 Xinyao Huang
Xinyao Huang Jianjun He2†
Jianjun He2† Renjun Gu
Renjun Gu- 1The First Clinical Medical College, Nanjing University of Chinese Medicine, Nanjing, China
- 2Department of Radiation Oncology, Jinling Hospital, Affiliated Hospital of Medical School, Nanjing University, Nanjing, China
- 3Department of Infectious Diseases and Hepatology, Jinling Hospital, Affiliated Hospital of Medical School, Nanjing University, Nanjing, China
- 4School of Chinese Medicine, Nanjing University of Chinese Medicine, Nanjing, China
- 5Department of Gastroenterology and Hepatology, Jinling Hospital, Affiliated Hospital of Medical School, Nanjing University, Nanjing, China
- 6School of Acupuncture-Moxibustion and Tuina, School of Health Preservation and Rehabilitation, Nanjing University of Chinese Medicine, Nanjing, China
Metabolic reprogramming is a central driving force in the malignant progression of digestive system tumors. It facilitates tumor proliferation, metastasis, and therapeutic resistance through aerobic glycolysis, disordered lipid metabolism, and altered amino acid metabolism. Pyruvate kinase M2 (PKM2) functions as a key regulator of tumor metabolism, promoting aerobic glycolysis and suppressing mitochondrial respiration via conformational changes and nuclear translocation. These processes are orchestrated by hypoxia-inducible factors and oncogenic signaling, ensuring a sustained energy supply and biosynthetic precursors for tumor growth. Additionally, PKM2 modulates lipid biosynthesis and amino acid metabolism by participating in epigenetic regulation and the organization of metabolic enzyme complexes. These functions contribute to tumor adaptation within the microenvironment and promote immune evasion. In digestive system tumors, the regulatory network of PKM2 demonstrates tissue specificity, mediated by non-coding RNAs, post-translational modifications, and crosstalk between metabolic and signaling pathways, collectively sustaining metabolic plasticity. Therapeutic strategies targeting PKM2 primarily aim to reverse the Warburg effect or inhibit compensatory metabolic pathways; however, their clinical translation remains challenging. The dual regulatory role of PKM2 may perturb immunometabolic homeostasis; the fluctuating nutrient landscape of the tumor microenvironment can drive adaptive resistance; and some inhibitors exhibit limited specificity or unacceptable toxicity. This review summarizes the molecular mechanisms through which PKM2 drives metabolic reprogramming in digestive system tumors, as well as the current therapeutic advances and clinical barriers.
1 Introduction
Metabolic reprogramming is a hallmark of cancer and plays a central role in tumor initiation, progression, and metastasis by coordinating intra- and extracellular signals to promote malignant phenotypes (1). To sustain rapid proliferation, tumor cells reshape their metabolic pathways to meet energy and biosynthetic demands and to adapt to microenvironmental stress (1). In digestive system cancers-including hepatocellular carcinoma (HCC), gastric cancer (GC), and colorectal cancer (CRC)-distinct metabolic alterations are observed, such as enhanced aerobic glycolysis, dysregulated glutamine utilization, and abnormal lipid synthesis. These changes not only support tumor growth but also promote invasion and metastasis (1, 2). Additionally, the accumulation of metabolites like lactate contributes to an immunosuppressive microenvironment, impairing immune cell function and facilitating immune escape and therapeutic resistance (3).
Pyruvate kinase M2 (PKM2), a rate-limiting enzyme in glycolysis, is a key driver of metabolic reprogramming in cancer by promoting the Warburg effect (4). It is frequently overexpressed in tumor tissues and correlates with poor prognosis (4). Even in the presence of sufficient oxygen, PKM2 favors aerobic glycolysis over mitochondrial respiration, leading to increased lactate production and supplying energy and biosynthetic substrates to support tumor growth (5). In addition to its enzymatic role, PKM2 regulates tumor proliferation, metastasis, and apoptosis through non-metabolic functions, including its activity as a protein kinase (6).
Beyond its metabolic role in tumor progression, PKM2 also participates in remodeling the tumor immune microenvironment. In digestive system malignancies, SUMOylated PKM2 can be secreted via exosomes and internalized by immune cells, where it activates STAT3 signaling and reprograms their metabolic and functional states (7). In pancreatic ductal adenocarcinoma, tumor-associated macrophage–derived TGF-β1 induces PKM2 nuclear translocation, which activates STAT1 signaling and modulates immune checkpoint pathways (8). These findings underscore the immunomodulatory potential of PKM2 within gastrointestinal tumors and highlight its multifaceted role beyond metabolism.
Building upon this dual role in metabolism and immune regulation, recent studies have further revealed that PKM2 is intricately involved in the development and therapy resistance of digestive system tumors. It exerts its effects by modulating structural conformation, engaging in epigenetic regulation, and interacting with diverse metabolic and signaling networks (5). Based on these insights, this review summarizes the molecular mechanisms by which PKM2 drives metabolic reprogramming, outlines tumor-specific regulatory networks across various digestive system malignancies, including hepatocellular carcinoma (HCC), gastric cancer (GC), and others, and discusses current therapeutic strategies targeting PKM2 along with challenges in clinical translation. Together, these perspectives provide a foundation for future research and the development of precision treatment approaches (Figure 1).
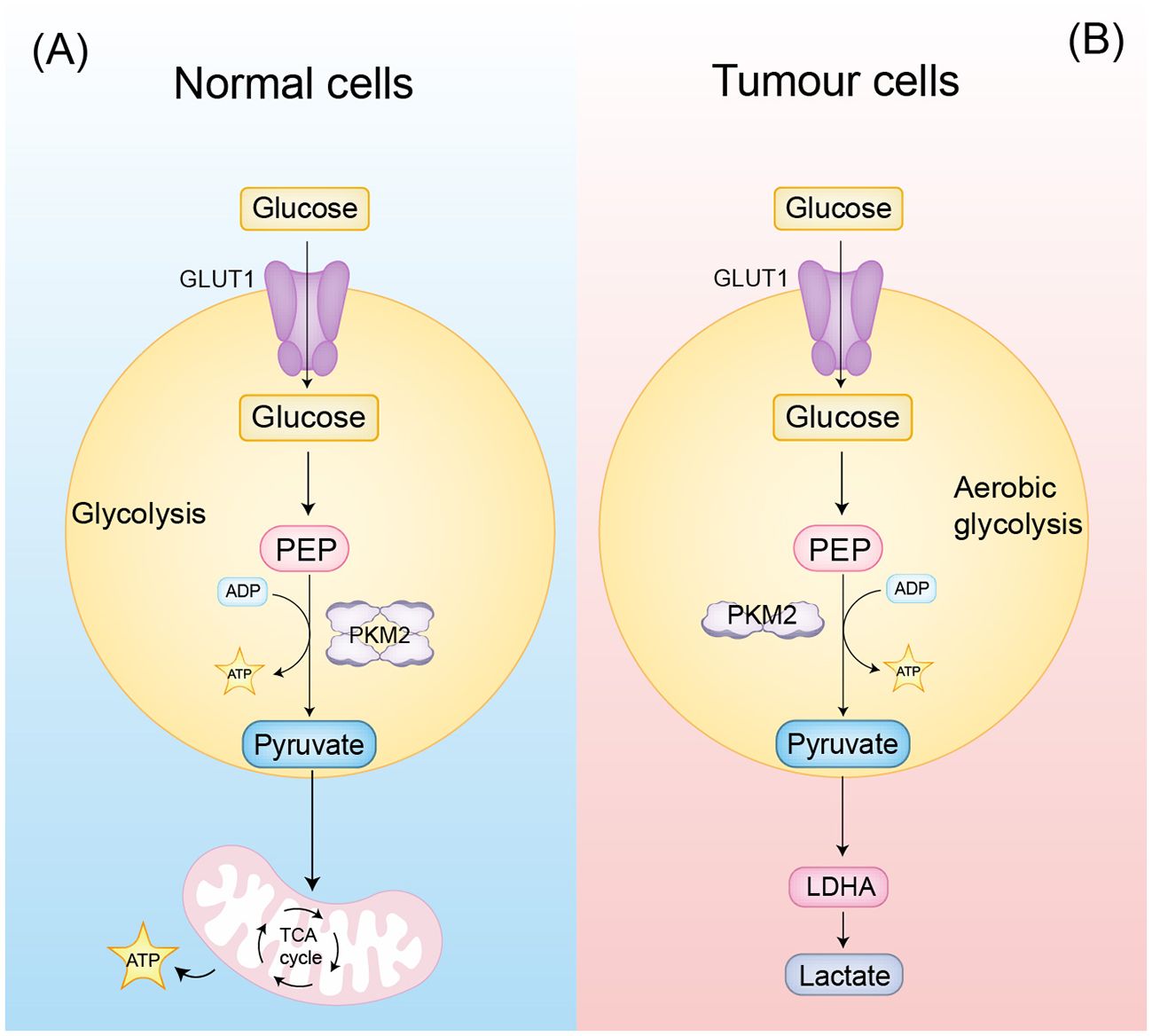
Figure 1. PKM2 classical function (A) In normal cells, PKM2 tetramers are involved in glycolysis. (B) In tumor cells, PKM2 dimers are involved in aerobic glycolysis, producing more lactate and further remodeling the tumor microenvironment.
2 The role of PKM2 multifunctional hub
2.1 Metabolic regulatory functions of PKM2
PKM2 primarily facilitates tumor-specific metabolic reprogramming by forming low-activity dimers that favor the final step of glycolysis, leading to pyruvate accumulation while suppressing mitochondrial respiration, thereby reinforcing the Warburg effect and promoting rapid cancer cell proliferation (9). It maintains metabolic homeostasis via several mechanisms: promoting Mitofusin 1/2 (MFN1/2)-mediated mitochondrial fusion to stabilize membrane potential (10), modulating the Nicotinamide Adenine Dinucleotide Phosphate (NADPH)/Glutathione (GSH) axis to maintain redox balance (11), and supporting Phosphoglycerate Dehydrogenase (PHGDH)-dependent serine synthesis to fulfill biosynthetic demands (12). PKM2 also contributes to therapy resistance by upregulating Death-Ligand 1 (PD-L1) expression through Signal Transducer and Activator of Transcription 3 (STAT3) phosphorylation, facilitating immune evasion (4, 13). Additionally, it promotes lactate-driven acidification of the tumor microenvironment and induces epigenetic modifications at the ATP-binding cassette sub-family B member 1 (ABCB1) promoter, maintaining cancer stemness and drug efflux capacity (4, 13). These metabolic functions, combined with regulatory roles in mitochondrial dynamics and redox homeostasis, establish PKM2 as a central metabolic hub in tumor adaptation.
2.2 Non-metabolic functions of PKM2 as a multifunctional hub
Beyond metabolism, PKM2 translocates to the nucleus under stress, where it phosphorylates histone H3 at threonine 11, activating c-Myc and Cyclin D1 expression and cooperating with HIF-1α to regulate the mTORC1 pathway, thus linking metabolic reprogramming to cell proliferation (4, 13, 14). In hepatocellular carcinoma, PKM2 is regulated via multi-level mechanisms including transcriptional activation by YAP through HIF-1α, post-translational modifications by HSP90 and GSK-3β that stabilize its dimeric conformation, and nuclear functions involving PRMT6 and STAT3 signaling to amplify aerobic glycolysis (15–18). PKM2’s SUMOylation promotes interaction with ARRDC1 and secretion via exosomes into the tumor microenvironment, activating STAT3 phosphorylation in monocytes and inducing their metabolic reprogramming and differentiation into macrophages (7). The exosomal circPETH-147aa further drives aerobic glycolysis via ALDOA-S36 phosphorylation, enhancing amino acid metabolic reprogramming and immune evasion (19). At the plasma membrane, TSP50 inhibits PKM2 activity via acetylation at K433, promoting HCC cell proliferation (20). PKM2 suppresses apoptosis by promoting Bim degradation, while its depletion stabilizes Bim and induces cell death (21). Moreover, PKM2 modulates immune escape by upregulating PD-L1 through STAT3 phosphorylation, recruiting HDAC3 to remodel chromatin accessibility, and sustaining oncogenic signaling through interactions with β-catenin and activation of CCND1 to accelerate the cell cycle (4, 14). These multifaceted non-metabolic functions position PKM2 as an integrative hub that connects metabolism, epigenetics, immune modulation, and cell cycle regulation in cancer progression.
3 Molecular mechanisms of PKM2-driven metabolic reprogramming
3.1 Glycolysis
PKM2 catalyzes the final, rate-limiting step of glycolysis by transferring a phosphate group from phosphoenolpyruvate (PEP) to adenosine diphosphate (ADP), generating pyruvate and adenosine triphosphate (ATP) (22). As a critical metabolic regulator, PKM2 undergoes dynamic structural transitions that enable dual functions. It facilitates the conversion of phosphoglycerate mutase (PGM)-derived intermediates into lactate and also acts as a protein kinase that participates in transcriptional regulation and metabolic reprogramming to sustain the Warburg effect (23). Post-translational modifications further modulate PKM2 activity. Phosphorylation at tyrosine 105 stabilizes the dimeric form and reduces pyruvate kinase activity, while hydroxylation at proline 403/408 enhances the expression of glucose transporter 1 (GLUT1) and lactate dehydrogenase A (LDHA) by activating hypoxia-inducible factor 1α (HIF-1α), reinforcing a positive feedback loop that maintains aerobic glycolysis (22, 24, 25). Nuclear translocation of PKM2, mediated by the extracellular signal-regulated kinase (ERK)/mitogen-activated protein kinase (MAPK) pathway, enables histone H3 phosphorylation and activation of c-Myc target genes. PKM2 also catalyzes phosphorylation of phosphoglycerate mutase 1 (PGAM1) at histidine 11, which enhances aerobic glycolysis and promotes tumor growth (6, 22, 26). The phosphoinositide 3-kinase (PI3K)/protein kinase B (Akt) pathway further supports this process by promoting LDHA-mediated conversion of pyruvate to lactate, contributing to tumor-specific accumulation of glycolytic end-products (27). In ovarian cancer, HIF-1α upregulates endothelial cell-specific molecule 1 (ESM1), which enhances PKM2 SUMOylation and stabilizes its dimeric form. This activates signal transducer and activator of transcription 5 (STAT5), forming a cycle that amplifies glucose uptake and lactate production (28). In lung cancer, PKM2 interacts with histone H2B and reduces its monoubiquitination (H2Bub1), thereby inhibiting the expression of mitochondrial respiration genes and promoting the Warburg effect (29). In triple-negative breast cancer, methyltransferase 14 (METTL14)-mediated N6-methyladenosine (m6A) modification facilitates PKM2 degradation through the miR-29c-3p/TRIM9 axis, shifting the balance toward the low-activity dimer (30). Meanwhile, crotonylation of polypyrimidine tract-binding protein 1 (PTBP1) at lysine 266 enhances heterogeneous nuclear ribonucleoproteins A1 and A2 (hnRNPA1/2) binding to PKM pre-mRNA, thereby promoting PKM2-specific splicing (31). In breast cancer, coactivator-associated arginine methyltransferase 1 (CARM1)-mediated methylation of PKM2 promotes its interaction with inositol 1,4,5-trisphosphate receptors (InsP3Rs), reducing endoplasmic reticulum to mitochondria Ca²+ flux and triggering aerobic glycolysis (32). In prostate cancer, long non-coding RNA (lncRNA) SNHG3 competitively binds to miR-139-5p and relieves its suppression of PKM2 mRNA, resulting in enhanced aerobic glycolysis (33). In non-small cell lung cancer (NSCLC), hypoxia-induced HIF-1α forms a complex with phosphorylated Smad3, which upregulates c-Myc and promotes PKM2 splicing, constructing a hypoxia-adaptive aerobic glycolysis network (34, 35). Notably, tyrosine phosphorylation at residues Y105 and Y148 exerts dual effects by maintaining PKM2 in its low-activity dimeric form while promoting Aldehyde Dehydrogenase-positive (ALDH+) cancer stem cell phenotypes (36). Despite its central role in glycolysis, the activity of PKM2 is modulated by cellular context through structural, epigenetic, and metabolic mechanisms. Structural remodeling, such as SUMOylation and O-linked β-N-acetylglucosamine (O-GlcNAcylation), stabilizes the dimeric conformation and adjusts enzymatic activity in response to environmental inputs (28, 37). Epigenetically, phosphorylation of hnRNPA1 at serine 6 enhances the recruitment of splicing factors, promoting the generation of the PKM2 isoform under specific regulatory cues (38–40). In parallel, PEP-dependent phosphorylation of PGAM1 facilitates the redirection of glycolytic intermediates into biosynthetic pathways, enabling cells to adjust to proliferative demands (26). In conclusion, these mechanisms allow PKM2 to serve as both a metabolic enzyme and a transcriptional modulator, with its function precisely tailored by tumor-specific microenvironmental signals and intracellular stress states.
These findings demonstrate that PKM2 serves as a central integrator of glycolytic regulation, shaped by structural remodeling, post-translational modifications, epigenetic control, and metabolic signaling. Multiple oncogenic pathways, including HIF-1α, MAPK, PI3K/Akt, and c-Myc, coordinately influence its oligomeric state, subcellular localization, and enzymatic activity. These regulatory inputs fine-tune PKM2 to sustain elevated aerobic glycolysis and enable cellular adaptation to hypoxia, nutrient fluctuations, and proliferative stress. By coupling metabolic output with transcriptional and post-transcriptional regulation, PKM2 functions not only as a metabolic enzyme but also as a signaling node that links energy metabolism to tumor progression (Figure 2).
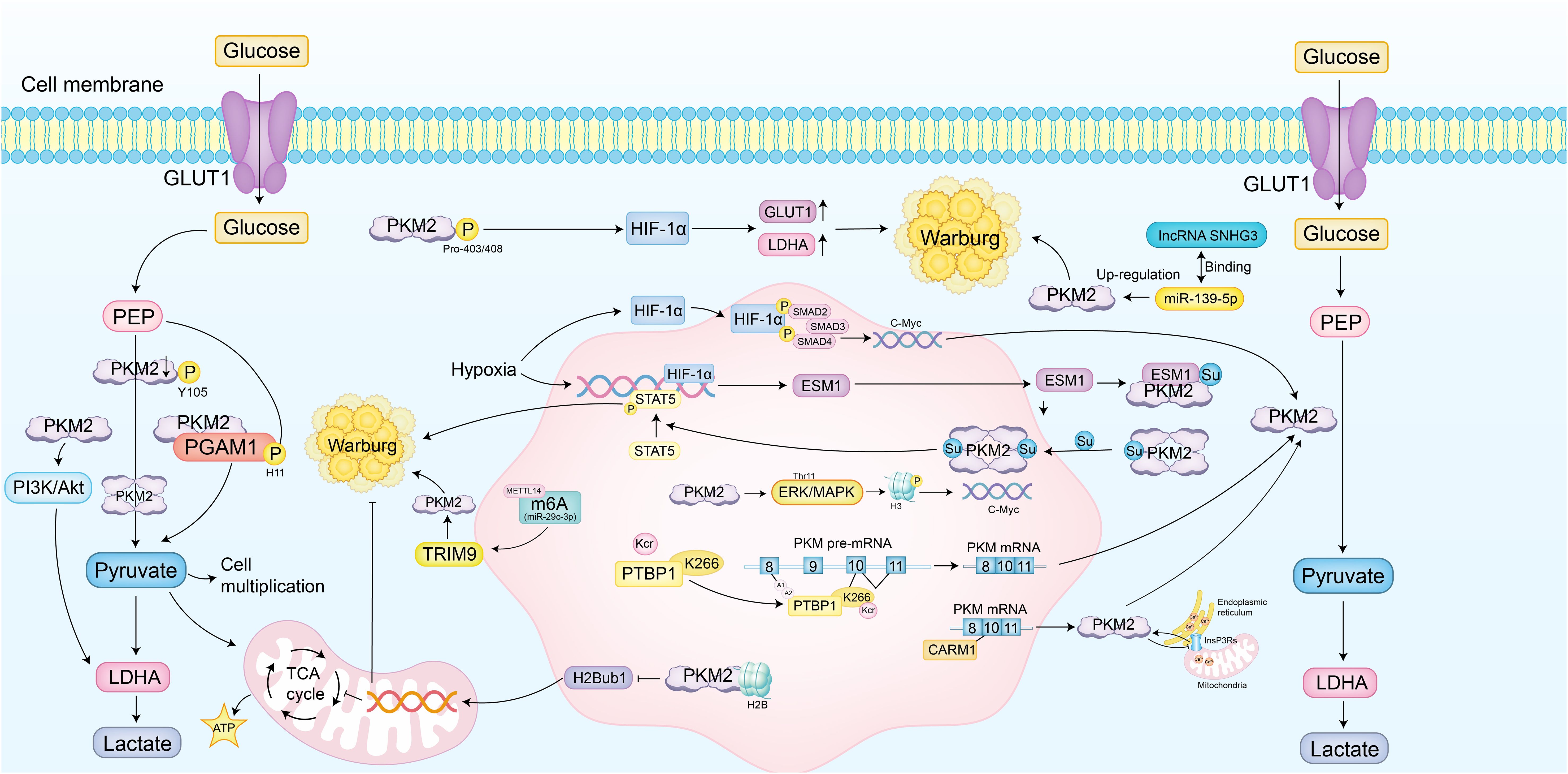
Figure 2. PKM2 drives metabolic reprogramming of glycolytic mechanisms. PKM2 catalyzes the final, rate-limiting step of glycolysis, converting phosphoenolpyruvate (PEP) to pyruvate and ATP. It also functions as a protein kinase involved in transcriptional regulation and metabolic reprogramming to sustain the Warburg effect. Post-translational modifications, such as phosphorylation at Tyr105, reduce its activity by stabilizing the dimeric form, while hydroxylation at Pro403/408 enhances the expression of glucose transporter 1 (GLUT1) and lactate dehydrogenase A (LDHA) via activation of hypoxia-inducible factor 1α (HIF-1α). Nuclear translocation of PKM2, mediated by the ERK/MAPK pathway, leads to histone H3 phosphorylation and activation of c-Myc target genes. PKM2 also phosphorylates phosphoglycerate mutase 1 (PGAM1) at His11, enhancing aerobic glycolysis. The PI3K/Akt pathway supports this process by promoting LDHA-mediated conversion of pyruvate to lactate. In ovarian cancer, HIF-1α upregulates endothelial cell-specific molecule 1 (ESM1), enhancing PKM2 SUMOylation and stabilizing its dimeric form, activating STAT5 and amplifying glucose uptake and lactate production. In lung cancer, PKM2 interacts with histone H2B, reducing its monoubiquitination (H2Bub1) and inhibiting mitochondrial respiration genes. In triple-negative breast cancer, N6-methyladenosine (m6A) modification by methyltransferase 14 (METTL14) facilitates PKM2 degradation, shifting towards the low-activity dimer. In breast cancer, methylation by coactivator-associated arginine methyltransferase 1 (CARM1) promotes PKM2 interaction with inositol 1,4,5-trisphosphate receptors (InsP3Rs), reducing endoplasmic reticulum to mitochondria Ca²+ flux. In prostate cancer, long non-coding RNA (lncRNA) SNHG3 relieves miR-139-5p suppression of PKM2 mRNA, enhancing aerobic glycolysis. In non-small cell lung cancer (NSCLC), hypoxia-induced HIF-1α forms a complex with phosphorylated Smad3, upregulating c-Myc and promoting PKM2 splicing. Structural remodeling, such as SUMOylation and O-GlcNAcylation, stabilizes the dimeric conformation and adjusts enzymatic activity. Epigenetic mechanisms, including phosphorylation of hnRNPA1, enhance the recruitment of splicing factors, promoting PKM2 isoform generation. PEP-dependent phosphorylation of PGAM1 redirects glycolytic intermediates into biosynthetic pathways, enabling cells to adjust to proliferative demands. These mechanisms allow PKM2 to serve as both a metabolic enzyme and a transcriptional modulator, with its function precisely tailored by tumor-specific microenvironmental signals and intracellular stress states.
3.2 Lipids
PKM2 orchestrates lipid metabolic reprogramming through structural transformation and signaling interactions that promote tumor progression. In the hypoxic microenvironment of ovarian cancer, HIF-1α upregulates endothelial cell-specific molecule 1 (ESM1), which induces PKM2 SUMOylation and stabilizes its dimeric conformation. This facilitates nuclear translocation of PKM2, enabling activation of fatty acid synthase (FASN) expression via STAT3 phosphorylation, thereby promoting de novo lipogenesis, tumor proliferation, and vasculogenic mimicry (28). This pro-oncogenic mechanism is amplified during peritoneal metastasis, where HIF-1α-induced PKM2 expression enhances both fatty acid uptake and lipid biosynthesis, establishing lipid metabolism as a core adaptive strategy in response to hypoxia and energy stress (41). In tumors associated with metabolic disorders, aerobic glycolysis-dominant PKM2 activity restricts pyruvate entry into mitochondria, suppressing oxidative phosphorylation (OXPHOS) and contributing to hepatic steatosis, which further reinforces a tumor-promoting metabolic niche (42). The lipid-regulatory role of PKM2 also exhibits context-dependent characteristics, particularly in response to therapeutic and metabolic stress. In cisplatin-resistant non-small cell lung cancer (NSCLC), PKM2 inhibition suppresses aerobic glycolysis and induces a compensatory increase in lipid metabolism through carnitine palmitoyltransferase 1A (CPT1A)-dependent fatty acid oxidation (FAO), sustaining cancer cell survival under chemotherapeutic pressure. This adaptive metabolic reprogramming can be reversed by Compound 3K, a PKM2 inhibitor that restores chemosensitivity (43, 44). In triple-negative breast cancer (TNBC), PKM2 upregulates acyl-CoA dehydrogenase very long chain (ACADVL) through the AMP-activated protein kinase–Krüppel-like factor 4 (AMPK–KLF4) axis, promoting lipid β-oxidation while depleting lipid droplet storage. This metabolic reprogramming concurrently suppresses aerobic glycolysis and enhances lipolysis, establishing a compensatory metabolic equilibrium that functions independently of BRCA mutation status (45). These findings highlight that PKM2 dynamically modulates lipid synthesis and degradation based on external stress conditions, underscoring its role in maintaining metabolic plasticity within diverse tumor microenvironments.
3.3 Amino acid
PKM2 reprograms amino acid metabolism by integrating enzymatic complex assembly with transcriptional control, thereby supporting tumor growth under metabolic stress. In triple-negative breast cancer (TNBC), the amino acid transporter SLC7A5 downregulates miR-152 and activates the E2F1/PTBP1 signaling axis, promoting alternative splicing of PKM pre-mRNA toward the PKM2 isoform. The increased expression of PKM2 enhances the uptake and utilization of essential amino acids, fueling biosynthetic demands and reinforcing tumor cell proliferation (46). This PKM2-driven shift also contributes to the emergence of drug-resistant metabolic phenotypes, highlighting its role in maintaining oncogenic adaptation through amino acid metabolic rewiring. Beyond its canonical glycolytic function, PKM2 exhibits a context-dependent regulatory role in response to amino acid deprivation. In the tumor microenvironment, where serine availability is limited, PKM2 coordinates with the c-Myc-responsive long non-coding RNA gLINC to assemble a metabolic enzyme complex comprising PGK1, PGAM1, ENO1, and LDHA. This complex significantly boosts aerobic glycolytic flux and enhances ATP production efficiency, enabling tumor cells to sustain energy output and survive serine-deficient stress (47, 48). These findings suggest that PKM2 facilitates metabolic flexibility not only through isoform control but also by structurally adapting to nutrient limitations, underscoring its dynamic role in amino acid-responsive metabolic reprogramming.
4 Specific regulatory network of PKM2 in digestive system tumors
While PKM2 is widely recognized for its role in glycolytic regulation, emerging evidence underscores a striking tissue-specific heterogeneity in its upstream modulation and functional outputs across digestive system tumors. In hepatocellular carcinoma, lncRNA DACT3-AS1 activates PKM2 via the HDAC2/FOXA3 axis, thereby promoting immune evasion and metastasis (49). In gastric cancer, the CCAT1–PTBP1 axis facilitates alternative splicing to favor PKM2 isoform dominance, promoting metabolic reprogramming and cancer stemness (50). In colorectal cancer, OTUB2-mediated deubiquitination prevents PKM2 degradation by interfering with Parkin, sustaining aerobic glycolysis under metabolic stress (51). In pancreatic ductal adenocarcinoma, TGF-β1 derived from tumor-associated macrophages induces PKM2 nuclear translocation and enhances STAT1-mediated PD-L1 transcription, linking glucose metabolism to immune escape (8). These findings reveal PKM2 as a tumor-context–sensitive integrator of metabolic, immunological, and epigenetic signals. Understanding these distinct regulatory circuits is critical for advancing tumor-specific metabolic therapies. The following sections will detail the regulatory networks of PKM2 in individual digestive cancers, highlighting both shared principles and unique adaptations.
4.1 Hepatocellular carcinoma
PKM2 exerts cancer-promoting effects in HCC through multi-level and multi-mechanistic regulation. In the hypoxic microenvironment, Yes-associated protein (YAP) maintains the stability of the interaction between HIF-1α and the PKM2 gene, directly activating PKM2 transcription and accelerating aerobic glycolysis (15). Glypican-3 promotes the metabolic reprogramming shift to aerobic glycolysis by upregulating PKM2 via HIF-1α (16). Nuclear-translocated PKM2 enhances aerobic glycolysis through Protein Arginine Methyltransferase 6 (PRMT6) and activates the STAT3 signaling pathway, persistently amplifying glycolytic flux (17). Post-translational modifications also strengthen the Warburg effect; specifically, Heat Shock Protein 90 (HSP90) and Glycogen Synthase Kinase 3 Beta (GSK-3β) cooperatively phosphorylate PKM2 at Thr-328, stabilizing its dimeric conformation (18). Under hypoxia, the RNA-binding protein HuR suppresses miR-199a, leading to increased PKM2 expression, which acts as a crucial switch for the Warburg effect (52). Likewise, the circMAT2B/miR-338-3p axis enhances PKM2 stability and expression under hypoxic conditions (53). Rhubarb extract and its active compound Rhein upregulate PKM2 expression, promoting aerobic glycolysis, though Rhein may exacerbate liver injury (54). A Methyltransferase-like 5 (METTL5) activates PKM2 transcription by upregulating Ubiquitin Specific Protease 5 (USP5) to inhibit c-Myc ubiquitin-mediated degradation (55), while downregulation of GATA6 drives metabolic reprogramming in HCC cells (56). Non-canonical functions of PKM2 include its SUMOylation, which promotes interaction with ARRDC1 and secretion via exosomes into the tumor microenvironment, activating STAT3 phosphorylation in monocytes and inducing their metabolic reprogramming and differentiation into macrophages (7). The exosomal circular RNA circPETH-147aa promotes aerobic glycolysis via ALDOA-S36 phosphorylation, driving amino acid metabolic reprogramming and immune evasion (19). At the plasma membrane, testis-specific protease 50 (TSP50) inhibits PKM2 activity through acetylation at K433, promoting HCC cell proliferation (20). Within metabolic interaction networks, Gankyrin activates the β-catenin/c-Myc axis to upregulate PKM2 expression, strengthening the connection between glucose and glutamine metabolism and accelerating tumor progression (57). In HCC associated with type Ia glycogen storage disease, G6Pase-α deficiency leads to PKM2 upregulation, promoting aerobic glycolysis and the hexose monophosphate shunt, thereby accelerating tumor development (58). HDAC8-mediated deacetylation of PKM2 at K62 facilitates its nuclear translocation, where it binds β-catenin and activates CCND1 to accelerate the cell cycle (59). Furthermore, PKM2 suppresses apoptosis by promoting Bim degradation; PKM2 depletion stabilizes Bim and induces cell death (21). Clinical studies confirm that PKM2 exerts its oncogenic role in HCC by downregulating MicroRNA-122 (miR-122); miR-122 directly targets the 3’UTR of PKM2, and restoration of miR-122 expression suppresses glucose uptake and tumor growth (60).
PKM2 also exhibits clear context-dependent effects in HCC. When Ser333 is unphosphorylated, PKM2 promotes tumor growth, whereas ULK1-mediated phosphorylation at Ser333 enhances PKM2 enzymatic activity, reduces nuclear localization, suppresses c-Myc expression, and attenuates the Warburg effect, demonstrating an inhibitory role dependent on context (61). High PKM2 expression predicts poor prognosis and inhibits apoptosis by promoting Bim degradation, while PKM2 knockdown stabilizes Bim and induces apoptosis, indicating that its cancer-promoting effect depends on its interaction with apoptotic regulators (21). Additionally, COX-2 and PKM2 are both elevated in HCC and correlate with poor prognosis. Knockdown of COX-2 reduces PKM2 and HIF-1α expression, inhibiting proliferation and increasing apoptosis. However, PKM2 inhibition increases apoptosis without altering COX-2 or HIF-1α levels, suggesting that PKM2’s effects rely on upstream COX-2/HIF-1α signaling (62) (Figure 3).
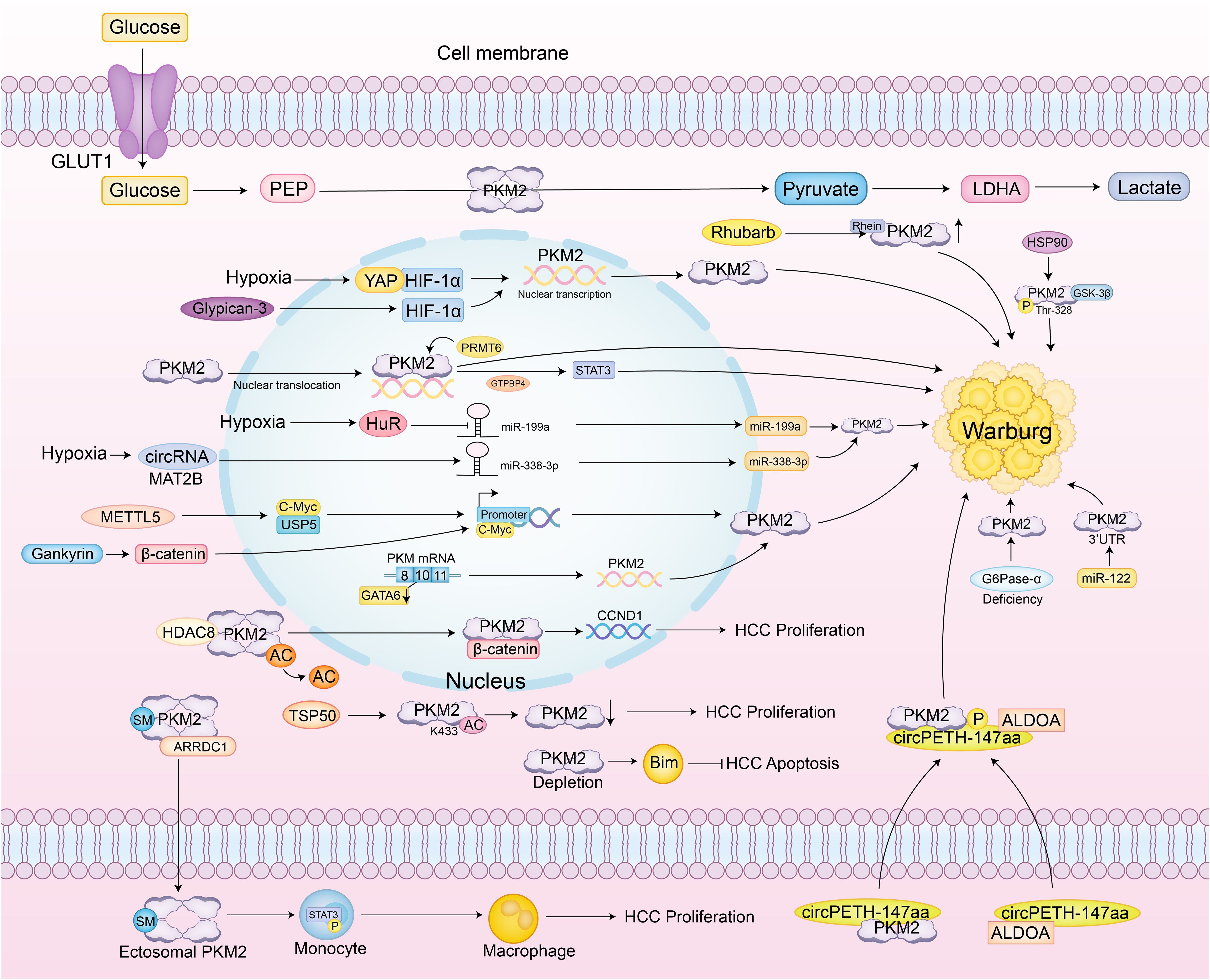
Figure 3. Specific regulatory network of PKM2 in hepatocellular carcinoma. PKM2 catalyzes the conversion of phosphoenolpyruvate (PEP) to pyruvate, which is further converted to lactate, a key aspect of the Warburg effect observed in cancer cells. Under hypoxic conditions, glycogen synthase kinase 3 beta (GSK-3β) and heat shock protein 90 (HSP90) phosphorylate PKM2 at Thr-328, stabilizing its dimeric form and enhancing aerobic glycolysis. Yes-associated protein (YAP) and hypoxia-inducible factor 1-alpha (HIF-1α) interact to directly activate PKM2 transcription, further promoting glycolysis. The RNA-binding protein HuR suppresses miR-199a under hypoxia, leading to increased PKM2 expression. Additionally, the circMAT2B/miR-338-3p axis enhances PKM2 stability and expression under hypoxic conditions. Methyltransferase-like 5 (METTL5) activates PKM2 transcription by upregulating Ubiquitin Specific Protease 5 (USP5), which inhibits c-Myc ubiquitin-mediated degradation. PKM2 also undergoes SUMOylation, promoting its interaction with ARRDC1 and secretion via exosomes into the tumor microenvironment, where it activates STAT3 phosphorylation in monocytes, inducing their metabolic reprogramming and differentiation into macrophages. The exosomal circular RNA circPETH-147aa promotes glycolysis via ALDOA-S36 phosphorylation, driving amino acid metabolic reprogramming and immune evasion. At the plasma membrane, testis-specific protease 50 (TSP50) inhibits PKM2 activity through acetylation at K433, promoting HCC cell proliferation. Gankyrin activates the β-catenin/c-Myc axis to upregulate PKM2 expression, strengthening the connection between glucose and glutamine metabolism and accelerating tumor progression. In HCC associated with type Ia glycogen storage disease, G6Pase-α deficiency leads to PKM2 upregulation, promoting aerobic glycolysis and the hexose monophosphate shunt, thereby accelerating tumor development. HDAC8-mediated deacetylation of PKM2 at K62 facilitates its nuclear translocation, where it binds β-catenin and activates CCND1 to accelerate the cell cycle. PKM2 also suppresses apoptosis by promoting Bim degradation; PKM2 depletion stabilizes Bim and induces cell death. Clinical studies confirm that PKM2 exerts its oncogenic role in HCC by downregulating MicroRNA-122 (miR-122); miR-122 directly targets the 3’UTR of PKM2, and restoration of miR-122 expression suppresses glucose uptake and tumor growth.
4.2 Gastric cancer
At the level of transcriptional splicing, the long non-coding RNA (lncRNA) CCAT1 facilitates alternative splicing of PKM pre-mRNA towards the low-activity PKM2 isoform by binding and stabilizing PTBP1 protein, resulting in dimeric PKM2 accumulation that lowers enzymatic activity. This causes glycolytic intermediates to accumulate, diverting metabolic flux toward aerobic glycolysis, thereby markedly increasing lactate production and glucose flux and establishing a pro-oncogenic metabolic reprogramming phenotype (50). Aerobic glycolysis is also precisely regulated at the epigenetic level-histone lysine methyltransferase SETD1A enhances HIF-1α recruitment to the PKM2 promoter through Histone H3 Lysine 4 (H3K4) methylation, forming a HIF-1α/SETD1A positive feedback loop that persistently amplifies glycolytic flux and sustains the continuous proliferation of gastric cancer cells (63). At the enzymatic regulation level, PKM2’s role is more nuanced and context-dependent. β-Arrestin 1 (ARRB1) directly binds PKM2 and inhibits its tetramer assembly, maintaining a low-activity dimeric state that promotes the Warburg effect. In contrast, LIM Homeobox 9 (LHX9) activates PKM2’s catalytic function, driving metabolic reprogramming and malignant phenotypes in gastric cancer stem cells, effects that can be reversed by LHX9 knockdown (64, 65). Additionally, cytoplasmic PKM2 exhibits non-canonical functions; reduced PKM2 expression may weaken PI3K-Akt-mTOR signaling, activating autophagy and reducing the migratory capacity of gastric cancer cells (66, 67). Overexpression of miR-let-7a suppresses proliferation, migration, and invasion of gastric cancer cells by downregulating PKM2, further illustrating the context-dependent regulation of PKM2’s oncogenic potential (68) (Figure 4).
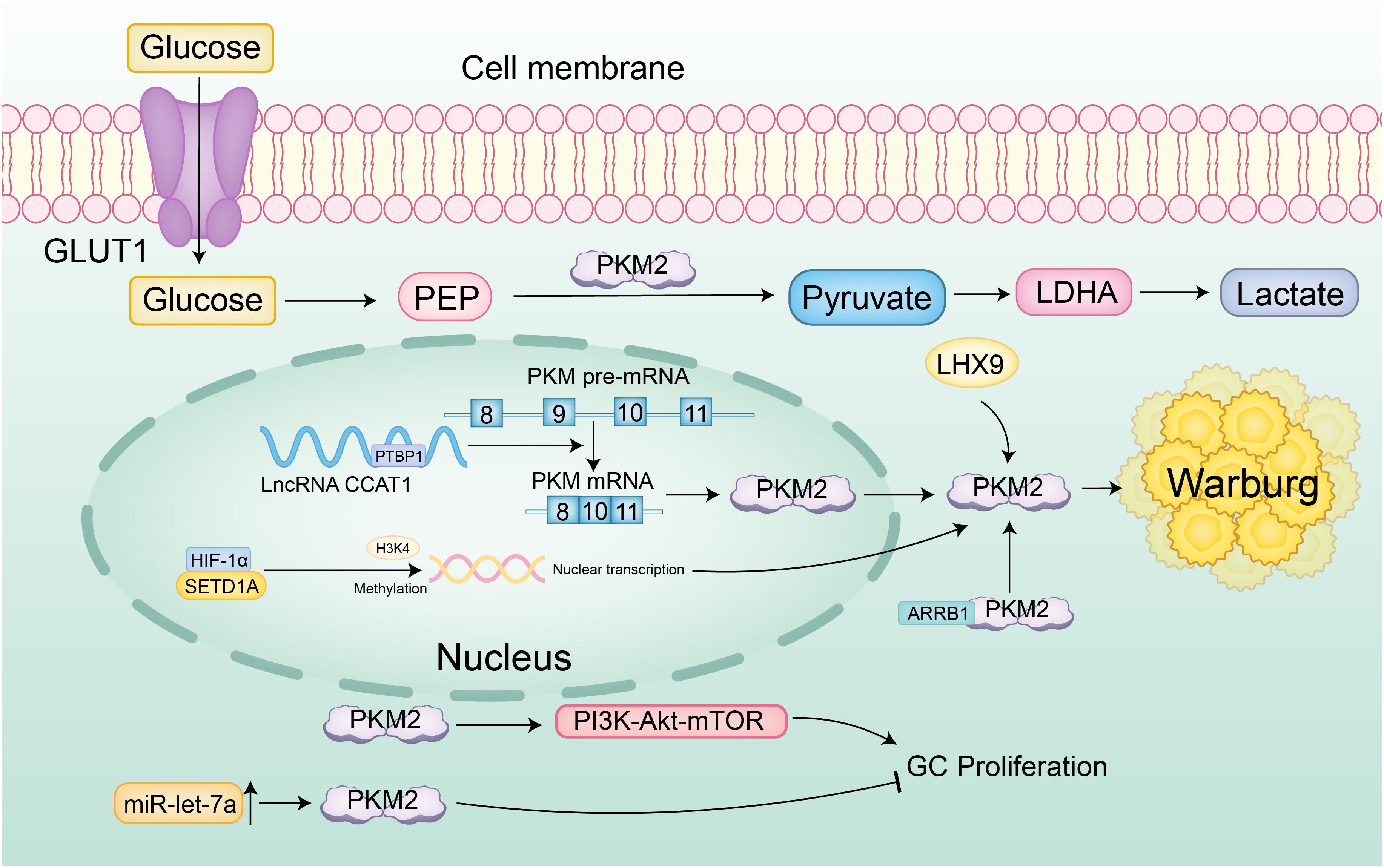
Figure 4. Specific regulatory network of PKM2 in gastric cancer. Glucose is transported into cells via GLUT1 and converted to pyruvate by PKM2, which is then metabolized to lactate, a hallmark of the Warburg effect. The long non-coding RNA (lncRNA) CCAT1 promotes the alternative splicing of PKM pre-mRNA towards the low-activity PKM2 isoform by stabilizing PTBP1, resulting in the accumulation of dimeric PKM2 and reduced enzymatic activity. Additionally, SETD1A enhances HIF-1α recruitment to the PKM2 promoter through H3K4 methylation, forming a positive feedback loop that persistently amplifies aerobic glycolysis. β-Arrestin 1 (ARRB1) binds PKM2, inhibiting its tetramer assembly and maintaining a low-activity dimeric state that promotes the Warburg effect. LIM Homeobox 9 (LHX9) activates PKM2’s catalytic function, driving metabolic reprogramming and malignant phenotypes in gastric cancer stem cells. Reduced PKM2 expression may weaken PI3K-Akt-mTOR signaling, activating autophagy and reducing the migratory capacity of gastric cancer cells. Furthermore, overexpression of miR-let-7a suppresses proliferation, migration, and invasion of gastric cancer cells by downregulating PKM2, highlighting the context-dependent regulation of PKM2’s oncogenic potential.
4.3 Colorectal cancer
In colorectal cancer, PKM2 plays a prominent cancer-promoting role by enhancing glycolytic flux and supporting tumor progression. The deubiquitinase OTUB2 directly binds to PKM2, preventing its interaction with the E3 ubiquitin ligase Parkin, thereby stabilizing PKM2 and enhancing its enzymatic activity by blocking ubiquitination and degradation. Activated PKM2 promotes aerobic glycolysis, markedly increasing glucose consumption and lactate production, particularly under glucose-starved conditions, where this pathway reinforces tumor cell dependence on aerobic glycolysis (51). At the transcriptional level, the 53-amino-acid peptide encoded by HOXB-AS3 binds to the RGG motif of hnRNP A1, preventing its interaction with PKM exon 9 and thereby inhibiting the splicing of PKM into the PKM2 isoform, reducing PKM2 levels and suppressing glucose metabolic reprogramming (69). In early tumorigenesis, PKM2 is already overexpressed and cooperates with HIF-1α and GLUT1 to activate the glycolytic program, establishing an early Warburg effect axis that provides metabolic advantages to emerging tumor cells (70). Epigenetically, PRL-3 promotes primary tumor proliferation and metastatic capacity by upregulating PKM2 and glycolytic enzymes such as Glut1, HK2, and LDHA, collectively enhancing glucose uptake and lactate production (71).
The functional output of PKM2 in colorectal cancer also exhibits notable context-dependent effects. The tumor suppressor NDRG2 inhibits metabolic reprogramming through a dual mechanism: it directly reduces PKM2 expression, limiting pyruvate production, while concurrently inhibiting c-Myc transcriptional activity, which in turn suppresses GLUT1- and HK2-mediated glucose uptake and phosphorylation (72). In metastasis regulation, activated YAP drives Glut3 expression and recruits PKM2 to synergistically enhance the transcriptional activation of glycolytic genes, promoting tumor invasiveness and stem-like properties in a feed-forward loop (73). However, during liver metastasis, this aerobic glycolysis-driven phenotype is modulated by PKLR, which reprograms glutathione metabolism to maintain redox homeostasis by negatively regulating PKM2 activity. Inhibition of PKLR disrupts this adaptive metabolic balance and significantly impairs the liver colonization ability of colorectal cancer cells (74).
4.4 Pancreatic cancer
At the transcriptional level, the lncRNA MIR210HG enhances glycolytic flux and promotes cancer cell proliferation and invasion by sponging miR-125b-5p to relieve its suppression on PKM2 and HK2. Knockout of MIR210HG reverses this phenotype, confirming the regulatory significance of the MIR210HG/miR-125b-5p/PKM2 axis (75). PKM2 also participates in energy production through atypical lactate metabolic pathways, as indicated by the abnormal upregulation of lactate dehydrogenase-B, reflecting broader metabolic reprogramming (76). In signaling pathway interactions, PKM2 activates the NF-κB/p65 pathway to upregulate HIF-1α expression and transcriptional activity and induces VEGF-A secretion to promote tumor angiogenesis. PKM2 deficiency impairs NF-κB signaling, reduces angiogenesis, and increases apoptosis (77). Functionally, PKM2 contributes to chemoresistance by suppressing p38-mediated phosphorylation of p53 at serine 46 and by inhibiting caspase 3/7 and PARP cleavage in response to gemcitabine treatment (78). In pancreatic ductal adenocarcinoma, PKM2 knockdown markedly reduces proliferation, migration, and tumorigenic potential, supporting its role as a core oncogenic driver (79). In metastasis regulation, PKM2 enhances cancer cell migration by stabilizing PAK2 protein through phosphorylation. Silencing PKM2 accelerates PAK2 degradation, disrupts tumor–stellate cell interactions, and inhibits the epithelial–mesenchymal transition process (80, 81).
The oncogenic functions of PKM2 in pancreatic cancer also demonstrate context-dependent characteristics, particularly under microenvironmental and metabolic constraints. Tumor-associated macrophage-derived TGF-β1 induces nuclear translocation of PKM2 and promotes its interaction with STAT1, which activates the PD-L1 promoter and drives immune checkpoint expression. PKM2 knockdown restores natural killer cell cytotoxicity and reverses immune evasion, indicating that its immunomodulatory effects depend on inflammatory signals within the tumor microenvironment (8). Under metabolic stress conditions, PKM2 sustains the Warburg effect by maintaining glucose uptake and lactate production. Its silencing suppresses aerobic glycolysis and activates caspase-3/7, thereby impairing cell survival and invasive capacity. These findings highlight that PKM2-mediated metabolic advantages are tightly linked to environmental nutrient availability (82).
4.5 Esophageal cancer
At the fundamental metabolic level of esophageal squamous cell carcinoma, ESRRG inhibits the transcriptional expression of PKM2 by directly binding to its promoter, thereby reducing glycolytic activity and blocking cell proliferation. Downregulation of ESRRG expression releases this inhibitory effect, resulting in abnormal PKM2 upregulation and enhanced lactate metabolism, which collectively establish a pro-cancer metabolic phenotype (83). In the context of treatment resistance, high PKM2 expression reduces cisplatin sensitivity by sustaining the activity of the pentose phosphate pathway, and its inhibition disrupts pyruvate kinase function, leading to a surge in ROS levels and imbalance of the NADPH/NADP ratio, thereby reversing chemotherapy resistance (84). In the oxidative stress response, the oncogenic effect of PKM2 is closely shaped by the surrounding regulatory state. Activation of Nrf2 promotes PKM2 oligomerization by inducing its glycosylation modification, driving metabolic reprogramming that supports tumor progression. However, this effect remains dependent on a functional glycolytic program, as specific glycolysis inhibition can effectively block the proliferation of esophageal squamous cell carcinoma cells exhibiting high Nrf2 activity, suggesting that PKM2 functions in a context-dependent manner under redox-sensitive conditions (85).
4.6 Oral squamous cell carcinoma
In oral squamous cell carcinoma, the circadian rhythm gene TIMELESS promotes tumor progression through a SIRT1-mediated metabolic axis. TIMELESS upregulates SIRT1, which activates glycolytic enzymes including PKM2, HK2, LDHA, and GLUT1. This pathway enhances glycolytic activity by increasing glucose uptake and lactate production, ensuring a continuous energy supply under hypoxic conditions and supporting tumor cell proliferation and survival (86) (Table 1).
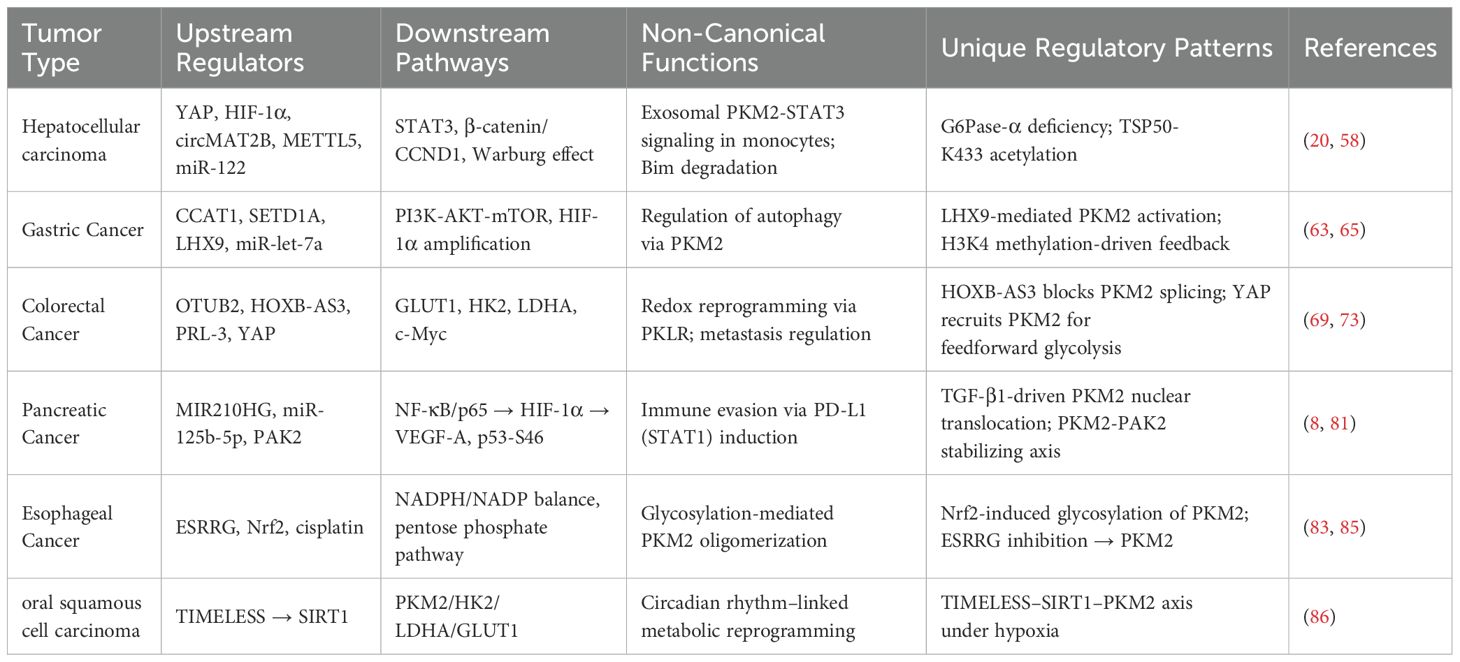
Table 1. Specific regulatory network of PKM2 in digestive system tumors.
5 Innovative therapeutic strategies targeting PKM2
5.1 Small molecule inhibitors and other metabolic interventions
PKM2-specific small molecule inhibitors exert antitumor effects by directly targeting its enzymatic activity or regulating its structural conformation. Isoacteoside binds to the PKM2 active site, inhibits its catalytic function, and synergistically enhances the antitumor efficacy of sorafenib in hepatocellular carcinoma (87). Among natural compounds, euphorbia factor L3 and ellagic acid act as competitive inhibitors, while curcumin and resveratrol function as non-competitive inhibitors by disrupting metabolic complex formation. Ellagic acid demonstrates the strongest anticancer activity among these (88). An irreversible inhibitor, N-(4-(3-(3-(methylamino)-3-oxo-propyl)-5-(4′-(trifluoromethyl)-[1,1’-biphenyl]-4-yl)-1H-pyrazol-1-yl)phenyl)propionamide, covalently binds to PKM2 at Cys326/317, selectively inhibiting its kinase activity and destabilizing the protein, thus suppressing glycolysis without affecting PKM1 (89). Shikonin reverses chemotherapy resistance caused by SIRT1 deficiency and restores oxaliplatin sensitivity in colorectal cancer by targeting PKM2 (90, 91). Tanshinone II.A upregulates miR-122, downregulates PKM2, blocks glycolysis, and induces cell cycle arrest in esophageal cancer (92). In non-small cell lung cancer, casticin and Coenzyme Q0 (CoQ0) suppress HIF-1α signaling, leading to reduced PKM2 expression, inhibition of glucose metabolism, and reversal of the Warburg effect (93, 94). These two mechanisms share similarities with the HIF-1α/PKM2 positive feedback loop observed in liver cancer.
Therapeutic modulation of PKM2 function can also be achieved through structural reprogramming. In gastric cancer, the PKM2 activator DASA-58 overcomes ARRB1-mediated tetramerization inhibition, restoring pyruvate kinase activity and suppressing tumor growth (64). Butyrate promotes PKM2 dephosphorylation and tetramer formation, suppresses the Warburg effect, and alters nucleotide metabolism to restore homeostasis in colorectal cancer (95). Metformin inhibits PKM2 via both AMP-activated protein kinase (AMPK)-dependent and -independent mechanisms, reducing FASN/HK2 expression through c-Myc suppression and directly impairing ATP production (96). Targeting splicing regulators provides an alternative strategy. The HOXB-AS3 peptide blocks hnRNPA1 from binding PKM pre-mRNA, suppressing PKM2 isoform generation (69). Similarly, miRNAs modulate splicing factor activity to promote PKM1-dominant expression, reversing glycolytic phenotypes (97). In hepatocellular carcinoma, ZFP91 promotes hnRNPA1 ubiquitination and inhibits PKM2 splicing (98), while SIRT1/6 inhibitors regulate hnRNPA1 acetylation to control PKM2 expression (99). Several compounds target PKM2-interacting proteins to modulate its activity. TRIM35 inhibits Y105 phosphorylation, thereby suppressing the Warburg effect (100); SULT2B1 inhibitors block the AKT/PKM2 axis to reduce glycolysis (101); and PRDX2 inhibitors prevent PKM2 nuclear translocation, attenuating STAT3 signaling activation (102). PKM2 agonists that bind allosteric pockets distal from the FBP site divert metabolic intermediates away from serine biosynthesis, indirectly promoting serine generation to support tumor proliferation (103) (Tables 2, 3).
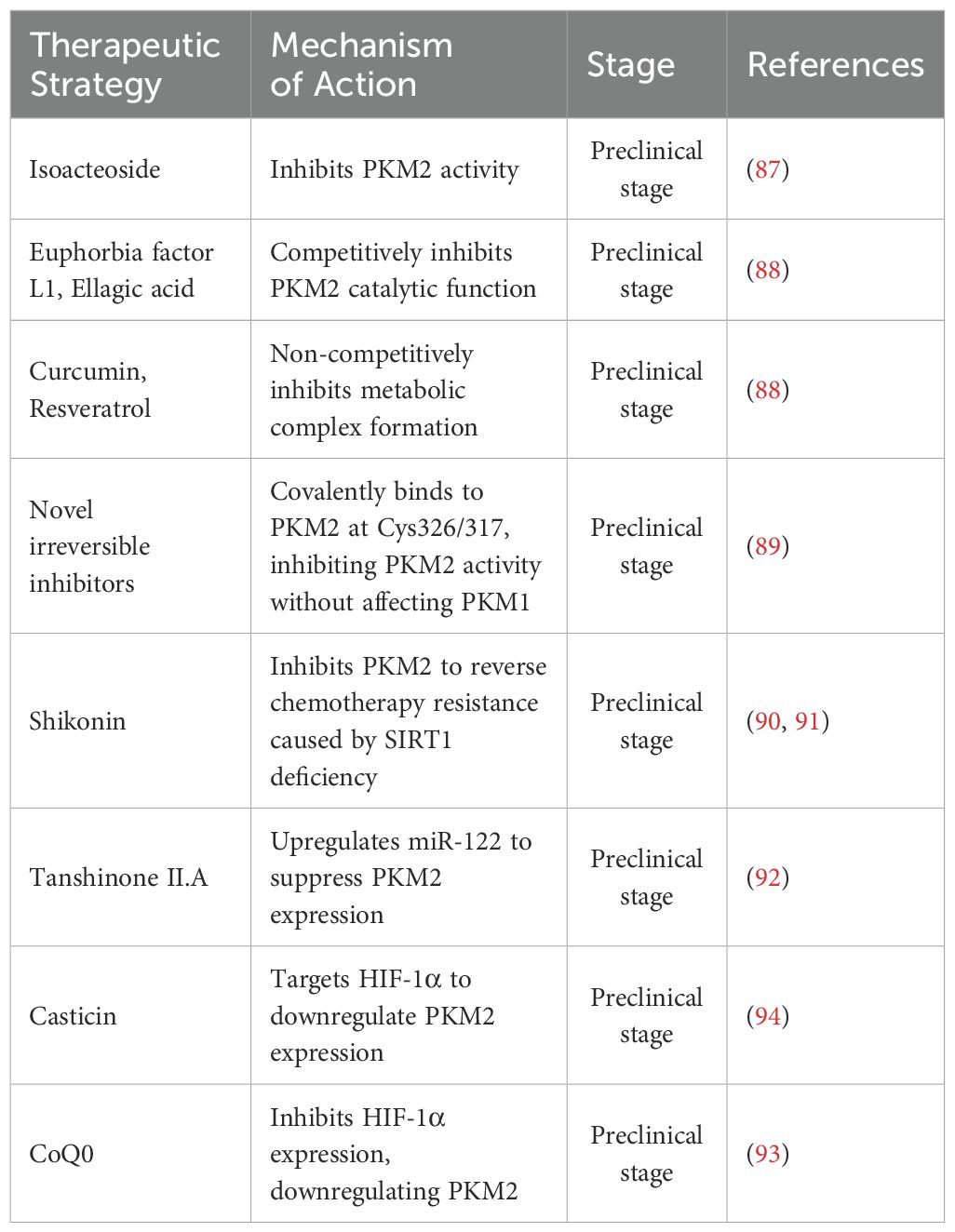
Table 2. Small molecule inhibitors.
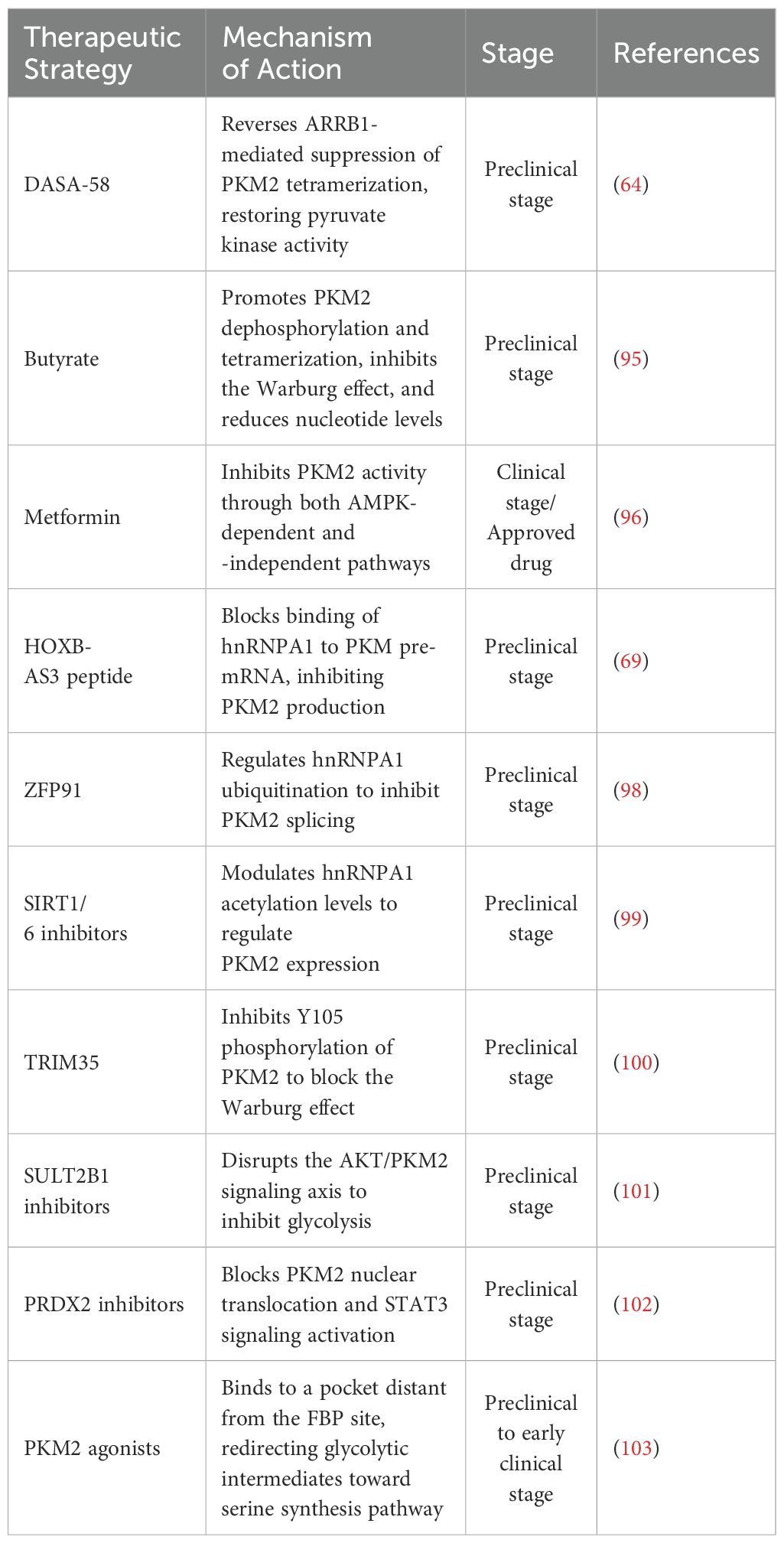
Table 3. Other metabolic interventions.
5.2 Combination therapeutic strategies
Combination therapies targeting PKM2 alongside other metabolic or signaling pathways have shown enhanced efficacy. In pancreatic cancer, inhibition of mitochondrial uncoupling protein 2 (UCP2) by genipin enhances the efficacy of 2-deoxyglucose (2-DG), suggesting that co-targeting the UCP2–PKM2 metabolic axis disrupts mitochondrial bioenergetics and glycolysis simultaneously (104). In 5-fluorouracil (5-FU)-resistant colorectal cancer, suppression of PKM2 leads to upregulation of pyruvate kinase M1 (PKM1) and impairment of the pentose phosphate pathway (PPP), resulting in decreased nicotinamide adenine dinucleotide phosphate (NADPH) production and weakened antioxidant defenses. Combined inhibition of OXPHOS further blocks energy compensation, suppresses cancer stemness, and restores drug sensitivity (105). Targeting redox balance through dual metabolic inhibition is also effective. The glyoxalase I (GLO I) inhibitor TLSC702 increases cellular respiratory dependence, while shikonin inhibits PKM2 activity. This combination induces methylglyoxal accumulation, ATP depletion, and apoptosis, effectively blocking the glycolysis–OXPHOS metabolic switch (106). In colorectal cancer, combined inhibition of estrogen signaling and PKM2 reduces glucose uptake, increases reactive oxygen species (ROS) levels, and triggers apoptotic cell death (107). Other combinations interfere with PKM2-dependent transcriptional signaling. DASA-58 promotes the tetrameric conformation of PKM2, thereby preventing its nuclear translocation. When used with metformin, an OXPHOS inhibitor, this approach disrupts metastasis driven by cancer-associated fibroblasts (CAFs) and targets both glycolytic flux and mitochondrial respiration (108). In tumors retaining wild-type tumor protein p53 (TP53), activation of circular RNA FRMD4A (circFRMD4A) suppresses PKM2 expression. Co-treatment with the copper ionophore elesclomol induces copper-dependent cell death (cuproptosis), offering a mechanism-based strategy for overcoming chemotherapy resistance in digestive system tumors (109).
Although no clinical trials directly targeting PKM2 have yet advanced to the registration stage in digestive system tumors, multiple preliminary mechanistic studies and animal model validations have provided a solid foundation for subsequent human trial design. This warrants further exploration and advancement across various digestive system malignancies in future research (Table 4).
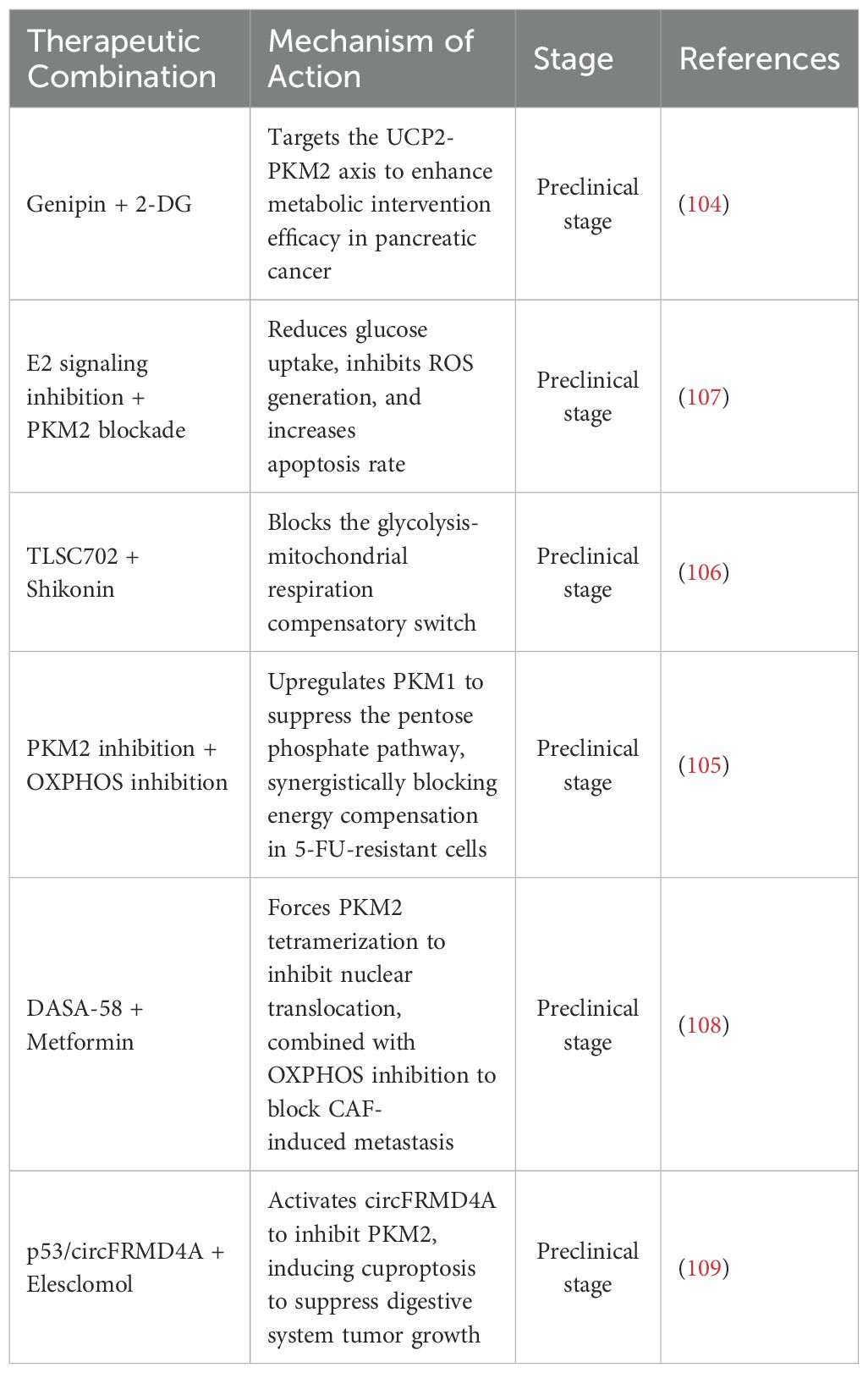
Table 4. Combination therapy strategies.
5.3 Clinical translation challenges
The clinical translation of PKM2-targeted therapies faces several multilayered challenges spanning biological, pharmacological, and regulatory domains. At the mechanistic level, the dual roles of PKM2 in cancer and immune regulation complicate therapeutic development. Beyond its function in tumor metabolism, PKM2 modulates TCF1+ CD8+ T cell activity via the PKM2–pentose phosphate pathway (PPP) axis. This immunometabolic crosstalk necessitates careful therapeutic design to balance tumor suppression with the preservation of immune effector function (110). The tumor microenvironment further complicates therapeutic outcomes. In pancreatic cancer, PKM2 inhibition under glucose-limited conditions paradoxically enhances cell survival, suggesting that nutrient availability can reprogram cellular responses to PKM2-targeted therapies (111). The tumor microenvironment further complicates therapeutic outcomes. In pancreatic cancer, PKM2 inhibition under glucose-limited conditions paradoxically enhances cell survival, suggesting that nutrient availability can reprogram cellular responses to PKM2-targeted therapies. Therefore, metabolic context must be considered to prevent adaptive resistance (112). Diagnostic-therapeutic integration also remains limited. The PKM2-targeted PET tracer Fluorine F 18 DASA-23 shows potential as an activator in glioblastoma imaging; however, its restricted blood-brain barrier permeability limits both diagnostic sensitivity and potential therapeutic extension (113).In conclusion, these challenges emphasize the need for context-dependent intervention strategies, rigorous preclinical validation, and early-phase clinical trial designs that integrate tumor metabolism, immune modulation, and pharmacodynamics into comprehensive evaluation frameworks.
Tumor cells often exploit metabolic plasticity to evade therapeutic interventions targeting PKM2. In glucose-limited microenvironments, digestive system tumors may switch from glycolysis to mitochondrial OXPHOS or FAO, thereby diminishing the efficacy of PKM2 inhibitors and paradoxically enhancing cell survival (43). For instance, in 5-FU-resistant colorectal cancer, PKM2 suppression leads to upregulation of PKM1 and impairment of the pentose phosphate pathway, which reduces NADPH and weakens redox defenses. However, cells compensate via increased OXPHOS dependency, a vulnerability that can be exploited through combined PKM2 and mitochondrial inhibition (64). Similarly, shikonin-mediated PKM2 inhibition induces glycolytic collapse, but cancer cells activate mitochondrial respiration unless this pathway is simultaneously blocked by agents such as TLSC702 (64). In cisplatin-resistant tumors, PKM2 inhibition activates CPT1A-dependent FAO, maintaining ATP production and conferring chemoresistance, which is reversible by dual-targeting metabolic regulators (44). These insights underscore the importance of developing therapeutic strategies that account for metabolic compensation by concurrently targeting glycolytic and compensatory energy pathways to circumvent resistance.
PKM2 plays a paradoxical role in immune regulation by both promoting tumor immune evasion and influencing T cell fate. On the one hand, nuclear PKM2 upregulates PD-L1 expression through STAT3 phosphorylation and remodels chromatin accessibility via HDAC3 recruitment, contributing to immunosuppressive tumor microenvironments (4). Lactate accumulation further reinforces this state by impairing T cell function and sustaining regulatory macrophage phenotypes (4). On the other hand, PKM2 also modulates CD8+ T cell differentiation. Its deficiency activates the pentose phosphate pathway and promotes the expansion of TCF1+ progenitor CD8+ T cells, which are essential for durable responses to immune checkpoint blockade (110). This duality presents a therapeutic dilemma: while PKM2 inhibition may benefit anti-tumor immunity via T cell reprogramming, it might concurrently impair metabolic homeostasis or drive adaptive resistance in the tumor. Additionally, agents such as PRDX2 inhibitors selectively block PKM2 nuclear translocation without abolishing its cytosolic functions, while preserving glycolytic support for T cells (102).
6 Conclusion
PKM2 functions as a central regulator of tumor metabolic reprogramming, orchestrating glycolysis, lipid synthesis, and amino acid metabolism to promote cancer cell proliferation, metastasis, and therapeutic resistance. Its regulatory influence spans the full spectrum of tumor initiation and progression, particularly in digestive system malignancies.
In carbohydrate metabolism, PKM2 inhibits mitochondrial oxidative phosphorylation via dynamic structural transitions and reinforces aerobic glycolysis by interacting with HIF-1α and c-Myc, thereby amplifying the Warburg effect (9, 13, 25). In lipid metabolism, it promotes de novo fatty acid synthesis by activating FASN through SUMOylation, contributing to tumor vascularization (28, 41). Within amino acid networks, PKM2 enhances serine biosynthesis by assembling enzyme complexes, supporting anabolic demands during rapid tumor growth (12, 47). Beyond metabolic regulation, PKM2 also establishes immunosuppressive microenvironments via STAT3 phosphorylation and HDAC3 recruitment, and mediates resistance to chemotherapy (4). These multifaceted roles identify PKM2 as a promising therapeutic target, particularly in tumors characterized by high metabolic plasticity. For instance, EZH2- and PKM2-mediated co-silencing of SLC16A9 in triple-negative breast cancer (TNBC) induces a metabolic shift from glycolysis to fatty acid oxidation, highlighting potential for synthetic lethality via dual-targeting strategies (114).
Although PKM2 is widely recognized for its oncogenic functions in digestive system malignancies, recent findings in other tumor types suggest that PKM2 may exhibit context-dependent tumor-suppressive roles. Notably, in head and neck squamous cell carcinoma, PKM2 has been reported to exert tumor-inhibitory effects, highlighting a bidirectional regulatory capacity (115, 116). This contrasts with the predominantly pro-tumorigenic role described in hepatocellular carcinoma, gastric cancer, colorectal cancer, and pancreatic cancer within the present review. Nevertheless, no direct experimental evidence within current digestive system tumor studies confirms comparable tumor-suppressive functions. Some observations imply inhibitory roles under particular modifications, but these findings are isolated and lack comprehensive mechanistic validation. For example, in HCC, ULK1-mediated phosphorylation at Ser333 enhances PKM2 enzymatic activity, limits its nuclear localization, and suppresses c-Myc expression, collectively attenuating the Warburg effect (61). In contrast, substantial evidence supports PKM2-mediated metabolic reprogramming, immune evasion, and drug resistance through STAT3 activation, lactate accumulation, and PD-L1 upregulation in gastrointestinal malignancies (4, 8, 13, 14). This suggests a predominantly pro-tumorigenic role in these tissues. The absence of confirmed suppressive functions may reflect tissue-specific regulatory inputs, differences in upstream signaling, or distinct metabolic dependencies. Future research should investigate whether PKM2 exhibits functional plasticity in digestive cancers through isoform-specific regulation, nutrient stress responses, and interactions with immune checkpoints or tumor suppressors (61, 110, 116).
Despite its multifaceted regulatory potential in metabolic reprogramming, current PKM2 research still faces limitations. Most mechanistic studies focus on PKM2’s role as an enzyme or kinase, while its function as a protein interaction hub involved in epigenetic regulation and RNA splicing remains underexplored, restricting an integrated understanding of its role in metabolic phenotype transitions. Clinically, PKM2’s dual regulatory properties may suppress T-cell function when inhibited, potentially leading to immune tolerance; thus, the therapeutic window and dosing control need further clarification (110). Additionally, dynamic nutrient fluctuations within the tumor microenvironment may trigger compensatory metabolic responses. For example, under low-glucose conditions, PKM2 inhibition paradoxically promotes cell survival, indicating that PKM2-targeted strategies must be evaluated in the context of microenvironmental status (111).
In response to these challenges, this paper systematically outlines the PKM2-mediated networks of glycolysis, lipid synthesis, and amino acid metabolism, summarizing its tissue-specific regulatory mechanisms across various digestive system tumors. These insights lay the groundwork for precision-targeted therapies and call for future research into PKM2’s context-specific roles, epigenetic functions, and immunological consequences.
Building on this foundation, a key area that warrants further investigation is the development of isoform-specific therapeutic strategies. Current inhibitors rarely distinguish between PKM2 and PKM1, risking disruption of physiological pyruvate flux and energy homeostasis in normal cells (4, 89). Given that PKM2-specific functions are largely dictated by alternative splicing and post-translational modifications, future research should prioritize the design of agents that modulate splicing regulators such as hnRNPA1 or lncRNAs including HOXB-AS3 to selectively suppress PKM2 isoform expression without impairing PKM1 activity (38, 69). Meanwhile, the dynamic interconversion between PKM2 tetramers and dimers offers an additional therapeutic axis that remains underutilized in drug development.
In parallel, the immunological implications of PKM2-targeted therapies demand careful re-evaluation. PKM2 not only drives tumor-intrinsic immune evasion by enhancing PD-L1 transcription and facilitating lactate accumulation but also regulates the metabolic programming of T cells, especially CD8+ TCF1+ subsets, through the pentose phosphate pathway (4, 8, 110). These dual and potentially opposing roles complicate the integration of PKM2 inhibition into immunotherapy regimens. Whether combinatorial strategies involving immune checkpoint blockade and PKM2 suppression would synergize or antagonize in gastrointestinal malignancies is currently unknown. Furthermore, how PKM2 impacts myeloid-derived suppressor cells, tumor-associated macrophages, or regulatory T cells within the digestive tumor microenvironment remains insufficiently characterized and warrants systematic exploration.
Beyond its metabolic and immunoregulatory capacities, PKM2 may also serve as a scaffold for chromatin and RNA regulatory complexes, functioning in ways that transcend enzymatic activity. Its reported interactions with HDAC3, PRMT6, and circular RNAs such as circMAT2B suggest that PKM2 plays a role in shaping epigenetic landscapes and transcriptomic plasticity under oncogenic stress (7, 19, 53). However, the spatial-temporal dynamics of these interactions, and their integration with metabolic cues such as nutrient depletion, hypoxia, or ROS accumulation, are not well defined. Future investigations into PKM2’s role in chromatin accessibility, alternative splicing, and long-range transcriptional regulation may yield novel insights into its non-canonical oncogenic functions.
Finally, the metabolic heterogeneity of the tumor microenvironment introduces additional complexity into PKM2-directed strategies. In glucose-limited conditions, digestive system tumor cells may shift toward lipid oxidation or glutamine catabolism, diminishing the efficacy of glycolysis-targeting agents and, paradoxically, rendering PKM2 inhibition survival-promoting (43, 82). Such context-dependent adaptations highlight the need for real-time metabolic profiling and companion diagnostics to stratify responsive tumor subsets. PKM2-targeted PET tracers or metabolomic signatures reflecting Warburg activity may aid in predicting treatment efficacy and guiding dosing (113).
In conclusion, as a central hub in tumor metabolic reprogramming, future research on PKM2 should focus on isoform-specific regulation, epigenetic functions, and immune modulation mechanisms. Beyond its metabolic roles, PKM2 participates in protein interactions, chromatin remodeling, and RNA splicing, necessitating deeper investigation of its spatial-temporal dynamics to uncover noncanonical oncogenic mechanisms. Designing precise inhibitors targeting PKM2 isoforms and modulating alternative splicing factors and post-translational modifications will enhance therapeutic specificity while minimizing effects on normal metabolism. The metabolic heterogeneity of the tumor microenvironment drives adaptive energy pathway shifts under nutrient stress, highlighting the need to integrate metabolomics and companion diagnostics for dynamic monitoring and patient stratification toward personalized therapies. Additionally, the dual role of PKM2 in immune cell metabolism and its interplay with immune checkpoint regulation calls for combinational strategies that pair immunotherapy with PKM2 inhibition. Integrating metabolic, immune, and epigenetic regulatory axes will offer novel insights and accelerate clinical translation of PKM2-targeted therapies in digestive system cancers, opening new frontiers for anticancer treatment.
Author contributions
XH: Writing – review & editing, Writing – original draft. JH: Writing – original draft, Writing – review & editing. HS: Writing – review & editing, Writing – original draft. YW: Writing – original draft, Writing – review & editing. RG: Writing – original draft, Writing – review & editing. ZL: Project administration, Writing – review & editing, Writing – original draft.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. 1) The first batch of the “Double Hundred Plan” (Academic Experience Inheritance Project for Famous Traditional Chinese Medicine Experts) of Nanjing University of Traditional Chinese Medicine (Nan Zhong Hua Da Ren Zi (2024) No. 35). 2) 75th Batch of General Projects of China Postdoctoral Science Foundation (2024M754279). 3) Natural Science Foundation of Jiangsu Province (BK20240738); 4) General Project of Basic Science (Natural Science) Research in Higher Education Institutions in Jiangsu Province (24KJB36004) 5) Jiangsu Province Traditional Chinese Medicine Science and Technology Development Plan Youth Talent Project (QN202206).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Nenkov M, Ma Y, Gaßler N, and Chen Y. Metabolic reprogramming of colorectal cancer cells and the microenvironment: implication for therapy. Int J Mol Sci. (2021) 22:6262. doi: 10.3390/ijms22126262
2. Pavlova NN and Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metab. (2016) 23:27–47. doi: 10.1016/j.cmet.2015.12.006
3. Wang X, Zhou L, Wang H, Chen W, Jiang L, Ming G, et al. Metabolic reprogramming, autophagy, and ferroptosis: Novel arsenals to overcome immunotherapy resistance in gastrointestinal cancer. Cancer Med. (2023) 12:20573–89. doi: 10.1002/cam4.6623
4. Zhu S, Guo Y, Zhang X, Liu H, Yin M, Chen X, et al. Pyruvate kinase M2 (PKM2) in cancer and cancer therapeutics. Cancer Lett. (2021) 503:240–8. doi: 10.1016/j.canlet.2020.11.018
5. He X, Du S, Lei T, Li X, Liu Y, Wang H, et al. PKM2 in carcinogenesis and oncotherapy. Oncotarget. (2017) 8:110656–70. doi: 10.18632/oncotarget.22529
6. Chunlian Z, Qi W, and Rui Z. The role of pyruvate kinase M2 posttranslational modification in the occurrence and development of hepatocellular carcinoma. Cell Biochem Funct. (2024) 42:e4125. doi: 10.1002/cbf.4125
7. Hou P-P, Luo L-J, Chen H-Z, Chen Q-T, Bian X-L, Wu S-F, et al. Ectosomal PKM2 promotes HCC by inducing macrophage differentiation and remodeling the tumor microenvironment. Mol Cell. (2020) 78:1192–1206.e10. doi: 10.1016/j.molcel.2020.05.004
8. Xia Q, Jia J, Hu C, Lu J, Li J, Xu H, et al. Tumor-associated macrophages promote PD-L1 expression in tumor cells by regulating PKM2 nuclear translocation in pancreatic ductal adenocarcinoma. Oncogene. (2022) 41:865–77. doi: 10.1038/s41388-021-02133-5
9. Stone OA, El-Brolosy M, Wilhelm K, Liu X, Romão AM, Grillo E, et al. Loss of pyruvate kinase M2 limits growth and triggers innate immune signaling in endothelial cells. Nat Commun. (2018) 9:4077. doi: 10.1038/s41467-018-06406-8
10. Li T, Han J, Jia L, Hu X, Chen L, and Wang Y. PKM2 coordinates glycolysis with mitochondrial fusion and oxidative phosphorylation. Protein Cell. (2019) 10:583–94. doi: 10.1007/s13238-019-0618-z
11. Yu W, Yang Z, Huang R, Min Z, and Ye M. SIRT6 promotes the Warburg effect of papillary thyroid cancer cell BCPAP through reactive oxygen species. Onco Targets Ther. (2019) 12:2861–8. doi: 10.2147/OTT.S194256
12. Chaneton B, Hillmann P, Zheng L, Martin ACL, Maddocks ODK, Chokkathukalam A, et al. Serine is a natural ligand and allosteric activator of pyruvate kinase M2. Nature. (2012) 491:458–62. doi: 10.1038/nature11540
13. Dai H, Zeng W, and Luo H. C-MET-dependent signal transduction mediates retinoblastoma growth by regulating PKM2 nuclear translocation. Cell Biochem Funct. (2020) 38:204–12. doi: 10.1002/cbf.3464
14. Angiari S, Runtsch MC, Sutton CE, Palsson-McDermott EM, Kelly B, Rana N, et al. Pharmacological activation of pyruvate kinase M2 inhibits CD4+ T cell pathogenicity and suppresses autoimmunity. Cell Metab. (2020) 31:391–405.e8. doi: 10.1016/j.cmet.2019.10.015
15. Zhang X, Li Y, Ma Y, Yang L, Wang T, Meng X, et al. Yes-associated protein (YAP) binds to HIF-1α and sustains HIF-1α protein stability to promote hepatocellular carcinoma cell glycolysis under hypoxic stress. J Exp Clin Cancer Research : CR. (2018) 37:216. doi: 10.1186/s13046-018-0892-2
16. Yao G, Yin J, Wang Q, Dong R, and Lu J. Glypican-3 enhances reprogramming of glucose metabolism in liver cancer cells. BioMed Res Int. (2019) 2019:2560650. doi: 10.1155/2019/2560650
17. Qu H, Liu J, Zhang D, Xie R, Wang L, and Hong J. Glycolysis in chronic liver diseases: mechanistic insights and therapeutic opportunities. Cells. (2023) 12:1930. doi: 10.3390/cells12151930
18. Xu Q, Tu J, Dou C, Zhang J, Yang L, Liu X, et al. HSP90 promotes cell glycolysis, proliferation and inhibits apoptosis by regulating PKM2 abundance via Thr-328 phosphorylation in hepatocellular carcinoma. Mol Cancer. (2017) 16:178. doi: 10.1186/s12943-017-0748-y
19. Lan T, Gao F, Cai Y, Lv Y, Zhu J, Liu H, et al. The protein circPETH-147aa regulates metabolic reprogramming in hepatocellular carcinoma cells to remodel immunosuppressive microenvironment. Nat Commun. (2025) 16:333. doi: 10.1038/s41467-024-55577-0
20. Gao F, Zhang X, Wang S, Zheng L, Sun Y, Wang G, et al. TSP50 promotes the Warburg effect and hepatocyte proliferation via regulating PKM2 acetylation. Cell Death Dis. (2021) 12:517. doi: 10.1038/s41419-021-03782-w
21. Hu W, Lu S-X, Li M, Zhang C, Liu L-L, Fu J, et al. Pyruvate kinase M2 prevents apoptosis via modulating Bim stability and associates with poor outcome in hepatocellular carcinoma. Oncotarget. (2015) 6:6570–83. doi: 10.18632/oncotarget.3262
22. Fukushi A, Kim H-D, Chang Y-C, and Kim C-H. Revisited metabolic control and reprogramming cancers by means of the warburg effect in tumor cells. Int J Mol Sci. (2022) 23:10037. doi: 10.3390/ijms231710037
23. Puckett DL, Alquraishi M, Chowanadisai W, and Bettaieb A. The role of PKM2 in metabolic reprogramming: insights into the regulatory roles of non-coding RNAs. Int J Mol Sci. (2021) 22:1171. doi: 10.3390/ijms22031171
24. Wang Y, Xu N, Ndzie Noah ML, Chen L, and Zhan X. Pyruvate kinase M1/2 proteoformics for accurate insights into energy metabolism abnormity to promote the overall management of ovarian cancer towards predictive, preventive, and personalized medicine approaches. Metabolites. (2025) 15:203. doi: 10.3390/metabo15030203
25. Luo W, Hu H, Chang R, Zhong J, Knabel M, O’Meally R, et al. Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell. (2011) 145:732–44. doi: 10.1016/j.cell.2011.03.054
26. Wang Y, Shu H, Qu Y, Jin X, Liu J, Peng W, et al. PKM2 functions as a histidine kinase to phosphorylate PGAM1 and increase glycolysis shunts in cancer. EMBO J. (2024) 43:2368–96. doi: 10.1038/s44318-024-00110-8
27. Fontana F, Giannitti G, Marchesi S, and Limonta P. The PI3K/akt pathway and glucose metabolism: A dangerous liaison in cancer. Int J Biol Sci. (2024) 20:3113–25. doi: 10.7150/ijbs.89942
28. Zhang J, Ouyang F, Gao A, Zeng T, Li M, Li H, et al. ESM1 enhances fatty acid synthesis and vascular mimicry in ovarian cancer by utilizing the PKM2-dependent warburg effect within the hypoxic tumor microenvironment. Mol Cancer. (2024) 23:94. doi: 10.1186/s12943-024-02009-8
29. Jing Y-Y, Cai F-F, Zhang L, Han J, Yang L, Tang F, et al. Epigenetic regulation of the Warburg effect by H2B monoubiquitination. Cell Death Differ. (2020) 27:1660–76. doi: 10.1038/s41418-019-0450-2
30. Wu H, Jiao Y, Guo X, Wu Z, and Lv Q. METTL14/miR-29c-3p axis drives aerobic glycolysis to promote triple-negative breast cancer progression though TRIM9-mediated PKM2 ubiquitination. J Cell Mol Med. (2024) 28:e18112. doi: 10.1111/jcmm.18112
31. Hou J-Y, Wang X-L, Chang H-J, Wang X-X, Hao S-L, Gao Y, et al. PTBP1 crotonylation promotes colorectal cancer progression through alternative splicing-mediated upregulation of the PKM2 gene. J Transl Med. (2024) 22:995. doi: 10.1186/s12967-024-05793-5
32. Liu F, Ma F, Wang Y, Hao L, Zeng H, Jia C, et al. PKM2 methylation by CARM1 activates aerobic glycolysis to promote tumorigenesis. Nat Cell Biol. (2017) 19:1358–70. doi: 10.1038/ncb3630
33. Yao Y, Chen X, Wang X, Li H, Zhu Y, Li X, et al. Glycolysis related lncRNA SNHG3/miR-139-5p/PKM2 axis promotes castration-resistant prostate cancer (CRPC) development and enzalutamide resistance. Int J Biol Macromol. (2024) 260:129635. doi: 10.1016/j.ijbiomac.2024.129635
34. Huang Y, Chen Z, Lu T, Bi G, Li M, Liang J, et al. HIF-1α switches the functionality of TGF-β signaling via changing the partners of smads to drive glucose metabolic reprogramming in non-small cell lung cancer. J Exp Clin Cancer Res. (2021) 40:398. doi: 10.1186/s13046-021-02188-y
35. Hua Q, Mi B, Xu F, Wen J, Zhao L, Liu J, et al. Hypoxia-induced lncRNA-AC020978 promotes proliferation and glycolytic metabolism of non-small cell lung cancer by regulating PKM2/HIF-1α axis. Theranostics. (2020) 10:4762–78. doi: 10.7150/thno.43839
36. Arundhathi JRD, Mathur SR, Gogia A, Deo SVS, Mohapatra P, and Prasad CP. Metabolic changes in triple negative breast cancer-focus on aerobic glycolysis. Mol Biol Rep. (2021) 48:4733–45. doi: 10.1007/s11033-021-06414-w
37. Wang Y, Shu H, Liu J, Jin X, Wang L, Qu Y, et al. EGF promotes PKM2 O-GlcNAcylation by stimulating O-GlcNAc transferase phosphorylation at Y976 and their subsequent association. J Biol Chem. (2022) 298:102340. doi: 10.1016/j.jbc.2022.102340
38. Li Y, Zhang S, Li Y, Liu J, Li Q, Zang W, et al. The regulatory network of hnRNPs underlying regulating PKM alternative splicing in tumor progression. Biomolecules. (2024) 14:566. doi: 10.3390/biom14050566
39. Sun Y, Luo M, Chang G, Ren W, Wu K, Li X, et al. Phosphorylation of Ser6 in hnRNPA1 by S6K2 regulates glucose metabolism and cell growth in colorectal cancer. Oncol Lett. (2017) 14:7323–31. doi: 10.3892/ol.2017.7085
40. Lan Z, Yao X, Sun K, Li A, Liu S, and Wang X. The Interaction Between lncRNA SNHG6 and hnRNPA1 Contributes to the Growth of Colorectal Cancer by Enhancing Aerobic Glycolysis Through the Regulation of Alternative Splicing of PKM. Front Oncol. (2020) 10:363. doi: 10.3389/fonc.2020.00363
41. Archid R, Solass W, Tempfer C, Königsrainer A, Adolph M, Reymond MA, et al. Cachexia anorexia syndrome and associated metabolic dysfunction in peritoneal metastasis. Int J Mol Sci. (2019) 20:5444. doi: 10.3390/ijms20215444
42. Gjorgjieva M, Calderaro J, Monteillet L, Silva M, Raffin M, Brevet M, et al. Dietary exacerbation of metabolic stress leads to accelerated hepatic carcinogenesis in glycogen storage disease type Ia. J Hepatol. (2018) 69:1074–87. doi: 10.1016/j.jhep.2018.07.017
43. Zhao Q-Y, Liu W-J, Wang J-G, Li H, Lv J-L, Wang Y, et al. Increasing cisplatin exposure promotes small-cell lung cancer transformation after a shift from glucose metabolism to fatty acid metabolism. J Cancer Res Clin Oncol. (2025) 151:126. doi: 10.1007/s00432-025-06164-3
44. Wang Y-N, Zeng Z-L, Lu J, Wang Y, Liu Z-X, He M-M, et al. CPT1A-mediated fatty acid oxidation promotes colorectal cancer cell metastasis by inhibiting anoikis. Oncogene. (2018) 37:6025–40. doi: 10.1038/s41388-018-0384-z
45. Zhang L, Cheng L, Ma Y, Li J, Zhong Y, Zhu X, et al. PKM2 knockout facilitates the activation of the AMPK/KLF4/ACADVL pathway, leading to increased oxidative degradation of fatty acids in TNBC. Med Oncol. (2025) 42:102. doi: 10.1007/s12032-025-02671-y
46. Jiang C, Qian Y, Bai X, Li S, Zhang L, Xie Y, et al. SLC7A5/E2F1/PTBP1/PKM2 axis mediates progression and therapy effect of triple-negative breast cancer through the crosstalk of amino acid metabolism and glycolysis pathway. Cancer Lett. (2025) 617:217612. doi: 10.1016/j.canlet.2025.217612
47. Zhu Y, Jin L, Shi R, Li J, Wang Y, Zhang L, et al. The long noncoding RNA glycoLINC assembles a lower glycolytic metabolon to promote glycolysis. Mol Cell. (2022) 82:542–554.e6. doi: 10.1016/j.molcel.2021.11.017
48. Anastasiou D. Tumour microenvironment factors shaping the cancer metabolism landscape. Br J Cancer. (2017) 116:277–86. doi: 10.1038/bjc.2016.412
49. Wang L, Li B, Bo X, Yi X, Xiao X, and Zheng Q. Hypoxia-induced LncRNA DACT3-AS1 upregulates PKM2 to promote metastasis in hepatocellular carcinoma through the HDAC2/FOXA3 pathway. Exp Mol Med. (2022) 54:848–60. doi: 10.1038/s12276-022-00767-3
50. Zhang C, Wang H, Liu Q, Dai S, Tian G, Wei X, et al. LncRNA CCAT1 facilitates the progression of gastric cancer via PTBP1-mediated glycolysis enhancement. J Exp Clin Cancer Res. (2023) 42:246. doi: 10.1186/s13046-023-02827-6
51. Yu S, Zang W, Qiu Y, Liao L, and Zheng X. Deubiquitinase OTUB2 exacerbates the progression of colorectal cancer by promoting PKM2 activity and glycolysis. Oncogene. (2022) 41:46–56. doi: 10.1038/s41388-021-02071-2
52. Zhang L-F, Lou J-T, Lu M-H, Gao C, Zhao S, Li B, et al. Suppression of miR-199a maturation by HuR is crucial for hypoxia-induced glycolytic switch in hepatocellular carcinoma. EMBO J. (2015) 34:2671–85. doi: 10.15252/embj.201591803
53. Li Q, Pan X, Zhu D, Deng Z, Jiang R, and Wang X. Circular RNA MAT2B promotes glycolysis and Malignancy of hepatocellular carcinoma through the miR-338-3p/PKM2 axis under hypoxic stress. Hepatology. (2019) 70:1298–316. doi: 10.1002/hep.30671
54. Zhao A, Liu X, Chen X, Na S, Wang H, Peng X, et al. Aqueous extract of rhubarb promotes hepatotoxicity via facilitating PKM2-mediated aerobic glycolysis in a rat model of diethylnitrosamine-induced liver cancer. Drug Des Devel Ther. (2024) 18:4497–510. doi: 10.2147/DDDT.S476273
55. Xia P, Zhang H, Lu H, Xu K, Jiang X, Jiang Y, et al. METTL5 stabilizes c-Myc by facilitating USP5 translation to reprogram glucose metabolism and promote hepatocellular carcinoma progression. Cancer Commun (Lond). (2023) 43:338–64. doi: 10.1002/cac2.12403
56. Tan H-W, Leung CO-N, Chan KK-S, Ho DW-H, Leung M-S, Wong C-M, et al. Deregulated GATA6 modulates stem cell-like properties and metabolic phenotype in hepatocellular carcinoma. Int J Cancer. (2019) 145:1860–73. doi: 10.1002/ijc.32248
57. Liu R, Li Y, Tian L, Shi H, Wang J, Liang Y, et al. Gankyrin drives metabolic reprogramming to promote tumorigenesis, metastasis and drug resistance through activating β-catenin/c-Myc signaling in human hepatocellular carcinoma. Cancer Lett. (2019) 443:34–46. doi: 10.1016/j.canlet.2018.11.030
58. Cho J-H, Kim G-Y, Mansfield BC, and Chou JY. Hepatic glucose-6-phosphatase-α deficiency leads to metabolic reprogramming in glycogen storage disease type Ia. Biochem Biophys Res Commun. (2018) 498:925–31. doi: 10.1016/j.bbrc.2018.03.083
59. Zhang R, Shen M, Wu C, Chen Y, Lu J, Li J, et al. HDAC8-dependent deacetylation of PKM2 directs nuclear localization and glycolysis to promote proliferation in hepatocellular carcinoma. Cell Death Dis. (2020) 11:1036. doi: 10.1038/s41419-020-03212-3
60. Wong CC-L, Au SL-K, Tse AP-W, Xu IM-J, Lai RK-H, Chiu DK-C, et al. Switching of pyruvate kinase isoform L to M2 promotes metabolic reprogramming in hepatocarcinogenesis. PLoS One. (2014) 9:e115036. doi: 10.1371/journal.pone.0115036
61. Zhou Z, Zheng X, Zhao J, Yuan A, Lv Z, Shao G, et al. ULK1-dependent phosphorylation of PKM2 antagonizes O-GlcNAcylation and regulates the Warburg effect in breast cancer. Oncogene. (2024) 43:1769–78. doi: 10.1038/s41388-024-03035-y
62. Wang Q, Lu D, Fan L, Li Y, Liu Y, Yu H, et al. COX-2 induces apoptosis-resistance in hepatocellular carcinoma cells via the HIF-1α/PKM2 pathway. Int J Mol Med. (2019) 43:475–88. doi: 10.3892/ijmm.2018.3936
63. Wu J, Chai H, Xu X, Yu J, and Gu Y. Histone methyltransferase SETD1A interacts with HIF1α to enhance glycolysis and promote cancer progression in gastric cancer. Mol Oncol. (2020) 14:1397–409. doi: 10.1002/1878-0261.12689
64. Yu H, Wang M, Zhang T, Cao L, Li Z, Du Y, et al. Dual roles of β-arrestin 1 in mediating cell metabolism and proliferation in gastric cancer. Proc Natl Acad Sci U.S.A. (2022) 119:e2123231119. doi: 10.1073/pnas.2123231119
65. Zhao H, Jiang R, Feng Z, Wang X, and Zhang C. Transcription factor LHX9 (LIM Homeobox 9) enhances pyruvate kinase PKM2 activity to induce glycolytic metabolic reprogramming in cancer stem cells, promoting gastric cancer progression. J Transl Med. (2023) 21:833. doi: 10.1186/s12967-023-04658-7
66. Wang C, Jiang J, Ji J, Cai Q, Chen X, Yu Y, et al. PKM2 promotes cell migration and inhibits autophagy by mediating PI3K/AKT activation and contributes to the Malignant development of gastric cancer. Sci Rep. (2017) 7:2886. doi: 10.1038/s41598-017-03031-1
67. Liu Y, Zhang Z, Wang J, Chen C, Tang X, Zhu J, et al. Metabolic reprogramming results in abnormal glycolysis in gastric cancer: a review. Onco Targets Ther. (2019) 12:1195–204. doi: 10.2147/OTT.S189687
68. Tang R, Yang C, Ma X, Wang Y, Luo D, Huang C, et al. MiR-let-7a inhibits cell proliferation, migration, and invasion by down-regulating PKM2 in gastric cancer. Oncotarget. (2016) 7:5972–84. doi: 10.18632/oncotarget.6821
69. Huang J-Z, Chen M, Chen D, Gao X-C, Zhu S, Huang H, et al. A peptide encoded by a putative lncRNA HOXB-AS3 suppresses colon cancer growth. Mol Cell. (2017) 68:171–184.e6. doi: 10.1016/j.molcel.2017.09.015
70. Cruz MD, Ledbetter S, Chowdhury S, Tiwari AK, Momi N, Wali RK, et al. Metabolic reprogramming of the premalignant colonic mucosa is an early event in carcinogenesis. Oncotarget. (2017) 8:20543–57. doi: 10.18632/oncotarget.16129
71. Xu H, Zeng Y, Liu L, Gao Q, Jin S, Lan Q, et al. PRL-3 improves colorectal cancer cell proliferation and invasion through IL-8 mediated glycolysis metabolism. Int J Oncol. (2017) 51:1271–9. doi: 10.3892/ijo.2017.4090
72. Xu X, Li J, Sun X, Guo Y, Chu D, Wei L, et al. Tumor suppressor NDRG2 inhibits glycolysis and glutaminolysis in colorectal cancer cells by repressing c-Myc expression. Oncotarget. (2015) 6:26161–76. doi: 10.18632/oncotarget.4544
73. Kuo C-C, Ling H-H, Chiang M-C, Chung C-H, Lee W-Y, Chu C-Y, et al. Metastatic colorectal cancer rewrites metabolic program through a glut3-YAP-dependent signaling circuit. Theranostics. (2019) 9:2526–40. doi: 10.7150/thno.32915
74. Nguyen A, Loo JM, Mital R, Weinberg EM, Man FY, Zeng Z, et al. PKLR promotes colorectal cancer liver colonization through induction of glutathione synthesis. J Clin Invest. (2016) 126:681–94. doi: 10.1172/JCI83587
75. Yu T, Li G, Wang C, Gong G, Wang L, Li C, et al. MIR210HG regulates glycolysis, cell proliferation, and metastasis of pancreatic cancer cells through miR-125b-5p/HK2/PKM2 axis. RNA Biol. (2021) 18:2513–30. doi: 10.1080/15476286.2021.1930755
76. Zhou W, Liotta LA, and Petricoin EF. Cancer metabolism and mass spectrometry-based proteomics. Cancer Lett. (2015) 356:176–83. doi: 10.1016/j.canlet.2013.11.003
77. Azoitei N, Becher A, Steinestel K, Rouhi A, Diepold K, Genze F, et al. PKM2 promotes tumor angiogenesis by regulating HIF-1α through NF-κB activation. Mol Cancer. (2016) 15:3. doi: 10.1186/s12943-015-0490-2
78. Kim DJ, Park YS, Kang MG, You Y-M, Jung Y, Koo H, et al. Pyruvate kinase isoenzyme M2 is a therapeutic target of gemcitabine-resistant pancreatic cancer cells. Exp Cell Res. (2015) 336:119–29. doi: 10.1016/j.yexcr.2015.05.017
79. Yokoyama M, Tanuma N, Shibuya R, Shiroki T, Abue M, Yamamoto K, et al. Pyruvate kinase type M2 contributes to the development of pancreatic ductal adenocarcinoma by regulating the production of metabolites and reactive oxygen species. Int J Oncol. (2018) 52:881–91. doi: 10.3892/ijo.2018.4258
80. Cheng T-Y, Yang Y-C, Wang H-P, Tien Y-W, Shun C-T, Huang H-Y, et al. Pyruvate kinase M2 promotes pancreatic ductal adenocarcinoma invasion and metastasis through phosphorylation and stabilization of PAK2 protein. Oncogene. (2018) 37:1730–42. doi: 10.1038/s41388-017-0086-y
81. Masamune A, Hamada S, Yoshida N, Nabeshima T, and Shimosegawa T. Pyruvate kinase isozyme M2 plays a critical role in the interactions between pancreatic stellate cells and cancer cells. Dig Dis Sci. (2018) 63:1868–77. doi: 10.1007/s10620-018-5051-2
82. Li C, Zhao Z, Zhou Z, and Liu R. PKM2 promotes cell survival and invasion under metabolic stress by enhancing warburg effect in pancreatic ductal adenocarcinoma. Dig Dis Sci. (2016) 61:767–73. doi: 10.1007/s10620-015-3931-2
83. Wang T, Zhu Y, Chen L, Zhang W, Qi H, Shi X, et al. ESRRG-PKM2 axis reprograms metabolism to suppress esophageal squamous carcinoma progression and enhance anti-PD-1 therapy efficacy. J Transl Med. (2023) 21:605. doi: 10.1186/s12967-023-04347-5
84. Fukuda S, Miyata H, Miyazaki Y, Makino T, Takahashi T, Kurokawa Y, et al. Pyruvate kinase M2 modulates esophageal squamous cell carcinoma chemotherapy response by regulating the pentose phosphate pathway. Ann Surg Oncol. (2015) 22 Suppl 3:S1461–1468. doi: 10.1245/s10434-015-4522-3
85. Fu J, Xiong Z, Huang C, Li J, Yang W, Han Y, et al. Hyperactivity of the transcription factor Nrf2 causes metabolic reprogramming in mouse esophagus. J Biol Chem. (2019) 294:327–40. doi: 10.1074/jbc.RA118.005963
86. Chen Y, Han Z, Zhang L, Gao C, Wei J, Yang X, et al. TIMELESS promotes reprogramming of glucose metabolism in oral squamous cell carcinoma. J Transl Med. (2024) 22:21. doi: 10.1186/s12967-023-04791-3
87. Zhao L, Qi H, Liu W, Lv H, Li P, Liu W, et al. Isoacteoside alleviates hepatocellular carcinoma progression by inhibiting PDHB-mediated reprogramming of glucose metabolism. Commun Biol. (2025) 8:205. doi: 10.1038/s42003-025-07622-x
88. Sarfraz I, Rasul A, Jabeen F, Sultana T, and Adem Ş. Identification of natural compounds as inhibitors of pyruvate kinase M2 for cancer treatment. Molecules. (2022) 27:7113. doi: 10.3390/molecules27207113
89. Hsieh I-S, Gopula B, Chou C-C, Wu H-Y, Chang G-D, Wu W-J, et al. Development of novel irreversible pyruvate kinase M2 inhibitors. J Med Chem. (2019) 62:8497–510. doi: 10.1021/acs.jmedchem.9b00763
90. Niu Y-R, Xiang M-D, Yang W-W, Fang Y-T, Qian H-L, and Sun Y-K. NAD+/SIRT1 pathway regulates glycolysis to promote oxaliplatin resistance in colorectal cancer. World J Gastroenterol. (2025) 31:100785. doi: 10.3748/wjg.v31.i11.100785
91. Martin SP, Fako V, Dang H, Dominguez DA, Khatib S, Ma L, et al. PKM2 inhibition may reverse therapeutic resistance to transarterial chemoembolization in hepatocellular carcinoma. J Exp Clin Cancer Res. (2020) 39:99. doi: 10.1186/s13046-020-01605-y
92. Zhang H-S, Zhang F-J, Li H, Liu Y, Du G-Y, and Huang Y-H. Tanshinone II A inhibits human esophageal cancer cell growth through miR-122-mediated PKM2 down-regulation. Arch Biochem Biophys. (2016) 598:50–6. doi: 10.1016/j.abb.2016.03.031
93. Yang H-L, Chang C-W, Vadivalagan C, Pandey S, Chen S-J, Lee C-C, et al. Coenzyme Q0 inhibited the NLRP3 inflammasome, metastasis/EMT, and Warburg effect by suppressing hypoxia-induced HIF-1α expression in HNSCC cells. Int J Biol Sci. (2024) 20:2790–813. doi: 10.7150/ijbs.93943
94. Wei J, Lei G, Chen Q, Huang W, Ning H, Yang M, et al. Casticin inhibits proliferation of Non-small cell lung cancer cells through regulating reprogramming of glucose metabolism. Phytomedicine. (2025) 136:156278. doi: 10.1016/j.phymed.2024.156278
95. Li Q, Cao L, Tian Y, Zhang P, Ding C, Lu W, et al. Butyrate suppresses the proliferation of colorectal cancer cells via targeting pyruvate kinase M2 and metabolic reprogramming. Mol Cell Proteomics. (2018) 17:1531–45. doi: 10.1074/mcp.RA118.000752
96. Zhang C, Hu J, Sheng L, Yuan M, Wu Y, Chen L, et al. Metformin delays AKT/c-Met-driven hepatocarcinogenesis by regulating signaling pathways for de novo lipogenesis and ATP generation. Toxicol Appl Pharmacol. (2019) 365:51–60. doi: 10.1016/j.taap.2019.01.004
97. Sun Y, Zhao X, Zhou Y, and Hu Y. miR-124, miR-137 and miR-340 regulate colorectal cancer growth via inhibition of the Warburg effect. Oncol Rep. (2012) 28:1346–52. doi: 10.3892/or.2012.1958
98. Chen D, Wang Y, Lu R, Jiang X, Chen X, Meng N, et al. E3 ligase ZFP91 inhibits Hepatocellular Carcinoma Metabolism Reprogramming by regulating PKM splicing. Theranostics. (2020) 10:8558–72. doi: 10.7150/thno.44873
99. Yang H, Zhu R, Zhao X, Liu L, Zhou Z, Zhao L, et al. Sirtuin-mediated deacetylation of hnRNP A1 suppresses glycolysis and growth in hepatocellular carcinoma. Oncogene. (2019) 38:4915–31. doi: 10.1038/s41388-019-0764-z
100. Chen Z, Wang Z, Guo W, Zhang Z, Zhao F, Zhao Y, et al. TRIM35 Interacts with pyruvate kinase isoform M2 to suppress the Warburg effect and tumorigenicity in hepatocellular carcinoma. Oncogene. (2015) 34:3946–56. doi: 10.1038/onc.2014.325
101. Ma J, Sun F, Li W, Du R, Liu M, Wei Q, et al. SULT2B1: a novel therapeutic target in colorectal cancer via modulation of AKT/PKM2-mediated glycolysis and proliferation. J Transl Med. (2024) 22:1093. doi: 10.1186/s12967-024-05910-4
102. Zhou Y, Wang M, Qian Y, Yu D, Zhang J, Fu M, et al. PRDX2 promotes gastric cancer progression by forming a feedback loop with PKM2/STAT3 axis. Cell Signal. (2025) 127:111586. doi: 10.1016/j.cellsig.2024.111586
103. Zahra K, Dey T, Ashish, Mishra SP, and Pandey U. Pyruvate kinase M2 and cancer: the role of PKM2 in promoting tumorigenesis. Front Oncol. (2020) 10:159. doi: 10.3389/fonc.2020.00159
104. Brandi J, Cecconi D, Cordani M, Torrens-Mas M, Pacchiana R, Dalla Pozza E, et al. The antioxidant uncoupling protein 2 stimulates hnRNPA2/B1, GLUT1 and PKM2 expression and sensitizes pancreas cancer cells to glycolysis inhibition. Free Radic Biol Med. (2016) 101:305–16. doi: 10.1016/j.freeradbiomed.2016.10.499
105. Denise C, Paoli P, Calvani M, Taddei ML, Giannoni E, Kopetz S, et al. 5-fluorouracil resistant colon cancer cells are addicted to OXPHOS to survive and enhance stem-like traits. Oncotarget. (2015) 6:41706–21. doi: 10.18632/oncotarget.5991
106. Shimada N, Takasawa R, and Tanuma S-I. Interdependence of GLO I and PKM2 in the Metabolic shift to escape apoptosis in GLO I-dependent cancer cells. Arch Biochem Biophys. (2018) 638:1–7. doi: 10.1016/j.abb.2017.12.008
107. Zamer BA, Cui Z-G, Eladl MA, Hamad M, and Muhammad JS. Estrogen treatment in combination with pyruvate kinase M2 inhibition precipitate significant cumulative antitumor effects in colorectal cancer. J Biochem Mol Toxicol. (2024) 38:e23799. doi: 10.1002/jbt.23799
108. Giannoni E, Taddei ML, Morandi A, Comito G, Calvani M, Bianchini F, et al. Targeting stromal-induced pyruvate kinase M2 nuclear translocation impairs oxphos and prostate cancer metastatic spread. Oncotarget. (2015) 6:24061–74. doi: 10.18632/oncotarget.4448
109. Liao Q, Deng J, Tong J, Gan Y, Hong W, Dong H, et al. p53 induces circFRMD4A to suppress cancer development through glycolytic reprogramming and cuproptosis. Mol Cell. (2025) 85:132–149.e7. doi: 10.1016/j.molcel.2024.11.013
110. Markowitz GJ, Ban Y, Tavarez DA, Yoffe L, Podaza E, He Y, et al. Deficiency of metabolic regulator PKM2 activates the pentose phosphate pathway and generates TCF1+ progenitor CD8+ T cells to improve immunotherapy. Nat Immunol. (2024) 25:1884–99. doi: 10.1038/s41590-024-01963-1
111. Li X, Deng S, Liu M, Jin Y, Zhu S, Deng S, et al. The responsively decreased PKM2 facilitates the survival of pancreatic cancer cells in hypoglucose. Cell Death Dis. (2018) 9:133. doi: 10.1038/s41419-017-0158-5
112. Rihan M, Nalla LV, Dharavath A, Shard A, Kalia K, and Khairnar A. Pyruvate kinase M2: a metabolic bug in re-wiring the tumor microenvironment. Cancer Microenviron. (2019) 12:149–67. doi: 10.1007/s12307-019-00226-0
113. Porporato PE, Dhup S, Dadhich RK, Copetti T, and Sonveaux P. Anticancer targets in the glycolytic metabolism of tumors: a comprehensive review. Front Pharmacol. (2011) 2:49. doi: 10.3389/fphar.2011.00049
114. Zhang Y, Wu M-J, Lu W-C, Li Y-C, Chang CJ, and Yang J-Y. Metabolic switch regulates lineage plasticity and induces synthetic lethality in triple-negative breast cancer. Cell Metab. (2024) 36:193–208.e8. doi: 10.1016/j.cmet.2023.12.003
115. Chen F, Tang C, Yang F, Ekpenyong A, Qin R, Xie J, et al. HSP90 inhibition suppresses tumor glycolytic flux to potentiate the therapeutic efficacy of radiotherapy for head and neck cancer. Sci Adv. (2024) 10:eadk3663. doi: 10.1126/sciadv.adk3663
Keywords: PKM2, metabolic reprogramming, digestive system neoplasm, glycolysis, Warburg effect, oncotherapy
Citation: Huang X, He J, Sun H, Wu Y, Gu R and Li Z (2025) PKM2-driven metabolic reprogramming in digestive system tumors: mechanisms, therapeutic advances, and clinical challenges. Front. Immunol. 16:1634786. doi: 10.3389/fimmu.2025.1634786
Received: 25 May 2025; Accepted: 14 July 2025;
Published: 06 August 2025.
Edited by:
Lilong Zhang, Renmin Hospital of Wuhan University, ChinaReviewed by:
Xinghao Wang, Capital Medical University, ChinaJingyuan Ning, Chinese Academy of Medical Sciences and Peking Union Medical College, China
Weibiao Zeng, The First Affiliated Hospital of Soochow University, China
Copyright © 2025 Huang, He, Sun, Wu, Gu and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ziyun Li, eml5dW5saUBuanVjbS5lZHUuY24=; Renjun Gu, cmVuanVuZ3VAaG90bWFpbC5jb20=; Yi Wu, bnVyc2V3dXlpMDkxN0AxNjMuY29t
†These authors share first authorship