance between protective immunity mediated by macrophages and anti-inflammatory responses. Here, we will discuss strategies that exploit macrophage plasticity towards the extreme proinflammatory M1 or pro-infection M2 phenotypes to prevent the establishment of disseminated and chronic infection or to temper parasite-driven inflammatory responses. Immunomodulation of macrophage phenotypes has been tested in experimental models of protozoan infections through pharmacological approaches, synergy between pro-M1 cytokines, and targeting of pro-M2 macrophage functions, such as efferocytosis. We will address the cellular and molecular mechanisms underlying strategies designed to redirect macrophage activation towards M1 and M2 phenotypes, as well as the challenges and open questions.
1 Introduction
The pathogenic protozoans Trypanosoma cruzi (1) and Leishmania spp. (2) cause, respectively, Chagas disease and the Leishmaniasis spectrum, which challenge Public Health systems worldwide and afflict impoverished populations (3, 4). Vector-borne Leishmania parasites establish localized infection and lesion in the skin or reach mucosa and target organs, such as liver and spleen, or yet disseminate systemically, causing different pathologies referred to as Leishmaniasis (2). Although other host cells have been described (5), macrophages are the preferential host cells for Leishmania spp. and their ability to contain phagocytosed parasites or otherwise to fuel intracellular infection depends both on the host immune system and pathogen molecules that induce or subvert protective macrophage-mediated responses (6–8).
T. cruzi parasites spread from the initial focus of vector-transmitted infection through the blood to reach multiple tissues, where they invade cell cytoplasm, replicate and induce rupture of fibroblasts, myocytes, macrophages, and other cells (9, 10). In addition to host T. cruzi parasites, macrophages play multiple roles in the immune response, by inducing inflammation and by harvesting cell debris, apoptotic cells and parasites released by other cells (11–13). Therefore, how macrophages deal with infection determines the extension of parasite spread to other cells/tissues, leading to the development of chronic infection and Chagas disease after multiple rounds of parasite-driven inflammation, especially in the heart (10–12, 14).
Macrophages are functionally plastic in response to environmental stimuli, such as parasite PAMPs (pathogen-associated molecular patterns), cytokines, tissue-derived DAMPs (damage-associated molecular patterns), and apoptotic cells, by ranging from pro-inflammatory M1 macrophages, which fight infection, to pro-tissue repair M2 macrophages that eventually promote parasite replication (15–19) (Figure 1A). Here we will discuss how host-directed therapies can modulate the balance between M1 and M2 macrophages (20) to prevent the pathogenic outcomes of protozoan infections caused by Leishmania spp. and T. cruzi (Figure 1B).

Figure 1. Targeting macrophage plasticity in parasite infection. (A) Tissue environment shapes macrophage differentiation towards M1 and M2 phenotypes. Upon parasite infection, monocytes generate inflammatory macrophages which depending on environmental stimuli differentiate into M1 that express iNOS, NO, and kill parasites or into pro-tissue repair M2 that express Arg1 and host parasites. Parasite PAMPs and DAMPs from disrupted infected cells, PMNs, and NK cells induce M1, whereas apoptotic cells, eosinophils, and ILC2 lead to alternative activation of macrophages toward M2 responses. In addition to innate immunity, T cells modulate M1 and M2 phenotypes and establish T cell-macrophage crosstalk that involves both cell surface ligands and secreted cytokines, such as IFN-γ and IL-4 to promote adaptive immunity. (B) During Leishmania spp. and T. cruzi infection, classically activated (M1) macrophages control infection but also induce exacerbated inflammatory responses that lead to pathology, whereas alternatively activated (M2) macrophages promote intracellular infection and/or tissue repair. The possible outcomes of modulation targeting macrophage plasticity include but are not restricted to protective immunity, exacerbated infection, pathology, and tissue remodeling depending on the direction and intensity of environmental stimuli. Created with BioRender.com. Arg1, arginase 1; CCL, C-C motif chemokine ligand; DAMP, damage-associated molecular pattern; IL, interleukin; ILC2, type 2 innate lymphoid cells; iNOS, induced NO synthase; IFN-γ, interferon-γ; M1, Macrophage 1; M2, Macrophage 2; NK; Natural Killer cells; PAMP, pathogen-associated molecular pattern; PMN, polymorphonuclear cells; RANKL, the receptor activator of Nuclear Factor-κB ligand; Th, T helper cells; TNF-α, tumor necrosis factor-α.
2 The control of macrophage plasticity in protozoan infections
Experimental Leishmaniasis is the prototype model where Th1 and Th2 responses mediated by IFN-γ and IL-4 correlate with genetic resistance and susceptibility to Leishmania major in different mouse strains, i.e. C57BL/6 (B6) and BALB/c, respectively (21, 22). Macrophages exposed to Th1 cytokines and PAMPs were described as classically activated (M1) macrophages able to produce NO and fight infection, whereas Th2 cytokines, such as IL-4, IL-10 and IL-13 (23) induce alternatively activated (M2) macrophages, which express Arginase 1 (Arg1) and metabolize L-arginine towards the polyamine pathway (15, 16, 24–26). In addition to experimental models that develop Th1 or Th2 responses (22) and in vitro settings that generate polarized M1 or M2 macrophages (16), T cell and macrophage responses to protozoan infections show multiple/intermediate phenotypes between the extreme poles, especially within the M2 spectrum (18, 24). Here, we will not use the M1 and M2 terms to designate the strict phenotypes (16), but as a ‘compass’ to guide discussion on the immunomodulation towards M1 and M2 responses.
Both adaptive immunity (22, 23) and innate immunity (7, 27, 28) influence macrophage phenotype during infection and increase resistance or contribute to the development of disease. Leishmania braziliensis- but not L. major-recruited monocytes develop early M1 responses in the peritoneum of BALB/c mice (29). However, L. braziliensis induced a more efficient M1 response in B6 than in BALB/c mice, characterized by increased expression of the M1 hallmarks IL-12, induced NO synthase (iNOS), and NO production (29). These and other (30–32) experiments indicate that both parasite species and genetic backgrounds are relevant for macrophage responses during innate immunity. Exacerbated M1 responses may correlate with BALB/c resistance to L. braziliensis versus L. major infection (33) and the development of inflammatory disease underlying human mucocutaneous Leishmaniasis (34–36). Conversely, a series of studies support the deleterious role of M2-like monocytes and macrophages, which are better host cells for Leishmania parasites (37–43). Dermal-resident macrophages express M2 hallmarks and host Leishmania infection even in a mixed IFN-γ/IL-4 environment (27, 40, 44, 45). IL-4 from eosinophils contributes to maintenance of M2-like macrophages in a Leishmania infection model (44). Contrary to the Th1/Th2 paradigm, however, IFN-γ can increase the recruitment of M2-like monocytes that express Arg1 activity and promote parasite infection (41). Overall, M1 and M2 macrophages play a key role in resistance and susceptibility to Leishmania infection either in coordination with Th1 and Th2 responses or in a complete independent or unexpected fashion (15, 22, 23, 28, 41).
During T. cruzi infection, both innate and adaptive immunity induce M1 microbicidal macrophages that help to control infection, as evidenced by increased parasitemia and mortality in macrophage-depleted mice (46) or in mice bearing IFN-γ-signaling deficient macrophages (47). Natural Killer cells, CD4 and CD8 T cells produce IFN-γ (13, 48) and help macrophage activation into NO/iNOS-expressing M1 macrophages which are able to kill T. cruzi parasites and reduce further parasite-driven pathogenesis (11). The absence of M1 features, such as IL-12, leads to increased differentiation of M2 macrophages that propagate parasite infection and contribute to the development of Chagas disease (49). We previously discussed the role of M1 and M2 responses (11, 28) and their relevance in resistance and susceptibility to parasite-driven neglected diseases, where immunomodulation might add new therapeutic avenues to the insufficient treatment/vaccine portfolio (8, 50–52). Other discussions are available for comprehensive review (7, 11, 18, 28) and correlation with human diseases (53, 54). Here, we will focus on the experimental models that used host-directed therapies, such as mimicking T-cell macrophage cytokine crosstalk and synergy with Th1 and Th2 cytokines to induce M1 and M2 phenotypes, pharmacological interventions targeting induction/function of M1 and M2 macrophages, and identification of new pro-M2 molecular targets.
3 RANKL helps to induce M1 macrophages by mimicking T-cell macrophage crosstalk
In addition to the Th1/Th2 axis, the crosstalk between macrophages and T cells might involve other cytokines and ligands (23), such as IL-17, as discussed elsewhere (55) and the Receptor Activator of Nuclear Factor-κB Ligand (RANKL). RANKL, also known for its pro-osteoclastic properties, is a potential vaccine adjuvant that activate dendritic cells and macrophages to improve T cell proliferation and Th1 responses (56, 57). Moreover, RANKL may synergize with Th1 and Th2 environments to induce M1 and M2 macrophages, respectively (58, 59). In the context of Th1 macrophage crosstalk, T cells from L. major-infected B6 mice induce M1 responses in parasite-recruited monocytes in an antigen, RANKL and IFN-γ dependent manner (60). Whereas IFN-γ alone promotes TNF-α production in parasite-stimulated cocultures, neutralization of either IFN-γ or RANKL precludes IL-12 responses (60). To dissect how RANKL might promote M1 responses, we showed that thioglycolate-induced inflammatory macrophages express the receptor RANK and shift from M2 to M1 phenotype upon treatment with suboptimal IFN-γ concentration in the presence of RANKL (60). Low IFN-γ dose/RANKL-induced M1 macrophages express IL-12p35, iNOS, but reduced M2 features, such as Arg1, MR (mannose receptor) MGL (galactose-type lectin), and CCL17 (60). IFN-γ and RANKL synergism induces M1 responses, such as NO production and IL-12 secretion, through the NF-κB signaling pathway (60). Furthermore, low IFN-γ dose and RANKL promoted L. major control by macrophages in a ROS and NO-dependent fashion (60).
Multiple T-cell help mechanisms are probably redundant and CD40L deficient mice remain resistant to low numbers of L. major parasites in the B6 genetic background (61). However, blockade of RANKL in L. major-infected CD40L deficient mice prevented lesion healing, providing evidence that RANKL is necessary for T-cell DC crosstalk, IL-12 production, and Th1 responses (62). Accordingly, RANKL has been tested as an adjuvant for treating Ag-loaded DCs to improve Th1 responses (56) and as a vaccine-associated RANKL gene to induce anti-T. cruzi CD8 T cells (63). Interestingly, only a less virulent T. cruzi strain induced RANKL signaling pathway (64), which might contribute to M1 responses and control of infection, whereas more virulent strains subvert protective responses. Therefore, RANKL delivered locally is a safer prophylactic/therapeutic strategy that might help to improve immunity to protozoan parasites without disrupting bone homeostasis.
Other potential adjuvants, such as the cytokines APRIL (a proliferation-inducing ligand) and BAFF (B-cell activating factor), produced by DCs and monocytes, can improve M1 responses through interactions with their receptor TACI (transmembrane activator and a CAML interactor) (65). Although there are still open questions, such as how intracellular TACI receptor is mobilized to interact with the ligands, APRIL and BAFF signal through TACI receptor in macrophages to induce M1 responses and potentiate the control of Leishmania infection (65). Therefore, APRIL and BAFF are potential therapies and vaccine adjuvants to improve immunity in parasite infections.
4 Targeting M1 to M2 shift in protozoan infections
Exacerbated Th1/M1 responses underly or at least might contribute to severe outcomes in inflammatory diseases caused by protozoan parasites (36). In this sense, diversion from the proinflammatory M1 towards M2 phenotype is a potential therapeutic strategy. By dissecting the role of monocytes in Leishmania infection, we found that treatment with all-trans-retinoic acid (ATRA) promotes macrophage maturation at the cost of effective M1 responses (66). Whereas ATRA injection helps T cell proliferation by reducing immature myeloid cells-mediated suppression, early treatment with ATRA also reduced NO production and increased parasite load in lymph nodes of L. major-infected B6 mice (66). The effects of ATRA injection on monocyte phenotype can be adaptive immunity independent as showed in B6 or BALB/c mice treated with ATRA 24 h after i.p. L. major infection and analysed for immune responses 24 h later (29). Treatment with ATRA reduced M1 features, such as iNOS expression, IL-12 and TNF-α secretion, and increased parasite load within peritoneal macrophages (29). For comparing the direct effects of ATRA in BALB/c and B6 bone-marrow derived macrophages (BMDMs), we used an LPS (lipopolysaccharide)/cytokine setting that mimics a mixed Th1/Th2/infection environment (29). Treatment with ATRA reduced LPS-induced M1 hallmarks, such as secretion of TNF-α and CXCL9, and increased the M2 chemokines CCL17 and CXCL13. Moreover, ATRA downmodulated iNOS expression and NO production by LPS-stimulated macrophages (29). Whereas ATRA treatment might be deleterious by increasing susceptibility to L. major infection, it is reasonable to envision that ATRA could attenuate exacerbated M1 pathogenic responses (28, 36) and prevent parasite-driven inflammation and pathology upon pro-M1 L. braziliensis infection. More proof-of-principle studies are necessary for guiding further research and strategy development to treat human diseases.
Similar to ATRA that signals through intracellular receptors, lipids extracted from T. cruzi parasites induce alternative activation of macrophages and counteract inflammatory responses (67). The activation of PPAR (peroxisome proliferator activator receptor) γ signaling pathway by parasite lipids might reduce NF-κB pathway and prevent M1 responses (67). Likewise, the PPARα ligand fenofibrate induces a pro-repair M2 response during acute and chronic T. cruzi infection (68, 69). Furthermore, treatment with fenofibrate reduces inflammation, fibrosis and biomarkers of tissue damage, and improves heart functioning in experimental Chagas disease in a macrophage dependent fashion (68, 69). Interestingly, a short-term treatment of chronically infected mice with the betulinic acid derivative BA5 helped to prevent inflammation and fibrosis by inducing IL-10 and M2 polarization (70). Treatment with BA5 did not change parasite burden but could be associated to current anti-parasite drugs as an anti-inflammatory therapy (70). How to apply these new anti-inflammatory tools to prevent pathology in Chagas disease is a path yet to be explored.
5 Pharmacologically targeting M1 and M2 macrophages
M1 macrophages play a protective role during acute T. cruzi infection by phagocytosing parasites released from disrupted infected cells, followed by parasite killing within macrophages (11, 12). By contrast, M2-like macrophages harbor and fuel parasite infection, by diverting L-arginine metabolism towards the polyamine pathway (37, 71). Moreover, delayed induction of protective M1 responses can contribute to parasite dissemination and disease (12), whereas exacerbated inflammation ensues pathology. Therefore, the mechanisms that govern M1 and M2 macrophage phenotypes are potential targets for immunomodulation to improve immunity or downregulate pathogenic inflammatory responses (Figure 1B).
In T. cruzi infected B6 mice, PLA2 (phospholipase A2) and PI3K (phosphatidyl inositol 3 kinase) signaling pathways induce macrophage activation and protective immunity, while genetic ablation and pharmacological inhibition promote a shift to M2 macrophages and result in increased parasitemia and parasite load in the heart, associated with heart pathology/defective function (72, 73). By contrast, regulatory mechanisms such as SLAMF1 (signaling lymphocytic activation molecule) that reduces NADPH (nicotinamide adenine dinucleotide phosphate) oxidase and CD73 ectonucleotidase downregulate macrophage activation in susceptible BALB/c mice and are potential targets to improve macrophage-mediated immunity towards M1 responses (74, 75). Importantly, CD73 ablation and pharmacological inhibition prevented heart pathology and arrhythmia associated with parasite infection, tissue damage and inflammation (75, 76).
Association between M2 macrophages and susceptibility to Leishmania parasites (37–40, 42, 53) indicate that macrophage phenotypes might be targets for immunotherapy in Leishmaniasis. The L-arginine metabolism through the Arg1 activity is a hallmark of diffuse cutaneous Leishmaniasis in patients (37, 77, 78). In experimental models, susceptibility versus resistance to L. major infection correlates well with increased Arg1 expression and Th2 responses in BALB/c versus B6 mice (79). Inhibition of Arg1 activity helped both parasite and lesion control in L. major-infected BALB/c mice (79). Conversely, treatment with L-ornithine increased susceptibility in otherwise resistant B6 mice (79). In T. cruzi infection, IL-13-induced susceptibility is associated with enhanced M2 responses, such as Arg1 activity, whereas treatment with Arg1 inhibitors reduced mortality (80). Accordingly, infection of BMDMs with virulent but not less virulent T. cruzi parasites subverts parasite killing by inducing Arg1 expression and downmodulating iNOS expression (81).
In addition to L-arginine metabolism, other aspects of immunometabolism are potential targets for the control of macrophage plasticity and Leishmania infection (82, 83). Iron containing nanoparticles target host cell metabolism and improve protective M1 responses to fight Leishmania parasites (8). Induction versus inhibition of glucose-6-phosphate dehydrogenase (G6PDH) activity regulates NO-dependent resistance versus macrophage susceptibility to Leishmania parasites (84).
T. cruzi infection induces the metabolic check point mammalian Target of Rapamycin inhibition (mTOR) mTORC1 pathway in macrophages (85). Moreover, in vitro treatment with the mTOR inhibitor rapamycin reduced M2 responses, increased proinflammatory cytokines, and promoted parasite control in a NLRP3-dependent fashion (85). How to regulate immunometabolism in vivo in a cell specific fashion is a challenge to develop successful therapy that prevents homeostasis disruption.
T. cruzi infection modifies macrophage miRNA responses (86) and some miRNAs control macrophage plasticity to induce M1 and M2 phenotypes (87). In macrophages infected with antimony-resistant Leishmania parasites, certain miRNAs downmodulate iNOS expression and subvert Myd88 (myeloid differentiation primary response 88)-NFκB signaling to promote early IL-10 secretion that contributes to increased parasite burden and pathology in visceral Leishmaniasis (88). Remarkably, modulation of miRNAs can be used in vivo and are potential tools to shape macrophage phenotypes and ability to control Leishmania infection (89, 90).
6 Identifying new inhibitable pro-M2 molecular targets
During infection, M2 macrophages are parasite-permissive host cells that also play a role in anti-inflammatory responses, tissue remodeling, and fibrosis (17, 49). Macrophages respond to Th2 cytokines and to recognition and removal of apoptotic cells (efferocytosis) by turning off M1 and switching to pro-M2 signaling pathways (17, 71). A major goal on drug discovery and development of host-directed therapies is to identify new selective targets that show anti-parasite potential without enhancing pathology or disrupting host homeostasis. i.e. tissue repair (68, 69) (Figure 1B).
We previously showed that T cell apoptosis increases during T. cruzi infection and contributes to defective T cell responses that might underly parasite persistence (91). Molecular mechanisms such as ligands, death receptors, and the components of proapoptotic machinery were studied and tested in proof-of-concept experiments in acute T. cruzi infection (92). By summarizing, treatment with anti-FasL and the pan caspase inhibitor zVAD improved both T-cell and macrophage-mediated immunity and reduced parasitemia during acute infection (93–95). Nonetheless, we observed a timely regulated increase in Th1 and Th2 responses in FasL deficient or anti-FasL treated mice (93, 96), and that caspase-8 deficiency also upregulated Th2 responses to T. cruzi and L. major infections (97, 98). Therefore, whereas interesting as a hypothesis test, interrupting apoptosis-inducing signaling might disrupt homeostasis and bring considerable concern issues. Nonetheless, a vaccine strategy prevented the induction of Fas-expressing proapoptotic CD8 T cells after T. cruzi challenge (99), opening a safer prophylaxis avenue than pharmacological targeting of apoptosis signaling pathways. Importantly, vaccine-induced CD8 T cells exhibit effector responses and differ from exhausted/proapoptotic T cells generated during T. cruzi infection (99), which might fail to induce early macrophage activation to control infection (12).
Upon apoptosis, efferocytosis removes apoptotic cells and prevents the release of DAMPs and subsequent inflammation. Multiple receptors detect phosphatidylserine exposure or other apoptosis features and initiate phagocytosis of apoptotic cells and anti-inflammatory signaling to ensure homeostasis (100–103). During inflammation, however, macrophages might use a different set of efferocytosis receptors providing an opportunity for selective pharmacological intervention. Accordingly, anti-inflammatory versus inflammatory stimuli induce preferential expression of the TAM (Tyro Axl Mer) receptors Mer versus Axl in macrophages (104).
Efferocytosis of apoptotic cells promotes T. cruzi replication within macrophages in a TGF-β, prostaglandin E2, and polyamine dependent fashion (71). In peritoneal macrophages from infected mice, the integrin αvβ3 was identified as a putative efferocytosis receptor for apoptotic cell-inducing signaling that contributes to T. cruzi growth (71). For addressing the role of efferocytosis receptors during parasite infection, we used single Mer or Axl defective mice and BMDMs cultured with T cells from T. cruzi-infected mice, which provided both effector and pro-apoptotic cells able to impact on macrophage phenotypes (105). In vitro, Mer deficiency significantly reduced efferocytosis but had little impact on macrophage phenotype (105). Remarkably, Axl defective macrophages showed improved M1 responses, such as CXCL9 and IL-12p35 expression, iNOS expression and NO production, and increased ability to control T. cruzi infection despite only partial inhibition of efferocytosis (105).
Moreover, Axl-deficient mice had reduced peak parasitemia and less inflammation and fibrosis in their hearts compared to infected B6 WT and Mer-/- mice (105). Infected Axl-/- mice also showed increased M1 responses in the peritoneum and spleen and iNOS expression in the heart (105). These results indicate that Axl is a selective target to improve macrophage-mediated immunity without interfering with apoptosis or Mer-mediated homeostatic efferocytosis. Nonetheless, the accumulation of apoptotic cells in infected Axl-deficient mice (105) is a potential deleterious side effect that deserves caution in efferocytosis inhibition.
During Leishmania infection, the TAM receptor Mer plays a role in the efferocytosis of infected neutrophils by DCs and suppression of T cell responses (106). Furthermore, Mer-mediated efferocytosis of infected neutrophils transfers Leishmania parasites to macrophages (107). Dual Mer/Axl genetic ablation reduced the development of M2 macrophages and parasite infection (107). Nonetheless, increased lesions in infected double KO mice indicate that Mer and/or Axl play an essential anti-inflammatory role to prevent parasite-induced pathology (107). New studies in single-receptor defective mice might clarify the individual roles of TAM receptors in L. major infection.
7 Concluding remarks
Targeting immunoregulatory host mechanisms such as T-cell coinhibitory receptors (51, 108) can improve otherwise suppressed immune responses or upregulate immunity. Likewise, unveiling the mechanisms of macrophage plasticity (87) might translate into host-directed therapies to mitigate human diseases. New drug delivery systems by using liposomes or nanoparticles (8) and vaccine mRNA technology will foster the development of new drugs, vaccines, and therapeutic vaccines to fight infectious diseases. How these remarkable scientific and technological advances (Table 1) might translate into clinical trials for Chagas disease and Leishmaniasis and lead to effective solutions for tropical neglected diseases will demand major scientific, industrial, and political efforts.
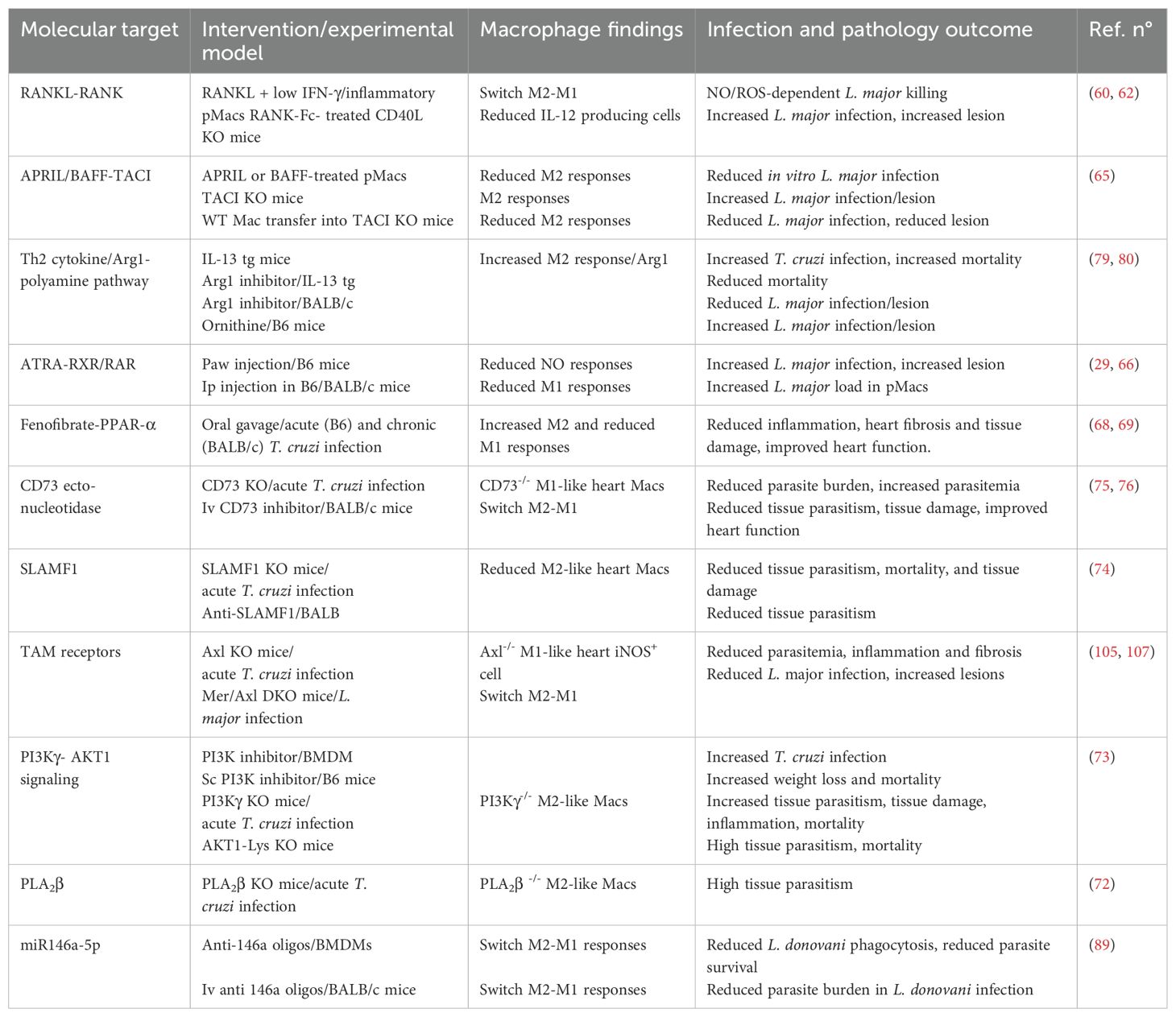
Table 1. Macrophage plasticity: molecular targets to shape M1 and M2 phenotypes.
Author contributions
NV: Writing – original draft, Writing – review & editing. TM-S: Writing – original draft, Writing – review & editing. ML: Conceptualization, Supervision, Writing – review & editing, Writing – original draft.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the Brazilian National Research Council (Conselho Nacional de Desenvolvimento Científico e Tecnológico, CNPq) and the Rio de Janeiro State Science Foundation (Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro, FAPERJ). ML is a research fellow at CNPq, Brazil. We also received fellowships from FAPERJ (NV and TM-S) and the American Association of Immunologists (NV and ML).
Acknowledgments
We acknowledge Jerson Lima Silva as the previous FAPERJ president for all the support for this research group and work.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Nunes MCP, Beaton A, Acquatella H, Bern C, Bolger AF, Echeverria LE, et al. Chagas cardiomyopathy: an update of current clinical knowledge and management: A scientific statement from the american heart association. Circulation. (2018) 138:e169–209. doi: 10.1161/CIR.0000000000000599
2. Burza S, Croft SL, and Boelaert M. Leishmaniasis. Lancet. (2018) 392:951–70. doi: 10.1016/S0140-6736(18)31204-2
3. Organization WH. Chagas disease (American trypanosomiasis) (2025). Available online at: https://www.who.int/health-topics/chagas-diseasetab=tab_1 (Accessed July 14, 2025).
4. Organization WH. Leishmaniasis (2025). Available online at: https://www.who.int/health-topics/leishmaniasistab=tab_1 (Accessed July 14, 2025).
5. Valigurova A and Kolarova I. Unrevealing the mystery of latent leishmaniasis: what cells can host leishmania? Pathogens. (2023) 12. doi: 10.3390/pathogens12020246
6. Bogdan C. Macrophages as host, effector and immunoregulatory cells in leishmaniasis: Impact of tissue micro-environment and metabolism. Cytokine X. (2020) 2:100041. doi: 10.1016/j.cytox.2020.100041
7. Carneiro MB, Vaz LG, Afonso LCC, Horta MF, and Vieira LQ. Regulation of macrophage subsets and cytokine production in leishmaniasis. Cytokine. (2021) 147:155309. doi: 10.1016/j.cyto.2020.155309
8. Palomino-Cano C, Moreno E, Irache JM, and Espuelas S. Targeting and activation of macrophages in leishmaniasis. A Focus iron Oxide nanoparticles Front Immunol. (2024) 15:1437430. doi: 10.3389/fimmu.2024.1437430
9. Bonney KM, Luthringer DJ, Kim SA, Garg NJ, and Engman DM. Pathology and pathogenesis of chagas heart disease. Annu Rev Pathol. (2019) 14:421–47. doi: 10.1146/annurev-pathol-020117-043711
10. De Alba-Alvarado MC, Torres-Gutierrez E, Reynoso-Ducoing OA, Zenteno-Galindo E, Cabrera-Bravo M, Guevara-Gomez Y, et al. Immunopathological mechanisms underlying cardiac damage in chagas disease. Pathogens. (2023) 12. doi: 10.3390/pathogens12020335
11. Vellozo NS, Matos-Silva TC, and Lopes MF. Immunopathogenesis in Trypanosoma cruzi infection: a role for suppressed macrophages and apoptotic cells. Front Immunol. (2023) 14:1244071. doi: 10.3389/fimmu.2023.1244071
12. Padilla AM, Rosenberg C, Cook P, Sanchez-Valdez F, McElhannon C, and Tarleton RL. Delayed Activation of T Cells at the Site of Infection Facilitates the Establishment of Trypanosoma cruzi in Both Naive and Immune Hosts. mSphere. (2023) 8:e0060122. doi: 10.1128/msphere.00601-22
13. Macaluso G, Grippi F, Di Bella S, Blanda V, Gucciardi F, Torina A, et al. A Review on the Immunological Response against Trypanosoma cruzi. Pathogens. (2023) 12. doi: 10.3390/pathogens12020282
14. Carvalho A, Ferraz IA, Hojo-Souza NS, Medeiros FAC, Viana LA, Bartholomeu DC, et al. Chagas cardiomyopathy is associated with a high susceptibility to T. cruzi infection in monocyte-derived macrophages and a predominance of CD4(+)CD45RO(+) T-cells with immunoregulatory patterns. Acta Trop. (2023) 237:106749. doi: 10.1016/j.actatropica.2022.106749
15. Mills CD, Kincaid K, Alt JM, Heilman MJ, and Hill AM. M-1/M-2 macrophages and the Th1/Th2 paradigm. J Immunol. (2000) 164:6166–73. doi: 10.4049/jimmunol.164.12.6166
16. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. (2014) 41:14–20. doi: 10.1016/j.immuni.2014.06.008
17. Bosurgi L, Cao YG, Cabeza-Cabrerizo M, Tucci A, Hughes LD, Kong Y, et al. Macrophage function in tissue repair and remodeling requires IL-4 or IL-13 with apoptotic cells. Science. (2017) 356:1072–6. doi: 10.1126/science.aai8132
18. Tomiotto-Pellissier F, Bortoleti B, Assolini JP, Goncalves MD, Carloto ACM, Miranda-Sapla MM, et al. Macrophage polarization in leishmaniasis: broadening horizons. Front Immunol. (2018) 9:2529. doi: 10.3389/fimmu.2018.02529
19. Almeida FS, Vanderley SER, Comberlang FC, Andrade AG, Cavalcante-Silva LHA, Silva EDS, et al. Leishmaniasis: immune cells crosstalk in macrophage polarization. Trop Med Infect Dis. (2023) 8. doi: 10.3390/tropicalmed8050276
20. Wang L, Lu Q, Gao W, and Yu S. Recent advancement on development of drug-induced macrophage polarization in control of human diseases. Life Sci. (2021) 284:119914. doi: 10.1016/j.lfs.2021.119914
21. Scott P, Natovitz P, Coffman RL, Pearce E, and Sher A. Immunoregulation of cutaneous leishmaniasis. T cell lines that transfer protective immunity or exacerbation belong to different T helper subsets and respond to distinct parasite antigens. J Exp Med. (1988) 168:1675–84. doi: 10.1084/jem.168.5.1675
22. Mosmann TR and Coffman RL. TH1 and TH2 cells: different patterns of lymphokine secretion lead to different functional properties. Annu Rev Immunol. (1989) 7:145–73. doi: 10.1146/annurev.iy.07.040189.001045
23. Osero BO, Aruleba RT, Brombacher F, and Hurdayal R. Unravelling the unsolved paradoxes of cytokine families in host resistance and susceptibility to Leishmania infection. Cytokine X. (2020) 2:100043. doi: 10.1016/j.cytox.2020.100043
24. Mosser DM and Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. (2008) 8:958–69. doi: 10.1038/nri2448
25. Locati M, Mantovani A, and Sica A. Macrophage activation and polarization as an adaptive component of innate immunity. Adv Immunol. (2013) 120:163–84. doi: 10.1016/B978-0-12-417028-5.00006-5
26. Mantovani A, Biswas SK, Galdiero MR, Sica A, and Locati M. Macrophage plasticity and polarization in tissue repair and remodelling. J Pathol. (2013) 229:176–85. doi: 10.1002/path.4133
27. Lee SH and Sacks DL. Resilience of dermis resident macrophages to inflammatory challenges. Exp Mol Med. (2024) 56:2105–12. doi: 10.1038/s12276-024-01313-z
28. Vellozo NS, Rigoni TS, and Lopes MF. New therapeutic tools to shape monocyte functional phenotypes in leishmaniasis. Front Immunol. (2021) 12:704429. doi: 10.3389/fimmu.2021.704429
29. Vellozo NS, Pereira-Marques ST, Cabral-Piccin MP, Filardy AA, Ribeiro-Gomes FL, Rigoni TS, et al. All-trans retinoic acid promotes an M1- to M2-phenotype shift and inhibits macrophage-mediated immunity to leishmania major. Front Immunol. (2017) 8:1560. doi: 10.3389/fimmu.2017.01560
30. Scott P and Sher A. A spectrum in the susceptibility of leishmanial strains to intracellular killing by murine macrophages. J Immunol. (1986) 136:1461–6. doi: 10.4049/jimmunol.136.4.1461
31. Tomiotto-Pellissier F, Miranda-Sapla MM, Silva TF, Bortoleti B, Goncalves MD, Concato VM, et al. Murine Susceptibility to Leishmania amazonensis Infection Is Influenced by Arginase-1 and Macrophages at the Lesion Site. Front Cell Infect Microbiol. (2021) 11:687633. doi: 10.3389/fcimb.2021.687633
32. Restrepo CM, Llanes A, Herrera L, Ellis E, Lleonart R, and Fernandez PL. Gene expression patterns associated with Leishmania panamensis infection in macrophages from BALB/c and C57BL/6 mice. PloS Negl Trop Dis. (2021) 15:e0009225. doi: 10.1371/journal.pntd.0009225
33. Rocha FJ, Schleicher U, Mattner J, Alber G, and Bogdan C. Cytokines, signaling pathways, and effector molecules required for the control of Leishmania (Viannia) Braziliensis in mice. Infect Immun. (2007) 75:3823–32. doi: 10.1128/IAI.01335-06
34. Giudice A, Vendrame C, Bezerra C, Carvalho LP, Delavechia T, Carvalho EM, et al. Macrophages participate in host protection and the disease pathology associated with Leishmania Braziliensis infection. BMC Infect Dis. (2012) 12:75. doi: 10.1186/1471-2334-12-75
35. Gaze ST, Dutra WO, Lessa M, Lessa H, Guimaraes LH, Jesus AR, et al. Mucosal leishmaniasis patients display an activated inflammatory T-cell phenotype associated with a nonbalanced monocyte population. Scand J Immunol. (2006) 63:70–8. doi: 10.1111/j.1365-3083.2005.01707.x
36. Carvalho AM, Bacellar O, and Carvalho EM. Protection and pathology in leishmania Braziliensis infection. Pathogens. (2022) 11. doi: 10.3390/pathogens11040466
37. Franca-Costa J, Van Weyenbergh J, Boaventura VS, Luz NF, Malta-Santos H, Oliveira MC, et al. polyamine, and prostaglandin E2 pathways suppress the inflammatory response and contribute to diffuse cutaneous leishmaniasis. J Infect Dis. (2015) 211:426–35. doi: 10.1093/infdis/jiu455
38. Hammami A, Abidin BM, Charpentier T, Fabie A, Duguay AP, Heinonen KM, et al. HIF-1alpha is a key regulator in potentiating suppressor activity and limiting the microbicidal capacity of MDSC-like cells during visceral leishmaniasis. PloS Pathog. (2017) 13:e1006616. doi: 10.1371/journal.ppat.1006616
39. Hu S, Marshall C, Darby J, Wei W, Lyons AB, and Korner H. Absence of Tumor Necrosis Factor Supports Alternative Activation of Macrophages in the Liver after Infection with Leishmania major. Front Immunol. (2018) 9:1. doi: 10.3389/fimmu.2018.00001
40. Lee SH, Charmoy M, Romano A, Paun A, Chaves MM, Cope FO, et al. Mannose receptor high, M2 dermal macrophages mediate nonhealing Leishmania major infection in a Th1 immune environment. J Exp Med. (2018) 215:357–75. doi: 10.1084/jem.20171389
41. Carneiro MB, Lopes ME, Hohman LS, Romano A, David BA, Kratofil R, et al. Th1-th2 cross-regulation controls early leishmania infection in the skin by modulating the size of the permissive monocytic host cell reservoir. Cell Host Microbe. (2020) 27:752–68 e7. doi: 10.1016/j.chom.2020.03.011
42. Moulik S, Karmakar J, Joshi S, Dube A, Mandal C, and Chatterjee M. Status of IL-4 and IL-10 driven markers in experimental models of Visceral Leishmaniasis. Parasite Immunol. (2021) 43:e12783. doi: 10.1111/pim.12783
43. Silva JMD, Silva H, Sarmento ALC, Hueb M, and Damazo AS. Analysis of clinical cure outcome, macrophages number, cytokines levels and expression of annexin-A1 in the cutaneous infection in patients with Leishmania Braziliensis. Rev Soc Bras Med Trop. (2024) 57:e00412. doi: 10.1590/0037-8682-0036-2024
44. Lee SH, Chaves MM, Kamenyeva O, Gazzinelli-Guimaraes PH, Kang B, Pessenda G, et al. M2-like, dermal macrophages are maintained via IL-4/CCL24-mediated cooperative interaction with eosinophils in cutaneous leishmaniasis. Sci Immunol. (2020) 5. doi: 10.1126/sciimmunol.aaz4415
45. Lee SH, Kang B, Kamenyeva O, Ferreira TR, Cho K, Khillan JS, et al. Dermis resident macrophages orchestrate localized ILC2 eosinophil circuitries to promote non-healing cutaneous leishmaniasis. Nat Commun. (2023) 14:7852. doi: 10.1038/s41467-023-43588-2
46. Kierszenbaum F, Knecht E, Budzko DB, and Pizzimenti MC. Phagocytosis: a defense mechanism against infection with Trypanosoma cruzi. J Immunol. (1974) 112:1839–44. doi: 10.4049/jimmunol.112.5.1839
47. Lykens JE, Terrell CE, Zoller EE, Divanovic S, Trompette A, Karp CL, et al. Mice with a selective impairment of IFN-gamma signaling in macrophage lineage cells demonstrate the critical role of IFN-gamma-activated macrophages for the control of protozoan parasitic infections in vivo. J Immunol. (2010) 184:877–85. doi: 10.4049/jimmunol.0902346
48. de Alba-Alvarado MC, Cabrera-Bravo M, Zenteno E, Salazar-Schetino PM, and Bucio-Torres MI. The functions of cytokines in the cardiac immunopathogenesis of chagas disease. Pathogens. (2024) 13. doi: 10.3390/pathogens13100870
49. Bastos KR, Alvarez JM, Marinho CR, Rizzo LV, and Lima MR. Macrophages from IL-12p40-deficient mice have a bias toward the M2 activation profile. J Leukoc Biol. (2002) 71:271–8. doi: 10.1189/jlb.71.2.271
50. Morillo CA, Marin-Neto JA, Avezum A, Sosa-Estani S, Rassi A Jr., Rosas F, et al. Randomized trial of benznidazole for chronic chagas' Cardiomyopathy. N Engl J Med. (2015) 373:1295–306. doi: 10.1056/NEJMoa1507574
51. Gannavaram S, Bhattacharya P, Ismail N, Kaul A, Singh R, and Nakhasi HL. Modulation of innate immune mechanisms to enhance leishmania vaccine-induced immunity: role of coinhibitory molecules. Front Immunol. (2016) 7:187. doi: 10.3389/fimmu.2016.00187
52. Pinazo MJ, Malchiodi E, Ioset JR, Bivona A, Gollob KJ, and Dutra WO. Challenges and advancements in the development of vaccines and therapies against Chagas disease. Lancet Microbe. (2024) 5:100972. doi: 10.1016/j.lanmic.2024.100972
53. Silva RL, Santos MB, Almeida PL, Barros TS, Magalhaes L, Cazzaniga RA, et al. sCD163 levels as a biomarker of disease severity in leprosy and visceral leishmaniasis. PloS Negl Trop Dis. (2017) 11:e0005486. doi: 10.1371/journal.pntd.0005486
54. Sandoval Pacheco CM, Araujo Flores GV, Gonzalez K, de Castro Gomes CM, Passero LFD, Tomokane TY, et al. Macrophage polarization in the skin lesion caused by neotropical species of leishmania sp. J Immunol Res. (2021) 2021:5596876. doi: 10.1155/2021/5596876
55. Morales-Primo AU, Becker I, Pedraza-Zamora CP, and Zamora-Chimal J. Th17 cell and inflammatory infiltrate interactions in cutaneous leishmaniasis: unraveling immunopathogenic mechanisms. Immune Netw. (2024) 24:e14. doi: 10.4110/in.2024.24.e14
56. Josien R, Li HL, Ingulli E, Sarma S, Wong BR, Vologodskaia M, et al. TRANCE, a tumor necrosis factor family member, enhances the longevity and adjuvant properties of dendritic cells in vivo. J Exp Med. (2000) 191:495–502. doi: 10.1084/jem.191.3.495
57. Anderson DM, Maraskovsky E, Billingsley WL, Dougall WC, Tometsko ME, Roux ER, et al. A homologue of the TNF receptor and its ligand enhance T-cell growth and dendritic-cell function. Nature. (1997) 390:175–9. doi: 10.1038/36593
58. Huang R, Wang X, Zhou Y, and Xiao Y. RANKL-induced M1 macrophages are involved in bone formation. Bone Res. (2017) 5:17019. doi: 10.1038/boneres.2017.19
59. Meng YH, Zhou WJ, Jin LP, Liu LB, Chang KK, Mei J, et al. RANKL-mediated harmonious dialogue between fetus and mother guarantees smooth gestation by inducing decidual M2 macrophage polarization. Cell Death Dis. (2017) 8:e3105. doi: 10.1038/cddis.2017.505
60. Rigoni TS, Vellozo NS, Cabral-Piccin M, Fabiano-Coelho L, Lopes UG, Filardy AA, et al. RANK ligand helps immunity to leishmania major by skewing M2-like into M1 macrophages. Front Immunol. (2020) 11:886. doi: 10.3389/fimmu.2020.00886
61. Padigel UM and Farrell JP. CD40-CD40 ligand costimulation is not required for initiation and maintenance of a Th1-type response to Leishmania major infection. Infect Immun. (2003) 71:1389–95. doi: 10.1128/IAI.71.3.1389-1395.2003
62. Padigel UM, Kim N, Choi Y, and Farrell JP. TRANCE-RANK costimulation is required for IL-12 production and the initiation of a Th1-type response to Leishmania major infection in CD40L-deficient mice. J Immunol. (2003) 171:5437–41. doi: 10.4049/jimmunol.171.10.5437
63. Miyahira Y, Akiba H, Katae M, Kubota K, Kobayashi S, Takeuchi T, et al. Cutting edge: a potent adjuvant effect of ligand to receptor activator of NF-kappa B gene for inducing antigen-specific CD8+ T cell response by DNA and viral vector vaccination. J Immunol. (2003) 171:6344–8. doi: 10.4049/jimmunol.171.12.6344
64. Herreros-Cabello A, Del Moral-Salmoral J, Morato E, Marina A, Barrocal B, Fresno M, et al. Quantitative proteomic analysis of macrophages infected with trypanosoma cruzi reveals different responses dependent on the SLAMF1 receptor and the parasite strain. Int J Mol Sci. (2024) 25. doi: 10.3390/ijms25137493
65. Allman WR, Dey R, Liu L, Siddiqui S, Coleman AS, Bhattacharya P, et al. TACI deficiency leads to alternatively activated macrophage phenotype and susceptibility to Leishmania infection. Proc Natl Acad Sci U S A. (2015) 112:E4094–103. doi: 10.1073/pnas.1421580112
66. Pereira WF, Ribeiro-Gomes FL, Guillermo LV, Vellozo NS, Montalvao F, Dosreis GA, et al. Myeloid-derived suppressor cells help protective immunity to Leishmania major infection despite suppressed T cell responses. J Leukoc Biol. (2011) 90:1191–7. doi: 10.1189/jlb.1110608
67. Penas FN, Bott E, Carneiro AB, Lopez SA, Torres Bozza P, Goren NB, et al. Modified lipids from Trypanosoma cruzi amastigotes down-regulate the pro-inflammatory response and increase the expression of alternative activation markers in macrophages. Microb Pathog. (2025) 198:107140. doi: 10.1016/j.micpath.2024.107140
68. Cevey AC, Pieralisi AV, Donato M, Rada J, Gelpi RJ, Mirkin GA, et al. Macrophages mediate healing properties of fenofibrate in experimental chagasic cardiomyopathy. ACS Infect Dis. (2023) 9:213–20. doi: 10.1021/acsinfecdis.2c00535
69. Luque JR, Cevey AC, Pieralisi AV, Poncini C, Díaz FE, Reis MVA, et al. Fenofibrate induces a resolving profile in heart macrophage subsets and attenuates acute chagas myocarditis. ACS Infect Diseases. (2024) 10:1793–807. doi: 10.1021/acsinfecdis.4c00125
70. Meira CS, Santos ES, Santo R, Vasconcelos JF, Orge ID, Nonaka CKV, et al. Betulinic acid derivative BA5, attenuates inflammation and fibrosis in experimental chronic chagas disease cardiomyopathy by inducing IL-10 and M2 polarization. Front Immunol. (2019) 10:1257. doi: 10.3389/fimmu.2019.01257
71. Freire-de-Lima CG, Nascimento DO, Soares MB, Bozza PT, Castro-Faria-Neto HC, de Mello FG, et al. Uptake of apoptotic cells drives the growth of a pathogenic trypanosome in macrophages. Nature. (2000) 403:199–203. doi: 10.1038/35003208
72. Sharma J, Blase JR, Hoft DF, Marentette JO, Turk J, and McHowat J. Mice with genetic deletion of group VIA phospholipase A2beta exhibit impaired macrophage function and increased parasite load in trypanosoma cruzi-induced myocarditis. Infect Immun. (2016) 84:1137–42. doi: 10.1128/IAI.01564-15
73. Silva MC, Davoli-Ferreira M, Medina TS, Sesti-Costa R, Silva GK, Lopes CD, et al. Canonical PI3Kgamma signaling in myeloid cells restricts Trypanosoma cruzi infection and dampens chagasic myocarditis. Nat Commun. (2018) 9:1513. doi: 10.1038/s41467-018-03986-3
74. Calderon J, Maganto-Garcia E, Punzon C, Carrion J, Terhorst C, and Fresno M. The receptor Slamf1 on the surface of myeloid lineage cells controls susceptibility to infection by Trypanosoma cruzi. PloS Pathog. (2012) 8:e1002799. doi: 10.1371/journal.ppat.1002799
75. Ponce NE, Sanmarco LM, Eberhardt N, Garcia MC, Rivarola HW, Cano RC, et al. CD73 inhibition shifts cardiac macrophage polarization toward a microbicidal phenotype and ameliorates the outcome of experimental chagas cardiomyopathy. J Immunol. (2016) 197:814–23. doi: 10.4049/jimmunol.1600371
76. Eberhardt N, Sanmarco LM, Bergero G, Theumer MG, Garcia MC, Ponce NE, et al. Deficiency of CD73 activity promotes protective cardiac immunity against Trypanosoma cruzi infection but permissive environment in visceral adipose tissue. Biochim Biophys Acta Mol Basis Dis. (2020) 1866:165592. doi: 10.1016/j.bbadis.2019.165592
77. Christensen SM, Belew AT, El-Sayed NM, Tafuri WL, Silveira FT, and Mosser DM. Host and parasite responses in human diffuse cutaneous leishmaniasis caused by L. amazonensis. PloS Negl Trop Dis. (2019) 13:e0007152. doi: 10.1371/journal.pntd.0007152
78. Malta-Santos H, Franca-Costa J, Macedo A, Queiroz ATL, Fukutani KF, Muxel SM, et al. Differential expression of polyamine biosynthetic pathways in skin lesions and in plasma reveals distinct profiles in diffuse cutaneous leishmaniasis. Sci Rep. (2020) 10:10543. doi: 10.1038/s41598-020-67432-5
79. Iniesta V, Carcelen J, Molano I, Peixoto PM, Redondo E, Parra P, et al. Arginase I induction during Leishmania major infection mediates the development of disease. Infect Immun. (2005) 73:6085–90. doi: 10.1128/IAI.73.9.6085-6090.2005
80. Abad Dar M and Holscher C. Arginase-1 is responsible for IL-13-mediated susceptibility to trypanosoma cruzi infection. Front Immunol. (2018) 9:2790. doi: 10.3389/fimmu.2018.02790
81. Wilkins-Rodriguez AA, Salazar-Schettino PM, Manning-Cela RG, and Gutierrez-Kobeh L. Differential Regulation of L-Arginine Metabolism through NOS2 and Arginases during Infection with Trypanosoma cruzi. Pathogens. (2024) 13. doi: 10.3390/pathogens13100878
82. Ferreira C, Estaquier J, and Silvestre R. Immune-metabolic interactions between Leishmania and macrophage host. Curr Opin Microbiol. (2021) 63:231–7. doi: 10.1016/j.mib.2021.07.012
83. Saunders EC and McConville MJ. Immunometabolism of leishmania granulomas. Immunol Cell Biol. (2020) 98:832–44. doi: 10.1111/imcb.12394
84. Zamani S, Hoseini AZ, and Namin AM. Glucose-6-phosphate dehydrogenase (G6PD) activity can modulate macrophage response to Leishmania major infection. Int Immunopharmacol. (2019) 69:178–83. doi: 10.1016/j.intimp.2019.01.028
85. Rojas Marquez JD, Ana Y, Baigorri RE, Stempin CC, and Cerban FM. Mammalian target of rapamycin inhibition in trypanosoma cruzi-infected macrophages leads to an intracellular profile that is detrimental for infection. Front Immunol. (2018) 9:313. doi: 10.3389/fimmu.2018.00313
86. Rego N, Libisch MG, Rovira C, Tosar JP, and Robello C. Comparative microRNA profiling of Trypanosoma cruzi infected human cells. Front Cell Infect Microbiol. (2023) 13:1187375. doi: 10.3389/fcimb.2023.1187375
87. Locati M, Curtale G, and Mantovani A. Diversity, mechanisms, and significance of macrophage plasticity. Annu Rev Pathol. (2020) 15:123–47. doi: 10.1146/annurev-pathmechdis-012418-012718
88. Mukherjee B, Paul J, Mukherjee S, Mukhopadhyay R, Das S, Naskar K, et al. Antimony-Resistant Leishmania donovani Exploits miR-466i To Deactivate Host MyD88 for Regulating IL-10/IL-12 Levels during Early Hours of Infection. J Immunol. (2015) 195:2731–42. doi: 10.4049/jimmunol.1402585
89. Das S, Mukherjee S, and Ali N. Super enhancer-mediated transcription of miR146a-5p drives M2 polarization during Leishmania donovani infection. PloS Pathog. (2021) 17:e1009343. doi: 10.1371/journal.ppat.1009343
90. Ganguly S, Ghoshal B, Banerji I, Bhattacharjee S, Chakraborty S, Goswami A, et al. Leishmania survives by exporting miR-146a from infected to resident cells to subjugate inflammation. Life Sci Alliance. (2022) 5. doi: 10.26508/lsa.202101229
91. Lopes MF, da Veiga VF, Santos AR, Fonseca ME, and DosReis GA. Activation-induced CD4+ T cell death by apoptosis in experimental Chagas' disease. J Immunol. (1995) 154:744–52. doi: 10.4049/jimmunol.154.2.744
92. Lopes MF, Guillermo LV, and Silva EM. Decoding caspase signaling in host immunity to the protozoan Trypanosoma cruzi. Trends Immunol. (2007) 28:366–72. doi: 10.1016/j.it.2007.06.004
93. Guillermo LV, Silva EM, Ribeiro-Gomes FL, De Meis J, Pereira WF, Yagita H, et al. The Fas death pathway controls coordinated expansions of type 1 CD8 and type 2 CD4 T cells in Trypanosoma cruzi infection. J Leukoc Biol. (2007) 81:942–51. doi: 10.1189/jlb.1006643
94. Silva EM, Guillermo LV, Ribeiro-Gomes FL, De Meis J, Nunes MP, Senra JF, et al. Caspase inhibition reduces lymphocyte apoptosis and improves host immune responses to Trypanosoma cruzi infection. Eur J Immunol. (2007) 37:738–46. doi: 10.1002/eji.200636790
95. Cabral-Piccin MP, Guillermo LV, Vellozo NS, Filardy AA, Pereira-Marques ST, Rigoni TS, et al. Apoptotic CD8 T-lymphocytes disable macrophage-mediated immunity to Trypanosoma cruzi infection. Cell Death Dis. (2016) 7:e2232. doi: 10.1038/cddis.2016.135
96. Lopes MF, Nunes MP, Henriques-Pons A, Giese N, Morse HC 3rd, Davidson WF, et al. Increased susceptibility of Fas ligand-deficient gld mice to Trypanosoma cruzi infection due to a Th2-biased host immune response. Eur J Immunol. (1999) 29:81–9. doi: 10.1002/(SICI)1521-4141(199901)29:01<81::AID-IMMU81>3.0.CO;2-Y
97. Silva EM, Guillermo LV, Ribeiro-Gomes FL, De Meis J, Pereira RM, Wu Z, et al. Caspase-8 activity prevents type 2 cytokine responses and is required for protective T cell-mediated immunity against Trypanosoma cruzi infection. J Immunol. (2005) 174:6314–21. doi: 10.4049/jimmunol.174.10.6314
98. Pereira-Manfro WF, Ribeiro-Gomes FL, Filardy AA, Vellozo NS, Guillermo LV, Silva EM, et al. Inhibition of caspase-8 activity promotes protective Th1- and Th2-mediated immunity to Leishmania major infection. J Leukoc Biol. (2014) 95:347–55. doi: 10.1189/jlb.0912463
99. Vasconcelos JR, Bruna-Romero O, Araujo AF, Dominguez MR, Ersching J, de Alencar BC, et al. Pathogen-induced proapoptotic phenotype and high CD95 (Fas) expression accompany a suboptimal CD8+ T-cell response: reversal by adenoviral vaccine. PloS Pathog. (2012) 8:e1002699. doi: 10.1371/journal.ppat.1002699
100. Henson PM. Cell removal: efferocytosis. Annu Rev Cell Dev Biol. (2017) 33:127–44. doi: 10.1146/annurev-cellbio-111315-125315
101. Trahtemberg U and Mevorach D. Apoptotic cells induced signaling for immune homeostasis in macrophages and dendritic cells. Front Immunol. (2017) 8:1356. doi: 10.3389/fimmu.2017.01356
102. Nagata S. Apoptosis and clearance of apoptotic cells. Annu Rev Immunol. (2018) 36:489–517. doi: 10.1146/annurev-immunol-042617-053010
103. Kourtzelis I, Hajishengallis G, and Chavakis T. Phagocytosis of apoptotic cells in resolution of inflammation. Front Immunol. (2020) 11:553. doi: 10.3389/fimmu.2020.00553
104. Zagorska A, Traves PG, Lew ED, Dransfield I, and Lemke G. Diversification of TAM receptor tyrosine kinase function. Nat Immunol. (2014) 15:920–8. doi: 10.1038/ni.2986
105. Rigoni TS, Vellozo NS, Guimaraes-Pinto K, Cabral-Piccin M, Fabiano-Coelho L, Matos-Silva TC, et al. Axl receptor induces efferocytosis, dampens M1 macrophage responses and promotes heart pathology in Trypanosoma cruzi infection. Commun Biol. (2022) 5:1421. doi: 10.1038/s42003-022-04401-w
106. Ribeiro-Gomes FL, Romano A, Lee S, Roffe E, Peters NC, Debrabant A, et al. Apoptotic cell clearance of Leishmania major-infected neutrophils by dendritic cells inhibits CD8(+) T-cell priming in vitro by Mer tyrosine kinase-dependent signaling. Cell Death Dis. (2015) 6:e2018. doi: 10.1038/cddis.2015.351
107. Chaves MM, Lee SH, Kamenyeva O, Ghosh K, Peters NC, and Sacks D. The role of dermis resident macrophages and their interaction with neutrophils in the early establishment of Leishmania major infection transmitted by sand fly bite. PloS Pathog. (2020) 16:e1008674. doi: 10.1371/journal.ppat.1008674
Keywords: ATRA, Axl, Chagas disease, efferocytosis, Leishmaniasis, M1 and M2 macrophages, RANKL, Th1 and Th2 cytokines
Citation: Vellozo NS, Matos-Silva TC and Lopes MF (2025) Targeting macrophage phenotypes to prevent diseases caused by Leishmania and Trypanosoma cruzi infections. Front. Immunol. 16:1595954. doi: 10.3389/fimmu.2025.1595954
Received: 18 March 2025; Accepted: 14 July 2025;
Published: 07 August 2025.
Edited by:
Joao Santana Silva, Oswaldo Cruz Foundation (Fiocruz), BrazilReviewed by:
Kathryn Marie Jones, Baylor College of Medicine, United StatesJunaid Jibran Jawed, Presidency University, India
Angel Ramos-Ligonio, Universidad Veracruzana, Mexico
Copyright © 2025 Vellozo, Matos-Silva and Lopes. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marcela F. Lopes, bWFyY2VsYWxAYmlvZi51ZnJqLmJy
†These authors have contributed equally to this work and share first authorship