 Xiaokang Wang
Xiaokang Wang Gengrui Xu1
Gengrui Xu1- 1Department of Pharmacy, Shenzhen Longhua District Central Hospital, Affiliated Longhua Hospital of Shenzhen University, Shenzhen, China
- 2Center of Community Health Service Management, Shenzhen Longhua District Central Hospital, Shenzhen, China
- 3Beijing University of Chinese Medicine Affiliated Shenzhen Hospital (Longgang), Shenzhen, China
Neuroblastoma (NB), the most prevalent extracranial solid malignancy in children, poses significant therapeutic challenges, particularly concerning high-risk subtypes characterized by an immunologically “cold” phenotype. These tumors evade immune surveillance through mechanisms such as impaired antigen presentation and immunosuppressive microenvironment formation. Despite the incorporation of immunotherapies (e.g., GD2 monoclonal antibodies) into international clinical guidelines, the 5-year survival rate of patients with NB persistently remains lower than 50%. Small-molecule targeted agents, distinguished by their low molecular weight and superior chemical stability, offer advantages over macromolecular antibody therapies by effectively penetrating cell membranes to engage intracellular targets. Epigenetic regulation, a DNA sequence-independent gene expression modulation system, plays a pivotal role in cell fate determination via dynamic DNA methylation, histone covalent modifications, chromatin spatial reorganization, and non-coding RNA-mediated pathways. Emerging evidence has highlighted a strong correlation between epigenetic dysregulation and NB progression, establishing a molecular rationale for novel therapeutic strategies. Current epigenetic research in NB primarily focuses on histone deacetylase inhibitors and DNA methyltransferase inhibitors. These drugs exhibit unique translational potential because of their accelerated development timelines and cost-effective production, significantly enhancing therapeutic accessibility. This review systematically examines the current landscape of epigenetic modulators in NB treatment and discusses their transformative potential in improving outcomes for high-risk patients with NB.
1 Introduction
Neuroblastoma (NB), the most prevalent extracranial solid tumor in children, accounts for 8%–10% of pediatric malignancies, is often termed the “king of childhood cancers” because of its aggressive behavior and dismal prognosis (1). Originating from neural crest-derived sympathetic ganglion cells, NB predominantly arises in the adrenal medulla (55%–60% of cases) and paravertebral sympathetic chains and less frequently arises in the mediastinum (20%) and pelvis (15%) (2, 3). Epidemiological studies reported an annual incidence of 6–8 cases per million children, highlighting its significant clinical variability and challenging therapeutic landscape (4).
Contemporary NB management confronts three principal challenges: diagnostic delays (60% of patients present with distant metastases to bone marrow, skeletal systems, or liver at diagnosis, resulting in persistently stagnant 5-year survival rates of 40%–50% in high-risk cohorts (5)); therapeutic limitations (even with multimodal intensive regimens combining surgical resection, high-dose chemotherapy, autologous stem cell transplantation, and radiotherapy, survival outcomes have not significantly improved (6)); and immune evasion mechanisms (including tumor microenvironment alterations such as MHC class I downregulation and PD-L1 overexpression, which compromise the efficacy of GD2 monoclonal antibody combined with retinoic acid immunotherapy (7)).
Molecular profiling has identified pivotal oncogenic drivers such as MYCN amplification (25%–30%) and ALK mutations (8%–10%) (8, 9). Although these discoveries have refined risk stratification systems, their translational potential remains unrealized. Consequently, the development of novel therapies targeting tumor stem cell eradication and epigenetic regulation has become an urgent priority in contemporary research.
Epigenetic therapeutics represent a promising intervention strategy to overcome chemoresistance and relapse in high-risk NB (HR-NB) (10). This approach focuses on modulating epigenetic regulators, which are critical functional proteins orchestrating dynamic chromatin remodeling (11). These regulators mediate multilayered control through DNA methylation (5mC/5hmC), histone modification (e.g., H3 lysine 27 methylation [H3K27me3], H3 lysine 9 acetylation [H3K9ac], and non-coding RNA (ncRNAs) networks (e.g., lncRNAs, miRNAs), enabling spatiotemporal gene expression regulation without altering DNA sequences (12). The inherent reversibility of epigenetic modifications renders them ideal therapeutic targets, with epigenetic drug targets constituting 18.7% of all cancer therapeutic targets (12). The key advantages of epigenetic drugs lie in their multipathway synergy enabling the coordinated modulation of MYCN signaling and p53 restoration through single-target interventions, bidirectional transcriptional control that simultaneously suppresses oncogene hyperactivation (e.g., ALK, PHOX2B) and reactivates epigenetically silenced tumor suppressors (e.g., CASZ1, CLU), and heritable chromatin remodeling effects ensuring sustained therapeutic outcomes through the stable transmission of modified chromatin states across cell divisions.
Recent genomic analyses have positioned NB as a biologically distinct solid tumor characterized by a remarkably low somatic mutation burden and the absence of dominant driver genes. This recognition has catalyzed a paradigm shift emphasizing epigenetic dysregulation as potentially central to NB pathogenesis. Whole-genome analyses have identified three hallmark epigenetic aberrations: DNA methylation landscape remodeling featuring genome-wide hypomethylation coupled with promoter-specific hypermethylation (e.g., >80% methylation at HOX gene clusters); histone modification imbalances exemplified by H3K27me3 depletion (observed in 62% of high-risk cases) and abnormal H3K4me3 accumulation; and chromatin remodeler dysfunction, including frequent subunit deletions in SWI/SNF complexes (up to 40%). Despite identifying characteristic alterations such as PRC2 overexpression and TET enzyme inactivation, clinically validated epigenetic biomarkers remain elusive for diagnostic or prognostic applications (13).
This comprehensive review systematically examines the molecular foundations of epigenetic dysregulation in NB, the clinical translation of existing epigenetic therapeutics, and rational combination therapy strategies.
2 Cellular origins and transformation mechanisms of NB
As the most prevalent pediatric extracranial solid tumor, NB arises from malignant transformation during the sympathetic–adrenal lineage differentiation of neural crest cells (NCCs). Tumorigenesis is initiated when NCC-derived chromaffin cell precursors undergo developmental arrest at critical differentiation checkpoints during the seventh gestational week, coinciding with their migration to the adrenal primordium. Accumulating evidence positions adrenergic lineage cells as the principal cellular origin of NB, with single-cell transcriptomic profiling demonstrating striking transcriptional congruence between NB tumor cells and fetal adrenal chromaffin progenitors (Figure 1). This molecular mimicry, preserved through the malignant reprogramming of developmental pathways, provides compelling evidence for the adrenal chromaffin origin hypothesis while revealing critical vulnerabilities in NB’s epigenomic regulatory architecture.
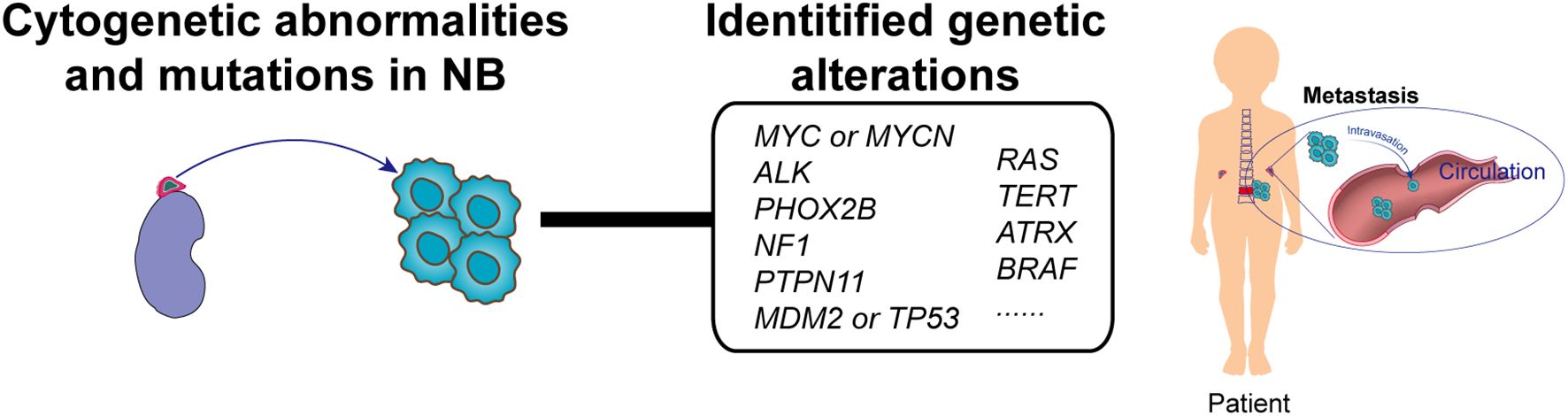
Figure 1. NB pathogenesis and key genetic alterations. Adrenergic lineage cells represent the predominant cellular origin of NB. NB pathogenesis is driven by the multilayered interplay of genomic aberrations. At the genomic level, recurrent somatic alterations include MYCN amplification, activating ALK mutations, and ATRX deletions.
NB pathogenesis is driven by the multilayered interplay of genomic and epigenetic aberrations. Genomically, recurrent somatic alterations include MYCN amplification (20% of cases, associated with 5-year survival rates of <50%), activating ALK F1174L mutations (8%), and ATRX deletions (11% in adolescents), with NF1 loss-of-function mutations synergizing with MYCN to drive tumorigenesis (14). Emerging evidence has further established clinical correlations of PHOX2B, TP53, RAS, and BRAF mutations with NB progression (Figure 1) (15, 16). Chromosomal instability manifests through pathognomonic chromothripsis events, which are detected in 19% of MYCN-amplified tumors. Epigenetically, coordinated dysregulation is typified by DNA methylation paradox (genome-wide hypomethylation coexisting with CpG island hypermethylation), EZH2 overexpression-mediated histone modification imbalance, and ncRNA networks governing proliferation-apoptosis homeostasis. These multilayered mechanisms converge to reshape developmental checkpoints and survival pathways, establishing NB’s unique oncogenic landscape.
3 The role of epigenetics in NB therapy
3.1 DNA methylation regulatory networks and therapeutic targeting
The dynamic equilibrium of DNA methylation is orchestrated by the antagonistic interplay between DNA methyltransferases (DNMTs) and ten-eleven translocation (TET) dioxygenases. Emerging pan-cancer analyses have revealed divergent TET family expression patterns: TET1 is transcriptionally silenced in 63% of hepatocellular carcinomas, whereas TET2 is mutated in 17% of gliomas (TCGA data). NB-specific epigenetic studies (17–19) identified TET3 as a potential prognostic biomarker, with its expression inversely correlating with the mitotic–karyorrhexis index. Elevated TET3 expression is correlated with improved 5-year survival (42%), and the tumor-suppressive role of TET3 is mechanistically linked to the 5mC hydroxylation-mediated maintenance of open chromatin states at neurodifferentiation-associated genes (e.g., PHOX2B). Contrastingly, TET1 drives oncogenesis through β2-adrenergic receptor pathway activation, inducing cAMP–PKA signaling that stabilizes MYCN protein (3.2-fold extended half-life). This isoform additionally partners with the histone demethylase KDM6B to form a transcriptional activation complex promoting tumor progression (20). The development of isoform-selective TET1 inhibitors and TET3 agonists represents a promising frontier for epigenetic therapy in NB (Figure 2A).
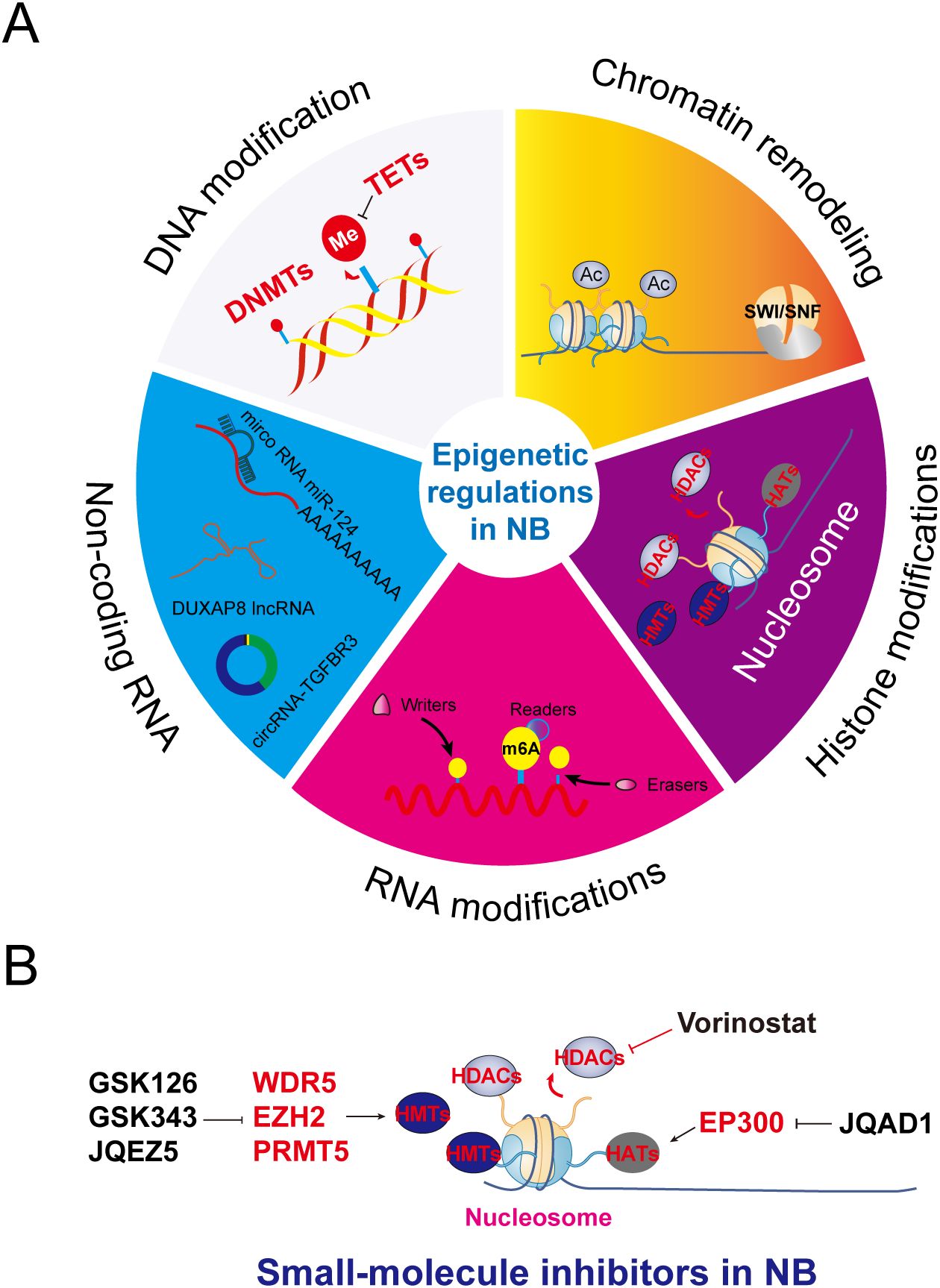
Figure 2. Epigenetic mechanisms and key examples of widely studied modifications and their modifying enzymes. (A) DNA modifications, chromatin remodeling, histone modifications, RNA modifications, and ncRNA-based regulation constitute the core content of epigenetics, being responsible for passing on heritable variations of genetic information independently of the DNA sequence. (B) Mechanistic schematic of epigenetic-targeting small-molecule inhibitors in NB.
Pioneering work by the Alaminos group (21) first elucidated the critical association among CpG island hypermethylation, MYCN amplification, and poor clinical outcomes in NB. Therapeutically, DNA methyltransferase inhibitors (e.g., decitabine) demonstrate dual efficacy, exhibiting standalone antiproliferative effects while synergistically enhancing conventional chemotherapeutics, a paradigm validated across multiple pediatric NB clinical trials. Notably, cisplatin-resistant models and high-risk NB subtypes exhibit marked upregulation of DNMT3A/B isoforms. Selective targeting of DNMT3B with nanaomycin A induces tumor-selective apoptosis through global methylation reduction (22), revealing a vulnerability in treatment-refractory disease. With the advancement of genome-wide methylation profiling technologies, high-resolution methylation mapping promises to resolve long-standing debates about the cellular origins of NB while informing precision epigenetic therapies (23).
3.2 Histone modification circuitry and therapeutic vulnerabilities in NB
Emerging evidence positions histone deacetylases (HDAC8/HDAC10) as central regulators of NB proliferation, differentiation, and chemosensitivity. Preclinical studies demonstrated that pan-HDAC inhibitors such as vorinostat suppress tumor growth through dual mechanisms: cell cycle arrest (G1 phase accumulation) and intrinsic apoptosis activation (caspase-3 cleavage) (24). These agents synergize with conventional chemotherapy or radiation, achieving enhanced tumor regression in xenograft models. Second-generation inhibitors (e.g., panobinostat, romidepsin) further exhibit blood–brain barrier penetration efficacy and display promise in central nervous system-metastasized NB subtypes (25), with six active HDAC-targeted clinical trials currently recruiting pediatric patients with NB. Paradoxically, although HDAC inhibition broadly suppresses oncogenic programs, context-dependent activation of differentiation pathways could underlie its therapeutic duality.
Beyond HDAC targeting, multilayered histone methylation networks involving WDR5 (H3K79me modulator), EZH2 (H3K27me3 writer), and PRTM5 (H3K4me3 eraser) orchestrate NB plasticity. WDR5–MYC complexes drive super-enhancer formation at oncogenic loci, whereas EZH2-mediated PRC2 activation silences tumor suppressors such as CLU and CADM1. PRMT5-centric arginine methylation sustains spliceosome integrity in MYCN-amplified cells. Pharmacological disruption of these nodes (e.g., EZH2 inhibitor tazemetostat, PRMT5 inhibitor GSK3326595) induces differentiation and reverses chemoresistance in preclinical models (26). These findings collectively map NB’s epigenetic vulnerabilities, facilitating the development of novel combinatorial strategies that simultaneously target histone-modifying enzymes and lineage-specific oncogenic drivers (Figure 2B).
The precise equilibrium between histone acetylation and deacetylation serves as a master epigenetic switch governing transcriptional plasticity, with its dysregulation representing a hallmark of tumorigenesis. In high-risk NB, HDAC8 and HDAC10 exhibit pathologic overexpression (27, 28), establishing them as actionable targets. Pharmacologic HDAC inhibition both suppresses tumor proliferation and chemosensitizes resistant cells to doxorubicin, potentially overcoming treatment barriers. Preclinical studies identified valproic acid as a broad-spectrum HDAC inhibitor capable of inducing a triad of effects: proliferation arrest, mitochondrial apoptosis, and neural differentiation.
Vorinostat, the first FDA-approved pan-HDAC inhibitor, demonstrates mechanistically distinct antitumor activity in NB. This drug triggers G2/M phase blockade through CDK1/cyclin B dysregulation and activates intrinsic apoptosis via Bim/PUMA transcriptional induction. Clinical translation has revealed striking efficacy. Combined with 131I-metaiodobenzylguanidine (MIBG), vorinostat elevates objective response rates in relapsed/refractory NB from 14% to 32% and extends median progression-free survival by 5.8 months (ASCO 2021 data) (29). Synergistic potential was further evidenced by its combined use with panobinostat, which extended survival in TH-MYCN transgenic mice by 62%, and with GD2-targeted immunotherapy, in which co-treatment enhanced antibody-dependent cellular cytotoxicity (30). These multimodal regimens exemplify the evolving paradigm of epigenetic-immune interplay in NB precision medicine.
Targeting SWI/SNF complex dysregulation represents a therapeutic mechanism in NB. Approximately 25% of human cancers harbor mutations in genes encoding mammalian SWI/SNF (mSWI/SNF) chromatin remodeling complexes (31). Core components include ATPase subunits (SMARCA4/BRG1 and SMARCA2/BRM) and structural subunits (e.g., SMARCC1/2, SMARCD1/2/3) (32). Mechanistically, SMARCB1 mutations induce the mislocalization of mSWI/SNF complexes at gene promoter regions accompanied by RNA polymerase II dysfunction and altered H3K27ac signatures. In NB, mutations in the ARID1A/ARID1B subunits of the SWI/SNF chromatin remodeling complex promote tumor progression and correlate with poor prognosis.
Emerging evidence has revealed the histone acetyltransferase EP300 as an epigenetic linchpin in NB pathogenesis, particularly through MYCN transcriptional regulation (33). Mechanistically, EP300 acetylates histones at MYCN super-enhancer regions, thereby sustaining oncogene addiction in MYCN-amplified tumors. Pioneering work by Durbin et al. (33) developed JQAD1, a PROTAC-based EP300 degrader that achieves tumor-selective depletion (90% reduction at 100 nM) through VHL E3 ligase recruitment. This agent induces rapid apoptosis (caspase-3 activation within 8 h) in MYCN-driven models while demonstrating exceptional safety margins (normal cell viability > 85%), establishing targeted protein degradation as a breakthrough paradigm.
Paralleling these advances, the histone methyltransferase EZH2 exhibits co-operative oncogenesis in MYCN-amplified NB. MYCN transcriptionally upregulates EZH2 (34) while physically interacting with its N-terminal domain to stabilize MYCN protein through PRC2-catalytic-independent mechanisms (35), creating a feed-forward malignancy loop. Pharmacologic disruption using catalytic inhibitors (GSK126, IC50 = 6 nM; JQEZ5, IC50 = 8 nM) induces tumor regression in orthotopic models (36), with current clinical efforts exploring combination regimens with BET inhibitors.
Despite these successes, structural biology limitations persist, as the domain structure has been resolved for fewer than 30% of epigenetic regulators, hampering the rational design of isoform-selective agents. Next-generation approaches currently prioritize covalent EZH2 inhibitors (e.g., MS1943) and dual EZH2/HDAC degraders to overcome compensatory resistance mechanisms, although tumor-selective delivery remains a critical translational barrier.
3.3 ncRNAs networks and therapeutic opportunities in NB
ncRNAs have emerged as master epigenetic regulators governing NB tumorigenesis, with distinct subclasses, namely miRNAs, lncRNAs, and circular RNAs (circRNAs), orchestrating malignant hallmarks through multilayered gene regulatory networks (37–39). Clinically, circulating miRNAs exhibit diagnostic potential through their exosome-mediated intercellular communication and stability in biofluids (40). For instance, miR-124 acts as a tumor suppressor by reversing therapy resistance in mesenchymal-type NB cells, and its targeted upregulation using PP121 (a tyrosine/PI3K kinase inhibitor) synergizes with BDNF-activated bufalin to induce neural differentiation and apoptosis (41, 42). This exemplifies the therapeutic potential of miRNA modulation in overcoming NB chemoresistance.
The lncRNA landscape reveals equally critical oncogenic drivers (37). DUXAP8, which is overexpressed in > 60% of high-risk NB tumors, accelerates tumor progression via dual mechanisms: sequestering miR-29 to derepress NOL4L-mediated cell cycle activation and enhancing metastatic potential through TWIST1 stabilization. CRISPR-mediated DUXAP8 silencing reduces xenograft tumor growth by 72%, validating its therapeutic candidacy.
The discovery of circRNAs has unveiled a new regulatory layer in NB pathogenesis, with MYCN amplification profoundly altering circRNA landscapes (43). Comparative deep sequencing analysis of five MYCN-amplified NB tumors versus matched normal tissues revealed 2242 significantly downregulated circRNAs, among which three tumor-suppressive circRNAs exhibited particularly promising therapeutic potential. Specifically, circTBC1D4 functions as a molecular sponge for oncogenic miR-21, thereby de-repressing PDCD4 expression and restoring apoptosis sensitivity in treatment-resistant cells. circNAALAD2 directly interacts with the PHLPP2 phosphatase to suppress AKT hyperphosphorylation, effectively inhibiting PI3K-driven survival pathways. circTGFBR3 structurally stabilizes the AXIN1–APC destruction complex, leading to β-catenin degradation and Wnt pathway suppression (44).
The primary mechanisms and functions of these epigenetic drugs in NB treatment are summarized in Table 1. These agents demonstrate multitarget regulatory capabilities in NB disease progression by modulating key nodes including apoptosis, proliferation, and epigenetic modifications, offering multifaceted mechanisms and potential therapeutic targets for neuroblastoma therapy. However, few epigenetic drugs have advanced to clinical trial phases.
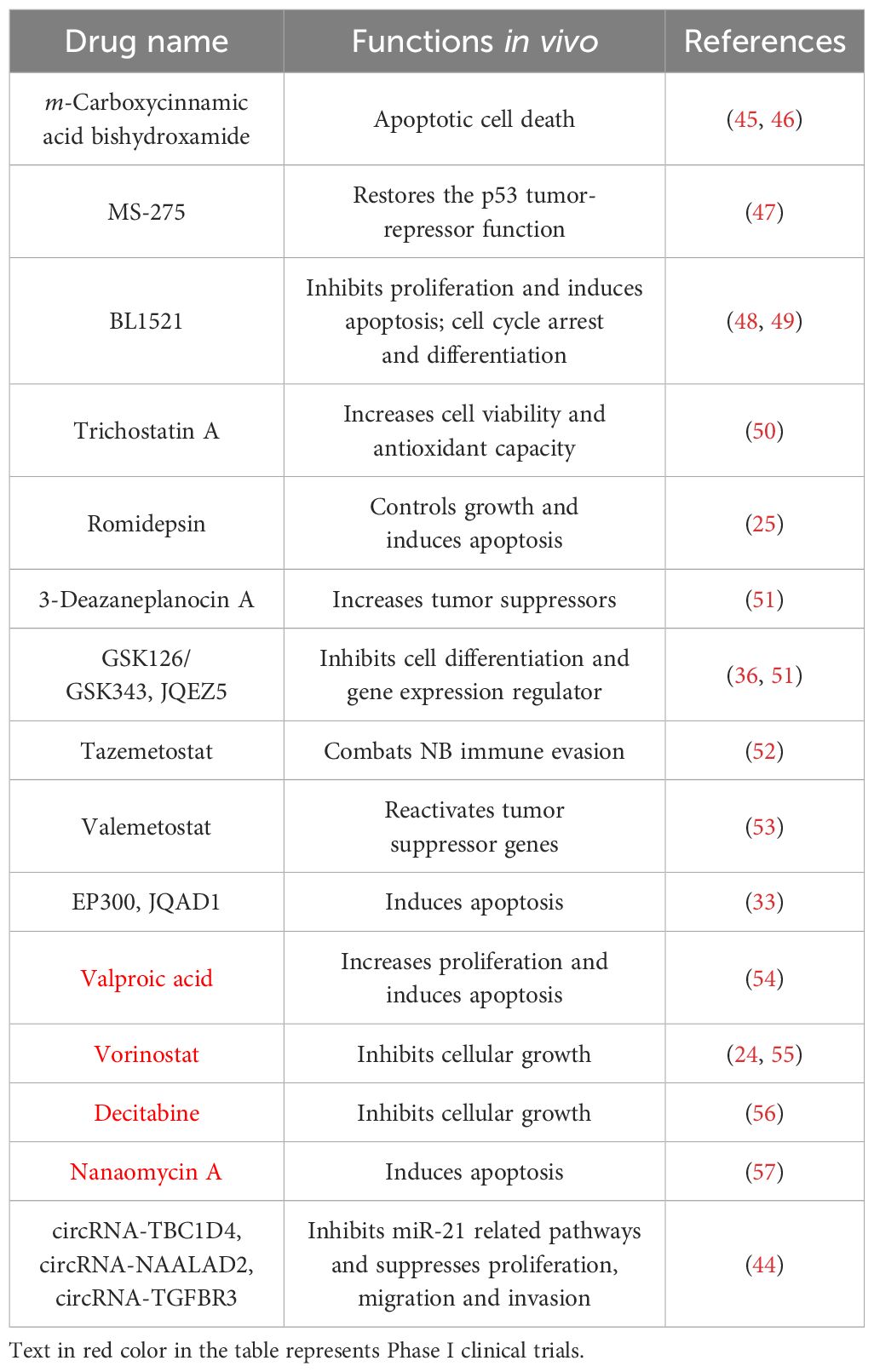
Table 1. Epigenetic drugs in NB.
Although the number of active clinical trials for epigenetic modifiers in NB remains limited, a substantial pool of potential novel epigenetic targets awaits exploration. The epigenetic regulatory genes with therapeutic potential for NB identified in current preclinical studies (Table 2) will facilitate the development of new compounds (epigenetic drugs).
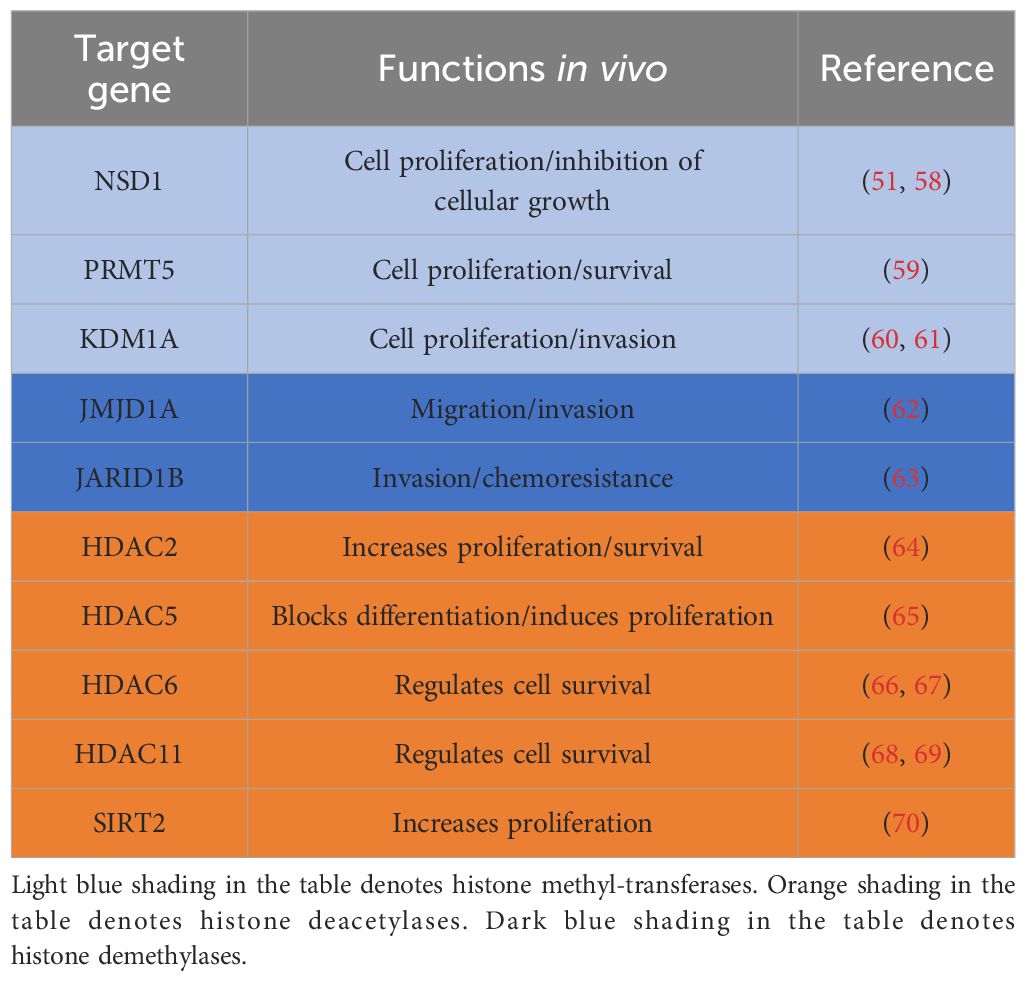
Table 2. Potential epigenetic targets in NB.
4 Diversified development in NB targeted therapy
Diversified clinical advances have been achieved in NB targeted therapy. Antibody-based immunotherapies (e.g., dinutuximab beta, naxitamab) are established frontline interventions, whereas small-molecule targeted agents offer advantages such as low molecular weight, oral bioavailability, and favorable cost-effectiveness. Eflornithine remains the only approved oral maintenance therapy, complementing preclinical-stage epigenetic modulators such as HDAC inhibitors. Mechanistic insights into NB pathogenesis have propelled the development of novel compounds such as IBL-302 (BRD4-targeting PROTAC) and APG-115 (MDM2 degrader), which are currently undergoing Phase I/II clinical trials after producing breakthrough efficacy in MYCN-driven models.
Notably, NB’s pronounced genomic heterogeneity poses significant therapeutic hurdles, as 40% of relapsed tumors exhibit ALK/RAS pathway co-activation. Future strategies require integrating multiomics platforms (single-cell epigenomics, spatial proteomics) to identify druggable targets, thereby addressing key obstacles in developing NB-targeted small molecules through precision target validation and pharmacological exploitation of genomic vulnerabilities.
Advanced ncRNA delivery systems have demonstrated transformative therapeutic potential. Specifically, nanoparticle encapsulation significantly enhances ncRNA stability and bioavailability, whereas naturally derived exosomes, with their inherent low immunogenicity and blood–brain barrier penetrance, enable targeted ncRNA delivery to NB cells without triggering immune responses. The field’s future development will strategically focus on three key directions: molecular therapeutics featuring MYCN/ALK inhibitors (lorlatinib), CDK4/6 inhibitors (ribociclib), and TRK inhibitors (entrectinib/larotrectinib) currently in clinical trials; advanced delivery platforms utilizing CRISPR-modified small extracellular vesicles that establish pre-metastatic niches through precision immune cell priming (71); and regimen optimization via chronologically coordinated combination therapies (e.g., 131I-MIBG radiotherapy with GD2-targeted immunotherapy or CAR-T regimens) to simultaneously enhance therapeutic efficacy and reduce long-term complications.
Author contributions
XW: Funding acquisition, Formal Analysis, Writing – review & editing, Methodology, Writing – original draft, Investigation. GX: Investigation, Formal Analysis, Methodology, Writing – original draft. HM: Writing – original draft, Investigation, Methodology. XD: Investigation, Writing – original draft, Data curation. GM: Methodology, Investigation, Funding acquisition, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the Guangdong Basic and Applied Basic Research Foundation (No. 2023A1515111116), the Shenzhen Foundation of Science and Technology (No. JCYJ20230807151308018), the Zhanjiang Science and Technology Project (2023B01176), Longgang District Science and Technology Innovation Bureau, Longgang District Medical and Health Technology Research Support Project (LGKCYLWS2024-22), Shenzhen Longhua District Science and Technology Innovation Fund Projects (Nos. 2022045, 2022051 and 2022056) and the Research Foundation of Shenzhen Longhua District Central Hospital (No. 202203).
Acknowledgments
We sincerely thank the reviewers for their valuable feedback on this paper. We also thank Medjaden Inc. for scientific editing of this manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Zhuo Z, Lin L, Miao L, Li M, and He J. Advances in liquid biopsy in neuroblastoma. Fundam Res. (2022) 2:903–17. doi: 10.1016/j.fmre.2022.08.005
2. Maris JM. Recent advances in neuroblastoma. N Engl J Med. (2010) 362:2202–11. doi: 10.1056/NEJMra0804577
3. Ghasempour A, Mohseni R, Sharif PM, and Hamidieh AA. Natural killer cell-based therapies in neuroblastoma. Cell Immunol. (2025) 407:104898. doi: 10.1016/j.cellimm.2024.104898
4. Kamihara J, Diller LR, Foulkes WD, Michaeli O, Nakano Y, Pajtler KW, et al. Neuroblastoma predisposition and surveillance-an update from the 2023 AACR childhood cancer predisposition workshop. Clin Cancer Res. (2024) 30:3137–43. doi: 10.1158/1078-0432.CCR-24-0237
5. Qiu B and Matthay KK. Advancing therapy for neuroblastoma. Nat Rev Clin Oncol. (2022) 19:515–33. doi: 10.1038/s41571-022-00643-z
6. Grossmann LD, Chen C-H, Uzun Y, Thadi A, Wolpaw AJ, Louault K, et al. Identification and characterization of chemotherapy-resistant high-risk neuroblastoma persister cells. Cancer Discov. (2024) 14:2387–406. doi: 10.1158/2159-8290.CD-24-0046
7. Tarannum M, Ding X, Barisa M, Hu S, Anderson J, Romee R, et al. Engineering innate immune cells for cancer immunotherapy. Nat Biotechnol. (2025) 43:516–33. doi: 10.1038/s41587-025-02629-5
8. Masserot C, Liu Q, Nguyen E, Gattolliat C-H, Valteau-Couanet D, Bénard J, et al. WT1 expression is inversely correlated with MYCN amplification or expression and associated with poor survival in non-MYCN-amplified neuroblastoma. Mol Oncol. (2016) 10:240–52. doi: 10.1016/j.molonc.2015.09.010
9. Borenäs M, Umapathy G, Lai W-Y, Lind DE, Witek B, Guan J, et al. ALK ligand ALKAL2 potentiates MYCN-driven neuroblastoma in the absence of ALK mutation. EMBO J. (2021) 40:e105784. doi: 10.15252/embj.2020105784
10. Nagarajan D, Parracho RT, Corujo D, Xie M, Kutkaite G, Olsen TK, et al. Epigenetic regulation of cell state by H2AFY governs immunogenicity in high-risk neuroblastoma. J Clin Invest. (2024) 134(21):e175310. doi: 10.1172/JCI175310
11. Tahghighi A, Seyedhashemi E, Mohammadi J, Moradi A, Esmaeili A, Pornour M, et al. Epigenetic marvels: exploring the landscape of colorectal cancer treatment through cutting-edge epigenetic-based drug strategies. Clin Epigenet. (2025) 17:34. doi: 10.1186/s13148-025-01844-w
12. Topper MJ, Vaz M, Marrone KA, Brahmer JR, and Baylin SB. The emerging role of epigenetic therapeutics in immuno-oncology. Nat Rev Clin Oncol. (2020) 17:75–90. doi: 10.1038/s41571-019-0266-5
13. Fetahu IS and Taschner-Mandl S. Neuroblastoma and the epigenome. Cancer Metastasis Rev. (2021) 40:173–89. doi: 10.1007/s10555-020-09946-y
14. Hackett CS, Quigley DA, Wong RA, Chen J, Cheng C, Song YK, et al. Expression quantitative trait loci and receptor pharmacology implicate Arg1 and the GABA-A receptor as therapeutic targets in neuroblastoma. Cell Rep. (2014) 9:1034–46. doi: 10.1016/j.celrep.2014.09.046
15. Berko ER, Naranjo A, Daniels AA, McNulty SN, Krytska K, Druley T, et al. Frequency and clinical significance of clonal and subclonal driver mutations in high-risk neuroblastoma at diagnosis: A children’s oncology group study. J Clin Oncol. (2025) 43:1673–84. doi: 10.1200/JCO-24-02407
16. Lin L, Miao L, Lin H, Cheng J, Li M, Zhuo Z, et al. Targeting RAS in neuroblastoma: Is it possible? Pharmacol Ther. (2022) 236:108054. doi: 10.1016/j.pharmthera.2021.108054
17. Dong Z-R, Ke A-W, Li T, Cai J-B, Yang Y-F, Zhou W, et al. CircMEMO1 modulates the promoter methylation and expression of TCF21 to regulate hepatocellular carcinoma progression and sorafenib treatment sensitivity. Mol Cancer. (2021) 20:75. doi: 10.1186/s12943-021-01361-3
18. Bai X, Zhang H, Zhou Y, Nagaoka K, Meng J, Ji C, et al. Ten-eleven translocation 1 promotes Malignant progression of cholangiocarcinoma with wild-type isocitrate dehydrogenase 1. Hepatology. (2021) 73:1747–63. doi: 10.1002/hep.31486
19. Li Z, Lyu C, Ren Y, and Wang H. Role of TET dioxygenases and DNA hydroxymethylation in bisphenols-stimulated proliferation of breast cancer cells. Environ Health Perspect. (2020) 128:27008. doi: 10.1289/EHP5862
20. Deng J, Jiang P, Yang T, Huang M, Xie J, Luo C, et al. β2−adrenergic receptor signaling promotes neuroblastoma cell proliferation by activating autophagy. Oncol Rep. (2019) 42:1295–306. doi: 10.3892/or.2019.7266
21. Mora J, Alaminos M, de Torres C, Illei P, Qin J, Cheung NKV, et al. Comprehensive analysis of the 9p21 region in neuroblastoma suggests a role for genes mapping to 9p21–23 in the biology of favourable stage 4 tumours. Br J Cancer. (2004) 91:1112–8. doi: 10.1038/sj.bjc.6602094
22. Penter L, Maier B, Frede U, Hackner B, Carell T, Hagemeier C, et al. A rapid screening system evaluates novel inhibitors of DNA methylation and suggests F-box proteins as potential therapeutic targets for high-risk neuroblastoma. Target Oncol. (2015) 10:523–33. doi: 10.1007/s11523-014-0354-5
23. Zhu T, Liu J, Beck S, Pan S, Capper D, Lechner M, et al. A pan-tissue DNA methylation atlas enables in silico decomposition of human tissue methylomes at cell-type resolution. Nat Methods. (2022) 19:296–306. doi: 10.1038/s41592-022-01412-7
24. Körholz K, Ridinger J, Krunic D, Najafi S, Gerloff XF, Frese K, et al. Broad-spectrum HDAC inhibitors promote autophagy through FOXO transcription factors in neuroblastoma. Cells. (2021) 10(5):1001. doi: 10.3390/cells10051001
25. Panicker J, Li Z, McMahon C, Sizer C, Steadman K, Piekarz R, et al. Romidepsin (FK228/depsipeptide) controls growth and induces apoptosis in neuroblastoma tumor cells. Cell Cycle. (2010) 9:1830–8. doi: 10.4161/cc.9.9.11543
26. Huang L, Zhang X-O, Rozen EJ, Sun X, Sallis B, Verdejo-Torres O, et al. PRMT5 activates AKT via methylation to promote tumor metastasis. Nat Commun. (2022) 13:3955. doi: 10.1038/s41467-022-31645-1
27. Oehme I, Deubzer HE, Lodrini M, Milde T, and Witt O. Targeting of HDAC8 and investigational inhibitors in neuroblastoma. Expert Opin Investig Drugs. (2009) 18:1605–17. doi: 10.1517/14728220903241658
28. Oehme I, Linke J-P, Böck BC, Milde T, Lodrini M, Hartenstein B, et al. Histone deacetylase 10 promotes autophagy-mediated cell survival. Proc Natl Acad Sci U S A. (2013) 110:E2592–601. doi: 10.1073/pnas.1300113110
29. DuBois SG, Granger MM, Groshen S, Tsao-Wei D, Ji L, Shamirian A, et al. Randomized phase II trial of MIBG versus MIBG, vincristine, and irinotecan versus MIBG and vorinostat for patients with relapsed or refractory neuroblastoma: A report from NANT consortium. J Clin Oncol. (2021) 39:3506–14. doi: 10.1200/JCO.21.00703
30. McNerney KO, Karageorgos S, Ferry GM, Wolpaw AJ, Burudpakdee C, Khurana P, et al. TH-MYCN tumors, but not tumor-derived cell lines, are adrenergic lineage, GD2+, and responsive to anti-GD2 antibody therapy. Oncoimmunology. (2022) 11:2075204. doi: 10.1080/2162402X.2022.2075204
31. Kadoch C, Hargreaves DC, Hodges C, Elias L, Ho L, Ranish J, et al. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human Malignancy. Nat Genet. (2013) 45:592–601. doi: 10.1038/ng.2628
32. Leung JY, Chiu HY, and Taneja R. Role of epigenetics in paediatric cancer pathogenesis & drug resistance. Br J Cancer. (2025) 132:757–69. doi: 10.1038/s41416-025-02961-2
33. Durbin AD, Wang T, Wimalasena VK, Zimmerman MW, Li D, Dharia NV, et al. EP300 selectively controls the enhancer landscape of MYCN-amplified neuroblastoma. Cancer Discov. (2022) 12:730–51. doi: 10.1158/2159-8290.CD-21-0385
34. Bate-Eya LT, Gierman HJ, Ebus ME, Koster J, Caron HN, Versteeg R, et al. Enhancer of zeste homologue 2 plays an important role in neuroblastoma cell survival independent of its histone methyltransferase activity. Eur J Cancer. (2017) 75:63–72. doi: 10.1016/j.ejca.2016.12.019
35. Wang L, Chen C, Song Z, Wang H, Ye M, Wang D, et al. EZH2 depletion potentiates MYC degradation inhibiting neuroblastoma and small cell carcinoma tumor formation. Nat Commun. (2022) 13:12. doi: 10.1038/s41467-021-27609-6
36. Gao J, Fosbrook C, Gibson J, Underwood TJ, Gray JC, and Walters ZS. Review: targeting EZH2 in neuroblastoma. Cancer Treat Rev. (2023) 119:102600. doi: 10.1016/j.ctrv.2023.102600
37. Wang X, Zhao J, Li Y, Rao J, and Xu G. Epigenetics and endoplasmic reticulum in podocytopathy during diabetic nephropathy progression. Front Immunol. (2022) 13:1090989. doi: 10.3389/fimmu.2022.1090989
38. Sethi SC, Singh R, Sahay O, Barik GK, and Kalita B. Unveiling the hidden gem: A review of long non-coding RNA NBAT-1 as an emerging tumor suppressor and prognostic biomarker in cancer. Cell Signal. (2025) 126:111525. doi: 10.1016/j.cellsig.2024.111525
39. Wang X, Tong Y, Xun T, Feng H, Lei Y, Li Y, et al. Functions, mechanisms, and therapeutic implications of noncoding RNA in acute myeloid leukemia. Fundam Res. (2025) 1797–811. doi: 10.1016/j.fmre.2023.04.012
40. Bhavsar SP. Recent advances in the roles of exosomal microRNAs in neuroblastoma. Front Oncol. (2022) 12:1091847. doi: 10.3389/fonc.2022.1091847
41. Pottoo FH, Barkat MA, Harshita, Ansari MA, Javed MN, Sajid Jamal QM, et al. Nanotechnological based miRNA intervention in the therapeutic management of neuroblastoma. Semin Cancer Biol. (2021) 69:100–8. doi: 10.1016/j.semcancer.2019.09.017
42. Nguyen LD, Sengupta S, Cho KI, Floru A, George RE, and Krichevsky AM. A drug that induces the microRNA miR-124 enables differentiation of retinoic acid-resistant neuroblastoma cells. Sci Signal. (2025) 18:eads2641. doi: 10.1126/scisignal.ads2641
43. Zhang L, Zhou H, Li J, Wang X, Zhang X, Shi T, et al. Comprehensive characterization of circular RNAs in neuroblastoma cell lines. Technol Cancer Res Treat. (2020) 19:1533033820957622. doi: 10.1177/1533033820957622
44. Lin W, Wang Z, Wang J, Yan H, Han Q, Yao W, et al. circRNA-TBC1D4, circRNA-NAALAD2 and circRNA-TGFBR3: Selected Key circRNAs in Neuroblastoma and Their Associations with Clinical Features. Cancer Manag Res. (2021) 13:4271–81. doi: 10.2147/CMAR.S297316
45. De los Santos M, Zambrano A, and Aranda A. Combined effects of retinoic acid and histone deacetylase inhibitors on human neuroblastoma SH-SY5Y cells. Mol Cancer Ther. (2007) 6:1425–32. doi: 10.1158/1535-7163.MCT-06-0623
46. Mühlethaler-Mottet A, Flahaut M, Bourloud KB, Auderset K, Meier R, Joseph J-M, et al. Histone deacetylase inhibitors strongly sensitise neuroblastoma cells to TRAIL-induced apoptosis by a caspases-dependent increase of the pro- to anti-apoptotic proteins ratio. BMC Cancer. (2006) 6:214. doi: 10.1186/1471-2407-6-214
47. Condorelli F, Gnemmi I, Vallario A, Genazzani AA, and Canonico PL. Inhibitors of histone deacetylase (HDAC) restore the p53 pathway in neuroblastoma cells. Br J Pharmacol. (2008) 153:657–68. doi: 10.1038/sj.bjp.0707608
48. de Ruijter AJM, Kemp S, Kramer G, Meinsma RJ, Kaufmann JO, Caron HN, et al. The novel histone deacetylase inhibitor BL1521 inhibits proliferation and induces apoptosis in neuroblastoma cells. Biochem Pharmacol. (2004) 68:1279–88. doi: 10.1016/j.bcp.2004.05.010
49. de Ruijter AJM, Leen R, Hoebink J, Caron HN, and van Kuilenburg ABP. Antagonistic effects of sequential administration of BL1521, a histone deacetylase inhibitor, and gemcitabine to neuroblastoma cells. Cancer Lett. (2006) 233:240–6. doi: 10.1016/j.canlet.2005.03.016
50. Li L-H, Peng W-N, Deng Y, Li J-J, and Tian X-R. Action of trichostatin A on Alzheimer’s disease-like pathological changes in SH-SY5Y neuroblastoma cells. Neural Regener Res. (2020) 15:293–301. doi: 10.4103/1673-5374.265564
51. Wang C, Liu Z, Woo C-W, Li Z, Wang L, Wei JS, et al. EZH2 Mediates epigenetic silencing of neuroblastoma suppressor genes CASZ1, CLU, RUNX3, and NGFR. Cancer Res. (2012) 72:315–24. doi: 10.1158/0008-5472.CAN-11-0961
52. Shaliman D, Takenobu H, Sugino RP, Ohira M, and Kamijo T. The PRC2 molecule EED is a target of epigenetic therapy for neuroblastoma. Eur J Cell Biol. (2022) 101:151238. doi: 10.1016/j.ejcb.2022.151238
53. Goleij P, Heidari MM, Tabari MAK, Hadipour M, Rezaee A, Javan A, et al. Polycomb repressive complex 2 (PRC2) pathway’s role in cancer cell plasticity and drug resistance. Funct Integr Genomics. (2025) 25:53. doi: 10.1007/s10142-025-01563-8
54. Dedoni S, Olianas A, Manconi B, Collu M, Tuveri B, Vincis ME, et al. Upregulation of p75NTR by histone deacetylase inhibitors sensitizes human neuroblastoma cells to targeted immunotoxin-induced apoptosis. Int J Mol Sci. (2022) 23(7):3849. doi: 10.3390/ijms23073849
55. van den Bijgaart RJE, Kroesen M, Brok IC, Reijnen D, Wassink M, Boon L, et al. Anti-GD2 antibody and Vorinostat immunocombination therapy is highly effective in an aggressive orthotopic neuroblastoma model. Oncoimmunology. (2020) 9:1817653. doi: 10.1080/2162402X.2020.1817653
56. Hou Y, Wang F, Cheng L, Luo T, Xu J, and Wang H. Expression profiles of SIRT1 and APP genes in human neuroblastoma SK-N-SH cells treated with two epigenetic agents. Neurosci Bull. (2016) 32:455–62. doi: 10.1007/s12264-016-0052-7
57. Izumi K, Aoki H, Kakita H, Takeshita S, Ueda H, Inoue Y, et al. The DNMT3B inhibitor nanaomycin A as a neuroblastoma therapeutic agent. Curr Cancer Drug Targets. (2023) 23:837–42. doi: 10.2174/1568009623666230522113645
58. Berdasco M, Ropero S, Setien F, Fraga MF, Lapunzina P, Losson R, et al. Epigenetic inactivation of the Sotos overgrowth syndrome gene histone methyltransferase NSD1 in human neuroblastoma and glioma. Proc Natl Acad Sci U S A. (2009) 106:21830–5. doi: 10.1073/pnas.0906831106
59. Park JH, Szemes M, Vieira GC, Melegh Z, Malik S, Heesom KJ, et al. Protein arginine methyltransferase 5 is a key regulator of the MYCN oncoprotein in neuroblastoma cells. Mol Oncol. (2015) 9:617–27. doi: 10.1016/j.molonc.2014.10.015
60. Althoff K, Beckers A, Odersky A, Mestdagh P, Köster J, Bray IM, et al. MiR-137 functions as a tumor suppressor in neuroblastoma by downregulating KDM1A. Int J Cancer. (2013) 133:1064–73. doi: 10.1002/ijc.28091
61. Yang H, Li Q, Zhao W, Yuan D, Zhao H, and Zhou Y. miR-329 suppresses the growth and motility of neuroblastoma by targeting KDM1A. FEBS Lett. (2014) 588:192–7. doi: 10.1016/j.febslet.2013.11.036
62. Tee AE, Ling D, Nelson C, Atmadibrata B, Dinger ME, Xu N, et al. The histone demethylase JMJD1A induces cell migration and invasion by up-regulating the expression of the long noncoding RNA MALAT1. Oncotarget. (2014) 5:1793–804. doi: 10.18632/oncotarget.1785
63. Kuo Y-T, Liu Y-L, Adebayo BO, Shih P-H, Lee W-H, Wang L-S, et al. JARID1B expression plays a critical role in chemoresistance and stem cell-like phenotype of neuroblastoma cells. PloS One. (2015) 10:e0125343. doi: 10.1371/journal.pone.0125343
64. Hsu C-L, Yin C-F, Chang Y-W, Fan Y-C, Lin S-H, Wu Y-C, et al. LncRNA SNHG1 regulates neuroblastoma cell fate via interactions with HDAC1/2. Cell Death Dis. (2022) 13:809. doi: 10.1038/s41419-022-05256-z
65. Sun Y, Liu PY, Scarlett CJ, Malyukova A, Liu B, Marshall GM, et al. Histone deacetylase 5 blocks neuroblastoma cell differentiation by interacting with N-Myc. Oncogene. (2014) 33:2987–94. doi: 10.1038/onc.2013.253
66. Tang H, Liang Y, Shen H, Cai S, Yu M, Fan H, et al. Discovery of a 2,6-diarylpyridine-based hydroxamic acid derivative as novel histone deacetylase 8 and tubulin dual inhibitor for the treatment of neuroblastoma. Bioorg Chem. (2022) 128:106112. doi: 10.1016/j.bioorg.2022.106112
67. Sixto-López Y, Gómez-Vidal JA, de Pedro N, Bello M, Rosales-Hernández MC, and Correa-Basurto J. Hydroxamic acid derivatives as HDAC1, HDAC6 and HDAC8 inhibitors with antiproliferative activity in cancer cell lines. Sci Rep. (2020) 10:10462. doi: 10.1038/s41598-020-67112-4
68. Thole TM, Lodrini M, Fabian J, Wuenschel J, Pfeil S, Hielscher T, et al. Neuroblastoma cells depend on HDAC11 for mitotic cell cycle progression and survival. Cell Death Dis. (2017) 8:e2635. doi: 10.1038/cddis.2017.49
69. Baselious F, Hilscher S, Hagemann S, Tripathee S, Robaa D, Barinka C, et al. Utilization of an optimized AlphaFold protein model for structure-based design of a selective HDAC11 inhibitor with anti-neuroblastoma activity. Arch Pharm (Weinheim). (2024) 357:e2400486. doi: 10.1002/ardp.202400486
70. Liu PY, Xu N, Malyukova A, Scarlett CJ, Sun YT, Zhang XD, et al. The histone deacetylase SIRT2 stabilizes Myc oncoproteins. Cell Death Differ. (2013) 20:503–14. doi: 10.1038/cdd.2012.147
Keywords: immunologically cold, small-molecule targeted agents, epigenetic regulation, pediatric neuroblastoma, therapeutic strategies
Citation: Wang X, Xu G, Ma H, Deng X and Ma G (2025) Emerging frontiers in epigenetic-targeted therapeutics for pediatric neuroblastoma. Front. Immunol. 16:1637626. doi: 10.3389/fimmu.2025.1637626
Received: 29 May 2025; Accepted: 08 July 2025;
Published: 25 July 2025.
Edited by:
Guan-Jun Yang, Ningbo University, ChinaCopyright © 2025 Wang, Xu, Ma, Deng and Ma. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaoyan Deng, ODcyMDY5NjI5QHFxLmNvbQ==; Guiping Ma, Nzg3MTA5MDA4QHFxLmNvbQ==