 Emilie Vernay1,2
Emilie Vernay1,2 Elisabeth Cerrato1,2
Elisabeth Cerrato1,2 François Santinon1,2
François Santinon1,2 Céline Monard3Pauline Perez3
Céline Monard3Pauline Perez3 Florence Allantaz1,2
Florence Allantaz1,2 Anne-Claire Lukaszewicz1,3
Anne-Claire Lukaszewicz1,3 Jean-François Llitjos1,2,3*
Jean-François Llitjos1,2,3*- 1EA 7426 “Pathophysiology of Injury-Induced Immunosuppression”, Joint Research Unit Université Claude Bernard Lyon 1 - Hospices Civils de Lyon - bioMérieux, Lyon, France
- 2Innovation and Partnership (I&P), bioMérieux S.A., Marcy L’Etoile, France
- 3Anaesthesia and Critical Care Medicine Department, Hospices Civils de Lyon, Edouard Herriot Hospital, Lyon, France
Owing to faster identification of sepsis and improvement of patient management, most septic patients now survive the early phase of sepsis. Therefore, one of the major challenges in sepsis management today is to identify those patients at risk and propose effective personalized therapy. The complexity of the mechanisms involved in the septic immune response and its dysregulation is reflected in the diversity of immune profiles among sepsis patients. It is now well recognized that this heterogeneity is a major obstacle to stratifying patients based on their susceptibility to secondary infections. Since sepsis can originate from different anatomical sites, some studies have investigated their impact to decipher the heterogeneity. They concluded that the site of infection affects patient outcomes and leads to different immune alterations. This narrative review focuses on pulmonary sepsis to highlight the importance of studying organ response directly with local immune cells. Understanding the persistent dysregulation within the lung, whether it involves pulmonary immune cells or other lung components, is critical. Some studies have already examined the remodeling and loss of functionality of alveolar macrophages after the initial insult. Ongoing research is also investigating the impact of imbalances in other lung players, such as epithelial cells or the microbiota, on susceptibility to pulmonary reinfection.
1 Introduction
Sepsis is defined as a dysregulated host response to infection, resulting in life-threatening organ dysfunction (1). In 2017, sepsis affected around 49 million people and was responsible for approximately 11 million deaths worldwide (2). In addition, sepsis is associated with significant long-term health issues, including higher rates of mortality after discharge, and physical and cognitive impairments (3). Due to its high incidence rate and associated complications, sepsis represents a major health concern (4).
Over the past decades, numerous studies have aimed to elucidate the pathophysiology of sepsis, with most data coming from the circulating compartment. Initially, the sepsis model was thought to be biphasic, starting with an initial phase of hyperinflammation including the secretion of pro-inflammatory cytokines and neutrophil recruitment (5). In this model, this pro-inflammatory phase was balanced by an immunosuppressive compensatory phase to restore homeostasis. The hallmarks of this anti-inflammatory phase comprise a decreased monocytic HLA-DR expression, T cell exhaustion and lymphocyte apoptosis (6). However, recent studies have challenged this model by identifying immunosuppressive features during the early phase of sepsis. Thus, the current consensus is that pro-inflammatory and anti-inflammatory processes occur simultaneously (7), with the early phase dominated by hyperinflammation and, in cases of unresolved sepsis, followed by a persistent immunosuppression (8).
Advances in understanding the early phase of sepsis and improved patient management have reduced the mortality of sepsis in the first 48 hours. However, patients remain at high risk of secondary infections, which have a serious impact on their short and medium-term prognosis (9). A retrospective study estimated that approximately one third of septic patients develop a nosocomial infection during their ICU stay, leading to an increased risk of mortality and prolonged hospitalization (10). Over the past years, a growing amount of clinical and experimental data has shown that this susceptibility to secondary infections is associated with persistent immunosuppression resulting from an imbalance in the host response (11).
Identifying patients at higher risk of secondary infections remains a challenge. Although several diagnostic solutions have been proposed, their performance does not provide a sufficient level of accuracy and does not allow their use in clinical routine (12–14). One of the reasons limiting their use is the wide range of responses that are observed, due to the heterogeneity of immune profile following initial inflammatory response. Several reasons have been suggested to explain this heterogeneity, such as host-related factors, nature or site of initial infection (15). Interestingly, data examining the host response in patients according to the source of sepsis suggest the existence of organ-specific immune alterations (16). Although scarcely evaluated to date, the heterogeneity of the immune response at the organ level, as well as the source of infection, represents an opportunity to advance our understanding of the mechanisms underlying sepsis. It may also improve the accuracy and relevance of diagnostic tools to identify patients at risk of secondary infection.
The aim of this review is to discuss the key role of local immune alterations in sepsis and to provide new perspectives for the development of immunomodulatory therapies to prevent secondary infections.
2 Heterogeneity of sepsis population
Although it is now well-accepted that septic patients are highly heterogeneous, the underlying mechanisms of this variability remain poorly understood. The sources of heterogeneity in the pathophysiology of sepsis are diverse and include the pathogens causing the infection (17), organ dysfunction, host responses, host factors and the primary site of infection. Hereafter, we discuss how the origin of the infection leading to sepsis influences outcomes and the organ-specific immune response.
2.1 Source of sepsis impacts clinical outcome and systemic host response
Sepsis is a syndrome defined by an uncontrolled immune response to infection, resulting in organ dysfunction. It can originate from a variety of anatomical sites, including the abdomen, lungs, central nervous system, skin and soft tissues, and genitourinary system. This definition suggests that the systemic immune response remains consistent, irrespective of the location of the infection.
Nevertheless, comparative analyses of patient outcomes across sepsis cohorts have demonstrated that clinical trajectories (particularly organ failure and length of stay) vary according to the initial source of infection (18). These differences were observed even when initial severity was similar between groups (19). In their retrospective study, Leligdowicz et al. (20), examined the association between initial site of infection and hospital mortality among sepsis patients, adjusting for predisposing factors such as comorbidities. Their findings suggested that there were differences in hospital mortality according to the initial site of infection. Specifically, intra-abdominal infections, lower respiratory tract infections, and biliary tract infections were associated with higher mortality rates. A similar conclusion was also drawn in a large cohort of sepsis patients in the UK (21). Furthermore, a longitudinal study demonstrated that the source of the initial infection in sepsis patients affects not only in-hospital mortality but also long-term outcomes (22).
Apart from mortality, anatomical sources of infection also influence the susceptibility to secondary infections. In a study focused on pulmonary sepsis, Llitjos et al. observed that an initial pulmonary infection increased susceptibility to a subsequent pulmonary infection (23). Similar results were also observed in another study (24).
To explore the systemic immune response, studies have compared several blood-based markers in septic patients, stratified by infection site. In the MARS cohort, transcriptomic analyses of peripheral blood revealed both shared and site-specific immune signatures (16). Furthermore, the levels of plasma cytokines involved in inflammation, endothelial cell activation, and coagulation activation vary according to the origin of sepsis. Stortz et al. corroborated these findings, demonstrating discrepancies in the plasma levels of pro-inflammatory markers among septic patients categorized by the initial infection site (22). Differences at cellular levels were also reported. A longitudinal evaluation of the absolute lymphocyte count (ALC) was conducted during the initial 14 days of intensive care unit (ICU) treatment. It revealed distinct recovery trajectories to normal ALC ranges among the study’s subgroups.
Taken together, these data demonstrate that different systemic immune responses are elicited depending on the location of the initial infection. A comprehensive analysis of sepsis, informed by the anatomical location of the infection, holds potential to elucidate the heterogeneity in immune responses and enhance the precision of clinical predictions, including mortality and length of hospital stay. Additionally, given that the primary sources of sepsis influence susceptibility to secondary infections, this suggests distinct biological immune alterations across organs.
2.2 Existence of a multilevel organ specific immune response
Previously cited studies have focused on the analysis of blood samples to investigate how the host immune response varies with the primary site of infection in sepsis. These differences may be attributed to the presence of organ-specific immune mechanisms. Murine models are frequently used to investigate these responses in depth due to the ease with which immune responses in different tissues can be evaluated. The most common murine models of sepsis involve two distinct approaches: cecal ligation and puncture (CLP), which mimics abdominal sepsis, and injection of lipopolysaccharide (LPS).
These models have enabled comparisons of transcriptomic dysregulation across organs (25–27). In particular, following sublethal injection of LPS, transcriptomic mapping of 13 different tissues unveiled organ-specific signatures of sepsis (26). The longitudinal assessments of gene expression in these tissues revealed different recovery capacities among organs. Similar results were obtained in a CLP model, where many dysregulated genes implicated in inflammatory responses, including interleukin-1β (IL-1β), reactive oxygen species (ROS) generation, and complement proteins, were differentially expressed across organs (27). These results were corroborated at the protein level. For instance, the production of tumor-necrosis factor alpha (TNF-α) in the peritoneum was found to be 16 times higher than in the blood using a CLP-induced sepsis model. While TNF-α levels normalized in the peritoneum within 24 hours, they remained elevated in the lungs for 5 days. This highlights differences in both the magnitude and duration of the inflammatory response between organs (28).
Beyond inflammatory mediators, organ-specific immune responses also involve other molecular pathways. In mice exposed to systemic LPS, lung and kidney tissues showed different expression levels of adhesion molecules and proteins involved in maintaining endothelial integrity (29). Since leukocyte recruitment to infected organs depends on their ability to cross the endothelial barrier, these findings demonstrate that organ-specific differences in endothelial function can influence immune cell infiltration.
Indeed following CLP, quantitative and functional variations in tissue macrophages are observed, with the magnitude and direction of these changes varying according to the tissues examined (30). Similarly, the intravenous injection of LPS into humans has been demonstrated to elicit a shift in the responsiveness of alveolar macrophages (AMs) in vivo (31). Within the first 6 hours, AMs exhibited enhanced functional responsiveness to secondary in vitro stimulation. Nonresident cells also undergo modifications in an organ-specific manner following systemic inflammation. A study of the response of natural killer (NK) cells in mice injected with endotoxin revealed differences in recruitment and activation across the lungs, spleen, and blood (32). Through adoptive transfer, Rasid et al. demonstrated that NK cells display organ-specific responses influenced not only by their tissue of origin but also by local remodeling within the organ microenvironment.
These data underscore organ-specific immune responses that vary in intensity and modify tissue-resident cell responsiveness. As different infectious sites elicit distinct immune responses, the organ of origin may undergo specific modulation following initial injury. Some human studies have investigated the impact of infection site on local inflammation. In experimental endotoxemia, comparing endobronchial to intravenous LPS administration, alveolar cytokine levels were measured in the hours following the challenge (33). Bronchoalveolar lavage from volunteers who received pulmonary LPS instillation showed higher levels of proinflammatory cytokines than those who received intravenous administration, confirming that the kinetics of the immune response depend on whether the organ is the primary site of infection. As the site of infection undergoes a rapid and intense inflammatory response, it may disrupt local immune balance. Indeed, AMs, from patients with pulmonary sepsis exhibited reduced HLA-DR expression in comparison to those with abdominal sepsis (34). As low HLA-DR expression is recognized as a hallmark of immunosuppression, it indicates that the site of infection undergoes suppressive modulation of its local immunity. Local immunosuppression in the days following the initial insult was confirmed in a mouse model of sepsis comparing pulmonary immune responses after abdominal or pulmonary infection (35). Comparison of murine models revealed distinct AM depletion and reduced bacterial clearance following pulmonary sepsis. This immune alteration was associated with reduced survival after a secondary pulmonary infection. In a mouse model with intravenous LPS instillation, it was confirmed that immunomodulation depends on whether the considered organ is the source of sepsis, with lung cells exhibiting lower endotoxin tolerance than circulating cells (36).
Sepsis affects organs differently, with immune responses being organ specific in terms of cellular response, transcriptomics and proteomics. In addition, the post-acute phase differs depending on whether the organ is the primary source of infection or not, with persistent modulation localized to the primary infected organ.
3 Compartmentalised immune response in sepsis: pneumonia as a use case
Given that organs exhibit unique immune responses, especially when they are the primary site of infection, a detailed examination of local immunity could help elucidate the heterogeneity of sepsis patients. We will focus on pneumonia as it is the most common source of community sepsis in high-income countries (21, 37, 38) and the most frequent source of healthcare-associated infections (39). Sepsis induced by pneumonia is associated with both increased mortality compared to other sites of infection and high risk of secondary infections (40). We will focus on bacterial infections, which account for the majority of severe lung infections (41).
3.1 Immune response to pneumonia differs between circulating and lung compartments
Most studies on sepsis focus on peripheral blood, regardless of the infection site, due to its accessibility. It is commonly assumed that the results obtained from peripheral blood are a pertinent surrogate for the local immune response. However, the diagnostic performance of circulating biomarkers is poor in both sepsis and pneumonia (42). For instance, circulating procalcitonin (PCT) and C-reactive protein (CRP) exhibit poor performance in identifying patients with positive culture among those suspected of pulmonary infection (43). Similarly, plasma levels of interleukin-6 (IL-6) and interleukin-8 (IL-8), two pro-inflammatory cytokines, were similar in mechanically ventilated patients with and without a nosocomial pneumonia (44).
Given the limitations of blood biomarkers, several studies have examined markers of the immune response in both blood and lung compartments (Figure 1). As the pulmonary immune response is associated with strong neutrophil infiltration in the alveolar space, the neutrophil chemoattractant IL-8 has been of particular interest (45). Conway et al. compared serum and alveolar levels of IL-8 and other pro-inflammatory cytokines in patients with and without ventilator-associated pneumonia (VAP) (46). None of the serum cytokines evaluated could discriminate between the two groups, unlike alveolar cytokines, all of which showed higher levels in infected patients. Similar results were obtained in COVID-19-related ARDS patients, with endotracheal aspirate/serum cytokines ratios ranging from approximately 2 to 30,000 (47). Bendib et al, confirmed higher levels of alveolar proinflammatory cytokines, particularly IL-8, and noted differences in cellular activation markers between blood and lung compartments (48). In pneumonia patients, TNFα concentrations in bronchoalveolar lavage fluid (BALF) were, on average, ten times higher than in blood (49). This variability is also found within the organs themselves, with a regionalization of the immune response. In patients with unilateral lobar pneumonia, the concentration of IL-6 is significantly higher in the affected lung lobe compared to the unaffected one (49).
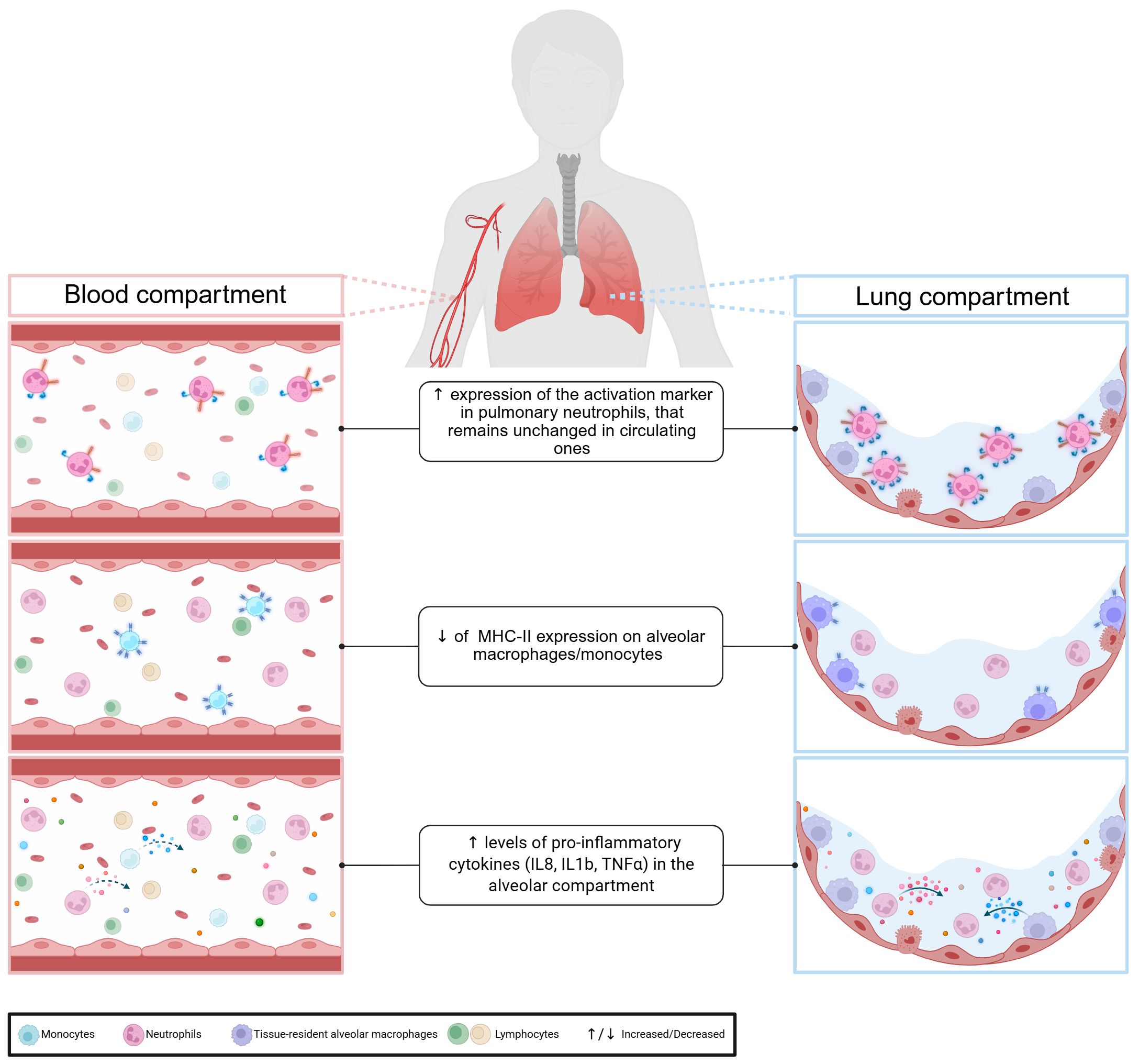
Figure 1. Compartmentalisation of pulmonary immune response.
Beyond quantitative differences, studies also reveal phenotypic and qualitative differences. In septic shock, monocytic HLA-DR expression differs significantly between blood and lung compartments (34). Another study reported that in a cohort of community acquired pneumonia, alveolar neutrophils expressed more activation markers than circulating neutrophils (50). Taken together, these data confirm that due to the compartmentalisation of the immune response, blood is a poor reflection of organ-specific immune responses. Therefore, to have a comprehensive view of the organ-specific immune response induced by pulmonary sepsis, we should focus on the immune response at the alveolar level.
3.2 Pulmonary inflammatory response in lower respiratory tract
3.2.1 Lower respiratory tract at homeostasis
At homeostasis the most abundant cells found in the lung are AMs. These tissue-resident cells are formed during prenatal stages (51) and act as patrolling cells to eliminate pathogens. AMs are a key component of pulmonary immunity as they are involved in inflammation but also in the return to homeostasis (52). AMs closely interact with alveolar epithelial cells (AEC), notably through cell-to-cell interactions involving CD200R, PD1, and SIRPa on macrophages and CD200, PDL1 and CD47 on alveolar epithelium, respectively (53). In addition to direct cell-to-cell interactions, AECs also secrete regulatory molecules, such as interleukin-10 (IL-10), granulocyte-macrophage colony-stimulating factor (GM-CSF), and transforming growth factor beta (TGF-β), that help maintain AMs in a steady state and prevent excessive inflammation (54, 55) (Figure 2). Recently, the study of the pulmonary immune response has included a less conventional player than immune cells: the microbiota. Its role in the development of respiratory infections and its interaction with the cells involved in the pulmonary immune response have been of particular interest (56, 57).
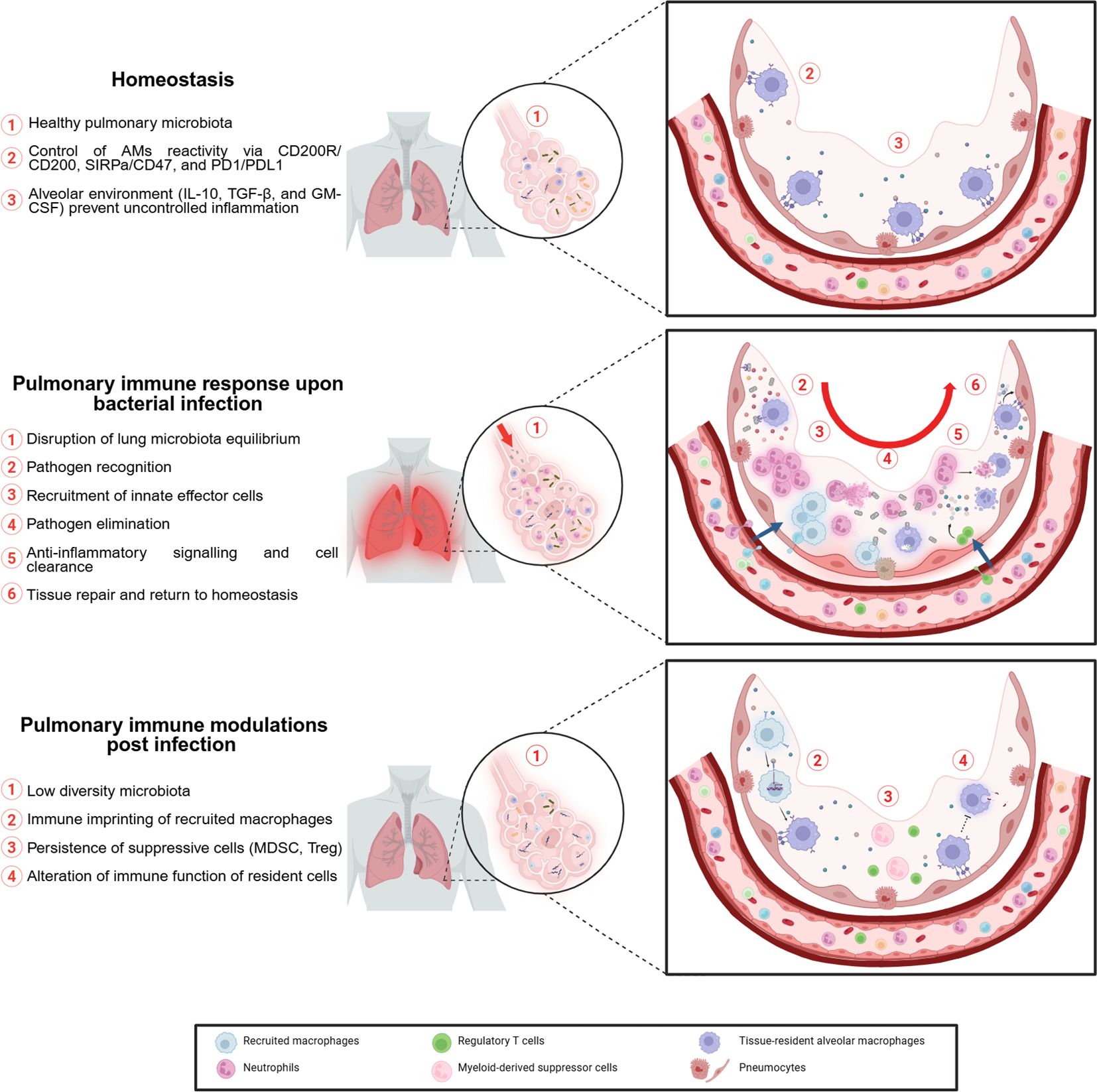
Figure 2. Impact of pulmonary bacterial infection on local environment.
3.2.2 Lung immune response to infection
The early phase of lung infection involves mainly resident AMs, which account for approximately 90% of patrolling cells in alveoli (58) and recruited neutrophils (59). Effector cells of the lung’s innate immune response eliminate bacteria through mechanisms such as phagocytosis (60), release of cytolytic molecules (e.g., reactive oxygen species, lysozyme, defensins) (61), and formation of neutrophil extracellular traps (NETs) (62). Alveolar epithelial cells also help initiate the immune response by secreting pro-inflammatory cytokines, including interleukin-33 (IL-33) (63, 64). This alarmin enhances host defense during pulmonary infection (65). Furthermore, through crosstalk, alveolar epithelial cells serve as gatekeepers for AMs responsiveness. Thus, their activation is critical for an efficient macrophage immune response (66). Innate immunity not only eliminates pathogens but also initiates the adaptive immune response. This process is mediated by antigen-presenting cells (APCs), including monocytes, macrophages, and dendritic cells (DCs). Although AMs have high phagocytic activity, their role as APCs remains unclear (67). APCs activate T cell response, which in the case of pulmonary bacterial infection is mainly mediated by Th17 cells (68). These polarized T cells secrete interleukin-17 (IL-17), which enhances the innate response, for example, by improving neutrophil recruitment (69).
To prevent tissue damage from excessive inflammation, the lung relies on a feedback loop. Resolution involves cell death of immune cells recruited during infection and their clearance to restore normal lung composition. Cell death occurs through various mechanisms, including apoptosis, necrosis, and pyroptosis (70, 71). These processes are triggered either by intracellular programming or by extracellular signals released from activated innate immune cells (72). Most mechanisms trigger the release of proteins and cellular debris identified by immune cells as damage-associated molecular patterns (DAMPs), which in turn amplify the inflammatory response. For example, when neutrophils undergo cell death, vesicles contained in their cytoplasm release cytolytic enzymes, that can cause tissue damage (73). To prevent this, phagocytic cells and particularly AMs are responsible for eliminating dying cells by efferocytosis (74). Dying cells display ‘eat-me’ signals, either soluble or membrane-bound, that help AMs recognize and clear them. For instance, through the TYRO3, AXL and MERTK (TAM) family of receptors, AMs can recognize apoptotic cells by binding to phosphatidylserine on their surface (75). Removal of dying cells by AMs induces a shift (52) from pro- to an anti-inflammatory profile promoting return to homeostasis and tissue repair. Noteworthy, murine AMs have demonstrated self-induction of immunosuppressive signaling following infection (76). To facilitate the return to homeostasis AMs secrete immunosuppressive cytokines that promote the dampening of inflammation (77). Tissue repair is promoted by AMs through activation of AECII proliferation (78), which can transdifferentiate into type I cells to rebuild the alveolar-capillary barrier (79). In addition, AMs are responsible for the homing of memory lymphocytes to the mucosa through prostanglandin E2 (PGE2) signaling, providing long-term immunity (80).
AMs are not the only cell type involved in the pulmonary feedback loop. Zhang et al, demonstrated that instillation of apoptotic cells into LPS-infected mouse lungs enhanced resolution of inflammation (81), in particular by inducing recruitment of functional lymphocyte T regulators (T reg). The role of Treg in the resolution of pulmonary inflammation is multifaceted. They are important sources of suppressive cytokines but are also capable of enhancing anti-inflammatory mechanisms such as AM efferocytosis (82). They also act as suppressors of immune function, notably by regulating IL-17 secretion by Th17 cells (83) and gamma delta (γδ) T cells (84), to prevent uncontrolled secretion that can lead to chronic disease (85). Among lymphoid cell types, unconventional lymphocytes, which include innate lymphoid cells (ILCs) and γδ T cells, also participate in the anti-inflammatory response. They favor cell clearance through cytotoxic activity (86) and support tissue repair through secretion of GM-CSF, which is involved in maintaining and replenishing the pool of AMs (87).
In summary, host defense involves a coordinated process, from pathogen recognition and elimination to restoration of homeostasis (Figure 2). This fragile balance between pro- and anti-inflammatory mechanisms can be disturbed by uncontrolled inflammation during sepsis. Thus, unraveling the pulmonary modulation resulting from the immune response could help explain susceptibility to lung reinfection and provide new approaches to boost immunity and prevent reinfection.
4 Increased susceptibility to secondary infections due to imbalanced pulmonary immune response
Similarly to the systemic response, the pulmonary immune response combines pro- and anti-inflammatory signals that are tightly regulated to provide sufficient and balanced inflammation to resolve infection without harming the host. In the context of pulmonary sepsis, immune dysfunction characterized by an uncontrolled inflammatory response is associated with tissue damage leading to lung injury or development of acute respiratory distress syndrome (ARDS) (88). Susceptibility to secondary infections is generally associated with persistent immunosuppression (89). In peripheral blood, it is characterized by uncontrolled apoptosis of immune cells, altered cell functionality and high levels of immunosuppressive factors (6). In parallel with these systemic alterations, we next examine how the pulmonary immune environment is modified following a primary infection, with particular attention to local immune dysfunction (Table 1).
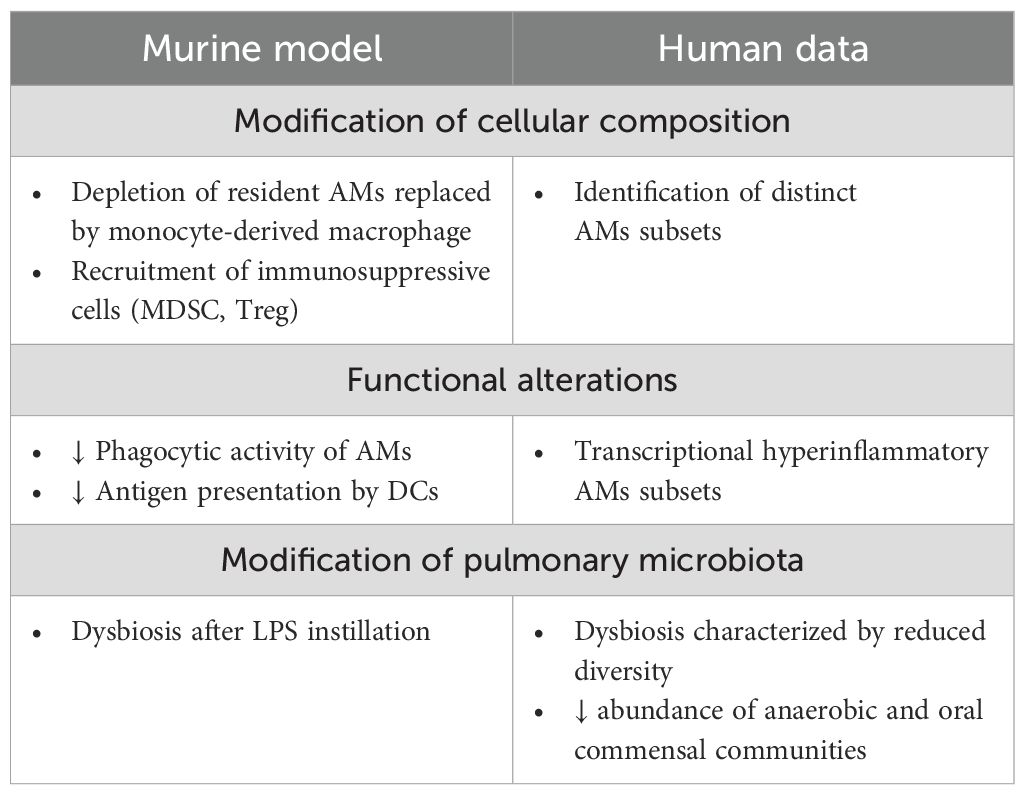
Table 1. Alteration post-infection of lung environment.
4.1 Post-infection cells remodeling
As tissue-resident cells, the maintenance and replenishment of the AMs pool during infection are critical. After infection with S.pneumonia, mouse AMs are depleted due to antimicrobial activity (90, 91). However, 35 days after infection, the absolute number of AMs was equivalent to the basal level. Using a mouse model of LPS pulmonary instillation, characterization of replenished AMs revealed that they were mainly newly recruited and differentiated from circulating monocytes (92), suggesting monocyte recruitment rather than proliferation.
In addition to phenotypic changes, infection also induces functional remodeling of AMs. In mice, both resident and recruited macrophages were transcriptionally activated up to 12 days after the onset of infection compared to baseline (93). However, the activated pathway and cytokine secretion differed between resident and recruited macrophages. In a similar model, 7 days after intratracheal instillation of LPS, recruited macrophages showed enrichment in pathways related to immune response and signal transduction. In contrast resident macrophages were enriched in PPAR signaling and fatty acid metabolism and degradation signaling (94). Functional assays of both type of macrophages identified that efferocytosis ability was higher in resident AMs.
To explain these functional differences, it has been proposed that the alveolar niche plays a pivotal role in regulating AMs activity through a process known as “tissue immune imprinting” (95). According to this model, resident AMs are shaped by the alveolar microenvironment, which limits their plasticity and promotes a hyporesponsive phenotype, helping to prevent excessive immune activation and facilitates rapid resolution. In contrast, during inflammatory episodes, circulating monocytes are recruited to the lung and differentiate into macrophages that retain a pro-inflammatory epigenetic program. Thus, newly recruited macrophages exhibit greater plasticity and are more prone to mounting exaggerated inflammatory responses. Therefore, alveolar niche imprinting is essential for maintaining the homeostatic function of AMs and preventing dysregulated immune responses.
While murine models of infection have identified distinct phenotypes for AMs and characterized the kinetics of each subset, little is known about this heterogeneity in humans. Recently, the heterogeneity of the AMs population at homeostasis was confirmed by single cell RNA-seq, which identified distinct subsets of AMs in BALF from healthy volunteers (96). In patients with mild or severe pneumonia, similar analyses revealed the emergence of hyperinflammatory subsets of neutrophils and AMs that were absent in healthy controls (97). Morell et al. further identified up to six transcriptionally distinct AM clusters in BALF from patients with acute hypoxemic respiratory failure, with one subset correlating with the severity of acute respiratory distress syndrome (ARDS). These studies pave the way for further functional and clinical characterization of AMs subsets, as their specific roles in pulmonary immune responses and disease progression remain to be fully elucidated.
4.2 Impact of alveolar remodeling on susceptibility to infections
Following primary infection, not only is the cellular composition of resident cells modified, but their functionality is also impaired. Using a mouse model of sequential pulmonary bacterial infection, Roquilly et al. investigated the functional changes induced by a first infection on alveolar cells (98). They observed reduced bacterial clearance in the lungs of mice infected twice compared to once. Evaluation of different immune functions of AMs from infected mice showed reduced phagocytic capacity without changes in major histocompatibility complex II (MHCII) expression or LPS responsiveness. Using adoptive transfer of naive AMs, the authors found that the alteration in phagocytic capacity depends on early signal regulatory protein α (SIRPα) signaling, which modulates the alveolar environment after infection. Thus, AMs paralysis results from an unbalanced immune response that perturbs the local immune environment. Consistent with these findings, another study showed that alterations in phagocytic capacity occurred only when the initial infection was pulmonary (35). Alteration of cell functionality is not limited to AMs. In a double pneumonia mouse model, it was observed that dendritic cells (DCs) developing in the lung after resolution of primary pneumonia had a reduced capacity to present antigens and secrete immunostimulatory cytokines (99). However, no DC alteration was observed in the spleen, highlighting the compartmentalisation of immune changes to infected tissues.
In another aspect, studies have reported recruitment of immunosuppressive cells associated with impaired immune response. Repeated pulmonary infection with A.Baumannii is associated with an increase in the Treg population in the lung altering the pro-inflammatory response (100). Myeloid-derived suppressor cells (MDSCs), suppressive cells derived from the myeloid lineage, have been reported to be recruited into the alveoli during bacterial infections (101). However, their potential beneficial or detrimental effects remain unclear. In K.pneumoniae infection, they were found to participate in resolving lung inflammation and have reparative function (102, 103). On the contrary, they promoted lung injury in a mouse model of severe tuberculosis (104). While suppression of T cell functions by MDSCs is well documented, their impact on AMs functions remains to be determined. For example, MDSCs induced by fungal infection impair AMs phagocytosis (105). As all these studies are based on the mouse model, the role of MDSCs in pulmonary sepsis on alveolar cells remains to be determined in humans.
This highlights the importance of murine models in providing evidence of profound modulation of pulmonary immunity following initial infection. Notably, pneumonia-induced remodeling of AMs subsets has been shown to be long-lasting, with phenotypic and transcriptional changes in murine AMs lasting up to six months after infection (106). These alterations include changes in the phenotype of resident lung cells as well as their functionality. The unbalanced immune response is also reflected by modification of the lung environment including immunosuppressive features. This underscores the importance of understanding how infection sequelae can disrupt mechanisms that maintain the balance of the pulmonary microcosm. Indeed, the existence of a pulmonary microbiota implies the presence of tolerance mechanisms towards commensal bacteria by resident pulmonary immune cells. However, the paralysis of their microbicidal mechanisms could create a favorable environment for commensal pathogenic bacteria to proliferate, leading to secondary infections.
4.3 The importance of the lung microbiota and its modulation
Contrary to the old belief that the lung is a sterile environment, it harbors a rich microbiota consisting of bacteria, viruses and fungi (56). The microbiota is continuously renewed, but the core lung microbiome includes genera such as Pseudomonas, Streptococcus, Proteus, Clostridium, Haemophilus, Veillonella, and Porphyromonas. Compared to the upper respiratory tract, the lung microbiota harbors the same bacterial populations but with a lower biomass (107). So far, the dynamics of the microbiota in pulmonary infections remain poorly characterized.
4.3.1 Pulmonary infection and microbiota dysbiosis
Because mechanical ventilation provides easier access to lung samples, most studies have examined the microbiota in ventilated patients either at the time of diagnosis of pulmonary infection or through longitudinal assessment after intubation. Fenn et al. found that the microbiota in patients with ventilator-acquired pneumonia was characterized by lower diversity with a predominance of pathogenic bacteria (108). Dysbiosis of the lung microbiota was also observed in diverse clinical context of pulmonary infection (109, 110). To identify if specific communities were impacted by microbiota dysbiosis, Kitsios et al. conducted a longitudinal study of the microbiome during the first two weeks of intubation in patients with acute respiratory failure (111). They observed a reduction in the abundance of anaerobic bacteria and oral commensal bacteria. They also showed that patients who maintained low microbiome diversity over the two-week period had worse survival outcomes, linking disease severity to microbiome dynamics. All studies concluded to a shift from a “healthy,” well-balanced polymicrobial microbiome to an “unhealthy,” restricted, and less diverse microbiota. These observations were confirmed in murine models of LPS-induced lung injury (112). However, due to the timing of sampling, often at the point of infection diagnosis, and the bacterial origin of the infection, it remains unclear whether dysbiosis is a cause or a consequence of secondary pneumonia.
4.3.2 Pulmonary microbiota dynamics
The dynamics of the lung results from three mechanisms: microbial immigration, microbial elimination, and the proliferation rates of its members. Dickinson et al. proposed that the microbiome in the healthy lung is determined by the balance between immigration and elimination mechanisms (113). Microbes originating from the air and upper respiratory tract migrate to the lower respiratory tract (114) and their numbers are regulated by numerous mechanisms such as coughing or mucociliary clearance. To date, the dysbiosis characteristic of pulmonary infection is thought to result from changes in endogenous growth conditions through mechanisms that are not yet fully understood, despite various hypotheses (115). Modification of the lung environment can result from abiotic or biotic factors. Abiotic factors include temperature, pressure, nutrient accessibility, but also mucus or pulmonary surfactant secretion, which are modified during the host response. It can also be modified by external interventions. For example, mechanical ventilation has been associated with a decrease in bacterial alpha diversity (116). The microbiota is also modulated by surrounding cells. As the lung is colonized with bacteria, at homeostasis, host-microbiota interactions exhibit symbiotic relationships to promote tolerance. Upon infection, lung cells are mobilized to mount an immune response, disrupting their basal state of activation.
4.3.3 Host-microbiota crosstalk
The interaction between microbiota and lung cells is a two-way crosstalk. At homeostasis, microbiota diversity influences the cytokine lung environment and cell composition (117, 118). AMs from microbiome-depleted mice show impaired ROS production in response to in vitro infection with K.pneumoniae (119). Moreover inflammatory signaling was impaired in mice lacking microbiota after infection with K.pneumoniae or S.pneumoniae, in particular, with lower levels of GM-CSF and IL-17A, two cytokines participating in host defense (120). However, most studies have investigated the global impact of microbiota on the immune response, rather than the importance of individual species. One study, identified Prevotella as an innate response enhancer that provides protection against infection in mice through pattern recognition receptor signaling (121). Further studies are needed to determine the impact of different species depletion on host immune cells, particularly to identify mechanisms of host-microbiota crosstalk that may provide new potential targets for enhancing the immune response.
Despite growing evidence that pulmonary infection is associated with microbiota dysbiosis, host-microbe interactions are complex and bidirectional, complicating the understanding of microbiota dynamics during infection (115). To elucidate the role of dysbiosis in susceptibility to secondary infection, either by favoring the outgrowth of certain bacteria or by altering immune cell function, several limitations in current research need to be addressed. These include overcoming of technical limitations (e.g low biomass, anaerobic microorganisms), expanding sequencing libraries to include viruses and microbial eukaryotes, describing the healthy dynamics of the microbiome, and accounting for interpersonal variability and confounding by prioritizing longitudinal studies.
5 New approaches for immunomodulatory therapies
Sepsis is characterized by an unbalanced immune response, making immunotherapy a promising approach to regulate the immune response in patients. Despite promising results in animal models, all clinical trials have failed to demonstrate a beneficial effect of immunomodulatory therapy. The limitations and reasons for these failures have been extensively reviewed, highlighting the importance of addressing the heterogeneity of immune profiles (122–124). To address this, several avenues for future research have been proposed, all pointing towards a comprehensive description of sepsis-induced alterations. Considering the compartmentalisation of the immune response will not only help to decipher this heterogeneity but will also provide a complete picture of all sepsis-induced local modulations, including all local actors involved in maintaining homeostasis. Identification of key mechanisms favoring the onset of nosocomial infections thus provides new potential targets for novel immunomodulatory strategies. Here, we will focus on the post-infection phase of pulmonary sepsis and how the dysregulation between pro-inflammatory signaling and compensatory mechanisms could be modulated to prevent secondary infection, either by directly targeting the immune system or indirectly by modulating the microbiota (Table 2).
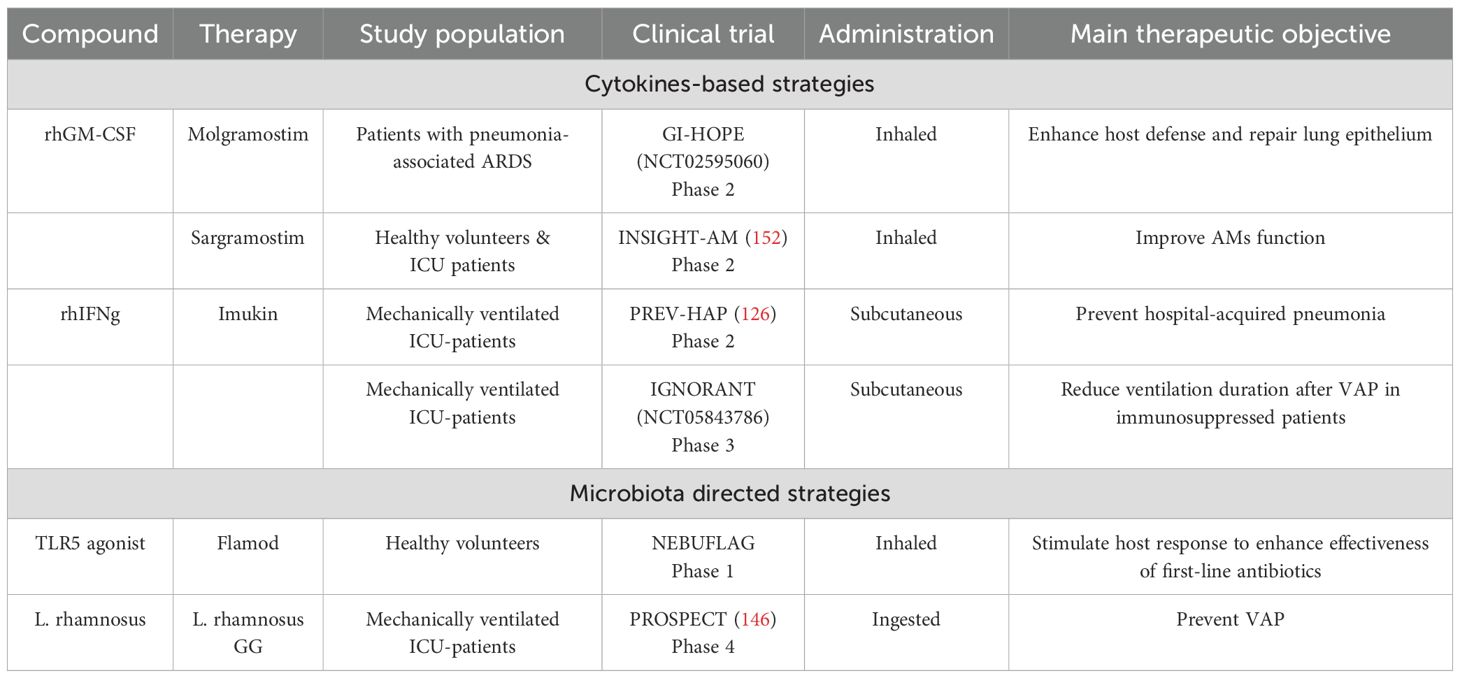
Table 2. Immunomodulatory strategies to improve management of pulmonary infection in ICU.
5.1 Immunotherapy to modulate locally sepsis-induced alterations
Immunomodulatory therapy aims to reverse defective immune function and boost the immune system. In the context of sepsis, several cytokines have been tested as immunomodulatory therapies, including IL-7 (125), GM-CSF and interferon-γ (IFN-γ) (126). In particular, in the context of pulmonary infection, a correlation has been found between lack of GM-CSF and IFN-γ and AMs paralysis (127). After in vivo LPS pulmonary instillation and depletion (or not) of alveolar GM-CSF and IFN-γ, AMs from mice were harvested and challenged with LPS in vitro. Surprisingly, AMs did not show endotoxin tolerance except in mice lacking alveolar GM-CSF or IFN-γ making these two cytokines particularly interesting as potent immunotherapies.
IFN-γ has pleiotropic effects on the innate and adaptive immune system. In sepsis clinical trial, recombinant IFN-γ was proved to restore HLA-DR expression on monocytes. Since reduced expression is considered a hallmark of sepsis-induced immunosuppression, this makes IFN-γ a good candidate for reversing immunosuppression in critically ill patients. However, the use of peripheral administration of recombinant IFN-γ did not reduce the incidence of hospital-acquired pneumonia among critically ill patients (126). With the growing importance of considering organ-specific immune responses, new studies are evaluating the effect of local administration. For example, prophylactic treatment with intranasal administration of recombinant IFN-γ provides protection against SARS-CoV-2 infection in a mouse model (128).
Unlike IFN-γ, which is known for its role in enhancing the immune response, GM-CSF is involved in maintaining alveolar homeostasis, recruiting myeloid cells, and differentiating newly recruited macrophages into resident AMs (129). As a result, GM-CSF is emerging as a promising therapeutic agent for modulating pulmonary immunity. Clinical trials in sepsis patients have shown that peripheral administration of recombinant GM-CSF can reverse the hyporesponsiveness of circulating monocytes (130). Local administration of GM-CSF has been shown to improve survival rates in mice with secondary pneumonia (131). In support of these findings in animal models, preliminary data from a limited number of patients have provided initial insights into the effects of this therapy on pneumonia-associated ARDS, suggesting enhanced activation of AMs (132). To further elucidate the potential beneficial effects of inhaled GM-CSF on AMs, several ongoing studies are investigating its effects in critically ill patients (ISRCTN78203402, NCT02595060).
Advances in understanding sepsis-induced changes have identified novel pathways as potential therapeutic targets. IL-33, an alarmin produced by injured tissues, has been shown to contribute to persistent immunosuppression during sepsis (133). Therefore, targeting the IL-33/ST2 axis may help to restore an effective immune response. Immunotherapy strategies aimed at restoring immune function may also focus on blocking immune checkpoints (134) such as SIRPα. This protein, which inhibits phagocytosis through its interaction with CD47, is implicated in the disruption of phagocytic function in AMs. Thus, targeting SIRPα may prevent AMs paralysis (98).
5.2 Targeting microbiota to indirectly restore immune balance
With an increasing number of studies aiming to decipher the role of the microbiota in the onset of pulmonary infections, strategies based on microbiota modulation to indirectly enhance pulmonary immunity are emerging as promising therapeutic approaches (110, 135–137). As reviewed, infected lung display reduced abundance of genera belonging to the healthy lung core microbiome. In addition, murine models have demonstrated that host–microbiota crosstalk contributes to the maintenance of mucosal immunity. Therefore, restoring the initial diversity of the airway microbiome could help reestablish an effective host response. In a murine model, oral aspiration of three human oral anaerobic bacteria reduced susceptibility to subsequent Streptococcus pneumoniae pulmonary infection (138). Interestingly, two of these anaerobic bacteria have been identified as reduced in the microbiome of patients with hospital-acquired pneumonia (139). Rather than using whole bacteria, an alternative strategy involves identifying specific microbial components or conserved molecular patterns that modulate host immunity (140, 141). This approach relies on immunomodulation by targeting the crosstalk between commensal bacteria and immune cells, which can trigger protective effects against infection. For example, stimulation of Toll-like receptor (TLR) 5 (142) or TLR2 (121) has been associated with protection against S. pneumoniae infection. Similarly prophylactic treatment with TLR4 agonist was demonstrated to enhanced immune response to K.pneumoniae in mouse model (143). Of note, the use of TLR agonists in combination with antibiotic has been proposed to improve antibiotic efficacy (144). In mice, therapeutic synergy between antibiotics and TLR5 stimulation with FLAMOD has already been observed in both antibiotic-sensitive and -resistant pneumonia. A clinical study is currently underway to evaluate this drug in humans.
Modulation of the microbiota can also target symbiotic relationships between different microbial communities. One such strategy, inspired by gut microbiota modulation, is the modulation through microbial consortium (145). This approach focuses on identifying microorganisms that naturally cooperate symbiotically and can collectively influence immune processes. Microbial cooperation can also occur between different compartments. Since modifications of gut microbiota have been identified during pulmonary infections, recent studies have explored how interactions between the microbiota of both compartments may modulate each other. Targeting the gut–lung axis can be achieved through dietary interventions or ingestion of targeted substrates (prebiotics), as well as through the administration of one or two specific bacterial strains (probiotics). A clinical trial has already investigated the impact of a probiotic treatment composed of L.rhamnosus on the onset of ventilator-associated pneumonia, but failed to demonstrate a beneficial effect (146).
Despite the diversity of these approaches, including direct or indirect modulation of pulmonary environment, only a few clinical trials have been conducted. This is partly due to the difficulty to translate from experimental models to clinical.
5.3 Limitations of current evidence and research gaps
Indeed, immunomodulation in critically ill patients continues to face significant challenges in translation from bench to bedside. This is partly due to the lack of human-based data. Although pulmonary cells are relatively accessible, the invasive procedures required to obtain them, limit ex vivo research on human alveolar cells. While animal models have advanced our understanding of immunological alterations, particularly in organ-specific contexts, their translational relevance remains limited (147). These models are typically designed for homogeneity and reproducibility, which fails to capture the clinical heterogeneity of ICU patients, including variations in medical history and environmental exposures. Even with humanized animal models, inherent anatomical (148) and immunological (149) differences between species cannot be eliminated and must be carefully considered when extrapolating findings to humans. In the context of bacterial pneumonia, numerous murine models have been used to decipher the immune response. Still while neutrophils are central to the pulmonary defense, their phenotype, circulating levels, and immune-related protein expression differ markedly between mice and humans (150).
Beyond the limitations of animal models, research gaps in immunomodulatory strategies also stem from the difficulty of implementing experimental protocols in clinical settings. These challenges include technical constraints imposed by routine clinical practice and the high heterogeneity of patient profiles. Microbiota-targeted therapies exemplify these complexities (151). As previously discussed, microbiome analysis is technically complex and difficult to adapt to clinical workflow. For example, the core pulmonary microbiota includes anaerobic bacteria, which require oxygen-free conditions for sampling and storage. Moreover, the host–microbiome environment is highly dynamic and influenced by numerous confounding factors, complicating the identification of causal relationships or mechanisms underlying dysbiosis, an essential step in defining therapeutic targets. Longitudinal studies would help evaluate temporal dynamics but require repeated sampling, which is logistically challenging in critically ill patients.
Therapeutic strategies involving microorganisms face an additional major challenge in the context of personalized medicine: the high heterogeneity of patient profiles. Given our limited understanding of host–microbe interactions, microbiota modulation, particularly through the administration of whole microorganisms, while potentially beneficial, carries the risk of unintended and possibly harmful outcomes. This highlights both the need for more human data and for experimental approaches that are better aligned with clinical constraints.
6 Conclusion
Sepsis is a complex dysregulated host response to infection leading to organ dysfunction that may be heterogeneous and asynchronous at the organ level. This heterogeneity of the immune response may explain differences in susceptibility to secondary infections and mortality. In pneumonia-related sepsis, compartmentalisation of the immune response in the lung is associated with quantitative and phenotypic alterations of key partners such as AMs, polymorphonuclear cells and microbiota. Immune modulation with local delivery of cytokines such as IFN-γ or growth factors such as GM-CSF could help restore normal alveolar antibacterial defenses. In addition, modulation of the lung microbiota after an infection appears to be a promising way to improve the regional lung immune response to further infection.
Author contributions
EV: Writing – original draft, Writing – review & editing. EC: Writing – original draft, Writing – review & editing. FS: Writing – original draft, Writing – review & editing. CM: Writing – review & editing. PP: Writing – review & editing. FA: Writing – review & editing. ACL: Writing – review & editing. JFL: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
FA, FS, EC, JFL and EV are employees of bioMérieux, an in vitro diagnostic company.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA. (2016) 315:801. doi: 10.1001/jama.2016.0287
2. Rudd KE, Johnson SC, Agesa KM, Shackelford KA, Tsoi D, Kievlan DR, et al. Global, regional, and national sepsis incidence and mortality, 1990–2017: analysis for the Global Burden of Disease Study. Lancet. (2020) 395:200–11. doi: 10.1016/S0140-6736(19)32989-7
3. Shankar-Hari M and Rubenfeld GD. Understanding long-term outcomes following sepsis: implications and challenges. Curr Infect Dis Rep. (2016) 18:37. doi: 10.1007/s11908-016-0544-7
4. World Health Organization. (2020). Available online at: https://iris.who.int/bitstream/handle/10665/334216/9789240010789-eng.pdf?sequence=1 (Accessed March 2025).
5. Hotchkiss RS and Karl IE. The pathophysiology and treatment of sepsis. N Engl J Med. (2003) 348:138–50. doi: 10.1056/NEJMra021333
6. Cao C, Yu M, and Chai Y. Pathological alteration and therapeutic implications of sepsis-induced immune cell apoptosis. Cell Death Dis. (2019) 10:782. doi: 10.1038/s41419-019-2015-1
7. Tamayo E, Fernández A, Almansa R, Carrasco E, Heredia M, Lajo C, et al. Pro- and anti-inflammatory responses are regulated simultaneously from the first moments of septic shock. Eur Cytokine Netw. (2011) 22:82–7. doi: 10.1684/ecn.2011.0281
8. Hotchkiss RS, Monneret G, and Payen D. Sepsis-induced immunosuppression: from cellular dysfunctions to immunotherapy. Nat Rev Immunol. (2013) 13:862–74. doi: 10.1038/nri3552
9. Otto GP, Sossdorf M, Claus RA, Rödel J, Menge K, Reinhart K, et al. The late phase of sepsis is characterized by an increased microbiological burden and death rate. Crit Care. (2011) 15:R183. doi: 10.1186/cc10332
10. Chen Y, Hu Y, Zhang J, Shen Y, Huang J, Yin J, et al. Clinical characteristics, risk factors, immune status and prognosis of secondary infection of sepsis: a retrospective observational study. BMC Anesthesiol. (2019) 19:185. doi: 10.1186/s12871-019-0849-9
11. Van Vught LA, Klein Klouwenberg PMC, Spitoni C, Scicluna BP, Wiewel MA, Horn J, et al. Incidence, risk factors, and attributable mortality of secondary infections in the intensive care unit after admission for sepsis. JAMA. (2016) 315:1469. doi: 10.1001/jama.2016.2691
12. Tremblay JA, Peron F, Kreitmann L, Textoris J, Brengel-Pesce K, Lukaszewicz AC, et al. A stratification strategy to predict secondary infection in critical illness-induced immune dysfunction: the REALIST score. Ann Intensive Care. (2022) 12:76. doi: 10.1186/s13613-022-01051-3
13. Peronnet E, Terraz G, Cerrato E, Imhoff K, Blein S, Brengel-Pesce K, et al. Use of Immune Profiling Panel to assess the immune response of septic patients for prediction of worsening as a composite endpoint. Sci Rep. (2024) 14:11305. doi: 10.1038/s41598-024-62202-z
14. Bodinier M, Monneret G, Casimir M, Fleurie A, Conti F, Venet F, et al. Identification of a sub-group of critically ill patients with high risk of intensive care unit-acquired infections and poor clinical course using a transcriptomic score. Crit Care. (2023) 27:158. doi: 10.1186/s13054-023-04436-3
15. Wang W and Liu CF. Sepsis heterogeneity. World J Pediatr. (2023) 19:919–27. doi: 10.1007/s12519-023-00689-8
16. Peters-Sengers H, Butler JM, Uhel F, Schultz MJ, Bonten MJ, Cremer OL, et al. Source-specific host response and outcomes in critically ill patients with sepsis: a prospective cohort study. Intensive Care Med. (2022) 48:92–102. doi: 10.1007/s00134-021-06574-0
17. Sun P, Cui M, Jing J, Kong F, Wang S, Tang L, et al. Deciphering the molecular and cellular atlas of immune cells in septic patients with different bacterial infections. J Transl Med. (2023) 21:777. doi: 10.1186/s12967-023-04631-4
18. Jeganathan N, Yau S, Ahuja N, Otu D, Stein B, Fogg L, et al. The characteristics and impact of source of infection on sepsis-related ICU outcomes. J Crit Care. (2017) 41:170–6. doi: 10.1016/j.jcrc.2017.05.019
19. Volakli E, Spies C, Michalopoulos A, Groeneveld AJ, Sakr Y, and Vincent JL. Infections of respiratory or abdominal origin in ICU patients: what are the differences? Crit Care. (2010) 14(2). doi: 10.1186/cc8909
20. Leligdowicz A, Dodek PM, Norena M, Wong H, Kumar A, and Kumar A. Association between source of infection and hospital mortality in patients who have septic shock. Am J Respir Crit Care Med. (2014) 189:1204–13. doi: 10.1164/rccm.201310-1875OC
21. Chou EH, Mann S, Hsu TC, Hsu WT, Liu CCY, Bhakta T, et al. Incidence, trends, and outcomes of infection sites among hospitalizations of sepsis: A nationwide study. PloS One. (2020) 15:e0227752. doi: 10.1371/journal.pone.0227752
22. Stortz JA, Cox MC, Hawkins RB, Ghita GL, Brumback BA, Mohr AM, et al. Phenotypic heterogeneity by site of infection in surgical sepsis: a prospective longitudinal study. Crit Care. (2020) 24:203. doi: 10.1186/s13054-020-02917-3
23. Llitjos JF, Gassama A, Charpentier J, Lambert J, De Roquetaillade C, Cariou A, et al. Pulmonary infections prime the development of subsequent ICU-acquired pneumonia in septic shock. Ann Intensive Care. (2019) 9:39. doi: 10.1186/s13613-019-0515-x
24. Philippart F, Bouroche G, Timsit JF, Garrouste-Orgeas M, Azoulay E, Darmon M, et al. Decreased risk of ventilator-associated pneumonia in sepsis due to intra-abdominal infection. PloS One. (2015) 10:e0137262. doi: 10.1371/journal.pone.0137262
25. Rumienczyk I, Kulecka M, Ostrowski J, Mar D, Bomsztyk K, Standage SW, et al. Multi-organ transcriptome dynamics in a mouse model of cecal ligation and puncture-induced polymicrobial sepsis. JIR. (2021) 14:2377–88. doi: 10.2147/JIR.S307305
26. Takahama M, Patil A, Richey G, Cipurko D, Johnson K, Carbonetto P, et al. A pairwise cytokine code explains the organism-wide response to sepsis. Nat Immunol. (2024) 25(2):226–39. doi: 10.1038/s41590-023-01722-8
27. Chinnaiyan AM, Huber-Lang M, Kumar-Sinha C, Barrette TR, Shankar-Sinha S, Sarma VJ, et al. Molecular signatures of sepsis. Am J Pathol. (2001) 159:1199–209. doi: 10.1016/S0002-9440(10)62505-9
28. Hiruma T, Tsuyuzaki H, Uchida K, Trapnell BC, Yamamura Y, Kusakabe Y, et al. IFN-β Improves sepsis-related alveolar macrophage dysfunction and postseptic acute respiratory distress syndrome–related mortality. Am J Respir Cell Mol Biol. (2018) 59:45–55. doi: 10.1165/rcmb.2017-0261OC
29. Aslan A, Van Meurs M, Moser J, Popa ER, Jongman RM, Zwiers PJ, et al. Organ-specific differences in endothelial permeability-regulating molecular responses in mouse and human sepsis. Shock. (2017) 48:69–77. doi: 10.1097/SHK.0000000000000841
30. Hoyer FF, Naxerova K, Schloss MJ, Hulsmans M, Nair AV, Dutta P, et al. Tissue-specific macrophage responses to remote injury impact the outcome of subsequent local immune challenge. Immunity. (2019) 51:899–914.e7. doi: 10.1016/j.immuni.2019.10.010
31. Smith PD, Suffredini AF, Allen JB, Wahl LM, Parrillo JE, and Wahl SM. Endotoxin administration to humans primes alveolar macrophages for increased production of inflammatory mediators. J Clin Immunol. (1994) 14:141–8. doi: 10.1007/BF01541347
32. Rasid O, Ciulean IS, Fitting C, Doyen N, and Cavaillon JM. Local microenvironment controls the compartmentalization of NK cell responses during systemic inflammation in mice. J Immunol. (2016) 197:2444–54. doi: 10.4049/jimmunol.1601040
33. Plovsing RR, Berg RMG, Evans KA, Konge L, Iversen M, Garred P, et al. Transcompartmental inflammatory responses in humans: IV versus endobronchial administration of endotoxin*. Crit Care Med. (2014) 42:1658–65. doi: 10.1097/CCM.0000000000000320
34. Skirecki T, Mikaszewska-Sokolewicz M, Hoser G, and Zielińska-Borkowska U. The early expression of HLA-DR and CD64 myeloid markers is specifically compartmentalized in the blood and lungs of patients with septic shock. Mediators Inflamm. (2016) 2016:1–8. doi: 10.1155/2016/3074902
35. Llitjos JF, Auffray C, Péju E, Hamou ZA, Rousseau C, Durand A, et al. Pulmonary and nonpulmonary sepsis differentially modulate lung immunity toward secondary bacterial pneumonia: A critical role for alveolar macrophages. Am J Respir Cell Mol Biol. (2023) 68:689–701. doi: 10.1165/rcmb.2022-0281OC
36. Fitting C, Dhawan S, and Cavaillon J. Compartmentalization of tolerance to endotoxin. J Infect Dis. (2004) 189:1295–303. doi: 10.1086/382657
37. Angus DC and van der Poll T. Severe sepsis and septic shock. N Engl J Med. (2013) 369:840–51. doi: 10.1056/NEJMra1208623
38. Vincent JL. International study of the prevalence and outcomes of infection in intensive care units. JAMA. (2009) 302:2323. doi: 10.1001/jama.2009.1754
39. European Centre for Disease Prevention and Control. (2024). Available online at: https://www.ecdc.europa.eu/sites/default/files/documents/healthcare-associated-infections-intensive-care-units-annual-epidemiological-report-2020.pdf (Accessed March 2025).
40. Ren E, Xiao H, Li J, Yu H, Liu B, Wang G, et al. Clinical characteristics and predictors of mortality differ between pulmonary and abdominal sepsis. Shock. (2023) 60:42–50. doi: 10.1097/SHK.0000000000002151
41. Walter J, Haller S, Quinten C, Kärki T, Zacher B, Eckmanns T, et al. Healthcare-associated pneumonia in acute care hospitals in European Union/European Economic Area countries: an analysis of data from a point prevalence survey, 2011 to 2012. Eurosurveillance. (2018) 23:32. doi: 10.2807/1560-7917.ES.2018.23.32.1700843
42. Póvoa P, Coelho L, Dal-Pizzol F, Ferrer R, Huttner A, Conway Morris A, et al. How to use biomarkers of infection or sepsis at the bedside: guide to clinicians. Intensive Care Med. (2023) 49:142–53. doi: 10.1007/s00134-022-06956-y
43. Kronberger JF, Köhler TC, Lang CN, Jäckel M, Bemtgen X, Wengenmayer T, et al. Bronchoalveolar lavage and blood markers of infection in critically ill patients—A single center registry study. JCM. (2021) 10:486. doi: 10.3390/jcm10030486
44. Bonten MJM, Froon AHM, Gaillard CA, Greve JWM, de Leeuw PW, Drent M, et al. The systemic inflammatory response in the development of ventilator-associated pneumonia. Am J Respir Crit Care Med. (1997) 156:1105–13. doi: 10.1164/ajrccm.156.4.9610002
45. Craig A, Mai J, Cai S, and Jeyaseelan S. Neutrophil recruitment to the lungs during bacterial pneumonia. Infect Immun. (2009) 77:568–75. doi: 10.1128/IAI.00832-08
46. Conway Morris A, Kefala K, Wilkinson TS, Moncayo-Nieto OL, Dhaliwal K, Farrell L, et al. Diagnostic importance of pulmonary interleukin-1 and interleukin-8 in ventilator-associated pneumonia. Thorax. (2010) 65:201–7. doi: 10.1136/thx.2009.122291
47. Jouan Y, Baranek T, Si-Tahar M, Paget C, and Guillon A. Lung compartmentalization of inflammatory biomarkers in COVID-19-related ARDS. Crit Care. (2021) 25:120. doi: 10.1186/s13054-021-03513-9
48. Bendib I, Beldi-Ferchiou A, Schlemmer F, Surenaud M, Maitre B, Plonquet A, et al. Alveolar compartmentalization of inflammatory and immune cell biomarkers in pneumonia-related ARDS. Crit Care. (2021) 25:23. doi: 10.1186/s13054-020-03427-y
49. Dehoux MS, Boutten A, Ostinelli J, Seta N, Dombret MC, Crestani B, et al. Compartmentalized cytokine production within the human lung in unilateral pneumonia. Am J Respir Crit Care Med. (1994) 150:710–6. doi: 10.1164/ajrccm.150.3.8087341
50. Droemann D, Aries SP, Hansen F, Moellers M, Braun J, Katus HA, et al. Decreased apoptosis and increased activation of alveolar neutrophils in bacterial pneumonia. Chest. (2000) 117:1679–84. doi: 10.1378/chest.117.6.1679
51. Guilliams M, De Kleer I, Henri S, Post S, Vanhoutte L, De Prijck S, et al. Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. J Exp Med. (2013) 210:1977–92. doi: 10.1084/jem.20131199
52. Hussell T and Bell TJ. Alveolar macrophages: plasticity in a tissue-specific context. Nat Rev Immunol. (2014) 14:81–93. doi: 10.1038/nri3600
53. Bissonnette EY, Lauzon-Joset JF, Debley JS, and Ziegler SF. Cross-talk between alveolar macrophages and lung epithelial cells is essential to maintain lung homeostasis. Front Immunol. (2020) 11:583042. doi: 10.3389/fimmu.2020.583042
54. Bain CC and MacDonald AS. The impact of the lung environment on macrophage development, activation and function: diversity in the face of adversity. Mucosal Immunol. (2022) 15:223–34. doi: 10.1038/s41385-021-00480-w
55. Bonfield TL, Konstan MW, Burfeind P, Panuska JR, Hilliard JB, and Berger M. Normal bronchial epithelial cells constitutively produce the anti-inflammatory cytokine interleukin-10, which is downregulated in cystic fibrosis. Am J Respir Cell Mol Biol. (1995) 13:257–61. doi: 10.1165/ajrcmb.13.3.7544594
56. Li R, Li J, and Zhou X. Lung microbiome: new insights into the pathogenesis of respiratory diseases. Sig Transduct Target Ther. (2024) 9:19. doi: 10.1038/s41392-023-01722-y
57. Natalini JG, Singh S, and Segal LN. The dynamic lung microbiome in health and disease. Nat Rev Microbiol avr. (2023) 21:222–35. doi: 10.1038/s41579-022-00821-x
58. Neupane AS, Willson M, Chojnacki AK, Vargas E Silva Castanheira F, Morehouse C, Carestia A, et al. Patrolling alveolar macrophages conceal bacteria from the immune system to maintain homeostasis. Cell. (2020) 183:110–125.e11. doi: 10.1016/j.cell.2020.08.020
59. Korkmaz FT and Traber KE. Innate immune responses in pneumonia. Pneumonia. (2023) 15:4. doi: 10.1186/s41479-023-00106-8
60. Van Der Geest R, Fan H, Peñaloza HF, Bain WG, Xiong Z, Kohli N, et al. Phagocytosis is a primary determinant of pulmonary clearance of clinical Klebsiella pneumoniae isolates. Front Cell Infect Microbiol. (2023) 13:1150658. doi: 10.3389/fcimb.2023.1150658
61. Ziltener P, Reinheckel T, and Oxenius A. Neutrophil and Alveolar Macrophage-Mediated Innate Immune Control of Legionella pneumophila Lung Infection via TNF and ROS. PloS Pathog. (2016) 12:e1005591. doi: 10.1371/journal.ppat.1005591
62. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science. (2004) 303:1532–5. doi: 10.1126/science.1092385
63. Liu X, Boyer MA, Holmgren AM, and Shin S. Legionella-infected macrophages engage the alveolar epithelium to metabolically reprogram myeloid cells and promote antibacterial inflammation. Cell Host Microbe. (2020) 28:683–698.e6. doi: 10.1016/j.chom.2020.07.019
64. Thorley A, Ford P, Giembycz M, Goldstraw P, Young A, and Tetley T. Differential regulation of cytokine release and leukocyte migration by lipopolysaccharide-stimulated primary human lung alveolar type II epithelial cells and macrophages. J Immunol (Baltimore Md: 1950). (2007) 178:463–73. doi: 10.4049/jimmunol.178.1.463
65. Ramirez-Moral I, Blok DC, Bernink JH, Garcia-Laorden MI, Florquin S, Boon L, et al. Interleukin-33 improves local immunity during Gram-negative pneumonia by a combined effect on neutrophils and inflammatory monocytes. J Pathol. (2021) 253(4):374–83. doi: 10.1002/path.5601
66. Moon HG, Cao Y, Yang J, Lee JH, Choi HS, and Jin Y. Lung epithelial cell-derived extracellular vesicles activate macrophage-mediated inflammatory responses via ROCK1 pathway. Cell Death Dis. (2015) 6:e2016–6. doi: 10.1038/cddis.2015.282
67. Kawasaki T, Ikegawa M, and Kawai T. Antigen presentation in the lung. Front Immunol. (2022) 13:860915. doi: 10.3389/fimmu.2022.860915
68. Gao CA, Morales-Nebreda L, and Pickens CI. Gearing up for battle: Harnessing adaptive T cell immunity against gram-negative pneumonia. Front Cell Infect Microbiol. (2022) 12:934671/full. doi: 10.3389/fcimb.2022.934671/full
69. Ye P, Garvey PB, Zhang P, Nelson S, Bagby G, Summer WR, et al. Interleukin-17 and Lung Host Defense against Klebsiella pneumoniae Infection. Am J Respir Cell Mol Biol. (2001) 25:335–40. doi: 10.1165/ajrcmb.25.3.4424
70. Dąbrowska D, Jabłońska E, Iwaniuk A, and Garley M. Many ways–one destination: different types of neutrophils death. Int Rev Immunol. (2019) 38(1):18–32. doi: 10.1080/08830185.2018.1540616
71. Shotland AM, Fontenot AP, and McKee AS. Pulmonary macrophage cell death in lung health and disease. Am J Respir Cell Mol Biol. (2021) 64:547–56. doi: 10.1165/rcmb.2020-0420TR
72. Steinwede K, Henken S, Bohling J, Maus R, Ueberberg B, Brumshagen C, et al. TNF-related apoptosis-inducing ligand (TRAIL) exerts therapeutic efficacy for the treatment of pneumococcal pneumonia in mice. J Exp Med. (2012) 209:1937–52. doi: 10.1084/jem.20120983
73. Lee HH, Aslanyan L, Vidyasagar A, Brennan MB, Tauber MS, Carrillo-Sepulveda MA, et al. Depletion of alveolar macrophages increases pulmonary neutrophil infiltration, tissue damage, and sepsis in a murine model of acinetobacter baumannii pneumonia. Infect Immun. (2020) 88:e00128–20. doi: 10.1128/IAI.00128-20
74. Boada-Romero E, Martinez J, Heckmann BL, and Green DR. The clearance of dead cells by efferocytosis. Nat Rev Mol Cell Biol. (2020) 21:398–414. doi: 10.1038/s41580-020-0232-1
75. Huynh MLN, Fadok VA, and Henson PM. Phosphatidylserine-dependent ingestion of apoptotic cells promotes TGF-β1 secretion and the resolution of inflammation. J Clin Invest. (2002) 109:41–50. doi: 10.1172/JCI0211638
76. Westphalen K, Gusarova GA, Islam MN, Subramanian M, Cohen TS, Prince AS, et al. Sessile alveolar macrophages communicate with alveolar epithelium to modulate immunity. Nature. (2014) 506:503–6. doi: 10.1038/nature12902
77. Peñaloza HF, Nieto PA, Muñoz-Durango N, Salazar-Echegarai FJ, Torres J, Parga MJ, et al. Interleukin-10 plays a key role in the modulation of neutrophils recruitment and lung inflammation during infection by Streptococcus pneumoniae. Immunology. (2015) 146(1):100–12. doi: 10.1111/imm.12486
78. Hung LY, Sen D, Oniskey TK, Katzen J, Cohen NA, Vaughan AE, et al. Macrophages promote epithelial proliferation following infectious and non-infectious lung injury through a Trefoil factor 2-dependent mechanism. Mucosal Immunol. (2019) 12:64–76. doi: 10.1038/s41385-018-0096-2
79. Jansing NL, McClendon J, Henson PM, Tuder RM, Hyde DM, and Zemans RL. Unbiased Quantitation of Alveolar Type II to Alveolar Type I Cell Transdifferentiation during Repair after Lung Injury in Mice. Am J Respir Cell Mol Biol. (2017) 57:519–26. doi: 10.1165/rcmb.2017-0037MA
80. Feehan KT, Bridgewater HE, Stenkiewicz-Witeska J, De Maeyer RPH, Ferguson J, Mack M, et al. Post-resolution macrophages shape long-term tissue immunity and integrity in a mouse model of pneumococcal pneumonia. Nat Commun. (2024) 15:4326. doi: 10.1038/s41467-024-48138-y
81. Zhang A, Lacy-Hulbert A, Anderton S, Haslett C, and Savill J. Apoptotic cell–directed resolution of lung inflammation requires myeloid αv integrin–mediated induction of regulatory T lymphocytes. Am J Pathol. (2020) 190:1224–35. doi: 10.1016/j.ajpath.2020.02.010
82. Proto JD, Doran AC, Gusarova G, Yurdagul A, Sozen E, Subramanian M, et al. Regulatory T cells promote macrophage efferocytosis during inflammation resolution. Immunity. (2018) 49:666–677.e6. doi: 10.1016/j.immuni.2018.07.015
83. Crome SQ, Clive B, Wang AY, Kang CY, Chow V, Yu J, et al. Inflammatory effects of ex vivo human th17 cells are suppressed by regulatory T cells. J Immunol. (2010) 185:3199–208. doi: 10.4049/jimmunol.1000557
84. Xu R, Jacques LC, Khandaker S, Beentjes D, Leon-Rios M, Wei X, et al. TNFR2+ regulatory T cells protect against bacteremic pneumococcal pneumonia by suppressing IL-17A-producing γδ T cells in the lung. Cell Rep. (2023) 42:112054. doi: 10.1016/j.celrep.2023.112054
85. Gurczynski SJ and Moore BB. IL-17 in the lung: the good, the bad, and the ugly. Am J Physiol Lung Cell Mol Physiol. (2018) 314:L6–16. doi: 10.1152/ajplung.00344.2017
86. Kirby A, Newton D, Carding S, and Kaye P. Pulmonary dendritic cells and alveolar macrophages are regulated by γδ T cells during the resolution of S. pneumoniae -induced inflammation. J Pathol. (2007) 212:29–37. doi: 10.1002/path.2149
87. Murray MP, Crosby CM, Marcovecchio P, Hartmann N, Chandra S, Zhao M, et al. Stimulation of a subset of natural killer T cells by CD103+ DC is required for GM-CSF and protection from pneumococcal infection. Cell Rep. (2022) 38:110209. doi: 10.1016/j.celrep.2021.110209
88. Gong H, Chen Y, Chen M, Li J, Zhang H, Yan S, et al. Advanced development and mechanism of sepsis-related acute respiratory distress syndrome. Front Med. (2022) 9:1043859. doi: 10.3389/fmed.2022.1043859
89. Torres LK, Pickkers P, and van der Poll T. Sepsis-induced immunosuppression. Annu Rev Physiol. (2022) 84:157–81. doi: 10.1146/annurev-physiol-061121-040214
90. Preston JA, Bewley MA, Marriott HM, McGarry Houghton A, Mohasin M, Jubrail J, et al. Alveolar macrophage apoptosis–associated bacterial killing helps prevent murine pneumonia. Am J Respir Crit Care Med. (2019) 200:84–97. doi: 10.1164/rccm.201804-0646OC
91. Arafa EI, Shenoy AT, Barker KA, Etesami NS, Martin IMC, Lyon De Ana C, et al. Recruitment and training of alveolar macrophages after pneumococcal pneumonia. JCI Insight. (2022) 7:e150239. doi: 10.1172/jci.insight.150239
92. Maus UA, Janzen S, Wall G, Srivastava M, Blackwell TS, Christman JW, et al. Resident alveolar macrophages are replaced by recruited monocytes in response to endotoxin-induced lung inflammation. Am J Respir Cell Mol Biol. (2006) 35:227–35. doi: 10.1165/rcmb.2005-0241OC
93. Mould KJ, Barthel L, Mohning MP, Thomas SM, McCubbrey AL, Danhorn T, et al. Cell Origin Dictates Programming of Resident versus Recruited Macrophages during Acute Lung Injury. Am J Respir Cell Mol Biol. (2017) 57:294–306. doi: 10.1165/rcmb.2017-0061OC
94. Han W, Tanjore H, Liu Y, Hunt RP, Gutor SS, Serezani APM, et al. Identification and characterization of alveolar and recruited lung macrophages during acute lung inflammation. J Immunol. (2023) 210:1827–36. doi: 10.4049/jimmunol.2200694
95. Guilliams M and Svedberg FR. Does tissue imprinting restrict macrophage plasticity? Nat Immunol. (2021) 22(2):118–27. doi: 10.1038/s41590-020-00849-2
96. Mould KJ, Moore CM, McManus SA, McCubbrey AL, McClendon JD, Griesmer CL, et al. Airspace macrophages and monocytes exist in transcriptionally distinct subsets in healthy adults. Am J Respir Crit Care Med. (2021) 203:946–56. doi: 10.1164/rccm.202005-1989OC
97. Xiao K, Cao Y, Han Z, Zhang Y, Luu LDW, Chen L, et al. A pan-immune panorama of bacterial pneumonia revealed by a large-scale single-cell transcriptome atlas. Sig Transduct Target Ther. (2025) 10:5. doi: 10.1038/s41392-024-02093-8
98. Roquilly A, Jacqueline C, Davieau M, Mollé A, Sadek A, Fourgeux C, et al. Alveolar macrophages are epigenetically altered after inflammation, leading to long-term lung immunoparalysis. Nat Immunol. (2020) 21:636–48. doi: 10.1038/s41590-020-0673-x
99. Roquilly A, McWilliam HEG, Jacqueline C, Tian Z, Cinotti R, Rimbert M, et al. Local modulation of antigen-presenting cell development after resolution of pneumonia induces long-term susceptibility to secondary infections. Immunity. (2017) 47:135–147.e5. doi: 10.1016/j.immuni.2017.06.021
100. Xu J, He W, Xiao N, and Xie L. Repetitive Acinetobacter baumannii pneumonia induces infection tolerance in mice. Microb Pathogen. (2024) 197:107009. doi: 10.1016/j.micpath.2024.107009
101. Öz HH, Zhou B, Voss P, Carevic M, Schroth C, Frey N, et al. Pseudomonas aeruginosa airway infection recruits and modulates neutrophilic myeloid-derived suppressor cells. Front Cell Infect Microbiol. (2016) 6:167/full. doi: 10.3389/fcimb.2016.00167/full
102. Peñaloza HF, Noguera LP, Ahn D, Vallejos OP, Castellanos RM, Vazquez Y, et al. Interleukin-10 produced by myeloid-derived suppressor cells provides protection to carbapenem-resistant Klebsiella pneumoniae sequence type 258 by enhancing its clearance in the airways. Infect Immun. (2019) 87(5):e00665–18. doi: 10.1128/IAI.00665-18
103. Poe SL, Arora M, Oriss TB, Yarlagadda M, Isse K, Khare A, et al. STAT1-regulated lung MDSC-like cells produce IL-10 and efferocytose apoptotic neutrophils with relevance in resolution of bacterial pneumonia. Mucosal Immunol. (2013) 6:189–99. doi: 10.1038/mi.2012.62
104. Barbosa Bomfim CC, Pinheiro Amaral E, Santiago-Carvalho I, Almeida Santos G, MaChado Salles É, Hastreiter AA, et al. Harmful effects of granulocytic myeloid-derived suppressor cells on tuberculosis caused by hypervirulent mycobacteria. J Infect Dis. (2021) 223:494–507. doi: 10.1093/infdis/jiaa708
105. Lei GS, Zhang C, and Lee CH. Myeloid-Derived Suppressor Cells Impair Alveolar Macrophages through PD-1 Receptor Ligation during Pneumocystis Pneumonia. Infect Immun. (2015) 83:572–82. doi: 10.1128/IAI.02686-14
106. Guillon A, Arafa EI, Barker KA, Belkina AC, Martin I, Shenoy AT, et al. Pneumonia recovery reprograms the alveolar macrophage pool. JCI Insight. (2020) 5:e133042. doi: 10.1172/jci.insight.133042
107. Charlson ES, Bittinger K, Haas AR, Fitzgerald AS, Frank I, Yadav A, et al. Topographical continuity of bacterial populations in the healthy human respiratory tract. Am J Respir Crit Care Med. (2011) 184:957–63. doi: 10.1164/rccm.201104-0655OC
108. Fenn D, Abdel-Aziz MI, Van Oort PMP, Brinkman P, Ahmed WM, Felton T, et al. Composition and diversity analysis of the lung microbiome in patients with suspected ventilator-associated pneumonia. Crit Care. (2022) 26:203. doi: 10.1186/s13054-022-04068-z
109. Spottiswoode N, Tsitsiklis A, Chu VT, Phan HV, DeVoe C, Love C, et al. Microbial dynamics and pulmonary immune responses in COVID-19 secondary bacterial pneumonia. Nat Commun. (2024) 15:9339. doi: 10.1038/s41467-024-53566-x
110. Roquilly A, Torres A, Villadangos JA, Netea MG, Dickson R, Becher B, et al. Pathophysiological role of respiratory dysbiosis in hospital-acquired pneumonia. Lancet Respir Med. (2019) 7:710–20. doi: 10.1016/S2213-2600(19)30140-7
111. Kitsios GD, Sayed K, Fitch A, Yang H, Britton N, Shah F, et al. Longitudinal multicompartment characterization of host-microbiota interactions in patients with acute respiratory failure. Nat Commun. (2024) 15:4708. doi: 10.1038/s41467-024-48819-8
112. Poroyko V, Meng F, Meliton A, Afonyushkin T, Ulanov A, Semenyuk E, et al. Alterations of lung microbiota in a mouse model of LPS-induced lung injury. Am J Physiol Lung Cell Mol Physiol. (2015) 309:L76–83. doi: 10.1152/ajplung.00061.2014
113. Dickson RP, Erb-Downward JR, Freeman CM, McCloskey L, Beck JM, Huffnagle GB, et al. Spatial variation in the healthy human lung microbiome and the adapted island model of lung biogeography. Ann ATS. (2015) 12:821–30. doi: 10.1513/AnnalsATS.201501-029OC
114. Dickson RP, Erb-Downward JR, Freeman CM, McCloskey L, Falkowski NR, Huffnagle GB, et al. Bacterial topography of the healthy human lower respiratory tract. mBio. (2017) 8:e02287–16. doi: 10.1128/mBio.02287-16
115. Dickson RP, Erb-Downward JR, and Huffnagle GB. Towards an ecology of the lung: new conceptual models of pulmonary microbiology and pneumonia pathogenesis. Lancet Respir Med. (2014) 2:238–46. doi: 10.1016/S2213-2600(14)70028-1
116. Emonet S, Lazarevic V, Leemann Refondini C, Gaïa N, Leo S, Girard M, et al. Identification of respiratory microbiota markers in ventilator-associated pneumonia. Intensive Care Med. (2019) 45:1082–92. doi: 10.1007/s00134-019-05660-8
117. Dickson RP, Erb-Downward JR, Falkowski NR, Hunter EM, Ashley SL, and Huffnagle GB. The lung microbiota of healthy mice are highly variable, cluster by environment, and reflect variation in baseline lung innate immunity. Am J Respir Crit Care Med. (2018) 198:497–508. doi: 10.1164/rccm.201711-2180OC
118. Segal LN, Clemente JC, Tsay JCJ, Koralov SB, Keller BC, Wu BG, et al. Enrichment of the lung microbiome with oral taxa is associated with lung inflammation of a Th17 phenotype. Nat Microbiol. (2016) 1:16031. doi: 10.1038/nmicrobiol.2016.31
119. Clarke TB. Early innate immunity to bacterial infection in the lung is regulated systemically by the commensal microbiota via nod-like receptor ligands. Infect Immun. (2014) 82:4596–606. doi: 10.1128/IAI.02212-14
120. Brown RL, Sequeira RP, and Clarke TB. The microbiota protects against respiratory infection via GM-CSF signaling. Nat Commun. (2017) 8:1512. doi: 10.1038/s41467-017-01803-x
121. Horn KJ, Schopper MA, Drigot ZG, and Clark SE. Airway Prevotella promote TLR2-dependent neutrophil activation and rapid clearance of Streptococcus pneumoniae from the lung. Nat Commun. (2022) 13:3321. doi: 10.1038/s41467-022-31074-0
122. Cavaillon J, Singer M, and Skirecki T. Sepsis therapies: learning from 30 years of failure of translational research to propose new leads. EMBO Mol Med. (2020) 12:e10128. doi: 10.15252/emmm.201810128
123. Giamarellos-Bourboulis EJ, Aschenbrenner AC, Bauer M, Bock C, Calandra T, Gat-Viks I, et al. The pathophysiology of sepsis and precision-medicine-based immunotherapy. Nat Immunol. (2024) 25:19–28. doi: 10.1038/s41590-023-01660-5
124. Monneret G and Payen D. Immunostimulation in critically ill patients: do not forget to consider the timing, stratification, and monitoring. Ann Intensive Care. (2024) 14:75, s13613–024-01302-5. doi: 10.1186/s13613-024-01302-5
125. Francois B, Jeannet R, Daix T, Walton AH, Shotwell MS, Unsinger J, et al. Interleukin-7 restores lymphocytes in septic shock: the IRIS-7 randomized clinical trial. JCI Insight. (2018) 3:e98960. doi: 10.1172/jci.insight.98960
126. Roquilly A, Francois B, Huet O, Launey Y, Lasocki S, Weiss E, et al. Interferon gamma-1b for the prevention of hospital-acquired pneumonia in critically ill patients: a phase 2, placebo-controlled randomized clinical trial. Intensive Care Med. (2023) 49:530–44. doi: 10.1007/s00134-023-07065-0
127. Philippart F, Fitting C, and Cavaillon JM. Lung microenvironment contributes to the resistance of alveolar macrophages to develop tolerance to endotoxin*. Crit Care Med. (2012) 40:2987–96. doi: 10.1097/CCM.0b013e31825b8d57
128. Hilligan KL, Namasivayam S, Clancy CS, Baker PJ, Old SI, Peluf V, et al. Bacterial-induced or passively administered interferon gamma conditions the lung for early control of SARS-CoV-2. Nat Commun. (2023) 14:8229. doi: 10.1038/s41467-023-43447-0
129. Gschwend J, Sherman SPM, Ridder F, Feng X, Liang HE, Locksley RM, et al. Alveolar macrophages rely on GM-CSF from alveolar epithelial type 2 cells before and after birth. J Exp Med. (2021) 218:e20210745. doi: 10.1084/jem.20210745
130. Meisel C, Schefold JC, Pschowski R, Baumann T, Hetzger K, Gregor J, et al. Granulocyte–macrophage colony-stimulating factor to reverse sepsis-associated immunosuppression: A double-blind, randomized, placebo-controlled multicenter trial. Am J Respir Crit Care Med. (2009) 180:640–8. doi: 10.1164/rccm.200903-0363OC
131. Umstead TM, Hewage EK, Mathewson M, Beaudoin S, Chroneos ZC, Wang M, et al. Lower respiratory tract delivery, airway clearance, and preclinical efficacy of inhaled GM-CSF in a postinfluenza pneumococcal pneumonia model. Am J Physiol Lung Cell Mol Physiol. (2020) 318:L571–9. doi: 10.1152/ajplung.00296.2019
132. Herold S, Hoegner K, Vadász I, Gessler T, Wilhelm J, Mayer K, et al. Inhaled granulocyte/macrophage colony–stimulating factor as treatment of pneumonia-associated acute respiratory distress syndrome. Am J Respir Crit Care Med. (2014) 189:609–11. doi: 10.1164/rccm.201311-2041LE
133. Nascimento DC, Melo PH, Piñeros AR, Ferreira RG, Colón DF, Donate PB, et al. IL-33 contributes to sepsis-induced long-term immunosuppression by expanding the regulatory T cell population. Nat Commun. (2017) 8:14919. doi: 10.1038/ncomms14919
134. Gillis A, Ben Yaacov A, and Agur Z. A new method for optimizing sepsis therapy by nivolumab and meropenem combination: importance of early intervention and CTL reinvigoration rate as a response marker. Front Immunol. (2021) 12:616881. doi: 10.3389/fimmu.2021.616881
135. Li Z, Li Y, Sun Q, Wei J, Li B, Qiu Y, et al. Targeting the pulmonary microbiota to fight against respiratory diseases. Cells. (2022) 11:916. doi: 10.3390/cells11050916
136. Pérez-Cobas AE, Baquero F, De Pablo R, Soriano MC, and Coque TM. Altered ecology of the respiratory tract microbiome and nosocomial pneumonia. Front Microbiol. (2022) 12:709421. doi: 10.3389/fmicb.2021.709421
137. Pitashny M, Kesten I, Shlon D, Hur DB, and Bar-Yoseph H. The future of microbiome therapeutics. Drugs. (2025) 85:117–25. doi: 10.1007/s40265-024-02107-3
138. Wu BG, Sulaiman I, Tsay JCJ, Perez L, Franca B, Li Y, et al. Episodic aspiration with oral commensals induces a myD88-dependent, pulmonary T-helper cell type 17 response that mitigates susceptibility to Streptococcus pneumoniae. Am J Respir Crit Care Med. (2021) 203:1099–111. doi: 10.1164/rccm.202005-1596OC
139. Montassier E, Kitsios GD, Radder JE, Le Bastard Q, Kelly BJ, Panzer A, et al. Robust airway microbiome signatures in acute respiratory failure and hospital-acquired pneumonia. Nat Med. (2023) 29:2793–804. doi: 10.1038/s41591-023-02617-9
140. Clua P, Kanmani P, Zelaya H, Tada A, Kober AKMH, Salva S, et al. Peptidoglycan from immunobiotic lactobacillus rhamnosus improves resistance of infant mice to respiratory syncytial viral infection and secondary pneumococcal pneumonia. Front Immunol. (2017) 8:948. doi: 10.3389/fimmu.2017.00948
141. Ortiz Moyano R, Raya Tonetti F, Tomokiyo M, Kanmani P, Vizoso-Pinto MG, Kim H, et al. The ability of respiratory commensal bacteria to beneficially modulate the lung innate immune response is a strain dependent characteristic. Microorganisms. (2020) 8:727. doi: 10.3390/microorganisms8050727
142. Muñoz N, Van Maele L, Marqués JM, Rial A, Sirard JC, and Chabalgoity JA. Mucosal administration of flagellin protects mice from Streptococcus pneumoniae lung infection. Infect Immun. (2010) 78:4226–33. doi: 10.1128/IAI.00224-10
143. Hernandez A, Zhou J, Bohannon JK, McBride MA, Gibson-Corley KN, Patil NK, et al. Intrapulmonary treatment with a novel tlr4 agonist confers protection against klebsiella pneumonia. Shock. (2022) 58:295–303. doi: 10.1097/SHK.0000000000001977
144. Baldry M, Costa C, Zeroual Y, Cayet D, Pardessus J, Soulard D, et al. Targeted delivery of flagellin by nebulization offers optimized respiratory immunity and defense against pneumococcal pneumonia. Antimicrob Agents Chemother. (2024) 68:e00866–24. doi: 10.1128/aac.00866-24
145. Van Der Lelie D, Oka A, Taghavi S, Umeno J, Fan TJ, Merrell KE, et al. Rationally designed bacterial consortia to treat chronic immune-mediated colitis and restore intestinal homeostasis. Nat Commun. (2021) 12:3105. doi: 10.1038/s41467-021-23460-x
146. Johnstone J, Meade M, Lauzier F, Marshall J, Duan E, Dionne J, et al. Effect of probiotics on incident ventilator-associated pneumonia in critically ill patients: A randomized clinical trial. JAMA. (2021) 326:1024. doi: 10.1001/jama.2021.13355
147. Seok J, Warren HS, Cuenca AG, Mindrinos MN, Baker HV, Xu W, et al. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci USA. (2013) 110:3507–12. doi: 10.1073/pnas.1222878110
148. Basil MC and Morrisey EE. Lung regeneration: a tale of mice and men. Semin Cell Dev Biol. (2020) 100:88–100. doi: 10.1016/j.semcdb.2019.11.006
149. Mestas J and Hughes CCW. Of mice and not men: differences between mouse and human immunology. J Immunol. (2004) 172:2731–8. doi: 10.4049/jimmunol.172.5.2731
150. Nauseef WM. Human neutrophils ≠ murine neutrophils: does it matter? Immunol Rev. (2023) 314(1):442–56. doi: 10.1111/imr.13154
151. Chotirmall SH, Bogaert D, Chalmers JD, Cox MJ, Hansbro PM, Huang YJ, et al. Therapeutic targeting of the respiratory microbiome. Am J Respir Crit Care Med. (2022) 206:535–44. doi: 10.1164/rccm.202112-2704PP
152. Simpson J, Scott J, Davidson R, and General Enquiries OPA. Investigating the effects of a novel inhaled intervention on the function of alveolar macrophage cells in the lung. Available online at: https://www.isrctn.com/ISRCTN78203402.
Keywords: sepsis, compartmentalisation, alveolar macrophages (AMs), lung infection, pulmonary microbiota
Citation: Vernay E, Cerrato E, Santinon F, Monard C, Perez P, Allantaz F, Lukaszewicz A-C and Llitjos J-F (2025) Considering local immunity for innovative immunomodulatory approaches: pulmonary sepsis as a use case. Front. Immunol. 16:1627313. doi: 10.3389/fimmu.2025.1627313
Received: 12 May 2025; Accepted: 08 July 2025;
Published: 07 August 2025.
Edited by:
David M. Smadja, Université Paris Cité, FranceReviewed by:
Alberto Chocron, United States Department of Veterans Affairs, United StatesHaizam Oubari, Mass General Brigham, United States
Copyright © 2025 Vernay, Cerrato, Santinon, Monard, Perez, Allantaz, Lukaszewicz and Llitjos. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jean-François Llitjos, amVhbmZyYW5jb2lzLmxsaXRqb3NAYmlvbWVyaWV1eC5jb20=