 Ying-Cheng Chen
Ying-Cheng Chen Marxa Leão Figueiredo
Marxa Leão Figueiredo- Department of Basic Medical Sciences, College of Veterinary Medicine, Purdue University, West Lafayette, IN, United States
Cancer has remained the second leading cause of death worldwide for over a century. Despite significant advances, effectively targeting cancer cells and overcoming therapeutic challenges remain critical goals. In this review, we focus on advanced metastatic prostate tumors, where the patients’ five-year survival rate is less than 35%. While standard androgen deprivation therapy (ADT) has been effective for most prostate cancer patients, recurrence of aggressive tumors is common, emphasizing an urgent need for new treatment strategies. Immunotherapy has gained attention for its potential to harness the immune system against cancer cells. Among these, oncolytic virotherapy stands out for its tumor-specific tropism, its ability to transform or convert the immune-suppressive tumor microenvironment by enhancing immune cell infiltration, and its capacity for therapeutic gene delivery. This review explores the background of commonly used viruses, evaluation models (including cell culture, animal models, ex vivo platforms, and clinical trials), and the anticipated outcomes and challenges of oncolytic virotherapy. By addressing these aspects, we aim to provide a comprehensive overview of the current state and future directions of oncolytic virotherapy models in the treatment of advanced prostate cancer.
Introduction
Prostate cancer has increased in incidence by 3% annually since 2014 (1). With significant advances in disease detection and treatment, the five-year survival rate of prostate cancer patients has now surpassed 97% (2). However, once patients develop resistance to standard androgen-deprivation therapy (ADT) and progress to castration-resistant prostate cancer (CRPC) or metastatic tumors, survival rates plummet to 33% (3). This stark contrast highlights the urgent need for new, more effective treatments for advanced prostate cancer patients. To address these challenges, innovative approaches for developing models that better recapitulate tumor progression and reveal the underlying mechanisms are required to evaluate and validate new therapeutic strategies.
In preclinical studies, LNCaP, PC3, and DU145 are three commonly used human metastatic prostate cancer cell lines, each with unique characteristics influencing their behavior and responsiveness to ADT. For example, LNCaP cells, derived from a supraclavicular lymph node metastasis site, express both androgen receptor (AR) and prostate-specific antigen (PSA), making them a valuable model for studying ADT-sensitive prostate cancer. In contrast, PC3 cells, originating from a vertebral metastasis, and DU145, derived from a brain metastatic site, lack expression of AR and PSA, rendering these cells resistant to ADT (4) and thus more aggressive. The CWR22Rv1 (22Rv1) cell line, another human prostate cancer cell line derived from xenografts of human metastatic prostate tumors, exhibits an intermediate profile with a mutated (overactive) AR and PSA expression (5), making it a unique tool for studying partial androgen signaling.
Although these human-derived cell lines provide insights into prostate cancer biology and response to therapy, their preclinical use is limited owing to immune rejection arising from species specificity. Therefore, various murine prostate cancers also have been isolated to enable a more comprehensive evaluation of the effects of therapeutics within the context of an intact immune system (6). For instance, commonly used murine cell lines include AR-expressing TRAMP-C1, TRAMP-C2, and TRAMP-C3, derived from 32-week prostatic adenocarcinomas of the probasin-SV40 T antigen-Transgenic Adenocarcinoma Mouse Prostate model (TRAMP) (7–9). In addition, the murine cell lines RM1, RM2, and RM9, developed by the Thompson group and deposited with the American Type Culture Collection (ATCC) in 1995, are AR-expressing, mesenchymal-like mouse prostate cancer cell lines. These cells were derived from 17-day-old mouse fetal urogenital sinuses and were retrovirally-transformed with the oncogenes ras and myc for studying androgen sensitivity (10, 11).
Both human and mouse prostate cancer cell lines, in addition to their key roles in advancing our understanding of tumor biology, typically also serve as platforms for testing novel therapeutic modalities, including oncolytic virotherapy. The concept of using viruses as a treatment for cancer was proposed over a hundred years ago, building on the first reported cancer cell remission in a patient with a natural viral infection (12, 13). Correspondingly, leukemia or lymphoma patients with a later viral infection also showed a period of tumor regression (14, 15). Thereafter, with the recent innovations made possible with genetic engineering, modified oncolytic viruses have been developed to reinforce their selective replication ability within cancer cells, particularly when equipped with a variety of therapeutic genes, as well as integrated strategies to disguise these viruses from the immune system (16, 17). For example, the oncolytic adenovirus H101, approved by Chinese regulatory agencies for patients with head and neck cancer as early as 2005, had deletions of the anti-apoptotic gene E1B and the immune evasion gene E3 (18). Similarly, T-VEC (herpes simplex virus 1; HSV-1), armed with human GM-CSF and deleted neurovirulence factors (ICP34.5 and ICP47), was the first documented modified oncolytic virus for glioma treatment and the first FDA-approved oncolytic therapy for unresectable stage III advanced melanoma patients in 2015 (19). At present, various viruses have been investigated in diverse cancer types, building on extensive studies that initially utilized cancer cell lines, demonstrating the potential of oncolytic virotherapy (including metastatic prostate cancer) for delivering different therapeutic genes and enabling combination therapies.
In this review, we explore the latest achievements in oncolytic virotherapy, a promising therapeutic approach that leverages viruses to selectively target and destroy cancer cells. We discuss commonly used oncolytic viruses in advanced prostate cancer from June 2019 to date, examining their mechanisms of action, therapeutic potential, and the challenges they face moving forward. Our focus is primarily on evaluation models for prostate cancer oncolytic virotherapy, with insights that may extend to other solid tumor types, aiming to showcase developments in the field that are likely to be adapted for prostate cancer.
Key characteristics and mechanisms of oncolytic virus therapy
Common types of viruses in prostate cancer treatment
Fourteen viruses, including RNA and DNA viruses, have been assessed in preclinical prostate cancer treatments over the past five years (Table 1). Among these, adenovirus, herpes simplex virus (HSV), and vesicular stomatitis virus (VSV) have been the most studied due to their genetic flexibility and high infectivity. While adenovirus, unlike HSV, lacks an envelope, both viruses consist of double-stranded DNA (dsDNA) and serve as mainstays in virotherapy because of their well-characterized genetic backgrounds and large capacity for carrying transgenes (52, 53). VSV is another frequently used oncolytic virus, composed of single-stranded RNA (ssRNA), for its rapid replication and abundant viral protein production that elicits robust immune responses (54). The main difference between DNA and RNA viruses is their mechanism of replication within host cells: DNA viruses require entry into the host cell nucleus, whereas positive-stranded RNA viruses can directly translate their proteins in the cytoplasm. In contrast, negative-stranded RNA viruses require an additional reverse transcription step in order to synthesize positive-stranded RNA before its proteins can be translated (55). Despite these differences, oncolytic viruses generally exert their cancer cell cytotoxicity through two main mechanisms: direct cell lysis (oncolysis), and indirect activation of anti-cancer immunity (immune-mediated) (Figure 1), with efficacy verified in both in vitro and in vivo studies, which have enabled these immune virotherapies to enter clinical trials (56).
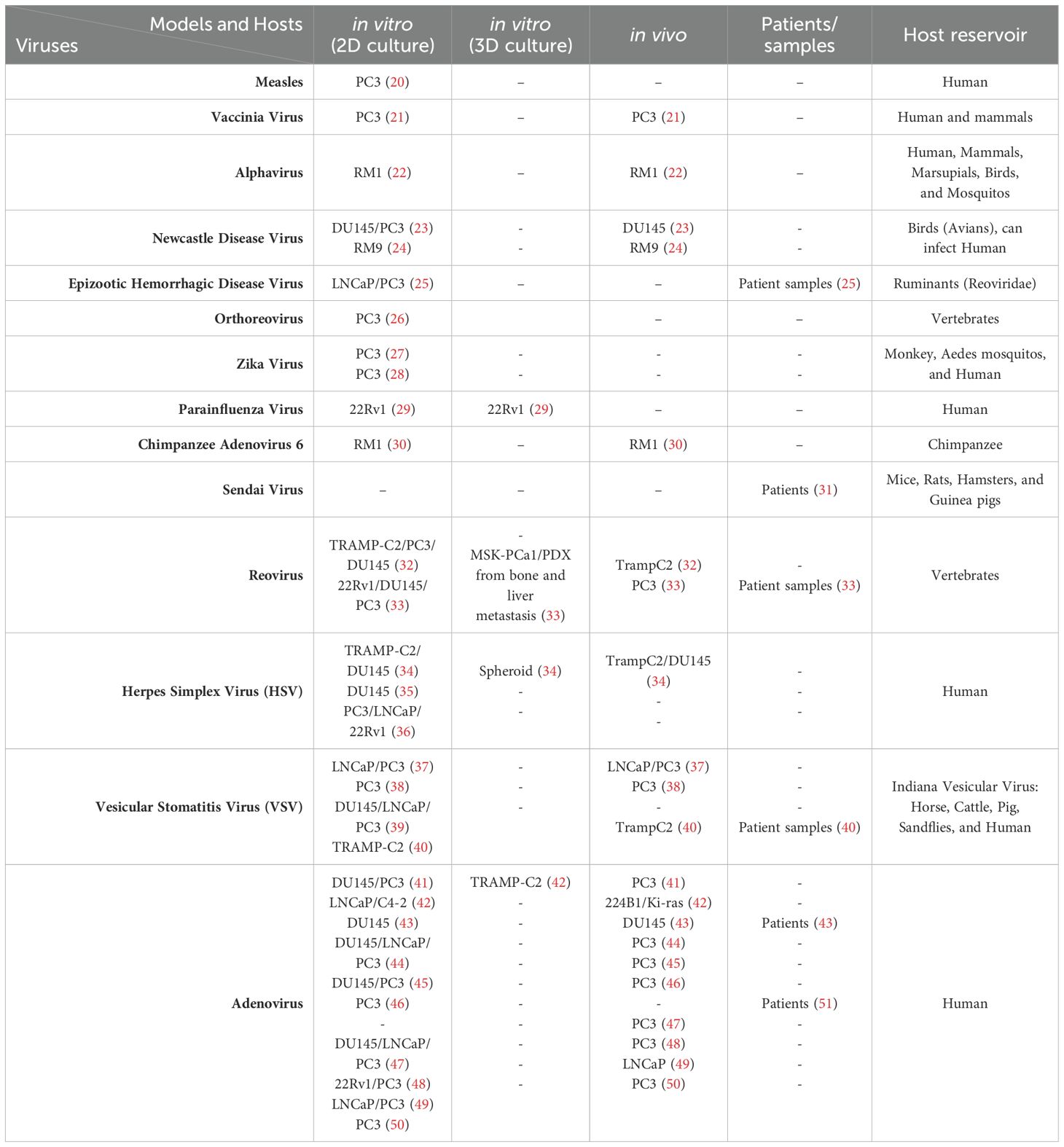
Table 1. Organized oncolytic virus publication on prostate cancer within 5 years.
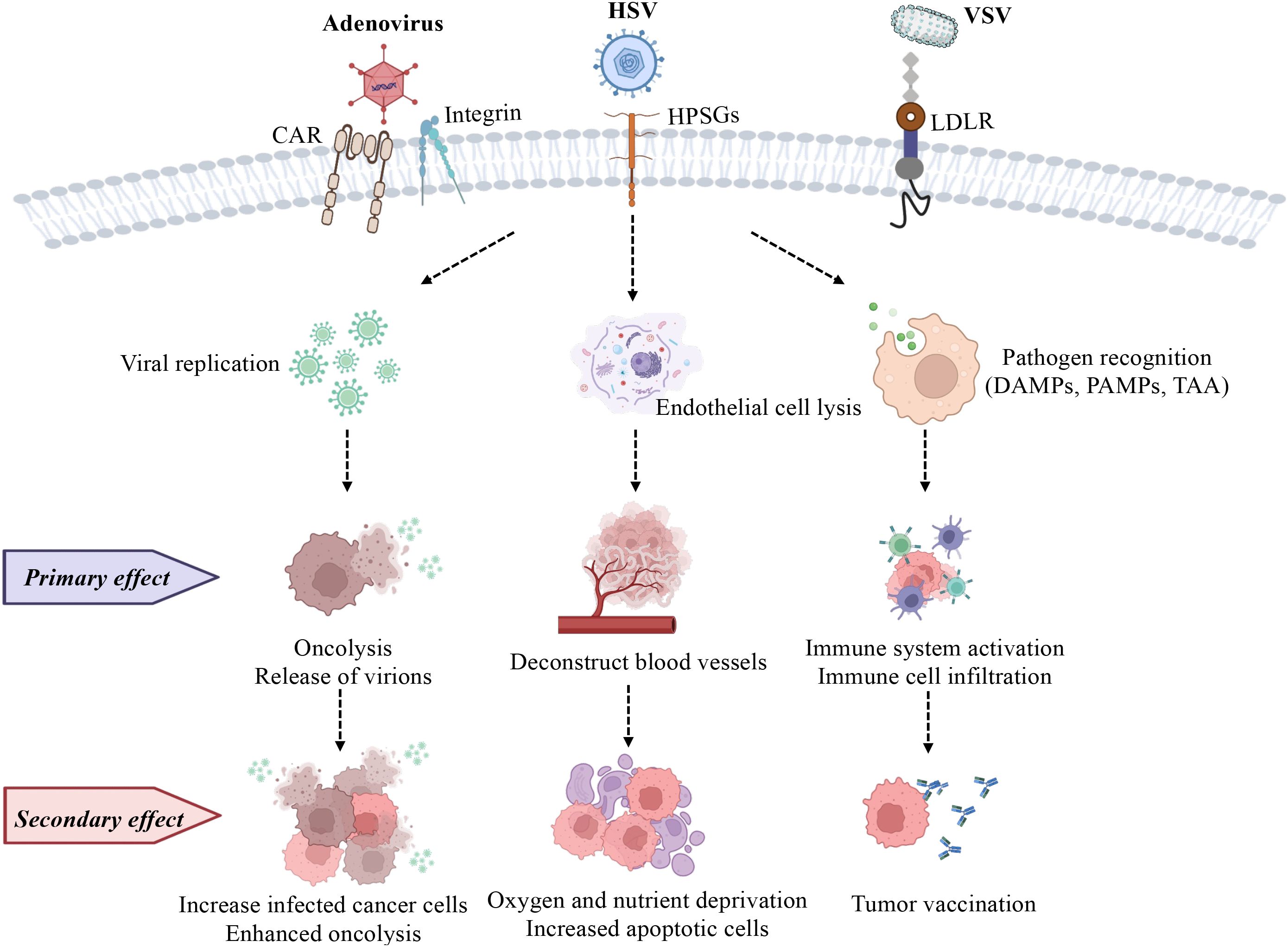
Figure 1. Commonly used oncolytic viruses (adenovirus, HSV, and VSV) are used to depict their effects on cancer cells. (1) Selective replication of OVs in cancer cells causes oncolysis (primary effect), and they further infect more cancer cells (secondary effect). (2) Endothelial cell death (anti-angiogenesis; primary effect) is a strategy for OVs to evade the immune system, meanwhile, the reduced vasculature decreases immune cell infiltration, and oxygen and nutrient supply (secondary effect), inhibiting tumor growth. (3) OVs alter the microenvironment by inducing both innate (primary effect) and adaptive immunity (secondary effect) as they respond to PAMPs, TAAs, and DAMPs. HPSGs, Heparan Sulfate Proteoglycans; LDLR, Low-Density Lipoprotein Receptor. Image created with BioRender.
Cytotoxicity of oncolytic viruses
The cytotoxicity of oncolytic viruses has been found to affect cancer cells through several mechanisms, including direct oncolysis, immune-mediated toxicity, and the disruption of tumor-associated blood vessels, all of which are processes that can assist in cancer cell elimination (Figure 2) (59). Upon viral infection, some key differences between healthy cells and tumor cells rather facilitate preferential virus replication. In normal cells, viral replication usually slows down metabolism and triggers the recruitment of leukocytes for viral clearance (60). Cancer cells, on the other hand, have developed ways to evade the immune system and avoid apoptosis, thereby supporting viral multiplication. These distinct cellular responses form the concept of oncolytic virotherapy, where viruses exhibit a natural tropism for replicating in cancer cells. Active multiplication drains the nutrients and energy of the host cells (61), causing direct oncolysis, a cell lysis process for virion release. Subsequently, tumor-associated antigens (TAAs), virus-associated antigens, and danger-associated molecular pattern molecules (DAMPs) are exposed, enhancing the recognition probability for phagocytosis by macrophages and dendritic cells (DCs), and facilitating antigen presentation to lymphocytes in lymph nodes. This process activates immune responses, resulting in immune-mediated indirect oncolysis (12). Additionally, to further promote replication and spread within tumor cells, some oncolytic viruses develop mechanisms to disrupt the host cells’ access to blood vessels, thus reducing nutrient supply and limiting the migration of immune cells to the tumor. Consequently, oncolytic viruses provide novel ways to robustly eliminate cancer cells, regardless of whether the cells were infected, by disrupting essential support systems such as the nutrient supply and migration routes (62, 63).
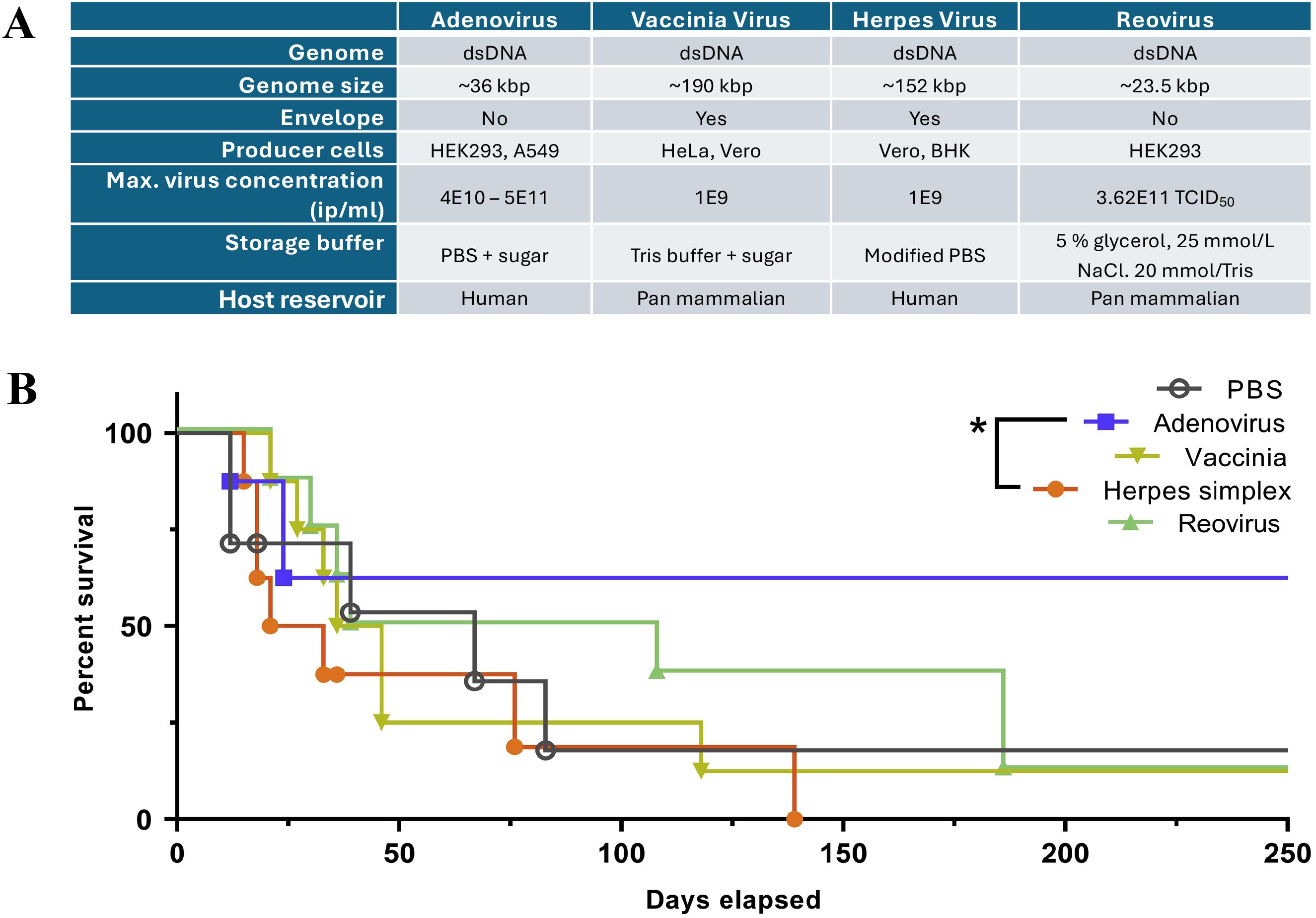
Figure 2. A compilation of oncolytic virus features for a brief comparison across OVs in (A), modified from Ungerechts et al. (57), 2016 (https://pmc.ncbi.nlm.nih.gov/articles/PMC4822647/) and used under the Creative Commons Attribution (CC BY-NC-SA 4.0 International License). (B) A comparison of the survival of pancreatic adenocarcinoma-bearing hamsters treated with different OVs, including Adenovirus, Vaccinia Virus, Herpes Simplex Virus, and Reovirus148. The graph is derived from the original article by Cervera-Carrascon et al. (58).
Optimizing oncolytic viruses for prostate cancer treatment
Viruses can exert cancer cell cytotoxicity as a monotherapy
In most OV immunotherapies, cancer cell death is directly caused by the susceptibility of cancer cells to OV (lytic viral replication) and their failure to respond to anti-pathogen signals (17, 64). In healthy cells, various defense mechanisms are present to fight against pathogens, including viruses (65). For instance, one commonly observed pathogenic defense pathway is through type I interferon (IFN), secreted after pathogen detection (66). Upon type I IFN exposure, downstream signals are conveyed to the JAK-STAT or PKR pathways, inducing the transcription of effector genes such as interferon-stimulated genes (IRFs), including PKR, which can lead to apoptosis to limit infection (67, 68) (Figure 3A). However, cancer cells lose some of these abilities and evade immune surveillance. In a study published by Owen et al. in 2020, the loss of intrinsic IFN expression in tumor cells enables immune evasion and thus promotes prostate tumor progression to a more advanced state. By further restoring the presence of IFN through activation of HDAC, a greater number of tumor cells are eliminated by immune cells (69). Altogether, the primary attack of viruses, which results in the exposure of damage-associated molecule patterns (DAMPs), pathogen-associated molecule patterns (PAMPs), and tumor-associated antigens (TAAs) released from nonviable or apoptotic cells, drastically increases the number of targets/antigens for immune effectors (i.e. CD8+ T cells and antibodies) to recognize. Thus, this reveals another distinct trait of OV: alteration of the tumor microenvironment leading to elevated exposure of cancer cells to immunocytes (70–72) (Figure 3B).
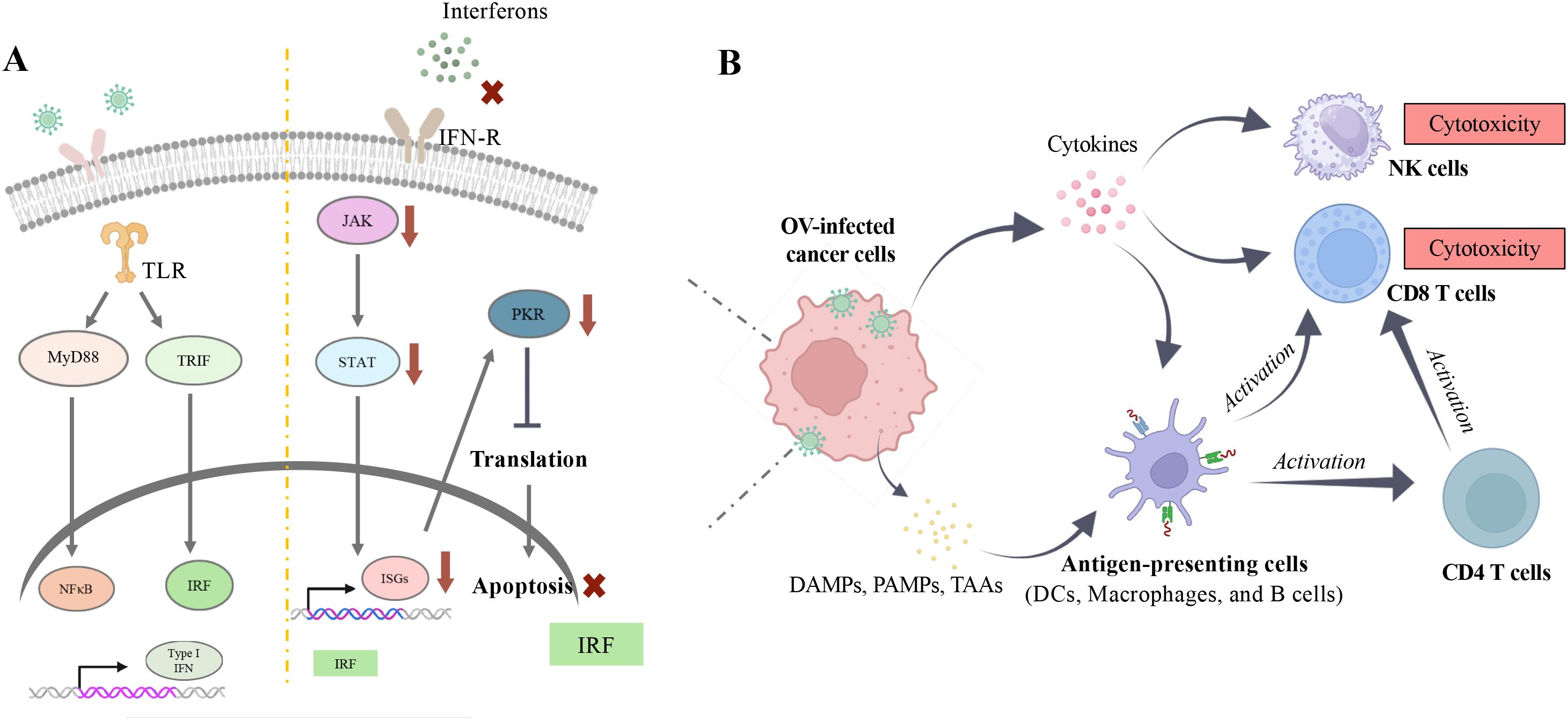
Figure 3. Immune modulation by oncolytic virus. (A) Upon viral infection, natural protective mechanisms in healthy cells can be activated once viral genetic material is recognized by Toll-like receptors (TLRs) on the endoplasmic reticulum (ER). This recognition triggers downstream signaling pathways, including MyD88/NFĸB and TRIF/IRF, which further lead to the active transcription of type I interferons (IFNs) (Left Panel). Under normal conditions, type I IFNs can initiate a cascade of immune responses through the JAK-STAT pathway to clear pathogens. However, in cancer cells, this response is impaired due to various mutations that allow them to evade detection by the immune system, such as becoming insensitive to IFN stimulation to avoid apoptosis (Right Panel). (B) Evasion of apoptosis facilitates viral replication within cancer cells and ultimately leads to oncolytic cell death. This process releases damage-associated molecular patterns (DAMPs), pathogen-associated molecular patterns (PAMPs), and tumor-associated antigens (TAAs), which enhance immune recognition. Images created with Biorender.
Viruses can serve as an independent monotherapy, exerting cytotoxic effects on prostate cancer cells. For example, mammalian orthoreovirus (MRV), one of the most promising oncolytic viruses, has completed phase I to III clinical trials for various cancers (73, 74). In a 2021 study by Bussiere and Miller, MRV infection was shown to reduce HIF-1α levels during the early-stage viral infection in prostate cancer cells (26). As HIF-1α contributes substantially to cancer cell aggressiveness (75, 76), this finding uncovered a potential mechanism of cancer cell inhibition mediated by MRV infection. Another notable oncolytic virus, the M protein mutated vesicular stomatitis virus (M51R-VSV), has shown the ability to infect several human cancer cell lines and exert cytotoxic effects through elevated apoptosis, as reported in 2008 by Ahmed et al. (77). However, this effect was absent in one human prostate cancer cell line, PC3, due to the consistent expression of interferon-stimulated genes (ISG) (78). To overcome this limitation of PC3 cells, Bayne et al. demonstrated that by silencing MAP3K and CHD1, PC3 cells could become highly susceptible to M51R-VSV infection. Similarly, in a mouse model, significant tumor growth inhibition was observed in PC3 cells expressing short hairpin RNA against mitogen-activated protein kinase 3 and chromodomain helicase DNA binding protein (shMAP3K/CHD1) after M51R-VSV treatment (38). Therefore, although oncolytic viruses alone can induce cancer cell death, there are still limitations to their therapeutic potential. These most recent studies suggest that combination therapies may be crucial in augmenting their efficacy.
Synergistic effects of modified oncolytic viruses and combination therapies
To enhance targeting specificity and therapeutic effects, the modification of oncolytic viruses with therapeutic genes or the use of combination therapies has been extensively studied with promising results. One example involves the oncolytic adenovirus DD3-ZD55-SPAG9, which uses a strategy of silencing sperm-associated antigen 9 (SPAG9), a protein involved in the MAPK signaling pathway and highly expressed in prostate cancer (79, 80), in the ZD55 backbone virus (an E1B55K-deleted adenovirus type serotype 5). Additionally, differential display code 3 (DD3) (81, 82), a promoter not expressed in normal prostate tissue, has been used to increase specificity in prostate cancer cell targeting (41). The results showed that DD3-ZD55-SPAG9 inhibited proliferation and migration in two human prostate cancer cell lines, PC3 and DU145. As combination therapies are widely used strategies in cancer therapeutics, DD3-ZD55-SPAG9 was combined with the chemotherapy drug docetaxel in both in vitro and in vivo systems, with results showing an even greater apoptotic effect on cancer cells. In this context, reovirus, another oncolytic virus demonstrating promising treatment outcomes, has also been investigated in combination with immune checkpoint blockade (PD-1) and immunomodulators (CD74). A study by Annels et al. demonstrated that unmodified reovirus, in combination with anti-PD-1 and anti-CD74 antibodies, significantly prolonged tumor growth inhibition (32). Immune profiling results showed an increase in chemokine receptors on the tumor cell surface, which could be recognized by T cells, NK cells, DCs, and B cells. This indicates that the reovirus infection altered the tumor microenvironment, facilitating greater immune cell infiltration. This change may further sensitize cancer cells to PD-1 and CD74 immuno-blockade therapies, leading to a significant reduction in tumor growth.
Prolonging the sustainability of oncolytic viruses
Despite promising results, oncolytic viruses still face the challenge of maintaining their stability within biological systems (83). Immunity, as the body’s primary defense against foreign pathogens, creates a barrier to viral therapy regimens. Therefore, several novel carriers have been proposed to help protect and deliver the virus as part of combined therapeutic strategies. For example, mesenchymal stromal/stem cells (MSCs) have been reported to express limited MHC class I and lack expression of MHC class II, allowing them to evade immune surveillance during their transit in circulation (84, 85). This characteristic makes MSCs promising candidates for delivering viruses into the system, which enables more viral particles to reach the target site to exert their cytolytic effects. In a 2019 study by Muhammad et al., MSCs were used as carriers for oncolytic adenoviruses and demonstrated a significant reduction in tumor growth in vivo, along with increased apoptosis of cancer cells within tumor tissues (42). Besides MSCs, nanoparticles have emerged as a popular field of investigation in recent years, as they can enhance the bioavailability of drugs and facilitate targeted delivery to tumor sites (86). For example, Anjum et al. demonstrated that a nano-formulated measles virus, combined with the chemotherapy drug vincristine, induced cancer cell death and G2/M cell cycle arrest (20). The results showed that the encapsulated virus and vincristine were released in a sustained manner for over 72 hours, offering benefits such as potentially less frequent treatments for patients and prolonged cytotoxic effects against cancer cells.
Platforms to determine oncolytic virus cytotoxicity in prostate cancer
In vitro models
Two-dimensional (2D) cell culture is one of the most accessible and commonly used tools to verify the direct cytotoxicity of OV in cancer cells. Multiple cancer cell lines allow researchers to test various genetically modified viruses for specific cell-type targeting, further revealing mechanisms of OV action. For example, Catharino’s team reported metabolic changes in the PC3 human prostate cancer cell line following Zika virus infection, which induced cancer cell death and attenuated proliferative ability (27). Infected PC3 cells showed an increase in eicosatetraenoic acid (FA 20:5), an omega-3 polyunsaturated fatty acid, and its derivatives, oxylipins, which inhibited the phosphorylation of PYK2 and ERK, key proteins involved in cell signaling. This inhibition led to the accumulation of reactive oxygen species (ROS) after a 5-day infection, ultimately reducing the number of viable cancer cells. In another study, evidence suggested that the Newcastle virus (NDV), usually found in avians, was able to infect and replicate in multiple human cancer cells, including prostate cancer cells (23, 87, 88). Wang et al. revealed that NDV infection in human prostate cancer cell lines PC3 and DU145 led to the release of DAMPs, which promoted apoptosis and enhanced immunogenic cell death (ICD). Further, combining this infection with STAT3 inhibition increased the cancer cell-killing effects, with a notable increase in ICD markers, including calreticulin, HSP70/90, and HMGB1 (23). Considering the complexity of the in vivo environment, more advanced cell culture models, such as spheroids, organoids, and patient-derived tissues, can also be used to complement 2D studies and to better assess OV cytotoxicity.
Along these lines, a three-dimensional (3D) cell culture system provides a more complex tumor microenvironment that closely resembles the in vivo environment (89), offering a better model for assessing OV treatment efficacy. Spheroids, which are suspended clusters of cancer cells in layers, increase the difficulty of oncolytic viral infection, mimicking a more in vivo-like topology. P/V/F, a modified parainfluenza virus with a mutation in the P/V viral gene (encoding P: phosphoprotein and V: accessory protein) and an additional viral fusion protein (F), can selectively target prostate cancer cells (22Rv1) depending on the levels of type I interferon (IFN-I) present in culture. Kedarinath and Parks showed that as low as a multiplicity of infection (MOI) of 0.05 of P/V/F was able to infect 22Rv1 cells in 2D cell culture, and while viral infection efficiency decreased in the spheroid model, the modified virus still induced cancer cell death after an 18-hour infection (29). Additionally, two modified HSV strains, G47Δ and MG18L, have shown strong therapeutic effects on prostate cancer stem cell spheres. G47Δ includes deletions in the virulence gene γ34.5, a lacZ insertion to inactivate UL39, and deletion within the α47 gene to enable ICP47 production for T cell recruitment, while MG18L has deletion of US3 to activate NF-κB signaling and inactivation of UL39; both achieved IC50 values as low as MOI 0.09 and 0.021, respectively (34). Greater reductions in cancer cell growth were observed when G47Δ was combined with BKM120, a pan-class I PI3K inhibitor, in both in vitro and in vivo models, suggesting a synergistic therapeutic effect. While 3D cultures provide valuable structural complexity for further evaluating OV treatments, they cannot replicate the systemic interactions and immune dynamics of living organisms. As such, pre-clinical animal models are essential for ultimately advancing the therapeutic evolution of OV toward clinical relevance.
Preclinical evaluation using animal models: insights and limitations
Pre-clinical animal models play a crucial role in providing a more comprehensive evaluation of therapeutics relative to the in vitro models discussed above. Among all species, rodent models are most frequently used in OV research, as they offer a broader perspective on treatment efficacy. This is particularly important since oncolytic virotherapies are highly dependent on stimulating the immune system to recruit effector cells.
Immune-competent mice can be especially useful, as they allow for detailed immune profiling and the assessment of synergistic effects with OVs, thereby shedding light on systemic responses. For example, Bai et al. demonstrated enhanced cytotoxicity using an oncolytic alphavirus combined with PD-L1-modulated Albendazole (ABZ) in immune-competent mice bearing RM-1 murine prostate tumors (22). Immune profiling and CD8+ T cell-deletion experiments showed that the increased cancer cell apoptosis was likely due to the greater CD8+ T cell infiltration detected in the tumor microenvironment. Additionally, the authors proposed that this combination therapy could sensitize tumors to immune checkpoint inhibitors such as anti-CTLA-4, thus promoting even more profound therapeutic outcomes. Similarly, McAusland et al. used RM-9 allograft C57BL/6 mice to evaluate the combination of oncolytic NDV and vanadyl sulfate, both of which possess anti-neoplastic properties, against melanoma (24). In these immune-competent mice, increased activation of NK cells and macrophages was detected. Interestingly, the combination treatment eliminated cancer cells through innate immunity, as no tumor-specific T cells were detected and the mice failed to respond when rechallenged with the same cancer cells. These mouse models are thus very valuable because they possess intact immune systems, enabling a more inclusive assessment of OV-based therapies. However, there are still notable genetic differences between mouse and human prostate cancer cells, which limit one’s ability to fully recapitulate human disease. Additionally, mice might not be permissive hosts for some oncolytic viruses, potentially leading to an underestimation of their therapeutic effects. To address these limitations, immune-compromised mice are frequently used in OV studies involving human cancer cell line implantation (90).
Immune-compromised mice are commonly used in studies that aim to mimic the human tumor microenvironment through xenografting of human prostate cancer cells. NCG mice, which lack T cells, B cells, and NK cells, are frequently used in OV studies as they greatly minimize graft-versus-host rejection. These mice are named for their genetic modifications: NOD (non-obese diabetic), CB17 background, and targeted deletion of the Gamma chain (common cytokine receptor γ-chain). In a study by Fang et al. (2023), NCG mice were implanted subcutaneously (s.c.) with DU145 human prostate cancer cells and treated with a combination therapy of Ad5Ki67-C3, an adenovirus serotype 5 (Ad5) driven by the Ki67 promoter to express CCL5, interleukin 12, and interferon-γ (43). This treatment, combined with radiation, synergized towards a prolonged survival rate and significant tumor size reduction over 60 days. Moreover, long-term immunity against the same cancer cells was observed when the mice were re-challenged. Nu/nu mice (nude mice or athymic mice), while deficient in T cells due to a homozygous mutation in Foxn1 causing thymic underdevelopment, are also frequently used in OV studies. One example is a study examining the efficacy of RCAd11pADP, an oncolytic adenovirus serotype 11b vector equipped with an adenovirus death protein (ADP). This virus demonstrated cytotoxic effects in both cell culture and PC3 xenograft mouse models (44). In BALB/c nude mice, increased apoptotic cancer cells were observed four weeks after two virus injections. In addition, elevated mRNA expression of viral E1A and hexon proteins indicated active viral replication within the tumor and its potential to suppress tumor growth. Similarly, Mao et al. (45) validated the apoptotic potential of ZD55-IL-24, an oncolytic adenovirus expressing the antitumor gene mda-7/interleukin-24, through both in vitro and in vivo studies. Elevated expression of Caspase-3 and Caspase-8 was detected 18 days post-ZD55-IL-24 treatment in prostate cancer cells and in PC3 tumor-bearing BALB/c nude mice, specially when combined with radiation. Although some immune-compromised mice retain partial immune function, such as innate immunity and innate-like T cells, the majority are athymic and lack diverse T cell populations (91). Given that T cells are crucial for the cytotoxic elimination of cancer cells, particularly in oncolytic virotherapy, results from these models might not fully capture the therapeutic potential observed in immune-competent systems.
With the continuous advancement of mouse models, humanized mice have emerged as some of the most relevant systems for mimicking key aspects of human biology. Humanized mice can be established using immune-deficient NSG mice transplanted with human hematopoietic stem cells (HSCs), human fetal thymic and liver tissue (BLT model), or through the injection of human peripheral blood mononuclear cells (PBMCs). These approaches temporarily allow researchers to analyze disease models and obtain human-relevant immunological responses (92). For example, Zafar et al. (2021) (46), used PBMC-humanized mice to evaluate a novel oncolytic adenovirus (Ad3-hTERT-CMV-hCD40L), equipped with human CD40L to stimulate dendritic cells (DCs). Mice treated with the hCD40L-expressing adenovirus showed significant tumor reduction and prolonged survival, with even greater effects observed in the presence of DCs. However, a key limitation of humanized mice is that they typically develop graft-versus-host disease (GvHD) around 40 days post-transplantation (93). This short experimental window poses challenges, especially in distinguishing immune responses induced by OV therapies from those caused by immune rejection.
In rodent systems, OV experiments often require multiple models to piece together a complete picture of how the virus interacts with both the tumor and the whole organism. This is largely due to species specific differences in viral susceptibility and compatibility with implanted cancer cells. While multiple mouse models may be needed to assess different aspects of OV function, a single hamster model can sometimes provide more comprehensive insights, especially for OVs that do not replicate efficiently in rodents, such as adenovirus species C (94). For instance, Li et al. observed glioma growth inhibition in hamsters following infection with Ad-TD-nsIL-12 (an Ad5 with three deleted genes and producing non-secreting interleukin 12). Viral E1A expression was detectable 12 days post-infection, indicating successful viral propagation in the hamster model, consistent with in vitro results (95). However, in the context of prostate cancer research, some limitations arise. For example, in a study by Koodie et al. (2019), a modified adenovirus with a chimeric fiber (Ad5/3) was used to improve viral entry into advanced prostate cancer cells, which often exhibit reduced expression of the coxsackie-adenovirus receptor (CAR), the primary receptor for adenovirus subgroup C (96). The results showed a decrease in Ad5/3 permissibility in hamsters (97), suggesting that this model may not be ideal for evaluating such vectors. Therefore, while the hamster model provides an immune-competent environment and supports viral propagation, making it advantageous over some mouse models, it might not fully apply to human prostate cancer studies. These limitations stem from phenotypic differences between human and hamster prostate cancer cell lines (98), as well as a lack of established hamster allograft disease models. To gain further insight into immune responses following OV infection, patient-derived samples might offer the most accurate and clinically relevant representation prior to human trials.
Ex vivo models using patient-derived tissues for OV research
Currently, no single model can fully recapitulate the complexity of prostate cancer tumors in patients, as preclinical systems have limited capacity to include all components of the tumor microenvironment. To overcome this limitation, patient-derived tumor tissues have become a valuable tool for more closely mirroring the biological characteristics of human cancers prior to clinical trials (99). In the same study mentioned earlier involving humanized mice, the authors further investigated the mechanisms underlying tumor reduction following synergistic OV and DC treatment using patient-derived prostate cancer tissues. Three days after viral infection, these tissue samples showed a notable elevation in DC maturation markers, including CD80, CD83, and CD86, along with a higher number of mature DCs in culture. In addition, significant upregulation of pro-inflammatory markers, such as IL-2, IL-12, TNF-α, and granzyme B, was detected in the culture media (46). A 2021 study by van de Merbel et al. further demonstrated the utility of patient-derived tissue slices and a xenograft mouse model to evaluate the therapeutic potential of jin-3, a reovirus variant with a spontaneous mutation in the Sigma-1 spike protein, allowing JAM-A (junction adhesion molecule A) independent infection of tumor cells (33). In tissue slices, time-course analysis confirmed viral replication, while in the patient-derived xenograft (PDX) mouse model, jin-3 treatment led to tumor shrinkage and widespread detection of the viral protein Sigma-3. Moreover, increases in apoptotic cell counts and decreases in proliferative markers were reported, supporting the virus’s cytotoxic activity. While human-derived samples might be the ideal testing platform for evaluating OV therapies before clinical trials, one key limitation remains, which is the scarcity and limited availability of these samples (100).
Focus on five-year clinical trials of OVs in prostate cancer
Various viruses have been designed and evaluated for prostate cancer treatment; however, due to the gaps in human-relevant research models mentioned above, few have progressed to clinical trials. Based on the latest updates from ClinicalTrials.gov, there have been only nine OV clinical trials in prostate cancer worldwide within the last five years. Four types of viruses have been included in these trials: coxsackievirus, vaccinia virus, reovirus, and adenovirus, which together comprise the majority of viruses utilized (Table 2). While not every clinical trial result is publicly available, only those with accessible data are discussed here, including clinical trials publications of oncolytic coxsackie virus, oncolytic adenovirus, and oncolytic vaccinia virus.
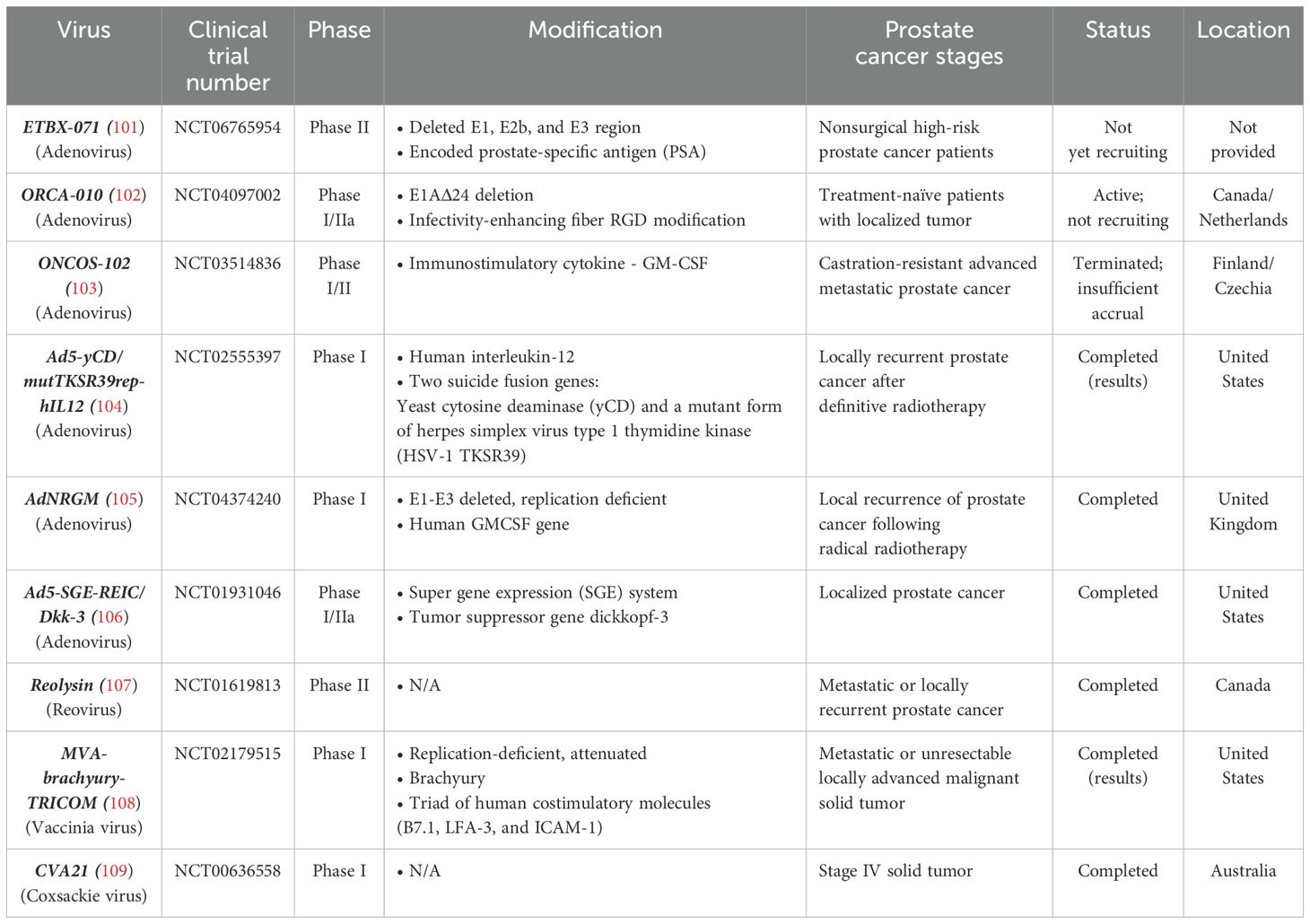
Table 2. 5-year clinical trials on prostate cancer.
The phase I clinical trial of unmodified oncolytic coxsackie virus, CVA21 (V937), completed in 2019, evaluated its effect on various solid tumors, including prostate cancer (110). Four patients with metastatic castration-resistant prostate cancer received CVA21 on days 1, 3, and 5 during the first 21-day cycle, followed by a single dose on day 1 of each of the next eight cycles. No dose-limiting toxicities (DLTs) were reported during monotherapy, and although no specific patient outcomes were indicated, an increase in V937 antibodies in serum was observed during the treatment cycles. This phase I clinical trial established the safety profile of CVA21 across several cancer types and provided data on the tolerance of repeated intravenous injections. However, as it was a pilot study assessing dosage tolerance, a larger cohort of prostate cancer patients would be needed to solidify the results and fully evaluate its therapeutic potential (109).
The modified oncolytic adenovirus Ad5-yCD/mutTKSR39rep-hIL-12 was evaluated for its dosage tolerance and safety in a phase I clinical trial involving 15 patients with localized recurrent prostate cancer. A single intraprostatic dose of the virus, ranging from 1 × 1010 to 1 × 1012 viral particles, was administered on the first day of the trial, then followed by seven days of 5-fluorocytosine (5-FC) and valganciclovir (vGCV) chemotherapy (51). The Ad was designed to express cytosine deaminase (CD) and HSV thymidine kinase (TK), which convert the pro-drugs 5-FC and GCV into toxic agents that eliminate the cancer cells by interfering with DNA synthesis. The safety of this treatment was confirmed, with no reported DLTs, and 92% of side effects were classified as either grade 1 (mild) or grade 2 (moderate). Also, elevated levels of CD3-CD56+ NK cells, CD3+CD4+ T helper cells, and CD3+CD8+ cytotoxic T cells were observed in patients, suggesting immune modulation in peripheral blood due to IL-12 expression from the Ad (104).
The modified oncolytic vaccinia Ankara (MVA) virus encoding TAA (brachyury) and a triad of T-cell co-stimulatory molecules (TRICOM), MVA-Brachyury-TRICOM, also completed a phase I clinical trial involving various cancers, including prostate cancer, to evaluate the safety of three different virus dosages (111). Preliminary results indicated changes in immune cell profiles, especially an increase in CD8+ T cells. Nonetheless, serious side effects were observed in patients receiving higher dosages. While only three prostate cancer patients were enrolled out of 38 total participants, no specific outcome data for these patients has been reported yet (108). Further studies focusing specifically on prostate cancer could benefit from larger patient enrollment and more detailed analyses, particularly regarding prognosis and response to treatment.
Trends of oncolytic viral therapy in cancer
Recent studies highlight the promising future of oncolytic viruses, with significant research focusing on adenovirus, herpes simplex virus, vaccinia virus, vesicular stomatitis virus, and reovirus. Here, a brief discussion covers the distinct traits of these oncolytic viruses and addresses several challenges that require attention: short therapeutic windows in vivo, insufficient accumulation in tumors, restricted delivery routes, and the need to better characterize their overall safety profiles. Regardless, advancements in oncolytic viral therapy research hold great promise for a wide variety of cancer types. By leveraging the unique properties of these viruses, the field can develop more effective immunotherapies, leading to improved treatment outcomes for patients, especially those whose tumors are resistant to immune checkpoint inhibitors.
Why adenovirus, herpes simplex virus, and more
Our literature review reveals that certain viruses are used more frequently than others due to their natural characteristics. For instance, reovirus, a dsRNA virus, naturally infects transformed cells, which synthesize viral proteins more efficiently (112, 113). Similarly, VSV, a negative-sense RNA virus, preferentially infects cancer cells with defective IFN pathways, which reduce their antiviral response and make them more permissive to infection (114). HSV also has a preference for cancer cells with mutated Ras pathways (115) and has the added advantage of a large genome capacity for further modification (19, 116). Likewise, adenovirus (117, 118) and vaccinia virus (VV) (119, 120) have relatively large genome capacities, allowing modifications to enhance their cytotoxicity against tumor cells.
However, therapeutic evaluation models can be challenging for these viruses. As previously mentioned, human adenovirus predominantly infects humans, making in vitro tests straightforward. However, when it comes to pre-clinical animal studies, limitations arise: there is no single model that can offer comprehensive validation of these treatments. Rodents, even across diverse strains, are generally non-permissive to infection or replication of these viruses, making it difficult to accurately assess therapeutic efficacy and safety in vivo. It has been reported that IFNs induced mouse Mx1 expression, which suppresses HSV replication in mice (121). In this context, immunity can act as a double-edged sword in oncolytic virotherapy. While a full immune response, including antiviral mechanisms, can recapitulate the human immune system, the inhibition of HSV replication also can reduce therapeutic effects. Therefore, careful consideration is needed when examining and choosing different models (122).
Neutralization and clearance of OV within the system
Throughout history, humans have co-evolved with viruses, and it may come as a surprise that over 50% of the human genome originates from viruses and transposable elements. These genetic materials, acquired through horizontal gene transfer, crossover and recombination, and transformation, have significantly shaped who we are today (123), including our immunity. Over time, the diversity of the major histocompatibility complex (MHC) has further impacted T cell and B cell specificity, as well as antibody production, thus strengthening our immune response to pathogens (124, 125). In a study by Alemany et al. in 2000 (10), it was reported that 1010 transducing units (t.u.) of adenovirus serotype 5 particles have a half-life of less than 2 minutes following vena cava injection, with viral sequences being cleared by Kupffer cells in the liver within 24 hours (126). To prolong circulation time, scientists have genetically engineered viruses, equipping them with inhibitors of CD8+ T cells (127) or NK cell activation (128). Similarly, disguising the virus using polyethylene glycol (PEG) (for Ad and VSV) or using mesenchymal stromal/stem cells (MSCs) as viral carriers has been shown also to prevent rapid clearance, thereby extending circulation time (126, 129–131). Protected virus not only extend their effective duration in the system but also enhance their migration toward target sites by reducing accumulation in the liver (129, 131). While rapid neutralization of oncolytic viruses might seem counter-intuitive as a therapeutic strategy, it could act to enhance immune cell infiltration into tumors, thereby improving therapeutic outcomes (132, 133).
To avoid virus loss during migration and inefficient OV accumulation at target sites (134), studies have focused on enhancing targeting toward cancer cells. For example, tumor-specific proteins have been expressed on the surface of tumor cells, and “tagging” the tumor surface or other aspects of its unique microenvironment can help direct the OV to target sites (135, 136). However, the majority of OVs are still lost during routing towards tumors, leading to an underestimated treatment effectiveness and a shift from clinically favorable intravenous injection to the more locally-limited intratumorally injection (24, 137–140). As a result, various delivery tools, including nanoparticles, vesicles, and cells (141), are being explored by several groups to attempt to overcome these challenges.
Safety of OV therapy and authorized OV therapy status
Another way to address delivery challenges is through administering a concentrated dosage of the virus. However, this approach could trigger an acute immune response (cytokine storm), resulting in immune reactivity, organ damage (142), and increased risk of infection depending on the type of virus (143). Hence, combination therapy provides a promising solution to enhance treatment efficacy (144) without increasing the virus dose injected. Encouraging results from preclinical studies using combination therapies have already positively influenced the number of clinical trials. For example, as of January 2025, there were 52 actively recruiting OV-related clinical trials across various phases worldwide. These trials include engineered OVs as monotherapies and as adjuvants in combination therapies for different types of cancer. Moreover, several oncolytic viruses have been approved by regulatory authorities, including T-VEC (HSV1 armed with human GM-CSF and deleted neurovirulence factors ICP34.5 and ICP47; Australia, Europe, Israel, and the USA), ECHO-7 (echovirus; Armenia, Georgia, and Latvia), Teserpaturev (Δ47, γ34.5, and ICP6 triple gene-mutated HSV1; Japan), and H101 (E1B- and E3- deleted adenovirus serotype 5; China) (145). However, due to partially unelucidated mechanisms of action against tumors, pharmacovigilance continues to be closely monitored for these clinically approved regimens. According to the latest publication utilizing the U.S. FDA Adverse Event Reporting System (FAERS) database to retrospectively analyze 1138 patients receiving T-VEC, the most commonly seen side effects matched prescribing information or previously reported cases. However, four unexpected adverse events (sepsis, encephalitis, syncope, and lymphadenopathy) were identified. Additionally, 10% of patients receiving T-VEC discontinued treatment, and 2% had life-threatening conditions (146), emphasizing the need for further comprehensive clinical studies to minimize potential harm to patients.
What types of cancer can benefit most from OV therapy?
The concept of “hot” and “cold” tumors was first introduced in 2006, whereby tumors can be classified based on the distribution of immune cells in the tumor microenvironment (147). With immunoscoring of tumor-infiltrating lymphocytes (TILs), “hot” tumors have been defined by the presence of TILs and a high incidence of tumor mutations, while “cold” tumors have been considered to be the opposite (148). Cold tumors develop mechanisms to evade immune surveillance, including the expression of PD-L1 to inhibit CD8+ T cell activation, expression of CD47 to escape from dendritic cell recognition (149), and decreased expression of MHC-I (150). While CAR-T (chimeric antigen receptor T cell) therapy, targeting CD19 and expressing co-stimulatory 4-1BB with CD3ζ signaling domain (151), has achieved great success in enhancing immune responses and tumor regression in hematologic cancers (lymphomas and leukemia), challenges remain in treating CD19-negative tumors, T cell malignancies, and solid tumors (152). Thus, various inflammatory cytokine-expressing CAR T cells have been studied in recent years to target solid tumors, with positive preclinical results (153). Yet, the immunosuppressive tumor microenvironment of cold tumors and the lack of distinct tumor-specific targets have remained as significant barriers (152).
Oncolytic viruses, with their unique ability to remodel the immunosuppressive tumor microenvironment (154, 155), have become promising candidates for treating challenging tumor types. Examples include increased IFNγ infiltration observed in head and neck squamous cell carcinoma (HNSCC) preclinical models treated with a TGFβ inhibitor-expressing VV, making the tumor more responsive to treatment (156). The oncolytic adenovirus Delta-24-RGDOX, also induced increased IDO in human and murine glioma cells (157). Additionally, Hirigoyen et al. reported enhanced extracellular vesicle (EV) secretion by VSV-infected human melanoma cells, which amplified CD8 T cell cytotoxicity when incubated with EVs (158). Thus, the potential and plasticity of oncolytic viruses are under investigation in various cancers, particularly in difficult-to-treat cancers where delivery of therapeutic agents is hindered by the blood-brain barrier (BBB), such as immune-cold glioblastomas (159). In these contexts, OVs have been considered revolutionary.
While some cancer types are identified as immune ‘hot’ or highly responsive to immune checkpoint inhibitors (ICIs), such as melanoma, it remains difficult to predict patient prognosis, as only about one-third of patients respond to ICIs (160). Vareki et al. have unveiled that tumor mutation profiles can further explain clinical responses, as tumors with high mutational loads may express more tumor-associated antigens, enhancing immunocyte recognition and recruiting more immune effectors (161). In contrast, immune “cold” malignancies, such as prostate cancer, pancreatic cancer, and neuroblastoma, typically have low mutational loads and poor responses to immunotherapy, prompting ongoing research seeking breakthroughs. The use of OVs to transform the tumor microenvironment (162–164) can enable better control of these cancers, with promising results reported from both pre-clinical (165, 166) and clinical studies (167, 168).
In summary, OVs excel as therapeutics across multiple dimensions, including selective targeting and replication in cancer cells, primary oncolytic activity, secondary immune-mediated lysis, and a high genomic capacity for delivering multiple therapeutic genes. Promising outcomes have been demonstrated in vitro, in vivo, ex vivo, and in clinical studies. However, several limitations remain to be addressed, including marginal virus accumulation at tumor sites (169), restricted administration routes, and incompletely elucidated cytotoxic mechanisms. We envision that continued research into optimizing delivery strategies, enhancing tumor specific accumulation, and clarifying mechanisms of action will be critical to fully harness the therapeutic potential of OVs in prostate cancer clinical oncology.
Author contributions
Y-CC: Conceptualization, Writing – original draft, Writing – review & editing, Software. MF: Conceptualization, Writing – original draft, Writing – review & editing, Resources.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Correction note
A correction has been made to this article. Details can be found at: 10.3389/fimmu.2025.1665709.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Van Blarigan EL, McKinley MA, Washington SL III, Cooperberg MR, Kenfield SA, Cheng I, et al. Trends in prostate cancer incidence and mortality rates. JAMA Network Open. (2025) 8:e2456825. doi: 10.1001/jamanetworkopen.2024.56825
2. Siegel RL, Giaquinto AN, and Jemal A. Cancer statistics, 2024. CA: A Cancer J Clin. (2024) 74:12–49. doi: 10.3322/caac.21820
3. Liu D, Kuai Y, Zhu R, Zhou C, Tao Y, Han W, et al. Prognosis of prostate cancer and bone metastasis pattern of patients: a SEER-based study and a local hospital based study from China. Sci Rep. (2020) 10:9104. doi: 10.1038/s41598-020-64073-6
4. Namekawa T, Ikeda K, Horie-Inoue K, and Inoue S. Application of prostate cancer models for preclinical study: advantages and limitations of cell lines, patient-derived xenografts, and three-dimensional culture of patient-derived cells. Cells. (2019) 8:74. doi: 10.3390/cells8010074
5. Saranyutanon S, Deshmukh SK, Dasgupta S, Pai S, Singh S, and Singh AP. Cellular and molecular progression of prostate cancer: models for basic and preclinical research. Cancers (Basel). (2020) 12:2651. doi: 10.3390/cancers12092651
6. Wu X, Gong S, Roy-Burman P, Lee P, and Culig Z. Current mouse and cell models in prostate cancer research. Endocr Relat Cancer. (2013) 20:50–170. doi: 10.1530/ERC-12–0285
7. Gingrich JR, Barrios RJ, Kattan MW, Nahm HS, Finegold MJ, and Greenberg NM. Androgen-independent prostate cancer progression in the TRAMP model. Cancer Res. (1997) 57:4687–91.
8. Greenberg NM. Transgenic models for prostate cancer research. Urol Oncol. (1996) 2:119–22. doi: 10.1016/S1078-1439(97)82844-X
9. Foster BA, Gingrich JR, Kwon ED, Madias C, and Greenberg NM. Characterization of prostatic epithelial cell lines derived from transgenic adenocarcinoma of the mouse prostate (TRAMP) model. Cancer Res. (1997) 57:3325–30.
10. Baley PA, Yoshida K, Qian W, Sehgal I, and Thompson TC. Progression to androgen insensitivity in a novelin vitro mouse model for prostate cancer. J Steroid Biochem Mol Biol. (1995) 52:403–13. doi: 10.1016/0960-0760(95)00001-G
11. Thompson TC, Southgate J, Kitchener G, and Land H. Multistage carcinogenesis induced by ras and myc oncogenes in a reconstituted organ. Cell. (1989) 56:917–30. doi: 10.1016/0092-8674(89)90625-9
12. Davola ME and Mossman KL. Oncolytic viruses: how “lytic” must they be for therapeutic efficacy? Oncoimmunology. (2019) 8:e1581528. doi: 10.1080/2162402X.2019.1596006
13. Dock G. THE INFLUENCE OF COMPLICATING DISEASES UPON LEUKAEMIA.: cases of tuberculosis and leukoemia. Miscellaneous infections. Changes in the red blood corpuscles. Qualitative changes in the blood, especially in the leukocytes. When does the change occur? The effects of various processes other than infection on leukoemia. BIBLIOGRAPHY. Am J Med Sci (1827-1924). (1904) 127:563. doi: 10.1097/00000441-190412740-00001
14. Bierman HR, Crile DM, Dod KS, Kelly KH, Petrakis NL, White LP, et al. Remissions in leukemia of childhood following acute infectious disease: staphylococcus and streptococcus, varicella, and feline panleukopenia. Cancer. (1953) 6:591–605. doi: 10.1002/1097-0142(195305)6:3<591::AID-CNCR2820060317>3.0.CO;2-M
15. Bluming AZ and Ziegler JL. Regression of Burkitt’s lymphoma in association with measles infection. Lancet. (1971) 2:105–6. doi: 10.1016/S0140-6736(71)92086-1
16. Cattaneo R, Miest T, Shashkova EV, and Barry MA. Reprogrammed viruses as cancer therapeutics: targeted, armed and shielded. Nat Rev Microbiol. (2008) 6:529–40. doi: 10.1038/nrmicro1927
17. Kaufman HL, Kohlhapp FJ, and Zloza A. Oncolytic viruses: a new class of immunotherapy drugs. Nat Rev Drug Discov. (2015) 14:642–62. doi: 10.1038/nrd4663
18. Garber K. China approves world’s first oncolytic virus therapy for cancer treatment. J Natl Cancer Inst. (2006) 98:298–300. doi: 10.1093/jnci/djj111
19. Aldrak N, Alsaab S, Algethami A, Bhere D, Wakimoto H, Shah K, et al. Oncolytic herpes simplex virus-based therapies for cancer. Cells. (2021) 10:1541. doi: 10.3390/cells10061541
20. Anjum S, Naseer F, Ahmad T, Liaquat A, Abduh MS, and Kousar K. Co-delivery of oncolytic virus and chemotherapeutic modality: Vincristine against prostate cancer treatment: A potent viro-chemotherapeutic approach. J Med Virol. (2024) 96:e29748. doi: 10.1002/jmv.29748
21. Cecil A, Gentschev I, Adelfinger M, Dandekar T, and Szalay AA. Vaccinia virus injected human tumors: oncolytic virus efficiency predicted by antigen profiling analysis fitted boolean models. Bioengineered. (2019) 10:190–6. doi: 10.1080/21655979.2019.1622220
22. Bai W, Tang X, Xiao T, Qiao Y, Tian X, Zhu B, et al. Enhancing antitumor efficacy of oncolytic virus M1 via albendazole-sustained CD8+ T cell activation. Mol Ther Oncol. (2024) 32:200813. doi: 10.1016/j.omton.2024.200813
23. Wang X, Shao X, Gu L, Jiang K, Wang S, Chen J, et al. Targeting STAT3 enhances NDV-induced immunogenic cell death in prostate cancer cells. J Cell Mol Med. (2020) 24:4286–97. doi: 10.1111/jcmm.15089
24. McAusland TM, van Vloten JP, Santry LA, Guilleman MM, Rghei AD, Ferreira EM, et al. Combining vanadyl sulfate with Newcastle disease virus potentiates rapid innate immune-mediated regression with curative potential in murine cancer models. Mol Ther - Oncolytics. (2021) 20:306–24. doi: 10.1016/j.omto.2021.01.009
25. Barer L, Schröder SK, Weiskirchen R, Bacharach E, and Ehrlich M. Lipocalin-2 regulates the expression of interferon-stimulated genes and the susceptibility of prostate cancer cells to oncolytic virus infection. Eur J Cell Biol. (2023) 102:151328. doi: 10.1016/j.ejcb.2023.151328
26. Bussiere LD and Miller CL. Inhibition of HIF-1α accumulation in prostate cancer cells is initiated during early stages of mammalian orthoreovirus infection. Virology. (2021) 558:38–48. doi: 10.1016/j.virol.2021.02.014
27. Delafiori J, Faria AV de S, de Oliveira AN, Sales GM, Dias-Audibert FL, and Catharino RR. Unraveling the metabolic alterations induced by zika infection in prostate epithelial (PNT1a) and adenocarcinoma (PC-3) cell lines. J Proteome Res. (2023) 22:193–203. doi: 10.1021/acs.jproteome.2c00630
28. Delafiori J, Lima E de O, Dabaja MZ, Dias-Audibert FL, de Oliveira DN, Melo CFOR, et al. Molecular signatures associated with prostate cancer cell line (PC-3) exposure to inactivated Zika virus. Sci Rep. (2019) 9:15351. doi: 10.1038/s41598-019-51954-8
29. Kedarinath K and Parks GD. Differential in vitro growth and cell killing of cancer versus benign prostate cells by oncolytic parainfluenza virus. Pathogens. (2022) 11:493. doi: 10.3390/pathogens11050493
30. Wang Q, Wang Y-H, Li Y, Xing M, and Zhou D-M. Anti-tumor effect of a novel oncolytic virus based on chimpanzee adenovirus type 6. Sichuan Da Xue Xue Bao Yi Xue Ban. (2022) 53:71–6.
31. Fujita K, Kato T, Hatano K, Kawashima A, Ujike T, Uemura M, et al. Intratumoral and s.c. injection of inactivated hemagglutinating virus of Japan envelope (GEN0101) in metastatic castration-resistant prostate cancer. Cancer Sci. (2020) 111:1692–8. doi: 10.1111/cas.v111.5
32. Annels NE, Simpson GR, Denyer M, Arif M, Coffey M, Melcher A, et al. Oncolytic reovirus-mediated recruitment of early innate immune responses reverses immunotherapy resistance in prostate tumors. Mol Ther - Oncolytics. (2021) 20:434–46. doi: 10.1016/j.omto.2020.09.010
33. van de Merbel AF, van der Horst G, van der Mark MH, Bots STF, van den Wollenberg DJM, de Ridder CMA, et al. Reovirus mutant jin-3 exhibits lytic and immune-stimulatory effects in preclinical human prostate cancer models. Cancer Gene Ther. (2022) 29:793–802. doi: 10.1038/s41417-021-00360-2
34. Wang L, Ning J, Wakimoto H, Wu S, Wu C, Humphrey MR, et al. Oncolytic herpes simplex virus and PI3K inhibitor BKM120 synergize to promote killing of prostate cancer stem-like cells. Mol Ther - Oncolytics. (2019) 13:58–66. doi: 10.1016/j.omto.2019.03.008
35. Clark CM, Jambunathan N, Collantes TMA, and Kousoulas KG. Inactivation of the UL37 deamidase enhances virus replication and spread of the HSV-1(VC2) oncolytic vaccine strain and secretion of GM-CSF. Viruses. (2023) 15:367. doi: 10.3390/v15020367
36. Vannini A, Parenti F, Bressanin D, Barboni C, Zaghini A, Campadelli-Fiume G, et al. Towards a Precision Medicine Approach and In Situ Vaccination against Prostate Cancer by PSMA-Retargeted oHSV. Viruses. (2021) 13:2085. doi: 10.3390/v13102085
37. Udayakumar TS, Betancourt DM, Ahmad A, Tao W, Totiger TM, Patel M, et al. Radiation attenuates prostate tumor antiviral responses to vesicular stomatitis virus containing IFNβ, resulting in pronounced antitumor systemic immune responses. Mol Cancer Res. (2020) 18:1232–43. doi: 10.1158/1541-7786.MCR-19-0836
38. Bayne RS, Puckett S, Rodrigues LU, Cramer SD, Lee J, Furdui CM, et al. MAP3K7 and CHD1 are novel mediators of resistance to oncolytic vesicular stomatitis virus in prostate cancer cells. Mol Ther - Oncolytics. (2020) 17:496–507. doi: 10.1016/j.omto.2020.05.004
39. Muscolini M, Castiello L, Palermo E, Zevini A, Ferrari M, Olagnier D, et al. SIRT1 modulates the sensitivity of prostate cancer cells to vesicular stomatitis virus oncolysis. J Virol. (2019) 93:e00626–19. doi: 10.1128/JVI.00626-19
40. Hodgins JJ, Abou-Hamad J, O’Dwyer CE, Hagerman A, Yakubovich E, Tanese de Souza C, et al. PD-L1 promotes oncolytic virus infection via a metabolic shift that inhibits the type I IFN pathway. J Exp Med. (2024) 221:e20221721. doi: 10.1084/jem.20221721
41. Lu M, Wei F, Wu C, Xu Z, Mao L, and Yang D. Oncolytic adenovirus with SPAG9 shRNA driven by DD3 promoter improved the efficacy of docetaxil for prostate cancer. J Oncol. (2022) 2022:7918067. doi: 10.1155/2022/7918067
42. Muhammad T, Sakhawat A, Khan AA, Ma L, Gjerset RA, and Huang Y. Mesenchymal stem cell-mediated delivery of therapeutic adenoviral vectors to prostate cancer. Stem Cell Res Ther. (2019) 10:190. doi: 10.1186/s13287-019-1268-z
43. Fang L, Yuan S, Wang M, Zhang C, Wang X, Li H, et al. Recombinant oncolytic adenovirus armed with CCL5, IL-12, and IFN-γ promotes CAR-T infiltration and proliferation in vivo to eradicate local and distal tumors. Cell Death Discov. (2023) 9:328. doi: 10.1038/s41420-023-01626-4
44. Wu H and Mei Y-F. An oncolytic adenovirus 11p vector expressing adenovirus death protein in the E1 region showed significant apoptosis and tumour-killing ability in metastatic prostate cells. Oncotarget. (2019) 10:1957–74. doi: 10.18632/oncotarget.v10i20
45. Mao L, Kan Y, Li B, Ma S, Liu Y, Yang D, et al. Combination therapy of prostate cancer by oncolytic adenovirus harboring interleukin 24 and ionizing radiation. Front Oncol. (2020) 10. doi: 10.3389/fonc.2020.00421
46. Zafar S, Basnet S, Launone I-M, Quixabeira DCA, Santos J, Hemminki O, et al. Oncolytic adenovirus type 3 coding for CD40L facilitates dendritic cell therapy of prostate cancer in humanized mice and patient samples. Hum Gene Ther. (2021) 32:192–202. doi: 10.1089/hum.2020.222
47. Zhang D, Jin T-T, Lu T-Y, and Zhou F-H. Relevance of specific oncolytic adenovirus in regulating PD-L1 expression in prostate cancer. Asian J Surg. (2023) 46:2252–3. doi: 10.1016/j.asjsur.2022.12.003
48. Man YKS, Aguirre-Hernández C, Fernandez A, Martin-Duque P, González-Pastor R, Halldén G, et al. Complexing the oncolytic adenoviruses adΔΔ and ad-3Δ-A20T with cationic nanoparticles enhances viral infection and spread in prostate and pancreatic cancer models. Int J Mol Sci. (2022) 23:8884. doi: 10.3390/ijms23168884
49. Iscaro A, Jones C, Forbes N, Mughal A, Howard FN, Janabi HA, et al. Targeting circulating monocytes with CCL2-loaded liposomes armed with an oncolytic adenovirus. Nanomedicine: Nanotechnology Biol Med. (2022) 40:102506. doi: 10.1016/j.nano.2021.102506
50. Cui C, Li Y, Sun Y, Zhu Y, Fang J, Bai B, et al. Antitumor effect of a dual cancer-specific oncolytic adenovirus on prostate cancer PC-3 cells. Urologic Oncology: Semin Original Investigations. (2019) 37:352.e1–352.e18. doi: 10.1016/j.urolonc.2018.12.012
51. Nyati S, Stricker H, Barton KN, Li P, Elshaikh M, Ali H, et al. A phase I clinical trial of oncolytic adenovirus mediated suicide and interleukin-12 gene therapy in patients with recurrent localized prostate adenocarcinoma. PloS One. (2023) 18:e0291315. doi: 10.1371/journal.pone.0291315
52. Taguchi S, Fukuhara H, Homma Y, and Todo T. Current status of clinical trials assessing oncolytic virus therapy for urological cancers. Int J Urol. (2017) 24:342–51. doi: 10.1111/iju.2017.24.issue-5
53. Lu M, Wei F, Ma S, Xu Z, Wang J, Yang C, et al. Oncolytic virus as a novel modality for the treatment of prostate cancer. Discov Med. (2021) 32:133–9.
54. Orzechowska BU, Jędryka M, Zwolińska K, and Matkowski R. VSV based virotherapy in ovarian cancer: the past, the present and … future? J Cancer. (2017) 8:2369–83. doi: 10.7150/jca.19473
55. Rampersad S and Tennant P. Replication and expression strategies of viruses. Viruses. (2018), 55–82. doi: 10.1016/B978-0-12-811257-1.00003-6
56. Wang G, Liu Y, Liu S, Lin Y, and Hu C. Oncolyic virotherapy for prostate cancer: lighting a fire in winter. Int J Mol Sci. (2022) 23:12647. doi: 10.3390/ijms232012647
57. Ungerechts G, Bossow S, Leuchs B, Holm PS, Rommelaere J, Coffey M, et al. Moving oncolytic viruses into the clinic: clinical-grade production, purification, and characterization of diverse oncolytic viruses. Mol Ther Methods Clin Dev. (2016) 3:16018. doi: 10.1038/mtm.2016.18
58. Cervera-Carrascon V, Quixabeira DCA, Havunen R, Santos JM, Kutvonen E, Clubb JHA, et al. Comparison of clinically relevant oncolytic virus platforms for enhancing T cell therapy of solid tumors. Mol Ther Oncolytics. (2020) 17:47–60. doi: 10.1016/j.omto.2020.03.003
59. Russell SJ and Peng K-W. Viruses as anticancer drugs. Trends Pharmacol Sci. (2007) 28:326–33. doi: 10.1016/j.tips.2007.05.005
60. Russell SJ, Peng K-W, and Bell JC. ONCOLYTIC VIROTHERAPY. Nat Biotechnol. (2012) 30:658–70. doi: 10.1038/nbt.2287
61. Thaker SK, Ch’ng J, and Christofk HR. Viral hijacking of cellular metabolism. BMC Biol. (2019) 17:59. doi: 10.1186/s12915-019-0678-9
62. Angarita FA, Acuna SA, Ottolino-Perry K, Zerhouni S, and McCart JA. Mounting a strategic offense: fighting tumor vasculature with oncolytic viruses. Trends Mol Med. (2013) 19:378–92. doi: 10.1016/j.molmed.2013.02.008
63. Ikeda Y, Kojima T, Kuroda S, Endo Y, Sakai R, Hioki M, et al. A novel antiangiogenic effect for telomerase-specific virotherapy through host immune system1. J Immunol. (2009) 182:1763–9. doi: 10.4049/jimmunol.182.3.1763
64. Alvarez-Breckenridge C, Kaur B, and Chiocca EA. Pharmacologic and chemical adjuvants in tumor virotherapy. Chem Rev. (2009) 109:3125–40. doi: 10.1021/cr900048k
65. Marshall JS, Warrington R, Watson W, and Kim HL. An introduction to immunology and immunopathology. Allergy Asthma Clin Immunol. (2018) 14:49. doi: 10.1186/s13223-018-0278-1
66. Janeway CA and Medzhitov R. Innate immune recognition. Annu Rev Immunol. (2002) 20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359
67. Raftery N and Stevenson NJ. Advances in anti-viral immune defence: revealing the importance of the IFN JAK/STAT pathway. Cell Mol Life Sci. (2017) 74:2525–35. doi: 10.1007/s00018-017-2520-2
68. Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol. (2005) 5:375–86. doi: 10.1038/nri1604
69. Owen KL, Gearing LJ, Zanker DJ, Brockwell NK, Khoo WH, Roden DL, et al. Prostate cancer cell-intrinsic interferon signaling regulates dormancy and metastatic outgrowth in bone. EMBO Rep. (2020) 21:e50162. doi: 10.15252/embr.202050162
70. Gulley JL, Madan RA, Pachynski R, Mulders P, Sheikh NA, Trager J, et al. Role of antigen spread and distinctive characteristics of immunotherapy in cancer treatment. J Natl Cancer Inst. (2017) 109:djw261. doi: 10.1093/jnci/djw261
71. Bommareddy PK, Shettigar M, and Kaufman HL. Integrating oncolytic viruses in combination cancer immunotherapy. Nat Rev Immunol. (2018) 18:498–513. doi: 10.1038/s41577-018-0014-6
72. Prestwich RJ, Errington F, Diaz RM, Pandha HS, Harrington KJ, Melcher AA, et al. The case of oncolytic viruses versus the immune system: waiting on the judgment of solomon. Hum Gene Ther. (2009) 20:1119–32. doi: 10.1089/hum.2009.135
73. Parakrama R, Fogel E, Chandy C, Augustine T, Coffey M, Tesfa L, et al. Immune characterization of metastatic colorectal cancer patients post reovirus administration. BMC Cancer. (2020) 20:569. doi: 10.1186/s12885-020-07038-2
74. Müller L, Berkeley R, Barr T, Ilett E, and Errington-Mais F. Past, present and future of oncolytic reovirus. Cancers (Basel). (2020) 12:3219. doi: 10.3390/cancers12113219
75. He J, Hu Y, Hu M, Zhang S, and Li B. The relationship between the preoperative plasma level of HIF-1α and clinic pathological features, prognosis in non-small cell lung cancer. Sci Rep. (2016) 6:20586. doi: 10.1038/srep20586
76. Luo Y, Yang Z, Yu Y, and Zhang P. HIF1α lactylation enhances KIAA1199 transcription to promote angiogenesis and vasculogenic mimicry in prostate cancer. Int J Biol Macromol. (2022) 222:2225–43. doi: 10.1016/j.ijbiomac.2022.10.014
77. Ahmed M, Cramer SD, and Lyles DS. Sensitivity of prostate tumors to wild type and M protein mutant vesicular stomatitis viruses. Virology. (2004) 330:34–49. doi: 10.1016/j.virol.2004.08.039
78. Carey BL, Ahmed M, Puckett S, and Lyles DS. Early steps of the virus replication cycle are inhibited in prostate cancer cells resistant to oncolytic vesicular stomatitis virus. J Virol. (2008) 82:12104–15. doi: 10.1128/JVI.01508-08
79. Xiao C, Li M, Huang Q, and Si-Tu J. SPAG9 promotes prostate cancer proliferation and metastasis via MAPK signaling pathway. Am J Transl Res. (2019) 11:5249–60.
80. Sun H-F, Wang W-D, and Feng L. Effect of SPAG9 on migration, invasion and prognosis of prostate cancer. Int J Clin Exp Pathol. (2017) 10:9468–74.
81. Schalken JA, Hessels D, and Verhaegh G. New targets for therapy in prostate cancer: differential display code 3 (DD3(PCA3)), a highly prostate cancer-specific gene. Urology. (2003) 62:34–43. doi: 10.1016/S0090-4295(03)00759-3
82. Mearini E, Antognelli C, Del Buono C, Cochetti G, Giannantoni A, Nardelli E, et al. The combination of urine DD3 PCA3 mRNA and PSA mRNA as molecular markers of prostate cancer. Biomarkers. (2009) 14:235–43. doi: 10.1080/13547500902807306
83. Kim J, Hall RR, Lesniak MS, and Ahmed AU. Stem cell-based cell carrier for targeted oncolytic virotherapy: translational opportunity and open questions. Viruses. (2015) 7:6200–17. doi: 10.3390/v7122921
84. Oh JY, Kim H, Lee HJ, Lee K, Barreda H, Kim HJ, et al. MHC class I enables MSCs to evade NK-cell-mediated cytotoxicity and exert immunosuppressive activity. Stem Cells. (2022) 40:870–82. doi: 10.1093/stmcls/sxac043
85. MaChado C, de V, and Telles PD. da S. & Nascimento, I. L. O. Immunological characteristics of mesenchymal stem cells. Rev Bras Hematol Hemoter. (2013) 35:62–7. doi: 10.5581/1516-8484.20130017
86. Ravindran S, Tambe AJ, Suthar JK, Chahar DS, Fernandes JM, and Desai V. Nanomedicine: Bioavailability, Biotransformation and Biokinetics. Curr Drug Metab. (2019) 20:542–555. Available online at: http://www.eurekaselect.com (Accessed May 4, 2025).
87. Meng C, Zhou Z, Jiang K, Yu S, Jia L, Wu Y, et al. Newcastle disease virus triggers autophagy in U251 glioma cells to enhance virus replication. Arch Virol. (2012) 157:1011–8. doi: 10.1007/s00705-012-1270-6
88. Bian J, Wang K, Kong X, Liu H, Chen F, Hu M, et al. Caspase- and p38-MAPK-dependent induction of apoptosis in A549 lung cancer cells by Newcastle disease virus. Arch Virol. (2011) 156:1335–44. doi: 10.1007/s00705-011-0987-y
89. Kapałczyńska M, Kolenda T, Przybyła W, Zajączkowska M, Teresiak A, Filas V, et al. 2D and 3D cell cultures – a comparison of different types of cancer cell cultures. Arch Med Sci. (2018) 14:910–9. doi: 10.5114/aoms.2016.63743
90. Olson B, Li Y, Lin Y, Liu ET, and Patnai A. Mouse models for cancer immunotherapy research. Cancer Discov. (2018) 8:1358–65. doi: 10.1158/2159-8290.CD-18-0044
91. Chulpanova DS, Kitaeva KV, Rutland CS, Rizvanov AA, and Solovyeva VV. Mouse tumor models for advanced cancer immunotherapy. Int J Mol Sci. (2020) 21:4118. doi: 10.3390/ijms21114118
92. Maurice Morillon Y, Sabzevari A, Schlom J, and Greiner JW. The development of next-generation PBMC humanized mice for preclinical investigation of cancer immunotherapeutic agents. Anticancer Res. (2020) 40:5329–41. doi: 10.21873/anticanres.14540
93. Lockridge JL, Zhou Y, Becker YA, Ma S, Kenney SC, Hematti P, et al. Mice engrafted with human fetal thymic tissue and hematopoietic stem cells develop pathology resembling chronic GVHD. Biol Blood Marrow Transplant. (2013) 19:1310–22. doi: 10.1016/j.bbmt.2013.06.007
94. Wold WSM and Toth K. Chapter three - Syrian hamster as an animal model to study oncolytic adenoviruses and to evaluate the efficacy of antiviral compounds. Adv Cancer Res. (2012) 115:69–92.
95. Li S, Guo Y, Ning W, Chen Y, Xu J, Zhao C, et al. Oncolytic virus Ad-TD-nsIL-12 inhibits glioma growth and reprograms the tumor immune microenvironment. Life Sci. (2024) 336:122254. doi: 10.1016/j.lfs.2023.122254
96. Sandberg L, Papareddy P, Silver J, Bergh A, and Mei Y-F. Replication-competent Ad11p vector (RCAd11p) efficiently transduces and replicates in hormone-refractory metastatic prostate cancer cells. Hum Gene Ther. (2009) 20:361–74. doi: 10.1089/hum.2007.124
97. Koodie L, Robertson MG, Chandrashekar M, Ruth G, Dunning M, Bianco RW, et al. Rodents versus pig model for assessing the performance of serotype chimeric ad5/3 oncolytic adenoviruses. Cancers (Basel). (2019) 11:198. doi: 10.3390/cancers11020198
98. Norris JS, Bowden C, and Kohler PO. Description of a new hamster ventral prostate cell line containing androgen receptors. In Vitro. (1977) 13:108–14. doi: 10.1007/BF02615074
99. Elbialy A, Kappala D, Desai D, Wang P, Fadiel A, Wang S-J, et al. Patient-derived conditionally reprogrammed cells in prostate cancer research. Cells. (2024) 13:1005. doi: 10.3390/cells13121005
100. Loewa A, Feng JJ, and Hedtrich S. Human disease models in drug development. Nat Rev Bioeng. (2023), 1–15. doi: 10.1038/s44222-023-00063-3
101. ImmunityBio, Inc. Open-label, phase 2 clinical trial of pre-radiation and post-radiation immunotherapy with N-803, ETBX-071, and M-CENK in combination with radiation for participants with high-risk prostate cancer. (2025). Available online at: https://clinicaltrials.gov/study/NCT06765954 (Accessed May 4, 2025).
102. Orca Therapeutics B.V. A phase I/IIa study evaluating the safety and tolerability of intratumoral administration of ORCA-010 in treatment-naïve patients with localized prostate cancer (2023). Available online at: https://clinicaltrials.gov/study/NCT04097002 (Accessed May 4, 2025).
103. SOTIO a.s. A phase I/II, clinical trial to evaluate the safety and immune activation of the combination of DCVAC/PCa, and ONCOS-102, in men with advanced metastatic castration-resistant prostate cancer. (2021). Available online at: https://clinicaltrials.gov/study/NCT03514836 (Accessed May 4, 2025).
104. Siddiqui F. Phase 1 trial of oncolytic adenovirus-mediated cytotoxic and interleukin 12 gene therapy for locally recurrent prostate cancer after definitive radiotherapy. (2024)Available online at: https://clinicaltrials.gov/study/NCT02555397 (Accessed May 4, 2025).
105. University of Birmingham. Phase I trial of replicative defective type 5 adenovirus vector expressing nitroreductase & GMCSF given via trans-perineal template-guided intra-prostatic injection followed by iv CB1954 in locally relapsed prostate cancer patients. (2021). Available online at: https://clinicaltrials.gov/study/NCT04374240 (Accessed May 4, 2025).
106. Momotaro-Gene Inc. A phase 1/2a study of in-situ REIC/dkk-3 therapy in patients with localized prostate cancer (MTG-REIC-PC003). (2020). Available online at: https://clinicaltrials.gov/study/NCT01931046 (Accessed May 4, 2025).
107. Canadian Cancer Trials Group. A randomized phase II study of reolysin in combination with docetaxel and prednisone or docetaxel and prednisone alone in patients with metastatic castration resistant prostate cancer (2023). Available online at: https://clinicaltrials.gov/study/NCT01619813 (Accessed May 4, 2025).
108. Gulley J. An open label phase I study to evaluate the safety and tolerability of a modified vaccinia ankara (MVA) based vaccine modified to express brachyury and T-cell costimulatory molecules (MVA brachyury-TRICOM) (2021). Available online at: https://clinicaltrials.gov/study/NCT02179515 (Accessed May 4, 2025).
109. Viralytics. A phase I, open-label, cohort study of multiple doses of cavatak™ (Coxsackie virus A21) given intravenously to stage IV solid tumour cancer patients bearing ICAM-1 with or without DAF expressing tumours (PSX-X04) (2019). Available online at: https://clinicaltrials.gov/study/NCT00636558 (Accessed May 4, 2025).
110. Rudin CM, Pandha HS, Zibelman M, Akerley WL, Harrington KJ, Day D, et al. Phase 1, open-label, dose-escalation study on the safety, pharmacokinetics, and preliminary efficacy of intravenous Coxsackievirus A21 (V937), with or without pembrolizumab, in patients with advanced solid tumors. J Immunother Cancer. (2023) 11:e005007. doi: 10.1136/jitc-2022-005007
111. Heery CR, Palena C, McMahon S, Donahue RN, Lepone LM, Grenga I, et al. Phase I study of a poxviral TRICOM-based vaccine directed against the transcription factor brachyury. Clin Cancer Res. (2017) 23:6833–45. doi: 10.1158/1078-0432.CCR-17-1087
112. Shmulevitz M, Marcato P, and Lee PWK. Unshackling the links between reovirus oncolysis, Ras signaling, translational control and cancer. Oncogene. (2005) 24:7720–8. doi: 10.1038/sj.onc.1209041
113. Cristi F, Gutiérrez T, Hitt MM, and Shmulevitz M. Genetic modifications that expand oncolytic virus potency. Front Mol Biosci. (2022) 9:831091. doi: 10.3389/fmolb.2022.831091
114. Stojdl DF, Lichty B, Knowles S, Marius R, Atkins H, Sonenberg N, et al. Exploiting tumor-specific defects in the interferon pathway with a previously unknown oncolytic virus. Nat Med. (2000) 6:821–5. doi: 10.1038/77558
115. Farassati F, Yang A-D, and Lee PWK. Oncogenes in Ras signalling pathway dictate host-cell permissiveness to herpes simplex virus 1. Nat Cell Biol. (2001) 3:745–50. doi: 10.1038/35087061
116. Scanlan H, Coffman Z, Bettencourt J, Shipley T, and Bramblett DE. Herpes simplex virus 1 as an oncolytic viral therapy for refractory cancers. Front Oncol. (2022) 12:940019. doi: 10.3389/fonc.2022.940019
117. Niemann J and Kühnel F. Oncolytic viruses: adenoviruses. Virus Genes. (2017) 53:700–6. doi: 10.1007/s11262-017-1488-1
118. Wang X, Zhong L, and Zhao Y. Oncolytic adenovirus: A tool for reversing the tumor microenvironment and promoting cancer treatment (Review). Oncol Rep. (2021) 45:1–9. doi: 10.3892/or.2021.8000
119. Tian Y, Xie D, and Yang L. Engineering strategies to enhance oncolytic viruses in cancer immunotherapy. Sig Transduct Target Ther. (2022) 7:1–21. doi: 10.1038/s41392-022-00951-x
120. Mirbahari SN, Da Silva M, Zuñ;iga AIM, Kooshki Zamani N, St-Laurent G, Totonchi M, et al. Recent progress in combination therapy of oncolytic vaccinia virus. Front Immunol. (2024) 15:1272351. doi: 10.3389/fimmu.2024.1272351
121. Tessema MB, Farrukee R, Andoniou CE, Degli-Esposti MA, Oates CV, Barnes JB, et al. Mouse mx1 inhibits herpes simplex virus type 1 genomic replication and late gene expression in vitro and prevents lesion formation in the mouse zosteriform model. J Virol. (2022) 96:e0041922. doi: 10.1128/jvi.00419-22
122. McCormack RM and Kaur B. Immune therapy, a double-edged sword for oncolytic viruses. Expert Opin Biol Ther. (2019) 19:1111–3. doi: 10.1080/14712598.2019.1650911
123. Broecker F and Moelling K. What viruses tell us about evolution and immunity: beyond Darwin? Ann N Y Acad Sci. (2019) 1447:53–68. doi: 10.1111/nyas.14097
124. Van Blerkom LM. Role of viruses in human evolution. Am J Phys Anthropol. (2003) 122:14–46. doi: 10.1002/(ISSN)1096-8644
125. Broecker F and Moelling K. Evolution of immune systems from viruses and transposable elements. Front Microbiol. (2019) 10:51. doi: 10.3389/fmicb.2019.00051
126. Alemany R, Suzuki K, and Curiel DT. Blood clearance rates of adenovirus type 5 in mice. J Gen Virol. (2000) 81:2605–9. doi: 10.1099/0022-1317-81-11-2605
127. Pourchet A, Fuhrmann SR, Pilones KA, Demaria S, Frey AB, Mulvey M, et al. CD8(+) T-cell immune evasion enables oncolytic virus immunotherapy. EBioMedicine. (2016) 5:59–67. doi: 10.1016/j.ebiom.2016.01.022
128. Xu B, Ma R, Russell L, Yoo JY, Han J, Cui H, et al. Amendments: Publisher Correction: An oncolytic herpesvirus expressing E-cadherin improves survival in mouse models of glioblastoma. Nat Biotechnol. (2019) 37:102. doi: 10.1038/nbt0119-102c
129. Doronin K, Shashkova EV, May SM, Hofherr SE, and Barry MA. Chemical modification with high molecular weight polyethylene glycol reduces transduction of hepatocytes and increases efficacy of intravenously delivered oncolytic adenovirus. Hum Gene Ther. (2009) 20:975–88. doi: 10.1089/hum.2009.028
130. Tesfay MZ, Kirk AC, Hadac EM, Griesmann GE, Federspiel MJ, Barber GN, et al. PEGylation of vesicular stomatitis virus extends virus persistence in blood circulation of passively immunized mice. J Virol. (2013) 87:3752–9. doi: 10.1128/JVI.02832-12
131. Na Y, Nam J-P, Hong J, Oh E, Shin HC, Kim HS, et al. Systemic administration of human mesenchymal stromal cells infected with polymer-coated oncolytic adenovirus induces efficient pancreatic tumor homing and infiltration. J Control Release. (2019) 305:75–88. doi: 10.1016/j.jconrel.2019.04.040
132. Gujar S, Pol JG, Kim Y, Lee PW, and Kroemer G. Antitumor benefits of antiviral immunity: an underappreciated aspect of oncolytic virotherapies. Trends Immunol. (2018) 39:209–21. doi: 10.1016/j.it.2017.11.006
133. Lin D, Shen Y, and Liang T. Oncolytic virotherapy: basic principles, recent advances and future directions. Sig Transduct Target Ther. (2023) 8:1–29. doi: 10.1038/s41392-023-01407-6
134. Osali A, Zhiani M, Ghaebi M, Meymanat M, and Esmaeilzadeh A. Multidirectional strategies for targeted delivery of oncolytic viruses by tumor infiltrating immune cells. Pharmacol Res. (2020) 161:105094. doi: 10.1016/j.phrs.2020.105094
135. Zhao Y, Liu Z, Li L, Wu J, Zhang H, Zhang H, et al. Oncolytic adenovirus: prospects for cancer immunotherapy. Front Microbiol. (2021) 12:707290. doi: 10.3389/fmicb.2021.707290
136. Chimeric oncolytic adenovirus armed chemokine rantes for treatment of breast cancer - PMC . Available online at: https://pmc.ncbi.nlm.nih.gov/articles/PMC9332706/.
137. Flores EB, Aksoy BA, and Bartee E. Initial dose of oncolytic myxoma virus programs durable antitumor immunity independent of. Vivo Viral replication. J Immunother Cancer. (2020) 8:e000804. doi: 10.1136/jitc-2020-000804
138. Naumenko V, Rajwani J, Turk M, Zhang C, Tse M, Davis RP, et al. Repeated dosing improves oncolytic rhabdovirus therapy in mice via interactions with intravascular monocytes. Commun Biol. (2022) 5:1–14. doi: 10.1038/s42003-022-04254-3
139. Eisenstein S, Coakley BA, Briley-Saebo K, Ma G, Chen H, Meseck M, et al. Myeloid-derived suppressor cells as a vehicle for tumor-specific oncolytic viral therapy. Cancer Res. (2013) 73:5003–15. doi: 10.1158/0008-5472.CAN-12-1597
140. Sakura K, Kuroyama M, Shintani Y, Funaki S, Atagi S, Kadota Y, et al. Dose-escalation, tolerability, and efficacy of intratumoral and subcutaneous injection of hemagglutinating virus of Japan envelope (HVJ-E) against chemotherapy-resistant Malignant pleural mesothelioma: a clinical trial. Cancer Immunol Immunother. (2024) 73:243. doi: 10.1007/s00262-024-03815-1
141. Macedo N, Miller DM, Haq R, and Kaufman HL. Clinical landscape of oncolytic virus research in 2020. J Immunother Cancer. (2020) 8:e001486. doi: 10.1136/jitc-2020-001486
142. Tang X-D, Ji T-T, Dong J-R, Feng H, Chen F-Q, Chen X, et al. Pathogenesis and treatment of cytokine storm induced by infectious diseases. Int J Mol Sci. (2021) 22:13009. doi: 10.3390/ijms222313009
143. Wu L, et al. rVSV(MΔ51)-M3 is an effective and safe oncolytic virus for cancer therapy. Hum Gene Ther. (2008) 19:635–47. doi: 10.1089/hum.2007.163
144. Chen F, et al. SMAC-armed oncolytic virotherapy enhances the anticancer activity of PD1 blockade by modulating PANoptosis. biomark Res. (2025) 13:8. doi: 10.1186/s40364-025-00726-w
145. Shalhout SZ, Miller DM, Emerick KS, and Kaufman HL. Therapy with oncolytic viruses: progress and challenges. Nat Rev Clin Oncol. (2023) 20:160–77. doi: 10.1038/s41571-022-00719-w
146. Hong Y, Cheng K, Qu H, Wang Y, Wang Y, Fan G, et al. Safety of talimogene laherparepvec: a real-world retrospective pharmacovigilance study based on FDA Adverse Event Reporting System (FAERS). J Pharm Health Care Sci. (2024) 10:79. doi: 10.1186/s40780-024-00388-0
147. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome.
148. Ouyang P, Wang L, Wu J, Tian Y, Chen C, Li D, et al. Overcoming cold tumors: a combination strategy of immune checkpoint inhibitors. Front Immunol. (2024) 15:1344272. doi: 10.3389/fimmu.2024.1344272
149. Feng M, Jiang W, Kim BYS, Cheng Zhang C, Fu Y-X, and Weissman IL. Phagocytosis checkpoints as new targets for cancer immunotherapy. Nat Rev Cancer. (2019) 19:568–86. doi: 10.1038/s41568-019-0183-z
150. Yamamoto K, Venida A, Yano J, Biancur DE, Kakiuchi M, Gupta S, et al. Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I. Nature. (2020) 581:100–5. doi: 10.1038/s41586-020-2229-5
151. Ahuja S and Zaheer S. The evolution of cancer immunotherapy: a comprehensive review of its history and current perspectives. Korean J Clin Oncol. (2024) 20:51–73. doi: 10.14216/kjco.24009
152. Mitra A, Barua A, Huang L, Ganguly S, Feng Q, He B, et al. From bench to bedside: the history and progress of CAR T cell therapy. Front Immunol. (2023) 14:1188049. doi: 10.3389/fimmu.2023.1188049
153. Labanieh L and Mackall CL. CAR immune cells: design principles, resistance and the next generation. Nature. (2023) 614:635–48. doi: 10.1038/s41586-023-05707-3
154. Wang L, Chard Dunmall LS, Cheng Z, and Wang Y. Remodeling the tumor microenvironment by oncolytic viruses: beyond oncolysis of tumor cells for cancer treatment. J Immunother Cancer. (2022) 10:e004167. doi: 10.1136/jitc-2021-004167
155. Gujar S, Pol JG, and Kroemer G. Heating it up: Oncolytic viruses make tumors ‘hot’ and suitable for checkpoint blockade immunotherapies. Oncoimmunology. (2018) 7:e1442169. doi: 10.1080/2162402X.2018.1442169
156. DePeaux K, Rivadeneira DB, Lontos K, Dean VG, Gunn WG, Watson MJ, et al. An oncolytic virus–delivered TGFβ inhibitor overcomes the immunosuppressive tumor microenvironment. J Exp Med. (2023) 220:e20230053. doi: 10.1084/jem.20230053
157. Nguyen TT, Shin DH, Sohoni S, Singh SK, Rivera-Molina Y, Jiang H, et al. Reshaping the tumor microenvironment with oncolytic viruses, positive regulation of the immune synapse, and blockade of the immunosuppressive oncometabolic circuitry. J Immunother Cancer. (2022) 10:e004935. doi: 10.1136/jitc-2022-004935
158. Hirigoyen U, Guilbaud C, Krejbich M, Fouet M, Fresquet J, Arnaud B, et al. Oncolytic viruses alter the biogenesis of tumor extracellular vesicles and influence their immunogenicity. Mol Therapy: Oncol. (2024) 32:200887. doi: 10.1016/j.omton.2024.200887
159. Asija S, Chatterjee A, Goda JS, Yadav S, Chekuri G, and Purwar R. Oncolytic immunovirotherapy for high-grade gliomas: A novel and an evolving therapeutic option. Front Immunol. (2023) 14:1118246. doi: 10.3389/fimmu.2023.1118246
160. Robert C, Ribas A, Schachter J, Arance A, Grob J-J, Mortier L, et al. Pembrolizumab versus ipilimumab in advanced melanoma (KEYNOTE-006): post-hoc 5-year results from an open-label, multicentre, randomised, controlled, phase 3 study. Lancet Oncol. (2019) 20:1239–51. doi: 10.1016/S1470-2045(19)30388-2
161. Maleki Vareki S. High and low mutational burden tumors versus immunologically hot and cold tumors and response to immune checkpoint inhibitors. J ImmunoTherapy Cancer. (2018) 6:157. doi: 10.1186/s40425-018-0479-7
162. Benoit A, Vogin G, Duhem C, Berchem G, and Janji B. Lighting up the fire in the microenvironment of cold tumors: A major challenge to improve cancer immunotherapy. Cells. (2023) 12:1787. doi: 10.3390/cells12131787
163. Galon J and Bruni D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat Rev Drug Discov. (2019) 18:197–218. doi: 10.1038/s41573-018-0007-y
164. Appleton E, Hassan J, Chan Wah Hak C, Sivamanoharan N, Wilkins A, Samson A, et al. Kickstarting immunity in cold tumours: localised tumour therapy combinations with immune checkpoint blockade. Front Immunol. (2021) 12:754436. doi: 10.3389/fimmu.2021.754436
165. Xuan Y, Yan W, Wang R, Wang X, Guo Y, Dun H, et al. GM-CSF and IL-21-armed oncolytic vaccinia virus significantly enhances anti-tumor activity and synergizes with anti-PD1 immunotherapy in pancreatic cancer. Front Immunol. (2024) 15:1506632. doi: 10.3389/fimmu.2024.1506632
166. Grimes JM, Ghosh S, Manzoor S, Li L X, Moran MM, Clements JC, et al. Oncolytic reprogramming of tumor microenvironment shapes CD4 T-cell memory via the IL6ra-Bcl6 axis for targeted control of glioblastoma. Nat Commun. (2025) 16:1095. doi: 10.1038/s41467-024-55455-9
167. Hajda J, Leuchs B, Angelova AL, Frehtman V, Rommelaere J, Mertens M, et al. Phase 2 trial of oncolytic H-1 parvovirus therapy shows safety and signs of immune system activation in patients with metastatic pancreatic ductal adenocarcinoma. Clin Cancer Res. (2021) 27:5546–56. doi: 10.1158/1078-0432.CCR-21-1020
168. Todo T, Ito H, Ino Y, Ohtsu H, Ota Y, Shibahara J, et al. Intratumoral oncolytic herpes virus G47Δ for residual or recurrent glioblastoma: a phase 2 trial. Nat Med. (2022) 28:1630–9. doi: 10.1038/s41591-022-01897-x
Keywords: oncolytic virus, metastatic, prostate cancer, immunotherapy, virotherapy, experimental, models
Citation: Chen Y-C and Figueiredo ML (2025) Experimental models for developing oncolytic virotherapy for metastatic prostate cancer. Front. Immunol. 16:1626432. doi: 10.3389/fimmu.2025.1626432
Received: 10 May 2025; Accepted: 17 June 2025;
Published: 10 July 2025; Corrected: 11 August 2025.
Edited by:
Pratik Bhojnagarwala, Wistar Institute, United StatesReviewed by:
Dhivya Sridaran, Washington University in St. Louis, United StatesMohammad Suhail Khan, The Wistar Institute, United States
Copyright © 2025 Chen and Figueiredo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marxa Leão Figueiredo, bWxmaWd1ZWlAcHVyZHVlLmVkdQ==