 Huan Gao
Huan Gao Zirui Zhang1
Zirui Zhang1 Jiayu Deng
Jiayu Deng Yanqing Song
Yanqing Song- 1College of Pharmacy, Jilin University, Changchun, Changchun, China
- 2Department of Clinical Pharmacy, the First Hospital of Jilin University, Changchun, China
- 3Department of Pharmacy, Lequn Branch, the First Hospital of Jilin University, Changchun, China
Cathepsin S (CTSS), a lysosomal cysteine protease predominantly expressed in immune cells, governs inflammatory and immunological cascades through proteolytic activity. Beyond maintaining lysosomal proteostasis through protein degradation, CTSS executes dual immunomodulatory functions: intracellularly processing antigen-presenting molecules and modulating inflammatory signaling cascades; extracellularly activating protease-activated receptors (PARs) and remodeling the extracellular matrix (ECM). Its dysregulation drives pathology in autoimmune disorders, chronic inflammation, and neoplasia, establishing CTSS as a multifaceted therapeutic target. This review comprehensively explores the contributions of CTSS signaling in immune-mediated inflammatory diseases, critically evaluates its therapeutic potential, highlighting its significance in the development of innovative treatment strategies.
1 Introduction
Inflammation represents a critical pathophysiological response to diverse stimuli including infections, tissue injury, cellular stress, and chemical agents. Immune cells orchestrate inflammatory processes through coordinated mechanisms: circulating leukocytes infiltrate damaged tissues, while resident macrophages modulate local inflammatory dynamics (1). Pathological inflammation underlies diseases, ranging from immune-mediated disorders (such as multiple sclerosis and Crohn’s disease) to cardiovascular pathologies (e.g., atherosclerosis), neurodegenerative conditions (e.g., Alzheimer’s disease), and psychiatric disorders (e.g., generalized anxiety disorder) (2–9). Within cellular microenvironments, inflammatory signaling is regulated by lysosome-dependent mechanisms, where macrophages and dendritic cells (DCs) employ lysosomal enzymes as key effectors (10). Among these, cathepsins (CTSs) mediate inflammatory signal transduction through their proteolytic processing of intracellular and extracellular proteins.
Cathepsin S (CTSS), a lysosomal cysteine protease, uniquely maintains enzymatic activity at neutral pH (≤7) due to its histidine-rich propeptide domain, distinguishing it from acid-dependent cathepsins (e.g., Cathepsin B/L/K) (11). This property enables CTSS to perform both intra- and extracellular functions. Intracellularly, lysosomal membrane permeabilization (LMP) releases CTSS into the cytosol, triggering lysosome-dependent cell death (12, 13). Extracellularly, lysosomal exocytosis facilitates CTSS secretion, enabling ECM remodeling, receptor activation (e.g., PAR2), and cytokine release (14, 15). Recent studies highlight CTSS as a central regulator of inflammatory pathways. CTSS is required for Major Histocompatibility Complex class II (MHC-II) maturation in antigen-presenting cells (including DCs), activating adaptive immunity (16–18). In addition, CTSS deficiency disrupts autophagic flux, leading to autophagosome accumulation and pro-inflammatory signaling that amplifies inflammatory responses (18–21). Crucially, extracellular CTSS activates Protease-activated receptor 2 (PAR2) and fractalkine (FKN) —key mediators in autoimmune disease pathogenesis (22, 23). The mechanisms contributing to anti-inflammatory effects are manifold. Despite these multifaceted roles, a systematic discussion of CTSS-mediated signaling networks remains lacking.
In this paper, we summarized upstream regulators of CTSS signaling, downstream inflammatory mediators and associated diseases. We then evaluate CTSS as a therapeutic target to inform novel treatment strategies for inflammatory disorders.
2 Stress factors upregulating CTSS expression
Stress conditions act as pathological factors which contribute to inflammation micro-environment and promote immunological diseases progression (Figure 1). CTSS overexpression induced by psychological, metabolic, or infectious stress correlates with disease progression, reinforcing its therapeutic relevance.
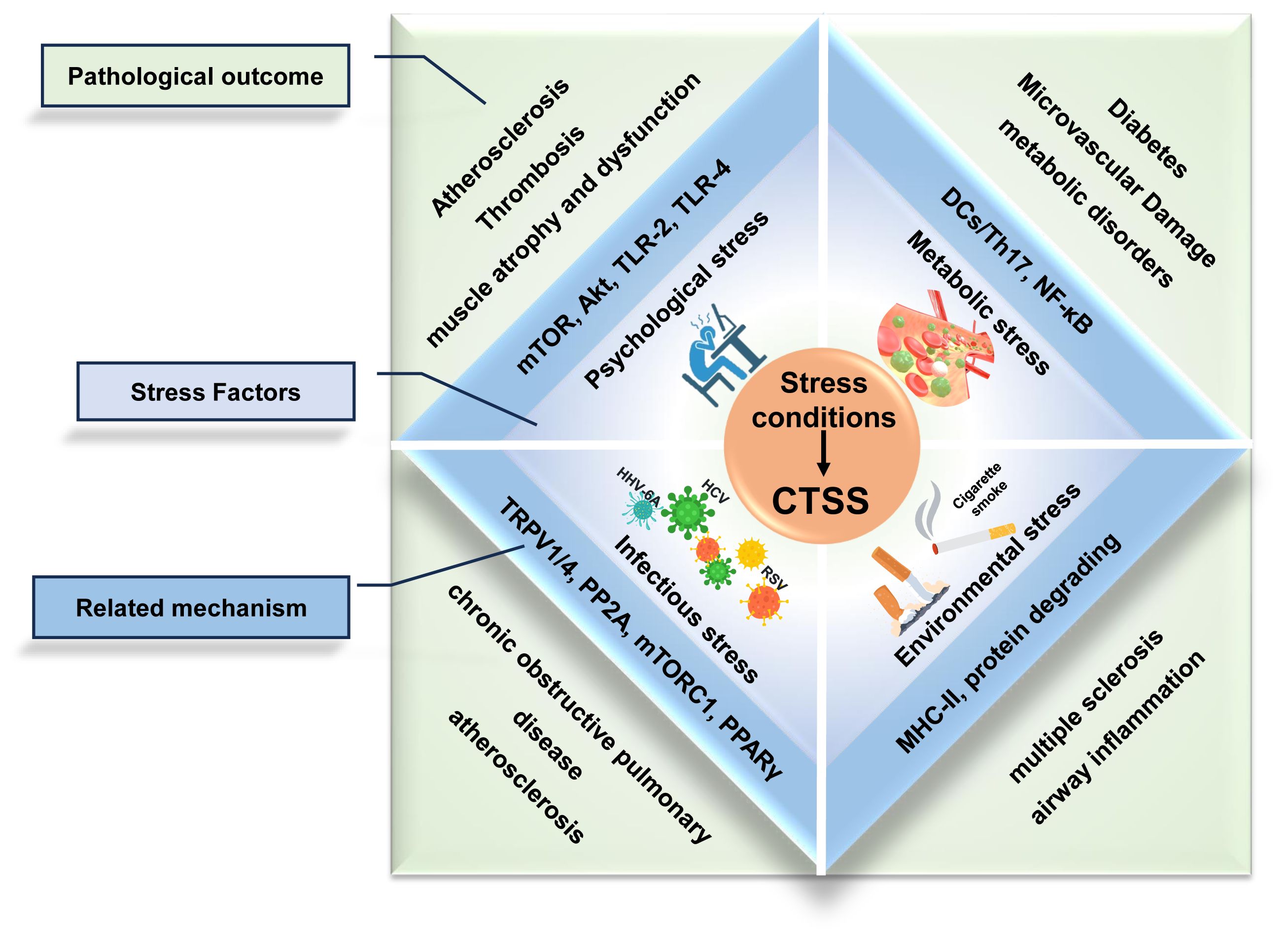
Figure 1. Stress-induced upregulation of CTSS drives inflammatory disease progression.
The major stress conditions (psychological, metabolic, environmental and infectious stress) converge to promote pathological overexpression of CTSS. This upregulation establishes a pro-inflammatory microenvironment characterized by dysregulated immune responses, protease activation, and tissue damage.
2.1 Psychological Stress
Chronic psychological stress (CPS) refers to persistent physiological dysregulation induced by exogenous stressors (e.g., occupational or social pressures), which can trigger systemic chronic inflammation (24, 25). CTSS upregulation correlates with CPS progression, driving pathologies like atherosclerosis and muscle atrophy. Notably, CTSS promotes upregulation of inflammatory factors (mTOR, Akt, TLR-2, TLR-4, Bcl-2, SOD, Caspase-3, MMP-2, MMP-9, MCP-1, and p-GSK3α/β), enhancing apoptosis, oxidative stress, and macrophage infiltration (26–28). Moreover, depending on the type of inflammation, CTSS can induce unique pathological changes. In stress-aggravated atherosclerosis, it promotes plaque elastin disruption, smooth muscle cell proliferation/migration, and neointimal hyperplasia. Conversely, stress-related thrombosis events involve endothelial loss and the activation of pro-coagulation factors (PAI-1, ADAMTS13, and vWF) (26, 27). For chronic stress-induced muscle atrophy and dysfunction, CTSS contributes to the loss of myotube myosin heavy chain content and the upregulation of Muscle RING-finger protein-1 (MuRF1) and Insulin Receptor Substrate 2 (IRS-2) (28). These distinct mechanisms demonstrate how CTSS upregulation advances CPS-related inflammation through pro-inflammatory effects and tissue-specific actions, establishing its role as a critical therapeutic target.
2.2 Metabolic stress
Hyperglycemia is a well-established risk factor for diabetes and its complications, including chronic inflammation and microvascular pathologies (29, 30). In diabetic models, it elevates CTSS levels in DCs within perivascular adipose tissue, which subsequently drive T helper 17 cell (Th17) differentiation and pro-inflammatory cytokine production (e.g., IL-6). Conversely, CTSS knockdown mitigates hyperglycemia-induced carotid restenosis, confirming immune cell-mediated CTSS involvement in microvascular complications (30). Hyperglycemia also directly upregulates CTSS in endothelial cells. CTSS activates nuclear factor kB (NF-κB) signaling and then triggering inflammatory cytokine release (TNF-α, IL-1β, IL-6), angiogenic factor overexpression (MCP-1, VEGFA, VCAM-1), and complement system activation (C3a, C5a) (29). These synergistic effects exacerbate endothelial injury, establishing CTSS as a dual mediator of hyperglycemia-induced vascular damage. Clinically, the association has been proved in a study of investigating CTSS and type 2 diabetes. Each baseline CTSS standard deviation increase correlates with 41- 48% higher diabetes risk across multivariable models (31). CTSS inhibition could alleviate hyperglycemia-induced endothelial inflammation in vitro, and reduce hepatic glucose production in murine models, while maintaining unaltered glucose metabolism in skeletal myotubes and adipocytes (29, 32). In summary, CTSS emerges as a critical molecular link between hyperglycemia and complications, positioning it as a promising target for both hyperglycemia prevention and complication management.
2.3 Environmental stress
Cigarette smoke contains multiple pro-inflammatory constituents, including nicotine, which induces systemic inflammatory damage across respiratory and cardiovascular systems, thereby promoting pathogenesis of chronic obstructive pulmonary disease (COPD) and atherosclerosis (33). Nicotine activates CTSS-dependent inflammatory pathways through distinct tissue-specific mechanisms. In pulmonary tissues, nicotine triggers Transient Receptor Potential Vanilloid 1/4 (TRPV1/4) receptors on alveolar macrophages, inducing P2X purinoceptor 7 (P2X7)-dependent intracellular calcium influx that stimulates p38/MAPK phosphorylation and enhances lysosomal CTSS production. The CTSS Overexpression amplifies oxidative stress, promotes macrophages recruitment, and disrupts proteases/anti-proteases balance, collectively damaging lung homeostasis to drive COPD onset and progression (34). CTSS-deficient mice demonstrated protection against smoke-induced pathologies including pulmonary inflammation, airway hyperresponsiveness, emphysema, and lung function decline, while Protein Phosphatase 2A (PP2A) activation suppresses CTSS-driven pulmonary disorders (35). Notably, endogenous nitrated fatty acids (NFAs) exhibit therapeutic potential by activating Peroxisome Proliferator-Activated Receptor γ (PPARγ) signaling and Cys25 S-alkylation modification of CTSS, effectively reversing nicotine-induced inflammation (36). In the cardiovascular system, nicotine inhibits the Mechanistic Target of Rapamycin Complex 1 (mTORC1) to promote Mitochondrial Transcription Elongation Factor B (TEFB) nuclear translocation and CTSS transcription. Conversely, nicotine-mediated mTORC1 inhibition also contributes to CTSS secretion by enhancing the Ras-related protein Rab-10-mTORC1 interactions to facilitate lysosomal exocytosis. CTSS overactivity disrupts vascular smooth muscle cell migration, contributing critically to atherosclerotic plaque formation (37). This mechanistic analysis identifies CTSS as a central mediator linking cigarette smoke to multi-organ inflammation. Though pharmacological CTSS inhibition represents a promising therapeutic strategy, smoking cessation remains the most effective preventive measure against CTSS-mediated systemic damage.
2.4 Infectious stress
CTSS drives viral-associated inflammatory diseases progression through pathogen-specific proteolytic mechanisms. In neuroinflammatory disorders, human herpesvirus-6A (HHV-6A) induces CTSS release from neural cells, directly degrading myelin basic protein to initiate demyelination cascades characteristic of multiple sclerosis (38). Respiratory syncytial virus (RSV) synergizes with cigarette smoke in COPD pathogenesis by coordinated upregulating CTSS expression, exacerbating airway inflammation via ECM remodeling (39). Similarly, hepatitis C virus (HCV) employs its NS5A core protein to disrupt Interferon-γ (IFN-γ)/(interferon regulatory factor 1 (IRF-1) signaling, inducing CTSS dysregulation that impairs MHC-II-mediated antigen presentation in hepatic DCs (40). These findings establish CTSS as a common pathogenic effector across diverse viral infections, with therapeutic implications for attenuating infection-associated immunopathology. Divergently, bacterial pathogens like Brucella abortus employ distinct mechanisms, suppressing MHC-II maturation through lipoprotein-dependent impairment of early antigen-presenting complex formation via CTSS-independent pathways (41). Collectively, CTSS emerges as a mediator of viral-associated inflammation and chronic disease transformation, though its pathogenic role in bacterial infections warrants systematic investigation.
3 CTSS mediated inflammatory signaling pathways
3.1 CTSS regulation in response to inflammatory transcriptional factors upstream
CTSS is a central mediator bridging upstream transcriptional regulators and downstream inflammatory outputs (Figure 2). Some transcription factors upregulate CTSS expression (IRF-1, PU.1, TFEB), while others perform as the downregulator (TGF-β/Smad). However, there are also some regulatory signals (Signal Transducer and Activator of Transcription 3, STAT3 etc.) with both the potential effects. The regulation of CTSS by these transcription factors depends on the pro/anti-inflammatory properties of upstream cytokines (IL-6, IL-10 etc.) or other signaling molecules (H2S etc.).
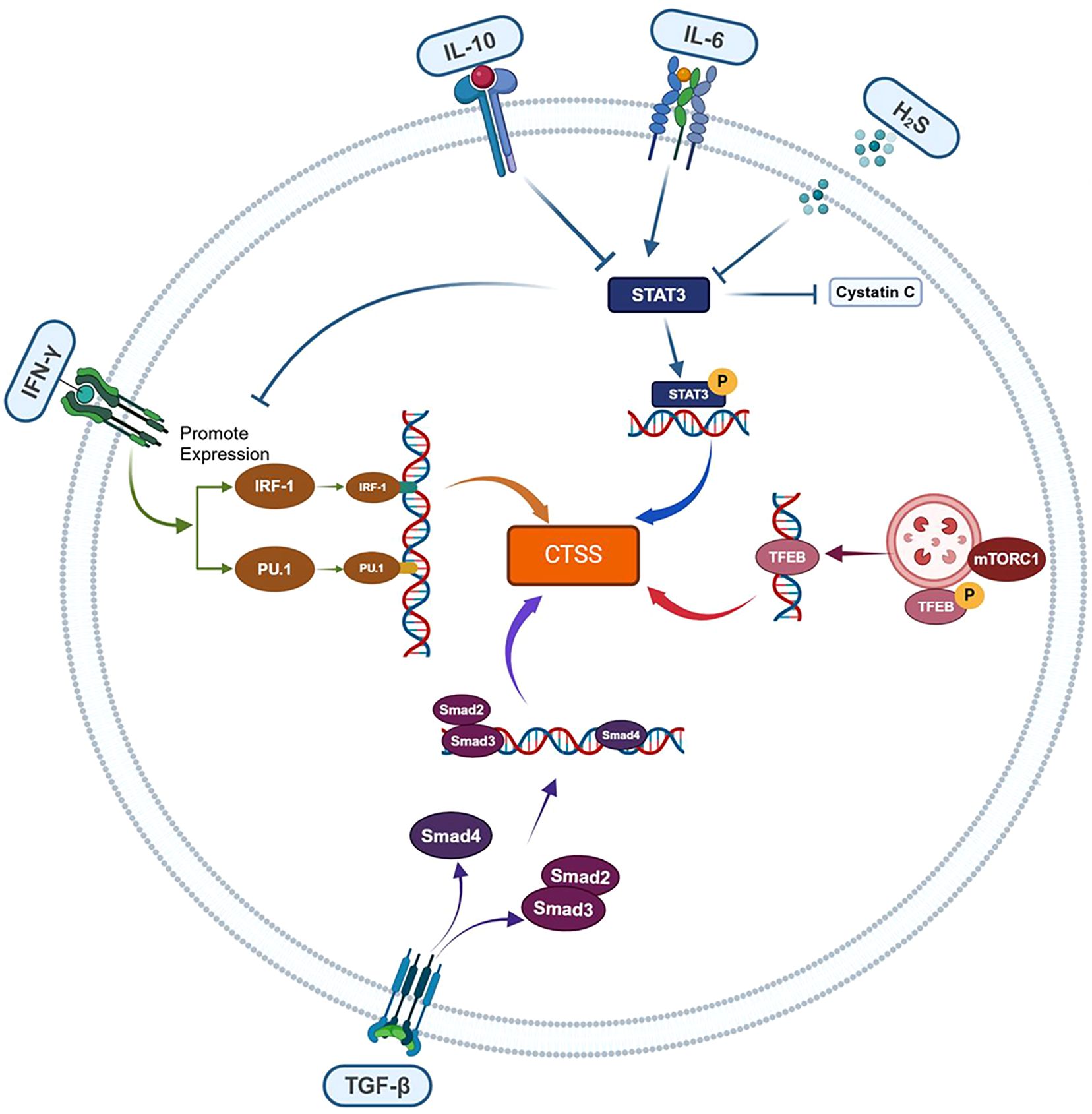
Figure 2. CTSS regulation by upstream inflammatory transcriptional factors. CTSS expression is dynamically modulated by inflammatory transcription factors with opposing regulatory effects: IRF-1, PU.1 and TFEB act as transcriptional activators, whereas TGF-β/Smad function as repressors. STAT3 exerts context-dependent effects, influenced by upstream signals (e.g., IL-6/IL-10 pro-/anti-inflammatory cytokines or H2S).
3.1.1 IRF-1
IRF-1 is a member of the interferon regulatory factor (IRF) family, critically promotes CTSS transcription (42). Its activity is tightly regulated by upstream cytokines, particularly IFN-γ. The IFN-γ/IRF-1/CTSS axis has been extensively studied in inflammatory pathologies including bronchial inflammation, radiation-induced oxidative stress, and hepatitis C (43). Specifically, IFN-γ upregulates IRF-1 expression, activating CTSS transcription via binding to the interferon-responsive sequence element (IRSE) promoter. Radiation-induced oxidative stress enhances IFN-γ release (44), whereas the HCV core protein N5SA suppresses IFN-γ activity (40), thereby modulating IRF-1/CTSS signaling. Conversely, in Brucella abortus-infected monocytes, bacterial lipoproteins downregulate IRF-1 through IL-6 signaling activation, reducing CTSS levels (41). Beyond cytokines, microRNAs have been implicated in the modulation of IRF-1 expression. In cystic fibrosis lung tissues, miR-31 expression is significantly reduced. Treatment with miR-31 analogs suppresses both IRF-1 expression and CTSS secretion, confirming miR-31-mediated negative regulation the IRF-1/CTSS signaling axis (45). Pharmacological agents also modulate this axis. Oxaliplatin upregulates IRF-1 expression in peripheral nerves, activating CTSS/store-operated calcium entry (SOCE) signaling to drive neuroinflammation (46). Thus, the IRF-1/CTSS pathway could be modulated by cytokines, microRNAs, and pharmaceuticals, which mediates inflammatory pathology in radiation injury, infections, genetic disorders, and pharmacogenetic diseases.
3.1.2 PU.1
PU.1, an E26 transformation-specific sequence (ETS) family transcription factor, is essential for the development and differentiation of immune cells, including B cells, macrophages, and neutrophils while disrupting fibrotic networks to promote multi-organ fibrosis regression (47). PU.1 enhances antigen presentation in macrophages, DCs, and B lymphocytes by upregulating CTSS expression, which is critical for MHC-II activity. In the periodontitis model, PU.1-mediated CTSS regulation in macrophages correlates with inflammatory pathway activation (e.g., p38 and NF-κB signaling) and elevated levels of inflammatory factors (e.g., IL-6) (48). PU.1 initiates CTSS transcription through direct binding to ETS motifs in its promoter (49), with all three protein domains driving promoter activity—explaining reduction in CTSS levels observed in PU.1-knockdown macrophages (50). Notably, PU.1 and IRF-1 exhibit co-regulatory behaviors in CTSS expression. Both transcription factors are upregulated by IFN-γ and possess binding sites on the CTSS promoter. Additionally, PU.1 can interact with other transcription factors, including IRF-1, IRF-4, and IRF-8, forming complexes that synergistically enhance CTSS expression (51). Thus, PU.1 drives CTSS expression both directly through promoter binding and cooperatively via transcription factor partnerships. Critically, PU.1-dependent CTSS induction mediates periodontitis progression, underscoring its pathogenic role in inflammatory disease mechanisms.
3.1.3 STAT3
STAT3 is a pivotal regulator of inflammatory pathologies, exerting context-dependent effects on disease progression. Under pro-inflammatory conditions (e.g., IL-6/JAK signaling), STAT3 phosphorylates and dimerizes (p-STAT3), translocating to the nucleus to upregulate CTSS expression while suppressing cystatin C transcription. This dual regulation amplifies CTSS activity, as demonstrated in DCs (52). Conversely, anti-inflammatory signals differentially modulate the STAT3/CTSS axis: IL-10 enhances STAT3 activation via a non-canonical pathway but inhibits IFN-γ-driven CTSS upregulation in macrophages. While both IL-6 and IL-10 upregulate STAT3 activity, they exert different effects on CTSS expression through distinct pathways (53). Notably, endogenous hydrogen sulfide (H2S) suppresses this axis through dual mechanisms (1): Cys-259 persulfidation of STAT3 impairs phosphorylation-dependent activation, indirectly reducing CTSS expression (2); Direct Cys-25 persulfidation of CTSS attenuates its enzymatic activity (54, 55). These regulatory networks are functionally validated in models spanning arterial calcification, elastin degradation, and neuroinflammation (54–56). In addition, the STAT3/CTSS signaling axis is implicated in diseases such as Alzheimer’s disease, diabetic nephropathy, and vascular calcification, highlighting its therapeutic potential in cardiovascular and neurological disorders (57–59).
3.1.4 Others
Lysosomes critically regulate CTSS activity. Nicotine impairs mTORC1-mediated autophagy-lysosomal pathway, triggering TFEB nuclear translocation that elevates CTSS transcription and promotes chronic inflammation in atherosclerosis (37). In lupus pathology, Blimp-1 suppresses CTSS expression and inhibits CTSS-mediated MHC-II activation in DCs, blocking antigen presentation to CD4+ follicular helper T cells (60). The TGF-β/Smad pathway exhibits different regulation of CTSS: Chen et al. demonstrated that TGF-β1/Smad2/Smad3/signaling promotes cardiac fibroblasts dedifferentiation via CTSS, upregulating ECM-associated proteins to exacerbate post-infarction fibrosis (61). Conversely, Zhang et al. found that TGF-β/Smad4 inhibition increases CTSS-dependent ECM remodeling, conferring protection against aortic aneurysms (62). This dual functionality highlights the TGF-β/Smad/CTSS axis complexity, warranting further mechanistic investigation.
3.2 CTSS passing inflammatory signal to targets downstream
CTSS exerts its hydrolysis ability to regulate the levels of proinflammatory transcription (NF-κB etc.), to expose the active domain or site of downstream signal proteins (PAR2, MHC-II etc.) or to liberate inflammatory factors (FKN etc.) as shown in Figure 3. Therefore, CTSS performs as an amplifier to transduce inflammatory signals.
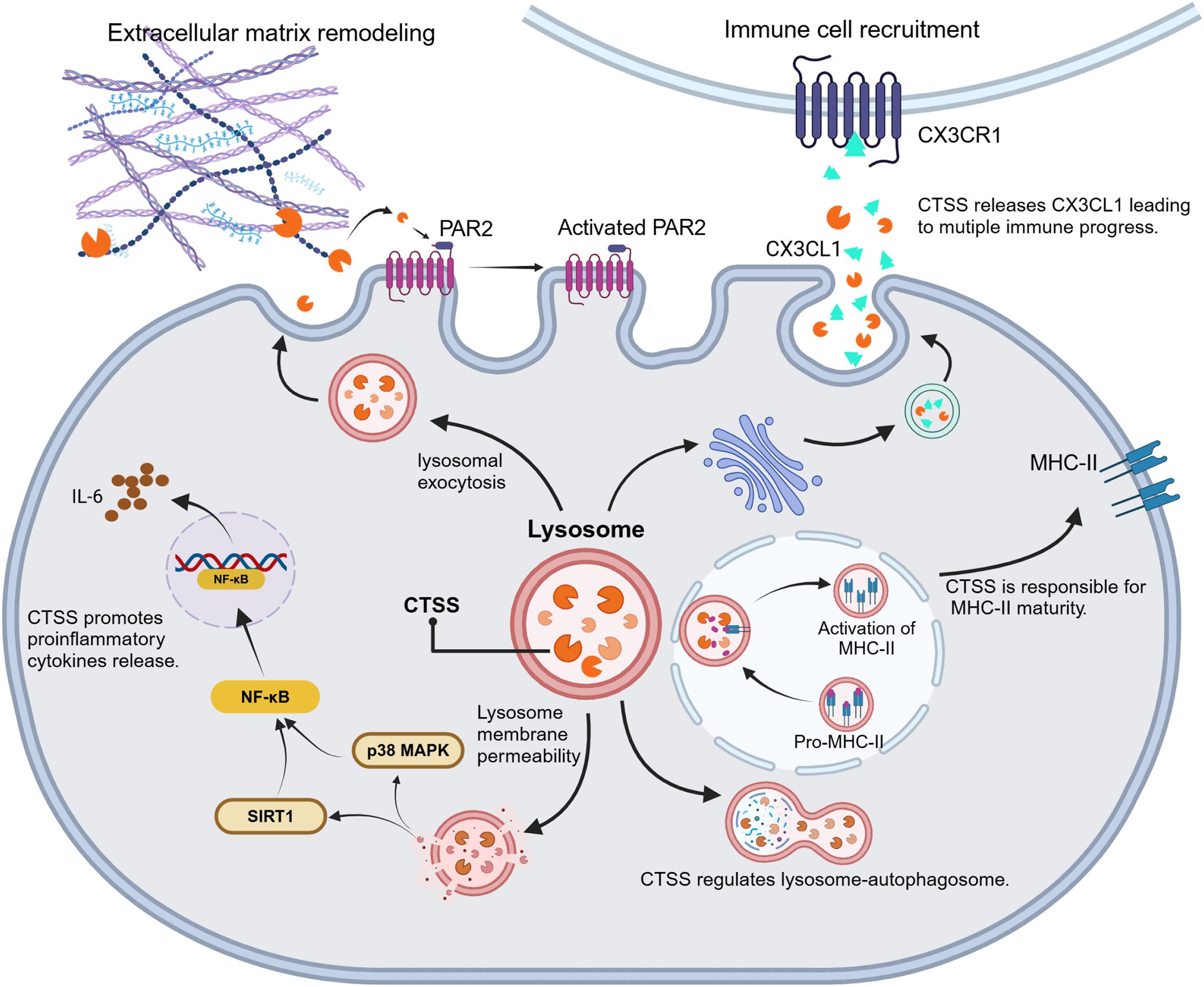
Figure 3. CTSS-driven inflammatory signal transduction. CTSS acts as a proteolytic hub to amplifies inflammatory responses through modulation of transcriptional activators (e.g., NF-κB activation), exposure of cryptic signaling domains (e.g., PAR2, MHC-II) and release of bioactive inflammatory factors (e.g., FKN/CX3CL1).
3.2.1 PAR2
PAR2, a G-protein-coupled receptor, drives inflammation progression, immune responses, and angiogenesis. CTSS activates PAR2 via cleavage at a specific site (E56-T57), distinct from the typical trypsin cleavage site (R36-S37) (63). This unique proteolysis generates signaling responses divergent from classical PAR2 pathways—calcium mobilization, ERK1/2 activation, β-arrestin recruitment, and endocytosis (64). The CTSS/PAR2 axis exacerbates inflammatory diseases via distinct molecular mechanisms. In colitis models, CTSS/PAR2 signaling activates the damage sensor transient receptor potential vanilloid 4 (TRPV4), resulting in visceral hypersensitivity (65). During atopic dermatitis, CTSS overexpression in DCs upregulates PAR2, enhancing secretion of inflammatory cytokines (IL-2, IL-4, IL-10, IFN-γ, and MCP-1) by Th1 cells and triggering scratching behavior (66). In desiccated ocular surfaces and cystic fibrosis lungs, CTSS/PAR2 signaling elevates pro-inflammatory mediators (IL-6, IL-8, TNF-α, IL-1β, and MMP-9) and mucins, respectively (67, 68). These findings collectively demonstrate that CTSS/PAR2 signaling amplifies inflammatory phenotypes through cytokine hypersecretion, receptor overexpression, and pathological protein accumulation.
3.2.2 FKN
FKN (CX3CL1), a transmembrane chemokine expressed in monocytes, NK cells, T cells, and vascular smooth muscle cells, undergoes CTSS-mediated proteolytic cleavage to generate soluble monomer (sFKN). The sFKN activates the G protein-coupled receptor CX3CR1, regulating immune homeostasis (69, 70). In rheumatoid arthritis models (collagen-induced arthritis, temporomandibular arthritis, and desiccation syndrome), CTSS is essential for sustaining inflammatory pain (23, 69, 71, 72). Mechanistically, peripheral nerve injury triggers microglial CTSS secretion. CTSS hydrolyzes membrane-anchored FKN from dorsal horn neurons, releasing sFKN that activates microglial CX3CR1 to induce p38/MAPK phosphorylation. This cascade upregulates nociceptive mediators (TRPV1, P2X4, BDNF, α2δ calcium channels, and activating transcription factor 3), amplifying pain signaling (57, 73, 74). Notably, P2X7 receptor-dependent CTSS upregulation establishes the P2X7/CTSS/FKN axis as a therapeutic target for inflammatory pain (75). In desiccation syndrome, CTSS/FKN signaling drives dacryoadenitis by recruiting T cells and macrophages into lacrimal gland—blocked by CTSS or FKN inhibition (76). Similarly, neuronal CTSS accelerates Alzheimer’s pathogenesis in murine models by driving microglial M1 polarization, activating the FKN-CX3CR1 axis, and enhancing JAK2-STAT3 signaling. Elevated CTSS in human Alzheimer brains and the therapeutic efficacy of CTSS inhibitor LY3000328 confirm its clinical relevance (57). In summary, the CTSS/FKN axis sustains chronic pain in arthritis and drives autoimmune inflammation through immune cell recruitment across multiple pathologies.
3.2.3 MHC-II
MHC-II complexes on antigen-presenting cells (APCs) enable immune recognition by presenting processed antigens to CD4+ T cells (77, 78). CTSS, the primary invariant chain (li)-processing enzyme, is highly expressed in MHC-II-positive APCs, including DCs, monocytes, lymphocytes, and splenocytes (79). This protease selectively cleaves the li chain within the MHC-II precursor complex (αβli trimer), generating mature αβ-CLIP complex that load exogenous antigens for CD4+ T cell activation and humoral immunity. Beyond antigen presentation, CTSS-mediated li degradation enhances DC motility by disrupting interactions with myosin II, thereby increasing migration speed and directional persistence for efficient antigen detection (18). Targeting CTSS has emerged as a therapeutic strategy for autoimmune diseases (e.g., rheumatoid arthritis, asthma, desiccation syndrome, multiple sclerosis) due to its dual role in MHC-II maturation and DC motility (80–83). Pharmacological inhibition like Clik60 block li degradation, causing accumulation of MHC-II-li intermediates and impairing MHC-II-peptide complex formation in APCs. This compromises antigen presentation while suppressing lymphocyte and eosinophil infiltration across multiple tissues. Consequently, CTSS inhibition alleviates autoimmune pathology through reduced autoantibody production and resolved tissue inflammation.
3.2.4 NF-κB
NF-κB, a master transcriptional regulator of inflammation and immunity, exhibits complex regulation with CTSS. Emerging evidence demonstrates CTSS promotes NF-κB activation in autoimmune encephalomyelitis, hepatitis, periodontitis, and hyperglycemia-induced endothelial inflammation (19, 29, 84). Notably, CTSS and MHC-II collaboratively regulate NF-κB activity—combination of both synergistically suppresses NF-κB-driven inflammation in encephalomyelitis (84). CTSS indirectly modulates NF-κB through distinct pathways (1): Degrading silent information regulator 1 (SIRT1) (an NF-κB suppressor), where CTSS inhibition stabilizes SIRT1 to repress NF-κB in hepatitis (19) (2); Activating PU.1/p38-NF-κB signaling in macrophages to drive IL-6-mediated periodontitis (48). Paradoxically, CTSS deficiency exacerbates angiotensin II-induced cardiac fibrosis by impairing lysosomal degradation. This disruption causes mitochondrial reactive oxygen species accumulation and subsequent NF-κB hyperactivation (85). Collectively, CTSS either amplifies or suppresses NF-κB signaling depending on tissue context and disease state.
3.2.5 Other
Recent studies indicate CTSS regulates additional inflammatory signaling pathways beyond established mechanisms, though some findings require further validation. In oxaliplatin-induced neuroinflammation, CTSS inhibition activates Stromal Interaction Molecule 1-mediated SOCE, which upregulates anti-inflammatory IL-10 through IRF-1 signaling to alleviate neuropathic pain (46). CTSS also contributes to hepatic fibrosis via ECM remodeling: Kupffer cell-derived CTSS cleaves collagen 18A1, liberating endothelial inhibitory peptides that activate integrin α5β1 on hepatic stellate cells and accelerate fibrogenesis (86). These findings position CTSS as a promising therapeutic target for both neuroinflammatory disorders and fibrotic diseases.
3.3 Clinical application of CTSS inhibitors
Small-molecule CTSS inhibitors have progressed to clinical trials across autoimmune and inflammatory disorders, demonstrating diverse therapeutic potential, as shown in Table 1. Petesicatib (RO5459072; Hoffmann-La Roche), a covalent reversible inhibitor, achieved phase II efficacy in Sjögren’s syndrome and phase I evaluation for celiac disease via a gluten challenge trial (NCT02679014), with ongoing exploration for idiopathic pulmonary fibrosis (IPF) (87). Another Roche compound, RO5461111, a competitive selective CTSS inhibitor, shows promise in systemic lupus erythematosus but awaits clinical entry (88, 89). Virobay’s oral inhibitors VBY-036 (phase I, neuropathic pain) and VBY-891 (phase II, psoriasis) exhibit tissue-specific targeting, with the latter favoring skin CTSS modulation, alongside preclinical VBY-825 for neuropathic pain and Alzheimer’s disease (90). Eli Lilly’s non-covalent inhibitor LY3000328 reduced plasma CTSS activity in phase I and advanced to phase II for aortic aneurysm; its derivatives also demonstrate immunomodulatory effects in bladder cancer by regulating T-cell activity (91). RWJ-445380 (Johnson & Johnson/Alza) achieved Phase II efficacy in rheumatoid arthritis (with methotrexate) and plaque psoriasis, though structural details remain undisclosed. Sanofi’s SAR114137 faced limitations due to cathepsin K cross-reactivity during phase I osteoarthritis pain trials. Celera’s CRA-028129 completed phase I for psoriasis but lacked further development (92). Preclinical candidates like AM-3840 (Amura; neuropathic pain/RA), CZ-007 (Merck; Chagas disease), and MIV-247 (Medivir; autoimmune/neuropathic pain) further expand the pipeline, alongside novel scaffolds in early discovery (90, 93). These efforts underscore the expanding applications of CTSS inhibition, from autoimmune diseases (Sjögren’s syndrome) to oncology and vascular remodeling, yet highlight critical challenges in optimizing selectivity and minimizing off-target effects for future candidates.
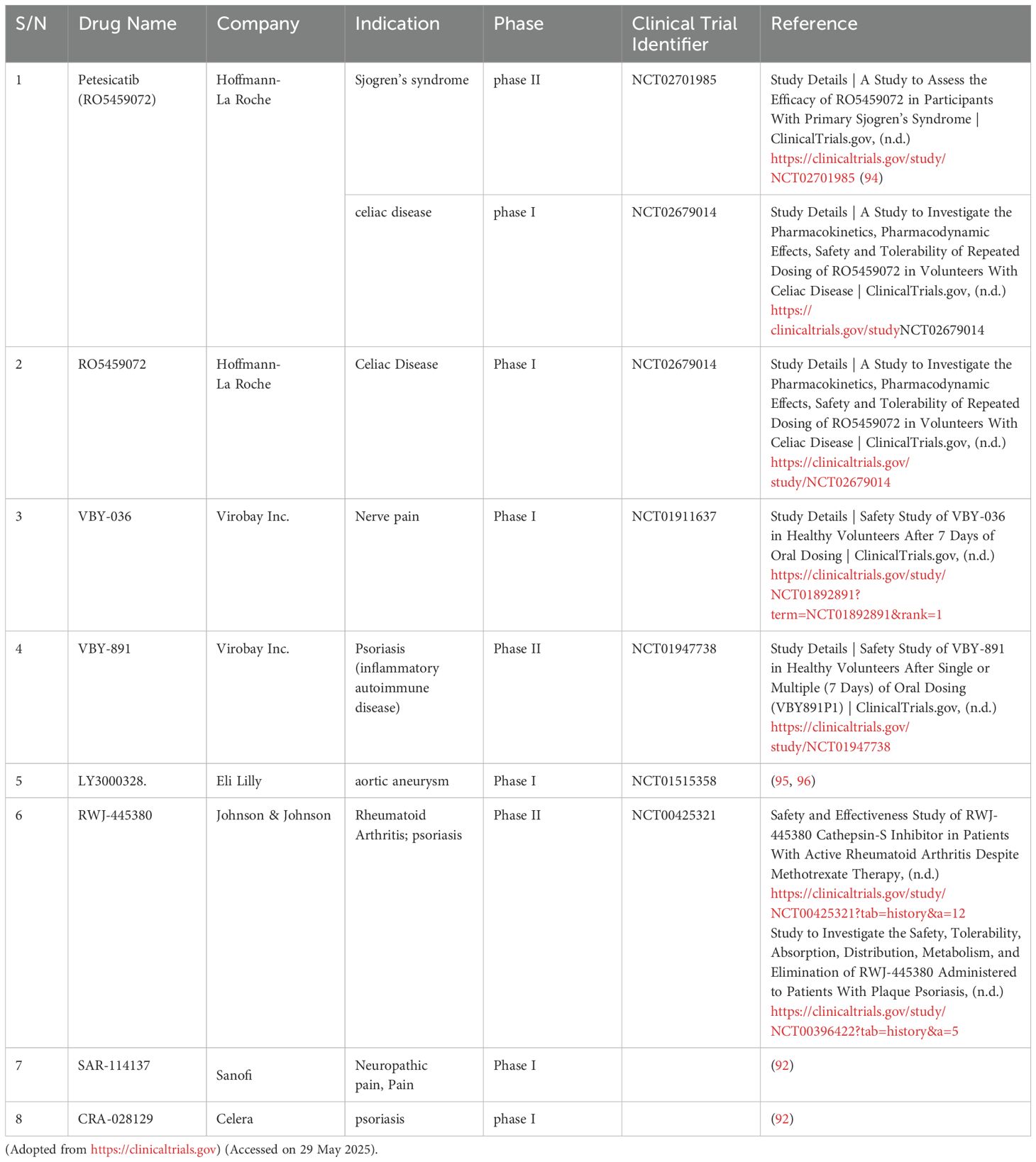
Table 1. A list of CTSS inhibitors at different stages of clinical trials.
4 Discussion
CTSS, a lysosomal cysteine protease with restricted immune cell expression, has evolved from a canonical proteinase to a master regulator of inflammatory and immune homeostasis. Unlike ubiquitously expressed cathepsins (B, C, F, H, L, O, X), CTSS exhibits restricted localization, primarily in antigen-presenting cells (B cells, macrophages, DCs) where it drives MHC-II-mediated immune responses through antigen processing. It also operates in epithelial, smooth muscle, endothelial cells, and neutrophils, enabling both intracellular proteolysis and extracellular modulation of inflammatory pathways. This unique tissue specificity and dual functionality underscore CTSS’s irreplaceable role in bridging lysosomal activity with systemic immune regulation. Its unique capacity to operate across pH gradients enables dual-compartment functionality: intracellularly, it processes antigen-presenting molecules and activates NF-κB/MAPK signaling cascades; extracellularly, it cleaves protease-activated receptors and remodels extracellular matrices. CTSS activity is regulated by microenvironment-responsive transcription factors (STAT3), distinguishing it from other cathepsins. It coordinates immune responses through the interconnected pathways: MHC-II antigen presentation, vesicular trafficking-dependent cytokine release, receptor proteolysis-driven signal amplification, and chemokine-mediated leukocyte recruitment. Additionally, it exhibits novel regulatory mechanisms, including pH-dependent enzymatic state switching, redox-sensitive zymogen activation, and miRNA-mediated expression tuning during immune differentiation. These pathways link CTSS to diverse clinical conditions spanning chronic inflammation, autoimmunity, and immunodeficiencies. Clinically, serum CTSS levels serve as biomarkers for interstitial lung disease and uveitis, while preclinical studies validate its therapeutic targeting in multi-organ pathologies affecting pulmonary, hepatic, cardiovascular, and neural systems.
While foundational insights into CTSS-disease associations have been established (97), there remain critical gaps in systematically mapping its molecular mechanisms across immune-inflammatory pathways. This review fills this gap by elucidating the CTSS-mediated signaling networks and highlighting CTSS as a central hub for therapeutic targeting in immune-related pathologies. The current clinical development of CTSS inhibitors shows promise in attenuating inflammatory lesions while preserving homeostatic functions, although further refinement is needed to optimize therapeutic efficacy. By positioning CTSS as a metabolic-inflammatory-immune signaling nexus, this review advances its characterization as a unique therapeutic node capable of coordinated multi-organ modulation, representing a paradigm shift from conventional protease-targeted approaches. Additionally, we propose a therapeutic strategy targeting CTSS through isoform-specific inhibition, subcellular localization control, and activity-state modulation, offering novel precision interventions for inflammation-related pathologies.
Author contributions
HG: Conceptualization, Funding acquisition, Writing – original draft. ZZ: Visualization, Writing – original draft. JD: Supervision, Writing – review & editing. YS: Conceptualization, Supervision, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This study was supported by the Talent Reserve Program (TRP) of The First Hospital of Jilin University (Grant number: JDYY-TRP-2024009), the education department of Jilin Province (Grant number: JJKH20231239KJ), the China International Medical Foundation (Grant number: Z-2021-46-2101) and the Wu Jieping Medical Foundation (Grant number: 320.6750.2022-20-4).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Medzhitov R. Origin and physiological roles of inflammation. Nature. (2008) 454:428–35. doi: 10.1038/nature07201
2. Michopoulos V, Powers A, Gillespie CF, Ressler KJ, and Jovanovic T. Inflammation in fear- and anxiety-based disorders: PTSD, GAD, and beyond. Neuropsychopharmacology. (2017) 42:254–70. doi: 10.1038/npp.2016.146
3. Hori H and Kim Y. Inflammation and post-traumatic stress disorder. Psychiatry Clin Neurosci. (2019) 73:143–53. doi: 10.1111/pcn.12820
4. Chen CC, Lin YA, Liu KT, Huang CY, Shih CM, Lee YT, et al. Navigating SARS-CoV-2-related immunopathology in Crohn’s disease: from molecular mechanisms to therapeutic challenges. Virol J. (2024) 21:288. doi: 10.1186/s12985-024-02529-1
5. Corrado A, Guadagni I, Picarelli G, and Variola A. Obesity and chronic inflammation: implications for rheumatoid arthritis, spondyloarthritis, and ulcerative colitis. Immun Inflammation Dis. (2025) 13:e70080. doi: 10.1002/iid3.70080
6. Jatczak-Pawlik I, Jurewicz A, Domowicz M, Ewiak-Paszyńska A, and Stasiołek M. CHI3L1 in multiple sclerosis-from bench to clinic. Cells. (2024) 13:2086. doi: 10.3390/cells13242086
7. Lamas A, Faria R, Marinho A, and Vasconcelos C. The mosaic of systemic lupus erythematosus: From autoimmunity to autoinflammation and immunodeficiency and back. Autoimmun Rev. (2024) 23:103675. doi: 10.1016/j.autrev.2024.103675
8. Soehnlein O and Libby P. Targeting inflammation in atherosclerosis - from experimental insights to the clinic. Nat Rev Drug Discov. (2021) 20:589–610. doi: 10.1038/s41573-021-00198-1
9. Glass CK, Saijo K, Winner B, Marchetto MC, and Gage FH. Mechanisms underlying inflammation in neurodegeneration. Cell. (2010) 140:918–34. doi: 10.1016/j.cell.2010.02.016
10. Deretic V. Autophagy in inflammation, infection, and immunometabolism. Immunity. (2021) 54:437–53. doi: 10.1016/j.immuni.2021.01.018
11. Vasiljeva O, Dolinar M, Pungercar JR, Turk V, and Turk B. Recombinant human procathepsin S is capable of autocatalytic processing at neutral pH in the presence of glycosaminoglycans. FEBS Lett. (2005) 579:1285–90. doi: 10.1016/j.febslet.2004.12.093
12. Vidak E, Javoršek U, Vizovišek M, and Turk B. Cysteine cathepsins and their extracellular roles: shaping the microenvironment. Cells. (2019) 8:264. doi: 10.3390/cells8030264
13. Lecaille F, Lalmanach G, and Andrault PM. Antimicrobial proteins and peptides in human lung diseases: A friend and foe partnership with host proteases. Biochimie. (2016) 122:151–68. doi: 10.1016/j.biochi.2015.08.014
14. Repnik U, Stoka V, Turk V, and Turk B. Lysosomes and lysosomal cathepsins in cell death. Biochim Biophys Acta. (2012) 1824:22–33. doi: 10.1016/j.bbapap.2011.08.016
15. Konjar S, Sutton VR, Hoves S, Repnik U, Yagita H, Reinheckel T, et al. Human and mouse perforin are processed in part through cleavage by the lysosomal cysteine proteinase cathepsin L. Immunology. (2010) 131:257–67. doi: 10.1111/j.1365-2567.2010.03299.x
16. Saegusa K, Ishimaru N, Yanagi K, Arakaki R, Ogawa K, Saito I, et al. Cathepsin S inhibitor prevents autoantigen presentation and autoimmunity. J Clin Invest. (2002) 110:361–9. doi: 10.1172/jci14682
17. Riese RJ, Mitchell RN, Villadangos JA, Shi GP, Palmer JT, Karp ER, et al. Cathepsin S activity regulates antigen presentation and immunity. J Clin Invest. (1998) 101:2351–63. doi: 10.1172/jci1158
18. Faure-André G, Vargas P, Yuseff MI, Heuzé M, Diaz J, Lankar D, et al. Regulation of dendritic cell migration by CD74, the MHC class II-associated invariant chain. Science. (2008) 322:1705–10. doi: 10.1126/science.1159894
19. de Mingo Á, de Gregorio E, Moles A, Tarrats N, Tutusaus A, Colell A, et al. Cysteine cathepsins control hepatic NF-κB-dependent inflammation via sirtuin-1 regulation. Cell Death Dis. (2016) 7:e2464. doi: 10.1038/cddis.2016.368
20. Tizon B, Sahoo S, Yu H, Gauthier S, Kumar AR, Mohan P, et al. Induction of autophagy by cystatin C: a mechanism that protects murine primary cortical neurons and neuronal cell lines. PloS One. (2010) 5:e9819. doi: 10.1371/journal.pone.0009819
21. Pires D, Bernard EM, Pombo JP, Carmo N, Fialho C, Gutierrez MG, et al. Mycobacterium tuberculosis Modulates miR-106b-5p to Control Cathepsin S Expression Resulting in Higher Pathogen Survival and Poor T-Cell Activation. Front Immunol. (2017) 8:1819. doi: 10.3389/fimmu.2017.01819
22. Cattaruzza F, Lyo V, Jones E, Pham D, Hawkins J, Kirkwood K, et al. Cathepsin S is activated during colitis and causes visceral hyperalgesia by a PAR2-dependent mechanism in mice. Gastroenterology. (2011) 141:1864–74. doi: 10.1053/j.gastro.2011.07.035
23. Clark AK, Yip PK, and Malcangio M. The liberation of fractalkine in the dorsal horn requires microglial cathepsin S. J Neurosci. (2009) 29:6945–54. doi: 10.1523/jneurosci.0828-09.2009
24. Vaccarino V and Bremner JD. Stress and cardiovascular disease: an update. Nat Rev Cardiol. (2024) 21:603–16. doi: 10.1038/s41569-024-01024-y
25. Niedhammer I, Bertrais S, and Witt K. Psychosocial work exposures and health outcomes: a meta-review of 72 literature reviews with meta-analysis. Scand J Work Environ Health. (2021) 47:489–508. doi: 10.5271/sjweh.3968
26. Xu S, Piao L, Wan Y, Huang Z, Meng X, Inoue A, et al. CTSS modulates stress-related carotid artery thrombosis in a mouse feCl(3) model. Arterioscler Thromb Vasc Biol. (2023) 43:e238–e53. doi: 10.1161/atvbaha.122.318455
27. Wang H, Meng X, Piao L, Inoue A, Xu W, Yu C, et al. Cathepsin S deficiency mitigated chronic stress-related neointimal hyperplasia in mice. J Am Heart Assoc. (2019) 8:e011994. doi: 10.1161/jaha.119.011994
28. Wan Y, Piao L, Xu S, Meng X, Huang Z, Inoue A, et al. Cathepsin S activity controls chronic stress-induced muscle atrophy and dysfunction in mice. Cell Mol Life Sci. (2023) 80:254. doi: 10.1007/s00018-023-04888-4
29. Sayed S, Faruq O, Preya UH, and Kim JT. Cathepsin S knockdown suppresses endothelial inflammation, angiogenesis, and complement protein activity under hyperglycemic conditions in vitro by inhibiting NF-κB signaling. Int J Mol Sci. (2023) 24:5428. doi: 10.3390/ijms24065428
30. Peng H, Lv Y, Li C, Cheng Z, He S, Wang C, et al. Cathepsin S inhibition in dendritic cells prevents Th17 cell differentiation in perivascular adipose tissues following vascular injury in diabetic rats. J Biochem Mol Toxicol. (2023) 37:e23419. doi: 10.1002/jbt.23419
31. Jobs E, Risérus U, Ingelsson E, Sundström J, Jobs M, Nerpin E, et al. Serum cathepsin S is associated with decreased insulin sensitivity and the development of type 2 diabetes in a community-based cohort of elderly men. Diabetes Care. (2013) 36:163–5. doi: 10.2337/dc12-0494
32. Karimkhanloo H, Keenan SN, Sun EW, Wattchow DA, Keating DJ, Montgomery MK, et al. Circulating cathepsin S improves glycaemic control in mice. J Endocrinol. (2021) 248:167–79. doi: 10.1530/joe-20-0408
33. Saint-André V, Charbit B, Biton A, Rouilly V, Possémé C, Bertrand A, et al. Smoking changes adaptive immunity with persistent effects. Nature. (2024) 626:827–35. doi: 10.1038/s41586-023-06968-8
34. Andrault PM, Schamberger AC, Chazeirat T, Sizaret D, Renault J, Staab-Weijnitz CA, et al. Cigarette smoke induces overexpression of active human cathepsin S in lungs from current smokers with or without COPD. Am J Physiol Lung Cell Mol Physiol. (2019) 317:L625–l38. doi: 10.1152/ajplung.00061.2019
35. Doherty DF, Nath S, Poon J, Foronjy RF, Ohlmeyer M, Dabo AJ, et al. Protein phosphatase 2A reduces cigarette smoke-induced cathepsin S and loss of lung function. Am J Respir Crit Care Med. (2019) 200:51–62. doi: 10.1164/rccm.201808-1518OC
36. Reddy AT, Lakshmi SP, Muchumarri RR, and Reddy RC. Nitrated fatty acids reverse cigarette smoke-induced alveolar macrophage activation and inhibit protease activity via electrophilic S-alkylation. PloS One. (2016) 11:e0153336. doi: 10.1371/journal.pone.0153336
37. Ni H, Xu S, Chen H, and Dai Q. Nicotine modulates CTSS (Cathepsin S) synthesis and secretion through regulating the autophagy-lysosomal machinery in atherosclerosis. Arterioscler Thromb Vasc Biol. (2020) 40:2054–69. doi: 10.1161/atvbaha.120.314053
38. Romeo MA, Gilardini Montani MS, Benedetti R, Arena A, Gaeta A, and Cirone M. The dysregulation of autophagy and ER stress induced by HHV-6A infection activates pro-inflammatory pathways and promotes the release of inflammatory cytokines and cathepsin S by CNS cells. Virus Res. (2022) 313:198726. doi: 10.1016/j.virusres.2022.198726
39. Foronjy RF, Dabo AJ, Taggart CC, Weldon S, and Geraghty P. Respiratory syncytial virus infections enhance cigarette smoke induced COPD in mice. PloS One. (2014) 9:e90567. doi: 10.1371/journal.pone.0090567
40. Kim H, Mazumdar B, Bose SK, Meyer K, Di Bisceglie AM, Hoft DF, et al. Hepatitis C virus-mediated inhibition of cathepsin S increases invariant-chain expression on hepatocyte surface. J Virol. (2012) 86:9919–28. doi: 10.1128/jvi.00388-12
41. Velásquez LN, Milillo MA, Delpino MV, Trotta A, Fernández P, Pozner RG, et al. Brucella abortus down-regulates MHC class II by the IL-6-dependent inhibition of CIITA through the downmodulation of IFN regulatory factor-1 (IRF-1). J Leukoc Biol. (2017) 101:759–73. doi: 10.1189/jlb.4A0416-196R
42. Rundberg Nilsson AJS, Xian H, Shalapour S, Cammenga J, and Karin M. IRF1 regulates self-renewal and stress responsiveness to support hematopoietic stem cell maintenance. Sci Adv. (2023) 9:eadg5391. doi: 10.1126/sciadv.adg5391
43. Lee H, Lee DH, Oh JH, and Chung JH. Skullcapflavone II suppresses TNF-α/IFN-γ-induced TARC, MDC, and CTSS production in haCaT cells. Int J Mol Sci. (2021) 22:6428. doi: 10.3390/ijms22126428
44. Seo HR, Bae S, and Lee YS. Radiation-induced cathepsin S is involved in radioresistance. Int J Cancer. (2009) 124:1794–801. doi: 10.1002/ijc.24095
45. Weldon S, McNally P, McAuley DF, Oglesby IK, Wohlford-Lenane CL, Bartlett JA, et al. miR-31 dysregulation in cystic fibrosis airways contributes to increased pulmonary cathepsin S production. Am J Respir Crit Care Med. (2014) 190:165–74. doi: 10.1164/rccm.201311-1986OC
46. Chen SJ, Chen LH, Yeh YM, Lin CK, Lin PC, Huang HW, et al. Targeting lysosomal cysteine protease cathepsin S reveals immunomodulatory therapeutic strategy for oxaliplatin-induced peripheral neuropathy. Theranostics. (2021) 11:4672–87. doi: 10.7150/thno.54793
47. Wohlfahrt T, Rauber S, Uebe S, Luber M, Soare A, Ekici A, et al. PU.1 controls fibroblast polarization and tissue fibrosis. Nature. (2019) 566:344–9. doi: 10.1038/s41586-019-0896-x
48. Zhang K, Wang S, Wang Z, Jiang Y, Huang M, Liu N, et al. Critical roles of PU.1/cathepsin S activation in regulating inflammatory responses of macrophages in periodontitis. J Periodontal Res. (2023) 58:939–47. doi: 10.1111/jre.13153
49. Zhang XY, Zhuo X, Cheng J, Wang X, Liang K, and Chen X. PU.1 regulates cathepsin S expression in large yellow croaker (Larimichthys crocea) macrophages. Front Immunol. (2021) 12:819029. doi: 10.3389/fimmu.2021.819029
50. Wang Y, Baron RM, Zhu G, Joo M, Christman JW, Silverman ES, et al. PU.1 regulates cathepsin S expression in professional APCs. J Immunol. (2006) 176:275–83. doi: 10.4049/jimmunol.176.1.275
51. Pang SH, Minnich M, Gangatirkar P, Zheng Z, Ebert A, Song G, et al. PU.1 cooperates with IRF4 and IRF8 to suppress pre-B-cell leukemia. Leukemia. (2016) 30:1375–87. doi: 10.1038/leu.2016.27
52. Weng C, Xu J, Ying X, Sun S, Hu Y, Wang X, et al. The PDIA3-STAT3 protein complex regulates IBS formation and development via CTSS/MHC-II pathway-mediated intestinal inflammation. Heliyon. (2024) 10:e36357. doi: 10.1016/j.heliyon.2024.e36357
53. Chan LL, Cheung BK, Li JC, and Lau AS. A role for STAT3 and cathepsin S in IL-10 down-regulation of IFN-gamma-induced MHC class II molecule on primary human blood macrophages. J Leukoc Biol. (2010) 88:303–11. doi: 10.1189/jlb.1009659
54. Zhou YB, Zhou H, Li L, Kang Y, Cao X, Wu ZY, et al. Hydrogen sulfide prevents elastin loss and attenuates calcification induced by high glucose in smooth muscle cells through suppression of stat3/cathepsin S signaling pathway. Int J Mol Sci. (2019) 20:4202. doi: 10.3390/ijms20174202
55. Cao L, Cao X, Zhou Y, Nagpure BV, Wu ZY, Hu LF, et al. Hydrogen sulfide inhibits ATP-induced neuroinflammation and Aβ(1-42) synthesis by suppressing the activation of STAT3 and cathepsin S. Brain Behav Immun. (2018) 73:603–14. doi: 10.1016/j.bbi.2018.07.005
56. Wang FZ, Zhou H, Wang HY, Dai HB, Gao Q, Qian P, et al. Hydrogen sulfide prevents arterial medial calcification in rats with diabetic nephropathy. BMC Cardiovasc Disord. (2021) 21:495. doi: 10.1186/s12872-021-02307-9
57. Liu PP, Liu XH, Ren MJ, Liu XT, Shi XQ, Li ML, et al. Neuronal cathepsin S increases neuroinflammation and causes cognitive decline via CX3CL1-CX3CR1 axis and JAK2-STAT3 pathway in aging and Alzheimer’s disease. Aging Cell. (2025) 24:e14393. doi: 10.1111/acel.14393
58. Xuan C, Chen D, Zhang S, Li C, Fang Q, Chen D, et al. Isoquercitrin alleviates diabetic nephropathy by inhibiting STAT3 phosphorylation and dimerization. Adv Sci (Weinh). (2025) 4:e2414587. doi: 10.1002/advs.202414587
59. Andrault PM, Panwar P, Mackenzie NCW, and Brömme D. Elastolytic activity of cysteine cathepsins K, S, and V promotes vascular calcification. Sci Rep. (2019) 9:9682. doi: 10.1038/s41598-019-45918-1
60. Kim SJ, Schätzle S, Ahmed SS, Haap W, Jang SH, Gregersen PK, et al. Increased cathepsin S in Prdm1(-/-) dendritic cells alters the T(FH) cell repertoire and contributes to lupus. Nat Immunol. (2017) 18:1016–24. doi: 10.1038/ni.3793
61. Chen H, Wang J, Xiang MX, Lin Y, He A, Jin CN, et al. Cathepsin S-mediated fibroblast trans-differentiation contributes to left ventricular remodelling after myocardial infarction. Cardiovasc Res. (2013) 100:84–94. doi: 10.1093/cvr/cvt158
62. Zhang P, Hou S, Chen J, Zhang J, Lin F, Ju R, et al. Smad4 deficiency in smooth muscle cells initiates the formation of aortic aneurysm. Circ Res. (2016) 118:388–99. doi: 10.1161/circresaha.115.308040
63. Rothmeier AS and Ruf W. Protease-activated receptor 2 signaling in inflammation. Semin Immunopathol. (2012) 34:133–49. doi: 10.1007/s00281-011-0289-1
64. Zhao P, Lieu T, Barlow N, Metcalf M, Veldhuis NA, Jensen DD, et al. Cathepsin S causes inflammatory pain via biased agonism of PAR2 and TRPV4. J Biol Chem. (2014) 289:27215–34. doi: 10.1074/jbc.M114.599712
65. Rondeau LE, Da Luz BB, Santiago A, Bermudez-Brito M, Hann A, De Palma G, et al. Proteolytic bacteria expansion during colitis amplifies inflammation through cleavage of the external domain of PAR2. Gut Microbes. (2024) 16:2387857. doi: 10.1080/19490976.2024.2387857
66. Kim N, Bae KB, Kim MO, Yu DH, Kim HJ, Yuh HS, et al. Overexpression of cathepsin S induces chronic atopic dermatitis in mice. J Invest Dermatol. (2012) 132:1169–76. doi: 10.1038/jid.2011.404
67. Klinngam W, Fu R, Janga SR, Edman MC, and Hamm-Alvarez SF. Cathepsin S alters the expression of pro-inflammatory cytokines and MMP-9, partially through protease-activated receptor-2, in human corneal epithelial cells. Int J Mol Sci. (2018) 19:3530. doi: 10.3390/ijms19113530
68. Small DM, Brown RR, Doherty DF, Abladey A, Zhou-Suckow Z, Delaney RJ, et al. Targeting of cathepsin S reduces cystic fibrosis-like lung disease. Eur Respir J. (2019) 53:1801523. doi: 10.1183/13993003.01523-2018
69. Clark AK, Staniland AA, and Malcangio M. Fractalkine/CX3CR1 signalling in chronic pain and inflammation. Curr Pharm Biotechnol. (2011) 12:1707–14. doi: 10.2174/138920111798357465
70. Szukiewicz D. CX3CL1 (Fractalkine)-CX3CR1 axis in inflammation-induced angiogenesis and tumorigenesis. Int J Mol Sci. (2024) 25:4679. doi: 10.3390/ijms25094679
71. Clark AK, Grist J, Al-Kashi A, Perretti M, and Malcangio M. Spinal cathepsin S and fractalkine contribute to chronic pain in the collagen-induced arthritis model. Arthritis Rheumatol. (2012) 64:2038–47. doi: 10.1002/art.34351
72. Clark AK, Yip PK, Grist J, Gentry C, Staniland AA, Marchand F, et al. Inhibition of spinal microglial cathepsin S for the reversal of neuropathic pain. Proc Natl Acad Sci U S A. (2007) 104:10655–60. doi: 10.1073/pnas.0610811104
73. Seo Y, Kim HS, Kang I, Choi SW, Shin TH, Shin JH, et al. Cathepsin S contributes to microglia-mediated olfactory dysfunction through the regulation of Cx3cl1-Cx3cr1 axis in a Niemann-Pick disease type C1 model. Glia. (2016) 64:2291–305. doi: 10.1002/glia.23077
74. Bolós M, Llorens-Martín M, Perea JR, Jurado-Arjona J, Rábano A, Hernández F, et al. Absence of CX3CR1 impairs the internalization of Tau by microglia. Mol Neurodegener. (2017) 12:59. doi: 10.1186/s13024-017-0200-1
75. Bonfante R, Napimoga MH, Macedo CG, Abdalla HB, Pieroni V, and Clemente-Napimoga JT. The P2X7 receptor, cathepsin S and fractalkine in the trigeminal subnucleus caudalis signal persistent hypernociception in temporomandibular rat joints. Neuroscience. (2018) 391:120–30. doi: 10.1016/j.neuroscience.2018.09.005
76. Fu R, Guo H, Janga S, Choi M, Klinngam W, Edman MC, et al. Cathepsin S activation contributes to elevated CX3CL1 (fractalkine) levels in tears of a Sjögren’s syndrome murine model. Sci Rep. (2020) 10:1455. doi: 10.1038/s41598-020-58337-4
77. Jurewicz MM and Stern LJ. Class II MHC antigen processing in immune tolerance and inflammation. Immunogenetics. (2019) 71:171–87. doi: 10.1007/s00251-018-1095-x
78. Roche PA and Furuta K. The ins and outs of MHC class II-mediated antigen processing and presentation. Nat Rev Immunol. (2015) 15:203–16. doi: 10.1038/nri3818
79. Bandola-Simon J, Ito Y, Wucherpfennig KW, and Roche PA. Defective removal of invariant chain peptides from MHC class II suppresses tumor antigen presentation and promotes tumor growth. Cell Rep. (2025) 44:115150. doi: 10.1016/j.celrep.2024.115150
80. Kwiatkowski AJ, Helm EY, Stewart JM, Drashansky TT, Cho JJ, Avram D, et al. Treatment with an antigen-specific dual microparticle system reverses advanced multiple sclerosis in mice. Proc Natl Acad Sci U S A. (2022) 119:e2205417119. doi: 10.1073/pnas.2205417119
81. Klinngam W, Janga SR, Lee C, Ju Y, Yarber F, Shah M, et al. Inhibition of cathepsin S reduces lacrimal gland inflammation and increases tear flow in a mouse model of sjögren’s syndrome. Sci Rep. (2019) 9:9559. doi: 10.1038/s41598-019-45966-7
82. Nakagawa TY, Brissette WH, Lira PD, Griffiths RJ, Petrushova N, Stock J, et al. Impaired invariant chain degradation and antigen presentation and diminished collagen-induced arthritis in cathepsin S null mice. Immunity. (1999) 10:207–17. doi: 10.1016/s1074-7613(00)80021-7
83. Wiendl H, Lautwein A, Mitsdörffer M, Krause S, Erfurth S, Wienhold W, et al. Antigen processing and presentation in human muscle: cathepsin S is critical for MHC class II expression and upregulated in inflammatory myopathies. J Neuroimmunol. (2003) 138:132–43. doi: 10.1016/s0165-5728(03)00093-6
84. Fissolo N, Kraus M, Reich M, Ayturan M, Overkleeft H, Driessen C, et al. Dual inhibition of proteasomal and lysosomal proteolysis ameliorates autoimmune central nervous system inflammation. Eur J Immunol. (2008) 38:2401–11. doi: 10.1002/eji.200838413
85. Pan L, Li Y, Jia L, Qin Y, Qi G, Cheng J, et al. Cathepsin S deficiency results in abnormal accumulation of autophagosomes in macrophages and enhances Ang II-induced cardiac inflammation. PloS One. (2012) 7:e35315. doi: 10.1371/journal.pone.0035315
86. Zuo T, Xie Q, Liu J, Yang J, Shi J, Kong D, et al. Macrophage-derived cathepsin S remodels the extracellular matrix to promote liver fibrogenesis. Gastroenterology. (2023) 165:746–61.e16. doi: 10.1053/j.gastro.2023.05.039
87. Parisis D, Chivasso C, Perret J, Soyfoo MS, and Delporte C. Current state of knowledge on primary sjögren’s syndrome, an autoimmune exocrinopathy. J Clin Med. (2020) 9:2299. doi: 10.3390/jcm9072299
88. Choi E, Jeon KH, Lee H, Mun GI, Kim JA, Shin JH, et al. Radiosensitizing effect of a novel CTSS inhibitor by enhancing BRCA1 protein stability in triple-negative breast cancer cells. Cancer Sci. (2024) 115:2036–48. doi: 10.1111/cas.16174
89. Kumar Vr S, Darisipudi MN, Steiger S, Devarapu SK, Tato M, Kukarni OP, et al. Cathepsin S cleavage of protease-activated receptor-2 on endothelial cells promotes microvascular diabetes complications. J Am Soc Nephrol. (2016) 27:1635–49. doi: 10.1681/asn.2015020208
90. Cianni L, Feldmann CW, Gilberg E, Gütschow M, Juliano L, Leitão A, et al. Can cysteine protease cross-class inhibitors achieve selectivity? J Med Chem. (2019) 62:10497–525. doi: 10.1021/acs.jmedchem.9b00683
91. Yan L, Ding S, Gu B, and Ma P. Clinical application of simultaneous detection of cystatin C, cathepsin S, and IL-1 in classification of coronary artery disease. J BioMed Res. (2017) 31:315–20. doi: 10.7555/jbr.31.20150152
92. Feth MP, Heyse W, Baumgartner B, Nagel N, Tappertzhofen C, Olpp T, et al. From laboratory to pilot plant: the solid-state process development of a highly potent cathepsin S/K inhibitor. Eur J Pharm Biopharm. (2013) 83:436–48. doi: 10.1016/j.ejpb.2012.11.007
93. Yoo Y, Choi E, Kim Y, Cha Y, Um E, Kim Y, et al. Therapeutic potential of targeting cathepsin S in pulmonary fibrosis. BioMed Pharmacother. (2022) 145:112245. doi: 10.1016/j.biopha.2021.112245
94. Bentley D, Fisher BA, Barone F, Kolb FA, and Attley G. A randomized, double-blind, placebo-controlled, parallel group study on the effects of a cathepsin S inhibitor in primary Sjögren’s syndrome. Rheumatol (Oxford). (2023) 62:3644–53. doi: 10.1093/rheumatology/kead092
95. Payne CD, Deeg MA, Chan M, Tan LH, LaBell ES, Shen T, et al. Pharmacokinetics and pharmacodynamics of the cathepsin S inhibitor, LY3000328, in healthy subjects. Br J Clin Pharmacol. (2014) 78:1334–42. doi: 10.1111/bcp.12470
96. Jadhav PK, Schiffler MA, Gavardinas K, Kim EJ, Matthews DP, Staszak MA, et al. Discovery of cathepsin S inhibitor LY3000328 for the treatment of abdominal aortic aneurysm. ACS Med Chem Lett. (2014) 5:1138–42. doi: 10.1021/ml500283g
Keywords: cathepsin S (CTSS), molecular mechanism, immunoregulation, inflammatory disease, therapeutic targets
Citation: Gao H, Zhang Z, Deng J and Song Y (2025) Cathepsin S: molecular mechanisms in inflammatory and immunological processes. Front. Immunol. 16:1600206. doi: 10.3389/fimmu.2025.1600206
Received: 26 March 2025; Accepted: 16 June 2025;
Published: 07 July 2025.
Edited by:
Iman Mamdouh Talaat, University of Sharjah, United Arab EmiratesReviewed by:
Eman Thabet, Alexandria University, EgyptMarwa Mahmoud Abdelaziz Mohamed Mady, Gulf Medical University, United Arab Emirates
Copyright © 2025 Gao, Zhang, Deng and Song. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yanqing Song, c29uZ3lhbnFAamx1LmVkdS5jbg==