 Ka Fai Leong
Ka Fai Leong Zihan Chen2
Zihan Chen2 Paolo Coghi
Paolo Coghi- 1Faculty of Chinese Medicine, Macau University of Science and Technology, Macau, Macau SAR, China
- 2School of Pharmacy, Macau University of Science and Technology, Macau, Macau SAR, China
Immunotherapy has revolutionized cancer treatment by leveraging the body’s immune system to recognize and eliminate tumor cells. While monoclonal antibodies and checkpoint inhibitors have shown dramatic clinical successes, small molecules are increasingly recognized for their potential to modulate the immune system with improved pharmacokinetics and oral bioavailability. The incorporation of fluorine atoms into small molecule structures has become a widely used strategy to enhance therapeutic efficacy. Fluorine’s unique chemical properties such as high electronegativity, metabolic stability, and ability to modulate lipophilicity make fluorinated small molecules especially attractive for immunotherapeutic applications. This minireview highlights recent advances in fluorinated small molecules that target key immune pathways, including immune checkpoints, STING agonists, IDO inhibitors, and kinase pathways involved in immune regulation. We explore the chemical rationale, mechanisms of action, and therapeutic outcomes of fluorinated compounds currently in preclinical and clinical development. The discussion also addresses challenges such as immunotoxicity, resistance, and design strategies to overcome them. Together, these findings underscore the growing relevance of fluorinated small molecule immunotherapeutics in cancer treatment.
1 Introduction
The advent of immunotherapy has marked a paradigm shift in oncology, offering long-term remission for cancers that were once considered untreatable (1). Immune checkpoint inhibitors, such as those targeting PD-1, PD-L1, and CTLA-4, have led to breakthrough responses in several cancers. However, many patients fail to respond or develop resistance, underscoring the need for novel therapeutic modalities (2).
Small molecules are emerging as valuable alternatives or complements to antibody-based immunotherapies. Their advantages include oral administration, better tumor penetration, lower costs, and ability to target intracellular sites inaccessible to biologics (3–5). In this context, halogen plays a critical role in optimizing their performance.
Halogenation, particularly the replacement of hydrogen atoms with halogens, is a widely used strategy in compound optimization. Halogen substitution can sometimes enhance potency by orders of magnitude (6). Among halogens, fluorine substitution is especially notable for its profound impact on key physicochemical properties such as pKa, lipophilicity, metabolic stability, and molecular conformation. For instance, fluorination can alter the pKa exceed one log unit.
Fluorination usually increases lipophilicity, but may reduce it at saturated alkyl groups. Fluorine’s van der Waals radius, close to hydrogen’s, minimally affects conformation when monosubstitution. However, larger fluorinated groups such as trifluoromethyl, whose steric volume is comparable to that of an ethyl group, can significantly alter molecular geometry by changing bond angles (7, 8).
Fluorine substitution at chiral centers prone to in vivo racemization—such as that seen in thalidomide—has been shown to prevent such stereochemical interconversion. From a molecular interaction standpoint, fluorine forms weaker hydrogen bonds than hydrogen but exhibits stronger electrostatic interactions. Importantly, fluorine substitution at sites susceptible to metabolism by enzymes can confer metabolic resistance, primarily due to increased steric hindrance. This strategy is used in designing analog inhibitors, including nucleoside analogues (9–16).
This minireview focuses on the expanding role of fluorinated small molecule derivatives in cancer immunotherapy in last 5 years. We categorize recent developments into key target areas and highlight representative examples that illustrate the therapeutic promise of these compounds.
Here we present an integrated schematic (Figure 1) summarizing the key immunometabolic and signaling pathways that shape tumor–immune interactions and highlight therapeutic targets within the tumor microenvironment (TME).
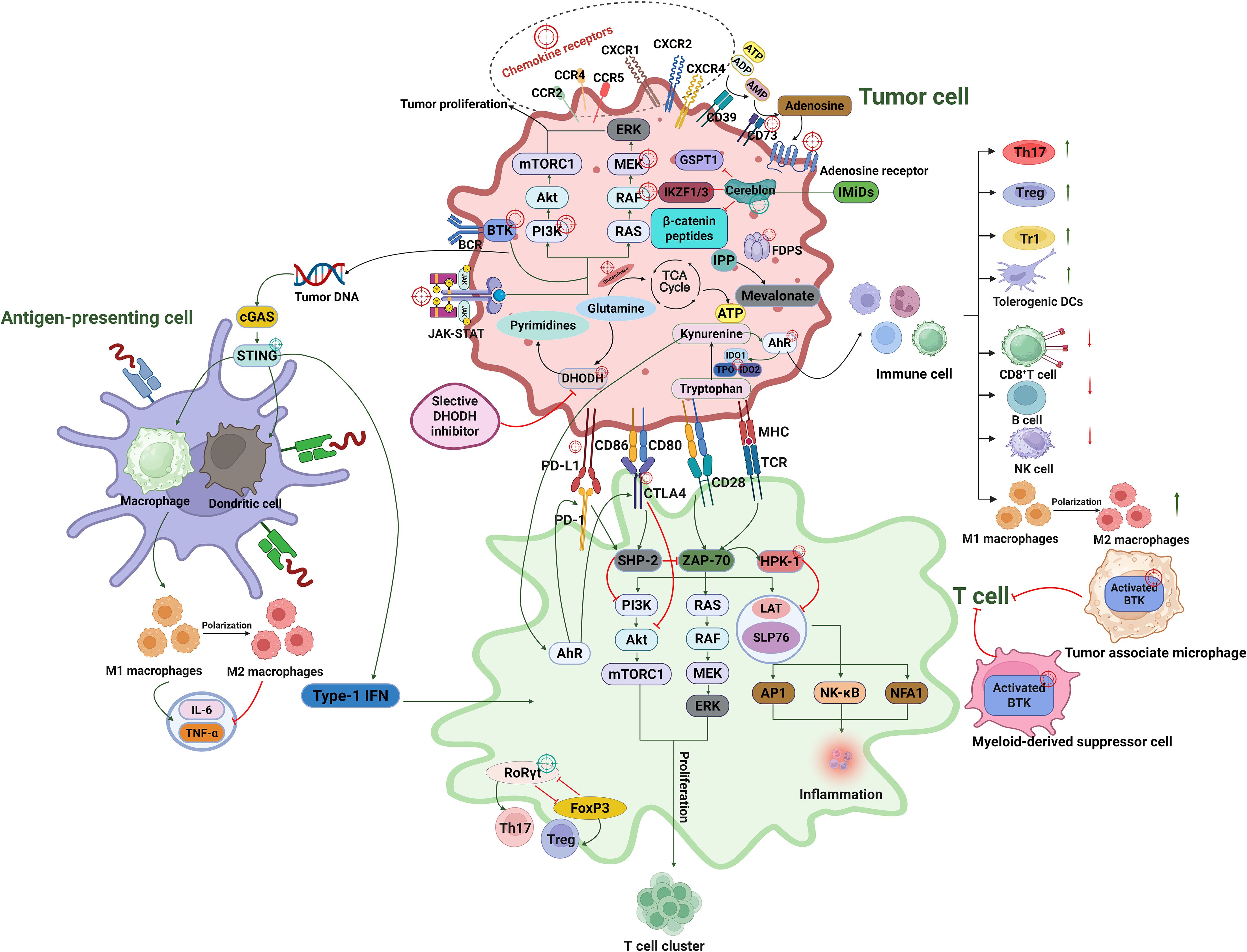
Figure 1. Integrated schematic of tumor immunomodulatory mechanisms involving metabolic, signaling, and checkpoint pathways across tumor cells, T cells, and antigen-presenting cells. The red crosshairs represent the suppression targets, and the green crosshairs represent the activation targets.
This diagram illustrates key molecular pathways regulating the tumor-immune interface. In tumor cells, metabolic regulators such as DHODH, glutamine, pyrimidine synthesis, the TCA cycle, and tryptophan catabolism (via IDO/TDO) modulate immune escape and support proliferation. Transcriptional and signaling pathways including PI3K/AKT/mTOR, RAS/RAF/MEK/ERK, BTK, and β-catenin converge to sustain tumor cell survival, EMT, and resistance. Immunomodulatory protein degradation by cereblon–recruited E3 ligases targets substrates such as IKZF1/3, GSPT1, and β-catenin fragments, influencing immune regulation and tumor viability. Tumor-derived chemokines (CCL2, CCL5, CCL22, CXCL8, CXCL12) recruit suppressive immune cells including tumor-associated macrophages (TAMs), myeloid-derived suppressor cells (MDSCs), and regulatory T cells (Treg) via chemokine receptors (CCR2, CCR4, CCR5, CXCR1/2, CXCR4), reinforcing an immunosuppressive TME. The STING–cGAS pathway in antigen-presenting cells, macrophages, dendritic cells (DCs), activates type I interferon responses that prime cytotoxic T cell activity. T cell activation is mediated through TCR/CD28 costimulation, engaging PI3K, LAT, and MAPK cascades; this is negatively regulated by immune checkpoints (PD-1/PD-L1, CTLA-4/CD80) and intracellular suppressors such as HPK1. T cell lineage commitment is modulated by transcriptional regulators including FoxP3 and RORγt, influencing Treg and Th17 differentiation, respectively. This figure synthesizes multiple molecular interactions to illustrate how tumor cells co-opt immune regulatory pathways and highlights potential therapeutic targets across immune metabolism, checkpoint inhibition, signal transduction, and transcriptional control.
2 Class of compounds
We identified over 80 fluorinated small molecules with reported immunomodulatory activity in the past five years. Their chemical structures are illustrated in Figure 2 and are categorized according to their pharmacological mechanisms.
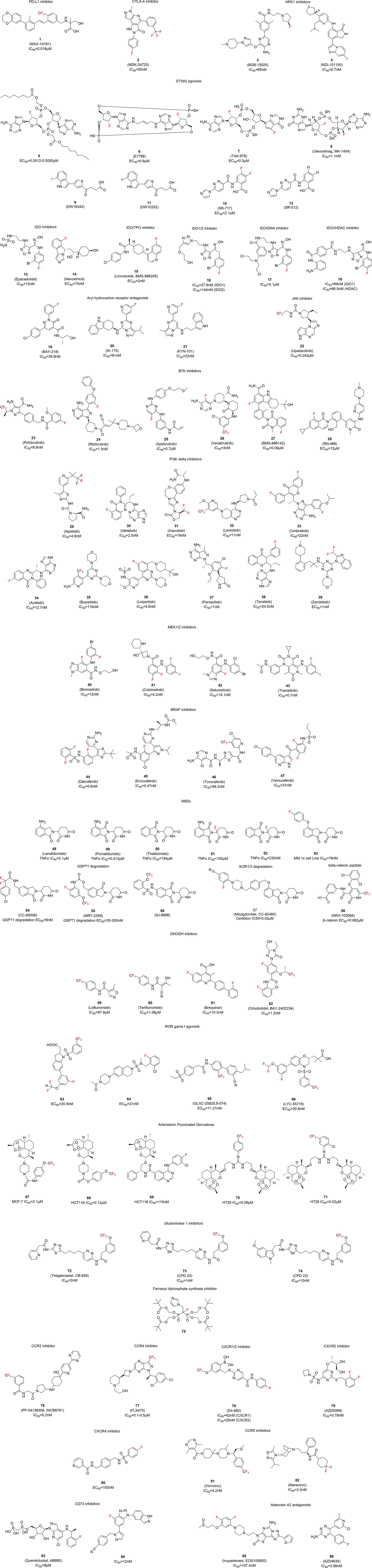
Figure 2. Fluorinated compounds with immunomodulatory activity.
2.1 Fluorinated immune checkpoint inhibitors
Immune checkpoints serve as critical regulators that suppress T cell activity to maintain immune homeostasis. However, tumors hijack these pathways to evade immune surveillance. Checkpoint blockade therapies, such as anti-PD-1 and anti-CTLA-4 antibodies, have revolutionized cancer treatment by reactivating antitumor immunity. Nonetheless, challenges such as immune-related toxicities, poor tumor penetration, and therapeutic resistance necessitate the exploration of alternative strategies.
Small molecule immune checkpoint inhibitors (ICIs) offer potential advantages, including oral bioavailability, improved tissue penetration, and cost-effective production (17, 18). Their development is crucial for broadening the scope of immunotherapy across different cancer types and patient populations.
2.1.1 Small molecule PD-1/PD-L1 inhibitors
While most approved PD-1/PD-L1 inhibitors are monoclonal antibodies, small molecule alternatives are under development to overcome limitations such as poor tissue penetration and immune-related adverse events (irAEs) (19, 20).
Fluorinated aromatic groups have been critical in enhancing the binding affinity of small molecule PD-L1 inhibitors. Compounds such as compound 1 (21, 22), and others utilize fluoroaryl moieties to improve hydrophobic interactions within the PD-L1 binding pocket. For instance, a fluorophenyl group in the BMS series increases π-stacking and binding site complementarity, enhancing antagonistic activity.
2.1.2 Small molecule CTLA-4/CD80 inhibitors
While less explored than PD-1/PD-L1, fluorinated derivatives compound 2 targeting the CTLA-4/CD80 axis are being investigated (23). These molecules often feature fluorinated indole or quinazoline cores to mimic protein-protein interaction surfaces.
2.1.3 Small molecule HKP1 inhibitors
Hematopoietic progenitor kinase 1 (HPK1) is an immunosuppressive regulatory factor that is initially expressed at low levels in lymphoid progenitor cells but becomes highly expressed in mature immune cells, such as T cells, B cells, DCs, and M2 macrophages (24, 25). Inhibiting HPK1 has emerged as a promising strategy to enhance antitumor immunity.
Several fluorinated small molecule HPK1 inhibitors have already progressed into clinical trials, including compound 3 and 4 (26–29). These inhibitors aim to counteract HPK1-mediated suppression of T cell receptor (TCR) signaling, thus potentiating T cell activation and improving the efficacy of cancer immunotherapies.
Fluorine in HPK1 inhibitors improves binding, stability, and pharmacokinetics, enabling synergy with checkpoint inhibitors for cancer therapy.
2.2 STING agonists with fluorinated enhancements
The STING (Stimulator of Interferon Genes) pathway activates innate immunity via type I interferon responses, making it a promising target for cancer immunotherapy. While many STING agonists are locally administered due to systemic toxicity, fluorinated analogs have shown potential for oral or systemic use by improving pharmacokinetic profiles.
2.2.1 Cyclic dinucleotide mimetics
Natural STING agonists, such as cyclic dinucleotides (CDNs), suffer from poor membrane permeability and metabolic degradation. Fluorinated analogs have been synthesized to address these limitations (30).
For example, fluorinated deoxyribose and indole-based STING agonists have demonstrated improved potency, cellular uptake, and resistance to enzymatic hydrolysis. Strategic fluorine placement stabilizes the molecule and enhances cGAS-STING pathway activation in tumor-associated immune cells (31).
Dejmek and colleagues substituted fluorine atoms for the free hydroxyl groups on the pentose ring of cyclic dinucleotides. Remarkably, this modification increased the agonistic activity by 10- to 100-fold. The enhanced efficacy is attributed to better cell permeability, greater anti-degradation properties, and stronger binding to the STING protein.
To further improve cellular permeability and metabolic stability, long-chain alkyl groups were conjugated to the phosphate hydroxyl groups to create prodrugs. For instance, compound 5, modified with an n-octanoyl group, exhibited an EC50 of less than 1 nM, representing more than a 10,000-fold improvement in potency compared to the parent CDN (32).
Several fluorinated CDN analogs, such as compound 6-8 (33–39), have advanced into phase I clinical trials, further underscoring the translational potential of this approach.
2.2.2 Non-nucleoside STING activation
Non-nucleoside drugs are also a research hotspots. Compared with traditional CDN STING agonists, non-nucleoside drugs have better metabolic stability and cell permeability. Some compounds are orally bioavailable. However, their development is hindered by structural complexity and species-specific STING binding differences between humans and models like mice.
Compound 9 (40) and 10 (41) are particularly noteworthy. Compared with its non-fluorinated analog compound 11, compound 9 incorporates fluorine at the 7-position of the indole ring, significantly improving binding stability, metabolic resistance, and, to a lesser extent, agonistic activity. Similarly, compound 10 demonstrates the crucial role of fluorine in modulating bioactivity: its non-fluorinated precursor, compound 12, exhibits no STING activation, whereas the fluorine-substituted compound 10 shows strong thermal stability and induces a closed conformation of the STING protein, effectively triggering downstream immune responses.
2.3 IDO and TDO pathway inhibitors
Indoleamine 2,3-dioxygenase (IDO1) and tryptophan 2,3-dioxygenase (TDO) enzymes mediate immune suppression in the TME by depleting tryptophan and producing immunosuppressive kynurenines (42–44). These pathways impair T cell proliferation, promote Treg differentiation, and support tumor immune escape.
2.3.1 Fluorinated IDO1 inhibitors
Fluorinated derivatives of IDO1 inhibitors have demonstrated improved pharmacological properties, including enhanced metabolic stability, target specificity, and bioavailability. Compounds such as compound 13 and 14 (45–47) incorporate fluoroaryl groups to fine-tune physicochemical properties such as pKa and lipophilicity, optimizing enzyme inhibition under physiological conditions. These modifications improve selectivity and reduce off-target toxicity, critical for clinical success.
2.3.2 Dual inhibition
To overcome redundancy between the IDO and TDO pathways, dual inhibitors are under development. Many of these compounds incorporate fluorinated aromatic rings that span both enzyme binding pockets, enhancing dual target engagement. One notable example is compound 15 (47, 48), a fluorinated dual IDO/TDO inhibitor with promising clinical activity.
Beyond IDO/TDO dual inhibitors, other bifunctional molecules have been designed to target IDO1 along with additional oncogenic or epigenetic pathways. For example, compound 16, a derivative of epacadostat 13, inhibits both IDO1 and IDO2 (49), compound 17 possesses both IDO1 inhibitory and DNA alkylation activities (50), and compound 18 simultaneously inhibits IDO1 and histone deacetylases (HDACs), which modulate chromatin architecture and gene expression (51). These inhibitors may enhance antitumor efficacy via synergy.
2.3.3 Aryl hydrocarbon receptor antagonists
Kynurenine binds to the aryl hydrocarbon receptor (AhR) and further suppresses antitumor immunity (52, 53). Fluorinated AhR antagonists—such as compound 19-21 (54–57)—can block this interaction, thereby restoring immune activation and reprogramming the TME. Fluorine incorporation into these molecules enhances their metabolic stability and binding affinity, which are essential for disrupting the kynurenine-AhR axis and achieving therapeutic benefit.
2.4 Kinase inhibitors with immunomodulatory functions
Many oncogenic kinases also regulate immune evasion and cytokine production, making kinase inhibitors effective immunomodulatory agents.
2.4.1 JAK/STAT pathway
Fluorinated Janus kinase (JAK) inhibitors, such as compound 22 (approved in 2019), modulate interferon signaling and T-cell responses. Fluorination enhances selectivity among JAK isoforms and reduces hematologic toxicity (58).
2.4.2 BTK and PI3K inhibitors
Bruton’s tyrosine kinase (BTK) and phosphoinositide 3-kinase delta (PI3K-δ) inhibitors influence B-cell malignancies and Treg populations. Fluorinated analogs exhibit improved target occupancy and immune reprogramming effects.
As of April 2025, five BTK inhibitors have been FDA-approved (59–63), two of which—compound 23,24—are fluorinated (64, 65). Other fluorinated BTK inhibitors, such as compound 25-28 (66–74), are under clinical or preclinical investigation.
Regarding PI3K-δ inhibitors, seven have received FDA approval (75–81), five of which—compound 29-33—are fluorinated (82–86). Additional fluorinated PI3K-δ inhibitors under clinical development include compound 34-39 (86–96) with most having completed phase II trials.
2.4.3 BRAF and MEK1/2 inhibitors
The RAF/MEK/ERK pathway regulates tumor cell proliferation, differentiation, survival, and migration, and it also modulates immune responses. ERK activation increases immunosuppressive factors, such as interleukin-10 (IL-10), promotes Th2 cell differentiation, and reduces CD8+ T-cell and B-cell activity (97).
Currently, five MEK1/2 inhibitors are FDA-approved (98–106), four of which——compound 40-43——are fluorinated. Additionally, all approved BRAF inhibitors, compound 44-47, are fluorinated agents (107–111).
2.5 Fluorinated immunomodulators on hematopoietic system and immune cell differentiation
2.5.1 Fluorinated derivatives of thalidomide and lenalidomide
Glutarimide-based immunomodulatory drugs (IMiDs), such as lenalidomide 48, pomalidomide 49 and thalidomide 50, modulate cereblon, a component of the E3 ubiquitin ligase complex, to induce selective protein degradation (112, 113). IMiDs enhance T cell and NK cell activities by promoting IL-2, CD28, and IFN-γ expression. Their combination with monoclonal antibodies like daratumumab or elotuzumab is recommended as first-line therapy for multiple myeloma (114). IMiDs are also under investigation in combination with CAR-T therapy and cancer vaccines such as PVX-410 (115). Fluorination of the phthalimide or glutarimide ring, compound 51-53, can fine-tune cereblon binding and degradation selectivity, thus providing a stronger immunomodulation effect (116, 117).
New-generation IMiDs have improved specificity to minimize side effects. Examples include compound 54,55 targeting GSPT1, compound 56 targeting GSPT1/2, compound 57 targeting IKZF1/3, and compound 58 targeting β-catenin peptides (118–124). G1 to S phase transition 1/2 (GSPT1/2) are a key factor in cell proliferation and Ikaros zing-finger family transcription factors (IKZF TFs) are a regulators play an important role in lymphocyte development and differentiation (125). In the other hand, β-catenin peptide is a key transducer in Wnt signal pathway, has a very close relationship with tumor occurrence (126).
2.5.2 DHODH inhibitor
Dihydroorotate dehydrogenase (DHODH) is a mitochondrial enzyme responsible for the fourth step of pyrimidine biosynthesis and is essential for cellular proliferation. Currently available fluorinated DHODH inhibitors include leflunomide 59 and its active metabolite teriflunomide 60, primarily used to treat rheumatoid arthritis and multiple sclerosis (127–129). Although these drugs inhibit T and B cell proliferation, many cancer cells upregulate DHODH expression to sustain rapid growth. Compounds such as compound 61,62 (129–132) have entered early-phase clinical trials targeting acute myeloid leukemia.
2.5.3 Immune cell differentiation
Retinoic acid receptor-related orphan receptor gamma t (RORγt) is a nuclear transcription factor specific to immune cells. Its activation induces the differentiation of T cells into Th17 cells, thereby enhancing antitumor immunity (133). Several fluorinated small molecules targeting RORγt have been reported, including Compound 63-66 (134–138), the latter having advanced into clinical trials.
2.6 Fluorinated molecules in tumor microenvironment modulation
The tumor microenvironment (TME) plays a central role in immune suppression. Fluorinated small molecules can modulate several aspects of the TME:
2.6.1 pH sensitive and iron sensitive fluorinated compound
pH-responsive fluorinated carriers enhance drug release in acidic tumor conditions. Fluorination improves nanoparticle stability and pH sensitivity without impairing self-assembly (139). These nanoparticles preferentially release drugs in acidic TME (pH 5.0–6.5), reducing systemic toxicity at physiological pH (~7.4).
Additionally, acidic conditions promote iron release, and tumor cells upregulate transferrin receptor 1 (TfR1) to meet proliferative demands (140). Artemisinin, leveraging its endoperoxide bridge, generates reactive oxygen species (ROS) in high-iron environments, inducing tumor cell death. Fluorinated derivatives of artemisinin, compounds 67-71, show improved cell penetration, ROS generation, stability, and tumor selectivity (141–144).
2.6.2 Metabolic inhibitors
Fluorinated metabolic inhibitors block lactate production or glutaminolysis, reprogramming immune cell metabolism. Metabolic reprogramming in tumors, such as increased lactate production and glutaminolysis, contributes to an immunosuppressive microenvironment. Targeting these pathways can modulate immune responses (145).
While specific fluorinated inhibitors are under investigation, the general strategy involves disrupting lactate dehydrogenase activity and glutamine metabolism to reprogram immune cell function within tumors (146). Notable drug candidates under clinical investigation include compound 72 and its preclinical analogues compound 73,74 (147–149).
In addition to targeting lactate and glutamine, inhibition of farnesyl diphosphate synthase (FDPS) is another emerging area of research. FDPS is a key enzyme in the mevalonate pathway, responsible for depleting isopentenyl pyrophosphate (IPP), thereby reducing T cell activation and impairing antigen presentation by DCs and macrophages. FDPS also activates osteoclasts, promoting tumor metastasis. Bisphosphonates like zoledronate target FDPS to reduce bone destruction. Fluorination of bisphosphonates, compound 75, significantly enhances their inhibitory potency, increases T cell activity—particularly Vγ2Vδ2 T cells—improves antigen presentation, and reduces bone metastasis (150).
2.6.3 Chemokine receptor
Chemokine receptors play critical roles in tumor growth, metastasis, and immune suppression (151), Several fluorinated chemokine receptor antagonists have been developed, targeting CCR2 (152), CCR4 (153), CCR5 (154), CXCR1 (155), CXCR2 (156), and CXCR4 (157, 158), effectively blocking immune cell trafficking that promotes tumor growth.
Multiple chemokine receptor antagonists have advanced to clinical trials, including compound 76 targeting CCR2 (159, 160), compound 77 targeting CCR4 (161–164), compound 78, a dual CXCR1/2 inhibitor (165, 166), compound 79 targeting CXCR2 (156, 167), Compound 80 targeting CXCR4 (157), and the repurposed HIV drugs vicriviroc 81 and maraviroc 82 (168–170) as CCR5 antagonists.
2.6.4 Fluorinated CD39/CD73/A2AR antagonists
Fluorinated CD39/CD73/A2AR antagonists can block tumors from exploiting high levels of ATP in the TME to establish an immune-cold shield and promote tumor growth. CD39 converts extracellular ATP and ADP into AMP, while CD73 further converts AMP into adenosine, which activates the adenosine 2A receptor (A2AR), promoting the expansion of immunosuppressive cell types such as Treg and Th2 cells (171).
Reported fluorinated CD73 inhibitors include compound 83 (172, 173) and a compound 84 discovered by Beatty et al. (174). Among A2AR antagonists, compound 85,86 (175–178) have entered clinical trials.
3 Discussion and future prospective
Fluorine incorporation in small molecule immunotherapeutics enhances potency, selectivity, and durability. In checkpoint inhibition, it improves PD-L1 and CTLA-4 antagonist properties. Fluorinated STING agonists show better uptake and activity, while IDO1 and TDO inhibitors gain improved pharmacokinetics and immune modulation, advancing cancer immunotherapy strategies.
Though promising for immunotherapy due to enhanced pharmacokinetic properties and specificity, fluorinated small molecules also present challenges, notably the risk of irAEs stemming from systemic immune activation (179). Although direct studies linking fluorinated compounds to irAEs are limited, insights can be drawn from the broader context of immunotherapies, particularly ICIs, which have been extensively studied for their irAE profiles (180, 181).
Researches indicate that ICIs disrupt immune balance, causing irAEs via multiple mechanisms. For instance, a comprehensive review highlights that the activation or reactivation of T cells is a dominant factor in the development of ICI-related irAEs, with enhanced Th17 cell responses contributing to the production of proinflammatory cytokines like IL-17A, IL-21, and IL-22 (179). These findings underscore the delicate balance required in immunotherapy: enhancing anti-tumor immunity while minimizing collateral immune activation that can lead to adverse events.
Fluorinated small molecules for immunotherapy may trigger irAEs. Mitigation strategies include dose optimization, immune pathway modulation, and thorough preclinical testing. Off-target effects and metabolite toxicity also warrant attention. Future designs should enhance selectivity, reduce toxicity, and address resistance mechanisms like immune suppression or compensatory signaling.
To overcome these challenges, future design strategies include:
Structure-guided drug design incorporating fluorine in metabolically vulnerable regions to enhance stability.
Proteolysis targeting chimera and molecular glues with fluorinated ligands to achieve targeted protein degradation (119).
Nanoformulations and prodrugs using fluorinated linkers to control release and reduce systemic exposure.
Companion diagnostics, such as fluorine-18 labeled PET tracers, to visualize immune response and guide therapy.
The integration of AI-driven molecule optimization, high-throughput screening, and deep learning models for ADMET prediction will further accelerate the development of next-generation fluorinated immunotherapeutics (182).
In summary, fluorinated small molecules represent a dynamic and growing class of agents in cancer immunotherapy. By merging the principles of fluorine chemistry with immuno-oncology, researchers can design novel agents capable of overcoming existing limitations and ushering in a new era of precision immunotherapy.
Author contributions
KFL: Conceptualization, Writing – original draft, Writing – review & editing. ZC: Writing – review & editing, Writing – original draft. PC: Conceptualization, Funding acquisition, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This research was funded by FDCT grants from Macao Science and Technology University to PC (Project Code: 0005-2023-RIA1).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Alderton GK and Bordon Y. Tumor immunotherapy–leukocytes take up the fight. Nat Rev Immunol. (2012) 12:237. doi: 10.1038/nri3197
2. Tan S, Li D, and Zhu X. Cancer immunotherapy: Pros, cons and beyond. BioMed Pharmacother. (2020) 124:109821. doi: 10.1016/j.biopha.2020.109821
3. Wu Y, Yang Z, Cheng K, Bi H, and Chen J. Small molecule-based immunomodulators for cancer therapy. Acta Pharm Sin B. (2022) 12:4287–308. doi: 10.1016/j.apsb.2022.11.007
4. Singh S, Barik D, Arukha AP, Prasad S, Mohapatra I, Singh A, et al. Small molecule targeting immune cells: A novel approach for cancer treatment. Biomedicines. (2023) 11:2621. doi: 10.3390/biomedicines11102621
5. Chattopadhyay S, Hazra R, Mallick A, Gayen S, and Roy S. Small-molecule in cancer immunotherapy: Revolutionizing cancer treatment with transformative, game-changing breakthroughs. Biochim Biophys Acta Rev Cancer. (2024) 1879:189170. doi: 10.1016/j.bbcan.2024.189170
6. Chiodi D and Ishihara Y. Magic Chloro”: Profound effects of the chlorine atom in drug discovery. J Med Chem. (2023) 66:5305–31. doi: 10.1021/acs.jmedchem.2c02015
7. Purser S, Moore PR, Swallow S, and Gouverneur V. Fluorine in medicinal chemistry. Chem Soc Rev. (2008) 37:320–30. doi: 10.1039/B610213C
8. Gillis EP, Eastman KJ, Hill MD, Donnelly DJ, and Meanwell NA. Applications of fluorine in medicinal chemistry. J Med Chem. (2015) 58:8315–59. doi: 10.1021/acs.jmedchem.5b00258
9. Chandra G, Singh DV, Mahato GK, and Patel S. Fluorine-a small magic bullet atom in the drug development: perspective to FDA approved and COVID-19 recommended drugs. Chem Pap. (2023) 77:4085–106. doi: 10.1007/s11696-023-02804-5
10. Mei H, Han J, Klika KD, Izawa K, Sato T, Meanwell NA, et al. Applications of fluorine-containing amino acids for drug design. Eur J Med Chem. (2020) 186:111826. doi: 10.1016/j.ejmech.2019.111826
11. Sheikhi N, Bahraminejad M, Saeedi M, and Mirfazli SS. A review: FDA-approved fluorine-containing small molecules from 2015 to 2022. Eur J Med Chem. (2023) 260:115758. doi: 10.1016/j.ejmech.2023.115758
12. Han J, Remete AM, Dobson LS, Kiss L, Izawa K, Moriwaki H, et al. Next generation organofluorine containing blockbuster drugs. J Fluor Chem. (2020) 239:109639. doi: 10.1016/j.jfluchem.2020.109639
13. Shabir G, Saeed A, Zahid W, Naseer F, Riaz Z, Khalil N, et al. Chemistry and pharmacology of fluorinated drugs approved by the FDA (2016-2022). Pharm (Basel). (2023) 16:1162. doi: 10.3390/ph16081162
14. Zhang J, Zhang Y, Qu B, Yang H, Hu S, and Dong X. If small molecules immunotherapy comes, can the prime be far behind? Eur J Med Chem. (2021) 218:113356. doi: 10.1016/j.ejmech.2021.113356
15. Wang F, Fu K, Wang Y, Pan C, Wang X, Liu Z, et al. Small-molecule agents for cancer immunotherapy. Acta Pharm Sin B. (2024) 14:905–52. doi: 10.1016/j.apsb.2023.12.010
16. Inoue M, Sumii Y, and Shibata N. Contribution of organofluorine compounds to pharmaceuticals. ACS Omega. (2020) 5:10633–40. doi: 10.1021/acsomega.0c00830
17. Topalian SL, Taube JM, Anders RA, and Pardoll DM. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat Rev Cancer. (2016) 16:275–87. doi: 10.1038/nrc.2016.36
18. Zhang H, Dai Z, Wu W, Wang Z, Zhang N, Zhang L, et al. Regulatory mechanisms of immune checkpoints PD-L1 and CTLA-4 in cancer. J Exp Clin Cancer Res. (2021) 40:184. doi: 10.1186/s13046-021-01987-7
19. Pla-López A, Castillo R, Cejudo-Marín R, García-Pedrero O, Bakir-Laso M, Falomir E, et al. Synthesis and biological evaluation of small molecules as potential anticancer multitarget agents. Int J Mol Sci. (2022) 23:7049. doi: 10.3390/ijms23137049
20. Xia Y, Zhang H, Du H, Huang L, Yu C, Wu H, et al. Design, synthesis, and antitumor activity evaluation of 1,2,3-triazole derivatives as potent PD-1/PD-L1 inhibitors. Bioorg Chem. (2024) 153:107813. doi: 10.1016/j.bioorg.2024.107813
21. Maxinovel Pty., Ltd. A phase I study to evaluate the safety, tolerability and pharmacokinetic characteristics of MAX-10181 in patients with advanced solid tumor (2022). Available online at: https://clinicaltrials.gov/study/NCT05196360 (Accessed April 29, 2025).
22. Wang Y, Xu Z, Wu T, He M, and Zhang N. Aromatic acetylene or aromatic ethylene compound, intermediate, preparation method, pharmaceutical composition and use thereof (2018). Available online at: https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018006795&_cid=P20-MBX35Q-59319–1 (Accessed June 15, 2025).
23. Green NJ, Xiang J, Chen J, Chen L, Davies AM, Erbe D, et al. Structure–activity studies of a series of dipyrazolo[3,4-b:3′,4′-d]pyridin-3-ones binding to the immune regulatory protein B7.1. Bioorg Med Chem. (2003) 11:2991–3013. doi: 10.1016/S0968-0896(03)00183-4
24. Hu MC, Qiu WR, Wang X, Meyer CF, and Tan TH. Human HPK1, a novel human hematopoietic progenitor kinase that activates the JNK/SAPK kinase cascade. Genes Dev. (1996) 10:2251–64. doi: 10.1101/gad.10.18.2251
25. Ahn MJ, Kim EH, Choi Y, Chae CH, Kim P, and Kim SH. Novel hematopoietic progenitor kinase 1 inhibitor KHK-6 enhances T-cell activation. PloS One. (2024) 19:e0305261. doi: 10.1371/journal.pone.0305261
26. BeiGene. A phase 2, open-label, randomized, multi-arm study of BGB-A445 in combination with investigational agents in non-small cell lung cancer patients previously treated with anti-PD-(L)1 antibody (2025). Available online at: https://clinicaltrials.gov/study/NCT06029127 (Accessed April 29, 2025).
27. Xu S, Li J, and Wang Z. 3-[(1h-Pyrazol-4-Yl)oxy]pyrazin-2-Amine Compounds as HPK1 inhibitor and use thereof (2022). Available online at: https://patentscope.wipo.int/search/en/WO2022068848 (Accessed June 12, 2025).
28. Noel MS, Demel KC, Srivastava B, Daigle SR, Boiko S, Hoerres A, et al. Phase 1/2 trial of the HPK1 inhibitor NDI-101150 as monotherapy and in combination with pembrolizumab: Clinical update. JCO. (2024) 42:3083–3. doi: 10.1200/JCO.2024.42.16_suppl.3083
29. Kaila N. Discovery of NDI-101150, a highly potent and selective HPK1 inhibitor for the treatment of cancer, through structure-based drug design (2024). Available online at: https://www.nimbustx.com/wp-content/uploads/Nimbus-HPK1-NDI-101150.pdf (Accessed June 12, 2025).
30. Amouzegar A, Chelvanambi M, Filderman JN, Storkus WJ, and Luke JJ. STING agonists as cancer therapeutics. Cancers. (2021) 13:2695. doi: 10.3390/cancers13112695
31. England DB, Langston SP, Lee HM, Ma L, Shi Z, Vyskocil S, et al. Antibody drug conjugates (2020). Available online at: https://patentscope.wipo.int/search/en/WO2020229982 (Accessed June 12, 2025).
32. Dejmek M, Šála M, Brazdova A, Vanekova L, Smola M, Klíma M, et al. Discovery of isonucleotidic CDNs as potent STING agonists with immunomodulatory potential. Structure. (2022) 30:1146–1156.e11. doi: 10.1016/j.str.2022.05.012
33. Luke JJ, Pinato DJ, Juric D, LoRusso P, Hosein PJ, Desai AM, et al. Phase I dose-escalation and pharmacodynamic study of STING agonist E7766 in advanced solid tumors. J Immunother Cancer. (2025) 13:e010511. doi: 10.1136/jitc-2024-010511
34. Takeda. An open-label, dose escalation, phase 1/2 study to evaluate the safety, tolerability, pharmacokinetics, and pharmacodynamics of TAK-676 as a single agent and in combination with pembrolizumab in adult patients with advanced or metastatic solid tumors (2025). Available online at: https://clinicaltrials.gov/study/NCT04420884 (Accessed April 25, 2025).
35. Takeda. An open-label, phase 1, dose-escalation study to evaluate the safety and preliminary antitumor activity of TAK-676 with pembrolizumab following radiation therapy in the treatment of non-small-cell lung cancer, triple-negative breast cancer, or squamous-cell carcinoma of the head and neck that has progressed on checkpoint inhibitors (2024). Available online at: https://clinicaltrials.gov/study/NCT04879849 (Accessed April 25, 2025).
36. Merck Sharp & Dohme LLC. A phase 2 study in first line metastatic or unresectable, recurrent head and neck squamous cell carcinoma to evaluate intratumoral MK-1454 in combination with IV pembrolizumab vs IV pembrolizumab monotherapy (2023). Available online at: https://clinicaltrials.gov/study/NCT04220866 (Accessed April 26, 2025).
37. Carideo Cunniff E, Sato Y, Mai D, Appleman VA, Iwasaki S, Kolev V, et al. TAK-676: A novel stimulator of interferon genes (STING) agonist promoting durable IFN-dependent antitumor immunity in preclinical studies. Cancer Res Commun. (2022) 2:489–502. doi: 10.1158/2767-9764.CRC-21-0161
38. Kim D-S, Fang F, Endo A, Choi H-W, Hao M-H, Bao X, et al. Cyclic di-nucleotides compounds for the treatment of cancer (2018). Available online at: https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018152450&_cid=P20-MBX3BU-63682–1 (Accessed June 15, 2025).
39. Chang W, Altman MD, Lesburg CA, Perera SA, Piesvaux JA, Schroeder GK, et al. Discovery of MK-1454: A potent cyclic dinucleotide stimulator of interferon genes agonist for the treatment of cancer. J Med Chem. (2022) 65:5675–89. doi: 10.1021/acs.jmedchem.1c02197
40. Wang X, Zhan Z, Wang Z, Zhang Y, Zhao K, Li H, et al. Discovery of a non-nucleotide stimulator of interferon genes (STING) agonist with systemic antitumor effect. MedComm (2020). (2025) 6:e70001. doi: 10.1002/mco2.70001
41. Chin EN, Yu C, Vartabedian VF, Jia Y, Kumar M, Gamo AM, et al. Antitumor activity of a systemic STING-activating non-nucleotide cGAMP mimetic. Science. (2020) 369:993–9. doi: 10.1126/science.abb4255
42. Yentz S and Smith D. Indoleamine 2,3-dioxygenase (IDO) inhibition as a strategy to augment cancer immunotherapy. BioDrugs. (2018) 32:311–7. doi: 10.1007/s40259-018-0291-4
43. Wujieti B, Feng X, Liu E, Li D, Hao M, Zhou L, et al. A theoretical study on the activity and selectivity of IDO/TDO inhibitors. Phys Chem Chem Phys. (2024) 26:16747–64. doi: 10.1039/D3CP06036E
44. Hou X, Gong X, Mao L, Zhao J, and Yang J. Discovery of novel 1,2,3-triazole derivatives as IDO1 inhibitors. Pharm (Basel). (2022) 15:1316. doi: 10.3390/ph15111316
45. Panfili E, Mondanelli G, Orabona C, Gargaro M, Volpi C, Belladonna ML, et al. The catalytic inhibitor epacadostat can affect the non-enzymatic function of IDO1. Front Immunol. (2023) 14:1134551. doi: 10.3389/fimmu.2023.1134551
46. Kumar S, Waldo JP, Jaipuri FA, Marcinowicz A, Van Allen C, Adams J, et al. Discovery of clinical candidate (1R,4R)-4-((R)-2-((S)-6-fluoro-5H-imidazo[5,1-a]isoindol-5-yl)-1-hydroxyethyl)cyclohexan-1-ol (Navoximod), a potent and selective inhibitor of indoleamine 2,3-dioxygenase 1. J Med Chem. (2019) 62:6705–33. doi: 10.1021/acs.jmedchem.9b00662
47. Prendergast GC, Malachowski WP, DuHadaway JB, and Muller AJ. Discovery of IDO1 inhibitors: From bench to bedside. Cancer Res. (2017) 77:6795–811. doi: 10.1158/0008-5472.CAN-17-2285
48. Balog A, Lin T, Maley D, Gullo-Brown J, Kandoussi EH, Zeng J, et al. Preclinical characterization of linrodostat mesylate, a novel, potent, and selective oral indoleamine 2,3-dioxygenase 1 inhibitor. Mol Cancer Ther. (2021) 20:467–76. doi: 10.1158/1535-7163.MCT-20-0251
49. He X, He G, Chu Z, Wu H, Wang J, Ge Y, et al. Discovery of the first potent IDO1/IDO2 dual inhibitors: A promising strategy for cancer immunotherapy. J Med Chem. (2021) 64:17950–68. doi: 10.1021/acs.jmedchem.1c01305
50. Fang K, Dong G, Wang H, He S, Wu S, Wang W, et al. Improving the potency of cancer immunotherapy by dual targeting of IDO1 and DNA. ChemMedChem. (2018) 13:30–6. doi: 10.1002/cmdc.201700666
51. Fang K, Dong G, Li Y, He S, Wu Y, Wu S, et al. Discovery of novel indoleamine 2,3-dioxygenase 1 (IDO1) and histone deacetylase (HDAC) dual inhibitors. ACS Med Chem Lett. (2018) 9:312–7. doi: 10.1021/acsmedchemlett.7b00487
52. Shinde R and McGaha TL. The Aryl hydrocarbon receptor: Connecting immunity to the microenvironment. Trends Immunol. (2018) 39:1005–20. doi: 10.1016/j.it.2018.10.010
53. Hezaveh K, Shinde RS, Klötgen A, Halaby MJ, Lamorte S, Ciudad MT, et al. Tryptophan-derived microbial metabolites activate the aryl hydrocarbon receptor in tumor-associated macrophages to suppress anti-tumor immunity. Immunity. (2022) 55:324–340.e8. doi: 10.1016/j.immuni.2022.01.006
54. Schmees N, Gutcher I, Roehn U, Irlbacher H, Stoeckigt D, Bader B, et al. Abstract 4454: Identification of BAY-218, a potent and selective small-molecule AhR inhibitor, as a new modality to counteract tumor immunosuppression. Cancer Res. (2019) 79:4454. doi: 10.1158/1538-7445.AM2019-4454
55. Schmees N, Gutcher I, Irlbacher H, Bader B, Zhao N, Platten M, et al. 3-oxo-2,6-diphenyl-2,3-dihydropyridazine-4-carboxamides (2017). Available online at: https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017202816&_cid=P20-MBX3DK-64912–1 (Accessed June 15, 2025).
56. McGovern K, Castro AC, Cavanaugh J, Coma S, Walsh M, Tchaicha J, et al. Discovery and characterization of a novel aryl hydrocarbon receptor inhibitor, IK-175, and its inhibitory activity on tumor immune suppression. Mol Cancer Ther. (2022) 21:1261–72. doi: 10.1158/1535-7163.MCT-21-0984
57. Campesato LF, Budhu S, Tchaicha J, Weng C-H, Gigoux M, Cohen IJ, et al. Blockade of the AHR restricts a Treg-macrophage suppressive axis induced by L-Kynurenine. Nat Commun. (2020) 11:4011. doi: 10.1038/s41467-020-17750-z
58. Mohamed M-EF, Bhatnagar S, Parmentier JM, Nakasato P, and Wung P. Upadacitinib: Mechanism of action, clinical, and translational science. Clin Transl Sci. (2024) 17:e13688. doi: 10.1111/cts.13688
59. Research C for DE. FDA grants accelerated approval to zanubrutinib for relapsed or refractory follicular lymphoma. Silver Spring, MD, USA: FDA (2024). Available at: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-zanubrutinib-relapsed-or-refractory-follicular-lymphoma.
60. Research C for DE. FDA grants accelerated approval to pirtobrutinib for chronic lymphocytic leukemia and small lymphocytic lymphoma. Silver Spring, MD, USA: FDA (2024). Available at: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-pirtobrutinib-chronic-lymphocytic-leukemia-and-small-lymphocytic.
61. Press Release: Rilzabrutinib granted orphan drug designation in the US for two rare diseases with no approved medicines . Available online at: https://www.sanofi.com/en/media-room/press-releases/2025/2025-04-03-05-00-00–3054815 (Accessed April 25, 2025).
62. Research C for DE. FDA approves ibrutinib for pediatric patients with chronic graft versus host disease, including a new oral suspension. Silver Spring, MD, USA: FDA (2024). Available at: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-ibrutinib-pediatric-patients-chronic-graft-versus-host-disease-including-new-oral.
63. Research C for DE. Project Orbis: FDA approves acalabrutinib for CLL and SLL. Silver Spring, MD, USA: FDA (2024). Available at: https://www.fda.gov/drugs/resources-information-approved-drugs/project-orbis-fda-approves-acalabrutinib-cll-and-sll.
64. Gomez EB, Ebata K, Randeria HS, Rosendahl MS, Cedervall EP, Morales TH, et al. Pirtobrutinib preclinical characterization: a highly selective, non-covalent (reversible) BTK inhibitor. Blood. (2023) 142(issue 1):62-72. doi: 10.1182/blood.2022018674
65. Langrish CL, Bradshaw JM, Francesco MR, Owens TD, Xing Y, Shu J, et al. Preclinical efficacy and anti-inflammatory mechanisms of action of the bruton tyrosine kinase inhibitor rilzabrutinib for immune-mediated disease. J Immunol. (2021) 206:1454–68. doi: 10.4049/jimmunol.2001130
66. Celgene. A phase 1b, multi-center, open-label study of novel combinations of CC-122, CC-223, CC-292, and rituximab in diffuse large B-cell lymphoma and follicular lymphoma (2024). Available online at: https://clinicaltrials.gov/study/NCT02031419 (Accessed April 25, 2025).
67. The Lymphoma Academic Research Organization. A phase Ib study of the BTKi CC-292 combined with lenalidomide in adults patients with relapsed/refractory B-cell lymphoma (2018). Available online at: https://clinicaltrials.gov/study/NCT01766583 (Accessed April 25, 2025).
68. Allan JN, Pinilla-Ibarz J, Gladstone DE, Patel K, Sharman JP, Wierda WG, et al. Phase Ib dose-escalation study of the selective, non-covalent, reversible Bruton’s tyrosine kinase inhibitor vecabrutinib in B-cell Malignancies. Hematologica. (2022) 107:984–7. doi: 10.3324/hematol.2021.280061
69. Conaghan PG, Nowak M, Du S, Luo Y, Landis J, Pachai C, et al. Evaluation of BMS-986142, a reversible Bruton’s tyrosine kinase inhibitor, for the treatment of rheumatoid arthritis: a phase 2, randomized, double-blind, dose-ranging, placebo-controlled, adaptive design study. Lancet Rheumatol. (2023) 5:e263–73. doi: 10.1016/S2665-9913(23)00089-9
70. Dong X-D, Zhang M, Ma X, Wang J-Q, Lei Z-N, Teng Q-X, et al. Bruton’s tyrosine kinase (BTK) inhibitor RN486 overcomes ABCB1-mediated multidrug resistance in cancer cells. Front Cell Dev Biol. (2020) 8:865. doi: 10.3389/fcell.2020.00865
71. Schafer PH, Kivitz AJ, Ma J, Korish S, Sutherland D, Li L, et al. Spebrutinib (CC-292) affects markers of B cell activation, chemotaxis, and osteoclasts in patients with rheumatoid arthritis: Results from a mechanistic study. Rheumatol Ther. (2020) 7:101–19. doi: 10.1007/s40744-019-00182-7
72. Aslan B, Hubner SE, Fox JA, Taverna P, Wierda WG, Kornblau SM, et al. Vecabrutinib inhibits B-cell receptor signal transduction in chronic lymphocytic leukemia cell types with wild-type or mutant Bruton tyrosine kinase. hematol. (2021) 107:292–7. doi: 10.3324/hematol.2021.279158
73. Lee SK, Xing J, Catlett IM, Adamczyk R, Griffies A, Liu A, et al. Safety, pharmacokinetics, and pharmacodynamics of BMS-986142, a novel reversible BTK inhibitor, in healthy participants. Eur J Clin Pharmacol. (2017) 73:689–98. doi: 10.1007/s00228-017-2226-2
74. Dong X-D, Lu Q, Li Y-D, Cai C-Y, Teng Q-X, Lei Z-N, et al. RN486, a Bruton’s Tyrosine Kinase inhibitor, antagonizes multidrug resistance in ABCG2-overexpressing cancer cells. J Trans Internal Med. (2024) 12:288–98. doi: 10.2478/jtim-2024-0011
75. Research C for DE. FDA approval of lymphoma medicine Ukoniq (umbralisib) is withdrawn due to safety concerns. Silver Spring, MD, USA: FDA (2025). Available at: https://www.fda.gov/drugs/drug-safety-and-availability/fda-approval-lymphoma-medicine-ukoniq-umbralisib-withdrawn-due-safety-concerns.
76. Research C for DE. FDA approves first treatment for activated phosphoinositide 3-kinase delta syndrome. Silver Spring, MD, USA: FDA (2023). Available at: https://www.fda.gov/drugs/news-events-human-drugs/fda-approves-first-treatment-activated-phosphoinositide-3-kinase-delta-syndrome.
77. Research C for DE. FDA approves inavolisib with palbociclib and fulvestrant for endocrine-resistant, PIK3CA-mutated, HR-positive, HER2-negative, advanced breast cancer. Silver Spring, MD, USA: FDA (2024). Available at: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-inavolisib-palbociclib-and-fulvestrant-endocrine-resistant-pik3ca-mutated-hr-positive.
78. Miller BW, Przepiorka D, de Claro RA, Lee K, Nie L, Simpson N, et al. Fda approval: Idelalisib monotherapy for the treatment of patients with follicular lymphoma and small lymphocytic lymphoma. Clin Cancer Res. (2015) 21:1525–9. doi: 10.1158/1078-0432.CCR-14-2522
79. Research C for DE. duvelisib (COPIKTRA, Verastem, Inc.) for adult patients with relapsed or refractory chronic lymphocytic leukemia (CLL) or small lymphocytic lymphoma (SLL). Silver Spring, MD, USA: FDA (2019). Available at: https://www.fda.gov/drugs/resources-information-approved-drugs/duvelisib-copiktra-verastem-inc-adult-patients-relapsed-or-refractory-chronic-lymphocytic-leukemia.
80. Research C for DE. FDA grants accelerated approval to copanlisib for relapsed follicular lymphoma. Silver Spring, MD, USA: FDA (2024). Available at: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-copanlisib-relapsed-follicular-lymphoma.
81. Research C for DE. FDA approves alpelisib for metastatic breast cancer. Silver Spring, MD, USA: FDA (2024). Available at: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-alpelisib-metastatic-breast-cancer.
82. Markham A. Alpelisib: First global approval. Drugs. (2019) 79:1249–53. doi: 10.1007/s40265-019-01161-6
83. Ikeda H, Hideshima T, Fulciniti M, Perrone G, Miura N, Yasui H, et al. PI3K/p110δ is a novel therapeutic target in multiple myeloma. Blood. (2010) 116:1460–8. doi: 10.1182/blood-2009-06-222943
84. Hanan EJ, Braun M-G, Heald RA, MacLeod C, Chan C, Clausen S, et al. Discovery of GDC-0077 (inavolisib), a highly selective inhibitor and degrader of mutant pi3kα. J Med Chem. (2022) 65:16589–621. doi: 10.1021/acs.jmedchem.2c01422
85. De SK. Leniolisib: a novel treatment for activated phosphoinositide-3 kinase delta syndrome. Front Pharmacol. (2024) 15:1337436. doi: 10.3389/fphar.2024.1337436
86. Lampson BL and Brown JR. PI3Kδ-selective and PI3Kα/δ-combinatorial inhibitors in clinical development for B-cell non-Hodgkin lymphoma. Expert Opin Investigational Drugs. (2017) 26:1267–79. doi: 10.1080/13543784.2017.1384815
87. Kater AP, Tonino SH, Spiering M, Chamuleau MED, Liu R, Adewoye AH, et al. Final results of a phase 1b study of the safety and efficacy of the PI3Kδ inhibitor acalisib (GS-9820) in relapsed/refractory lymphoid Malignancies. Blood Cancer J. (2018) 8:1–4. doi: 10.1038/s41408-018-0055-x
88. Soulieres D, Faivre SJ, Dreyer K, and Licitra LF. The BURAN study of buparlisib (AN2025) in combination with paclitaxel compared to paclitaxel alone, in patients with recurrent or metastatic head and neck squamous cell carcinoma. J Clin Oncol. (2021) 39:TPS6090–TPS6090. doi: 10.1200/JCO.2021.39.15_suppl.TPS6090
89. Wang T, Sun X, Qiu L, Su H, Cao J, Li Z, et al. The oral PI3Kδ inhibitor linperlisib for the treatment of relapsed and/or refractory follicular lymphoma: A phase II, single-arm, open-label clinical trial. Clin Cancer Res. (2023) 29:1440–9. doi: 10.1158/1078-0432.CCR-22-2939
90. Yacoub A, Borate U, Rampal RK, Ali H, Wang ES, Gerds AT, et al. Phase 2 study of add-on parsaclisib for patients with myelofibrosis and suboptimal response to ruxolitinib: final results. Blood Adv. (2024) 8:1515–28. doi: 10.1182/bloodadvances.2023011620
91. Makharadze T, Kiladze I, Dzagnidze G, Semionova N, Katselashvili L, Vekua N, et al. Efficacy and safety of tenalisib, a PI3K δ/γ and SIK3 inhibitor, in patients with locally advanced or metastatic breast cancer: Updated results from an on-going phase II study. J Clin Oncol. (2023) 41:1064–4. doi: 10.1200/JCO.2023.41.16_suppl.1064
92. Munakata W, Kumode T, Goto H, Fukuhara N, Shimoyama T, Takeuchi M, et al. A phase II study of zandelisib in patients with relapsed or refractory indolent non-Hodgkin lymphoma: ME-401-K02 study. Br J Hematol. (2025) 206:541–50. doi: 10.1111/bjh.19994
93. Maira S-M, Pecchi S, Huang A, Burger M, Knapp M, Sterker D, et al. Identification and characterization of NVP-BKM120, an orally available pan-class I PI3-Kinase inhibitor. Mol Cancer Ther. (2012) 11:317–28. doi: 10.1158/1535-7163.MCT-11-0474
94. Jiang B, Qi J, Song Y, Li Z, Tu M, Ping L, et al. Phase 1 clinical trial of the PI3Kδ inhibitor YY-20394 in patients with B-cell hematological Malignancies. J Hematol Oncol. (2021) 14:130. doi: 10.1186/s13045-021-01140-z
95. Yue EW, Li Y-L, Douty B, He C, Mei S, Wayland B, et al. INCB050465 (parsaclisib), a novel next-generation inhibitor of phosphoinositide 3-kinase delta (PI3Kδ). ACS Med Chem Lett. (2019) 10:1554–60. doi: 10.1021/acsmedchemlett.9b00334
96. Moreno O and Wood J. Absorption, distribution, and binding profile of ME-401, a potent and selective oral small-molecule inhibitor of phosphatidylinositol 3-kinase δ (PI3Kδ) in animal and B-cell lymphoma models. Targ Oncol. (2019) 14:603–11. doi: 10.1007/s11523-019-00668-y
97. Ullah R, Yin Q, Snell AH, and Wan L. RAF-MEK-ERK pathway in cancer evolution and treatment. Semin Cancer Biol. (2022) 85:123–54. doi: 10.1016/j.semcancer.2021.05.010
98. Research C for DE. FDA grants accelerated approval to dabrafenib in combination with trametinib for unresectable or metastatic solid tumors with BRAF V600E mutation. Silver Spring, MD, USA: FDA (2024). Available at: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-dabrafenib-combination-trametinib-unresectable-or-metastatic-solid.
99. Research C for DE. FDA approves selumetinib for neurofibromatosis type 1 with symptomatic, inoperable plexiform neurofibromas. Silver Spring, MD, USA: FDA (2024). Available at: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-selumetinib-neurofibromatosis-type-1-symptomatic-inoperable-plexiform-neurofibromas.
100. Research C for DE. FDA approves mirdametinib for adult and pediatric patients with neurofibromatosis type 1 who have symptomatic plexiform neurofibromas not amenable to complete resection. Silver Spring, MD, USA: FDA (2025). Available at: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-mirdametinib-adult-and-pediatric-patients-neurofibromatosis-type-1-who-have-symptomatic.
101. Fala L. Cotellic (cobimetinib) FDA approved as part of a combination for patients with advanced melanoma and a BRAF mutation. Langhorne, PA, USA: Am Health Drug Benefits (2016). Available at: https://ahdbonline.com/issues/2016/march-2016-vol-9-seventh-annual-payers-guide/cotellic-cobimetinib-fda-approved-as-part-of-a-combination-for-patients-with-advanced-melanoma-and-a-braf-mutation.
102. Research C for DE. FDA approves encorafenib with binimetinib for metastatic non-small cell lung cancer with a BRAF V600E mutation. Silver Spring, MD, USA: FDA (2023). Available at: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-encorafenib-binimetinib-metastatic-non-small-cell-lung-cancer-braf-v600e-mutation.
103. Lee PA, Wallace E, Marlow A, Yeh T, Marsh V, Anderson D, et al. Abstract 2515: Preclinical development of ARRY-162, a potent and selective MEK 1/2 inhibitor. Cancer Res. (2010) 70:2515–5. doi: 10.1158/1538-7445.AM10-2515
104. Andrlová H, Zeiser R, and Meiss F. Cobimetinib (GDC-0973, XL518). In: Martens UM, editor. Small molecules in oncology. Recent results in cancer research. Springer International Publishing, Cham (2018). p. 177–86. doi: 10.1007/978-3-319-91442-8_12
105. Campagne O, Yeo KK, Fangusaro J, and Stewart CF. Clinical pharmacokinetics and pharmacodynamics of selumetinib. Clin Pharmacokinet. (2021) 60:283–303. doi: 10.1007/s40262-020-00967-y
106. Gilmartin AG, Bleam MR, Groy A, Moss KG, Minthorn EA, Kulkarni SG, et al. GSK1120212 (JTP-74057) is an inhibitor of MEK activity and activation with favorable pharmacokinetic properties for sustained In Vivo pathway inhibition. Clin Cancer Res. (2011) 17:989–1000. doi: 10.1158/1078-0432.CCR-10-2200
107. Roskoski R. Properties of FDA-approved small molecule protein kinase inhibitors: A 2025 update. Pharmacol Res. (2025) 216:107723. doi: 10.1016/j.phrs.2025.107723
108. Kainthla R, Kim KB, and Falchook GS. Dabrafenib. In: Martens UM, editor. Small molecules in oncology. Recent results in cancer research. Springer Berlin Heidelberg, Berlin, Heidelberg (2014). p. 227–40. doi: 10.1007/978-3-642-54490-3_14
109. Shirley M. Encorafenib and binimetinib: First global approvals. Drugs. (2018) 78:1277–84. doi: 10.1007/s40265-018-0963-x
110. Tkacik E, Li K, Gonzalez-Del Pino G, Ha BH, Vinals J, Park E, et al. Structure and RAF family kinase isoform selectivity of type II RAF inhibitors tovorafenib and naporafenib. J Biol Chem. (2023) 299:104634. doi: 10.1016/j.jbc.2023.104634
111. Bollag G, Hirth P, Tsai J, Zhang J, Ibrahim PN, Cho H, et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature. (2010) 467:596–9. doi: 10.1038/nature09454
112. Marriott JB, Muller G, Stirling D, and Dalgleish AG. Immunotherapeutic and antitumor potential of thalidomide analogues. Expert Opin Biol Ther. (2001) 1:675–82. doi: 10.1517/14712598.1.4.675
113. Muller GW, Corral LG, Shire MG, Wang H, Moreira A, Kaplan G, et al. Structural modifications of thalidomide produce analogs with enhanced tumor necrosis factor inhibitory activity. J Med Chem. (1996) 39:3238–40. doi: 10.1021/jm9603328
114. Kumar SK, Callander NS, Adekola K, Anderson LD, Baljevic M, Baz R, et al. Multiple myeloma, version 2.2024, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw. (2023) 21(12):1281–301. doi: 10.6004/jnccn.2023.0061
115. Colley A, Brauns T, Sluder AE, Poznansky MC, and Gemechu Y. Immunomodulatory drugs: a promising clinical ally for cancer immunotherapy. Trends Mol Med. (2024) 30:765–80. doi: 10.1016/j.molmed.2024.05.001
116. Man H-W, Corral LG, Stirling DI, and Muller GW. α-Fluoro-substituted thalidomide analogues. Bioorg Med Chem Lett. (2003) 13:3415–7. doi: 10.1016/s0960-894x(03)00778-9
117. Wang Y, Mi T, Li Y, Kan W, Xu G, Li J, et al. Design, synthesis and biological evaluation of thioether-containing lenalidomide and pomalidomide derivatives with anti-multiple myeloma activity. Eur J Medicinal Chem. (2021) 209:112912. doi: 10.1016/j.ejmech.2020.112912
118. Tiedt R, Schillo M, Osmont A, Bonenfant D, Narayan R, Wallace O, et al. Abstract 3294: The GSPT1 molecular glue degrader MRT-2359 is active against prostate cancer. Cancer Res. (2024) 84:3294. doi: 10.1158/1538-7445.AM2024-3294
119. Dong G, Ding Y, He S, and Sheng C. Molecular glues for targeted protein degradation: From serendipity to rational discovery. J Med Chem. (2021) 64:10606–20. doi: 10.1021/acs.jmedchem.1c00895
120. Chang Y, Keramatnia F, Ghate PS, Nishiguchi G, Gao Q, Iacobucci I, et al. The orally bioavailable GSPT1/2 degrader SJ6986 exhibits in vivo efficacy in acute lymphoblastic leukemia. Blood. (2023) 142:629–42. doi: 10.1182/blood.2022017813
121. Hansen JD, Correa M, Alexander M, Nagy M, Huang D, Sapienza J, et al. CC-90009: A cereblon E3 ligase modulating drug that promotes selective degradation of GSPT1 for the treatment of acute myeloid leukemia. J Med Chem. (2021) 64:1835–43. doi: 10.1021/acs.jmedchem.0c01489
122. Fasching B, Ryckmans T, Flohr A, Ritzén A, Harvey F, and Mcallister L. Isoindolinone compounds (2022). Available online at: https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2022152821&_cid=P20-MBX4AS-91393–1 (Accessed June 15, 2025).
123. Hansen JD, Correa M, Nagy MA, Alexander M, Plantevin V, Grant V, et al. Discovery of CRBN E3 ligase modulator CC-92480 for the treatment of relapsed and refractory multiple myeloma. J Med Chem. (2020) 63:6648–76. doi: 10.1021/acs.jmedchem.9b01928
124. Simonetta KR, Taygerly J, Boyle K, Basham SE, Padovani C, Lou Y, et al. Prospective discovery of small molecule enhancers of an E3 ligase-substrate interaction. Nat Commun. (2019) 10:1402. doi: 10.1038/s41467-019-09358-9
125. Cippitelli M, Stabile H, Kosta A, Petillo S, Gismondi A, Santoni A, et al. Role of Aiolos and Ikaros in the antitumor and immunomodulatory activity of IMiDs in multiple myeloma: Better to lose than to find them. Int J Mol Sci. (2021) 22:1103. doi: 10.3390/ijms22031103
126. Zhang X, Wang L, and Qu Y. Targeting the β-catenin signaling for cancer therapy. Pharmacol Res. (2020) 160:104794. doi: 10.1016/j.phrs.2020.104794
127. Zhou Y, Tao L, Zhou X, Zuo Z, Gong J, Liu X, et al. DHODH and cancer: promising prospects to be explored. Cancer Metab. (2021) 9:22. doi: 10.1186/s40170-021-00250-z
128. Sykes DB, Kfoury YS, Mercier FE, Wawer MJ, Law JM, Haynes MK, et al. Inhibition of dihydroorotate dehydrogenase overcomes differentiation blockade in acute myeloid leukemia. Cell. (2016) 167:171–186.e15. doi: 10.1016/j.cell.2016.08.057
129. Knecht W and Löffler M. Species-related inhibition of human and rat dihydroorotate dehydrogenase by immunosuppressive isoxazol and cinchoninic acid derivatives. Biochem Pharmacol. (1998) 56:1259–64. doi: 10.1016/S0006-2952(98)00145-2
130. Clear Creek Bio, Inc. A phase 1b/2a open-label, multi-center study to assess the safety, efficacy and pharmacokinetics of intrapatient dose-adjusted brequinar and inhibition of dihydroorotate dehydrogenase (DHODH) in adult subjects with AML. Bethesda, MD, USA: clinicaltrials.gov (2022). Available at: https://clinicaltrials.gov/study/NCT03760666.
131. Bayer. An open-label, multicenter phase 1 study to characterize the safety, tolerability, preliminary antileukemic activity, pharmacokinetics, and maximum tolerated dose or pharmacological active dose of BAY2402234 in patients with advanced myeloid Malignancies. Bethesda, MD, USA: clinicaltrials.gov (2021). Available at: https://clinicaltrials.gov/study/NCT03404726.
132. Christian S, Merz C, Evans L, Gradl S, Seidel H, Friberg A, et al. The novel dihydroorotate dehydrogenase (DHODH) inhibitor BAY 2402234 triggers differentiation and is effective in the treatment of myeloid Malignancies. Leukemia. (2019) 33:2403–15. doi: 10.1038/s41375-019-0461-5
133. He Y-W, Deftos ML, Ojala EW, and Bevan MJ. RORγt, a novel isoform of an orphan receptor, negatively regulates fas ligand expression and IL-2 production in T cells. Immunity. (1998) 9:797–806. doi: 10.1016/S1074-7613(00)80645-7
134. Zhu Y, Sun N, Yu M, Guo H, Xie Q, and Wang Y. Discovery of aryl-substituted indole and indoline derivatives as RORγt agonists. Eur J Med Chem. (2019) 182:111589. doi: 10.1016/j.ejmech.2019.111589
135. Ma X, Sun N, Li X, and Fu W. Discovery of novel N-sulfonamide-tetrahydroisoquinolines as potent retinoic acid receptor-related orphan receptor γt agonists. Eur J Med Chem. (2021) 222:113585. doi: 10.1016/j.ejmech.2021.113585
136. Xia L, Tian E, Yu M, Liu C, Shen L, Huang Y, et al. RORγt agonist enhances anti-PD-1 therapy by promoting monocyte-derived dendritic cells through CXCL10 in cancers. J Exp Clin Cancer Res. (2022) 41:155. doi: 10.1186/s13046-022-02289-2
137. Lycera Corp. A phase 1/2a multicenter, open-label study of LYC-55716 in adult subjects with locally advanced or metastatic cancer (2019). Available online at: https://clinicaltrials.gov/study/NCT02929862 (Accessed April 28, 2025).
138. Zhu D, Wang Y, Xie Q, Tian E, Xia L, and Yu M. ROR gamma agonist and application thereof in preparation of medicine for treating tumor diseases and promotion of Type17 cell differentiation. (2022). (Accessed June 15, 2025)
139. Wallat JD, Harrison JK, and Pokorski JK. pH responsive doxorubicin delivery by fluorous polymers for cancer treatment. Mol Pharm. (2018) 15:2954–62. doi: 10.1021/acs.molpharmaceut.7b01046
140. Sacco A, Battaglia AM, Botta C, Aversa I, Mancuso S, Costanzo F, et al. Iron metabolism in the tumor microenvironment—Implications for anti-cancer immune response. Cells. (2021) 10:303. doi: 10.3390/cells10020303
141. Li S, Li G, Yang X, Meng Q, Yuan S, He Y, et al. Design, synthesis and biological evaluation of artemisinin derivatives containing fluorine atoms as anticancer agents. Bioorg Med Chem Lett. (2018) 28:2275–8. doi: 10.1016/j.bmcl.2018.05.035
142. Lin L, Lu W, Dai T, Chen H, Wang T, Yang L, et al. Novel artemisinin derivatives with potent anticancer activities and the anti-colorectal cancer effect by the mitochondria-mediated pathway. Bioorg Chem. (2021) 106:104496. doi: 10.1016/j.bioorg.2020.104496
143. Wang L-L, Kong L, Liu H, Zhang Y, Zhang L, Liu X, et al. Design and synthesis of novel artemisinin derivatives with potent activities against colorectal cancer in vitro and in vivo. Eur J Med Chem. (2019) 182:111665. doi: 10.1016/j.ejmech.2019.111665
144. Xie L, Zhao Y, Zhai X, Li P, Liu C, Li Y, et al. The application of tandem Aza-Wittig reaction to synthesize artemisinin–guanidine hybrids and their anti-tumor activity. Arch Pharm. (2011) 344:631–8. doi: 10.1002/ardp.201000363
145. Aden D, Sureka N, Zaheer S, Chaurasia JK, and Zaheer S. Metabolic reprogramming in cancer: Implications for immunosuppressive microenvironment. Immunology. (2025) 174:30–72. doi: 10.1111/imm.13871
146. Matés JM, Di Paola FJ, Campos-Sandoval JA, Mazurek S, and Márquez J. Therapeutic targeting of glutaminolysis as an essential strategy to combat cancer. Semin Cell Dev Biol. (2020) 98:34–43. doi: 10.1016/j.semcdb.2019.05.012
147. Riess JW, Frankel P, Shackelford D, Dunphy M, Badawi RD, Nardo L, et al. Phase 1 trial of MLN0128 (Sapanisertib) and CB-839 HCl (Telaglenastat) in patients with advanced NSCLC (NCI 10327): rationale and study design. Clin Lung Cancer. (2021) 22:67–70. doi: 10.1016/j.cllc.2020.10.006
148. Calithera Biosciences, Inc. A phase 1b/2, open label, dose escalation and expansion study of the glutaminase inhibitor telaglenastat (CB-839) in combination with CDK4/6 inhibitor palbociclib in patients with advanced or metastatic solid tumors (2022). Available online at: https://clinicaltrials.gov/study/NCT03965845 (Accessed April 29, 2025).
149. Chen Z, Li D, Xu N, Fang J, Yu Y, Hou W, et al. Novel 1,3,4-selenadiazole-containing kidney-type glutaminase inhibitors showed improved cellular uptake and antitumor activity. J Med Chem. (2019) 62:589–603. doi: 10.1021/acs.jmedchem.8b01198
150. Mizuta S, Tagod MSO, Iwasaki M, Nakamura Y, Senju H, Mukae H, et al. Synthesis and immunomodulatory activity of fluorine-containing bisphosphonates. ChemMedChem. (2019) 14:462–8. doi: 10.1002/cmdc.201800764
151. Kraus S, Kolman T, Yeung A, and Deming D. Chemokine receptor antagonists: Role in oncology. Curr Oncol Rep. (2021) 23:131. doi: 10.1007/s11912-021-01117-8
152. Sanford DE, Belt BA, Panni RZ, Mayer A, Deshpande AD, Carpenter D, et al. Inflammatory monocyte mobilization decreases patient survival in pancreatic cancer: A role for targeting the CCL2/CCR2 axis. Clin Cancer Res. (2013) 19:3404–15. doi: 10.1158/1078-0432.CCR-13-0525
153. Robles O, Jackson JJ, Marshall L, Talay O, Chian D, Cutler G, et al. Novel piperidinyl-azetidines as potent and selective CCR4 antagonists elicit antitumor response as a single agent and in combination with checkpoint inhibitors. J Med Chem. (2020) 63:8584–607. doi: 10.1021/acs.jmedchem.0c00988
154. Jiao X, Nawab O, Patel T, Kossenkov AV, Halama N, Jaeger D, et al. Recent advances targeting CCR5 for cancer and its role in immuno-oncology. Cancer Res. (2019) 79:4801–7. doi: 10.1158/0008-5472.CAN-19-1167
155. Sun L, Clavijo PE, Robbins Y, Patel P, Friedman J, Greene S, et al. Inhibiting myeloid-derived suppressor cell trafficking enhances T cell immunotherapy. JCI Insight. (2019) 4. doi: 10.1172/jci.insight.126853
156. Nicholls DJ, Wiley K, Dainty I, MacIntosh F, Phillips C, Gaw A, et al. Pharmacological characterization of AZD5069, a slowly reversible CXC chemokine receptor 2 antagonist. J Pharmacol Exp Ther. (2015) 353:340–50. doi: 10.1124/jpet.114.221358
157. Wu R, Yu W, Yao C, Liang Z, Yoon Y, Xie Y, et al. Amide-sulfamide modulators as effective anti-tumor metastatic agents targeting CXCR4/CXCL12 axis. Eur J Med Chem. (2020) 185:111823. doi: 10.1016/j.ejmech.2019.111823
158. Chatterjee S, Behnam Azad B, and Nimmagadda S. The intricate role of CXCR4 in cancer. Adv Cancer Res. (2014) 124:31–82. doi: 10.1016/b978-0-12-411638-2.00002-1
159. Pfizer. Phase 1b/2 study of PF-04136309 in combination with gemcitabine and nab-paclitaxel in patients with previously untreated metastatic pancreatic ductal adenocarcinoma (2019). Available online at: https://clinicaltrials.gov/study/NCT02732938 (Accessed April 29, 2025).
160. Xue C-B, Wang A, Han Q, Zhang Y, Cao G, Feng H, et al. Discovery of INCB8761/PF-4136309, a potent, selective, and orally bioavailable CCR2 antagonist. ACS Med Chem Lett. (2011) 2:913–8. doi: 10.1021/ml200199c
161. RAPT Therapeutics, Inc. Phase 1/2 dose-escalation and expansion study of FLX475 alone and in combination with pembrolizumab in advanced cancer (2025). Available online at: https://clinicaltrials.gov/study/NCT03674567 (Accessed April 29, 2025).
162. Hanmi Pharmaceutical Company Limited. A phase 2 study to assess the safety, efficacy of FLX475 combined with pembrolizumab in patients with advanced or metastatic gastric cancer (2024). Available online at: https://clinicaltrials.gov/study/NCT04768686 (Accessed April 29, 2025).
163. RAPT Therapeutics, Inc. Phase 2 study of FLX475 in combination with ipilimumab in advanced melanoma (2023). Available online at: https://clinicaltrials.gov/study/NCT04894994 (Accessed April 29, 2025).
164. Beck H, Biannic B, Bui MH, Hu D, Ketcham J, Powers J, et al. Chemokine receptor modulators and uses thereof (2018). Available online at: https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018022992&_cid=P20-MBX4MI-00041–1 (Accessed June 15, 2025).
165. Gulley J. Phase I/II trial investigating the safety, tolerability, pharmacokinetics, immune and clinical activity of SX-682 in combination with BinTrafusp Alfa (M7824 or TGF-beta “Trap”/PD-L1) with CV301 TRICOM in advanced solid tumors (STAT) (2025). Available online at: https://clinicaltrials.gov/study/NCT04574583 (Accessed April 29, 2025).
166. Zebala J, Maeda D, and Schuler A. 2- [5- [n- (4 -fluorophenyl) carbamoyl] pyrimidin- 2 - ylsulfanylmethyl] -4- (trifluoromet hoxy) phenyl] boronic acid (2015). Available online at: https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015016938&_cid=P20-MBX4MY-00400–1 (Accessed June 15, 2025).
167. AstraZeneca. A phase 1b/2, open-label, multicenter study assessing the safety, tolerability, pharmacokinetics, and preliminary anti-tumor activity of MEDI4736 in combination with AZD9150 or AZD5069 in patients with advanced solid Malignancies and subsequently comparing AZD9150 and AZD5069 both as monotherapy and in combination with MEDI4736 as second line treatment in patients with recurrent and/or metastatic squamous cell carcinoma of the head and neck (2025). Available online at: https://clinicaltrials.gov/study/NCT02499328 (Accessed April 29, 2025).
168. Merck Sharp & Dohme LLC. A phase 2 trial to evaluate the safety and efficacy of vicriviroc (MK-7690) in combination with pembrolizumab (MK-3475) in participants with advanced/metastatic microsatellite stable (MSS) colorectal cancer (CRC) (2024). Available online at: https://clinicaltrials.gov/study/NCT03631407 (Accessed April 29, 2025).
169. Halama N. LUMINESCENCE-001 ipilimumab, maraviroc and nivolumab in advanced metastatic colorectal and pancreatic cancer (2023). Available online at: https://clinicaltrials.gov/study/NCT04721301 (Accessed April 29, 2025).
170. Strizki JM, Tremblay C, Xu S, Wojcik L, Wagner N, Gonsiorek W, et al. Discovery and characterization of vicriviroc (SCH 417690), a CCR5 antagonist with potent activity against human immunodeficiency virus type 1. Antimicrob Agents Chemother. (2005) 49:4911–9. doi: 10.1128/AAC.49.12.4911-4919.2005
171. Jin H, Lee J-S, Kim D-C, Ko Y-S, Lee G-W, and Kim H-J. Increased extracellular adenosine in radiotherapy-resistant breast cancer cells enhances tumor progression through A2AR-Akt-β-Catenin signaling. Cancers. (2021) 13:2105. doi: 10.3390/cancers13092105
172. Lawson KV, Kalisiak J, Lindsey EA, Newcomb ET, Leleti MR, Debien L, et al. Discovery of AB680: A potent and selective inhibitor of CD73. J Med Chem. (2020) 63:11448–68. doi: 10.1021/acs.jmedchem.0c00525
173. Bowman CE, Da Silva RG, Pham A, and Young SW. An exceptionally potent inhibitor of human CD73. Biochemistry. (2019) 58:3331–4. doi: 10.1021/acs.biochem.9b00448
174. Beatty JW, Lindsey EA, Thomas-Tran R, Debien L, Mandal D, Jeffrey JL, et al. Discovery of potent and selective non-nucleotide small molecule inhibitors of CD73. J Med Chem. (2020) 63:3935–55. doi: 10.1021/acs.jmedchem.9b01713
175. iTeos Therapeutics. A multicenter, open-label, phase I clinical study to assess the safety, tolerability, pharmacokinetics and food-effect of inupadenant new formulations in participants with advanced solid tumors (2024). Available online at: https://clinicaltrials.gov/study/NCT05117177 (Accessed April 28, 2025).
176. AstraZeneca. A phase II, open-label, study to assess the efficacy, safety, and tolerability of AZD4635 in combination with durvalumab and in combination with cabazitaxel and durvalumab in patients who have progressive metastatic castrate-resistant prostate cancer (AARDVARC) (2023). Available online at: https://clinicaltrials.gov/study/NCT04495179 (Accessed April 28, 2025).
177. Crosignani S, Gomes B, and Houthuys E. 2-oxo-thiazole derivatives as A2a inhibitors and compounds for use in the treatment of cancers (2018). Available online at: https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018178338&_cid=P20-MBX4O2-01228–1 (Accessed June 15, 2025).
178. Borodovsky A, Barbon CM, Wang Y, Ye M, Prickett L, Chandra D, et al. Small molecule AZD4635 inhibitor of A2A R signaling rescues immune cell function including CD103+ dendritic cells enhancing anti-tumor immunity. J Immunother Cancer. (2020) 8:e000417. doi: 10.1136/jitc-2019-000417
179. Liu W, Luo Z, Liu Y, and Sun B. Current landscape and tailored management of immune-related adverse events. Front Pharmacol. (2023) 14:1078338. doi: 10.3389/fphar.2023.1078338
180. Das S and Johnson DB. Immune-related adverse events and anti-tumor efficacy of immune checkpoint inhibitors. J Immunother Cancer. (2019) 7:306. doi: 10.1186/s40425-019-0805-8
181. Khoja L, Day D, Chen TW-W, Siu LL, and Hansen AR. Tumor- and class-specific patterns of immune-related adverse events of immune checkpoint inhibitors: a systematic review. Ann Oncol. (2017) 28:2377–85. doi: 10.1093/annonc/mdx286
Keywords: fluorinated small molecules, cancer immunotherapy, immune checkpoint inhibitors, STING agonists, fluorine in drug design, IDO1/TDO inhibitors
Citation: Leong KF, Chen Z and Coghi P (2025) Fluorinated small molecule derivatives in cancer immunotherapy: emerging frontiers and therapeutic potential. Front. Immunol. 16:1622091. doi: 10.3389/fimmu.2025.1622091
Received: 02 May 2025; Accepted: 23 June 2025;
Published: 18 July 2025.
Edited by:
N. Gopalakrishna Iyer, National Cancer Center, SingaporeReviewed by:
Hongfeng Gu, China Pharmaceutical University, ChinaCopyright © 2025 Leong, Chen and Coghi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paolo Coghi, Y29naGlwc0BtdXN0LmVkdS5tbw==