 Maria Lteif
Maria Lteif Marc Pallardy
Marc Pallardy Isabelle Turbica
Isabelle Turbica- Université Paris-Saclay, INSERM, Inflammation, Microbiome and Immunosurveillance, Orsay, France
Therapeutic antibodies have revolutionized the treatment of many diseases. However, their safety and efficacy are often altered by their immunogenicity, as many patients frequently develop anti-drug antibodies. Dendritic cells (DCs) are the most potent antigen-presenting cells of the immune system. DCs initiate the immunogenic adaptive immune response by internalizing therapeutic antibodies using different pathways and receptors, leading to antigen presentation to T-cells. Recently, studies have shown that the uptake of antibodies by immune cells could contribute to their immunogenicity. This review will present in detail the different DC internalization mechanisms and then discuss the impact of therapeutic antibodies’ properties and aggregation on their uptake by DCs and, therefore, their immunogenicity. We will also highlight cellular models and strategies used to evaluate antibodies’ internalization. Addressing the uptake of antibodies by DCs could help to predict the risk of immunogenicity and to develop mitigation strategies.
1 Introduction
Therapeutic antibodies (Abs) have emerged as the fastest-growing class of biotherapeutics and are used to treat and detect many diseases (1). However, they can elicit unwanted immunogenicity in a subset of patients, leading to the production of anti-drug Abs (ADAs). This implicates the development of a T-cell-dependent immune response. This response is initiated by the internalization of Abs by antigen-presenting cells (APCs), such as dendritic cells (DCs), followed by the processing of these Abs into peptides for presentation on major histocompatibility (MHC) class II molecules on the cell surface. These peptides are recognized by cognate T cells, resulting in their activation, which in turn activates antigen-specific B cells that mature into Ab-secreting plasma cells (2). Recently, measurements of Ab internalization in DCs have been proposed as a tool for immunogenicity risk assessment (3–6). These assays have shown that the rate of Ab internalization can influence immunogenicity risk (4, 6–8). However, different factors contribute to ADA generation, including the intrinsic properties of the therapeutic protein, the therapeutic regimen, and the patient-specific characteristics such as its immune status or genetic background (9).
Understanding the factors influencing Ab internalization into DCs could help optimizing their design and reducing their immunogenic risk. This review aims to explore the structure and function of Abs, the mechanisms of Abs’ internalization by DCs, and the impact of Abs’ properties on this process. By examining these aspects, we will highlight how these factors influence the immune response and discuss the current methods for evaluating Abs’ internalization in vitro. Finally, we will propose an evidence-based strategy for evaluating the immunogenicity risk of therapeutic Abs candidates, including internalization assessment.
Immunoglobulin (Ig)-based therapeutic Abs have dominated the biotherapeutic field mainly due to their structural and functional properties. However, alternative Abs formats have been developed over the past two decades. These include smaller or engineered constructs designed to improve biodistribution, enhance specificity, or reduce side effects. Importantly, the current preclinical and clinical landscape now includes a wide array of structurally and functionally diverse Ab-based molecules such as single-domain Abs/nanobodies, multispecific Abs (e.g., bispecific, trispecifics), polymeric Abs such as IgA dimers and IgM pentamers, Ab-drug conjugates) (10). Numerous novel Abs are currently being investigated in clinical trials, and the number of monoclonal Abs receiving marketing authorization continues to grow. This evolving landscape is reviewed annually in the “Antibodies to Watch” series (1).
Abs or Igs mediate the humoral response and are most effective against extracellular pathogens (11). They are typically composed of light chains (LCs) and heavy chains (HCs). There are two LC isotypes, kappa (κ) and lambda (λ), and five HC isotypes, gamma (γ), alpha (α), mu (µ), delta (δ), and epsilon (ϵ) which define the Ab isotype (IgG: γ, IgA: α, IgM: µ, IgD: δ, IgE: ϵ) and confer distinct effector functions (12, 13). Importantly, the overall structure and composition of Ig classes differ significantly. While IgG, IgA (monomeric), IgD, and IgE exist as monomers composed of two LCs and two HCs, IgM typically forms a pentameric structure (and occasionally a hexamer), comprising five Ig units linked by disulfide bonds and a joining (J) chain. This results in a molecule containing 10 HC and LC and confers high valency and strong avidity for antigens. Dimeric forms of IgA also include a J chain and are predominant in mucosal secretions (12, 14). Both HC and LC are composed of constant and variable domains. The amino-terminal variable regions of the HC and LC (VH and VL, respectively) together form the antigen-binding site, shaped by three hypervariable loops known as complementarity-determining regions (CDRs 1, 2, and 3), which are separated by conserved beta-sheet framework regions (FRs). The constant region determines the Ab’s effector functions and varies by isotype. The γ, α, and δ heavy chains contain three constant domains (CH1, CH2, and CH3) and a hinge region, which provides flexibility and enhances antigen binding and cross-linking. In contrast, the µ and ϵ heavy chains include a fourth constant domain (CH4), which replaces the hinge region and contributes to a more rigid structure (15).
Based on enzymatic cleavage, the Ab can be divided into functionally distinct regions: two fragment antigen-binding (Fab) regions and one fragment crystallizable (Fc) region. The Fab region contains the VH, VL, CH1, and CL domains and mediates antigen recognition, while the Fc region—comprising CH2 and CH3 domains (and CH4 in IgM and IgE)—is responsible for interacting with immune effector molecules (16). These functions are determined by their interactions with different binding partners: antigens or therapeutic targets through their Fab region, Fc receptors (FcR) for IgG (FcγRs), the neonatal Fc receptor (FcRn), and the complement system via their Fc portion (17). The main mechanisms of action of Abs include antagonism or agonism for a soluble ligand or receptor and blockade of cell–cell interaction. Ab-receptor or target binding at the cell surface can thus lead to the endocytosis of the formed complex (18, 19). On the other hand, the Fc-mediated effector functions comprise Ab-dependent cell-mediated cytotoxicity (ADCC), Ab-dependent cellular phagocytosis (ADCP), and complement-dependent cytotoxicity (CDC) (20). NK cells and macrophages express FcγRIIIa (CD16a) on their surface, recognizing Abs’ Fc portion and are the major mediators of ADCC, resulting in target cell killing. This mechanism plays a key role in the mode of action of therapeutic monoclonal Abs such as rituximab (anti-CD20) and cetuximab (anti-EGFR) in immunotherapy (21).
2 Mechanisms of internalization by dendritic cells
The mechanisms of therapeutic Ab internalization by DCs depend on the physico-chemical properties of the Abs, which influence the rate of uptake and the subsequent immune responses. This internalization process is also significantly affected by the expression levels of the target antigens on the DC surface. Higher antigen density typically facilitates more efficient Ab binding and uptake, thereby enhancing antigen processing and presentation, critical steps for initiating effective immune responses. Recent studies suggested a link between mAb’s internalization rate and their immunogenicity (4, 22). Before considering the relevance of different endocytic pathways for Abs by APCs, and particularly DCs, we will first summarize the internalization pathways utilized by DCs.
Internalization pathways include macropinocytosis, clathrin-mediated endocytosis (CME), clathrin-independent dynamin-dependent endocytosis or fast endophilin-mediated endocytosis (FEME), clathrin-independent carrier/glycosylphosphatidylinositol-anchored protein-enriched early endocytic compartment (CLIC/GEEC), phagocytosis, and caveolae-dependent endocytosis. These pathways have been described in detail in a previous review (3). Macropinocytosis, CME, and phagocytosis, the three main mechanisms implicated in DC internalization of therapeutic Abs, are described below and summarized in Figure 1. The FEME, CLIC/GEEC, and caveolae-dependent pathways are less described in the literature as involved in Abs uptake. Studies suggest their role in oxidized low-density lipoprotein (23), polyomavirus particles (24), and Simian Virus 40 uptake (23) by immune cells.
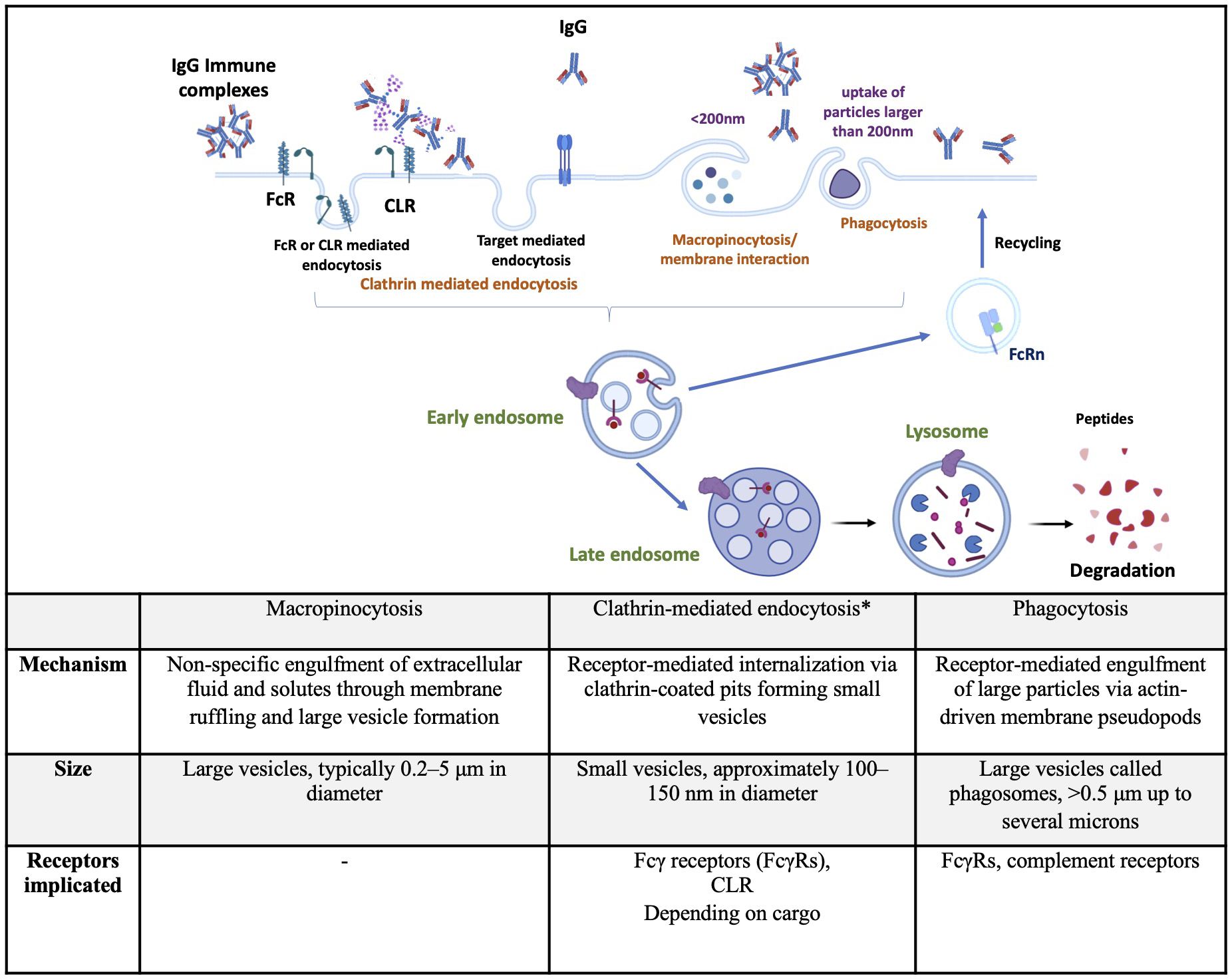
Figure 1. Summary of endocytic pathways implicated in therapeutic antibodies internalization by dendritic cells. Top: Schematic representation of major endocytic routes used by dendritic cells to internalize IgG or immune complexes, including clathrin-mediated endocytosis, macropinocytosis, and phagocytosis. Bottom: Tabular summary comparing the key features of each pathway, including the mechanism of internalization, vesicle size, and receptors involved. *includes target-mediated endocytosis. CLR, C-type lectine receptor; FcR, Fc receptor; FcRn, neonatal Fc receptor;IgG, immunoglobulin G. Created using BioRender.com.
Macropinocytosis occurs constitutively in DCs and mediates the non-specific uptake of soluble antigens. It is initiated by the polymerization of actin at the plasma membrane, leading to the formation of ruffles and large endocytic vesicles known as macropinosomes (25). This process is regulated by the small GTPases Rac1 and Cdc42, involved in actin polymerization, (26) modifications of the submembranous pH, and the activity of Na+/H+ exchangers (27). Macropinosomes destined for antigen presentation undergo a defined sequence of maturation steps involving acidification, fusion with endosomal compartments, and enzymatic degradation to process their contents, a series of events detailed later. Briefly, they might recycle back to the plasma membrane or undergo a series of changes that ultimately lead to their interaction and fusion with endolysosomal compartments, where their cargoes are degraded by hydrolytic enzymes (28). Since DCs are specialized in antigen capture, considerable research effort has been expanded to study the mechanisms of antigen uptake in these cells. Many of these studies use pharmacological inhibitors to study macropinocytosis. However, these inhibitors are notoriously non-specific, which limits the understanding of the mechanisms underlying constitutive macropinocytosis (29). For instance, amiloride derivatives inhibit macropinocytosis by impairing Na+/H+ exchanger activity, which is important for actin remodeling mediated by Rho family GTPases (30). Similarly, wortmannin, which inhibits PI3 kinase activity, also suppresses actin polymerization, a process involved in other internalization mechanisms, making the interpretation of these results more complex (25, 31).
CME is the most studied and well-characterized endocytic mechanism, as it constitutes the major endocytic pathway in mammalian cells. This pathway is essential for regulating cell signaling and mediates nutrient uptake (32). Upon binding, activated receptors are phosphorylated by G protein-receptor kinases, which in turn recruit adaptor proteins and initiate a cascade leading to the formation of clathrin-coated pits. These pits rapidly invaginate and pinch off to form clathrin-coated vesicles, then outline pathways of uncoating, transport, and fusion with early endosomes to deliver their cargo (33, 34). DCs express several receptors mediating CME, such as C-type lectin receptors (CLR), sialic acid binding immunoglobulin (Ig)-like lectins (Siglecs), death receptors (Fas, TNF, and TRAIL receptors), scavenger receptors (CD36, SR-A), and FcR (35–37). In this review, we will mainly focus on CLR and FcR, as the literature indicates that these are the most implicated receptors in the cellular uptake of therapeutic Abs. Siglecs recognize sialic acid via their extracellular domain (38). Scavenger receptors bind and internalize modified low-density lipoproteins, and death receptors and damage-associated molecular patterns that are released during different cell death mechanisms will not be further detailed in this review, as they are not implicated in Abs uptake. CLRs are a diverse family of soluble and transmembrane proteins that bind carbohydrates in a calcium-dependent manner using a conserved carbohydrate recognition domain (CRD) (39). Type I transmembrane CLRs possess various CRD and comprise the mannose receptor family, such as DEC-205 (CD205 or LY75) and the macrophage-mannose receptor (MMR or CD206), both implicated in antigen uptake. Type II transmembrane CLRs have a single CRD domain and include DC-associated C-type lectin 1 (Dectin-1 or CLCE7A), Dectin-2 (CLEC6A), macrophage-inducible C-type lectin (Mincle or CLEC4A), the dendritic-cell-specific ICAM3-grabbing nonintegrin (DC-SIGN or CD209), and DC NK lectin group receptor-1 (DNGR-1 or CLEC9A). These receptors are involved in pathogen recognition and the shaping of innate immune responses (40). MMR (CD206) and DC-SIGN (CD209) are some of the major mannose-binding CLRs in human DCs (41, 42). CD206 and CD205 recognize glycans that terminate in mannose, fucose, or N-acetylglucosamine (43, 44). Whereas these two receptors bind preferentially to a single residue, CD209 also binds terminal high-mannose (HM) glycans (45). Ig’s glycosylation (N-and O-glycosylation) is one of the major co‐translational modifications and/or post‐translational modifications (PTMs) (46). FcRs that recognize the Fc part of Abs are classified according to the Ig isotype: FcγR and neonatal FcRn bind IgG, FcαR binds IgA, FcϵR binds IgE, FcμR binds IgM, and FcδR binds IgD (47). Since IgG are the main used isotype among Abs in the therapeutic field (48), we will mainly focus on FcγR and FcRn. FcγRs are broadly classified as activating or inhibitory, depending on the signaling properties of their intracellular domain. In humans, activating FcγRs include FcγRI, FcγRIIa, FcγRIIc, and FcγRIIIa, signaling through immunoreceptor tyrosine activating motifs (ITAMs). FcγRIIb represents the only inhibitory FcγR, signaling through an immunoreceptor tyrosine inhibitory motif (ITIM) in its cytoplasmic region (49). In vitro, APC expresses a wide range of FcγRs with a potential role in antigen presentation. Human monocyte-derived DCs (moDCs)s express mainly the activating FcγRIIa and the inhibitory FcγRIIb receptors, and to a lesser extent, FcγRI and FcγRIIIa. Human in vitro generated macrophages express all FcγRs with high levels of FcγRIIa (50). FcγRs can also be classified by their binding affinities to human IgG: FcγRI binds monomeric IgG with high affinity, whereas FcγRII and FcγRIII bind multimeric IgG or immune complexes (ICs) with very high affinity (51), and also Abs aggregates. Binding of IgGs to activating FcγR leads to ITAM phosphorylation, activation of the Src-Syk pathway, FγR-IgG ICs’ internalization, and routing to lysosomes. Syk activates downstream signaling molecules, primarily increasing calcium flux and the activation of protein kinase C (52) These intracellular changes lead to activation of Rho GTPases and actin remodeling, which is critical for IC’s internalization (53). The studies on therapeutic Abs internalization through the FcRs are detailed in the next section.
The FcRn binds to the Fc portion at acidic pH in the early endosome and recycles IgGs to the plasma membrane to be released at neutral pH (54), rescuing them from lysosomal degradation and extending their serum half-life (55). At higher concentrations of IgG, FcRn becomes saturated, leading to a smaller proportion of IgG being routed to the lysosomal compartments. The latter trafficking also occurs when FcRn binds to multimeric IgG, such as aggregates or ICs (54). Importantly, FcRn saturation also affects the processing of Abs in DCs’ endolysosomal compartments (56). If the FcRn-mediated IgG recycling is saturated, a greater proportion of IgG molecules is directed to lysosomal degradation within DCs (54, 57). This enhances antigen presentation and may increase immunogenicity by promoting T-cell activation against the therapeutic Ab. This mechanism highlights a key consideration in clinical Ab therapy, where FcRn saturation can influence both pharmacokinetics and immune responses.
Phagocytosis consists of the recognition of particles larger than 0.5µm in diameter by phagocytic receptors and their uptake into a plasma membrane-derived vesicle, known as phagosomes. In humans, phagocytosis is restricted to specialized cells called phagocytes, including macrophages, neutrophils, and DCs (58). Plasma membrane receptors of phagocytes are divided into non-opsonic or opsonic receptors. Non-opsonic receptors recognize directly distinct molecular patterns on the particle and include C-type lectins, such as Dectin-1, Dectin-2, Mincle, or DC-SIGN. Opsonic phagocytic receptors include FcRs and complement receptors recognizing Ab- or complement-opsonized particles, respectively (59). C3bi is the major opsonin recognized by the complement receptor on APCs, thus leading to the opsonized particle’s phagocytosis (60). After ligand binding, phagocytic receptors initiate signaling pathways leading to modification in the membrane composition and regulation of the actin cytoskeleton, therefore resulting in the formation of pseudopods covering the particle. Additional pseudopods are generated around the target, forming a phagocytic cup that closes up at their distal margins to form phagosomes (61).
Phagocytosis is relevant to the immunogenicity of therapeutic Abs because it governs the uptake, processing, and presentation of Ab-derived peptides on MHC molecules, which in turn can activate T cells and start adaptive immune responses. Evidence supporting this comes from studies demonstrating that FcγR-mediated phagocytosis enhances antigen processing and cross-presentation by DCs, promoting robust CD4+ and CD8+ T cell responses (49). Furthermore, alterations in the Fc glycosylation pattern of Abs can modulate their interaction with FcγR and phagocytic uptake, thereby influencing their immunogenic potential (62).
Importantly, this process is also shaped by factors such as the size of the Ab or ICs, as well as the binding affinity of phagocytic receptors—topics that will be explored in the following section. Collectively, these observations highlight phagocytosis as a critical determinant in shaping the immunogenicity profile of therapeutic Abs.
3 Fate of internalized peptides
As for other proteins, there is strong evidence that the trafficking fate of internalized Abs is common for all major endocytosis pathways. Internalized protein particles are routed to the early endosomes, also called the sorting endosomes (63). In this sorting station, internalized material is either recycled back to the plasma membrane or routed from the early endosome to late endosomes and lysosomes for degradation for a subsequent presentation (64). Early endosomes are characterized by the presence of the GTPase Rab5, which regulates endocytic membrane trafficking by recruiting several effectors. They will become increasingly acidic as endosomes mature from early/recycling to late endosomes. This process requires the switch from Rab5 to Rab7 to drive the maturation of early endosomes into late endosomes, which can then fuse with lysosomes for cargo degradation (65). In the lysosomes, cathepsins are the most abundant proteases responsible for degrading macromolecules. They comprise 12 members, mainly endopeptidase-cleaving peptide bonds. Among them, cathepsin S, a cysteine protease, has a central role in degrading antigens in APCs and their presentation in association with MHC-II molecules to CD4+ T cells (66). In fact, MHC-II molecules bind peptides of around 13 to 25 amino acids generated by this proteolysis process (67). Efficient peptide binding requires the access of MHC-II molecules to lysosomal compartments. Newly synthesized MHC class II molecules associate with the invariant chain (Ii), preventing premature ligand binding in the ER and directing the complexes to the endocytic compartment (68). Ii is proteolyzed, and the resulting CLIP occupies the peptide-binding groove (69). HLA-DM facilitates the release of CLIP and stabilizes the empty MHC class II until an exogenous peptide is loaded. The MHC class II-peptide complex is then transported to the cell surface for recognition by CD4+ T cells (70). Activation of antigen-specific CD4+ Th cells helps activate cognate antigen-specific B cells to proliferate and differentiate into plasma cells producing high-affinity ADA (71).
4 Impact of antibodies properties on their internalization
The internalization of therapeutic Abs by APCs is influenced by their properties, such as size, post-translational modifications, charge distribution, hydrophobicity, degradation, and aggregation.
4.1 Size
The size of internalized protein particles mainly impacts the route of internalization. While small particles tend to be internalized via fluid-phase endocytosis, bigger complexes and aggregated peptides or proteins are probably uptaken by phagocytosis (72). These uptake pathways differ in how they deliver antigens to intracellular processing compartments, potentially influencing the efficiency and nature of antigen presentation to the immune system (73, 74). FcγRs generally exhibit low affinity for monomeric IgG, preferentially binding ICs that enable receptor cross-linking and subsequent internalization. Although FcγRI (CD64) can bind monomeric IgG with higher affinity and mediate Ab internalization in vitro (e.g., for avelumab (75)), most FcγRs require multivalent ICs or antigen bound IgGs to initiate uptake and signaling (15, 76). Endogenous IgGs are present at high concentrations in plasma (~10 mg/mL) (77), exceeding therapeutic Ab levels, leading to FcγRI saturation under physiological conditions (78). This saturation limits FcγR availability for free therapeutic Ab, which compete with endogenous IgGs for receptor binding (76). When Abs’ targets are soluble and circulate in the bloodstream, IC formation facilitates effective FcγR engagement (79), thereby influencing Ab clearance and immunogenicity. This competitive and dynamic interplay underscores that IC formation, rather than free Ab concentration alone, primarily governs FcγR-mediated uptake. Multimeric Abs and large ICs are primarily routed to lysosomes as they escape FcRn binding, resulting in their degradation into peptides that are later loaded onto MHC class II molecules (54). This increases their potential to trigger CD4+ T cell responses, a key step in the development of ADA. On the other hand, stress occurring during handling or administration can favor aggregation (80). Aggregated forms of Abs more readily uptaken by immune cells (81–83), as they engage low-affinity FcR more effectively than monomeric Abs (49). This enhanced engagement may promote immune recognition by facilitating antigen processing and presentation. Thus, both the size and aggregation state of therapeutic Abs can significantly influence their internalization route, intracellular fate, and ultimately, their immunogenicity risk.
4.2 Protein modifications and their impact on recognition by specific receptors
Glycosylation is a common post-translational modification that occurs during the production of Abs. A significant proportion of endogenous IgG Abs naturally exhibit N-glycosylated residues in their Fab regions (84, 85). However, Fab glycosylation is generally avoided in therapeutic Abs primarily for stability and manufacturability reasons. Despite this, certain studies have highlighted potential benefits of Fab glycosylation under specific contexts. For example, Fab sialylation has been shown to improve the serum half-life of Abs, such as cetuximab (an anti-EGFR monoclonal Ab (86). Courtois et al. showed that introducing glycans to shield aggregation-prone regions enhances Ab’s stability to a comparable extent as replacing hydrophobic amino acid residues with hydrophilic ones (87). Knowledge of the biological role of Fab-associated glycans in immunity remains limited, necessitating further investigation to elucidate their functional significance. On the other hand, IgGs contain a conserved N-glycosylation site at the asparagine 297 (Asn297 or N297) residue within their Fc region, which modulates their interactions with CLR and FcγRs. The type of oligosaccharides found in the conserved Asn297 glycosylation site of Ab Fc portions depends on the mode of production. Mammalian systems generally result in complex-type biantennary oligosaccharides in the Fc portion (62). The oligosaccharides in the Fc region comprise HM glycan, containing five to nine mannose residues that are recognized by mannose receptors (MRs), complex glycans in which “antennae” initiated by N-acetyl-d-glucosamine (GlcNAc) extend the core, and hybrid glycans, which are the combination of HM and complex glycans (88) (Figure 2). For biantennary N-glycans, additional fucosylation, galactosylation, and sialylation may occur (89). APCs expressing many CLR can recognize the glycans entities on therapeutic Abs. While endogenous human IgG1 contains relatively low percentages (0.1%) of HM glycans, recombinant IgG might contain up to 10% depending on the producing cell line (90). Studies have shown that HM has potentially advantageous biological activities. Zhou et al. demonstrated that mannose resulted in a higher ADCC activity and an increased binding affinity to FcγRIIIa (91). These properties could be explained by the lack of core fucose in addition to the presence of mannose-ending glycans (92). Similar results were found by Yu and collaborators but they demonstrated a negative impact on CDC (93). On the other hand, HM residues influence Ig’s pharmacokinetics with an increased serum clearance of oligomannose glycoforms, which are recognized by mannose receptors and rapidly eliminated from circulation (94). Fc’s HM glycans are recognized by DC-SIGN and MR,and serum mannose-binding lectin (95). These mannose-sensitive receptors are shown to be implicated in the internalization of viruses (96) and factor VIII procoagulant protein (FVIII) (97). Moreover, internalization of mannosylated antigens are associated with an enhanced uptake by DC and therefore with an enhanced presentation to CD4 T cells (97, 98). Few studies focused on therapeutic Abs’ uptake through mannose-sensitive receptors. Wolf et al. showed an increased internalization of mannosylated rituximab compared to the wild-type form which colocalized in the lysosome (99). Interestingly when therapeutic Abs are injected SC, the subcutaneous compartment lacks the serum alpha-mannosidase that trims the substrate Man9, and therefore, M9–6 glycans are not converted into M5 types and may interact with cutaneous DC through DC-SIGN (100). However, recognition by these specific CLRs can shape T-cell responses. For instance, mannosylated antigens internalized via the mannose receptor (MMR) enhance antigen presentation and T-cell activation (reviewed in (101)). This was also observed with mannosylated rituximab, which led to increased T-cell activation in 50% of tested donors compared to the wild-type Ab. Similar findings have been reported for antigens engaging DC-SIGN (102). Wawrzyniak et al. found that fucosylation of adalimumab does not influence its uptake by moDCs nor their activation (103). Human IgGs contain low levels of sialylated Fc (5%–10%). Studies have shown that sialylation of the N-linked glycan conveys Abs an anti-inflammatory activity. The dependency of this effect on the interaction between DC-SIGN and sialylated Fc remains under debate (104). Moreover, the uptake of sialic acid-modified antigens by DCs results in the initiation of a tolerogenic T-cell response (105). Wolf et al. showed that the hypersialylated variant of rituximab had a decreased internalization rate by DC and weak or nearly no co-localization in lysosomal compartments (99). Besides their recognition by CLR, Fc fragment glycosylation on Abs is also crucial for its interaction with FcR (106). The presence of fucose on IgG N-linked glycan is a modification of the human IgG1 Fc structure with many functional consequences. The core fucose causes a steric inhibition, limiting the interaction between FcγRIII-glycan and IgG-Fc and, therefore, leading to a suboptimal affinity (107). Hence, afucosylated IgG bind to FcγRIIIa and FcγRIIIb with a higher affinity (108). Studies have shown that a fucosylated IgG induces FcγRIIIa-dependent signaling and promotes ADCC (109, 110), the major mode of action to deplete tumor cells. Macrophages also express FcγRIII, which could mediate phagocytosis of opsonized IgG. However, many studies report FcγRI as the major implicated receptor (111, 112). Afucosylated therapeutic Abs are not generally reported to enhance phagocytosis significantly. On the other hand, Abs can be engineered to reduce or completely eliminate their interaction with FcγRs, thereby minimizing their effector functions such as ADCC and CDC. This approach is particularly applied to Abs used in oncology to reduce off-target toxicity and enhance therapeutic efficacy. One common strategy involves removing the N-linked glycosylation site at Asn297 as exemplified by atezolizumab, an anti-PDL1 (113), or introducing silencing mutations such as the so-called LALA mutation (Leu234Ala together with Leu235Ala) to reduce or abolish FcγR binding and effector functions like ADCC/CDC (114). Glycan residues on the Fc region of IgG critically influence the recruitment and binding affinity of FcγR on DCs, thereby modulating Ab internalization and directing their intracellular trafficking and immunological fate (78, 115). Jin et al. showed a higher internalization level of avelumab through FcγRI-mediated internalization in comparison to the non-glycosylated variant (75). The Fc-mediated internalization of ICs is associated with enhanced antigen uptake and presentation by DCs and macrophages (50, 116, 117). Amigorena et al. showed the dependency of this enhanced antigen presentation on tyrosine residues of the cytoplasmic domain of ITAM as the mutation of these residues motif inhibited both the internalization of IgG–antigen complexes and the presentation of the IgG-coated antigen (118). FcγR activation by ICs also induces their sequestration in intracellular vesicles for lysosomal degradation and antigen processing (119). However, beyond the internalization step, FcγRs vary in their intercellular trafficking capacities of ICs. ICs transported by the full-length FcγRIA are routed to MHC-II compartments, whereas those up taken by a truncated, tail-deleted FcγRIA are redirected to recycling compartments, resulting in reduced antigen presentation (120). Thus, the uptake pathway might significantly influence their functional response. FcγRIIb also plays a role in antigen capture. However, its inhibitory role is controversial; some studies have shown that FcγRIIb deficiency results in an enhanced potential to generate antigen-specific T cell responses, while others indicate that FcγRIIb-mediated uptake by DCs can elicit a weak T cell response (121, 122). Since both glycan recognition by CLR and FcR binding modulate immune cell activation and antigen presentation, these molecular modifications may play a crucial role in clinical immunogenicity by influencing the likelihood of immune responses. However, translating in vitro findings on FcγR-mediated internalization to clinical settings is challenging, and many aspects should be considered, such as the competitive receptor occupancy by endogenous IgGs, the dynamic formation of ICs in vivo and the receptor expression profiles on relevant cell types. The use of humanized Abs may therefore provide more predictive insights into FcγR interactions and therapeutic Ab fate in patients (76, 123).
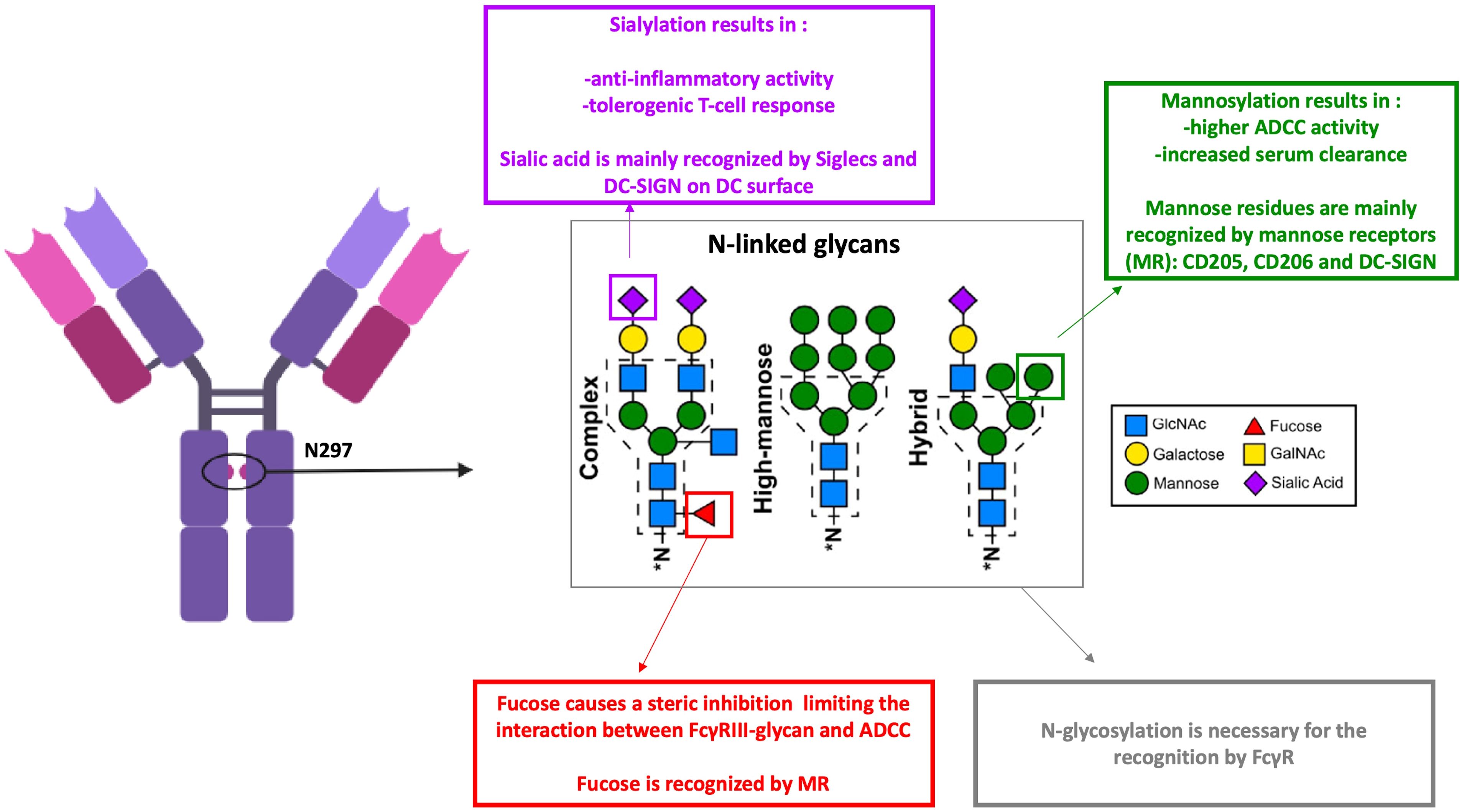
Figure 2. Antibodies N-glycans and their recognition by dendritic cells receptors. ADCC, antibody dependent cell-mediated cytotoxicity; FcγR, Fc gamma receptors; MR, mannose receptors. Monoclonal antibodies (mAbs) glycosylation modifies their functional properties and impacts their recognition by dendritic cell (DC) receptors through carbohydrate residues. Sialic acid is primarily recognized by Siglecs and DC-SIGN on the surface of DCs. In contrast, fucosylation of mAbs reduces their interaction with FcγRIII, leading to decreased antibody-dependent cellular cytotoxicity (ADCC). However, mannosylation of mAbs enhances ADCC activity and is associated with increased clearance. Mannose-sensitive receptors, such as CD205, CD206, and DC-SIGN, are capable of recognizing fucose and mannose residues. Created using BioRender.com.
Another modification that could influence therapeutic Abs receptor recognition is PEGylation. Polyethylene glycol (PEG) is a flexible, hydrophilic polymer that is chemically attached to therapeutic Abs after the purification process. PEG enhances the half-life of Abs by creating a steric barrier that reduces protein binding, thereby decreasing the frequency of administration. This technique, known as PEGylation, can resemble to post-translational modifications of proteins and is clinically proven as an effective strategy for extending the half-life of protein-based therapies (124). Sanchez and collaborators showed that partial coating of PEG reduces the macrophage uptake of particles (125). Other studies demonstrated a reduced internalization of nanocarriers with a longer PEG chain by macrophages (126, 127). Recently, de Bourayne et al. showed that pegylation of certolizumab reduces its uptake by DC, peptide presentation to T-cells, and T-cell priming (128). PEGylation is frequently associated with diminished immunogenicity by masking immune epitopes (129, 130). PEGylation shields therapeutic Abs from recognition and internalization by immune cells, thereby playing a key role in minimizing clinical immunogenicity.
4.3 Charge distribution and hydrophobicity
Abs’ behavior is influenced by their surface characteristics. Results have demonstrated that modifying the charged and hydrophobic regions can enhance the solution properties of Abs (131, 132). However, positive charge patches were also described as adversely affecting Abs pharmacokinetic (133). The presence of large positively charged patches in the CDR can lead to non specific binding (134). On the other hand, hydrophobicity as well as surface charges have been linked to undesirable aggregation in IgG (135, 136). Studies have shown a higher internalization rate of positively charged polymeric nanoparticles following an ionic interaction with negatively charged membranes (137, 138). Jin et al. observed in vitro a lower internalization of a variant of avelumab (anti-PD-L1) with lower isoelectric point (75). Moreover, bococizumab, an anti-PCSK9 Ab with excess positive charges, showed poor pharmacokinetics properties and high immunogenicity (139). Wen et al. showed its high internalization rate in vitro by moDCs in comparison to other Abs (8). Liu et al. also found that positively charged Abs exhibited enhanced cellular uptake (140). Thompson et al. observed an accumulation of transferrin and an endosomal maturation following positively charged proteins’ internalization, thus suggesting an alteration of peptide processing and presentation (141). Another recent study also showed that adding positive residues to therapeutics Abs resulted in their lysosomal accumulation and the increase of antigenic presentation (5), two critical steps of immune response initiation. Other studies showed a correlation between particle uptake and their hydrophobicity (142, 143) following an improvement of their interaction with the cellular membrane. This effect is interesting for enhancing Ab drug conjugates (ADC)s antitumor activity by favoring cell penetration (144). Together, these findings emphasize that both charge distribution and hydrophobicity can influence how therapeutic Abs are internalized, processed, and presented by immune cells.
4.4 Aggregation
Abs aggregation occurs during storage manufacturing, processing, storage, handling, and administration due to exposure to different stresses such as temperature variation, shaking, or light exposure (145). This stress results in Ab’s partial unfolding, leading to monomer-monomer association followed by nucleation and growth (80). Changes in hydrophobicity and charge, which are probably linked to an increased uptake, also promote aggregation (146). Even though it is now well accepted that Abs aggregates enhance immunogenicity, few data are published concerning their internalization by DCs. Ahmadi et al. showed that aggregated rituximab has a higher internalization rate compared to the native Abs, and it rapidly accumulates in late endosomes associated with antigen presentation (147). Another study identified the implication of FcγRs in ADC aggregates internalization into non-target cells compared to the control (148). Moreover, using MHC-Associated Peptide Proteomics assay (MAPPs), Rombach-Riegraf et al. identified a higher number of Ab-specific peptides associated with MHC class II molecules after loading moDCs of stir-stress aggregated Abs. According to the authors, these results suggest a higher uptake (149). The factors mentioned above impact mAb’s internalization rate and presentation by DCs. An increased presentation of internalized particles might allow the recruitment of T cells with a lower T cell receptor avidity. Moreover, modifications such aggregation can lead to structure and sequence modification, which could lead to the generation of neoepitopes priming T-cells. Therefore, evaluating Abs’ internalization should be considered part of in vitro testing of therapeutic Ab immunogenicity.
5 Target-mediated internalization
The target-mediated endocytosis is a process initiated by the binding of ligands to specific receptors on the cell surface.This leads to the formation of receptor-ligand complexes that initiate receptor-mediated endocytosis. During internalization, these complexes are internalized into early endosomes then either recycled back to the plasma membrane or degraded in lysosomes. The fate of these complexes is often determined by their pH-dependent dissociation: ligands that dissociate rapidly in acidic endosomal conditions tend to promote receptor recycling, whereas complexes that remain intact under low pH conditions are targeted for lysosomal degradation (150). Target expression by DCs facilitates therapeutic Abs’ uptake. For example, atezolizumab, binds to PD-L1 expressed on DCs as well as tumor cells, influencing immune checkpoint regulation and enhancing antitumor immunity (151). A study demonstrated that high uptake of radiolabeled anti-PD-L1 occurred in the liver and spleen, corresponding to its binding to PD-L1 receptors expressed on lymphocytes and DCs in these tissues (152). Jin et al. also showed that a variant of avelumab (an anti-PD-L1) deficient in PD-L1 binding exhibited significantly reduced internalization by immune cells, highlighting that PD-L1 engagement is critical for efficient receptor-mediated uptake of the Ab (75). Another group demonstrated that anti-TNF Abs undergo rapid target-mediated endocytosis following their binding to transmembrane TNF on the DC surface followed by its routing to degradative compartments (153). Internalization and processing of Ab-receptor complexes by DCs can potentially modulate immune responses by enhancing antigen presentation. The degradation of therapeutic Abs in lysosomes could lead to the generation of immunogenic peptides that might provoke ADA responses, impacting treatment efficacy and safety. Therefore, understanding target-mediated endocytosis is essential not only for optimizing therapeutic Ab design and delivery but also for anticipating and managing immunogenicity-related challenges in clinical applications.
6 Assays to evaluate internalization
As internalization gained interest in the last decade, different in vitro assay formats were developed (Table 1). Endocytosis mechanisms are commonly evaluated by flow cytometry and fluorescence microscopy (154), both requiring tracking of the molecule by direct or secondary labeling. Direct Abs labeling with a fluorescent dye is a widely used method to study their internalization (147, 155). Although this technique is straightforward, it is partially limited by the difficulty of discriminating between surface-bound and effective-internalized material and the lack of information concerning the intracellular trafficking. This could be completed by microscopic study or by adding more controls. For instance, incubating cells at 4°C inhibits the internalization, and the measured fluorescence corresponds to the surface binding signal. Washing with an acid buffer or using quenchers like trypan blue could also remove the cell surface binding signal (156). Alternative methods based on direct labeling with a pH-sensitive dye, either reactive to amine or directed to heavy chain N-linked glycans, are developed to evaluate the endocytosis and to assess lysosomal degradation. Using this technique, Deora et al. demonstrated the rapid internalization of anti-tumor necrosis factor (TNF) Abs following their formation of a complex with transmembrane TNF (tmTNF) (153). This complex is initially routed into early endosomes and subsequently transported to lysosomes, where it undergoes further degradation. Jin et al. also compared the internalization of different variant of avelumab using pHrodo labeling (75). Moreover, Siegel et al. also labeled different therapeutic Abs with a pH-sensitive fluorophore specifically directed to their Fc glycosylation site and compared their internalization rate in moDCs (5). Results revealed a linear correlation between the cellular accumulation of these Abs and their subsequent presentation by MHC-II molecules. Förster resonance energy transfer (FRET) is another method that allows the evaluation of internalized Abs. It requires the conjugation of the Ab with both a fluorophore and a quencher dye. At the cell surface, the fluorophore is masked by the quencher, and once internalized and degraded, the physical separation of the fluorophore and quencher results in a fluorescent signal detected by flow cytometry (157). This method was used in different studies to compare many Abs’ internalization rate and correlate with their immunogenic potential (8, 158, 159). However, since the FRET construct binds to the Fc region, Fc-mediated endocytosis could not be assessed, despite it being a potentially significant uptake pathway for many Abs. Recently, a group developed a new format for assessing Ab’s internalization based on cell surface staining and an intracellular staining of cells with an anti-human IgG F(ab’)2 following a 24 hours incubation with unlabeled mAb studies tested different Abs and compared their internalization rate and their immunogenic potential through different assay ( (4, 7).These different tools used to evaluate therapeutic Ab’s internalization are summarized in Table 1. Many in vitro assays are available based on different techniques to evaluate the role of a specific receptor in mAb’s internalization. Jin et al. demonstrate the implication of FcγR in avelumab’s uptake by testing and comparing it to FcγR binding–deficient variants (75). On the other hand, Xue et al. showed that an anti-IL21 receptor internalization by DCs is slightly dependent on FcγR as blocking these receptors by an Fc block did not modify the cellular binding (155). Other studies were based on competition or blocking experiments to identify a specific receptor. For example, Aoyama et al. identified the implication of FcγRs in ADC aggregates internalization by analyzing the uptake inhibition by flow cytometry after blocking FcγRIIa (148). Another study demonstrated the MMR-dependent endocytosis of factor VIII by blocking mannose-sensitive receptors after preincubating DCs with mannan (97). Another alternative consists of analyzing uptake by cellular model that overexpresses a particular receptor. After incubation with wild type or CD206-expressing Chinese hamster ovary (CHO) cells, a receptor-specific targeting and uptake was shown for nanocarriers by flow cytometry and confocal microscopy (160). Anderson et al. showed that using the same technique principle, the specific uptake of a glycopeptide in DC-SIGN CHO (+) was achieved (161).

Table 1. Tools to evaluate the internalization of therapeutic antibodies and their limitations.
7 Conclusion
The development of an immune response against therapeutic Abs requires their internalization by APCs, mainly DCs, and their processing into peptides. Understanding how these Abs are internalized by cells and the influence of their physico-chemical properties on their uptake is critical for evaluating their immunogenicity. Here, we focused on Abs’ properties and the aggregation impact on their uptake by DCs, presented a general overview of the current understanding of endocytosis, and discussed current experimental techniques. Aspects of Abs’ properties, such as size, PTM, surface chemistry, and their aggregation propensity, are thought to influence the route and the rate of internalization. The current assays evaluating Ab’s internalization present some limitations, such as the evaluation of one main endocytic route, which might lead to false output. Considerable research effort is still expanding to develop tools to evaluate Ab’s internalization and integrate it into the overall evaluation of risk of immunogenicity. An increased understanding of these processes could greatly improve the ability to predict the risk of immunogenicity and facilitate the development of effective mitigation strategies. To this end, an evidence-based strategy is proposed for assessing the immunogenicity risk of therapeutic Abs candidates. This strategy includes internalization assessments, along with other evaluation methods that address various stages of the immune response. It also incorporates multiple risk-mitigation approaches, allowing for a more comprehensive and predictive evaluation of potential immunogenic reactions. Such a framework is essential for optimizing Ab design, minimizing the risk of adverse immune responses, and improving the overall safety and efficacy of Ab-based therapies (Figure 3).
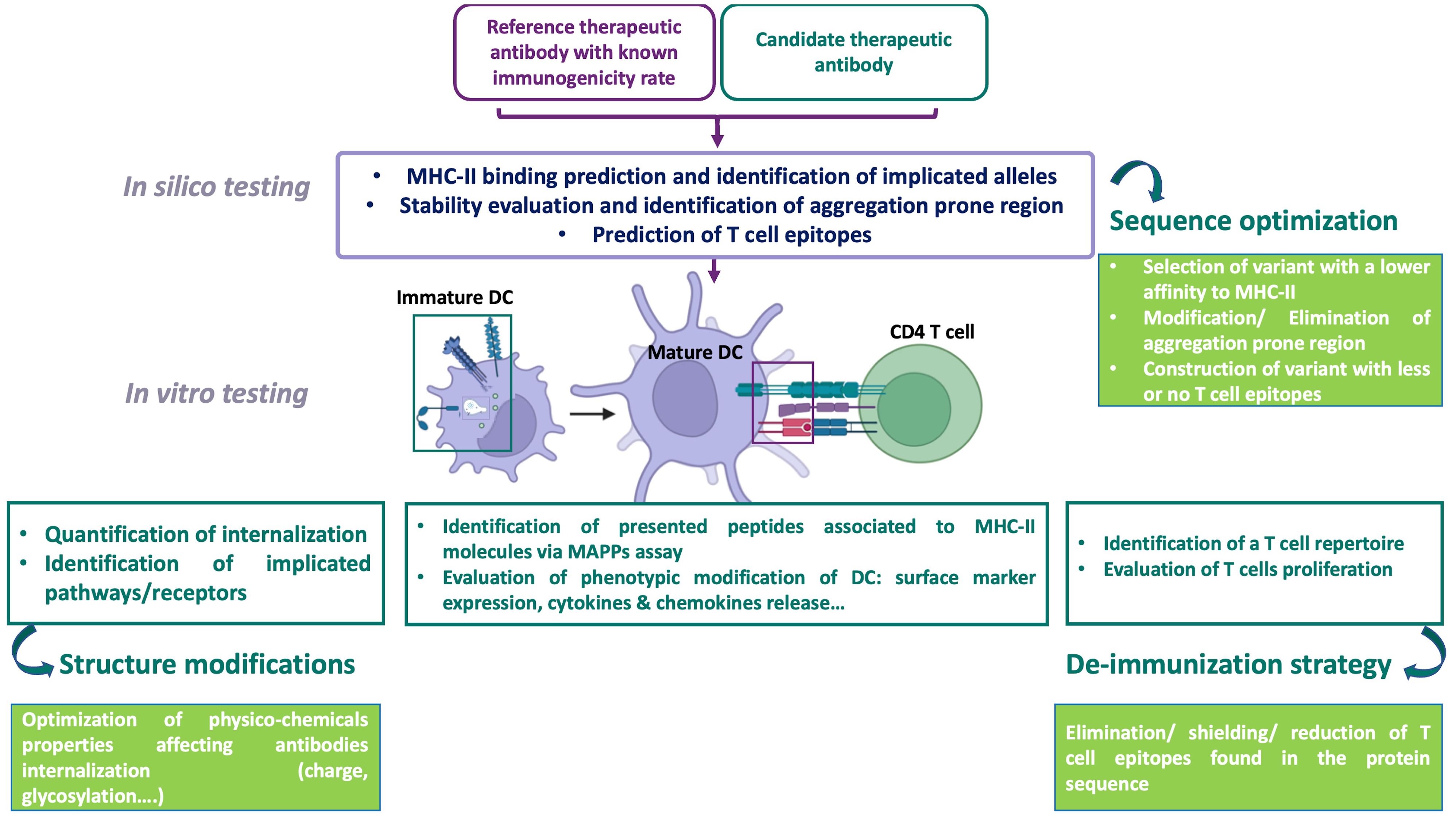
Figure 3. An evidence-based strategy for assessing the immunogenicity risk of therapeutic protein candidates, including an internalization assessment test. DC, dendritic cell; MAPPS, MHC-associated peptide proteomics; MHC, major histocompatibility complex, The immunogenicity of therapeutic antibodies is evaluated using complementary approaches. In silico evaluation, based on a database of therapeutic antibodies with known immunogenic risks, predicts MHC-II binding, identifies aggregation-prone regions (APRs), and assesses epitope-based risks and implicated alleles. This approach can guide antibody sequence optimization by selecting variants with lower MHC-II affinity, reduced APRs, and fewer T cell epitopes, while maintaining activity. The next step involves in vitro evaluation of the cellular mechanisms involved in T cell response initiation. Assessing the internalization and processing of antibodies by monocyte-derived dendritic cells (moDCs)—two key steps in this immune response—is important for quantifying antibody uptake and comparing it to a reference antibody with known immunogenicity. This is complemented by identifying internalization pathways and receptors using pharmacological inhibitors or receptor-overexpressing models, as antibody entry depends on properties such as glycosylation and charge. Optimization involves modifying the antibody’s physicochemical properties, such as testing different monoclonal antibodies (mAbs) with the same Fab but varying Fc portions (e.g., glycosylated versus non-glycosylated). The MAPPS assay complements internalization evaluation by identifying peptides presented on MHC-II molecules to CD4 T cells. Additionally, dendritic cell (DC) activation testing is crucial for assessing the biological activity of impurities or large aggregates. T cell assays encompass multiple stages of the immune response: internalization, antigen presentation, peptide recognition, and T cell activation. These assays either identify the presence of a T cell repertoire in response to an antibody, which is a prerequisite for T cell initiation, or assess T cell proliferation in response to the antibody. Based on these results, antibody sequence optimization may be considered to eliminate impurities and reduce immunogenicity by removing, masking, or reducing T cell epitopes. Creating using BioRender.com.
Author contributions
ML: Writing – original draft, Writing – review & editing. MP: Writing – review & editing. IT: Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Crescioli S, Kaplon H, Wang L, Visweswaraiah J, Kapoor V, and Reichert JM. Antibodies to watch in 2025. mAbs. (2025) 17. doi: 10.1080/19420862.2024.2443538
2. Jawa V, Terry F, Gokemeijer J, Mitra-Kaushik S, Roberts BJ, Tourdot S, et al. T-cell dependent immunogenicity of protein therapeutics pre-clinical assessment and mitigation–updated consensus and review 2020. Front Immunol. (2020) 11:1301. doi: 10.3389/fimmu.2020.01301
3. Lteif M, Pallardy M, and Turbica I. Antibodies internalization mechanisms by dendritic cells and their role in therapeutic antibody immunogenicity. Eur J Immunol. (2024) 54:2250340. doi: 10.1002/eji.202250340
4. Melendez R, Ordonia B, Guerrero J, Hassanzadeh A, Tran P, Low J, et al. Introducing dendritic cell antibody internalization as an immunogenicity risk assessment tool. Bioanalysis. (2022) 14:703–13. doi: 10.4155/bio-2022-0024
5. Siegel, Bolender, Ducret, Fraidling, Hartman, Looney, et al. Internalization of therapeutic antibodies into Dendritic cells as a risk factor for immunogenicity. Front Immunol. (2024) 15. doi: 10.3389/fimmu.2024.1406643
6. Siegel M, Padamsey A, Bolender A-L, Hargreaves P, Fraidling J, Ducret A, et al. Development and characterization of dendritic cell internalization and activation assays contributing to the immunogenicity risk evaluation of biotherapeutics. Front Immunol. (2024) 15:1406804. doi: 10.3389/fimmu.2024.1406804
7. Tsai W-TK, Li Y, Yin Z, Tran P, Phung Q, Zhou Z, et al. Nonclinical immunogenicity risk assessment for knobs-into-holes bispecific IgG 1 antibodies. mAbs. (2024) 16:2362789. doi: 10.1080/19420862.2024.2362789
8. Wen Y, Wang X, Cahya S, Anderson P, Velasquez C, Torres C, et al. Comparability study of monocyte derived dendritic cells, primary monocytes, and THP1 cells for innate immune responses. J Immunol Methods. (2021) 498:113147. doi: 10.1016/j.jim.2021.113147
9. Harris CT and Cohen S. Reducing immunogenicity by design: approaches to minimize immunogenicity of monoclonal antibodies. BioDrugs. (2024) 38:205–26. doi: 10.1007/s40259-023-00641-2
10. Lyu X, Zhao Q, Hui J, Wang T, Lin M, Wang K, et al. The global landscape of approved antibody therapies. Antibody Ther. (2022) 5:233–57. doi: 10.1093/abt/tbac021
11. Marshall JS, Warrington R, Watson W, and Kim HL. An introduction to immunology and immunopathology. Allergy Asthma Clin Immunol. (2018) 14:49. doi: 10.1186/s13223-018-0278-1
12. Chiu ML, Goulet DR, Teplyakov A, and Gilliland GL. Antibody structure and function: the basis for engineering therapeutics. Antibodies. (2019) 8:55. doi: 10.3390/antib8040055
13. Townsend CL, Laffy JMJ, Wu Y-CB, Silva O’Hare J, Martin V, Kipling D, et al. Significant differences in physicochemical properties of human immunoglobulin kappa and lambda CDR3 regions. Front Immunol. (2016) 7:388. doi: 10.3389/fimmu.2016.00388
15. Vidarsson G, Dekkers G, and Rispens T. IgG subclasses and allotypes: from structure to effector functions. Front Immunol. (2014) 5:520. doi: 10.3389/fimmu.2014.00520
16. Hussain A, Grimm A, Sheng W, Zhang C, Al-Rawe M, Bräutigam K, et al. Toward homogenous antibody drug conjugates using enzyme-based conjugation approaches. Pharmaceuticals. (2021) 14:343. doi: 10.3390/ph14040343
17. Forthal DN. Functions of antibodies. Microbiol Spectr. (2014) 2:1–17. doi: 10.1128/microbiolspec.AID-0019-2014
18. Castelli MS, McGonigle P, and Hornby PJ. The pharmacology and therapeutic applications of monoclonal antibodies. Pharmacol Res Perspect. (2019) 7:e00535. doi: 10.1002/prp2.535
19. Chan AC and Carter PJ. Therapeutic antibodies for autoimmunity and inflammation. Nat Rev Immunol. (2010) 10:301–16. doi: 10.1038/nri2761
20. Van Der Horst HJ, Nijhof IS, Mutis T, and Chamuleau MED. Fc-engineered antibodies with enhanced Fc-effector function for the treatment of B-cell Malignancies. Cancers. (2020) 12:3041. doi: 10.3390/cancers12103041
21. Narvekar A, Pardeshi A, Jain R, and Dandekar P. ADCC enhancement: A conundrum or a boon to mAb therapy? Biologicals. (2022) 79:10–8. doi: 10.1016/j.biologicals.2022.08.006
22. Wen Y, Cahya S, Zeng W, Lin J, Wang X, Liu L, et al. Development of a FRET-based assay for analysis of mAbs internalization and processing by dendritic cells in preclinical immunogenicity risk assessment. AAPS J. (2020) 22:68. doi: 10.1208/s12248-020-00444-1
23. Ewers H. GM1 structure determines SV40-induced membrane invagination and infection. Nature Cell Biology (2010) 12:20. doi: 10.1038/ncb1999
24. Sikorski M, Coulon F, Peltier C, Braudeau C, Garcia A, Giraud M, et al. Non-permissive human conventional CD1c+ dendritic cells enable trans-infection of human primary renal tubular epithelial cells and protect BK polyomavirus from neutralization. PloS Pathog. (2021) 17:e1009042. doi: 10.1371/journal.ppat.1009042
25. Rennick JJ, Johnston APR, and Parton RG. Key principles and methods for studying the endocytosis of biological and nanoparticle therapeutics. Nat Nanotechnology. (2021) 16:266–76. doi: 10.1038/s41565-021-00858-8
26. Egami Y, Taguchi T, Maekawa M, Arai H, and Araki N. Small GTPases and phosphoinositides in the regulatory mechanisms of macropinosome formation and maturation. Front Physiol. (2014) 5:374. doi: 10.3389/fphys.2014.00374
27. Recouvreux MV and Commisso C. Macropinocytosis: A metabolic adaptation to nutrient stress in cancer. Front Endocrinol. (2017) 8:261. doi: 10.3389/fendo.2017.00261
28. Buckley CM and King JS. Drinking problems: Mechanisms of macropinosome formation and maturation. FEBS J. (2017) 284:3778–90. doi: 10.1111/febs.14115
29. Ivanov AI. Pharmacological inhibition of endocytic pathways: is it specific enough to be useful? Methods in Molecular Biology (2008) 19:15–33. doi: 10.1007/978-1-59745-178-9_2
30. Canton J, Schlam D, Breuer C, Gütschow M, Glogauer M, and Grinstein S. Calcium-sensing receptors signal constitutive macropinocytosis and facilitate the uptake of NOD2 ligands in macrophages. Nat Commun. (2016) 7:11284. doi: 10.1038/ncomms11284
31. Lin XP, Mintern JD, and Gleeson PA. Macropinocytosis in different cell types: similarities and differences. Membranes. (2020) 10:177. doi: 10.3390/membranes10080177
32. Kaksonen M and Roux A. Mechanisms of clathrin-mediated endocytosis. Nat Rev Mol Cell Biol. (2018) 19:313–26. doi: 10.1038/nrm.2017.132
33. Mettlen M, Chen P-H, Srinivasan S, Danuser G, and Schmid SL. Regulation of clathrin-mediated endocytosis. Annu Rev Biochem. (2018) 87:871–96. doi: 10.1146/annurev-biochem-062917-012644
34. Moore CAC, Milano SK, and Benovic JL. Regulation of receptor trafficking by GRKs and arrestins. Annu Rev Physiol. (2007) 69:451–82. doi: 10.1146/annurev.physiol.69.022405.154712
35. Lamendour L, Deluce-Kakwata-Nkor N, Mouline C, Gouilleux-Gruart V, and Velge-Roussel F. Tethering innate surface receptors on dendritic cells: A new avenue for immune tolerance induction? Int J Mol Sci. (2020) 21:5259. doi: 10.3390/ijms21155259
36. Lu G, Janjic BM, Janjic J, Whiteside TL, Storkus WJ, and Vujanovic NL. Innate direct anticancer effector function of human immature dendritic cells. II. Role of TNF, lymphotoxin-α1β2, fas ligand, and TNF-related apoptosis-inducing ligand. J Immunol. (2002) 168:1831–9. doi: 10.4049/jimmunol.168.4.1831
37. Wang D, Sun B, Feng M, Feng H, Gong W, Liu Q, et al. Role of scavenger receptors in dendritic cell function. Hum Immunol. (2015) 76:442–6. doi: 10.1016/j.humimm.2015.03.012
38. Lenza MP, Atxabal U, Oyenarte I, Jiménez-Barbero J, and Ereño-Orbea J. Current status on therapeutic molecules targeting siglec receptors. Cells. (2020) 9:2691. doi: 10.3390/cells9122691
39. van der Zande HJP, Nitsche D, Schlautmann L, Guigas B, and Burgdorf S. The mannose receptor: from endocytic receptor and biomarker to regulator of (Meta)Inflammation. Front Immunol. (2021) 12:765034. doi: 10.3389/fimmu.2021.765034
40. Fischer S, Stegmann F, Gnanapragassam VS, and Lepenies B. From structure to function – Ligand recognition by myeloid C-type lectin receptors. Comput Struct Biotechnol J. (2022) 20:5790–812. doi: 10.1016/j.csbj.2022.10.019
41. García-Vallejo JJ and Van Kooyk Y. Endogenous ligands for C-type lectin receptors: The true regulators of immune homeostasis. Immunol Rev. (2009) 230:22–37. doi: 10.1111/j.1600-065X.2009.00786.x
42. Geijtenbeek TBH, Torensma R, Van Vliet SJ, Van Duijnhoven GCF, Adema GJ, Van Kooyk Y, et al. Identification of DC-SIGN, a novel dendritic cell–specific ICAM-3 receptor that supports primary immune responses. Cell. (2000) 100:575–85. doi: 10.1016/S0092-8674(00)80693-5
43. Feinberg H, Jégouzo SAF, Lasanajak Y, Smith DF, Drickamer K, Weis WI, et al. Structural analysis of carbohydrate binding by the macrophage mannose receptor CD206. J Biol Chem. (2021) 296:100368. doi: 10.1016/j.jbc.2021.100368
44. Shrimpton RE, Butler M, Morel A-S, Eren E, Hue SS, and Ritter MA. CD205 (DEC-205): A recognition receptor for apoptotic and necrotic self. Mol Immunol. (2009) 46:1229–39. doi: 10.1016/j.molimm.2008.11.016
45. Guo Y, Feinberg H, Conroy E, Mitchell DA, Alvarez R, Blixt O, et al. Structural basis for distinct ligand-binding and targeting properties of the receptors DC-SIGN and DC-SIGNR. Nat Struct Mol Biol. (2004) 11:591–8. doi: 10.1038/nsmb784
46. De Haan N, Falck D, and Wuhrer M. Monitoring of immunoglobulin N- and O-glycosylation in health and disease. Glycobiology. (2020) 30:226–40. doi: 10.1093/glycob/cwz048
47. Hamdan TA, Lang PA, and Lang KS. The diverse functions of the ubiquitous Fcγ Receptors and their unique constituent, FcRγ Subunit. Pathogens. (2020) 9:140. doi: 10.3390/pathogens9020140
48. Kang SH and Lee C-H. Development of therapeutic antibodies and modulating the characteristics of therapeutic antibodies to maximize the therapeutic efficacy. Biotechnol Bioprocess Eng. (2021) 26:295–311. doi: 10.1007/s12257-020-0181-8
49. Junker F, Gordon J, and Qureshi O. Fc gamma receptors and their role in antigen uptake, presentation, and T cell activation. Front Immunol. (2020) 11:1393. doi: 10.3389/fimmu.2020.01393
50. Guilliams M, Bruhns P, Saeys Y, Hammad H, and Lambrecht BN. The function of Fcγ receptors in dendritic cells and macrophages. Nat Rev Immunol. (2014) 14:94–108. doi: 10.1038/nri3582
51. Bournazos S, Gupta A, and Ravetch JV. The role of IgG Fc receptors in antibody-dependent enhancement. Nat Rev Immunol. (2020) 20:633–43. doi: 10.1038/s41577-020-00410-0
52. Lowell CA. Src-family and Syk kinases in activating and inhibitory pathways in innate immune cells: Signaling cross talk. Cold Spring Harbor Perspect Biol. (2011) 3:a002352. doi: 10.1101/cshperspect.a002352
53. Croisé P, Estay-Ahumada C, Gasman S, and Ory S. Rho GTPases, phosphoinositides, and actin: A tripartite framework for efficient vesicular trafficking. Small GTPases. (2014) 5:e29469. doi: 10.4161/sgtp.29469
54. Pyzik M, Kozicky LK, Gandhi AK, and Blumberg RS. The therapeutic age of the neonatal Fc receptor. Nat Rev Immunol. (2023) 23:415–32. doi: 10.1038/s41577-022-00821-1
55. Kontermann RE. Strategies for extended serum half-life of protein therapeutics. Curr Opin Biotechnol. (2011) 22:868–76. doi: 10.1016/j.copbio.2011.06.012
56. Baker K, Rath T, Pyzik M, and Blumberg RS. The role of fcRn in antigen presentation. Front Immunol. (2014) 5:408. doi: 10.3389/fimmu.2014.00408
57. Qiao S-W, Kobayashi K, Johansen F-E, Sollid LM, Andersen JT, Milford E, et al. Dependence of antibody-mediated presentation of antigen on FcRn. Proc Natl Acad Sci. (2008) 105:9337–42. doi: 10.1073/pnas.0801717105
58. Uribe-Querol E and Rosales C. Phagocytosis: our current understanding of a universal biological process. Front Immunol. (2020) 11:1066. doi: 10.3389/fimmu.2020.01066
59. Fu YL and Harrison RE. Microbial phagocytic receptors and their potential involvement in cytokine induction in macrophages. Front Immunol. (2021) 12:662063. doi: 10.3389/fimmu.2021.662063
60. Dunkelberger JR and Song W-C. Complement and its role in innate and adaptive immune responses. Cell Res. (2010) 20:34–50. doi: 10.1038/cr.2009.139
61. Jaumouillé V and Waterman CM. Physical constraints and forces involved in phagocytosis. Front Immunol. (2020) 11:1097. doi: 10.3389/fimmu.2020.01097
62. Reusch D and Tejada ML. Fc glycans of therapeutic antibodies as critical quality attributes. Glycobiology. (2015) 25:1325–34. doi: 10.1093/glycob/cwv065
63. Jovic, Sharma, Rahajeng, and Caplan. The early endosome: A busy sorting station for proteins at the crossroads. Histol Histopathology. (2009) 25:99–112. doi: 10.14670/HH-25.99
64. Placidi G, Mattu C, Ciardelli G, and Campa CC. Small molecules targeting endocytic uptake and recycling pathways. Front Cell Dev Biol. (2023) 11:1125801. doi: 10.3389/fcell.2023.1125801
65. Skjeldal FM, Haugen LH, Mateus D, Frei DM, Rødseth AV, Hu X, et al. De novo formation of early endosomes during Rab5-to-Rab7a transition. J Cell Sci. (2021) 134:jcs254185. doi: 10.1242/jcs.254185
66. Bonam SR, Wang F, and Muller S. Lysosomes as a therapeutic target. Nat Rev Drug Discov. (2019) 18:923–48. doi: 10.1038/s41573-019-0036-1
67. Wieczorek M, Abualrous ET, Sticht J, Álvaro-Benito M, Stolzenberg S, Noé F, et al. Major histocompatibility complex (MHC) class I and MHC class II proteins: conformational plasticity in antigen presentation. Front Immunol. (2017) 8:292. doi: 10.3389/fimmu.2017.00292
68. ten Broeke T, Wubbolts R, and Stoorvogel W. MHC class II antigen presentation by dendritic cells regulated through endosomal sorting. Cold Spring Harbor Perspect Biol. (2013) 5:a016873. doi: 10.1101/cshperspect.a016873
69. Fortin J-S, Cloutier M, and Thibodeau J. Exposing the specific roles of the invariant chain isoforms in shaping the MHC class II peptidome. Front Immunol. (2013) 4:443. doi: 10.3389/fimmu.2013.00443
70. Mellins ED and Stern LJ. HLA-DM and HLA-DO, key regulators of MHC-II processing and presentation. Curr Opin Immunol. (2014) 26:115–22. doi: 10.1016/j.coi.2013.11.005
71. Kurosaki T, Kometani K, and Ise W. Memory B cells. Nat Rev Immunol. (2015) 15:149–59. doi: 10.1038/nri3802
72. Couceiro JR, Gallardo R, De Smet F, De Baets G, Baatsen P, Annaert W, et al. Sequence-dependent internalization of aggregating peptides. J Biol Chem. (2015) 290:242–58. doi: 10.1074/jbc.M114.586636
73. Amigorena S and Savina A. Intracellular mechanisms of antigen cross presentation in dendritic cells. Curr Opin Immunol. (2010) 22:109–17. doi: 10.1016/j.coi.2010.01.022
74. Trombetta ES and Mellman I. Cell biology of antigen processing in vitro and in vivo. Annu Rev Immunol. (2005) 23:975–1028. doi: 10.1146/annurev.immunol.22.012703.104538
75. Jin H, D’Urso V, Neuteboom B, McKenna SD, Schweickhardt R, Gross AW, et al. Avelumab internalization by human circulating immune cells is mediated by both Fc gamma receptor and PD-L1 binding. OncoImmunology. (2021) 10:1958590. doi: 10.1080/2162402X.2021.1958590
76. Bruhns P. Properties of mouse and human IgG receptors and their contribution to disease models. Blood. (2012) 119:5640–9. doi: 10.1182/blood-2012-01-380121
77. Ryman JT and Meibohm B. Pharmacokinetics of monoclonal antibodies. CPT: Pharmacometrics Syst Pharmacol. (2017) 6:576–88. doi: 10.1002/psp4.12224
78. Nimmerjahn F and Ravetch JV. Fcγ receptors as regulators of immune responses. Nat Rev Immunol. (2008) 8:34–47. doi: 10.1038/nri2206
79. Chenoweth AM, Wines BD, Anania JC, and Mark Hogarth P. Harnessing the immune system via FcγR function in immune therapy: A pathway to next-gen mAbs. Immunol Cell Biol. (2020) 98:287–304. doi: 10.1111/imcb.12326
80. Pang KT, Yang YS, Zhang W, Ho YS, Sormanni P, Michaels TCT, et al. Understanding and controlling the molecular mechanisms of protein aggregation in mAb therapeutics. Biotechnol Adv. (2023) 67:108192. doi: 10.1016/j.bioteChadv.2023.108192
81. Kayser V, Chennamsetty N, Voynov V, Forrer K, Helk B, and Trout BL. Glycosylation influences on the aggregation propensity of therapeutic monoclonal antibodies. Biotechnol J. (2011) 6:38–44. doi: 10.1002/biot.201000091
82. Wang X, Kumar S, Buck PM, and Singh SK. Impact of deglycosylation and thermal stress on conformational stability of a full length murine igG2a monoclonal antibody: Observations from molecular dynamics simulations. Proteins: Structure Function Bioinf. (2013) 81:443–60. doi: 10.1002/prot.24202
83. Zheng K, Bantog C, and Bayer R. The impact of glycosylation on monoclonal antibody conformation and stability. mAbs. (2011) 3:568–76. doi: 10.4161/mabs.3.6.17922
84. Liu S and Liu X. IgG N-glycans. In: Advances in Clinical Chemistry, vol. 105. San Diego, CA, USA: Elsevier (2021). p. 1–47. doi: 10.1016/bs.acc.2021.02.001
85. Van De Bovenkamp FS, Hafkenscheid L, Rispens T, and Rombouts Y. The emerging importance of IgG fab glycosylation in immunity. J Immunol. (2016) 196:1435–41. doi: 10.4049/jimmunol.1502136
86. Van De Bovenkamp FS, Derksen NIL, Ooijevaar-de Heer P, Van Schie KA, Kruithof S, Berkowska MA, et al. Adaptive antibody diversification through N -linked glycosylation of the immunoglobulin variable region. Proc Natl Acad Sci. (2018) 115:1901–6. doi: 10.1073/pnas.1711720115
87. Courtois F, Agrawal NJ, Lauer TM, and Trout BL. Rational design of therapeutic mAbs against aggregation through protein engineering and incorporation of glycosylation motifs applied to bevacizumab. mAbs. (2016) 8:99–112. doi: 10.1080/19420862.2015.1112477
88. Meudt M, Baumeister J, Mizaikoff B, Ebert S, Rosenau F, Blech M, et al. Comprehensive analysis and characterization of glycan pairing in therapeutic antibodies and Fc-containing biotherapeutics: Addressing current limitations and implications for N-glycan impact. European Journal of Pharmaceutics and Biopharmaceutics. (2024) 200:114325. doi: 10.1016/j.ejpb.2024.114325
89. Shi HH and Goudar CT. Recent advances in the understanding of biological implications and modulation methodologies of monoclonal antibody N-linked high mannose glycans. Biotechnol Bioengineering. (2014) 111:1907–19. doi: 10.1002/bit.25318
90. Flynn GC, Chen X, Liu YD, Shah B, and Zhang Z. Naturally occurring glycan forms of human immunoglobulins G1 and G2. Mol Immunol. (2010) 47:2074–82. doi: 10.1016/j.molimm.2010.04.006
91. Zhou Q, Shankara S, Roy A, Qiu H, Estes S, McVie-Wylie A, et al. Development of a simple and rapid method for producing non-fucosylated oligomannose containing antibodies with increased effector function. Biotechnol Bioengineering. (2008) 99:652–65. doi: 10.1002/bit.21598
92. Qian J, Liu T, Yang L, Daus A, Crowley R, and Zhou Q. Structural characterization of N-linked oligosaccharides on monoclonal antibody cetuximab by the combination of orthogonal matrix-assisted laser desorption/ionization hybrid quadrupole-quadrupole time-of-flight tandem mass spectrometry and sequential enzymatic digestion. Analytical Biochem. (2007) 364:8–18. doi: 10.1016/j.ab.2007.01.023
93. Yu M, Brown D, Reed C, Chung S, Lutman J, Stefanich E, et al. Production, characterization, and pharmacokinetic properties of antibodies with N-linked mannose-5 glycans. mAbs. (2012) 4:475–87. doi: 10.4161/mabs.20737
94. Alessandri L, Ouellette D, Acquah A, Rieser M, Leblond D, Saltarelli M, et al. Increased serum clearance of oligomannose species present on a human IgG1 molecule. mAbs. (2012) 4:509–20. doi: 10.4161/mabs.20450
95. Boesch AW, Brown EP, Cheng HD, Ofori MO, Normandin E, Nigrovic PA, et al. Highly parallel characterization of IgG Fc binding interactions. mAbs. (2014) 6:915–27. doi: 10.4161/mabs.28808
96. Bermejo-Jambrina M, Eder J, Helgers LC, Hertoghs N, Nijmeijer BM, Stunnenberg M, et al. C-type lectin receptors in antiviral immunity and viral escape. Front Immunol. (2018) 9:590. doi: 10.3389/fimmu.2018.00590
97. Dasgupta S, Navarrete A-M, Bayry J, Delignat S, Wootla B, André S, et al. A role for exposed mannosylations in presentation of human therapeutic self-proteins to CD4+ T lymphocytes. Proc Natl Acad Sci. (2007) 104:8965–70. doi: 10.1073/pnas.0702120104
98. Sheng K-C, Kalkanidis M, Pouniotis DS, Esparon S, Tang CK, Apostolopoulos V, et al. Delivery of antigen using a novel mannosylated dendrimer potentiates immunogenicity in vitro and in vivo. Eur J Immunol. (2008) 38:424–36. doi: 10.1002/eji.200737578
99. Wolf B, Piksa M, Beley I, Patoux A, Besson T, Cordier V, et al. Therapeutic antibody glycosylation impacts antigen recognition and immunogenicity. Immunology. (2022) 166:380–407. doi: 10.1111/imm.13481
100. Menon S, Rosenberg K, Graham SA, Ward EM, Taylor ME, Drickamer K, et al. Binding-site geometry and flexibility in DC-SIGN demonstrated with surface force measurements. Proc Natl Acad Sci. (2009) 106:11524–9. doi: 10.1073/pnas.0901783106
101. Paurević M, Šrajer Gajdošik M, and Ribić R. Mannose ligands for mannose receptor targeting. Int J Mol Sci. (2024) 25:1370. doi: 10.3390/ijms25031370
102. Švajger U, Anderluh M, Jeras M, and Obermajer N. C-type lectin DC-SIGN: An adhesion, signalling and antigen-uptake molecule that guides dendritic cells in immunity. Cell Signalling. (2010) 22:1397–405. doi: 10.1016/j.cellsig.2010.03.018
103. Wawrzyniak M, Morsy Y, Mladenov R, Tontodonati G, Turgay Y, Herwig S, et al. Fucosylation and Sialylation of Fc-Fragment of anti-Tumour Necrosis Factor Alpha Antibodies do not Influence Their Immunogenicity in Monocyte-Derived Dendritic Cells. J Crohn’s Colitis. (2021) 15:1596–601. doi: 10.1093/ecco-jcc/jjab038
104. Vattepu R, Sneed SL, and Anthony RM. Sialylation as an important regulator of antibody function. Front Immunol. (2022) 13:818736. doi: 10.3389/fimmu.2022.818736
105. Perdicchio M, Ilarregui JM, Verstege MI, Cornelissen LAM, Schetters STT, Engels S, et al. Sialic acid-modified antigens impose tolerance via inhibition of T-cell proliferation and de novo induction of regulatory T cells. Proc Natl Acad Sci. (2016) 113:3329–34. doi: 10.1073/pnas.1507706113
106. Bruhns P and Jönsson F. Mouse and human FcR effector functions. Immunol Rev. (2015) 268:25–51. doi: 10.1111/imr.12350
107. Ferrara C, Grau S, Jäger C, Sondermann P, Brünker P, Waldhauer I, et al. Unique carbohydrate–carbohydrate interactions are required for high affinity binding between FcγRIII and antibodies lacking core fucose. Proc Natl Acad Sci. (2011) 108:12669–74. doi: 10.1073/pnas.1108455108
108. Golay J, Andrea AE, and Cattaneo I. Role of Fc core fucosylation in the effector function of IgG1 antibodies. Front Immunol. (2022) 13:929895. doi: 10.3389/fimmu.2022.929895
109. Shields RL, Lai J, Keck R, O’Connell LY, Hong K, Meng YG, et al. Lack of fucose on human IgG1 N-linked oligosaccharide improves binding to human FcγRIII and antibody-dependent cellular toxicity. J Biol Chem. (2002) 277:26733–40. doi: 10.1074/jbc.M202069200
110. Suzuki E, Niwa R, Saji S, Muta M, Hirose M, Iida S, et al. A nonfucosylated anti-HER2 antibody augments antibody-dependent cellular cytotoxicity in breast cancer patients. Clin Cancer Res. (2007) 13:1875–82. doi: 10.1158/1078-0432.CCR-06-1335
111. Leidi M, Gotti E, Bologna L, Miranda E, Rimoldi M, Sica A, et al. M2 macrophages phagocytose rituximab-opsonized leukemic targets more efficiently than M1 cells in vitro. J Immunol. (2009) 182:4415–22. doi: 10.4049/jimmunol.0713732
112. Nagelkerke SQ, Bruggeman CW, Den Haan JMM, Mul EPJ, Van Den Berg TK, Van Bruggen R, et al. Red pulp macrophages in the human spleen are a distinct cell population with a unique expression of Fc-γ receptors. Blood Adv. (2018) 2:941–53. doi: 10.1182/bloodadvances.2017015008
113. Damelang T, Brinkhaus M, Van Osch TLJ, Schuurman J, Labrijn AF, Rispens T, et al. Impact of structural modifications of IgG antibodies on effector functions. Front Immunol. (2024) 14:1304365. doi: 10.3389/fimmu.2023.1304365
114. Hale G. Living in LALA land? Forty years of attenuating Fc effector functions. Immunol Rev. (2024) 328:422–37. doi: 10.1111/imr.13379
115. Jefferis R. Glycosylation as a strategy to improve antibody-based therapeutics. Nat Rev Drug Discov. (2009) 8:226–34. doi: 10.1038/nrd2804
116. Amigorena S, Lankar D, Briken V, Gapin L, Viguier M, and Bonnerot C. Type II and III receptors for immunoglobulin G (IgG) control the presentation of different T cell epitopes from single IgG-complexed antigens. J Exp Med. (1998) 187:505–15. doi: 10.1084/jem.187.4.505
117. van Montfoort N, Mangsbo SM, Camps MGM, van Maren WWC, Verhaart IEC, Waisman A, et al. Circulating specific antibodies enhance systemic cross-priming by delivery of complexed antigen to dendritic cells in vivo. Eur J Immunol. (2012) 42:598–606. doi: 10.1002/eji.201141613
118. Amigorena S, Salamero J, Davoust J, Fridman WH, and Bonnerot C. Tyrosine-containing motif that transduces cell activation signals also determines internalization and antigen presentation via type III receptors for IgG. Nature. (1992) 358:337–41. doi: 10.1038/358337a0
119. Bournazos S, Wang TT, Dahan R, Maamary J, and Ravetch JV. Signaling by antibodies: recent progress. Annu Rev Immunol. (2017) 35:285–311. doi: 10.1146/annurev-immunol-051116-052433
120. Van Vugt MJ, Kleijmeer MJ, Keler T, Zeelenberg I, Van Dijk MA, Leusen JHW, et al. The FcγRIa (CD64) ligand binding chain triggers major histocompatibility complex class II antigen presentation independently of its associated FcR γ-chain. Blood. (1999) 94:808–17. doi: 10.1182/blood.V94.2.808
121. Kalergis AM and Ravetch JV. Inducing tumor immunity through the selective engagement of activating fcγ Receptors on dendritic cells. J Exp Med. (2002) 195:1653–9. doi: 10.1084/jem.20020338
122. Li F, Smith P, and Ravetch JV. Inhibitory Fcγ Receptor is required for the maintenance of tolerance through distinct mechanisms. J Immunol. (2014) 192:3021–8. doi: 10.4049/jimmunol.1302934
123. Casey E, Bournazos S, Mo G, Mondello P, Tan KS, Ravetch JV, et al. A new mouse expressing human Fcγ receptors to better predict therapeutic efficacy of human anti-cancer antibodies. Leukemia. (2018) 32:547–9. doi: 10.1038/leu.2017.293
124. Ebrahimi SB and Samanta D. Engineering protein-based therapeutics through structural and chemical design. Nat Commun. (2023) 14:2411. doi: 10.1038/s41467-023-38039-x
125. Sanchez L, Yi Y, and Yu Y. Effect of partial PEGylation on particle uptake by macrophages. Nanoscale. (2017) 9:288–97. doi: 10.1039/c6nr07353k
126. Brückner M, Fichter M, Da Costa Marques R, Landfester K, and Mailänder V. PEG spacer length substantially affects antibody-based nanocarrier targeting of dendritic cell subsets. Pharmaceutics. (2022) 14:1614. doi: 10.3390/pharmaceutics14081614
127. Quach QH, Kong RLX, and Kah JCY. Complement activation by PEGylated gold nanoparticles. Bioconjugate Chem. (2018) 29:976–81. doi: 10.1021/acs.bioconjchem.7b00793
128. De Bourayne M, Meunier S, Bitoun S, Correia E, Mariette X, Nozach H, et al. Pegylation reduces the uptake of certolizumab pegol by dendritic cells and epitope presentation to T-cells. Front Immunol. (2022) 13:808606. doi: 10.3389/fimmu.2022.808606
129. Gefen T, Vaya J, Khatib S, Harkevich N, Artoul F, Heller ED, et al. The impact of PEGylation on protein immunogenicity. Int Immunopharmacol. (2013) 15:254–9. doi: 10.1016/j.intimp.2012.12.012
130. Veronese FM and Mero A. The impact of PEGylation on biological therapies. BioDrugs. (2008) 22:315–29. doi: 10.2165/00063030-200822050-00004
131. Datta-Mannan A, Thangaraju A, Leung D, Tang Y, Witcher DR, Lu J, et al. Balancing charge in the complementarity-determining regions of humanized mAbs without affecting pI reduces non-specific binding and improves the pharmacokinetics. mAbs. (2015) 7:483–93. doi: 10.1080/19420862.2015.1016696
132. Nichols P, Li L, Kumar S, Buck PM, Singh SK, Goswami S, et al. Rational design of viscosity reducing mutants of a monoclonal antibody: Hydrophobic versus electrostatic inter-molecular interactions. mAbs. (2015) 7:212–30. doi: 10.4161/19420862.2014.985504
133. Jain T, Boland T, and Vásquez M. Identifying developability risks for clinical progression of antibodies using high-throughput in vitro and in silico approaches. mAbs. (2023) 15:2200540. doi: 10.1080/19420862.2023.2200540
134. Rabia LA, Zhang Y, Ludwig SD, Julian MC, and Tessier PM. Net charge of antibody complementarity-determining regions is a key predictor of specificity. Protein Engineering Design Selection. (2018) 31:409–18. doi: 10.1093/protein/gzz002
135. Das TK, Chou DK, Jiskoot W, and Arosio P. Nucleation in Protein Aggregation in Biotherapeutic Development: A look into the Heart of the Event. J Pharm Sci. (2022) 111:951–9. doi: 10.1016/j.xphs.2022.01.017
136. Esfandiary R, Parupudi A, Casas-Finet J, Gadre D, and Sathish H. Mechanism of reversible self-association of a monoclonal antibody: role of electrostatic and hydrophobic interactions. J Pharm Sci. (2015) 104:577–86. doi: 10.1002/jps.24237
137. Manzanares D and Ceña V. Endocytosis: the nanoparticle and submicron nanocompounds gateway into the cell. Pharmaceutics. (2020) 12:371. doi: 10.3390/pharmaceutics12040371
138. Sousa De Almeida M, Susnik E, Drasler B, Taladriz-Blanco P, Petri-Fink A, and Rothen-Rutishauser B. Understanding nanoparticle endocytosis to improve targeting strategies in nanomedicine. Chem Soc Rev. (2021) 50:5397–434. doi: 10.1039/D0CS01127D
139. Wang EQ, Kaila N, Plowchalk D, Gibiansky L, Yunis C, and Sweeney K. Population PK/PD modeling of low-density lipoprotein cholesterol response in hypercholesterolemic participants following administration of bococizumab, a potent anti-PCSK9 monoclonal antibody. CPT: Pharmacometrics Syst Pharmacol. (2023) 12:2013–26. doi: 10.1002/psp4.13050
140. Liu S, Verma A, Kettenberger H, Richter WF, and Shah DK. Effect of variable domain charge on in vitro and in vivo disposition of monoclonal antibodies. mAbs. (2021) 13:1993769. doi: 10.1080/19420862.2021.1993769
141. Thompson DB, Villaseñor R, Zerial M, and Liu DR. Cellular uptake mechanisms and endosomal trafficking of supercharged proteins. Chem Biol. (2012) 19:831–43. doi: 10.1016/j.chembiol.2012.06.014
142. Deng Y, Govers C, Teodorowicz M, Liobyte I, De Simone I, Hettinga K, et al. Hydrophobicity drives receptor-mediated uptake of heat-processed proteins by THP-1 macrophages and dendritic cells, but not cytokine responses. PloS One. (2020) 15:e0236212. doi: 10.1371/journal.pone.0236212
143. Sabourian P, Yazdani G, Ashraf SS, Frounchi M, Mashayekhan S, Kiani S, et al. Effect of physico-chemical properties of nanoparticles on their intracellular uptake. Int J Mol Sci. (2020) 21:8019. doi: 10.3390/ijms21218019
144. Su R, Cao X, Zhao J, and Wang F. A high hydrophobic moment arginine-rich peptide screened by a machine learning algorithm enhanced ADC antitumor activity. J Pept Sci. (2024) 30(2):e3628. doi: 10.1002/psc.3628
145. Jain K, Salamat-Miller N, and Taylor K. Freeze–thaw characterization process to minimize aggregation and enable drug product manufacturing of protein based therapeutics. Sci Rep. (2021) 11:11332. doi: 10.1038/s41598-021-90772-9
146. Hebditch M and Warwicker J. Charge and hydrophobicity are key features in sequence-trained machine learning models for predicting the biophysical properties of clinical-stage antibodies. PeerJ. (2019) 7:e8199. doi: 10.7717/peerj.8199
147. Ahmadi M, Bryson CJ, Cloake EA, Welch K, Filipe V, Romeijn S, et al. Small amounts of sub-visible aggregates enhance the immunogenic potential of monoclonal antibody therapeutics. Pharm Res. (2015) 32:1383–94. doi: 10.1007/s11095-014-1541-x
148. Aoyama M, Tada M, Yokoo H, Demizu Y, and Ishii-Watabe A. Fcγ Receptor-dependent internalization and off-target cytotoxicity of antibody-drug conjugate aggregates. Pharm Res. (2022) 39:89–103. doi: 10.1007/s11095-021-03158-x
149. Rombach-Riegraf V, Karle AC, Wolf B, Sordé L, Koepke S, Gottlieb S, et al. Aggregation of human recombinant monoclonal antibodies influences the capacity of dendritic cells to stimulate adaptive T-cell responses in vitro. PloS One. (2014) 9:e86322. doi: 10.1371/journal.pone.0086322
150. Sung Y, Choi Y, Kim ES, Ryu JH, and Kwon IC. Receptor-ligand interactions for optimized endocytosis in targeted therapies. J Controlled Release. (2025) 380:524–38. doi: 10.1016/j.jconrel.2025.01.060
151. Shah NJ, Kelly WJ, Liu SV, Choquette K, and Spira A. Product review on the Anti-PD-L1 antibody atezolizumab. Hum Vaccines Immunotherapeutics. (2018) 14:269–76. doi: 10.1080/21645515.2017.1403694
152. Zanello A, Bortolotti M, Maiello S, Bolognesi A, and Polito L. Anti-PD-L1 immunoconjugates for cancer therapy: Are available antibodies good carriers for toxic payload delivering? Front Pharmacol. (2022) 13:972046. doi: 10.3389/fphar.2022.972046
153. Deora A, Hegde S, Lee J, Choi C-H, Chang Q, Lee C, et al. Transmembrane TNF-dependent uptake of anti-TNF antibodies. mAbs. (2017) 9:680–95. doi: 10.1080/19420862.2017.1304869
154. Ecker M, Redpath GMI, Nicovich PR, and Rossy J. Quantitative visualization of endocytic trafficking through photoactivation of fluorescent proteins. Mol Biol Cell. (2021) 32:892–902. doi: 10.1091/mbc.E20-10-0669
155. Xue L, Hickling T, Song R, Nowak J, and Rup B. Contribution of enhanced engagement of antigen presentation machinery to the clinical immunogenicity of a human interleukin (IL)-21 receptor-blocking therapeutic antibody. Clin Exp Immunol. (2015) 183:102–13. doi: 10.1111/cei.12711
156. Kameyama S, Horie M, Kikuchi T, Omura T, Tadokoro A, Takeuchi T, et al. Acid wash in determining cellular uptake of Fab/cell-permeating peptide conjugates. Pept Sci. (2007) 88:98–107. doi: 10.1002/bip.20689
157. Verma AK, Noumani A, Yadav AK, and Solanki PR. FRET based biosensor: principle applications recent advances and challenges. Diagnostics. (2023) 13:1375. doi: 10.3390/diagnostics13081375
158. Walsh RE, Lannan M, Wen Y, Wang X, Moreland CA, Willency J, et al. Post-hoc assessment of the immunogenicity of three antibodies reveals distinct immune stimulatory mechanisms. mAbs. (2020) 12:1764829. doi: 10.1080/19420862.2020.1764829
159. Wen Y, Waltman A, Han H, and Collier JH. Switching the immunogenicity of peptide assemblies using surface properties. ACS Nano. (2016) 10:9274–86. doi: 10.1021/acsnano.6b03409
160. Scherger M, Bolli E, Antunes ARP, Arnouk S, Stickdorn J, Van Driessche A, et al. Transient multivalent nanobody targeting to CD206-expressing cells via PH-degradable nanogels. Cells. (2020) 9:2222. doi: 10.3390/cells9102222
Keywords: therapeutic antibodies, dendritic cells, internalization, immunogenicity, immunoglobulin
Citation: Lteif M, Pallardy M and Turbica I (2025) Antigen-presenting cell internalization is key for understanding and evaluating therapeutic antibodies’ immunogenicity. Front. Immunol. 16:1617395. doi: 10.3389/fimmu.2025.1617395
Received: 24 April 2025; Accepted: 13 August 2025;
Published: 05 September 2025.
Edited by:
Roger Y. Tam, Health Canada, CanadaReviewed by:
Swarali N. Kurle, National AIDS Research Institute, IndiaThomas Emanuel Kraft, Roche Diagnostics GmbH, Germany
Copyright © 2025 Lteif, Pallardy and Turbica. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Isabelle Turbica, aXNhYmVsbGUudHVyYmljYUB1bml2ZXJzaXRlLXBhcmlzLXNhY2xheS5mcg==
†These authors share last authorship