 Enrico Maggi1
Enrico Maggi1 Nadine Landolina1
Nadine Landolina1 Francesca Romana Mariotti1
Francesca Romana Mariotti1 Enrico Munari2
Enrico Munari2 Nicola Tumino3
Nicola Tumino3 Paola Vacca3
Paola Vacca3 Bruno Azzarone1
Bruno Azzarone1 Lorenzo Moretta1*
Lorenzo Moretta1*- 1Tumor Immunology Unit, Bambino Gesù Children’s Hospital, IRCCS, Rome, Italy
- 2Department of Pathology and Diagnostics, University and Hospital Trust Verona, Verona, Italy
- 3Innate Lymphoid Cells Unit, Bambino Gesù Children’s Hospital, IRCCS, Rome, Italy
Innate immunity is the first line of defense against infections, including the detection and response to SARS-CoV-2. Cells of the innate system are usually activated within hours after pathogen exposure and do not generate conventional immunological memory. In this review, the current knowledge of the innate immune cells and of pattern-recognition receptors in sensing and responding to SARS-CoV-2 to mount a protective response has been shortly reviewed. Subsequently, the evasion strategies of the virus, as the inhibition of IFN-I/III production and autophagic response, counteracting the innate cell activity (including NK cells), have been briefly outlined. In the course of the infection, these strategies are also capable of rendering dysfunctional most innate cells, thus deeply interfering with the onset and maintenance of adaptive immunity. Possible mechanism(s) for the maintenance of dysfunctional innate immune response are also discussed. In this context, the importance of a rapid and robust activation of innate immunity through toll-like receptor (TLR) 4 as a key paradigm central to host defense against COVID-19 pathogenesis is also illustrated. We also discuss how the viral excess plus inflammatory signals upregulating TLR4 on innate cells may initiate a vicious loop which maintains and improves hyperinflammation, leading to the most critical outcomes. Targeting the TLR4 or its signaling pathway may be a promising therapeutic strategy, offering the dual benefits of viral suppression and decreasing inflammation.
1 Introduction
COVID-19 pandemic has been caused by the β-coronavirus SARS-CoV-2, that had a dreadful impact, resulting in more than seven million documented deaths worldwide and four hundred million (underestimated) of Long-COVID cases as far as 2024 (1). Although originally defined as a respiratory viral infection, COVID-19 is now clearly recognized as a more complex, multistep, multi-organ immune-mediated disease. The virus infects primarily the cells of the upper- and then of the lower respiratory tract, triggering a wide spectrum of clinical manifestations, from asymptomatic, mild, and moderate to severe and critical symptoms (2). While most SARS-CoV-2 infections are mild, some patients develop uncontrolled inflammatory cell death, systemic inflammation with severe cytokine storm (a general term applied to maladaptive cytokine release in response to infection and other stimuli) (3), pneumonia, acute respiratory distress syndrome -ARDS-, thrombosis, and multiorgan failure with fatal outcomes (4–6).
The innate immune system plays a primary role against infections, including SARS-CoV-2. It is usually activated within hours after pathogen exposure and does not generate immunological memory. It can be distinguished in immediate or induced innate immunity. The former is rapidly activated (0–4 h) and relies on the activity of preformed soluble antimicrobial molecules, including antimicrobial enzymes and complement (C‘) system proteins. The induced innate immunity begins later (4–72 h), involves the activation and the recruitment of cells (as neutrophils, monocytes, macrophages and natural killer -NK cells), and lasts few days after the first exposure to pathogens. In this phase, innate cells can mount a process of resistance to reinfection, termed “trained immunity”, which involves structural chromatin modifications, alterations in DNA methylation, histone acetylation, upregulation of inflammation-related genes and changes in metabolic intermediates. This “trained immunity” provides the innate system with a memory-like activity, allowing to respond more effectively to re-exposure to pathogens (7).
In this review, we will briefly examine the current knowledge of innate immune cells and pattern-recognition receptors (PRRs) in sensing and responding to SARS-CoV-2 as well as evasion strategies of the virus counteracting the innate immunity. We will focus on mechanism(s) for maintenance of dysfunctional innate immune cells through the upregulation/activation of toll-like receptor (TLR) 4 and leading to the most severe outcomes.
2 SARS-CoV-2 structure and activation of the innate immune response
2.1 SARS-CoV-2 genome and virus cell cycle
SARS-CoV-2 is made up of an enveloped structure containing a genome of approximately 30 kb, constituted by single-stranded RNA (ss-RNA) encoding 29 proteins with diverse functions (8). Four are structural virion components such as the spike (S), envelope (E), membrane (M), and nucleocapsid (N) proteins (9). The S glycoprotein (SP), assembled into homotrimers on virion particles, mediates viral entry by attaching to and fusing with the host cell membrane (10). SP is cleaved by convertases (as transmembrane serine protease type II -TMPRSS2-, cathepsins, furin or metalloproteases), into a mature protein formed by the two non-covalently associated S1 and S2 subunits (11). S1 consists of the amino-terminal (NTD), the receptor-binding (RBD), and two carboxy-terminal (CTDs) domains protecting the inner S2 subunit.
S1 binds the receptor Angiotensin-Converting Enzyme 2 (ACE2) through the RBD, while S2 links the cell membrane allowing viral entry (12). Endocytosis is another viral entry modality involving ACE2 plus other co-factors (as HSPG, PS receptors, NRP, CD147, C-type lectins) whose mechanism is partially defined. ACE2 is scarcely expressed on circulating immune cells, while it is highly present on cells (as Monocytes, Dendritic cells -DC-, Epithelial cells, type 2 Pneumocytes, Alveolar macrophages, etc.) of tissues and organs, especially in the respiratory and digestive mucosa and myocardium (13), making these tissues more susceptible to infection.
Within the host cell, the viral genome encodes nine accessory proteins that promote viral shape. The genome is immediately translated by producing two long polyproteins (pp1a and pp1ab) which are cleaved by virus-encoded proteases to 16 nonstructural proteins (Nsp) that are devoted to assembly the replication–transcription complex, to modulate host cell compartments for generation of new virions and to release them through exocytosis. They are effective upon interaction with multiple genetically encoded PRRs (14).
2.2 Virus-mediated activation of immunity
2.2.1 PRRs engagement
Thanks to the expression of different PRRs most of innate cells sense pathogen-associated- and damage-associated molecular patterns (PAMPs DAMPs) along the infection. Activated receptors drive the expression of pro-inflammatory cytokines, chemokines, adhesion molecules and interferons (IFNs), which recruit and activate other innate immune cells. This further amplifies the immune response and cell death to eliminate infected cells, promotes pathogen clearance, and, if the infection is not eradicated, activates adaptive immune response (15, 16). Among different PRRs, SARS-CoV-2 engages and triggers retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs), TLRs, cGAS and stimulator of interferon genes (STING) pathway, the inflammasome nucleotide-binding oligomerization domain (NOD)-like- (NLRs)/absent in melanoma 2 like- AIM2 (ALRs) and C-type lectin (CLRs) receptors (17). SARS-CoV-2 proteins induce also mitochondria damage which release of mitDNA activating cGAS-STING pathway, contributing to IFN-β expression (18) and, in endothelial cells, to vascular damage and coagulopathy in patients with severe COVID-19 (19). Importantly, TLR, RLR, cGAS engagement have a prevalent anti-viral impact (beneficial in early clearing the virus but detrimental if stimulation persists), while NLRs/ALRs or CLRs triggering are essentially devoted to promote inflammation and apoptosis of infected cells (14, 20). Some extensive reviews on the PRRs and their signaling pathways activated by SARS-CoV-2 has been recently reported (14, 21–25).
2.2.2 Autophagy induction
Macro-autophagy is part of the antiviral innate response (26, 27) and can be activated upon viral infection, stress sensing kinases, or triggered PRRs (28). During autophagy, cytoplasmic cargo, including viruses, is engulfed by double-layered membrane vesicles, named autophagosomes, and degraded upon fusion with lysosomes in a tightly regulated process that involves more than 30 autophagy-related proteins (ATGs) (29, 30). Following DNA/RNA virus entry, autophagy is the most active promoter of innate immunity through DAMP release. The molecular interplay between SARS-CoV-2 proteins and the immune-related process of autophagy is described in the section 2. Viral peptides derived from autophagic degradation are subsequently presented on MHC class I/II antigens by lymph nodal DCs to CD8+ and CD4+ T cells to initiate the virus-specific adaptive response (31).
2.2.3 SARS-CoV-2 full protection
It involves a plethora of immune cells of both innate and adaptive immunity (32–34). Monocytes play the major role in the very early response being recruited into lymph nodes where they differentiate into immature-DCs and subsequently in mature DCs which present viral peptides to T cells (35). Primed CD8+ T cells home to the site of infection to directly kill infected cells or secrete antiviral cytokines as IFN-γ and tumor necrosis factor alpha (TNF-α). In the lymph nodal germinal center, CD4+ T follicular helper (Tfh) cells promote B cell affinity maturation and their activation into antibody-secreting cells (31). Monocytes, recruited into infected tissues as lung, differentiate into macrophages; when activated by the virus, they promote M1-like polarization and release of chemokines favoring homing/activation of circulating (NK/NKT cells and neutrophils) and other tissue effector cells as mucosal-associated invariant T -MAIT-, γδ T, innate lymphoid cells -ILCs-, T resident memory -Trm-. NK and virus-specific CD8+ T cells play the major role in limiting infection through their ability to lyse infected cells and produce antiviral cytokines. When infected cells and viral load are significantly reduced, monocytes/macrophages turn off inflammation by reducing effector cells through the production of anti-inflammatory/suppressive cytokines (IL-10, IL-27, IL-35, TGF.β) (32–34, 36).
2.3 SARS-CoV-2 variants of concern (VOCs)
VOCs are characterized by mutations in the viral genome, particularly in the SP playing a crucial role in the virus’s ability to infect host cells (Table 1). Variants such as Alpha (B.1.1.7), Beta (B.1.351), Gamma (P.1), Delta (B.1.617.2), and Omicron (B.1.1.529) have been identified and classified by the World Health Organization as VOCs due to their different transmissibility, virulence, pathogenicity, ability to induce Long-Covid-19 and resistance to neutralizing antibodies induced by vaccines (37). The Omicron variant demonstrated a substantial increase in transmissibility compared to previous strains, likely linked to the high number of SP mutations. Even though the Delta variant’s R0 was estimated to be very high (between 5 and 8), however, R0 of Omicron VOC was found to be even higher (approximately 3.19 times greater than that of Delta) (38, 39). Moreover, Omicron VOC displays lower pathogenicity, high immune escape potential and a significant reduction in Long COVID induction and vaccine efficacy for infection (37, 40). Table 1 summarizes the main features of VOCs, including their interaction/activation of SP of each variant with TLR4/MD-2 complex (41).
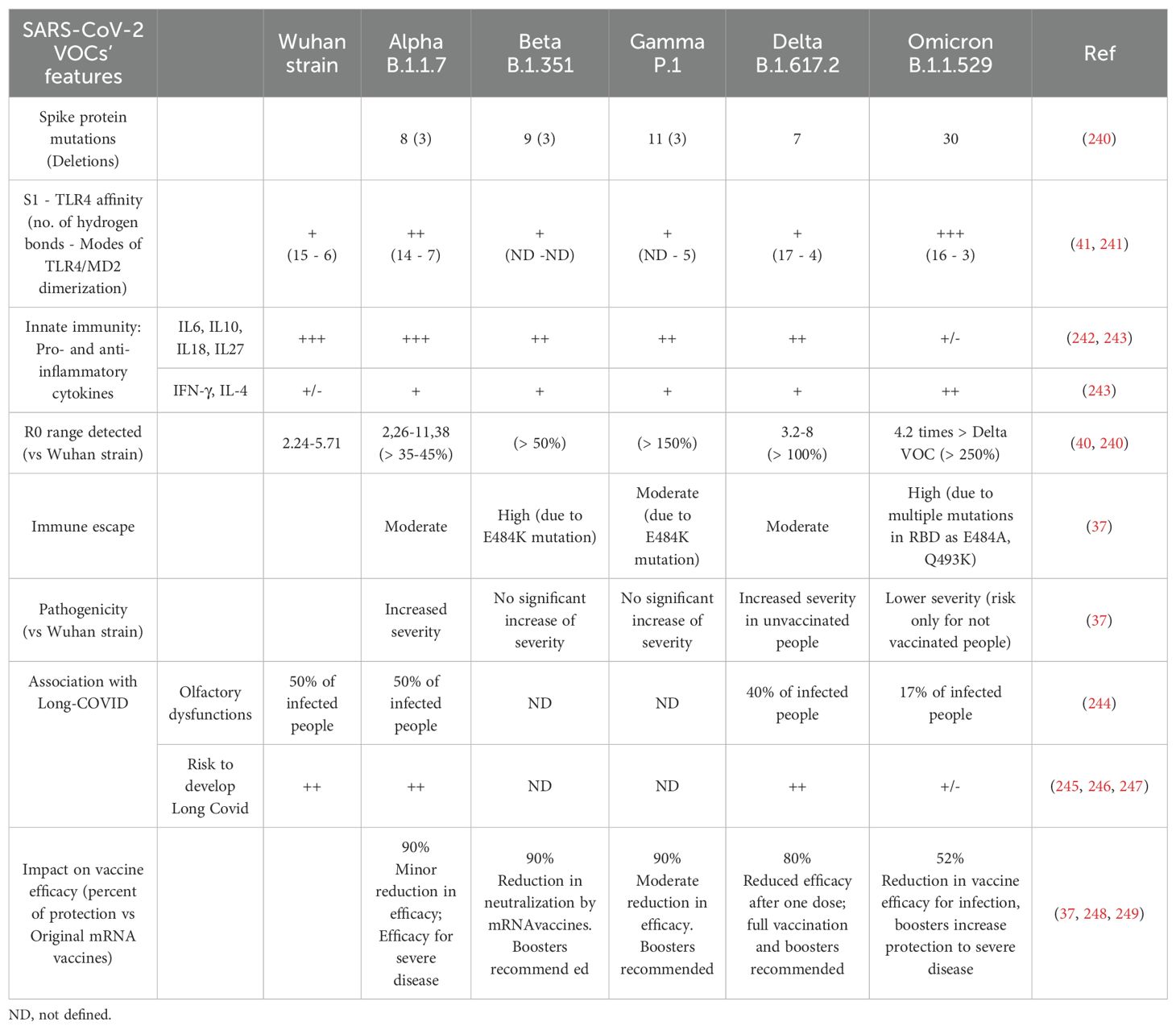
Table 1. Structural and pathophysiological features of the main SARS-CoV-2 VOCs.
3 Evasion strategies of SARS-CoV-2 counteracting innate immunity
The principal function of the innate system is to induce an inflammatory response devoted to limiting viral replication. However, the virus may evolve some evasion strategies to suppress host defense: of note, more than 50% of the SARS-CoV-2-encoded proteins may counteract innate response (42, 43). SARS-CoV-2 has evolved distinct, sometimes overlapping, mechanisms to antagonize IFN production and autophagy at multiple levels, all promoting viral replication.
The major IFN antagonists are Nsp1, Nsp3, ORF6 and ORF3a/b/c. Nsp1 was shown to block the mRNA channel of the ribosome, turning off the expression of cytokines or IFN-stimulated antiviral genes in infected cells (44). By harnessing its de-ubiquitinase activity exerted by papain-like protease (PLP) 2 domain, Nsp3 inhibits various steps of PRR signaling (45, 46), cleaving the ubiquitin-like ISG15 protein, thus antagonizing MDA5 and IRF3 activation (47, 48). SARS-CoV-2 induces multiple proteins encoded in the Open Reading Frame (ORF) loci. ORF3a reduces STAT1 phosphorylation, the main transcription factor of IFN (49), while ORF3b and ORF3c inhibit type I IFN production by targeting mitochondrial antiviral-signaling protein (MAVS) for cleavage by caspase-3 (50). ORF6 disrupts nucleocytoplasmic trafficking by binding the nuclear pore Nup98-Rae1 complex, inhibiting STAT1 nuclear translocation (51, 52). Even though ORF6 alone is not sufficient to antagonize IFN pathway (53), its high levels could increase IFN resistance of SARS-CoV-2 VOCs compared to original strain (54–57).
SARS-CoV-2 infection leads to an incomplete/dysfunctional autophagy with a higher turnover of autophagosomes. Pharmacological activation of autophagy reduces replication of human Coronaviruses (huCoVs) and spreading (58).
ORF3a and ORF7a are the key viral components causing impaired autophagy (59–64), by preventing viral clearance and favoring viral replication by the accumulation of autophagosomes.
ORF3a interacts with autophagy process at different levels.: it prevents the fusion between autophagosomes and lysosomes, decreasing the autophagic flux and providing an immune escape from autophagy (42, 61, 63). In addition, it shows activity on endo-lysosomal compartments promoting lysosomal exocytosis and improving viral release (65). Lastly, ORF3a has been reported to counteract the flux, modulating the antiviral effect of the non-canonical STING1-mediated autophagy (66).
ORF7a dysregulates the late stages of autophagy by inhibiting the acidification of lysosomes (42, 67) and prevents autophagosome-lysosome fusion by promoting the degradation of the SNARE protein SNAP29 (62).
SARS-CoV-2 can also block autophagy turnover through the structural proteins M and E, which leads to the accumulation of autophagosomes and p62 in the cell (42, 67). Similarly, M and ORF10 are able to counteract innate immunity by promoting the autophagic degradation of MAVS (through mitophagy), which are important antiviral elements associated with mitochondria, leading to a reduction of type I IFN (IFNI) production (68, 69). Like ORF3a ORF10 also counteracts non-canonical autophagy by inhibiting STING1 activation (70).
Non-structural proteins also are able to downregulate autophagy: SARS-CoV-2 papain-like protease Nsp3 reduces the starvation-induced autophagy and disrupts the formation of the initiation complex that involves ULK1 and ATG13 (71). Nsp4 promotes accumulation of autophagosomes (42), while Nsp6 inhibits the autophagy initiation by preventing the formation of pre-autophagosomal structures (72). The helicase Nsp13 mediates the autophagic degradation of TBK1, impairing IFN-I production and reducing innate immunity (53), while Nsp15, as ORF3a alters early phases of autophagy with the reduction of autophagosome formation (42).
The interplay beween SARS-CoV-2 proteins and the process of autophagy are extensively reviewed in three recent reports (73–75).
Overall, the principal effect of SARS-CoV-2 evasion strategies is the increased viremia which favors the persistent viral stimulation with two main consequences: an increased programmed cell death (PCD) and an increased dysfunction/unbalance of innate immunity.
3.1 Increased programmed cell death
Some PCD pathways are upstream of inflammatory processes, playing a critical role in favoring severe outcomes. Cytokines, PAMPs, and DAMPs promote some lytic forms of inflammatory cell death, contributing to fatal evolution of COVID-19 (76–79). For instance, the combination of IFN-γ and TNF-α induces PANoptosis (77), an inflammatory lytic cell death pathway of innate immunity driven by caspases and receptor-interacting protein kinases (RIPKs) that are regulated by multiprotein PANoptosome complexes. The IFN signaling molecules STAT1, IRF-1, NOS2 also promote activation of caspase-8-dependent complex inducing PANoptosis (77, 79, 80).
Proteins induced by IFN-α-activated IFN Signature Genes (ISG) (as ZBP1, AIM2, and ISG-15) may sense viral components forming similar multiprotein complexes leading to PANoptosis (76). In addition, AIM2 recognizing mitochondrial DNA, cell-free DNA, or endogenous DNA, forms another multiprotein complex (AIM2-PANoptosome) leading to PANoptosis. NLR pirin domain containing 1 and 3 (NLRP1/3) and AIM2 bind cytosolic DAMPs and PAMPs and activate the inflammasome, leading to pyroptosis.
PANoptosis induces the death of cells which, in turn, release DAMPs and alarmins engaging PRRs resulting in amplification of inflammation (77, 81). PCD is induced during the entire SARS-CoV-2 progression: initially, the virus infects the upper-airways, sensitizing epithelial cells to cell death (82, 83). Subsequently, it may spreads to alveoli infecting type II pneumocytes and trigger innate cells (84, 85) that undergo pyroptosis (61), releasing PAMPs/DAMPs and cytokines further recruiting and activating other cells (86, 87). Neutrophils mainly undergo neutrophil-extracellular traps (NETs) PCD (82, 88–91). Similarly, PANoptosis contributes to endothelial cell death and organ damage in adults with severe COVID-19 and children with Multisystem inflammatory syndrome (MIS-C) (92), possibly resulting in abnormal blood clots, lung damage, myocardial infarction, and stroke (93).
3.2 Dysfunction of innate cells
The virus and soluble S1 can interact with some active receptors/molecules (membrane binding lectins – MBL-, CD26, CD147, CD209, Histamine receptor H1, TLRs) expressed on many cell types by directly or indirectly interfering with functions of the majority of innate- and, subsequently, adaptive cells (94, 95). It also displays sequences with super antigenic- and/or self-antigen-like activity inducing activation of polyclonal- or autoreactive T cells.
Herein we will briefly examine how Virus/S1 protein-receptors interplay can modify the features of infected- and not infected cells of innate immunity during SARS-CoV-2 infection.
Thus, the number of circulating monocytes is reduced, showing an activated immature phenotype that does not result in production of excess of cytokines (96). In contrast, PB DCs appear much more dysfunctional expressing reduced anti-viral ISGs, MHC-Class II antigens and cytolytic activity (97, 98). Lung alveolar macrophages are replaced by inflammatory CD163+monocyte-derived (not tissue resident) macrophages sharing some regulatory activities with myeloid-derived suppressor cells (MDSCs), overexpressing inflammasome-, pro-fibrotic- and C’-related genes and producing pro-inflammatory chemokines and cytokines (99, 100). These molecules start and amplify a vicious loop further promoting lung homing of blood activated monocytes and T (Th17 and cytolytic CD8+) cells, that improve DAMP release and tissue damage, further activation of macrophages with increase of cytokine release (97).
It is not clear if neutrophils can be directly infected (89, 91, 101): TLR engagement activates downstream NF-kB and interferon regulatory factor 7 (IRF7) with the production of proteases, cationic polypeptides and pro-inflammatory cytokines/chemokines. Virus engagement of neutrophil TLR7/8, activates protein arginase deiminase 4 (PAD4), inducing chromatin de-condensation and NET formation (102, 103). NETs trigger a positive loop with macrophages that are activated, produce IL-1β, CCL1, CCL2, IL-6 and TNF-α, and further recruit neutrophils. Extracellular histones from NETs cause cell cytotoxicity promoting ARDS, sepsis and organ failure, while extracellular DNA, favors autoimmunity and, through mucus hyperproduction, bacteria superinfection with respiratory failure. Lastly, NETs release fibrinogen, Von Willebrand Factor (VWF) causing thrombosis in lung, kidney, liver and peripheral vessels (104). Endothelium damage and thrombosis can also be improved by SARS-CoV-2-mediated C’ activation through all three C’ pathways: they are started up S1 and N proteins, producing anaphylatoxins C3a and C5a, whose receptors are present on endothelial cells, platelets and most leukocytes, inducing a prothrombotic state frequently triggering thrombo-inflammatory events (105–107).
Increased mononuclear (M-) (108) and polymorphonuclear (PMN) MDSCs (109, 110) have been reported in COVID-19 patients (111). The MDSC gene signature is predominant in PB from severe patients: M-MDSC number is higher in severe vs mild patients (112), is related to viral load (113) and associated with secondary infections and mortality (114). The reduced/delayed IFN production associates with enhanced chemokines that recruit MDSCs into the lung, while high IL-6 may favor MDSC proliferation (115). PMN-MDSCs use reactive oxygen species (ROS) and L-arginase, whereas M-MDSCs use inducible nitric oxide synthase (iNOS) and L-arginase to suppress bystander immune cells (116). Regulatory functions of MDSCs include i. Induction of high PD-L1 expression decreasing antigen-specific T-cells through interaction with PD-1+ T-cells, ii. increased signaling through Galectin-9 and Tim-3 pathways inhibiting Th1 and CD8+T cells, iii. enhanced TGF-β and IL-10 enhancing suppressive function of M2-macrophages and proliferation of Treg cells, iv. upregulation of TGF-β, ROS and L-arginase inhibiting NK and CD8+ T cells, v. elevated pro-inflammatory cytokines contributing to cytokine storm (117).
Few reports on helper Innate Lymphoid Cells (ILCs) indicated decreased ILC- and ILC precursor subsets in all COVID-19 patients: however, the percentage of ILC2 upon total ILCs is increased in patients vs not infected controls (118, 119). CD117low ILC2, a subset secreting more type 2 cytokines, is expanded in COVID-19 patients as also confirmed by single cell RNA sequencing (scRNAseq) (118, 119). ILC2 and ILC precursors display enhanced CD69 and NKG2D and reduced CXCR3 and CCR4, CD25 and KLRG1 expression (118, 119). At present it is not clear whether ILC changes are related to worse or improved outcomes or may be considered a simple epiphenomenon (101).
The NK cell dysfunction in early infected- or severe patients has been repeatedly reported (118–120), the absolute number being reduced during infection (121) and restored after recovery (122). Patients with severe disease usually show at the early onset increased pro-inflammatory cytokines, including IFN−γ produced by NK cells (123). These cells are strongly activated (106) and highly express inhibitory checkpoint- (LAG3, PD-1, TIM-3) or inhibiting receptors mainly in CD56dim subset, suggesting a dysfunctional/exhausted profile (124), favoring the pathogenesis rather than limiting infection (125). Even though some reports indicated that NK alterations are due to enviromental signals (94, 123, 126), we recently demonstrated that the virus can directly activate NK cells till their exhaustion (127). HLA-E-binding S1 peptide(s) expressed by infected epithelial cells may favor lung homing and recognition of inhibitory CD94/NKG2A+ NK cells (128). NK cell dysfunction also associates with low NK-stimulating cytokines (as IL-12, IL-15) from APCs which, instead, produce IL-10 and TGF-β (129), as virus-stimulated fibroblasts, epithelial and endothelial cells (109). The IL-6 overproduction inhibits in vitro NK cell cytotoxicity (130) indirectly confirmed by the in vivo treatment with anti-IL-6R mAb which increases NK cell function in COVID-19 patients (131).
Dysfunctional innate cells have a great impact also on the upgrowth of altered adaptive immunity (110). Modified function of APCs impairs the induction of virus-specific T cells which also display lower cytotoxicity and IFN-γ production. M2-type macrophages and MDSCs lead to expanded non cytolytic type 2 cells in tissues (Th2, ILC2, Tfh2, etc) that, at lymph nodal level, stimulate antibody- and, sometimes, autoantibody production by B cells. Increased viral components (expressing superantigens or autoantigens) favor the expansion of non-specific polyclonal- or autoreactive T cells. Lastly, the cytokine milieu (TGF-β, IL-1β, IL-6, IL-23) of tissue inflamed cells favors the development and expansion of Th17 and Treg cells which can switch each other along the infection (126).
4 TLR4-SARS-CoV-2 interaction: role for inflammation maintenance
The maintenance of inflammation evolving to critical outcomes is essentially due to vicious circles involving the virus, dysfunctional innate cells and soluble molecules released from cells of the inflamed tissues. SARS-CoV-2 and its soluble proteins activate innate sensors, as TLRs, mostly expressed in innate cells and detecting not only pathogens but also DAMPs (132). TLR engagement initiates downstream signaling cascade, leading to the release of effector molecules such as inflammatory cytokines/chemokines (132). In the chapter below, we will discuss how, among extracellular TLRs, TLR4 plays a crucial role in maintaining SARS-CoV-2 infection.
4.1 The virus preferentially activates TLR4 signaling
Several viruses such as the respiratory syncytial-, vesicular stomatitis- and Ebola virus, can directly engage and activate TLR4 through their surface glycoproteins (133). Although the precise mechanism(s) by which these viruses activate TLR4 remains partially unknown, some authors emphasize the role of glycosylation or hydrophobic (hydrophobic pocket of MD-2 linked to TLR4) interactions. In silico studies have demonstrated that soluble S1 protein can bind TLR2 and TLR4, with a higher affinity for TLR4 (134, 135). This prediction was further validated by in vitro, ex vivo, and in vivo experiments clearly indicating that TLR4 is a high-affinity (~300 nM) cognate receptor for the trimeric S glycoprotein, suggesting its role as a mediator of the proinflammatory response in COVID-19 (127, 136–138).
4.1.1 Direct or endotoxin-mediated S1-TLR4 interactions
The S1-TLR4 direct interaction was debated for long time since a computational modeling analysis revealed a high affinity also between LPS and S1 (139), suggesting that endotoxin contamination in recombinant S1 preparations (produced in E. coli or human cells) might be responsible for TLR4 engagement and human macrophage activation. According to these authors, the compound Spike/LPS should act synergistically to induce cell activation, while individual components do not (140). Indeed, some evidence suggests that LPS may be involved in the hyperinflammation of SARS-CoV-2 infection: hospitalized severe COVID-19 patients exhibit elevated levels of LPS in circulation, which increase as the disease progresses (141). Moreover, subclinical infections with Gram-negative bacteria or low levels of LPS from the gut could contribute to interactions between LPS and the S protein in infected patients (142).
In odds with these findings, however, many reports underlined that endotoxin contamination is unlikely to be the sole driver for proinflammatory responses reiterating the direct activation of TLR4 by S1 (136). In agreement, our results indicate that a large spectrum of doses of exogeneous LPS did not potentiate the NK cell functions induced by ultrapure S1, but, rather, resulted in a decrease (127). In addition, S1 induces the NK cell release of TNF-α and IFN-γ that enhance the transcription of CD40 which, interacting with its ligand, stabilizes the membrane expression of TLR4 that serves as a receptor of S1 (143). Omicron S protein which exerts stronger binding affinity for TLR4 than the other VOCs, has been reported to bind LPS with reduced affinity compared to other variants (41, 144). Finally, taking into account that LPS directly binds CD14 and MD2, but not TLR4 (145), the recent in silico definition of the fine hydrophobic bonds between residues of S1 and TLR4 and their ability to induce TLR4 dimerization, strongly suggests that, at least in part, S protein binds TLR4 and triggers subsequent signaling (146). Further insights are, however, mandatory to define the exact role of LPS in S1-TLR4 interaction mainly in severe COVID19 patients.
4.1.2 The TLR4 structure and signaling
The structure of TLR4 includes an extracellular leucine rich repeat (LRR) domain, a transmembrane domain, and an intracellular Toll/Interleukin-1 receptor like (TIR) domain interacting with adaptor proteins TIR domain-containing adaptor protein (TIRAP) and TRIF-related adaptor molecule (TRAM) (147). The TLR4 signaling complex consists of cluster of differentiation 14 (CD14), myeloid differentiation factor-2 (MD-2), TLR4, and TIRAP or TRAM that initiate downstream signaling pathways in a dynamic manner. S1 protein from SARS-CoV-2 triggers two pathways, starting with TLR4 transformation and binding with Myeloid differentiation primary response 88 (MyD88) and TIR domain-containing adaptor protein inducing interferon beta (TRIF) proteins. The first pathway (MyD88-dependent) leads to inflammation through the activation of the IRAK4-IRAK1/IRAK2 complex and of TAK1, allowing the degradation of IκBα and favoring the entry of NF-κB into the nucleus to start the transcription of proinflammatory cytokine genes. TAK1 also triggers MAPK pathways with AP1 activation, which is crucial for cell survival and proliferation. The second pathway (via TRIF, MyD88-independent pathway), essential for the antiviral response, induces the activation of TRAF3 and TRAF6 and later of TBK1 of IKKϵ, two enzymes phosphorylating IRF3, which enters the nucleus and begins the transcription of type I IFN genes (ISG) (132).
4.1.3 TLR4 activation by soluble Spike protein
The entire virions, soluble S1 proteins and S1-bound exosomes are involved in TLR4 activation. The presence of soluble S1 is a relatively frequent event in SARS-CoV-2 inflamed environment due to its release from virus-infected and apoptotic cells. The cleavage of SP from each viral particle can produce about 50–100 molecules/virion of soluble S1, that can elicit multiple biological activities (148). High serum S1 levels have been reported during early onset and severe outcomes (124, 125). In post-acute sequelae of SARS-CoV-2 infection (PASC or Long-COVID19) high levels of soluble S1 and TLR4 expression have been described: importantly, in mice and humans, soluble SP may induce TLR4-mediated long-term cognitive dysfunction (135, 149–151). However, there is no evidence that TLR4 activation in PASC persists independently of detectable viral antigens. Clinical studies indicate that antagonizing TLR4 signaling dampens the cytokine storm of severe COVID-19, reduces mortality rates (152, 153), and has therapeutic effects in PASC patients (154).
High serum S1 levels have been also reported in post-vaccination side effects (155): indeed, the S1-coding mRNA, established by replacement of uridine with N1-methylpseudouridine (156) and packaged into lipid nanoparticles, is able to accumulate at the injection site and transported to lymph nodes through DCs. The unprocessed residual vaccine particles are spilled into bloodstream and high S1 levels may persist in blood and tissues for a long time after vaccination. The persistence of soluble S1 in some vaccinated individuals may induce pathogenic processes, being associated with increased expression of TLR4 in specific target cells (157–161).
SARS-CoV-2 can also indirectly activate TLR4 and hyperinflammatory pathways through high plasma levels of DAMPs or alarmins mostly produced by increased apoptosis of infected cells and causing cytokine storm in severe COVID-19 patients (162–165). DAMPs include i. Heat Shock Protein 70 which triggers inflammatory responses during chronic stress through TLR4 (166); ii. S100A8/A9, calcium-binding proteins that activate TLR4–MyD88 pathway (167, 168), thus favoring the output of cells as MDSCs (168); iii. Fibrinogen which stimulates the production of pro-inflammatory cytokines/chemokines in macrophages via TLR4 (169); iv. Secreted Protein, Acidic and Rich in Cysteines-like 1 (SPARCL1) that, through TLR4, induces lung inflammation inducing M1-macrophages and activating the NF-κB pathway (170); v. High Mobility Group Box 1 (HMGB1), released upon necrotic or hypoxic conditions, which promotes inflammation through TLR4 (171). HMGB1 serum level is higher in COVID-19 patients admitted to ICU compared to mild infection (172), whereas SPARCL1 plasma levels are increased in fatal COVID-19 compared to survivors (170).
4.2 SARS-CoV-2 infection contributes to upregulation of TLR4
Since peripheral blood mononuclear cells (PBMCs) from COVID-19 patients show enhanced TLR4 expression and phosphorylated NF-κB in circulating monocytes compared to healthy donors (HD) (173, 174), it has been hypothesized that some viral proteins or inflammatory signals may upregulate TLR4 during infection, thus facilitating the amplification of inflammatory circuits. Even though there is no evidence that soluble S1 or S1-bound exosomes can directly upregulate TLR4 after triggering innate cells, other mechanisms indirectly due to SARS-CoV-2 infection have been shown to enhance TLR4 expression.
The first signal is constituted by the decrease of surfactant proteins: the alveolar type II (ATII) pneumocytes represent the targets for SARS-CoV-2 infection due to high co-expression of ACE2 and TMPRSS2 (175, 176). ATII cells are also the sole producers of surfactants, a group of molecules selectively downregulating TLR4 expression on many cell types (177). Surfactant is a mixture of lipids (90%) and proteins (10%), exclusively produced by ATII. Palmitoyl-oleolyl-phosphatidylglycerols (POPG) present in extracellular compartment of alveoli are the molecules most implicated in blocking TLR4 in the lung to avoid untowards proinflammatory effects. The viral induced-ATII apoptosis leads to the decrease of surfactant and the contemporary break of suppressed TLR4 expression (22, 178, 179). Indeed, several reports indicated that reduced levels of POPG in the lung might contribute to develop, or cause, lung diseases as ARDS, because of insufficient suppression of inflammation due to high TLR4 expression and activation (180, 181). The increased TLR4 expression upregulates its interaction with S1, further increasing proinflammatory signaling pathways with enhancement of ISG and ACE2 expression (177). This favors viral replication and innate cell infiltration with a bias towards the signaling pathway leading to hyperinflammation within the alveoli (21). Engagement of overexpressed TLR4 can be considered a critical factor for the severity and mortality of COVID-19 patients, mainly those with comorbidities (177, 182), and for long-COVID-19 cardiac abnormalities (183).
Another signal upregulating TLR4 expression is related to platelet activation (184). Platelets expressing ACE2 and TMPRSS2 are activated by S1 proteins, favoring the release of Tissue Factor converting prothrombin to thrombin and starting clot formation (185). Some reports showed an increased expression of TLR4 on the surface of thrombin-activated platelets of severe COVID-19 patients. Indeed, thrombin can signal through the protease-activated (PAR1 or PAR4) receptors expressed on platelets to induce a phospholipase C (PLC)-dependent intracellular calcium mobilization, which activates calpain and favors intracellular α-granules-containing TLR4 trafficking towards the surface of platelets (186). This event may have relevant inflammatory consequences, since S1 and TLR4 co-localize on platelets isolated from COVID-19 patients with aggregated platelets and thrombus growth (186). Additionally, SARS-CoV-2-containing S100A8/A9+megakaryocytes, exhibit high TLR4 surface expression that correlates with NF-κB activation and the levels of released IL-6 and IL-1β (187). These cells are considered significant risk factors for mortality and multiorgan injury in COVID-19 patients (187). The platelet activation by S1 and the subsequent TLR4 overexpression are responsible for the thrombophilia state associated with severe outcomes (184).
A third mechanism upregulating TLR4 expression is the direct consequence of S1-ACE2 interaction which is usually associated with downregulation of ACE2 via different mechanisms (188). ACE2 reduction favors the increase of angiotensin II (AngII) which, by interacting with AngII type 1 receptors (AT1-R), induces vasoconstriction, hypoxemia, increased endothelial injury, and tissue necrosis which further contribute to increase both inflammation and diffuse thromboembolic effects (189, 190). Downstream signaling as receptor tyrosine kinases, NF-κB, MAP Kinases and, above all, the upregulation of TLR4 are linked to the AngII-AT1-R interaction, especially in SARS-CoV-2-related cardiovascular diseases (191). Interestingly, the increase of TLR4/NF-κB signaling and cytokines by M1 macrophages following AngII-AT1-R interaction have been largely confirmed in animal models (192).
Finally, the elevated testosterone levels and some TLR4 polymorphisms could also be implicated in the upregulation of TLR4.
4.3 TLR4 activation of immune- and non Immune cells
4.3.1 Immune cells
The viral infection has a deep impact on the increase of TLR4 expression on innate (macrophages, DC, neutrophils, NK) immune and non-immune (neurons, epithelial) cells, which, in turn, upgrow inflammatory responses through the same receptor (137).
4.3.2 Macrophages/DCs
S1-TLR4 interplay can primarily activate macrophages/DCs in murine models leading to the release of inflammatory mediators such as IL-1β, TNF-α, IL-6 and nitric oxide through the NF-kB and JNK signaling pathways, which are deeply suppressed with specific antagonists. In agreement, the proinflammatory response to S1 was abrogated in macrophages from TLR4−/− mice (136) or with siRNA targeting TLR4 (193). TLR4 engagement favors ACE2 expression, viral entry and hyperinflammation of macrophages, playing the major role in amplifying inflammatory circuits during severe COVID-19 (194).
4.3.3 Neutrophils
TLR4 also contributes to the formation of neutrophil extracellular traps (NETs) that exacerbates and prolongs the deleterious proinflammatory environment in severe patients (195). TLR4-induced expression of ACE2 was higher in the myeloid cells of severe COVID-19 patients and was associated with elevated levels of the immune checkpoint molecule PD-L1 that can suppress antiviral T cell response upon interaction with PD-1 on effector cells (196, 197).
4.3.4 NK cells
Furthermore, S1-TLR4 interaction involves directly also NK cells. We have recently shown that S1 from the Wuhan strain and other VOCs bind and activate TLR4 (and less TLR2) on purified PB NK cells by increasing phosphorylation of NFkB, activation marker expression, cytokine release, and cytolytic activity (127). Of note, S1-TLR2/4 interaction does not trigger ACE2 on NK cells or their activating receptors (DNAM1, NKGD2 Nkp30/44, etc) (127). Recently recovered patients displayed a higher proportion of circulating NK cells (vs HD) which can be stimulated in vitro by S1. This likely explains why NK cells are currently highly activated in vivo during infection and recovery. In addition, S1 significantly amplified in vitro NKG2C+CD56dim NK cells, a phenotype typical of “trained cells” (127). In agreement, some studies observed that increased adaptive response is followed by expansion of exhausted NKG2C+CD57+NK cells (198, 199). Notably, since signals inducing trained immunity, such as BCG, initiate their activity via TLR2/TLR4 engagement, it is conceivable that S1 induces expansion of trained NK cells through a similar mechanism (200). It is likely that S1-driven NK cell activation can induce initially the amplification of protective trained (NKG2C+CD56dim) cells favoring cytolytic activity of infected cells and upregulation of IFN-γ activating a response characterized by M1- and Th1 cells. Persistent viral load and chronic S1 stimulation may, however, lead to exhaustion of NK cells downregulating cytotoxicity and IFN-γ production and favoring sustained inflammation and viral spread (Figures 1A, B).
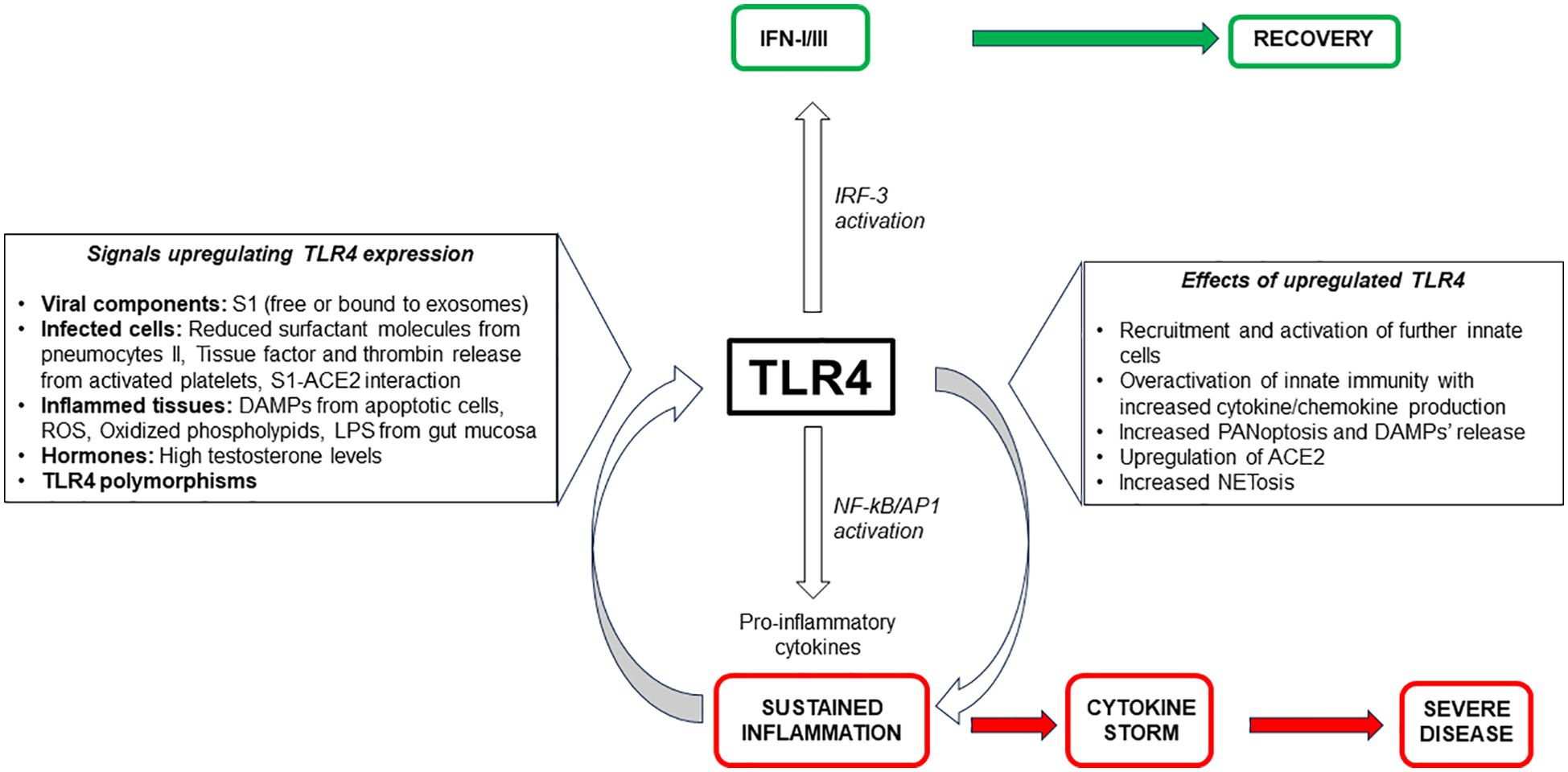
Figure 1. (A) Protective Immunity loops to fight SARS-CoV-2 infection. (B) Persistent viral load and high soluble S1 protein impair innate immunity leading to detrimental effects.
4.3.5 Non-immune cells
Beyond the effects on endothelial cells, TLR4 triggering has also been shown to directly mediate neuroinflammation or renal tubular epithelial cell damages.
Indeed, in murine models S1 induces neuro-inflammation and memory dysfunction in post-COVID-19 syndrome through TLR4 pathway (149, 201). Other in vivo studies have revealed the possibility that S1 may induce neuroinflammation and memory dysfunction through TLR4-expressing microglial cells and neurons (149). The pediatric cerebral cortical neuronal cell line (HCN-2) lacking ACE2 exhibited elevated TLR4 transcript levels alongside increased secretion of proinflammatory cytokines and chemokines, indicating that TLR4 can mediate neurological effects (202). Remarkably, by crossing the blood–brain barrier S1 triggers neuroinflammation thus supporting the hypothesis that the virus may produce comparable effects in human Long COVID-19 patients experiencing cognitive dysfunction (203).
Lastly, it has been shown that TLR4 is one of the risk genes associated with immune-inflammation-promoting renal injury in severe COVID-19 patients (204). By using human kidney cell lines, it has been demonstrated that SARS-CoV-2 directly induces damage of renal tubular epithelial cells via TLR4 and IL-1R signaling (205). Moreover, TLR4 contributes to kidney damage favoring SARS-CoV-2-induced inhibition of albumin endocytosis through decreased Akt activity in proximal tubule epithelial cells (206).
4.4 Population genetics and comorbilities
TLR4 polymorphisms can also play a role in the pathogenesis of COVID-19. Human TLR4 presents two notable single nucleotide polymorphisms (SNPs)—896 A/G and 1196 C/T—favoring COVID-19 severity (207). On the other hand, the 896 A/G variant has recently been identified as a protective factor against COVID-19 progression among younger individuals without cardiovascular abnormalities (208). Patients carrying the 1196 C/T SNP develop more frequently pneumonia, leading to critical manifestations (209), whereas the rs4986790 (896) GG genotype displays a defective TLR4 signaling leading to cellular dysfunction, associated with severe disease (210). Other TLR4 polymorphisms linked to COVID-19 severity as SNP -2604G>A has been associated with increased neuroinflammation and cognitive dysfunction (149). TLR4 polymorphism is likely to be involved also in Long-COVID-19: 52% of severe long-COVID patients carried at least one disease signature variant in TLR4 (154). Lastly, beyond genetic polymorphisms, TLR4 expression is higher in men vs women due to testosterone levels, correlating with elevated pro-inflammatory cytokine levels, mainly in COVID-19 (195).
Some recent reports on the quantitative analysis of public transcriptomic datasets on TLR4 expression levels in COVID 19 have been published, even though they do not relate it to the disease severity. By comparing two or three gene expression datasets and performing bioinformatic methods to construct protein-protein interaction (PPI) networks, in three separate reports TLR4 always resulted among the hub genes more expressed (211–213). By contrast, metanalyses on TLR4 expression in COVID-19 are at present lacking. The role of TLR4 hyperexpression and signaling on innate immune cells is summarized on Figure 2.
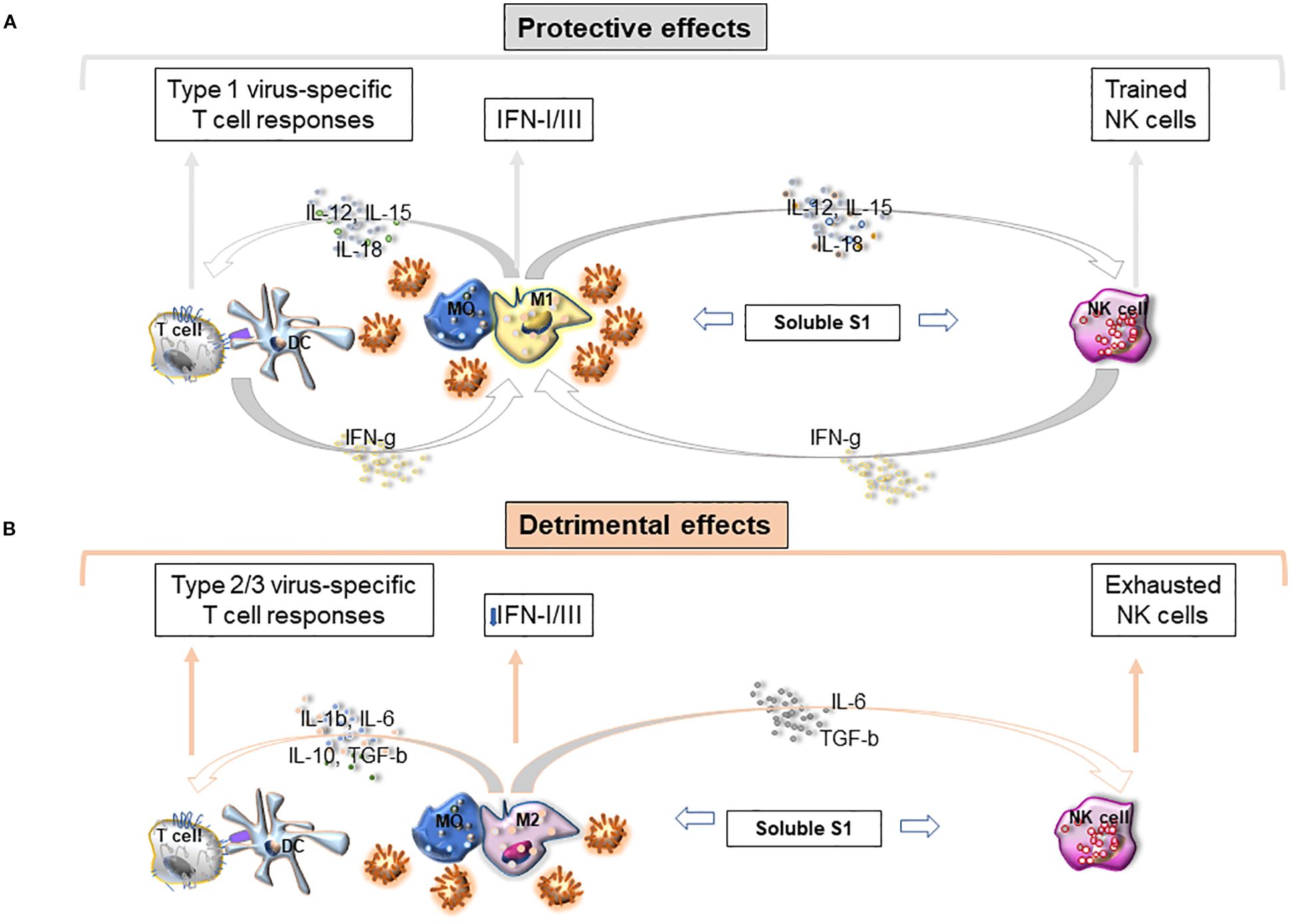
Figure 2. Role of TLR4 expression and signaling on innate cells in the pathogenesis of severe COVID-19.
4.5 Clinical relevance of S1-TLR4 interplay
Clearcut results indicated that TLR4 is broadly upregulated in COVID-19 patients and participates in various COVID-19-related pathologies. TLR4 (and less TLR2) expression is upregulated in PBMCs or BALF from severe patients compared to mild cases or HD (173, 214). Critical COVID-19 patients exhibit higher levels of TLR4 and phosphorylated NF-κB in CD14+ HLA-DRhigh circulating monocytes, with increased NF-κB p65 phosphorylation in the CD14+ HLA-DRlow monocyte subset (174, 215). Moreover, severe patients exhibited a two-fold increase of TLR4 expression in nasopharyngeal cell samples as compared to patients with mild disease (216) and TLR4 plasma levels correlated positively with COVID-19 severity (217). Lastly, in the autopsy of COVID-19 patients a massive TLR4 upregulation in the lung was associated with increased macrophage infiltration, presenting a shift from GAL-3+ alveolar macrophages to CD163+ myeloid-derived monocyte-macrophages: even though these results do not show any direct cause/effect relationship, however, they indicate that TLR4 expression may induce a persistent inflammation, with inefficient resolution, and pathological macrophage shift which could be one of the mechanisms of lethal COVID-19 (218). Notably, COVID-19 non-survivors have higher plasma levels of LPS (the most usual TLR4 ligand) than survivors, due to virus-related intestinal permeabilization and translocation into the blood of enteric pathogens or their products (141). Hospitalized severe COVID-19 patients also display elevated LPS levels increasing with the disease progression, thus confirming that the endotoxin itself may play a role in SARS-CoV-2 hyperinflammation (141). Finally, TLR4 activation also exhibited reduced cytokine secretion from monocytes of convalescent COVID-19 patients (219). These data suggest that following SARS-CoV-2 infection, chronically stimulated monocytes exhibit exhausted steady-state gene expression and reduced responsiveness. This may be also due to the increased levels of soluble TLR4 and CD14 acting as decoy receptors (probably released upon an excess of TLR4 activation) which decrease TLR4-mediated signaling and inflammatory responses (220). A clinical consequence of a sustained decrease in the response of these PRRs could also be an increased susceptibility to other unrelated infections or superinfection with other pathogens (221, 222).
Lastly, as previously underlined, TLR4 is also involved in SARS-CoV-2-associated extra-pulmonary immune-pathologies as the kidney injury and the neurological symptoms (149, 206).
4.6 Central role of TLR4 to orient the outcomes of infection
TLR4 does not fully encompass the disease’s complex and intricate immune mechanisms of SARS-CoV-2 infection, involving a wide network of immune sensors and pathways. However, the TLR4 pathway plays a significant role in severe inflammation since, as described, the inflammatory signals enhancing TLR4 expression make TLR4-bearing cells more susceptible to triggering by viral components, thus increasing and maintaining inflammation (22). Thus, it is possible to speculate on a dual role of TLR4, both protective and deleterious, depending on the phase of the disease. As long as the type I IFN induced by the TLR4 signaling pathway remains unmodified, the antiviral response is effective; the viral load is at low levels and clinical remission occurs. In this phase S1-activated TLR4+ NK cells favor the antiviral effects by increasing their function, mounting a “trained immunity” response and contributing to protection towards the virus. However, when the evasion strategies of the virus mainly impairing IFN release and activity, predominates, the viral load increases and the pro-inflammatory responses induced by TLR4 signaling greatly prevail. Under these conditions a vicious circle is established essentially due to multiple mechanisms increasing TLR4 expression and its active signaling. In the absence of a valid IFN response, the upregulation of TLR4, in turn, stimulates ACE2 expression, NETosis and PANoptosis, with a substantial increase of cytokines/chemokines recruiting new circulating cells. In this phase persistent S1 activation of NK cells through TLR4 leads to cell exhaustion and consequentially to the enhancement of viral load. Such events facilitate stimulation of macrophages by viral S1 and DAMPs which further upregulate TLR4, thus creating a feedback loop, where heightened TLR4 levels increase accessibility to S protein leading to the inflammation maintenance and favoring severe outcomes. According with some authors TLR4 can be considered a critical “fate-deciding molecule” for the pathogenesis of severe COVID-19 (22, 153, 179) (Figure 2).
5 Targeting TLR4 is a novel therapeutic option for SARS-CoV-2 infection
By considering the TLR4 role in immune response and disease pathogenesis, molecules or vaccines targeting TLR4 may provide a therapeuticl option for SARS-CoV-2 and for the majority of huCoV infections (22). A complete and exhaustive review on TLR4 agonists and antagonists and drugs interfering with TLR4-S1 interaction has been recently reported (21). It is important to underline, however, that few papers are at present available on TLR4 inhibition in animal models of severe SARS-CoV-1 infection and COVID-19 (223, 224). In addition, the majority of these approaches often failed when employed in other types of diseases such as sepsis and ischemic stroke (225–227).
A variety of natural products, particularly biomacromolecules (LPS from the bacterium Rhodobacter sphaeroides and TLR4-binding peptide derived from Bacillus-fermented soybean), have been investigated as alternative options to block TLR4 or disrupt downstream signaling pathways (228). Phytochemicals, such as jacareubin, cajastelebenic acid, andrographolide, cannabidiol, and berberine, have been documented as potent blockers of TLR4, some also being validated with clinical trials (229). A series of chemically synthesized compounds and peptides have been identified for their ability to interfere with TLR4 activity that might help in combating COVID-19. For example, TLR4 antagonists such as Eritoran sulfate (E5564) and FP7 have shown efficacy in reducing lethal damage associated with severe influenza and sepsis (230).
Other potentially useful compounds are Opioids (naloxone, naltrexone, and tramadol), which exhibit TLR4-antagonizing properties. Many small molecules (as Disulfiram, dimethyl fumarate, fluoroquinolone antibiotics) that directly or indirectly antagonize TLR4 are also in development or undergoing preclinical validation. Some TLR4 agonists and antagonists have reached various phases of clinical trials, including peptides (EC-18), chemical compounds like imiquimod, hydroxychloroquine, and artesunate, DPP4 inhibitors or small molecules (as PUL-042) (153).
Moreover, the administration of probiotic bacterial strains has emerged as a promising approach to impair the harmful effects of TLR4 activation with potential benefit in COVID-19 patients. Genetically engineered probiotic (bacterium Lactobacillus paracasei F19 producing palmitoyl-ethanolamide) has been intranasal administered resulting effective in reducing SARS-CoV-2-associated lung injury by blocking TLR4- mediated NLRP3 activation and decreasing pro- inflammatory cytokines (231).
Alternative strategies targeting TLR4 include monoclonal antibodies (mAbs) with inhibitory activity (232). For instance, paridiprubart (EB05) prevents TLR4 dimer formation, thereby blocking the response to TLR4 agonists such as S1. Importantly, this compound resulted in 100% survival in coronavirus mouse model (168). It has potential for yielding similar beneficial effects in impairing the extreme inflammatory response observed in Interstitial lung fibrosis and ARDS. Even though approved for advanced phase 3 trials, unfortunately they were suspended or ended inconclusively for the lack of patient recruitment (https://clinicaltrials.gov/study/NCT04401475, https://clinicaltrials.gov/study/NCT05293236). Since many VOCs and their subvariants have developed resistance to mAb treatments, the design of chimeric mAbs incorporating complementarity-determining regions (CDRs) from regdanivimab and sotrovimab, or from bebtelovimab and adintrevimab, has been proposed (233–235).
Aptamers are a further innovative approach to target TLR4 in COVID-19 since they: i. are small size, single-stranded DNA or RNA, molecules folded into unique 3D structures, ii. are specifically bound to target molecules with high affinity (236), usually displaying less steric hindrance and better access to binding sites compared to antibodies, iii. are of easy synthesis, low immunogenicity, and useful in detection and therapy. APToll is a notable aptamer currently in clinical use for cerebral ischemic events, demonstrating the potential benefit of aptamer-based therapies (236).
The emergence of new VOCs often evading the protection provided by the antibody-induced response elicited by Spike-based vaccines imposes to develop new type of vaccines based on pathogenetic, poorly mutagenic molecular structures (153). In the AbhiSCoVac vaccine the constructed peptide is designed to stably engage major immune sensors like TLR4, TLR2, MHC class I and II (237). Due the Spike-TLR4 interaction a common feature of huCVs, studies are presently focused to identify specific sequences responsible for this interaction, to develop a multi-epitope multi-target chimeric vaccine effective against not only SARS-CoV-2 VOCs but also virtually all huCVs (237).
Table 2 summarizes the most recent therapeutic approaches antagonizing TLR4 engagement and signaling.
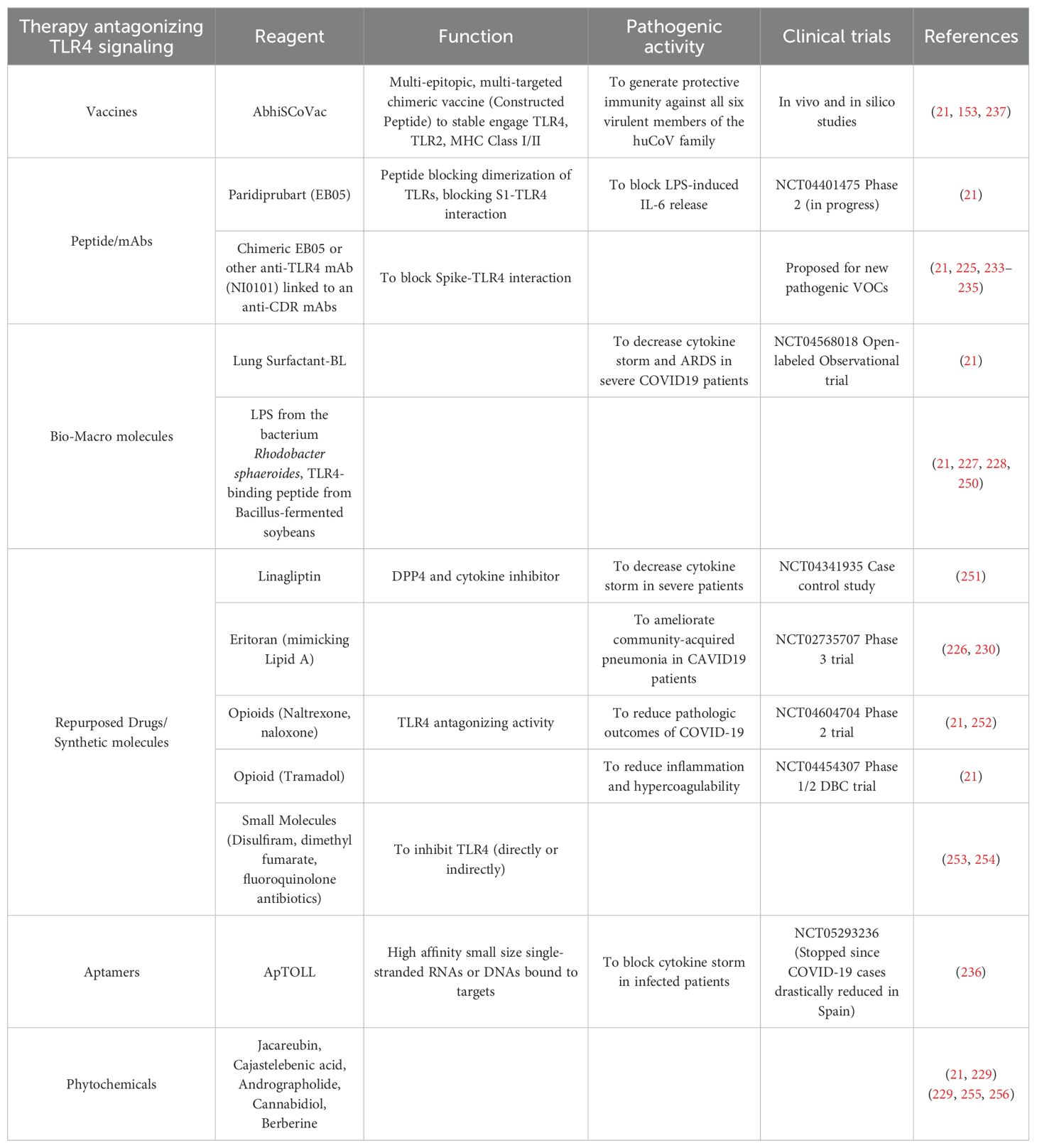
Table 2. Therapeutical compounds to inhibit TLR4 engagement or its signaling.
In conclusion, even though some data are highly promising, more studies are needed to define how any proposed TLR4-related therapeutic strategies would be relevant in real world practice, due to the complexities (as host-genetic polymorphisms, VOC-specific immune engagement, correct timing and duration of treatment) and risks (potential losing antiviral and even a general immune protection) to interfering with such an innate fundamental receptor.
6 Conclusions
The innate immune system is the first line of defense against infections, including SARS-CoV-2. In this review we have examined cells and molecules of the innate immunity playing a critical role in the early phase of infection and conditioning the subsequent adaptive response and clinical outcome towards recovery. In this phase, TLRs has a beneficial effect, since the early and strong protective TLR-mediated innate immune response against viruses or viral components is essential for viral clearance, though the secretion of antiviral cytokines, chemokines and type I IFNs.
However, the virus exploits evasion strategies to counteract the innate response prevalently through the inhibition of type I/III IFN and autophagic mechanisms: this leads to C’ overactivation, hyperinflammation, pan-apoptosis and increased viral load which exert a deep dysfunctional impact on cells of both innate- and adaptive immunity.
The maintenance of SARS-CoV-2-related inflammation evolving into critical outcomes is essentially sustained by vicious loops involving dysfunctional innate cells and signals from inflamed environmental cells. In this phase TLRs may be harmful for SARS-CoV-2 infection eliciting dysregulated immune signaling: the excessive TLR activation due to overstimulation by viral proteins or DAMPs released from apoptotic cells can lead to the untoward production of proinflammatory cytokines and chemokines, resulting in severe disease.
In this context the principal vicious circle involves in particular TLR4, which is selectively engaged by S1 protein. These receptors are triggered not only on all APC but also on NK cells: S1 protein strongly increases the function of these cells, selectively expanding initially NKG2C+NK (trained) cells. The persistent S1 stimulation by soluble protein which may be elevated along the infection and in Long-COVID-19, turns this protective mechanism into a progressive exhaustion which increases inflammation and favors virus persistence. The viral excess (S1 protein and Nsps blocking IFN and protective mechanisms) plus various inflammatory signals upregulating TLR4 on innate cells creates a vicious circle maintaining and further enhancing hyperinflammation mediated primarily by monocytes and macrophages, leading to severe or even fatal outcomes. Thus, TLR-dependent anti-viral response or excessive inflammation may tip the balance towards the former or the latter, altering the equilibrium that drives the severity of disease (216). For these reasons, therapeutics targeting the TLR4 signaling pathway may be a promising strategy, potentially offering the dual benefits of viral suppression and inflammation shutdown. Persistent inflammation and immune dysregulation sustained by TLR4 involvement are thought to play an important role also in the case of Long-Covid-19 (238). Pharmacologic agents targeting TLR4 could help in rebalancing the immune system, reducing the likelihood of autoimmune-like conditions observed in these patients (239). Thus, TLR4 inhibitors not only offer a means to mitigate the acute inflammatory response during the initial infection but also provide an option to address the long-term sequelae of COVID-19, mitigating symptoms and accelerating patient recovery.
Author contributions
EMa: Visualization, Validation, Conceptualization, Writing – review & editing, Writing – original draft. NL: Writing – review & editing. FM: Writing – review & editing. EMu: Writing – review & editing. NT: Writing – review & editing. PV: Writing – review & editing. BA: Writing – review & editing. LM: Visualization, Funding acquisition, Validation, Writing – review & editing, Conceptualization, Writing – original draft.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This work was supported by Associazione Italiana per la Ricerca sul Cancro (project no. 5x1000–2018 Id 21147 to LM) and by the Italian Ministry of Health with “Current Research funds”. NL is supported by Fondazione Umberto Veronesi.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Kronvall G and Nordenfelt E. On the history of human coronaviruses. APMIS. (2021) 129:381–3. doi: 10.1111/apm.13109
2. Wu F, Zhao S, Yu B, Chen YM, Wang W, Song ZG, et al. A new coronavirus associated with human respiratory disease in China. Nature. (2020) 579:265–9. doi: 10.1038/s41586-020-2008-3
3. Ye Q, Wang B, and Mao J. The pathogenesis and treatment of the;Cytokine Storm’ in COVID-19. J Infect. (2020) 80:607–13. doi: 10.1016/j.jinf.2020.03.037
4. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. (2020) 395:497–506. doi: 10.1016/S0140-6736(20)30183-5
5. Del Valle DM, Kim-Schulze S, Huang HH, Beckmann ND, Nirenberg S, Wang B, et al. An inflammatory cytokine signature predicts COVID-19 severity and survival. Nat Med. (2020) 26:1636–43. doi: 10.1038/s41591-020-1051-9
6. Karki R and Kanneganti TD. The ‘cytokine storm’: molecular mechanisms and therapeutic prospects. Trends Immunol. (2021) 42:681–705. doi: 10.1016/j.it.2021.06.001
7. Dagenais A, Villalba-Guerrero C, and Olivier M. Trained immunity: A “new” weapon in the fight against infectious diseases. Front Immunol. (2023) 14:1147476. doi: 10.3389/fimmu.2023.1147476
8. Cao C, Cai Z, Xiao X, Rao J, Chen J, Hu N, et al. The architecture of the SARS-CoV-2 RNA genome inside virion. Nat Commun. (2021) 12:3917. doi: 10.1038/s41467-021-22785-x
9. Troyano-Hernaez P, Reinosa R, and Holguin A. Evolution of SARS-coV-2 envelope, membrane, nucleocapsid, and spike structural proteins from the beginning of the pandemic to september 2020: A global and regional approach by epidemiological week. Viruses. (2021) 13:243. doi: 10.3390/v13020243
10. Jackson CB, Farzan M, Chen B, and Choe H. Mechanisms of SARS-CoV-2 entry into cells. Nat Rev Mol Cell Biol. (2022) 23:3–20. doi: 10.1038/s41580-021-00418-x
11. Hoffmann M, Kleine-Weber H, and Pohlmann S. A multibasic cleavage site in the spike protein of SARS-coV-2 is essential for infection of human lung cells. Mol Cell. (2020) 78:779–84 e5. doi: 10.1016/j.molcel.2020.04.022
12. Nejat R, Torshizi MF, and Najafi DJ. S protein, ACE2 and host cell proteases in SARS-coV-2 cell entry and infectivity; is soluble ACE2 a two blade sword? A narrative review. Vaccines (Basel). (2023) 11:204. doi: 10.3390/vaccines11020204
13. Ashraf UM, Abokor AA, Edwards JM, Waigi EW, Royfman RS, Hasan SA, et al. SARS-CoV-2, ACE2 expression, and systemic organ invasion. Physiol Genomics. (2021) 53:51–60. doi: 10.1152/physiolgenomics.00087.2020
14. Malireddi RKS, Sharma BR, and Kanneganti TD. Innate immunity in protection and pathogenesis during coronavirus infections and COVID-19. Annu Rev Immunol. (2024) 42:615–45. doi: 10.1146/annurev-immunol-083122-043545
15. Diamond MS and Kanneganti TD. Innate immunity: the first line of defense against SARS-CoV-2. Nat Immunol. (2022) 23:165–76. doi: 10.1038/s41590-021-01091-0
16. Karki R and Kanneganti TD. Innate immunity, cytokine storm, and inflammatory cell death in COVID-19. J Transl Med. (2022) 20:542. doi: 10.1186/s12967-022-03767-z
17. Yamada T and Takaoka A. Innate immune recognition against SARS-CoV-2. Inflammation Regen. (2023) 43:7. doi: 10.1186/s41232-023-00259-5
18. Singh KK, Chaubey G, Chen JY, and Suravajhala P. Decoding SARS-CoV-2 hijacking of host mitochondria in COVID-19 pathogenesis. Am J Physiol Cell Physiol. (2020) 319:C258–C67. doi: 10.1152/ajpcell.00224.2020
19. Liu Y, Jesus AA, Marrero B, Yang D, Ramsey SE, Sanchez GAM, et al. Activated STING in a vascular and pulmonary syndrome. N Engl J Med. (2014) 371:507–18. doi: 10.1056/NEJMoa1312625
20. Lu Q, Liu J, Zhao S, Gomez Castro MF, Laurent-Rolle M, Dong J, et al. SARS-CoV-2 exacerbates proinflammatory responses in myeloid cells through C-type lectin receptors and Tweety family member 2. Immunity. (2021) 54:1304–19 e9. doi: 10.1016/j.immuni.2021.05.006
21. Mukherjee S and Bayry J. The yin and yang of TLR4 in COVID-19. Cytokine Growth Factor Rev. (2025) 82:70–85. doi: 10.1016/j.cytogfr.2024.10.001
22. Asaba CN, Ekabe CJ, Ayuk HS, Gwanyama BN, Bitazar R, and Bukong TN. Interplay of TLR4 and SARS-coV-2: unveiling the complex mechanisms of inflammation and severity in COVID-19 infections. J Inflammation Res. (2024) 17:5077–91. doi: 10.2147/JIR.S474707
23. Gambari R and Finotti A. Interplay of TLR4 and SARS-coV-2: possible involvement of microRNAs [Letter. J Inflammation Res. (2024) 17:7963–4. doi: 10.2147/JIR.S501862
24. Behzadi P, Chandran D, Chakraborty C, Bhattacharya M, Saikumar G, Dhama K, et al. The dual role of toll-like receptors in COVID-19: Balancing protective immunity and immunopathogenesis. Int J Biol Macromol. (2025) 284:137836. doi: 10.1016/j.ijbiomac.2024.137836
25. Stegeman SK, Kourko O, Amsden H, Pellizzari Delano IE, Mamatis JE, Roth M, et al. RNA viruses, toll-like receptors, and cytokines: the perfect storm? J Innate Immun. (2025) 17:126–53. doi: 10.1159/000543608
26. Levine B, Mizushima N, and Virgin HW. Autophagy in immunity and inflammation. Nature. (2011) 469:323–35. doi: 10.1038/nature09782
27. Choi Y, Bowman JW, and Jung JU. Autophagy during viral infection - a double-edged sword. Nat Rev Microbiol. (2018) 16:341–54. doi: 10.1038/s41579-018-0003-6
28. Dunlop EA and Tee AR. mTOR and autophagy: a dynamic relationship governed by nutrients and energy. Semin Cell Dev Biol. (2014) 36:121–9. doi: 10.1016/j.semcdb.2014.08.006
29. He C and Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. (2009) 43:67–93. doi: 10.1146/annurev-genet-102808-114910
30. Wesselborg S and Stork B. Autophagy signal transduction by ATG proteins: from hierarchies to networks. Cell Mol Life Sci. (2015) 72:4721–57. doi: 10.1007/s00018-015-2034-8
31. Manfrini N, Notarbartolo S, Grifantini R, and Pesce E. SARS-coV-2: A glance at the innate immune response elicited by infection and vaccination. Antibodies (Basel). (2024) 13:13. doi: 10.3390/antib13010013
32. Silverstein NJ, Wang Y, Manickas-Hill Z, Carbone C, Dauphin A, Boribong BP, et al. Innate lymphoid cells and COVID-19 severity in SARS-CoV-2 infection. Elife. (2022) 11:11. doi: 10.7554/eLife.74681
33. Schultze JL and Aschenbrenner AC. COVID-19 and the human innate immune system. Cell. (2021) 184:1671–92. doi: 10.1016/j.cell.2021.02.029
34. Nguyen THO, Rowntree LC, Chua BY, Thwaites RS, and Kedzierska K. Defining the balance between optimal immunity and immunopathology in influenza virus infection. Nat Rev Immunol. (2024) 24:720–35. doi: 10.1038/s41577-024-01029-1
35. Chang T, Yang J, Deng H, Chen D, Yang X, and Tang ZH. Depletion and dysfunction of dendritic cells: understanding SARS-coV-2 infection. Front Immunol. (2022) 13:843342. doi: 10.3389/fimmu.2022.843342
36. Guilliams M, Mildner A, and Yona S. Developmental and functional heterogeneity of monocytes. Immunity. (2018) 49:595–613. doi: 10.1016/j.immuni.2018.10.005
37. Alhamlan FS and Al-Qahtani AA. SARS-coV-2 variants: genetic insights, epidemiological tracking, and implications for vaccine strategies. Int J Mol Sci. (2025) 26:1263. doi: 10.3390/ijms26031263
38. Abudunaibi B, Liu W, Guo Z, Zhao Z, Rui J, Song W, et al. A comparative study on the three calculation methods for reproduction numbers of COVID-19. Front Med (Lausanne). (2023) 9:1079842. doi: 10.3389/fmed.2022.1079842
39. Ito K, Piantham C, and Nishiura H. Estimating relative generation times and reproduction numbers of Omicron BA.1 and BA.2 with respect to Delta variant in Denmark. Math Biosci Eng. (2022) 19:9005–17. doi: 10.3934/mbe.2022418
40. Karimizadeh Z, Dowran R, Mokhtari-Azad T, and Shafiei-Jandaghi NZ. The reproduction rate of severe acute respiratory syndrome coronavirus 2 different variants recently circulated in human: a narrative review. Eur J Med Res. (2023) 28:94. doi: 10.1186/s40001-023-01047-0
41. Chakraborty C, Mallick B, Bhattacharya M, and Byrareddy SN. SARS-CoV-2 Omicron Spike shows strong binding affinity and favourable interaction landscape with the TLR4/MD2 compared to other variants. J Genet Eng Biotechnol. (2024) 22:100347. doi: 10.1016/j.jgeb.2023.100347
42. Hayn M, Hirschenberger M, Koepke L, Nchioua R, Straub JH, Klute S, et al. Systematic functional analysis of SARS-CoV-2 proteins uncovers viral innate immune antagonists and remaining vulnerabilities. Cell Rep. (2021) 35:109126. doi: 10.1016/j.celrep.2021.109126
43. Lee JH, Koepke L, Kirchhoff F, and Sparrer KMJ. Interferon antagonists encoded by SARS-CoV-2 at a glance. Med Microbiol Immunol. (2023) 212:125–31. doi: 10.1007/s00430-022-00734-9
44. Thoms M, Buschauer R, Ameismeier M, Koepke L, Denk T, Hirschenberger M, et al. Structural basis for translational shutdown and immune evasion by the Nsp1 protein of SARS-CoV-2. Science. (2020) 369:1249–55. doi: 10.1126/science.abc8665
45. Freitas BT, Durie IA, Murray J, Longo JE, Miller HC, Crich D, et al. Characterization and noncovalent inhibition of the deubiquitinase and deISGylase activity of SARS-coV-2 papain-like protease. ACS Infect Dis. (2020) 6:2099–109. doi: 10.1021/acsinfecdis.0c00168
46. Armstrong LA, Lange SM, Dee Cesare V, Matthews SP, Nirujogi RS, Cole I, et al. Biochemical characterization of protease activity of Nsp3 from SARS-CoV-2 and its inhibition by nanobodies. PloS One. (2021) 16:e0253364. doi: 10.1371/journal.pone.0253364
47. Liu G, Lee JH, Parker ZM, Acharya D, Chiang JJ, van Gent M, et al. ISG15-dependent activation of the sensor MDA5 is antagonized by the SARS-CoV-2 papain-like protease to evade host innate immunity. Nat Microbiol. (2021) 6:467–78. doi: 10.1038/s41564-021-00884-1
48. Shin D, Mukherjee R, Grewe D, Bojkova D, Baek K, Bhattacharya A, et al. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature. (2020) 587:657–62. doi: 10.1038/s41586-020-2601-5
49. Yang X, Chen X, Bian G, Tu J, Xing Y, Wang Y, et al. Proteolytic processing, deubiquitinase and interferon antagonist activities of Middle East respiratory syndrome coronavirus papain-like protease. J Gen Virol. (2014) 95:614–26. doi: 10.1099/vir.0.059014-0
50. Stewart H, Lu Y, O’Keefe S, Valpadashi A, Cruz-Zaragoza LD, Michel HA, et al. The SARS-CoV-2 protein ORF3c is a mitochondrial modulator of innate immunity. iScience. (2023) 26:108080. doi: 10.1016/j.isci.2023.108080
51. Li T, Wen Y, Guo H, Yang T, Yang H, and Ji X. Molecular mechanism of SARS-coVs orf6 targeting the rae1-nup98 complex to compete with mRNA nuclear export. Front Mol Biosci. (2021) 8:813248. doi: 10.3389/fmolb.2021.813248
52. Kehrer T, Cupic A, Ye C, Yildiz S, Bouhaddou M, Crossland NA, et al. Impact of SARS-CoV-2 ORF6 and its variant polymorphisms on host responses and viral pathogenesis. Cell Host Microbe. (2023) 31:1668–84 e12. doi: 10.1016/j.chom.2023.08.003
53. Li M, Ayyanathan K, Dittmar M, Miller J, Tapescu I, Lee JS, et al. SARS-CoV-2 ORF6 protein does not antagonize interferon signaling in respiratory epithelial Calu-3 cells during infection. mBio. (2023) 14:e0119423. doi: 10.1128/mbio.01194-23
54. Nchioua R, Schundner A, Klute S, Koepke L, Hirschenberger M, Noettger S, et al. Reduced replication but increased interferon resistance of SARS-CoV-2 Omicron BA.1. Life Sci Alliance. (2023) 6:e202201745. doi: 10.26508/lsa.202201745
55. Bouhaddou M, Reuschl AK, Polacco BJ, Thorne LG, Ummadi MR, Ye C, et al. SARS-CoV-2 variants evolve convergent strategies to remodel the host response. Cell. (2023) 186:4597–614 e26. doi: 10.1016/j.cell.2023.08.026
56. Sacchi A, Giannessi F, Sabatini A, Percario ZA, and Affabris E. SARS-coV-2 evasion of the interferon system: can we restore its effectiveness? Int J Mol Sci. (2023) 24:9353. doi: 10.3390/ijms24119353
57. Ahmadi S, Bazargan M, Elahi R, and Esmaeilzadeh A. Immune evasion of severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2); molecular approaches. Mol Immunol. (2023) 156:10–9. doi: 10.1016/j.molimm.2022.11.020
58. Li M, Ferretti M, Ying B, Descamps H, Lee E, Dittmar M, et al. Pharmacological activation of STING blocks SARS-CoV-2 infection. Sci Immunol. (2021) 6:eabi9007. doi: 10.1126/sciimmunol.abi9007
59. Su WQ, Yu XJ, and Zhou CM. SARS-coV-2 ORF3a induces incomplete autophagy via the unfolded protein response. Viruses. (2021) 13:2467. doi: 10.3390/v13122467
60. Qu Y, Wang X, Zhu Y, Wang W, Wang Y, Hu G, et al. ORF3a-mediated incomplete autophagy facilitates severe acute respiratory syndrome coronavirus-2 replication. Front Cell Dev Biol. (2021) 9:716208. doi: 10.3389/fcell.2021.716208
61. Zhang Y, Sun H, Pei R, Mao B, Zhao Z, Li H, et al. The SARS-CoV-2 protein ORF3a inhibits fusion of autophagosomes with lysosomes. Cell Discov. (2021) 7:31. doi: 10.1038/s41421-021-00268-z
62. Hou P, Wang X, Wang H, Wang T, Yu Z, Xu C, et al. The ORF7a protein of SARS-CoV-2 initiates autophagy and limits autophagosome-lysosome fusion via degradation of SNAP29 to promote virus replication. Autophagy. (2023) 19:551–69. doi: 10.1080/15548627.2022.2084686
63. Miao G, Zhao H, Li Y, Ji M, Chen Y, Shi Y, et al. ORF3a of the COVID-19 virus SARS-CoV-2 blocks HOPS complex-mediated assembly of the SNARE complex required for autolysosome formation. Dev Cell. (2021) 56:427–442.e5. doi: 10.1016/j.devcel.2020.12.010
64. Li S, Li X, Liang H, Yu K, Zhai J, Xue M, et al. SARS-CoV-2 ORF7a blocked autophagy flux by intervening in the fusion between autophagosome and lysosome to promote viral infection and pathogenesis. J Med Virol. (2023) 95:e29200. doi: 10.1002/jmv.29200
65. Chen D, Zheng Q, Sun L, Ji M, Li Y, Deng H, et al. ORF3a of SARS-CoV-2 promotes lysosomal exocytosis-mediated viral egress. Dev Cell. (2021) 56:3250–3263.e5. doi: 10.1016/j.devcel.2021.10.006
66. Su J, Shen S, Hu Y, Chen S, Cheng L, Cai Y, et al. SARS-CoV-2 ORF3a inhibits cGAS-STING-mediated autophagy flux and antiviral function. J Med Virol. (2023) 95:e28175. doi: 10.1002/jmv.28175
67. Koepke L, Hirschenberger M, Hayn M, Kirchhoff F, and Sparrer KM. Manipulation of autophagy by SARS-CoV-2 proteins. Autophagy. (2021) 17:2659–61. doi: 10.1080/15548627.2021.1953847
68. Li X, Hou P, Ma W, Wang X, Wang H, Yu Z, et al. SARS-CoV-2 ORF10 suppresses the antiviral innate immune response by degrading MAVS through mitophagy. Cell Mol Immunol. (2022) 19:67–78. doi: 10.1038/s41423-021-00807-4
69. Hui X, Zhang L, Cao L, Huang K, Zhao Y, Zhang Y, et al. SARS-CoV-2 promote autophagy to suppress type I interferon response. Signal Transduct Target Ther. (2021) 6:180. doi: 10.1038/s41392-021-00574-8
70. Han L, Zheng Y, Deng J, Nan ML, Xiao Y, Zhuang MW, et al. SARS-CoV-2 ORF10 antagonizes STING-dependent interferon activation and autophagy. J Med Virol. (2022) 94:5174–88. doi: 10.1002/jmv.27965
71. Mohamud Y, Xue YC, Liu H, Ng CS, Bahreyni A, Jan E, et al. The papain-like protease of coronaviruses cleaves ULK1 to disrupt host autophagy. Biochem Biophys Res Commun. (2021) 540:75–82. doi: 10.1016/j.bbrc.2020.12.091
72. Kumar S, Javed R, Mudd M, Pallikkuth S, Lidke KA, Jain A, et al. Mammalian hybrid pre-autophagosomal structure HyPAS generates autophagosomes. Cell. (2021) 184:5950–5969.e22. doi: 10.1016/j.cell.2021.10.017
73. Yu J, Ge S, Li J, Zhang Y, Xu J, Wang Y, et al. Interaction between coronaviruses and the autophagic response. Front Cell Infect Microbiol. (2024) 14:1457617. doi: 10.3389/fcimb.2024.1457617
74. Shao Q, Liu T, Hu B, and Chen L. Interplay between autophagy and apoptosis in human viral pathogenesis. Virus Res. (2025) 359:199611. doi: 10.1016/j.virusres.2025.199611
75. Zhou H, Hu Z, and Castro-Gonzalez S. Bidirectional interplay between SARS-CoV-2 and autophagy. mBio. (2023) 14:e0102023. doi: 10.1128/mbio.01020-23
76. Karki R, Lee S, Mall R, Pandian N, Wang Y, Sharma BR, et al. ZBP1-dependent inflammatory cell death, PANoptosis, and cytokine storm disrupt IFN therapeutic efficacy during coronavirus infection. Sci Immunol. (2022) 7:eabo6294. doi: 10.1126/sciimmunol.abo6294
77. Karki R, Sharma BR, Tuladhar S, Williams EP, Zalduondo L, Samir P, et al. Synergism of TNF-alpha and IFN-gamma triggers inflammatory cell death, tissue damage, and mortality in SARS-coV-2 infection and cytokine shock syndromes. Cell. (2021) 184:149–68 e17. doi: 10.1016/j.cell.2020.11.025
78. Sefik E, Qu R, Junqueira C, Kaffe E, Mirza H, Zhao J, et al. Inflammasome activation in infected macrophages drives COVID-19 pathology. bioRxiv. (2022), 2021.09.27.461948. doi: 10.1101/2021.09.27.461948. 2021.09.27.461948.
79. Simpson DS, Pang J, Weir A, Kong IY, Fritsch M, Rashidi M, et al. Interferon-gamma primes macrophages for pathogen ligand-induced killing via a caspase-8 and mitochondrial cell death pathway. Immunity. (2022) 55:423–41 e9. doi: 10.1016/j.immuni.2022.01.003
80. Peng R, Wang CK, Wang-Kan X, Idorn M, Kjaer M, Zhou FY, et al. Human ZBP1 induces cell death-independent inflammatory signaling via RIPK3 and RIPK1. EMBO Rep. (2022) 23:e55839. doi: 10.15252/embr.202255839
81. Schifanella L, Anderson J, Wieking G, Southern PJ, Antinori S, Galli M, et al. The defenders of the alveolus succumb in COVID-19 pneumonia to SARS-coV-2 and necroptosis, pyroptosis, and PANoptosis. J Infect Dis. (2023) 227:1245–54. doi: 10.1093/infdis/jiad056
82. Abbate A, Toldo S, Marchetti C, Kron J, Van Tassell BW, and Dinarello CA. Interleukin-1 and the inflammasome as therapeutic targets in cardiovascular disease. Circ Res. (2020) 126:1260–80. doi: 10.1161/CIRCRESAHA.120.315937
83. Varga Z, Flammer AJ, Steiger P, Haberecker M, Andermatt R, Zinkernagel AS, et al. Endothelial cell infection and endotheliitis in COVID-19. Lancet. (2020) 395:1417–8. doi: 10.1016/S0140-6736(20)30937-5
84. Wu A, Peng Y, Huang B, Ding X, Wang X, Niu P, et al. Genome composition and divergence of the novel coronavirus (2019-nCoV) originating in China. Cell Host Microbe. (2020) 27:325–8. doi: 10.1016/j.chom.2020.02.001
85. Hou YJ, Okuda K, Edwards CE, Martinez DR, Asakura T, Dinnon KH 3rd, et al. SARS-coV-2 reverse genetics reveals a variable infection gradient in the respiratory tract. Cell. (2020) 182:429–46 e14. doi: 10.1016/j.cell.2020.05.042
86. Saichi M, Ladjemi MZ, Korniotis S, Rousseau C, Ait Hamou Z, Massenet-Regad L, et al. Single-cell RNA sequencing of blood antigen-presenting cells in severe COVID-19 reveals multi-process defects in antiviral immunity. Nat Cell Biol. (2021) 23:538–51. doi: 10.1038/s41556-021-00681-2
87. Shi CS, Nabar NR, Huang NN, and Kehrl JH. SARS-Coronavirus Open Reading Frame-8b triggers intracellular stress pathways and activates NLRP3 inflammasomes. Cell Death Discov. (2019) 5:101. doi: 10.1038/s41420-019-0181-7
88. Koupenova M, Corkrey HA, Vitseva O, Tanriverdi K, Somasundaran M, Liu P, et al. SARS-coV-2 initiates programmed cell death in platelets. Circ Res. (2021) 129:631–46. doi: 10.1161/CIRCRESAHA.121.319117
89. Veras FP, Pontelli MC, Silva CM, Toller-Kawahisa JE, de Lima M, Nascimento DC, et al. SARS-CoV-2-triggered neutrophil extracellular traps mediate COVID-19 pathology. J Exp Med. (2020) 217:e20201129. doi: 10.1084/jem.20201129
90. Zuo Y, Yalavarthi S, Shi H, Gockman K, Zuo M, Madison JA, et al. Neutrophil extracellular traps in COVID-19. JCI Insight. (2020) 5:e138999. doi: 10.1172/jci.insight.138999
91. Arcanjo A, Logullo J, Menezes CCB, de Souza Carvalho Giangiarulo TC, Dos Reis MC, de Castro GMM, et al. The emerging role of neutrophil extracellular traps in severe acute respiratory syndrome coronavirus 2 (COVID-19). Sci Rep. (2020) 10:19630. doi: 10.1038/s41598-020-76781-0
92. Rowley AH. Understanding SARS-CoV-2-related multisystem inflammatory syndrome in children. Nat Rev Immunol. (2020) 20:453–4. doi: 10.1038/s41577-020-0367-5
93. Leentjens J, van Haaps TF, Wessels PF, Schutgens REG, and Middeldorp S. COVID-19-associated coagulopathy and antithrombotic agents-lessons after 1 year. Lancet Haematol. (2021) 8:e524–e33. doi: 10.1016/S2352-3026(21)00105-8
94. Maggi E, Azzarone BG, Canonica GW, and Moretta L. What we know and still ignore on COVID-19 immune pathogenesis and a proposal based on the experience of allergic disorders. Allergy. (2022) 77:1114–28. doi: 10.1111/all.15112
95. Yu F, Liu X, Ou H, Li X, Liu R, Lv X, et al. The histamine receptor H1 acts as an alternative receptor for SARS-CoV-2. mBio. (2024) 15:e0108824. doi: 10.1128/mbio.01088-24
96. Wilk AJ, Rustagi A, Zhao NQ, Roque J, Martinez-Colon GJ, McKechnie JL, et al. A single-cell atlas of the peripheral immune response in patients with severe COVID-19. Nat Med. (2020) 26:1070–6. doi: 10.1038/s41591-020-0944-y
97. Knoll R, Schultze JL, and Schulte-Schrepping J. Monocytes and macrophages in COVID-19. Front Immunol. (2021) 12:720109. doi: 10.3389/fimmu.2021.720109
98. Leon J, Michelson DA, Olejnik J, Chowdhary K, Oh HS, Hume AJ, et al. A virus-specific monocyte inflammatory phenotype is induced by SARS-CoV-2 at the immune-epithelial interface. Proc Natl Acad Sci U.S.A. (2022) 119:e2116853118. doi: 10.1073/pnas.2116853118
99. Wauters E, Van Mol P, Garg AD, Jansen S, Van Herck Y, Vanderbeke L, et al. Discriminating mild from critical COVID-19 by innate and adaptive immune single-cell profiling of bronchoalveolar lavages. Cell Res. (2021) 31:272–90. doi: 10.1038/s41422-020-00455-9
100. Grassi G, Notari S, Gili S, Bordoni V, Casetti R, Cimini E, et al. Myeloid-derived suppressor cells in COVID-19: the paradox of good. Front Immunol. (2022) 13:842949. doi: 10.3389/fimmu.2022.842949
101. Khan Z, Shen XZ, Bernstein EA, Giani JF, Eriguchi M, Zhao TV, et al. Angiotensin-converting enzyme enhances the oxidative response and bactericidal activity of neutrophils. Blood. (2017) 130:328–39. doi: 10.1182/blood-2016-11-752006
102. Silva CMS, Wanderley CWS, Veras FP, Sonego F, Nascimento DC, Goncalves AV, et al. Gasdermin D inhibition prevents multiple organ dysfunction during sepsis by blocking NET formation. Blood. (2021) 138:2702–13. doi: 10.1182/blood.2021011525
103. Thiam HR, Wong SL, Wagner DD, and Waterman CM. Cellular mechanisms of NETosis. Annu Rev Cell Dev Biol. (2020) 36:191–218. doi: 10.1146/annurev-cellbio-020520-111016
104. Radermecker C, Detrembleur N, Guiot J, Cavalier E, Henket M, d’Emal C, et al. Neutrophil extracellular traps infiltrate the lung airway, interstitial, and vascular compartments in severe COVID-19. J Exp Med. (2020) 217:e20201012. doi: 10.1084/jem.20201012
105. Gupta A and Gupta GS. Status of mannose-binding lectin (MBL) and complement system in COVID-19 patients and therapeutic applications of antiviral plant MBLs. Mol Cell Biochem. (2021) 476:2917–42. doi: 10.1007/s11010-021-04107-3
106. Conway EM and Pryzdial ELG. Complement contributions to COVID-19. Curr Opin Hematol. (2022) 29:259–65. doi: 10.1097/MOH.0000000000000724
107. Xiao MT, Ellsworth CR, and Qin X. Emerging role of complement in COVID-19 and other respiratory virus diseases. Cell Mol Life Sci. (2024) 81:94. doi: 10.1007/s00018-024-05157-8
108. Kvedaraite E, Hertwig L, Sinha I, Ponzetta A, Hed Myrberg I, Lourda M, et al. Major alterations in the mononuclear phagocyte landscape associated with COVID-19 severity. Proc Natl Acad Sci U S A. (2021) 118:e2018587118. doi: 10.1073/pnas.2018587118
109. Silvin A, Chapuis N, Dunsmore G, Goubet AG, Dubuisson A, Derosa L, et al. Elevated calprotectin and abnormal myeloid cell subsets discriminate severe from mild COVID-19. Cell. (2020) 182:1401–1418.e18. doi: 10.1016/j.cell.2020.08.002
110. LaSalle TJ, Gonye ALK, Freeman SS, Kaplonek P, Gushterova I, Kays KR, et al. Longitudinal characterization of circulating neutrophils uncovers phenotypes associated with severity in hospitalized COVID-19 patients. Cell Rep Med. (2022) 3:100779. doi: 10.1016/j.xcrm.2022.100779
111. Schulte-Schrepping J, Reusch N, Paclik D, Bassler K, Schlickeiser S, Zhang B, et al. Severe COVID-19 is marked by a dysregulated myeloid cell compartment. Cell. (2020) 182:1419–1440.e23. doi: 10.1016/j.cell.2020.08.001
112. Thompson EA, Cascino K, Ordonez AA, Zhou W, Vaghasia A, Hamacher-Brady A, et al. Metabolic programs define dysfunctional immune responses in severe COVID-19 patients. Cell Rep. (2021) 34:108863. doi: 10.1016/j.celrep.2021.108863
113. Xue G, Jiang M, Zhao R, Le A, and Li J. Elevated frequencies of CD14(+)HLA-DR(lo/neg) MDSCs in COVID-19 patients. Aging (Albany NY). (2021) 13:6236–46. doi: 10.18632/aging.202571
114. Marais C, Claude C, Semaan N, Charbel R, Barreault S, Travert B, et al. Myeloid phenotypes in severe COVID-19 predict secondary infection and mortality: a pilot study. Ann Intensive Care. (2021) 11:111. doi: 10.1186/s13613-021-00896-4
115. Reyes M, Filbin MR, Bhattacharyya RP, Sonny A, Mehta A, Billman K, et al. Plasma from patients with bacterial sepsis or severe COVID-19 induces suppressive myeloid cell production from hematopoietic progenitors in vitro. Sci Transl Med. (2021) 13:eabe9599. doi: 10.1126/scitranslmed.abe9599
116. Beliakova-Bethell N, Maruthai K, Xu R, Salvador LCM, and Garg A. Monocytic-myeloid derived suppressor cells suppress T-cell responses in recovered SARS coV2-infected individuals. Front Immunol. (2022) 13:894543. doi: 10.3389/fimmu.2022.894543
117. Len JS, Koh CWT, and Chan KR. The functional roles of MDSCs in severe COVID-19 pathogenesis. Viruses. (2023) 16:27. doi: 10.3390/v16010027
118. Garcia M, Kokkinou E, Carrasco Garcia A, Parrot T, Palma Medina LM, Maleki KT, et al. Innate lymphoid cell composition associates with COVID-19 disease severity. Clin Transl Immunol. (2020) 9:e1224. doi: 10.1002/cti2.1224
119. Gomez-Cadena A, Spehner L, Kroemer M, Khelil MB, Bouiller K, Verdeil G, et al. Severe COVID-19 patients exhibit an ILC2 NKG2D(+) population in their impaired ILC compartment. Cell Mol Immunol. (2021) 18:484–6. doi: 10.1038/s41423-020-00596-2
120. Di Vito C, Calcaterra F, Coianiz N, Terzoli S, Voza A, Mikulak J, et al. Natural killer cells in SARS-coV-2 infection: pathophysiology and therapeutic implications. Front Immunol. (2022) 13:888248. doi: 10.3389/fimmu.2022.888248
121. Giamarellos-Bourboulis EJ, Netea MG, Rovina N, Akinosoglou K, Antoniadou A, Antonakos N, et al. Complex immune dysregulation in COVID-19 patients with severe respiratory failure. Cell Host Microbe. (2020) 27:992–1000.e3. doi: 10.1016/j.chom.2020.04.009
122. Maucourant C, Filipovic I, Ponzetta A, Aleman S, Cornillet M, Hertwig L, et al. Natural killer cell immunotypes related to COVID-19 disease severity. Sci Immunol. (2020) 5:eabd6832. doi: 10.1126/sciimmunol.abd6832
123. Kramer B, Knoll R, Bonaguro L, ToVinh M, Raabe J, Astaburuaga-Garcia R, et al. Early IFN-alpha signatures and persistent dysfunction are distinguishing features of NK cells in severe COVID-19. Immunity. (2021) 54:2650–2669.e14. doi: 10.1016/j.immuni.2021.09.002
124. Demaria O, Carvelli J, Batista L, Thibult ML, Morel A, Andre P, et al. Identification of druggable inhibitory immune checkpoints on Natural Killer cells in COVID-19. Cell Mol Immunol. (2020) 17:995–7. doi: 10.1038/s41423-020-0493-9
125. Gallardo-Zapata J and Maldonado-Bernal C. Natural killer cell exhaustion in SARS-CoV-2 infection. Innate Immun. (2022) 28:189–98. doi: 10.1177/17534259221077750
126. Maggi E, Canonica GW, and Moretta L. COVID-19: Unanswered questions on immune response and pathogenesis. J Allergy Clin Immunol. (2020) 146:18–22. doi: 10.1016/j.jaci.2020.05.001
127. Landolina N, Ricci B, Veneziani I, Alicata C, Mariotti FR, Pelosi A, et al. TLR2/4 are novel activating receptors for SARS-CoV-2 spike protein on NK cells. Front Immunol. (2024) 15:1368946. doi: 10.3389/fimmu.2024.1368946
128. Bortolotti D, Gentili V, Rizzo S, Rotola A, and Rizzo R. SARS-coV-2 spike 1 protein controls natural killer cell activation via the HLA-E/NKG2A pathway. Cells. (2020) 9:1975. doi: 10.3390/cells9091975
129. Osman M, Faridi RM, Sligl W, Shabani-Rad MT, Dharmani-Khan P, Parker A, et al. Impaired natural killer cell counts and cytolytic activity in patients with severe COVID-19. Blood Adv. (2020) 4:5035–9. doi: 10.1182/bloodadvances.2020002650
130. Cifaldi L, Prencipe G, Caiello I, Bracaglia C, Locatelli F, De Benedetti F, et al. Inhibition of natural killer cell cytotoxicity by interleukin-6: implications for the pathogenesis of macrophage activation syndrome. Arthritis Rheumatol. (2015) 67:3037–46. doi: 10.1002/art.39295
131. Mazzoni A, Salvati L, Maggi L, Capone M, Vanni A, Spinicci M, et al. Impaired immune cell cytotoxicity in severe COVID-19 is IL-6 dependent. J Clin Invest. (2020) 130:4694–703. doi: 10.1172/JCI138554
132. Kawasaki T and Kawai T. Toll-like receptor signaling pathways. Front Immunol. (2014) 5:461. doi: 10.3389/fimmu.2014.00461
133. Olejnik J, Hume AJ, and Muhlberger E. Toll-like receptor 4 in acute viral infection: Too much of a good thing. PloS Pathog. (2018) 14:e1007390. doi: 10.1371/journal.ppat.1007390
134. Bhattacharya M, Sharma AR, Mallick B, Sharma G, Lee SS, and Chakraborty C. Immunoinformatics approach to understand molecular interaction between multi-epitopic regions of SARS-CoV-2 spike-protein with TLR4/MD-2 complex. Infect Genet Evol. (2020) 85:104587. doi: 10.1016/j.meegid.2020.104587
135. Choudhury A and Mukherjee S. In silico studies on the comparative characterization of the interactions of SARS-CoV-2 spike glycoprotein with ACE-2 receptor homologs and human TLRs. J Med Virol. (2020) 92:2105–13. doi: 10.1002/jmv.25987
136. Zhao Y, Kuang M, Li J, Zhu L, Jia Z, Guo X, et al. SARS-CoV-2 spike protein interacts with and activates TLR41. Cell Res. (2021) 31:818–20. doi: 10.1038/s41422-021-00495-9
137. Shirato K and Kizaki T. SARS-CoV-2 spike protein S1 subunit induces pro-inflammatory responses via toll-like receptor 4 signaling in murine and human macrophages. Heliyon. (2021) 7:e06187. doi: 10.1016/j.heliyon.2021.e06187
138. Sahanic S, Hilbe R, Dunser C, Tymoszuk P, Loffler-Ragg J, Rieder D, et al. SARS-CoV-2 activates the TLR4/MyD88 pathway in human macrophages: A possible correlation with strong pro-inflammatory responses in severe COVID-19. Heliyon. (2023) 9:e21893. doi: 10.1016/j.heliyon.2023.e21893
139. Petruk G, Puthia M, Petrlova J, Samsudin F, Stromdahl AC, Cerps S, et al. SARS-CoV-2 spike protein binds to bacterial lipopolysaccharide and boosts proinflammatory activity. J Mol Cell Biol. (2020) 12:916–32. doi: 10.1093/jmcb/mjaa067
140. Cinquegrani G, Spigoni V, Iannozzi NT, Parello V, Bonadonna RC, and Dei Cas A. SARS-CoV-2 Spike protein is not pro-inflammatory in human primary macrophages: endotoxin contamination and lack of protein glycosylation as possible confounders. Cell Biol Toxicol. (2022) 38:667–78. doi: 10.1007/s10565-021-09693-y
141. Teixeira PC, Dorneles GP, Santana Filho PC, da Silva IM, Schipper LL, Postiga IAL, et al. Increased LPS levels coexist with systemic inflammation and result in monocyte activation in severe COVID-19 patients. Int Immunopharmacol. (2021) 100:108125. doi: 10.1016/j.intimp.2021.108125
142. Violi F, Cammisotto V, Bartimoccia S, Pignatelli P, Carnevale R, and Nocella C. Gut-derived low-grade endotoxaemia, atherothrombosis and cardiovascular disease. Nat Rev Cardiol. (2023) 20:24–37. doi: 10.1038/s41569-022-00737-2
143. Niu C, Liang T, Chen Y, Zhu S, Zhou L, Chen N, et al. SARS-CoV-2 spike protein induces the cytokine release syndrome by stimulating T cells to produce more IL-2. Front Immunol. (2024) 15:1444643. doi: 10.3389/fimmu.2024.1444643
144. Samsudin F, Raghuvamsi P, Petruk G, Puthia M, Petrlova J, MacAry P, et al. SARS-CoV-2 spike protein as a bacterial lipopolysaccharide delivery system in an overzealous inflammatory cascade. J Mol Cell Biol. (2023) 14:mjac058. doi: 10.1093/jmcb/mjac058
145. Kawai T and Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity. (2011) 34:637–50. doi: 10.1016/j.immuni.2011.05.006
146. Kircheis R. In silico analyses indicate a lower potency for dimerization of TLR4/MD-2 as the reason for the lower pathogenicity of omicron compared to wild-type virus and earlier SARS-coV-2 variants. Int J Mol Sci. (2024) 25:5451. doi: 10.3390/ijms25105451
147. Kuzmich NN, Sivak KV, Chubarev VN, Porozov YB, Savateeva-Lyubimova TN, and Peri F. TLR4 signaling pathway modulators as potential therapeutics in inflammation and sepsis. Vaccines (Basel). (2017) 5:34. doi: 10.3390/vaccines5040034
148. Ayyubova G, Gychka SG, Nikolaienko SI, Alghenaim FA, Teramoto T, Shults NV, et al. The role of furin in the pathogenesis of COVID-19-associated neurological disorders. Life (Basel). (2024) 14:279. doi: 10.3390/life14020279
149. Fontes-Dantas FL, Fernandes GG, Gutman EG, De Lima EV, Antonio LS, Hammerle MB, et al. SARS-CoV-2 Spike protein induces TLR4-mediated long-term cognitive dysfunction recapitulating post-COVID-19 syndrome in mice. Cell Rep. (2023) 42:112189. doi: 10.1016/j.celrep.2023.112189
150. Azzarone B, Landolina N, Mariotti FR, Moretta L, and Maggi E. Soluble SARS-CoV-2 Spike glycoprotein: considering some potential pathogenic effects. Front Immunol. (2025) 16:1616106. doi: 10.3389/fimmu.2025.1616106
151. Yang OO. The immunopathogenesis of SARS-CoV-2 infection: Overview of lessons learned in the first 5 years. J Immunol. (2025) 214:1095–104. doi: 10.1093/jimmun/vkaf033
152. Liu ZM, Yang MH, Yu K, Lian ZX, and Deng SL. Toll-like receptor (TLRs) agonists and antagonists for COVID-19 treatments. Front Pharmacol. (2022) 13:989664. doi: 10.3389/fphar.2022.989664
153. Mukherjee S. Toll-like receptor 4 in COVID-19: friend or foe? Future Virol. (2022), 10.2217/fvl-2021-0249. doi: 10.2217/fvl-2021-0249
154. Taylor K, Pearson M, Das S, Sardell J, Chocian K, and Gardner S. Genetic risk factors for severe and fatigue dominant long COVID and commonalities with ME/CFS identified by combinatorial analysis. J Transl Med. (2023) 21:775. doi: 10.1186/s12967-023-04588-4
155. Bellavite P, Ferraresi A, and Isidoro C. Immune response and molecular mechanisms of cardiovascular adverse effects of spike proteins from SARS-coV-2 and mRNA vaccines. Biomedicines. (2023) 11:451. doi: 10.3390/biomedicines11020451
156. Ouranidis A, Vavilis T, Mandala E, Davidopoulou C, Stamoula E, Markopoulou CK, et al. mRNA therapeutic modalities design, formulation and manufacturing under pharma 4.0 principles. Biomedicines. (2021) 10:50. doi: 10.3390/biomedicines10010050
157. Fertig TE, Chitoiu L, Marta DS, Ionescu VS, Cismasiu VB, Radu E, et al. Vaccine mRNA Can Be Detected in Blood at 15 Days Post-Vaccination. Biomedicines. (2022) 10:1538. doi: 10.3390/biomedicines10071538
158. Liang F, Lindgren G, Lin A, Thompson EA, Ols S, Rohss J, et al. Efficient Targeting and Activation of Antigen-Presenting Cells In Vivo after Modified mRNA Vaccine Administration in Rhesus Macaques. Mol Ther. (2017) 25:2635–47. doi: 10.1016/j.ymthe.2017.08.006
159. Bellamkonda N, Lambe UP, Sawant S, Nandi SS, Chakraborty C, and Shukla D. Immune response to SARS-coV-2 vaccines. Biomedicines. (2022) 10:1464. doi: 10.3390/biomedicines10071464
160. Roltgen K, Nielsen SCA, Silva O, Younes SF, Zaslavsky M, Costales C, et al. Immune imprinting, breadth of variant recognition, and germinal center response in human SARS-CoV-2 infection and vaccination. Cell. (2022) 185:1025–1040.e14. doi: 10.1016/j.cell.2022.01.018
161. Ogata AF, Cheng CA, Desjardins M, Senussi Y, Sherman AC, Powell M, et al. Circulating severe acute respiratory syndrome coronavirus 2 (SARS-coV-2) vaccine antigen detected in the plasma of mRNA-1273 vaccine recipients. Clin Infect Dis. (2022) 74:715–8. doi: 10.1093/cid/ciab465
162. Fraser DD, Cepinskas G, Slessarev M, Martin C, Daley M, Miller MR, et al. Inflammation profiling of critically ill coronavirus disease 2019 patients. Crit Care Explor. (2020) 2:e0144. doi: 10.1097/CCE.0000000000000144
163. Chen L, Long X, Xu Q, Tan J, Wang G, Cao Y, et al. Elevated serum levels of S100A8/A9 and HMGB1 at hospital admission are correlated with inferior clinical outcomes in COVID-19 patients. Cell Mol Immunol. (2020) 17:992–4. doi: 10.1038/s41423-020-0492-x
164. Wang Z, Yang X, Zhou Y, Sun J, Liu X, Zhang J, et al. COVID-19 severity correlates with weaker T-cell immunity, hypercytokinemia, and lung epithelium injury. Am J Respir Crit Care Med. (2020) 202:606–10. doi: 10.1164/rccm.202005-1701LE
165. Mapelli M, Salvioni E, Mattavelli I, Banfi C, Ghilardi S, Greco A, et al. Surfactant-derived protein type B: a new biomarker linked to respiratory failure and lung damage in mild to moderate SARS-CoV-2 pneumonia. ERJ Open Res. (2024) 10:00301–2024. doi: 10.1183/23120541.00301-2024
166. Fang H, Wu Y, Huang X, Wang W, Ang B, Cao X, et al. Toll-like receptor 4 (TLR4) is essential for Hsp70-like protein 1 (HSP70L1) to activate dendritic cells and induce Th1 response. J Biol Chem. (2011) 286:30393–400. doi: 10.1074/jbc.M111.266528
167. Mellett L and Khader SA. S100A8/A9 in COVID-19 pathogenesis: Impact on clinical outcomes. Cytokine Growth Factor Rev. (2022) 63:90–7. doi: 10.1016/j.cytogfr.2021.10.004
168. Guo Q, Zhao Y, Li J, Liu J, Yang X, Guo X, et al. Induction of alarmin S100A8/A9 mediates activation of aberrant neutrophils in the pathogenesis of COVID-19. Cell Host Microbe. (2021) 29:222–235.e4. doi: 10.1016/j.chom.2020.12.016
169. Smiley ST, King JA, and Hancock WW. Fibrinogen stimulates macrophage chemokine secretion through toll-like receptor 4. J Immunol. (2001) 167:2887–94. doi: 10.4049/jimmunol.167.5.2887
170. Zhao G, Gentile ME, Xue L, Cosgriff CV, Weiner AI, Adams-Tzivelekidis S, et al. Vascular endothelial-derived SPARCL1 exacerbates viral pneumonia through pro-inflammatory macrophage activation. Nat Commun. (2024) 15:4235. doi: 10.1038/s41467-024-48589-3
171. Yang H, Wang H, Ju Z, Ragab AA, Lundback P, Long W, et al. MD-2 is required for disulfide HMGB1-dependent TLR4 signaling. J Exp Med. (2015) 212:5–14. doi: 10.1084/jem.20141318
172. Passos FRS, Heimfarth L, Monteiro BS, Correa CB, Moura TR, Araujo AAS, et al. Oxidative stress and inflammatory markers in patients with COVID-19: Potential role of RAGE, HMGB1, GFAP and COX-2 in disease severity. Int Immunopharmacol. (2022) 104:108502. doi: 10.1016/j.intimp.2021.108502
173. Sohn KM, Lee SG, Kim HJ, Cheon S, Jeong H, Lee J, et al. COVID-19 patients upregulate toll-like receptor 4-mediated inflammatory signaling that mimics bacterial sepsis. J Korean Med Sci. (2020) 35:e343. doi: 10.3346/jkms.2020.35.e343
174. Dorneles GP, Teixeira PC, Peres A, Rodrigues Junior LC, da Fonseca SG, Monteiro MC, et al. Endotoxin tolerance and low activation of TLR-4/NF-kappaB axis in monocytes of COVID-19 patients. J Mol Med (Berl). (2023) 101:183–95. doi: 10.1007/s00109-023-02283-x
175. Carcaterra M and Caruso C. Alveolar epithelial cell type II as main target of SARS-CoV-2 virus and COVID-19 development via NF-Kb pathway deregulation: A physio-pathological theory. Med Hypotheses. (2021) 146:110412. doi: 10.1016/j.mehy.2020.110412
176. Lukassen S, Chua RL, Trefzer T, Kahn NC, Schneider MA, Muley T, et al. SARS-CoV-2 receptor ACE2 and TMPRSS2 are primarily expressed in bronchial transient secretory cells. EMBO J. (2020) 39:e105114. doi: 10.15252/embj.20105114
177. Frolova EI, Palchevska O, Lukash T, Dominguez F, Britt W, and Frolov I. Acquisition of furin cleavage site and further SARS-coV-2 evolution change the mechanisms of viral entry, infection spread, and cell signaling. J Virol. (2022) 96:e0075322. doi: 10.1128/jvi.00753-22
178. Watson A, Madsen J, and Clark HW. SP-A and SP-D: dual functioning immune molecules with antiviral and immunomodulatory properties. Front Immunol. (2021) 11:622598. doi: 10.3389/fimmu.2020.622598
179. Voelker DR and Numata M. Phospholipid regulation of innate immunity and respiratory viral infection. J Biol Chem. (2019) 294:4282–9. doi: 10.1074/jbc.AW118.003229
180. Milad N and Morissette MC. Revisiting the role of pulmonary surfactant in chronic inflammatory lung diseases and environmental exposure. Eur Respir Rev. (2021) 30:210077. doi: 10.1183/16000617.0077-2021
181. Tlatelpa-Romero B, Cazares-Ordonez V, Oyarzabal LF, and Vazquez-de-Lara LG. The role of pulmonary surfactant phospholipids in fibrotic lung diseases. Int J Mol Sci. (2022) 24:326. doi: 10.3390/ijms24010326
182. Russell CD, Lone NI, and Baillie JK. Comorbidities, multimorbidity and COVID-19. Nat Med. (2023) 29:334–43. doi: 10.1038/s41591-022-02156-9
183. Choudhury A and Mukherjee S. Taming the storm in the heart: exploring different therapeutic choices against myocardial inflammation in COVID-19. Recent Adv Antiinfect Drug Discov. (2021) 16:89–93. doi: 10.2174/2772434416666210616124505
184. Carnevale R, Cammisotto V, Bartimoccia S, Nocella C, Castellani V, Bufano M, et al. Toll-like receptor 4-dependent platelet-related thrombosis in SARS-coV-2 infection. Circ Res. (2023) 132:290–305. doi: 10.1161/CIRCRESAHA.122.321541
185. Zhang S, Liu Y, Wang X, Yang L, Li H, Wang Y, et al. SARS-CoV-2 binds platelet ACE2 to enhance thrombosis in COVID-19. J Hematol Oncol. (2020) 13:120. doi: 10.1186/s13045-020-00954-7
186. Tsai JC, Lin YW, Huang CY, Lin CY, Tsai YT, Shih CM, et al. The role of calpain-myosin 9-Rab7b pathway in mediating the expression of Toll-like receptor 4 in platelets: a novel mechanism involved in alpha-granules trafficking. PloS One. (2014) 9:e85833. doi: 10.1371/journal.pone.0085833
187. Fortmann SD, Patton MJ, Frey BF, Tipper JL, Reddy SB, Vieira CP, et al. Circulating SARS-CoV-2+ megakaryocytes are associated with severe viral infection in COVID-19. Blood Adv. (2023) 7:4200–14. doi: 10.1182/bloodadvances.2022009022
188. Mousavizadeh L and Ghasemi S. Genotype and phenotype of COVID-19: Their roles in pathogenesis. J Microbiol Immunol Infect. (2021) 54:159–63. doi: 10.1016/j.jmii.2020.03.022
189. Calabrese C, Annunziata A, Coppola A, Pafundi PC, Guarino S, Di Spirito V, et al. ACE gene I/D polymorphism and acute pulmonary embolism in COVID19 pneumonia: A potential predisposing role. Front Med (Lausanne). (2021) 7:631148. doi: 10.3389/fmed.2020.631148
190. Iniguez M, Perez-Matute P, Villoslada-Blanco P, Recio-Fernandez E, Ezquerro-Perez D, Alba J, et al. ACE gene variants rise the risk of severe COVID-19 in patients with hypertension, dyslipidemia or diabetes: A spanish pilot study. Front Endocrinol (Lausanne). (2021) 12:688071. doi: 10.3389/fendo.2021.688071
191. Okamoto H and Ichikawa N. The pivotal role of the angiotensin-II-NF-kappaB axis in the development of COVID-19 pathophysiology. Hypertens Res. (2021) 44:126–8. doi: 10.1038/s41440-020-00560-7
192. Yang XF, Wang H, Huang Y, Huang JH, Ren HL, Xu Q, et al. Myeloid angiotensin II type 1 receptor mediates macrophage polarization and promotes vascular injury in DOCA/salt hypertensive mice. Front Pharmacol. (2022) 13:879693. doi: 10.3389/fphar.2022.879693
193. Olajide OA, Iwuanyanwu VU, Adegbola OD, and Al-Hindawi AA. SARS-coV-2 spike glycoprotein S1 induces neuroinflammation in BV-2 microglia. Mol Neurobiol. (2022) 59:445–58. doi: 10.1007/s12035-021-02593-6
194. Aboudounya MM and Heads RJ. COVID-19 and toll-like receptor 4 (TLR4): SARS-coV-2 may bind and activate TLR4 to increase ACE2 expression, facilitating entry and causing hyperinflammation. Mediators Inflamm. (2021) 2021:8874339. doi: 10.1155/2021/8874339
195. Cicco S, Cicco G, Racanelli V, and Vacca A. Neutrophil extracellular traps (NETs) and damage-associated molecular patterns (DAMPs): two potential targets for COVID-19 treatment. Mediators Inflamm. (2020) 2020:7527953. doi: 10.1155/2020/7527953
196. Hashimoto M, Kamphorst AO, Im SJ, Kissick HT, Pillai RN, Ramalingam SS, et al. CD8 T cell exhaustion in chronic infection and cancer: opportunities for interventions. Annu Rev Med. (2018) 69:301–18. doi: 10.1146/annurev-med-012017-043208
197. Retnakumar SV, Chauvin C, and Bayry J. The implication of anti-PD-1 therapy in cancer patients for the vaccination against viral and other infectious diseases. Pharmacol Ther. (2023) 245:108399. doi: 10.1016/j.pharmthera.2023.108399
198. Claus M, Pieris N, Urlaub D, Brode P, Schaaf B, Durak D, et al. Early expansion of activated adaptive but also exhausted NK cells during acute severe SARS-CoV-2 infection. Front Cell Infect Microbiol. (2023) 13:1266790. doi: 10.3389/fcimb.2023.1266790
199. Zheng M, Gao Y, Wang G, Song G, Liu S, Sun D, et al. Functional exhaustion of antiviral lymphocytes in COVID-19 patients. Cell Mol Immunol. (2020) 17:533–5. doi: 10.1038/s41423-020-0402-2
200. Brueggeman JM, Zhao J, Schank M, Yao ZQ, and Moorman JP. Trained immunity: an overview and the impact on COVID-19. Front Immunol. (2022) 13:837524. doi: 10.3389/fimmu.2022.837524
201. Frank MG, Nguyen KH, Ball JB, Hopkins S, Kelley T, Baratta MV, et al. SARS-CoV-2 spike S1 subunit induces neuroinflammatory, microglial and behavioral sickness responses: Evidence of PAMP-like properties. Brain Behav Immun. (2022) 100:267–77. doi: 10.1016/j.bbi.2021.12.007
202. Gugliandolo A, Chiricosta L, Calcaterra V, Biasin M, Cappelletti G, Carelli S, et al. SARS-coV-2 infected pediatric cerebral cortical neurons: transcriptomic analysis and potential role of toll-like receptors in pathogenesis. Int J Mol Sci. (2021) 22:8059. doi: 10.3390/ijms22158059
203. Etter MM, Martins TA, Kulsvehagen L, Possnecker E, Duchemin W, Hogan S, et al. Severe Neuro-COVID is associated with peripheral immune signatures, autoimmunity and neurodegeneration: a prospective cross-sectional study. Nat Commun. (2022) 13:6777. doi: 10.1038/s41467-022-34068-0
204. Chen Z, Chen C, Chen F, Lan R, Lin G, and Xu Y. Bioinformatics analysis of potential pathogenesis and risk genes of immunoinflammation-promoted renal injury in severe COVID-19. Front Immunol. (2022) 13:950076. doi: 10.3389/fimmu.2022.950076
205. Nakazawa D, Takeda Y, Kanda M, Tomaru U, Ogawa H, Kudo T, et al. Inhibition of Toll-like receptor 4 and Interleukin-1 receptor prevent SARS-CoV-2 mediated kidney injury. Cell Death Discov. (2023) 9:293. doi: 10.1038/s41420-023-01584-x
206. Silva-Aguiar RP, Teixeira DE, Peruchetti DB, Peres RAS, Alves SAS, Calil PT, et al. Toll like receptor 4 mediates the inhibitory effect of SARS-CoV-2 spike protein on proximal tubule albumin endocytosis. Biochim Biophys Acta Mol Basis Dis. (2024) 1870:167155. doi: 10.1016/j.bbadis.2024.167155
207. Taha SI, Shata AK, Baioumy SA, Fouad SH, Anis SG, Mossad IM, et al. Toll-like receptor 4 polymorphisms (896A/G and 1196C/T) as an indicator of COVID-19 severity in a convenience sample of Egyptian patients. J Inflammation Res. (2021) 14:6293–303. doi: 10.2147/JIR.S343246
208. Zacher C, Schonfelder K, Rohn H, Siffert W, and Mohlendick B. The single nucleotide polymorphism rs4986790 (c.896A>G) in the gene TLR4 as a protective factor in corona virus disease 2019 (COVID-19). Front Immunol. (2024) 15:1355193. doi: 10.3389/fimmu.2024.1355193
209. Bakaros E, Voulgaridi I, Paliatsa V, Gatselis N, Germanidis G, Asvestopoulou E, et al. Innate immune gene polymorphisms and COVID-19 prognosis. Viruses. (2023) 15:1784. doi: 10.3390/v15091784
210. Flores-Gonzalez J, Chavez-Galan L, Falfan-Valencia R, Roldan IB, Fricke-Galindo I, Veronica-Aguilar A, et al. Variant rs4986790 of toll-like receptor 4 affects the signaling and induces cell dysfunction in patients with severe COVID-19. Int J Infect Dis. (2024) 138:102–9. doi: 10.1016/j.ijid.2023.11.032
211. Li Y, Liu Y, Duo M, Wu R, Jiang T, Li P, et al. Bioinformatic analysis and preliminary validation of potential therapeutic targets for COVID-19 infection in asthma patients. Cell Commun Signal. (2022) 20:201. doi: 10.1186/s12964-022-01010-2
212. Ma YN, Ma SR, Yang L, Wu J, Wang YR, Bao LJ, et al. Diagnostic biomarkers and immune infiltration profiles common to COVID-19, acute myocardial infarction and acute ischaemic stroke using bioinformatics methods and machine learning. BMC Neurol. (2025) 25:201. doi: 10.1186/s12883-025-04212-6
213. Ceylan H. A bioinformatics approach for identifying potential molecular mechanisms and key genes involved in COVID-19 associated cardiac remodeling. Gene Rep. (2021) 24:101246. doi: 10.1016/j.genrep.2021.101246
214. Menezes MCS, Veiga ADM, Martins de Lima T, Kunimi Kubo Ariga S, Vieira Barbeiro H, de Lucena Moreira C, et al. Lower peripheral blood Toll-like receptor 3 expression is associated with an unfavorable outcome in severe COVID-19 patients. Sci Rep. (2021) 11:15223. doi: 10.1038/s41598-021-94624-4
215. Alturaiki W, Alkadi H, Alamri S, Awadalla ME, Alfaez A, Mubarak A, et al. Association between the expression of toll-like receptors, cytokines, and homeostatic chemokines in SARS-CoV-2 infection and COVID-19 severity. Heliyon. (2023) 9:e12653. doi: 10.1016/j.heliyon.2022.e12653
216. Mantovani S, Oliviero B, Varchetta S, Renieri A, and Mondelli MU. TLRs: innate immune sentries against SARS-coV-2 infection. Int J Mol Sci. (2023) 24:8065. doi: 10.3390/ijms24098065
217. Mohamed HA, Abdelkafy AE, Khairy RMM, Abdelraheim SR, Kamel BA, and Marey H. MicroRNAs and cytokines as potential predictive biomarkers for COVID-19 disease progression. Sci Rep. (2023) 13:3531. doi: 10.1038/s41598-023-30474-6
218. Pedicillo MC, De Stefano IS, Zamparese R, Barile R, Meccariello M, Agostinone A, et al. The role of toll-like receptor-4 in macrophage imbalance in lethal COVID-19 lung disease, and its correlation with galectin-3. Int J Mol Sci. (2023) 24:13259. doi: 10.3390/ijms241713259
219. Unterberger S, Terrazzini N, and Sacre S. Convalescent COVID-19 monocytes exhibit altered steady-state gene expression and reduced TLR2, TLR4 and RIG-I induced cytokine expression. Hum Immunol. (2025) 86:111249. doi: 10.1016/j.humimm.2025.111249
220. Houssen ME, Elmaria MO, Badr D, El-Mahdy R, Ghannam MA, El-Ashwah S, et al. Serum soluble toll-like receptor 4 and risk for clinical severity in COVID-19 patients. Pneumonia (Nathan). (2024) 16:1. doi: 10.1186/s41479-023-00121-9
221. Buckner CM, Kardava L, El Merhebi O, Narpala SR, Serebryannyy L, Lin BC, et al. Interval between prior SARS-CoV-2 infection and booster vaccination impacts magnitude and quality of antibody and B cell responses. Cell. (2022) 185:4333–4346.e14. doi: 10.1016/j.cell.2022.09.032
222. Musuuza JS, Watson L, Parmasad V, Putman-Buehler N, Christensen L, and Safdar N. Prevalence and outcomes of co-infection and superinfection with SARS-CoV-2 and other pathogens: A systematic review and meta-analysis. PloS One. (2021) 16:e0251170. doi: 10.1371/journal.pone.0251170
223. Totura AL, Whitmore A, Agnihothram S, Schafer A, Katze MG, Heise MT, et al. Toll-like receptor 3 signaling via TRIF contributes to a protective innate immune response to severe acute respiratory syndrome coronavirus infection. mBio. (2015) 6:e00638–15. doi: 10.1128/mBio.00638-15
224. Shamseldin MM, Read KA, Hall JM, Tuazon JA, Brown JM, Guo M, et al. The adjuvant BcfA activates antigen presenting cells through TLR4 and supports T(FH) and T(H)1 while attenuating T(H)2 gene programming. Front Immunol. (2024) 15:1439418. doi: 10.3389/fimmu.2024.1439418
225. Monnet E, Choy EH, McInnes I, Kobakhidze T, de Graaf K, Jacqmin P, et al. Efficacy and safety of NI-0101, an anti-toll-like receptor 4 monoclonal antibody, in patients with rheumatoid arthritis after inadequate response to methotrexate: a phase II study. Ann Rheum Dis. (2020) 79:316–23. doi: 10.1136/annrheumdis-2019-216487
226. Shirey KA, Lai W, Scott AJ, Lipsky M, Mistry P, Pletneva LM, et al. The TLR4 antagonist Eritoran protects mice from lethal influenza infection. Nature. (2013) 497:498–502. doi: 10.1038/nature12118
227. Choudhury A, Mukherjee G, and Mukherjee S. Chemotherapy vs. Immunotherapy in combating nCOVID19: An update. Hum Immunol. (2021) 82:649–58. doi: 10.1016/j.humimm.2021.05.001
228. Padhi S, Sanjukta S, Chourasia R, Labala RK, Singh SP, and Rai AK. A multifunctional peptide from bacillus fermented soybean for effective inhibition of SARS-coV-2 S1 receptor binding domain and modulation of toll like receptor 4: A molecular docking study. Front Mol Biosci. (2021) 8:636647. doi: 10.3389/fmolb.2021.636647
229. Huang Z, Ye B, Han J, Kong F, Shan P, Lu Z, et al. NACHT, LRR and PYD domains-containing protein 3 inflammasome is activated and inhibited by berberine via toll-like receptor 4/myeloid differentiation primary response gene 88/nuclear factor-kappaB pathway, in phorbol 12-myristate 13-acetate-induced macrophages. Mol Med Rep. (2018) 17:2673–80. doi: 10.3892/mmr.2017.8189/
230. Patra R, Chandra Das N, and Mukherjee S. Targeting human TLRs to combat COVID-19: A solution? J Med Virol. (2021) 93:615–7. doi: 10.1002/jmv.26387
231. Zamora-Pineda J, Kalinina O, Sperling AI, and Knight KL. Mechanism of TLR4-mediated anti-inflammatory response induced by exopolysaccharide from the probiotic bacillus subtilis. J Immunol. (2023) 211:1232–9. doi: 10.4049/jimmunol.2200855
232. Zhou S, Wang G, and Zhang W. Effect of TLR4/MyD88 signaling pathway on sepsis-associated acute respiratory distress syndrome in rats, via regulation of macrophage activation and inflammatory response. Exp Ther Med. (2018) 15:3376–84. doi: 10.3892/etm.2018.5815
233. Cox M, Peacock TP, Harvey WT, Hughes J, Wright DW, Consortium C-GU, et al. SARS-CoV-2 variant evasion of monoclonal antibodies based on in vitro studies. Nat Rev Microbiol. (2023) 21:112–24. doi: 10.1038/s41579-022-00809-7
234. Das NC, Chakraborty P, Bayry J, and Mukherjee S. Comparative binding ability of human monoclonal antibodies against omicron variants of SARS-coV-2: an in silico investigation. Antibodies (Basel). (2023) 12:17. doi: 10.3390/antib12010017
235. Das NC, Chakraborty P, Bayry J, and Mukherjee S. In silico analyses on the comparative potential of therapeutic human monoclonal antibodies against newly emerged SARS-coV-2 variants bearing mutant spike protein. Front Immunol. (2022) 12:782506. doi: 10.3389/fimmu.2021.782506
236. Mahmoudi A, Alavizadeh SH, Hosseini SA, Meidany P, Doagooyan M, Abolhasani Y, et al. Harnessing aptamers against COVID-19: A therapeutic strategy. Drug Discov Today. (2023) 28:103663. doi: 10.1016/j.drudis.2023.103663
237. Choudhury A, Sen Gupta PS, Panda SK, Rana MK, and Mukherjee S. Designing AbhiSCoVac - A single potential vaccine for all ‘corona culprits’: Immunoinformatics and immune simulation approaches. J Mol Liq. (2022) 351:118633. doi: 10.1016/j.molliq.2022.118633
238. Akbar AN and Gilroy DW. Aging immunity may exacerbate COVID-19. Science. (2020) 369:256–7. doi: 10.1126/science.abb0762
239. van der Made CI, Simons A, Schuurs-Hoeijmakers J, van den Heuvel G, Mantere T, Kersten S, et al. Presence of genetic variants among young men with severe COVID-19. JAMA. (2020) 324:663–73. doi: 10.1001/jama.2020.13719
240. Verma A, Manojkumar A, Dhasmana A, Tripathi MK, Jaggi M, Chauhan SC, et al. Recurring SARS-CoV-2 variants: an update on post-pandemic, co-infections and immune response. Nanotheranostics. (2024) 8:247–69. doi: 10.7150/ntno.91910. eCollection 2024.
241. Yousefbeigi S and Marsusi F. Structural insights into ACE2 interactions and immune activation of SARS-CoV-2 and its variants: an in-silico study. J Biomol Struct Dyn. (2025) 43:665–78. doi: 10.1080/07391102.2023.2283158
242. Korobova ZR, Arsentieva NA, Liubimova NE, Batsunov OK, Dedkov VG, Gladkikh AS, et al. Cytokine profiling in different SARS-coV-2 genetic variants. Int J Mol Sci. (2022) 23:14146. doi: 10.3390/ijms232214146
243. Barh D, Tiwari S, Gomes LGR, Pinto CHR, Andrade BS, Ahmad S, et al. SARS-coV-2 variants show a gradual declining pathogenicity and pro-inflammatory cytokine stimulation, an increasing antigenic and anti-inflammatory cytokine induction, and rising structural protein instability: A minimal number genome-based approach. Inflammation. (2023) 46:297–312. doi: 10.1007/s10753-022-01734-w
244. Anastassopoulou C, Davaris N, Ferous S, Siafakas N, Boufidou F, Anagnostopoulos K, et al. The molecular basis of olfactory dysfunction in COVID-19 and long COVID. Lifestyle Genom. (2024) 17:42–56. doi: 10.1159/000539292
245. Demirhan S, Goldman DL, and Herold BC. Differences in the clinical manifestations and host immune responses to SARS-coV-2 variants in children compared to adults. J Clin Med. (2023) 13:128. doi: 10.3390/jcm13010128
246. Yildirim Arslan S, Avcu G, Sahbudak Bal Z, Arslan A, Ozkinay FF, and Kurugol Z. Evaluation of post-COVID symptoms of the SARS-CoV-2 Delta and Omicron variants in children: A prospective study. Eur J Pediatr. (2023) 182:4565–71. doi: 10.1007/s00431-023-05134-6
247. Lokanuwatsatien T, Satdhabudha A, Tangsathapornpong A, Bunjoungmanee P, Sinlapamongkolkul P, Chaiyakulsil C, et al. Prevalence and associating factors of long COVID in pediatric patients during the Delta and the Omicron variants. Front Pediatr. (2023) 11:1127582. doi: 10.3389/fped.2023.1127582
248. Planas D, Veyer D, Baidaliuk A, Staropoli I, Guivel-Benhassine F, Rajah MM, et al. Reduced sensitivity of SARS-CoV-2 variant Delta to antibody neutralization. Nature. (2021) 596:276–80. doi: 10.1038/s41586-021-03777-9
249. Shinde V, Bhikha S, Hoosain Z, Archary M, Bhorat Q, Fairlie L, et al. Efficacy of NVX-CoV2373 Covid-19 vaccine against the B. 1.351 variant. N Engl J Med. (2021) 384:1899–909. doi: 10.1056/NEJMoa2103055
250. Abdelmageed ME, El-Awady MS, Abdelrahim M, and Suddek GM. LPS-RS attenuation of lipopolysaccharide-induced acute lung injury involves NF-κB inhibition. Can J Physiol Pharmacol. (2016) 94:140–6. doi: 10.1139/cjpp-2015-0219
251. Mani S, Kaur A, Jakhar K, Kumari G, Sonar S, Kumar A, et al. Targeting DPP4- RBD interactions by sitagliptin and linagliptin delivers a potential host-directed therapy against pan-SARS-CoV-2 infections. Int J Biol Macromol. (2023) 245:125444. doi: 10.1016/j.ijbiomac.2023.125444
252. Eagleton M, Stokes S, Fenton F, and Keenan E. Therapeutic potential of long-acting opioids and opioid antagonists for SARS-CoV-2 infection. Br J Anaesth. (2021) 127:e212–4. doi: 10.1016/j.bja.2021.08.022
253. Bai Y, Min R, Chen P, Mei S, Deng F, Zheng Z, et al. Disulfiram blocks inflammatory TLR4 signaling by targeting MD-2. Proc Natl Acad Sci U S A. (2023) 120:e2306399120. doi: 10.1073/pnas.2306399120
254. Gold R, Linker RA, and Stangel M. Fumaric acid and its esters: an emerging treatment for multiple sclerosis with antioxidative mechanism of action. Clin Immunol. (2012) 142:44–8. doi: 10.1016/j.clim.2011.02.017
255. Das NC, Labala RK, Patra R, Chattoraj A, and Mukherjee S. In silico identification of new anti-SARS-CoV-2 agents from bioactive phytocompounds targeting the viral spike glycoprotein and human TLR4. Lett Drug Des Discov. (2021) 19:175–91. doi: 10.2174/1570180818666210901125519
256. Corpetti C, Del Re A, Seguella L, Palenca I, Rurgo S, De Conno B, et al. Cannabidiol inhibits SARS-Cov-2 spike (S) protein-induced cytotoxicity and inflammation through a PPARγ-dependent TLR4/NLRP3/Caspase-1 signaling suppression in Caco-2 cell line. Phytother Res. (2021) 35:6893–903. doi: 10.1002/ptr.7302
Keywords: innate immunity, NK cells, SARS-CoV2 infection, spike glycoprotein, TLR4
Citation: Maggi E, Landolina N, Mariotti FR, Munari E, Tumino N, Vacca P, Azzarone B and Moretta L (2025) The innate immune response in SARS-CoV2 infection: focus on toll-like receptor 4 in severe disease outcomes. Front. Immunol. 16:1658396. doi: 10.3389/fimmu.2025.1658396
Received: 02 July 2025; Accepted: 17 September 2025;
Published: 07 October 2025.
Edited by:
Annalisa Del Prete, University of Brescia, ItalyReviewed by:
Carlos Bueno, National Scientific and Technical Research Council (CONICET), ArgentinaHelena Stabile, Sapienza University of Rome, Italy
Copyright © 2025 Maggi, Landolina, Mariotti, Munari, Tumino, Vacca, Azzarone and Moretta. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lorenzo Moretta, bG9yZW56by5tb3JldHRhQG9wYmcubmV0