 Qi Wang
Qi Wang He Xu1
He Xu1 Lothar Jänsch
Lothar Jänsch Senlin Li
Senlin Li- 1School of Life Sciences, Zhengzhou University, Zhengzhou, China
- 2Cellular Proteomics, Helmholtz Centre for Infection Research, Braunschweig, Germany
Autophagy is a conserved cellular degradative pathway that has been demonstrated to play a crucial role in the innate immune response to combat infection with a range of pathogenic bacteria via xenophagy. Although this process has been well-described in terrestrial animals, the extent to which autophagy contributes to aquatic animal-bacteria interactions remains poorly understood. Autophagy can directly eliminate intracellular pathogens by acting as a conduit for their lysosomes delivery. Consequently, bacteria have evolved a variety of tactics to evade autophagy. This is accomplished by interfering with autophagy signaling or the autophagy machinery itself. In certain instances, bacteria even utilize autophagy as a means of promoting their growth. This review discusses canonical and non-canonical autophagy pathways and current knowledge of autophagy in aquatic animals. This review illuminates the intricate relationship between autophagy components and intracellular bacteria. It explores how the autophagic machinery senses these bacteria directly or indirectly, the interaction between autophagy and effectors/toxins secreted by bacteria, and how some of these bacterial pathogens evade autophagy.
1 Introduction
Autophagy constitutes a highly conserved “clean-up” process in eukaryotic organisms, essential for maintaining intracellular homeostasis through the break down and recycle unnecessary or damaged materials like long-lived proteins, aggregated cellular components, and superfluous or dysfunctional organelles, including mitochondria and peroxisomes. This process is categorized into three primary subtypes: macroautophagy (where the cell wraps debris in a membrane-bound sac), microautophagy (where the lysosome directly engulfs material), and chaperone-mediated autophagy (which uses helper proteins to shuttle specific cargo to the lysosome). Furthermore, emerging evidence highlights autophagy as an integral component of innate immunity, particularly through xenophagy, a specialized form of selective autophagy, which enabling the recognition, encapsulation, and lysosomal degradation of harmful bacteria, viruses, or other invaders inside the cell, thereby limiting their proliferation and mitigating infection. This interplay underscores autophagy’s dual role in cellular maintenance and protect organisms by preventing pathogens from hijacking cellular resources (1, 2). Growing evidence has elucidated that bacterial pathogens have evolved sophisticated counterstrategies to subvert autophagy (3) (as shown in Figure 1).
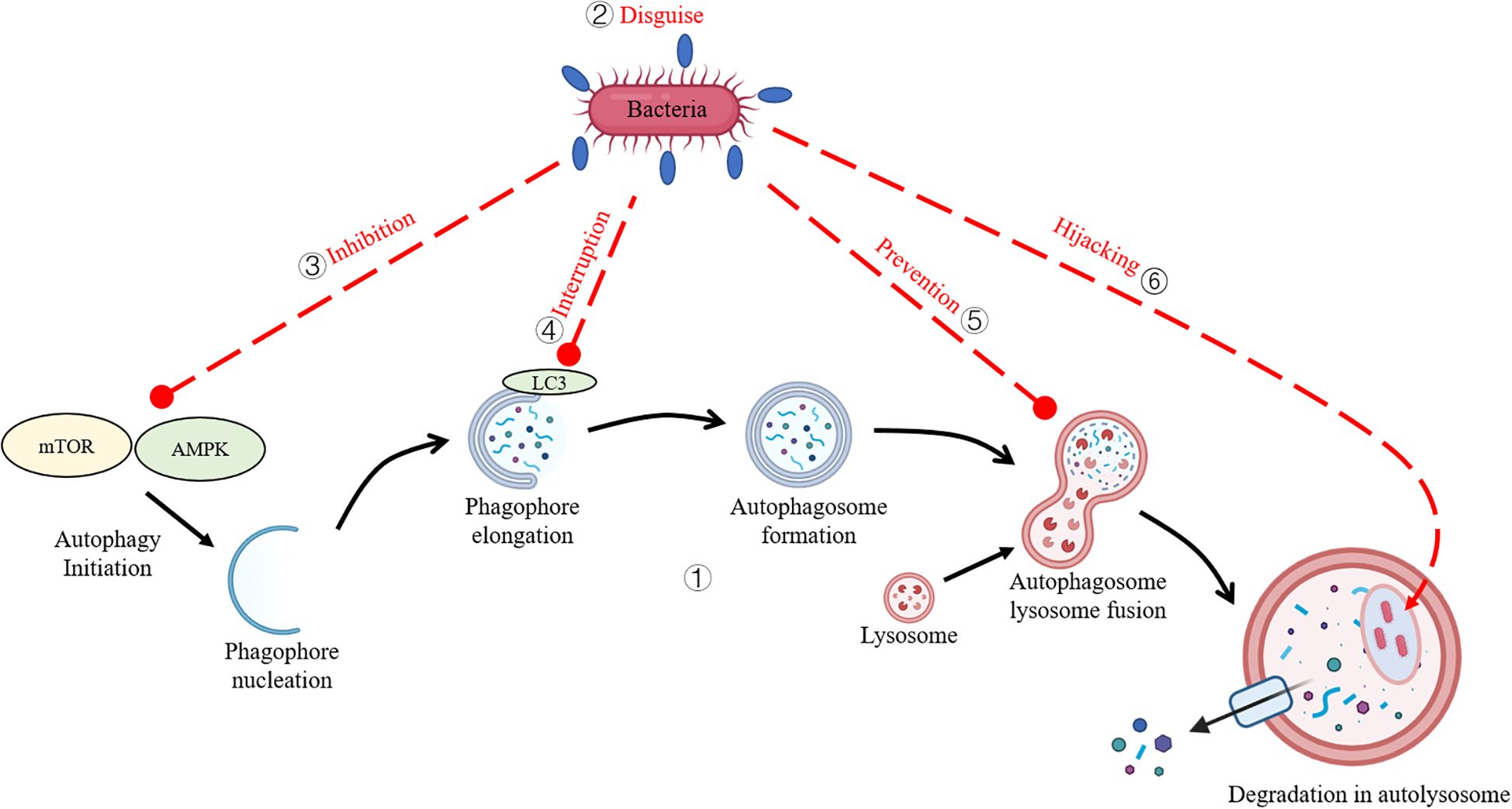
Figure 1. Bacterial Strategies to Evade Autophagy. (1) The core autophagy pathway in teleost species. (2) Bacteria disguise their surface antigens by coating them with the host’s own proteins. (3) Bacteria subvert host autophagy by inhibiting upstream signaling cascades. (4) Bacteria interfere with autophagic proteins to evade capture. (5) Bacteria inhibit autophagosome-lysosome fusion. (6) Bacteria exploit autophagy machinery to create intracellular replicative niches.
For example, some bacteria block the signals that trigger autophagy by inhibiting upstream signaling cascades (e.g., mTOR or AMPK pathways) (4, 5), disguise their surface antigens using the host’s own proteins to evade being flagged for destruction, or mess with the proteins (e.g., ATG5, LC3) responsible for autophagy to dodge capture (6–8). Additionally, certain species obstruct autophagosome-lysosome fusion, thereby preventing pathogen degradation (9). Shockingly, a few bacteria hijack parts of the autophagy system to establish replicative niches, enhancing intracellular survival (10). While extensive research has illuminated the interplay between autophagy and bacterial pathogens in mammalian systems, this topic hasn’t gotten nearly as much attention in aquatic organisms. Notably, review addressing autophagy-pathogen interactions in aquatic species are scarce, reflecting a critical gap in the field. This review dives into the molecular interplay between autophagy and intracellular pathogens, elucidating the mechanisms by which xenophagic pathways target invasive bacteria and the sneaky tactics pathogens employ to evade detection, avoid autophagic capture, or inhibit lysosomal degradation. This review compiles publications from 1997 to 2025, focusing on bacterial interactions with host autophagy and their evasion strategies in teleost. Additionally, some bacteria known to infect mammals are included to provide broader insights into bacterial evasion mechanisms. By piecing together emerging evidence, this work sheds light on the evolutionary adaptations of pathogens, offering insights into potential therapeutic avenues for aquatic disease management.
2 Autophagy
2.1 Autophagy classification
Canonical autophagy, mediated by a conserved suite of autophagy-related proteins, involves double-membrane autophagosome formation. A hallmark of this process is the recruitment of microtubule-associated protein 1 light chain 3 (LC3), a canonical autophagosome marker. Phagosomes—vesicles mediating extracellular particle engulfment—recruit LC3 via LC3-associated phagocytosis (LAP) (11), a distinct pathway sharing lysosomal degradation aims but diverging in structure and origin. While autophagosomes sequester cytoplasmic cargo within double membranes, LAP involves LC3 conjugation to single-membrane phagosomes (12). Notably, Rubicon, essential for LAP, is dispensable in canonical autophagy targeting intracellular pathogens, highlighting the functional plasticity of autophagy machinery in homeostasis and immunity (13). Autophagy operates via non-selective and selective modalities. Non-selective autophagy indiscriminately engulfs cytoplasmic components en masse for energy recycling. In contrast, selective autophagy employs molecular tagging (e.g., ubiquitination) and adaptor-mediated cargo recognition to precision-target substrates—including mitochondria (mitophagy), ribosomes (ribophagy), peroxisomes (pexophagy), and pathogens (xenophagy)—for lysosomal degradation (14) (as shown in Table 1). This review focuses on xenophagy, delineating its antimicrobial mechanisms and bacterial evasion tactics.
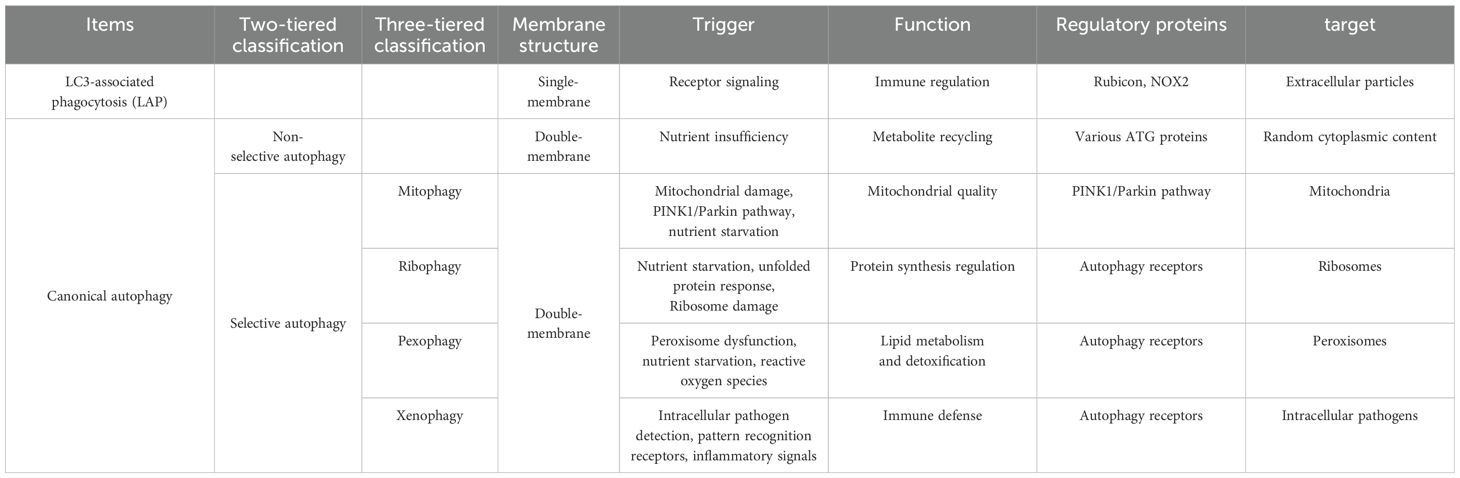
Table 1. Classification and key differences between autophagy pathways.
2.2 Xenophagy targeting mechanisms
Host cells deploy cytosolic surveillance systems to detect invasive pathogens via pathogen- and damage-associated molecular patterns (PAMPs/DAMPs), which activate pattern recognition receptors (PRRs) such as NOD-like receptors. These receptors trigger innate immune responses, enabling detection of both microbial presence and infection-induced cellular stress (15, 16). And then, xenophagy, a selective autophagy pathway, targets intracellular pathogens for lysosomal degradation, serving as a key antimicrobial mechanism (17, 18). Pathogens are categorized by their intracellular niches: those confined within vesicles or replicating freely in the cytosol (19). Typically, Rapid bacterial clearance occurs unless pathogens evolve evasion strategies (20). Among autophagy-mediated targeting mechanisms, those involving protein ubiquitination remain the most extensively characterized. Ubiquitin chains play a pivotal role in autophagy, functioning as molecular signals that direct bacterial clearance. During infection, ubiquitin orchestrates the autophagic sequestration of invasive bacteria by mediating interactions between pathogen-containing endosomes and the autophagic machinery upstream of LC3 recruitment. Ubiquitination is central to autophagy, with E3 ligases (e.g., PRKN, SMURF1) depositing ubiquitin chains on pathogens to recruit autophagy adaptors (SQSTM1/p62, NDP52, optineurin), which bridge cargo to nascent autophagosomes. These adaptors bridge ubiquitinated cargo to LC3-decorated autophagosomes via ubiquitin-binding and LC3-interacting domains (21, 22). TBK1-mediated phosphorylation of autophagy receptors enhances their recruitment to bacteria and autophagosome formation during autophagy (23). Emerging evidence underscores ubiquitin’s direct interaction with Atg16L1, bypassing LC3 to streamline pathogen recognition and degradation, reinforcing ubiquitin’s pivotal role in autophagy (24–26).
3 Evasion of xenophagy by intracellular bacteria
Xenophagy primarily functions to eliminate intracellular pathogens by detecting and tag them within autophagosomes, which subsequently fuse with lysosomes to mediate pathogen degradation. However, numerous bacterial pathogens have developed counterstrategies to subvert xenophagic clearance, enabling their survival and replication within host cells.
3.1 Avoidance of autophagy recognition
Xenophagy targets cytosolic bacteria via host ubiquitin machinery, which deposits polyubiquitin coats on pathogens, enabling recognition by autophagy receptors (e.g., p62, NDP52) (27). Autophagosome formation is initiated by BECLIN 1, with LC3 lipidation via the ATG5–12–16L1 complex enabling bacterial capture (28, 29). Subsequent lysosomal fusion mediates degradation. During infection, intracellular bacteria prompt the formation of ubiquitinated aggregates (e.g., vacuolar remnants, bacterial debris, aggresome-like structures), which recognized by ubiquitin-binding proteins (30). But, a hallmark of many intracellular pathogens, including Salmonella enterica, who replicates within a membrane-bound compartment, the Salmonella-containing vacuole (SCV), that counteracts this by deploying the SPI-2 type III secretion system (T3SS) effector SseL, a deubiquitinase, reducing autophagic marker recruitment and enhancing bacterial replication within SCVs (31, 32).
The Gram-positive bacterium Listeria monocytogenes (L. monocytogenes) is a facultative intracellular pathogen capable of invading and replicating within mammalian or teleost cells (33). Upon entering the cytosol, it employs the surface protein ActA to evade autophagic recognition. ActA’s amino-terminal domain binds and activates the host Arp2/3 complex to drive actin nucleation, while its central proline-rich region recruits Ena/VASP proteins to enhance bacterial motility (34). By recruits host cytoskeletal components (e.g., Arp2/3, VASP, Actin), ActA orchestrates actin polymerization, disguising the bacterium as a host organelle and preventing detection by autophagy markers such as polyubiquitin, p62, and LC3 (35). Additionally, the protein InlK further shields L. monocytogenes by interacting with the host major vault protein, blocking ubiquitin tagging by E3 ligases and autophagy receptor recruitment (36).
Shigella flexneri (S. flexneri), a Gram-negative bacterium causing shigellosis, evades xenophagy via its T3SS effector IcsB and surface protein IcsA. Autophagy protein ATG5 binds IcsA, which normally targets pathogens for degradation, and promotes actin polymerization. This indirectly supports the formation of septin cages—cytoskeletal structures that, alongside autophagy proteins, trap Shigella to limit spread (37). Ubiquitin binding adaptor proteins p62 and NDP52 then direct the bacteria to septin- and actin-dependent autophagy pathways (38). However, IcsB competitively blocks ATG5 binding to IcsA by occupying the same IcsA domain (amino acids 320–433) in a dose-dependent manner, preventing autophagosome recognition (39).
Rickettsia species are Gram-negative, obligate intracellular bacteria that infect vascular endothelial cells, dendritic cells, and macrophages. R. parkeri evades xenophagy recognition primarily via its outer membrane protein B (OmpB), which blocks polyubiquitylation of bacterial surface proteins. Unlike pathogens that manipulate host actin to avoid ubiquitylation, OmpB acts locally to protect OmpA—a common target of host ubiquitin machinery—from autophagic recognition (35). Potential mechanisms include OmpB’s deubiquitylase activity, its abundance (over 10% of bacterial protein mass) enabling surface camouflage, recruitment of host proteins to cleave ubiquitin chains, or enzymatic modification of the bacterial surface required for recognition by the host ubiquitin machinery (40).
3.2 Interference with autophagy initiation
Autophagy, marked by autophagosome formation, is regulated by the ULK1 complex (ULK1, FIP200, ATG101), controlled by mTORC1 and AMPK (41). While AMPK activates ULK1, mTORC1 suppresses it via phosphorylation of Ser757 (41). Salmonella enterica, a facultative intracellular pathogen, employs its T3SS to inject effectors like SopB into host cells (42). In B cells, SopB’s phosphatase activity elevates PIP3 levels, activating the PI3K/AKT pathway (43). This triggers mTORC1, which inhibits ULK1 and blocks autophagosome formation. By suppressing autophagy initiation via this PI3K/AKT/mTORC1 axis, enabling S. enterica prolonged survival within B cells (44).
Tuberculosis, caused by Mycobacterium tuberculosis (Mtb), remains a global health threat, particularly in low- and middle-income countries (45). Mtb could evades xenophagy to survive in macrophages. Autophagy initiation relies on the ULK1 complex (ULK1, FIP200, ATG13, ATG101), which recruits the VPS34 complex (VPS34, BECLIN-1, VPS15, and ATG14L) to generate phosphatidylinositol 3-phosphate (PI3P), driving autophagosome formation. The produced PI3P then binds with PI3P-binding proteins such as WIPI2B and DFCP1, together with other proteins, leads to the formation and expansion of the autophagosome, eventually forming the complete autophagosome (46). AMPK activates ULK1 (via phosphorylation of Ser317/Ser777), while mTORC1 suppresses it by phosphorylating ULK1 Ser757, blocking the interaction between ULK1 and AMPK. This coordinated phosphorylation is important for ULK1 in autophagy induction (41). Mtb disrupts this balance using secreted acid phosphatase SapM (Rv3310). SapM enhances mTORC1 activity by dephosphorylating Raptor (a key mTORC1 component) at Ser792, countering AMPK’s inhibitory effect (47, 48). This sustains mTORC1 activation, preventing autophagy initiation and aiding bacterial survival (45).
3.3 Manipulation of autophagy machinery
Staphylococcus aureus, an opportunistic pathogen colonizing human skin and nares, subverts xenophagy to persist intracellularly (49). Its cell wall components can be detected as PAMPs and trigger autophagy via ubiquitination and receptor recruitment (e.g., p62, NDP52) in non-phagocytic cells (50, 51), while Galectin-8 targets damaged endosomes for autophagic clearance (52). In phagocytes, S. aureus initially resides in vesicles before lysosomal fusion (53), but certain strains block autophagy flux—disrupting LC3-II and p62—and alkalinise autolysosomes to create replication niches (54). Central to this evasion is the accessory gene regulatory (AGR) quorum-sensing system, which regulates toxins like α-toxin and phenol-soluble modulins (PSMα) (55). S. aureus secreted α-toxin to inhibit autophagosome-lysosome fusion, while PSMα aids phagosomal escape (56). AGR also dysregulates S. aureus-containing phagosomes maturation, preventing autophagosome acidification and LAMP-2 acquisition (57). In polymorphonuclear neutrophils, AGR-driven p53 accumulation driving transcriptional activation of pro-autophagic membrane protein damage-regulated autophagy monitor (DRAM), inducing autophagosome buildup to sustain survival niche for S. aureus (58). Additionally, S. aureus phosphorylates mitogen-activated protein kinase 14 (MAPK14) and ATG5, further blocking autophagosome maturation (50). By manipulating autophagy through AGR-dependent and -independent mechanisms, S. aureus evades lysosomal degradation, ensuring intracellular persistence.
Salmonella enterica serovar Typhimurium (S. Typhimurium), a Gram-negative intracellular pathogen, employs two virulence gene clusters—Salmonella Pathogenicity Island 1 (SPI1) and SPI2—to invade host cells and survive intracellularly (59). SPI1’s T3SS enables entry into nonphagocytic cells, while SPI2’s T3SS facilitates survival within SCVs by secreting effectors that block lysosomal fusion (60). These SPI2 effectors manipulate host signalling pathways, including sustained activation of AKT and mTOR, which suppress autophagy—a cellular recycling process controlled by mTOR, a nutrient-sensing kinase, activation of which forms two multiprotein complexes, mTORC1 and mTORC2 (61). Notably, the SPI2 effector SopB activate AKT at Ser473 via mTORC2 early in infection (62). Additionally, S. Typhimurium disrupts AMPK, a key energy sensor activated during ATP depletion. Although infection initially triggers AMPK activity due to low ATP levels, SPI2 targets the AMPK-activation complex (including Sirtuin-1 [SIRT1] and liver kinase B1) for lysosomal degradation, blunting AMPK’s role in autophagy. AMPK normally promotes autophagy by inhibiting mTOR or directly phosphorylating autophagy-related proteins like ULK1 (41). SIRT1 further regulates autophagy by deacetylating components of the autophagic machinery (e.g., ATG5, LC3) and interacting with AMPK in a feedback loop (63, 64). By hijacking these pathways—activating AKT/mTOR, degrading AMPK components, and disrupting SIRT1—SPI2-dependent mechanisms reduce autophagic flux in macrophages (65). This allows S. Typhimurium to evade host defenses, replicate within SCVs, and propagate systemic infection.
Listeria monocytogenes (L. monocytogenes), a Gram-positive intracellular pathogen, causes listeriosis—a severe foodborne illness. It enters the host via the gastrointestinal epithelium, either through M cells in Peyer’s patches or alternative routes. Once inside, dendritic cells and macrophages transport the bacteria to mesenteric lymph nodes and tissues, enabling systemic spread (66). To survive, L. monocytogenes rapidly escapes phagosomes using listeriolysin O (LLO), a pore-forming toxin, aided by phospholipases C enzymes PLCA and PLCB (67). LLO punctures the phagosomal membrane, creating pores that expand over time, allowing bacterial release into the cytosol (68, 69). These enzymes also disrupt autophagy by depleting PI3P, a key molecule for autophagosome formation (70, 71). Host factors like GILT (activating LLO via thiol reduction) and CFTR (altering phagosomal chloride levels) aid bacterial escape (72). Inefficient LLO activity leads to Spacious Listeria-containing Phagosomes (SLAPs)—enlarged, non-maturing compartments where bacteria persist and replicate slowly. By subverting xenophagy and hijacking host pathways, L. monocytogenes evades degradation, ensuring intracellular survival and systemic infection (73).
Burkholderia pseudomallei (B. pseudomallei), a Gram-negative intracellular pathogen, invades both phagocytic and non-phagocytic cells. Within 15 minutes of entry, it escapes endocytic vesicles via its TTSS3 and effector protein BOPA, avoiding lysosomal degradation (74, 75). BOPA contains two functional domains: a Rho GTPase inactivation domain and a cholesterol-binding domain (76, 77). The latter may disrupt lysosomal fusion by accumulating cholesterol on phagosomal membranes, akin to mechanisms seen in Mycobacterium avium infections (78). TTSS structural genes like BSAZ and effector BIPD are critical for timely vesicle escape, as mutants show delayed evasion of LAMP-1-positive vacuoles (79). Metabolic genes (PURM, PURN, HISF, PABB) and BPSL1528 further support intracellular replication (80). To survive, B. pseudomallei subverts xenophagy by upregulating miR-146a, which inhibits lysosomal acid lipase A, blocking autophagosome-lysosome fusion (81). It also suppresses ATG10 (essential for autophagy) via miRNAs (MIR4458, MIR4667-5p, MIR4668-5p), dampening autophagic activity. These strategies enable the pathogen to persist and thrive within host cells (82).
Brucella, a Gram-negative intracellular pathogen, comprises six species (B. melitensis, B. abortus, B. suis, B. ovis, B. canis, B. neotoma) with genetic similarity but varying host preferences and virulence (83). It survives within macrophages and non-phagocytic cells by hijacking xenophagy, forming Brucella-containing vacuoles (BCVs) that mature into endoplasmic reticulum-derived replicative niches (84). The T4SS secretion system delivers effectors (e.g., RICA, VCEA, BtpB) to manipulate host processes. VCEA and BTPB directly regulate autophagy, while host proteins like WIPI, ATG9, BECLIN1, and ATG14L aid BCV formation (85, 86). However, how Brucella precisely exploits autophagy machinery—or which bacterial factors control intracellular trafficking—remains unclear. By subverting lysosomal fusion and leveraging autophagosome-like structures, the pathogen ensures survival and proliferation within host cells.
3.4 Hijacking autophagy for replication
Francisella species thrive in the cytosol of diverse host cells and organisms. After internalization, the bacteria transiently occupy a vacuole marked by early and late endosome markers before escaping into the cytosol—a critical step for rapid replication, mediated by genes in the Francisella pathogenicity island (homologous to TVISS) (87). Autophagy, triggered by infection via an ATG5-independent pathway, supplies amino acids and carbon for bacterial metabolism. Inhibiting autophagy stalls Francisella growth, but adding non-essential amino acids or pyruvate restores replication, confirming their role as nutrients (88). During infection, bacteria localize near autophagosomes in host cells, suggesting exploitation of autophagy for resources. Interestingly, late-stage infection in murine macrophages reveals large autophagic Francisella-containing vacuoles (FCVs), dependent on bacterial protein synthesis (89). However, FCVs form only in mouse cells, not human macrophages, leaving their biological relevance uncertain (90). Identifying bacterial factors that manipulate autophagy could clarify how Francisella hijacks this process for nutrition.
Brucella spp. persist within membrane-bound BCVs, hijacking the host secretory pathway to transform their initial endosomal BCV into an ER-derived replicative BCV. Critical to this process is the VIRB T4SS, which injects bacterial effectors to manipulate host functions (91, 92). Following internalization, BCVs acquire early and late endosomal markers (e.g., LAMP-1) and transiently interact with lysosomes via RAB7 (93). However, the BCV eventually diverts to ER exit sites (ERES), excluding endosomal markers. This shift relies on Sar1 and Rab2 GTPases, which regulate ERES integrity and Golgi-ER transport (94, 95). Brucella infection also activates the unfolded protein response (UPR), a stress pathway restoring ER homeostasis (96). While B. melitensis triggers all three UPR arms (IRE1α, protein kinase RNA-like ER kinase, and activating transcription factor 6-dependent pathways) via effector TCPB. B. abortus and B. suis primarily activate IRE1α, linked to host detection of T4SS activity (97, 98). IRE1α activation—dependent on host protein YIP1A—upregulates Sar1 and COPII components, enhancing ERES function and ER membrane acquisition for BCV biogenesis (99). This process intersects with autophagy: IRE1α-driven UPR stimulates ATG9- and WIPI-dependent autophagosome formation, which Brucella exploits to establish its replicative BCV (99). Despite progress, the exact interplay between UPR, autophagy, and BCV formation remains unclear, particularly how ER stress benefits Brucella’s intracellular survival.
The Chlamydiaceae family comprises Gram-negative, obligate intracellular bacteria that infect humans and animals (100). Among these, Chlamydia trachomatis (C. trachomatis) primarily targets the human female genital tract. During infection, the pathogen establishes a protective vacuole called the inclusion, where it replicates (101). Evidence suggests C. trachomatis hijacks autophagosomes—either as nutrient sources or transport vehicles—to support its survival (102). A key player here is LC3, a protein central to autophagy. Normally, LC3-I (cytosolic) converts to lipid-bound LC3-II, marking autophagosomal membranes. However, C. trachomatis repurposes LC3 in an autophagy-independent manner: the protein stabilizes the host cell’s microtubule network, which is critical for bacterial inclusion stability and movement. Depleting LC3—even in autophagy-deficient cells—severely hampers C. trachomatis growth, underscoring its exploitation of LC3 beyond conventional autophagy pathways. Essentially, the pathogen co-opts LC3’s structural role to anchor inclusions to the cytoskeleton, ensuring its replication niche remains intact (103).
Coxiella burnetii, the Gram-negative bacterium behind Q fever, thrives by hijacking host xenophagy to form its replication niche, the Coxiella-containing vacuole (CCV) (104). Stimulating autophagy—via nutrient deprivation or rapamycin—boosts bacterial replication and enlarges the CCV, while blocking autophagy disrupts CCV development (105). Key autophagy genes (TFEB/TFE3, ATG proteins, STX17) enable homotypic vacuole fusion, forming expansive CCVs (106). The pathogen delays lysosomal protease delivery (e.g., cathepsin D) and exploits autophagy machinery, increasing lipidated LC3B-II and stabilizing p62—a cargo receptor degraded during normal autophagy (107, 108). This manipulation relies on bacterial effectors secreted via the T4SS (109). Critical effectors like CVPB (CIG2) label CCVs, recruit autophagosomal LC3, and sustain phosphatidylinositol 3-phosphate to maintain autolysosomal maturation (110, 111). CVPB, alongside CVPC-E, ensures CCV-LAMP1 vesicle fusion, vital for replication (112). Similarly, CVPF interacts with RAB26—a GTPase regulating autophagy—by binding its inactive form, possibly acting as a GEF/GDF to recruit RAB26 to CCVs. Active RAB26 collaborates with ATG16L1 to stimulate LC3 lipidation, enhancing autophagosome-CCV fusion (113). Depleting RAB26 reduces LC3 recruitment and CCV size, impairing bacterial growth. Another effector, CIG57, recruits clathrin to CCVs, indirectly supporting LC3B association (108). Together, these effectors orchestrate RAB GTPase activity and autophagy flux, diverting host membranes and nutrients to expand CCVs (112). In short, C. burnetii’s T4SS effectors co-opt autophagy regulators (e.g., RAB26, clathrin) to stabilize its niche, illustrating how intracellular pathogens rewire vesicular trafficking for survival.
Bacteria of the genus Yersinia cause illnesses ranging from enteritis to plague. Y. enterocolitica, a species with diverse strains classified by biochemical profiles and O-antigen serotyping, manipulates autophagy to create a protective niche. After invading host cells, it occupies vacuoles resembling autophagosomes but actively blocks lysosomal fusion and acidification, enabling survival and replication (114, 115). Studies show Yersinia persists in intestinal macrophages during early infection and replicates within autophagosomes in macrophages by halting maturation (116, 117). Disrupting autophagy forces bacteria into acidic compartments for degradation, confirming autophagy’s role in sustaining their niche (116). Similarly, Y. pseudotuberculosis exploits arrested autophagosomes, likely fueling replication via nutrient-rich autophagosomal membranes. Staphylococcus aureus also hijacks xenophagy. In HeLa cells, it replicates within LC3-positive autophagosomes for 3–12 hours before escaping into the cytosol to trigger apoptosis. Autophagy-deficient cells (e.g., ATG5 knockouts) prevent bacterial replication, as phagosome-lysosome fusion resumes. Notably, AGR mutants—which lack virulence gene expression—fail to induce autophagy and cannot survive intracellularly (57).
4 Xenophagy in aquaculture
Xenophagy, a selective form of autophagy targeting intracellular pathogens, is particularly critical in combating bacterial infections prevalent in aquatic animals, such as those caused by Aeromonas hydrophila, Edwardsiella, Mycobacterium, and so on (as shown in Table 2).
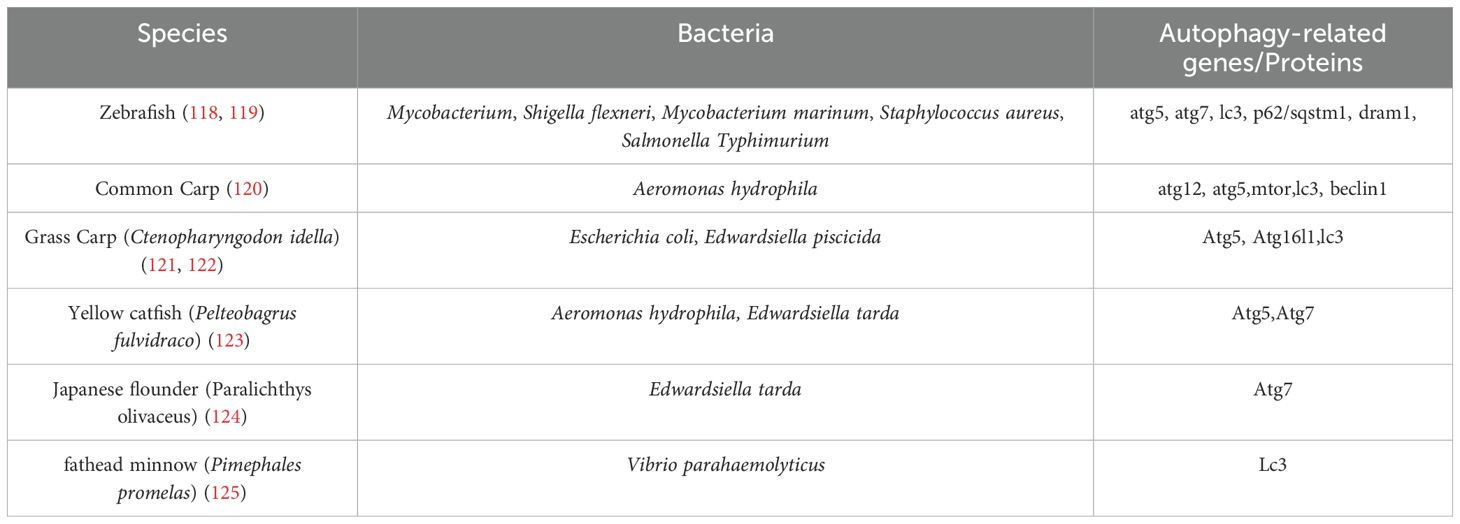
Table 2. Bacteria regulated autophagy genes/proteins in teleost species.
In teleosts, xenophagy is integral to innate immunity. For instance, zebrafish macrophages utilize xenophagic pathways to degrade intracellular pathogens like Shigella flexneri and Salmonella Typhimurium, with autophagy-related genes (ATG5, p62, DRAM1) being essential for bacterial clearance (126). However, certain pathogens subvert these mechanisms (127). Edwardsiella piscicida, a Gram-negative bacterium, induces mitophagy in fish monocytes/macrophages, promoting its intracellular survival by degrading damaged mitochondria and evading antimicrobial ROS (128). This pathogen downregulates host autophagy regulators (e.g., ATG16L1) and PRRs such as NOD1, impairing immune detection and facilitating replication (129, 130). Similarly, Legionella pneumophila manipulates autophagosome maturation, exploiting endoplasmic reticulum (ER)-derived vacuoles to avoid lysosomal degradation, while Mycobacterium marinum recruits autophagy proteins (e.g., LC3) via its ESX-1 secretion system to create replication-permissive compartments (131–133). Pathogen effector proteins further illustrate this subversion. Vibrio parahaemolyticus secretes VopQ, which blocks autophagic flux, impairing inflammasome activation and enabling immune evasion (134). Conversely, Vibrio harveyi exploits host eukaryotic translation initiation factor 3k to degrade MyD88, a key mediator of NF-κB signalling, thereby suppressing inflammatory responses (135, 136). Such strategies highlight the complex interplay between xenophagy and bacterial survival, where pathogens either inhibit autophagic degradation or co-opt its machinery to establish infection niches. Notably, the role of xenophagy varies contextually. While it restricts pathogens like Edwardsiella tarda via the pol-miR-3p-2-p53-BECLIN1 axis in Japanese flounder (137), it paradoxically aids Staphylococcus aureus replication in zebrafish neutrophils by forming non-acidified LC3-positive phagosomes (138). These findings underscore the dual nature of xenophagy in aquatic immunity and pathogenicity. Advancing disease management in aquaculture requires elucidating molecular mechanisms underlying xenophagy-pathogen interactions, including effector protein functions, PRR modulation, and organelle-specific autophagy (e.g., mitophagy). Targeted therapeutic strategies, such as enhancing autophagic flux or blocking pathogen-mediated subversion, hold promise for mitigating infections. Advancing autophagy research in aquatic species requires specialized tools. While DNA, RNA, and protein-based assays adapted from mammalian systems exist, most are only validated in zebrafish. Key RNA techniques include CRISPR/Cas9, TALENs, and morpholino knockdowns, while transcript analysis uses qRT-PCR and sequencing, and protein studies employ WB and TEM (139–141). However, species-specific reagents and protocols for non-model teleosts are lacking, and dynamic autophagy studies face financial and technical hurdles. Developing standardized assays for diverse teleosts remains a critical need. Future research should prioritize in vivo models and multi-omics approaches to unravel these complexities, ultimately informing sustainable aquaculture practices.
5 Conclusion and future perspectives
Xenophagy, a conserved eukaryotic mechanism, functions dually in host defense and pathogen exploitation. While it typically degrades intracellular microbes, certain bacteria subvert xenophagy via effector proteins to inhibit lysosomal fusion, hijacking autophagosomes as replicative niches. This interplay is pathogen- and cell type-dependent, highlighting xenophagy’s complex role in infection dynamics—critical for developing targeted therapies amid rising antibiotic resistance. In teleost, autophagy regulates physiological and pathological processes through pathways analogous to mammals, offering potential as a biomolecular marker or therapeutic target in teleosts. Although research on autophagy in teleost remains limited, progress has been made in identifying autophagy-inducing conditions, genes, and pathogen-triggered autophagic responses. Transgenic zebrafish and cell lines have been instrumental in studying autophagy regulation and its role in anti-pathogen defense. With advancing genetic and imaging tools, zebrafish will continue to enhance our understanding of autophagy in bacterial immunity. Additionally, due to their aquatic environment, teleost lysosome-autophagy systems serve as sensitive biomarkers for ecosystem health monitoring (142). Beyond theoretical significance, autophagy research in teleost holds practical value, offering insights into disease mechanisms and potential applications for aquaculture benefits.
Author contributions
QW: Writing – original draft. HX: Writing – review & editing. JJ: Writing – review & editing. YY: Writing – review & editing. LJ: Writing – review & editing. SL: Supervision, Writing – review & editing, Conceptualization, Writing – original draft, Funding acquisition.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the Key R&D and Promotion Projects in Henan Province (242102111146), the fellowship of China Postdoctoral Science Foundation (2022M712898). International Postdoctoral Exchange Fellowship Program 2024 by the Office of China Postdoctoral Council (ZD2024007).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that Generative AI was used in the creation of this manuscript.
check and correct the spelling and grammar mistakes.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Glossary
ActA: Actin assembly-inducing protein
Agr: accessory gene regulatory
AMPK: adenosine monophosphate-activated protein kinase
AKT: serine/threonine protein kinase B
Arp2/3 complex: Actin-related protein 2/3 complex
ATG5: autophagy related 5
Atg16L1: Autophagy related 16 like 1
BCVs: Brucella-containing vacuoles
BORC: BLOC-one-related complex
B. pseudomallei: Burkholderia pseudomallei
CCV: Coxiella-containing vacuole
CFTR: cystic fibrosis transmembrane conductance regulator
CRISPR/Cas9: Clustered regularly interspaced palindromic repeats associated protein 9;C. trachomatis, Chlamydia trachomatis
CvpB: Coxiella vacuolar protein B
DAMPs: damage-associated molecular patterns
DFCP1: Double FYVE containing protein 1
DRAM: damage-regulated autophagy monitor
ERES: ER exit sites
ERK: extracellular signal-regulated kinase
ESAT-6: early secretory antigenic target 6
ESX-1: ESAT-6 system 1
ESPB: ESX-1 secretion-associated protein B
FCVs: Francisella-containing vacuoles
FIP200: focal adhesion kinase family interacting protein of 200 kD
GAS: Group A Streptococcus
GILT: Gamma-interferon-inducible lysosomal thiol reductase
IcsB: N-epsilon-fatty acyltransferase IcsB
IFN-γR1: IFN-γ receptor 1
InlK: Internalin K
IRE1α: Inositol-requiring transmembrane kinase/endonuclease-1
LAP: LC3-associated phagocytosis
LAMP-2: Lysosome-associated membrane protein-2
LC3: microtubule-associated protein 1 light chain 3
L. monocytogenes: Listeria monocytogenes
LLO: listeriolysin O
L. monocytogenes: Listeria monocytogenes
MAPK14: mitogen-activated protein kinase 14
Mitophagy: mitochondria autophagy
Mtb: Mycobacterium tuberculosis
mTOR: mechanistic target of rapamycin kinase
mTORC1: mechanistic target of rapamycin complex 1
NBR1: neighbor of BRCA1 gene 1
NDP52: nuclear dot protein, 52 kDa
OmpB: outer membrane protein B
PAMPs: pathogen-associated molecular patterns
Pexophagy: peroxisomes autophagy
PI3P: phosphatidylinositol 3-phosphate
PIP3: Phosphatidylinositol (3,4,5)-trisphosphate
PSMα: phenol-soluble modulins α
PRRs: pattern recognition receptors
PRKN: parkin RBR E3 ubiquitin protein ligase
PTPA: Protein phosphatase 2A phosphatase activator
R. parkeri: Rickettsia parkeri
Ribophagy: ribophagy autophagy
SapM: Secretion of an Acid Phosphatase of M. tuberculosis
SCV: Salmonella -containing vacuole
S. flexneri: Shigella flexneri
SLAPs: Spacious Listeria-containing Phagosomes
SMURF1: SMAD specific E3 ubiquitin protein ligase 1
SpeB: Streptococcal pyrogenic exotoxin B
S. Typhimurium: Salmonella enterica serovar Typhimurium
SPI1: Salmonella Pathogenicity Island 1
SIRT1: Sirtuin-1
SQSTM1: sequestosome 1
STAT1: signal transducer and activator of transcription 1
STX17: syntaxin 17
TALENs: transcription activator-like effector nucleases
TBK1: TANK-binding kinase 1
TEM: Transmission electron microscopy
TFEB: transcription factor EB
T3SS: type III secretion system
TLR2: Toll-like receptors 2
ULK1: Unc-51-like kinase 1
UPR: unfolded protein response
VASP: vasodilator-stimulated phosphoprotein
VPS34: vacuolar protein sorting 34
WB: western blot
WIPI: WD-repeat protein interacting with phosphoinositides
Xenophagy: pathogens autophagy.
References
1. Mostowy S, Boucontet L, Mazon Moya MJ, Sirianni A, Boudinot P, Hollinshead M, et al. The zebrafish as a new model for the in vivo study of Shigella flexneri interaction with phagocytes and bacterial autophagy. PloS Pathog. (2013) 9:e1003588. doi: 10.1371/journal.ppat.1003588
2. Shahnazari S and Brumell JH. Mechanisms and consequences of bacterial targeting by the autophagy pathway. Curr Opin Microbiol. (2011) 14:68–75. doi: 10.1016/j.mib.2010.11.001
3. Baxt LA, Garza-Mayers AC, and Goldberg MB. Bacterial subversion of host innate immune pathways. Science. (2013) 340:697–701. doi: 10.1126/science.1235771
4. Shahnazari S, Namolovan A, Mogridge J, Kim PK, and Brumell JH. Bacterial toxins can inhibit host cell autophagy through cAMP generation. Autophagy. (2011) 7:957–65. doi: 10.4161/auto.7.9.16435
5. Tattoli I, Sorbara MT, Vuckovic D, Ling A, Soares F, Carneiro LA, et al. Amino acid starvation induced by invasive bacterial pathogens triggers an innate host defense program. Cell Host Microbe. (2012) 11:563–75. doi: 10.1016/j.chom.2012.04.012
6. Choy A, Dancourt J, Mugo B, O’Connor TJ, Isberg RR, Melia TJ, et al. The Legionella effector RavZ inhibits host autophagy through irreversible Atg8 deconjugation. Science. (2012) 338:1072–6. doi: 10.1126/science.1227026
7. Dong N, Zhu Y, Lu Q, Hu L, Zheng Y, and Shao F. Structurally distinct bacterial TBC-like GAPs link Arf GTPase to Rab1 inactivation to counteract host defenses. Cell. (2012) 150:1029–41. doi: 10.1016/j.cell.2012.06.050
8. Dortet L, Mostowy S, Samba-Louaka A, Gouin E, Nahori MA, Wiemer EA, et al. Recruitment of the major vault protein by InlK: a Listeria monocytogenes strategy to avoid autophagy. PloS Pathog. (2011) 7:e1002168. doi: 10.1371/journal.ppat.1002168
9. Chargui A, Cesaro A, Mimouna S, Fareh M, Brest P, Naquet P, et al. Subversion of autophagy in adherent invasive Escherichia coli-infected neutrophils induces inflammation and cell death. PloS One. (2012) 7:e51727. doi: 10.1371/journal.pone.0051727
10. Niu H, Yamaguchi M, and Rikihisa Y. Subversion of cellular autophagy by Anaplasma phagocytophilum. Cell Microbiol. (2008) 10:593–605. doi: 10.1111/j.1462-5822.2007.01068.x
11. Martinez J, Almendinger J, Oberst A, Ness R, Dillon CP, Fitzgerald P, et al. Microtubule-associated protein 1 light chain 3 alpha (LC3)-associated phagocytosis is required for the efficient clearance of dead cells. Proc Natl Acad Sci USA. (2011) 108:17396–401. doi: 10.1073/pnas.1113421108
12. Keller MD, Torres VJ, and Cadwell K. Autophagy and microbial pathogenesis. Cell Death Differentiation. (2020) 27:872–86. doi: 10.1038/s41418-019-0481-8
13. Martinez J, Malireddi RS, Lu Q, Cunha LD, Pelletier S, Gingras S, et al. RETRACTED ARTICLE: Molecular characterization of LC3-associated phagocytosis reveals distinct roles for Rubicon, NOX2 and autophagy proteins. Nat Cell Biol. (2015) 17:893–906. doi: 10.1038/ncb3192
14. Zhang L, Sung JJ, Yu J, Ng SC, Wong SH, Cho CH, et al. Xenophagy in Helicobacter pylori- and Epstein-Barr virus-induced gastric cancer. J Pathol. (2014) 233:103–12. doi: 10.1002/path.4351
15. Deretic V. Autophagy in infection. Curr Opin Cell Biol. (2010) 22:252–62. doi: 10.1016/j.ceb.2009.12.009
16. Vance RE, Isberg RR, and Portnoy DA. Patterns of pathogenesis: discrimination of pathogenic and nonpathogenic microbes by the innate immune system. Cell Host Microbe. (2009) 6:10–21. doi: 10.1016/j.chom.2009.06.007
17. Sui X, Liang X, Chen L, Guo C, Han W, Pan H, et al. Bacterial xenophagy and its possible role in cancer: A potential antimicrobial strategy for cancer prevention and treatment. Autophagy. (2017) 13:237–47. doi: 10.1080/15548627.2016.1252890
18. Wileman T. Autophagy as a defence against intracellular pathogens. Essays Biochem. (2013) 55:153–63. doi: 10.1042/bse0550153
19. Goebel W and Kuhn M. Bacterial replication in the host cell cytosol. Curr Opin Microbiol. (2000) 3:49–53. doi: 10.1016/s1369-5274(99)00050-8
20. Huang J and Brumell JH. Bacteria-autophagy interplay: a battle for survival. Nat Rev Microbiol. (2014) 12:101–14. doi: 10.1038/nrmicro3160
21. Sharma V, Verma S, Seranova E, Sarkar S, and Kumar D. Selective autophagy and xenophagy in infection and disease. Front Cell Dev Biol. (2018) 6:147. doi: 10.3389/fcell.2018.00147
22. Yin Z, Popelka H, Lei Y, Yang Y, and Klionsky DJ. The roles of ubiquitin in mediating autophagy. Cells. (2020) 9. doi: 10.3390/cells9092025
23. Nozawa T, Sano S, Minowa-Nozawa A, Toh H, Nakajima S, Murase K, et al. TBC1D9 regulates TBK1 activation through Ca(2+) signaling in selective autophagy. Nat Commun. (2020) 11:770. doi: 10.1038/s41467-020-14533-4
24. Cemma M, Kim PK, and Brumell JH. The ubiquitin-binding adaptor proteins p62/SQSTM1 and NDP52 are recruited independently to bacteria-associated microdomains to target Salmonella to the autophagy pathway. Autophagy. (2011) 7:341–5. doi: 10.4161/auto.7.3.14046
25. Fujita N and Yoshimori T. Ubiquitination-mediated autophagy against invading bacteria. Curr Opin Cell Biol. (2011) 23:492–7. doi: 10.1016/j.ceb.2011.03.003
26. Fujita N, Morita E, Itoh T, Tanaka A, Nakaoka M, Osada Y, et al. Recruitment of the autophagic machinery to endosomes during infection is mediated by ubiquitin. J Cell Biol. (2013) 203:115–28. doi: 10.1083/jcb.201304188
27. Zheng YT, Shahnazari S, Brech A, Lamark T, Johansen T, and Brumell JH. The adaptor protein p62/SQSTM1 targets invading bacteria to the autophagy pathway. J Immunol. (2009) 183:5909–16. doi: 10.4049/jimmunol.0900441
28. Hanada T, Noda NN, Satomi Y, Ichimura Y, Fujioka Y, Takao T, et al. The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J Biol Chem. (2007) 282:37298–302. doi: 10.1074/jbc.C700195200
29. Ichimura Y, Kirisako T, Takao T, Satomi Y, Shimonishi Y, Ishihara N, et al. A ubiquitin-like system mediates protein lipidation. Nature. (2000) 408:488–92. doi: 10.1038/35044114
30. Collins CA, De Mazière A, van Dijk S, Carlsson F, Klumperman J, and Brown EJ. Atg5-independent sequestration of ubiquitinated mycobacteria. PloS Pathogens. (2009) 5:e1000430. doi: 10.1371/journal.ppat.1000430
31. Mesquita FS, Thomas M, Sachse M, Santos AJ, Figueira R, and Holden DW. The Salmonella deubiquitinase SseL inhibits selective autophagy of cytosolic aggregates. PloS Pathogens. (2012) 8:e1002743. doi: 10.1371/journal.ppat.1002743
32. Rytkönen A and Holden DW. Bacterial interference of ubiquitination and deubiquitination. Cell Host Microbe. (2007) 1:13–22. doi: 10.1016/j.chom.2007.02.003
33. Zakrzewski AJ, Chajęcka-Wierzchowska W, Zadernowska A, and Podlasz P. Virulence characterization of Listeria monocytogenes, Listeria innocua, and Listeria welshimeri isolated from fish and shrimp using in vivo early zebrafish larvae models and molecular study. Pathogens. (2020) 9:1028. doi: 10.3390/pathogens9121028
34. Welch MD, Iwamatsu A, and Mitchison TJ. Actin polymerization is induced by Arp 2/3 protein complex at the surface of Listeria monocytogenes. Nature. (1997) 385:265–9. doi: 10.1038/385265a0
35. Cheng MI, Chen C, Engström P, Portnoy DA, and Mitchell G. Actin-based motility allows Listeria monocytogenes to avoid autophagy in the macrophage cytosol. Cell Microbiol. (2018) 20:e12854. doi: 10.1111/cmi.12854
36. Cemma M, Lam GY, Stöckli M, Higgins DE, and Brumell JH. Strain-specific interactions of listeria monocytogenes with the autophagy system in host cells. PloS One. (2015) 10:e0125856. doi: 10.1371/journal.pone.0125856
37. Mostowy S, Bonazzi M, Hamon MA, Tham TN, Mallet A, Lelek M, et al. Entrapment of intracytosolic bacteria by septin cage-like structures. Cell Host Microbe. (2010) 8:433–44. doi: 10.1016/j.chom.2010.10.009
38. Mostowy S and Cossart P. Autophagy and the cytoskeleton: new links revealed by intracellular pathogens. Autophagy. (2011) 7:780–2. doi: 10.4161/auto.7.7.15593
39. Ogawa M, Yoshimori T, Suzuki T, Sagara H, Mizushima N, and Sasakawa C. Escape of intracellular Shigella from autophagy. Science. (2005) 307:727–31. doi: 10.1126/science.1106036
40. Engström P, Burke TP, Mitchell G, Ingabire N, Mark KG, Golovkine G, et al. Evasion of autophagy mediated by Rickettsia surface protein OmpB is critical for virulence. Nat Microbiol. (2019) 4:2538–51. doi: 10.1038/s41564-019-0583-6
41. Kim J, Kundu M, Viollet B, and Guan K-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. (2011) 13:132–41. doi: 10.1038/ncb2152
42. Rosales-Reyes R, Alpuche-Aranda C, Ramírez-Aguilar M, Castro-Eguiluz AD, and Ortiz-Navarrete V. Survival of Salmonella enterica serovar Typhimurium within late endosomal-lysosomal compartments of B lymphocytes is associated with the inability to use the vacuolar alternative major histocompatibility complex class I antigen-processing pathway. Infect Immun. (2005) 73:3937–44. doi: 10.1128/IAI.73.7.3937-3944.2005
43. Knodler LA, Finlay BB, and Steele-Mortimer O. The Salmonella effector protein SopB protects epithelial cells from apoptosis by sustained activation of Akt. J Biol Chem. (2005) 280:9058–64. doi: 10.1074/jbc.M412588200
44. Luis L-B, Ana G-T, Carlos G-E, Abraham G-G, Iris E-G, Martha M-L, et al. Salmonella promotes its own survival in B cells by inhibiting autophagy. Cells. (2022) 11:2061. doi: 10.3390/cells11132061
45. Zhang W, Dong C, and Xiong S. Mycobacterial SapM hampers host autophagy initiation for intracellular bacillary survival via dephosphorylating Raptor. Iscience. (2024) 27. doi: 10.1016/j.isci.2024.109671
46. Zachari M and Ganley IG. The mammalian ULK1 complex and autophagy initiation. Essays Biochem. (2017) 61:585–96. doi: 10.1042/EBC20170021
47. Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. (2008) 30:214–26. doi: 10.1016/j.molcel.2008.03.003
48. Van Nostrand JL, Hellberg K, Luo E-C, Van Nostrand EL, Dayn A, Yu J, et al. AMPK regulation of Raptor and TSC2 mediate metformin effects on transcriptional control of anabolism and inflammation. Genes Dev. (2020) 34:1330–44. doi: 10.1101/gad.339895.120
49. Jeon YJ, Gil CH, Won J, Jo A, and Kim HJ. Symbiotic microbiome Staphylococcus aureus from human nasal mucus modulates IL-33-mediated type 2 immune responses in allergic nasal mucosa. BMC Microbiol. (2020) 20:1–12. doi: 10.1186/s12866-020-01974-6
50. Neumann Y, Bruns SA, Rohde M, Prajsnar TK, Foster SJ, and Schmitz I. Intracellular Staphylococcus aureus eludes selective autophagy by activating a host cell kinase. Autophagy. (2016) 12:2069–84. doi: 10.1080/15548627.2016.1226732
51. Wu H-M, Wang J, Zhang B, Fang L, Xu K, and Liu R-Y. CpG-ODN promotes phagocytosis and autophagy through JNK/P38 signal pathway in Staphylococcus aureus-stimulated macrophage. Life Sci. (2016) 161:51–9. doi: 10.1016/j.lfs.2016.07.016
52. Soong G, Paulino F, Wachtel S, Parker D, Wickersham M, Zhang D, et al. Methicillin-resistant Staphylococcus aureus adaptation to human keratinocytes. MBio. (2015) 6. doi: 10.1128/mbio.00289-00215
53. Knodler LA and Celli J. Eating the strangers within: host control of intracellular bacteria via xenophagy. Cell Microbiol. (2011) 13:1319–27. doi: 10.1111/j.1462-5822.2011.01632.x
54. Cai J, Li J, Zhou Y, Wang J, Li J, Cui L, et al. Staphylococcus aureus facilitates its survival in bovine macrophages by blocking autophagic flux. J Cell Mol Med. (2020) 24:3460–8. doi: 10.1111/jcmm.15027
55. O’Keeffe KM, Wilk MM, Leech JM, Murphy AG, Laabei M, Monk IR, et al. Manipulation of autophagy in phagocytes facilitates Staphylococcus aureus bloodstream infection. Infect Immun. (2015) 83:3445–57. doi: 10.1128/IAI.00358-15
56. Grosz M, Kolter J, Paprotka K, Winkler AC, Schäfer D, Chatterjee SS, et al. Cytoplasmic replication of S taphylococcus aureus upon phagosomal escape triggered by phenol-soluble modulin α. Cell Microbiol. (2014) 16:451–65. doi: 10.1111/cmi.2014.16.issue-4
57. Schnaith A, Kashkar H, Leggio SA, Addicks K, Kronke M, and Krut O. Staphylococcus aureus subvert autophagy for induction of caspase-independent host cell death. J Biol Chem. (2007) 282:2695–706. doi: 10.1074/jbc.M609784200
58. Mulcahy ME, O’Brien EC, O’Keeffe KM, Vozza EG, Leddy N, and McLoughlin RM. Manipulation of autophagy and apoptosis facilitates intracellular survival of Staphylococcus aureus in human neutrophils. Front Immunol. (2020) 11:565545. doi: 10.3389/fimmu.2020.565545
59. de Jong HK, Parry CM, van der Poll T, and Wiersinga WJ. Host–pathogen interaction in invasive salmonellosis. PLoS Pathog. (2012) 8(10):e1002933. doi: 10.1371/journal.ppat.1002933
60. Chen J, Xavier S, Moskowitz-Kassai E, Chen R, Lu CY, Sanduski K, et al. Cathepsin cleavage of sirtuin 1 in endothelial progenitor cells mediates stress-induced premature senescence. Am J Pathol. (2012) 180:973–83. doi: 10.1016/j.ajpath.2011.11.033
61. Owen KA, Meyer CB, Bouton AH, and Casanova JE. Activation of focal adhesion kinase by Salmonella suppresses autophagy via an Akt/mTOR signaling pathway and promotes bacterial survival in macrophages. PloS Pathogens. (2014) 10:e1004159. doi: 10.1371/journal.ppat.1004159
62. Cooper KG, Winfree S, Malik-Kale P, Jolly C, Ireland R, Knodler LA, et al. Activation of Akt by the bacterial inositol phosphatase, SopB, is wortmannin insensitive. PloS One. (2011) 6:e22260. doi: 10.1371/journal.pone.0022260
63. Cantó C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, et al. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. (2009) 458:1056–60. doi: 10.1038/nature07813
64. Lan F, Cacicedo JM, Ruderman N, and Ido Y. SIRT1 modulation of the acetylation status, cytosolic localization, and activity of LKB1: possible role in AMP-activated protein kinase activation. J Biol Chem. (2008) 283:27628–35. doi: 10.1074/jbc.M805711200
65. Ganesan R, Hos NJ, Gutierrez S, Fischer J, Stepek JM, Daglidu E, et al. Salmonella Typhimurium disrupts Sirt1/AMPK checkpoint control of mTOR to impair autophagy. PloS Pathogens. (2017) 13:e1006227. doi: 10.1371/journal.ppat.1006227
66. Barbuddhe SB and Chakraborty T. Listeria as an enteroinvasive gastrointestinal pathogen. Curr Top Microbiol Immunol. (2009) 337:173–95. doi: 10.1007/978-3-642-01846-6_6
67. Cossart P. Illuminating the landscape of host–pathogen interactions with the bacterium Listeria monocytogenes. Proc Natl Acad Sci. (2011) 108:19484–91. doi: 10.1073/pnas.1112371108
68. Cong Z, Xiong Y, Lyu L, Fu B, Guo D, Sha Z, et al. The relationship between Listeria infections and host immune responses: Listeriolysin O as a potential target. BioMed Pharmacother. (2024) 171:116129. doi: 10.1016/j.biopha.2024.116129
69. Schnupf P and Portnoy DA. Listeriolysin O: a phagosome-specific lysin. Microbes Infect. (2007) 9:1176–87. doi: 10.1016/j.micinf.2007.05.005
70. Mitchell G, Ge L, Huang Q, Chen C, Kianian S, Roberts MF, et al. Avoidance of autophagy mediated by PlcA or ActA is required for Listeria monocytogenes growth in macrophages. Infect Immun. (2015) 83:2175–84. doi: 10.1128/IAI.00110-15
71. Tattoli I, Sorbara MT, Yang C, Tooze SA, Philpott DJ, and Girardin SE. Listeria phospholipases subvert host autophagic defenses by stalling pre-autophagosomal structures. EMBO J. (2013) 32:3066–78. doi: 10.1038/emboj.2013.234
72. Radtke AL, Anderson KL, Davis MJ, DiMagno MJ, Swanson JA, and O’Riordan MX. Listeria monocytogenes exploits cystic fibrosis transmembrane conductance regulator (CFTR) to escape the phagosome. Proc Natl Acad Sci. (2011) 108:1633–8. doi: 10.1073/pnas.1013262108
73. Birmingham CL, Higgins DE, and Brumell JH. Avoiding death by autophagy: interactions of Listeria monocytogenes with the macrophage autophagy system. Autophagy. (2008) 4:368–71. doi: 10.4161/auto.5594
74. Gong L, Cullinane M, Treerat P, Ramm G, Prescott M, Adler B, et al. The Burkholderia pseudomallei type III secretion system and BopA are required for evasion of LC3-associated phagocytosis. PloS One. (2011) 6:e17852. doi: 10.1371/journal.pone.0017852
75. Yu D, Yin Z, Jin Y, Zhou J, Ren H, Hu M, et al. Evolution of bopA gene in burkholderia: A case of convergent evolution as a mechanism for bacterial autophagy evasion. BioMed Res Int. (2016) 2016:6745028. doi: 10.1155/2016/6745028
76. Kayath CA, Hussey S, Nagra K, Philpott D, and Allaoui A. Escape of intracellular Shigella from autophagy requires binding to cholesterol through the type III effector, IcsB. Microbes Infect. (2010) 12:956–66. doi: 10.1016/j.micinf.2010.06.006
77. Pei J and Grishin NV. The Rho GTPase inactivation domain in Vibrio cholerae MARTX toxin has a circularly permuted papain-like thiol protease fold. Proteins: Structure Function Bioinf. (2009) 77:413–9. doi: 10.1002/prot.22447
78. De Chastellier C and Thilo L. Cholesterol depletion in Mycobacterium avium-infected macrophages overcomes the block in phagosome maturation and leads to the reversible sequestration of viable mycobacteria in phagolysosome-derived autophagic vacuoles. Cell Microbiol. (2006) 8:242–56. doi: 10.1111/j.1462-5822.2005.00617.x
79. Stevens MP, Wood MW, Taylor LA, Monaghan P, Hawes P, Jones PW, et al. An Inv/Mxi-Spa-like type III protein secretion system in Burkholderia pseudomallei modulates intracellular behaviour of the pathogen. Mol Microbiol. (2002) 46:649–59. doi: 10.1046/j.1365-2958.2002.03190.x
80. Pilatz S, Breitbach K, Hein N, Fehlhaber B, Schulze J, Brenneke B, et al. Identification of Burkholderia pseudomallei genes required for the intracellular life cycle and in vivo virulence. Infect Immun. (2006) 74:3576–86. doi: 10.1128/IAI.01262-05
81. Hu ZQ, Li Q, Hu ZH, Liu HC, Rao CL, Zhang MJ, et al. MicroRNA-146a inhibits autophagy to maintain the intracellular survival of Burkholderia pseudomallei by targeting LIPA. Microb Pathog. (2021) 158:104969. doi: 10.1016/j.micpath.2021.104969
82. Li Q, Fang Y, Zhu P, Ren CY, Chen H, Gu J, et al. Burkholderia pseudomallei survival in lung epithelial cells benefits from miRNA-mediated suppression of ATG10. Autophagy. (2015) 11:1293–307. doi: 10.1080/15548627.2015.1058474
83. Roop RM, Barton IS, Hopersberger D, and Martin DW. Uncovering the hidden credentials of Brucella virulence. Microbiol Mol Biol Rev. (2021) 85. doi: 10.1128/mmbr.00021-00019
84. Gorvel JP and Moreno E. Brucella intracellular life: from invasion to intracellular replication. Veterinary Microbiol. (2002) 90:281–97. doi: 10.1016/S0378-1135(02)00214-6
85. Li J, Qi L, Diao Z, Zhang M, Li B, Zhai Y, et al. Brucella btpB manipulates apoptosis and autophagic flux in RAW264. 7 cells. Int J Mol Sci. (2022) 23:14439. doi: 10.3390/ijms232214439
86. Zhang J, Li M, Li Z, Shi J, Zhang Y, Deng X, et al. Deletion of the type IV secretion system effector VceA promotes autophagy and inhibits apoptosis in Brucella-infected human trophoblast cells. Curr Microbiol. (2019) 76:510–9. doi: 10.1007/s00284-019-01651-6
87. Nano FE, Zhang N, Cowley SC, Klose KE, Cheung KK, Roberts MJ, et al. A Francisella tularensis pathogenicity island required for intramacrophage growth. J Bacteriol. (2004) 186:6430–6. doi: 10.1128/JB.186.19.6430-6436.2004
88. Steele S, Brunton J, Ziehr B, Taft-Benz S, Moorman N, and Kawula T. Francisella tularensis harvests nutrients derived via ATG5-independent autophagy to support intracellular growth. PloS Pathogens. (2013) 9:e1003562. doi: 10.1371/journal.ppat.1003562
89. Checroun C, Wehrly TD, Fischer ER, Hayes SF, and Celli J. Autophagy-mediated reentry of Francisella tularensis into the endocytic compartment after cytoplasmic replication. Proc Natl Acad Sci. (2006) 103:14578–83. doi: 10.1073/pnas.0601838103
90. Edwards JA, Rockx-Brouwer D, Nair V, and Celli J. Restricted cytosolic growth of Francisella tularensis subsp. tularensis by IFN-γ activation of macrophages. Microbiology. (2010) 156:327–39. doi: 10.1099/mic.0.031716-0
91. Myeni S, Child R, Ng TW, Kupko JJ III, Wehrly TD, Porcella SF, et al. Brucella modulates secretory trafficking via multiple type IV secretion effector proteins. PloS Pathogens. (2013) 9:e1003556. doi: 10.1371/journal.ppat.1003556
92. O’Callaghan D, Cazevieille C, Allardet-Servent A, Boschiroli ML, Bourg G, Foulongne V, et al. A homologue of the Agrobacterium tumefaciens VirB and Bordetella pertussis Ptl type IV secretion systems is essential for intracellular survival of Brucella suis. Mol Microbiol. (1999) 33:1210–20. doi: 10.1046/j.1365-2958.1999.01569.x
93. Starr T, Ng TW, Wehrly TD, Knodler LA, and Celli J. Brucella intracellular replication requires trafficking through the late endosomal/lysosomal compartment. Traffic. (2008) 9:678–94. doi: 10.1111/j.1600-0854.2008.00718.x
94. Celli J, Salcedo SP, and Gorvel J-P. Brucella coopts the small GTPase Sar1 for intracellular replication. Proc Natl Acad Sci. (2005) 102:1673–8. doi: 10.1073/pnas.0406873102
95. Fugier E, Salcedo SP, de Chastellier C, Pophillat M, Muller A, Arce-Gorvel V, et al. The glyceraldehyde-3-phosphate dehydrogenase and the small GTPase Rab 2 are crucial for Brucella replication. PloS Pathogens. (2009) 5:e1000487. doi: 10.1371/journal.ppat.1000487
96. Wu J and Kaufman R. From acute ER stress to physiological roles of the unfolded protein response. Cell Death Differentiation. (2006) 13:374–84. doi: 10.1038/sj.cdd.4401840
97. de Jong MF, Starr T, Winter MG, den Hartigh AB, Child R, Knodler LA, et al. Sensing of bacterial type IV secretion via the unfolded protein response. MBio. (2013) 4. doi: 10.1128/mbio.00418-00412
98. Smith JA, Khan M, Magnani DD, Harms JS, Durward M, Radhakrishnan GK, et al. Brucella induces an unfolded protein response via TcpB that supports intracellular replication in macrophages. PloS Pathogens. (2013) 9:e1003785. doi: 10.1371/journal.ppat.1003785
99. Taguchi Y, Imaoka K, Kataoka M, Uda A, Nakatsu D, Horii-Okazaki S, et al. Yip1A, a novel host factor for the activation of the IRE1 pathway of the unfolded protein response during Brucella infection. PloS Pathogens. (2015) 11:e1004747. doi: 10.1371/journal.ppat.1004747
100. Bachmann NL, Polkinghorne A, and Timms P. Chlamydia genomics: providing novel insights into chlamydial biology. Trends Microbiol. (2014) 22:464–72. doi: 10.1016/j.tim.2014.04.013
101. Stelzner K, Vollmuth N, and Rudel T. Intracellular lifestyle of Chlamydia trachomatis and host–pathogen interactions. Nat Rev Microbiol. (2023) 21:448–62. doi: 10.1038/s41579-023-00860-y
102. Al-Younes HM, Brinkmann V, and Meyer TF. Interaction of Chlamydia trachomatis serovar L2 with the host autophagic pathway. Infect Immun. (2004) 72:4751–62. doi: 10.1128/IAI.72.8.4751-4762.2004
103. Rucks EA. Type III secretion in chlamydia. Microbiol Mol Biol Rev. (2023) 87:e00034–00023. doi: 10.1128/mmbr.00034-23
104. Shaw EI and Voth DE. Coxiella burnetii: a pathogenic intracellular acidophile. Microbiology. (2019) 165:1–3. doi: 10.1099/mic.0.000707
105. Gutierrez MG, Vázquez CL, Munafó DB, Zoppino FC, Berón W, Rabinovitch M, et al. Autophagy induction favours the generation and maturation of the Coxiella-replicative vacuoles. Cell Microbiol. (2005) 7:981–93. doi: 10.1111/j.1462-5822.2005.00527.x
106. Padmanabhan B, Fielden LF, Hachani A, Newton P, Thomas DR, Cho H-J, et al. Biogenesis of the spacious Coxiella-containing vacuole depends on host transcription factors TFEB and TFE3. Infect Immun. (2020) 88. doi: 10.1128/iai.00534-00519
107. Larson CL, Sandoz KM, Cockrell DC, and Heinzen RA. Noncanonical inhibition of mTORC1 by Coxiella burnetii promotes replication within a phagolysosome-like vacuole. MBio. (2019) 10. doi: 10.1128/mbio.02816-02818
108. Latomanski EA and Newton HJ. Interaction between autophagic vesicles and the Coxiella-containing vacuole requires CLTC (clathrin heavy chain). Autophagy. (2018) 14:1710–25. doi: 10.1080/15548627.2018.1483806
109. Winchell CG, Graham JG, Kurten RC, and Voth DE. Coxiella burnetii type IV secretion-dependent recruitment of macrophage autophagosomes. Infect Immun. (2014) 82:2229–38. doi: 10.1128/IAI.01236-13
110. Kohler LJ, Reed SR, Sarraf SA, Arteaga DD, Newton HJ, and Roy CR. Effector protein Cig2 decreases host tolerance of infection by directing constitutive fusion of autophagosomes with the Coxiella-containing vacuole. MBio. (2016) 7. doi: 10.1128/mbio.01127-01116
111. Martinez E, Allombert J, Cantet F, Lakhani A, Yandrapalli N, Neyret A, et al. Coxiella burnetii effector CvpB modulates phosphoinositide metabolism for optimal vacuole development. Proc Natl Acad Sci. (2016) 113:E3260–9. doi: 10.1073/pnas.1522811113
112. Siadous FA, Cantet F, Van Schaik E, Burette M, Allombert J, Lakhani A, et al. Coxiella effector protein CvpF subverts RAB26-dependent autophagy to promote vacuole biogenesis and virulence. Autophagy. (2021) 17:706–22. doi: 10.1080/15548627.2020.1728098
113. Jin RU and Mills JC. RAB26 coordinates lysosome traffic and mitochondrial localization. J Cell Sci. (2014) 127:1018–32. doi: 10.1242/jcs.138776
114. Lemarignier M and Pizarro-Cerdá J. Autophagy and intracellular membrane trafficking subversion by pathogenic yersinia species. Biomolecules. (2020) 10:1637. doi: 10.3390/biom10121637
115. Valencia Lopez MJ, Schimmeck H, Gropengießer J, Middendorf L, Quitmann M, Schneider C, et al. Activation of the macroautophagy pathway by Yersinia enterocolitica promotes intracellular multiplication and egress of yersiniae from epithelial cells. Cell Microbiol. (2019) 21:e13046. doi: 10.1111/cmi.13046
116. Moreau K, Lacas-Gervais S, Fujita N, Sebbane F, Yoshimori T, Simonet M, et al. Autophagosomes can support Yersinia pseudotuberculosis replication in macrophages. Cell Microbiol. (2010) 12:1108–23. doi: 10.1111/j.1462-5822.2010.01456.x
117. Wong K-W and Isberg RR. Emerging views on integrin signaling via Rac1 during invasin-promoted bacterial uptake. Curr Opin Microbiol. (2005) 8:4–9. doi: 10.1016/j.mib.2004.12.009
118. Muñoz-Sánchez S, van der Vaart M, and Meijer AH. Autophagy and Lc3-associated phagocytosis in zebrafish models of bacterial infections. Cells. (2020) 9:2372.
119. van der Vaart M, Korbee CJ, Lamers GE, Tengeler AC, Hosseini R, Haks MC, et al. The DNA damage-regulated autophagy modulator DRAM1 links mycobacterial recognition via TLR-MYD88 to autophagic defense. Cell Host & Microbe. (2014) 15:753–67.
120. Chen J, Liu N, Zhang H, Zhao Y, and Cao X. The effects of Aeromonas hydrophila infection on oxidative stress, nonspecific immunity, autophagy, and apoptosis in the common carp. Dev Comp Immunol. (2020) 105:103587.
121. Yang M, Lu Z, Li F, Shi F, Zhan F, Zhao L, et al. Escherichia coli induced ferroptosis in red blood cells of grass carp (Ctenopharyngodon idella). Fish Shellfish Immunol. (2021) 112:159–167.
122. Yin L, Lv M, Qiu X, Wang X, Zhang A, Yang K, et al. IFN-γ manipulates NOD1-mediated interaction of autophagy and edwardsiella piscicida to augment intracellular clearance in fish. J Immunol. (2021) 207:1087–98.
123. Guo S, Zeng M, Wang Z, Zhao L, Fan Y, Shi Q, et al. Characterization and expression profiles of cGAS (cyclic GMP-AMP synthase) and STING (stimulator of interferon) genes in various immune tissues of hybrid yellow catfish under bacterial infections. Aquaculture Reports. (2024) 37:102238
124. Zhang Z, Guan X, and Sun L. A novel teleost microRNA regulates autophagy and NF-κB activation during bacterial infection. Fish Shellfish Immunol. (2023) 137:108778.
125. Zhao Z, Zhang L, Ren C, Zhao J, Chen C, Jiang X, et al. Autophagy is induced by the type III secretion system of Vibrio alginolyticus in several mammalian cell lines. Arch Microbiol. (2011) 193:53–61.
126. Al Azzaz J, Rieu A, Aires V, Delmas D, Chluba J, Winckler P, et al. Resveratrol-induced xenophagy promotes intracellular bacteria clearance in intestinal epithelial cells and macrophages. Front Immunol. (2019) 9:3149. doi: 10.3389/fimmu.2018.03149
127. Ogawa M and Sasakawa C. Bacterial evasion of the autophagic defense system. Curr Opin Microbiol. (2006) 9:62–8. doi: 10.1016/j.mib.2005.12.007
128. Liu J, Ren J, Wang D, Wang Z, Ma X, and Zhou H. Edwardsiella piscicida promotes mitophagy to escape autophagy-mediated antibacterial defense in teleost monocytes/macrophages. Aquaculture. (2025) 596:741784. doi: 10.1016/j.aquaculture.2024.741784
129. Chen W, Xu Q, Chang M, Nie P, and Peng K. Molecular characterization and expression analysis of nuclear oligomerization domain proteins NOD1 and NOD2 in grass carp Ctenopharyngodon idella. Fish Shellfish Immunol. (2010) 28:18–29. doi: 10.1016/j.fsi.2009.09.012
130. Rao PSS, Lim TM, and Leung KY. Opsonized virulent Edwardsiella tarda strains are able to adhere to and survive and replicate within fish phagocytes but fail to stimulate reactive oxygen intermediates. Infect Immun. (2001) 69:5689–97. doi: 10.1128/IAI.69.9.5689-5697.2001
131. Joshi AD and Swanson MS. Secrets of a successful pathogen: Legionella resistance to progression along the autophagic pathway. Front Microbiol. (2011) 2:138. doi: 10.3389/fmicb.2011.00138
132. Lerena MC and Colombo MI. Mycobacterium marinum induces a marked LC3 recruitment to its containing phagosome that depends on a functional ESX-1 secretion system. Cell Microbiol. (2011) 13:814–35. doi: 10.1111/j.1462-5822.2011.01581.x
133. Lopez-Jimenez AT, Cardenal-Munoz E, Leuba F, Gerstenmaier L, Barisch C, Hagedorn M, et al. The ESCRT and autophagy machineries cooperate to repair ESX-1-dependent damage at the Mycobacterium-containing vacuole but have opposite impact on containing the infection. PloS Pathogens. (2018) 14:e1007501. doi: 10.1371/journal.ppat.1007501
134. Higa N, Toma C, Koizumi Y, Nakasone N, Nohara T, Masumoto J, et al. Vibrio parahaemolyticus effector proteins suppress inflammasome activation by interfering with host autophagy signaling. PloS Pathogens. (2013) 9:e1003142. doi: 10.1371/journal.ppat.1003142
135. Chen G-H, Zhao T, Wei X-L, Zhang D-G, Zhuo M-Q, and Luo Z. miR-101b Regulates lipid deposition and metabolism of primary hepatocytes in teleost yellow catfish Pelteobagrus fulvidraco. Genes. (2020) 11:861. doi: 10.3390/genes11080861
136. Chen G, Wu K, Zhao T, Ling S, Liu W, and Luo Z. miR-144 mediates high fat–induced changes of cholesterol metabolism via direct regulation of C/EBPα in the liver and isolated hepatocytes of yellow catfish. J Nutr. (2020) 150:464–74. doi: 10.1093/jn/nxz282
137. Guan X-l, Zhang B-C, and Sun L. Japanese flounder pol-miR-3p-2 suppresses Edwardsiella tarda infection by regulation of autophagy via p53. Dev Comp Immunol. (2020) 103:103531. doi: 10.1016/j.dci.2019.103531
138. Prajsnar TK, Serba JJ, Dekker BM, Gibson JF, Masud S, Fleming A, et al. The autophagic response to Staphylococcus aureus provides an intracellular niche in neutrophils. Autophagy. (2021) 17:888–902. doi: 10.1080/15548627.2020.1739443
139. Fodor E, Sigmond T, Ari E, Lengyel K, Takács-Vellai K, Varga M, et al. Methods to study autophagy in zebrafish. Methods Enzymol. (2017) 588:467–96. doi: 10.1016/bs.mie.2016.10.028
140. Moss JJ, Hammond CL, and Lane JD. Zebrafish as a model to study autophagy and its role in skeletal development and disease. Histochem Cell Biol. (2020) 154:549–64. doi: 10.1007/s00418-020-01917-2
141. Varshney GK, Sood R, and Burgess SM. Understanding and editing the zebrafish genome. Adv Genet. (2015) 92:1–52. doi: 10.1016/bs.adgen.2015.09.002
Keywords: autophagy, xenophagy, immunology, teleost, bacteria
Citation: Wang Q, Xu H, Jin J, Yang Y, Jänsch L and Li S (2025) The battle between bacterial infection and autophagy in aquatic animals. Front. Immunol. 16:1614182. doi: 10.3389/fimmu.2025.1614182
Received: 18 April 2025; Accepted: 04 June 2025;
Published: 23 June 2025.
Edited by:
Quanyuan Wan, Lerner Research Institute, United StatesCopyright © 2025 Wang, Xu, Jin, Yang, Jänsch and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Senlin Li, c2VubGluX2xpQHp6dS5lZHUuY24=