 Jifeng Zhang1,2†
Jifeng Zhang1,2† Mei Long
Mei Long- 1Zibo Central Hospital, Zibo, China
- 2Department of Oncology, Zibo Central Hospital, Zibo, China
- 3Department of Hematology, Zibo Central Hospital, Zibo, China
- 4Department of Pediatrics, Zibo Central Hospital, Zibo, China
- 5Department of Internal Medicine, Zibo Central Hospital, Zibo, China
Immunogenic cell death (ICD) effectively triggers adaptive immune responses against cancer, yet its clinical application in solid tumors is hindered by tumor microenvironment (TME) barriers. These include immunosuppressive cell populations, dense extracellular matrix, abnormal vasculature, hypoxia, and metabolic suppression, which collectively impede immune infiltration and function. This review evaluates current therapeutic strategies to overcome these barriers, including vascular normalization (restoring abnormal tumor blood vessels to a more structured and functional state to improve perfusion and immune cell infiltration), extracellular matrix (ECM) modulation, alleviation of hypoxia, metabolic reprogramming, immunosuppressive cell targeting, physical remodeling, and nanoparticle-based drug delivery. Clinical evidence highlights the potential of these integrated approaches to enhance ICD-induced antitumor immunity, suggesting promising avenues for improving patient outcomes through combined modulation of the TME and ICD induction.
1 Introduction
Immunogenic cell death (ICD) has emerged as a therapeutic strategy that initiates adaptive immune responses against cancer (1, 2). By promoting dendritic cell (DC) activation, antigen presentation, and cytotoxic T-cell priming, ICD effectively turns dying tumor cells into an in situ vaccine, harnessing the patient’s own immune system to combat malignancies (3–5). However, despite advances in understanding ICD mechanisms and applications, the clinical translation of ICD-based therapies, particularly in solid tumors, remains limited due to the presence of multiple TME barriers (6).
The TME of solid tumors is immunosuppressive, structurally dense, and metabolically hostile. It comprises immunosuppressive cell populations, including regulatory T cells (Tregs), tumor-associated macrophages (TAMs), and myeloid-derived suppressor cells (MDSCs), which collectively suppress effector immune cell infiltration and function (7, 8). Additionally, physical constraints, such as dense extracellular matrix deposition, fibrosis, and abnormal tumor vasculature, further impede immune cell trafficking and infiltration, thereby reducing the therapeutic potential of ICD (9, 10). Furthermore, metabolic factors, including hypoxia, nutrient scarcity, and accumulation of immunosuppressive metabolites (adenosine, lactate), impair T-cell survival and function within the TME (11, 12).
Therefore, a crucial challenge—and equally significant opportunity—lies in targeting and overcoming these microenvironmental barriers to realize the therapeutic potential of ICD in solid tumors (2, 13). In this review article, we discuss the existing tumor microenvironmental obstacles that limit ICD efficacy, highlight emerging therapeutic strategies to dismantle these barriers, and underscore the clinical promise of strategically combining microenvironment-targeted therapies with ICD induction to significantly enhance cancer immunotherapy outcomes.
2 Tumor microenvironmental barriers impairing ICD effectiveness
Despite the promising therapeutic potential of ICD in activating antitumor immunity, its clinical effectiveness in solid tumors is severely hampered by complex microenvironmental barriers (14). These barriers encompass immunological, physical, and metabolic factors that collectively restrict immune cell infiltration, activation, and sustained function within tumor sites.
Firstly, the immunosuppressive cellular environment impairs ICD-induced immune activation. Solid tumors are populated by immunosuppressive cell types such as Tregs, TAMs polarized toward the anti-inflammatory M2 phenotype, myeloid-derived suppressor cells (MDSCs), and cancer-associated fibroblasts (CAFs) (15). These cells produce various immunosuppressive mediators, including transforming growth factor-beta (TGF-β), interleukin-10 (IL - 10), and vascular endothelial growth factor (VEGF), which collectively inhibit dendritic cell (DC) maturation, reduce cytotoxic T-cell proliferation, and dampen effector immune responses. Consequently, even when ICD successfully generates immunogenic signals, their translation into meaningful clinical responses remains suboptimal due to these immunological checkpoints within the TME (16–18).
Secondly, physical barriers posed by the TME also present significant hurdles to effective immune cell trafficking and infiltration. An extensively developed extracellular matrix (ECM, the network of structural proteins such as collagen and glycosaminoglycans that provides physical support to tissues), characterized by dense collagen fiber networks and high levels of hyaluronan, physically obstructs immune cell penetration and reduces the diffusion of therapeutic agents into tumor cores (19–21). Tumor fibrosis, driven largely by activated CAFs, further exacerbates this issue by enhancing ECM rigidity and reducing the functional perfusion of intratumoral vessels (22). Aberrant and tortuous tumor vasculature, featuring dysfunctional endothelial cells and compromised lymphatic drainage, further limits immune cell entry and migration, thereby maintaining the immune-excluded or immune-desert phenotype characteristic of many solid tumors.
Thirdly, metabolic alterations within the TME restrict immune cell function and survival, thereby limiting the effectiveness of ICD-induced immune responses. Hypoxia, resulting from poor vascular perfusion and rapid tumor cell proliferation, triggers tumor adaptation through hypoxia-inducible factors (HIFs), leading to increased anaerobic glycolysis (Warburg effect), excessive lactate production, and acidification of the tumor milieu (23). The accumulation of metabolites such as lactate, adenosine, and kynurenine directly suppresses cytotoxic T-cell activation, proliferation, and effector functions, while promoting the differentiation and function of immunosuppressive cells (24). Nutrient depletion (glucose, amino acids such as tryptophan and arginine) further impairs the metabolic fitness and persistence of infiltrating immune cells, ultimately compromising antitumor immunity following ICD induction.
Figure 1 illustrates the key tumor microenvironmental barriers—including immunosuppressive cells, dense extracellular matrix, abnormal tumor vasculature, hypoxia, and metabolic suppression—that significantly impede the effectiveness of ICD. The figure also outlines corresponding therapeutic strategies currently under clinical evaluation aimed at dismantling these barriers, thus enhancing immune infiltration, activation, and sustained antitumor immune responses. Specifically, checkpoint inhibitors and depletion agents are designed to overcome immunosuppressive cell populations such as Tregs and MDSCs; vascular normalization strategies (e.g., anti-VEGF therapy) target abnormal tumor vasculature to improve immune cell delivery; ECM-modulating agents (e.g., hyaluronidase-based therapies) reduce matrix density to facilitate T cell penetration; and metabolic modulators aim to restore nutrient and oxygen availability in hypoxic or metabolically suppressive regions of the TME. These immunological, physical, and metabolic barriers within the solid tumor microenvironment substantially limit the potential of ICD to induce effective and sustained antitumor immune responses (25). Therefore, therapeutic strategies specifically designed to address and mitigate these barriers are urgently required to enhance the clinical impact and effectiveness of ICD-based cancer immunotherapies. In the following sections, we discuss how these identified barriers inform the design of therapeutic strategies, emphasizing the direct connections between each obstacle and the corresponding interventions currently under preclinical or clinical evaluation.
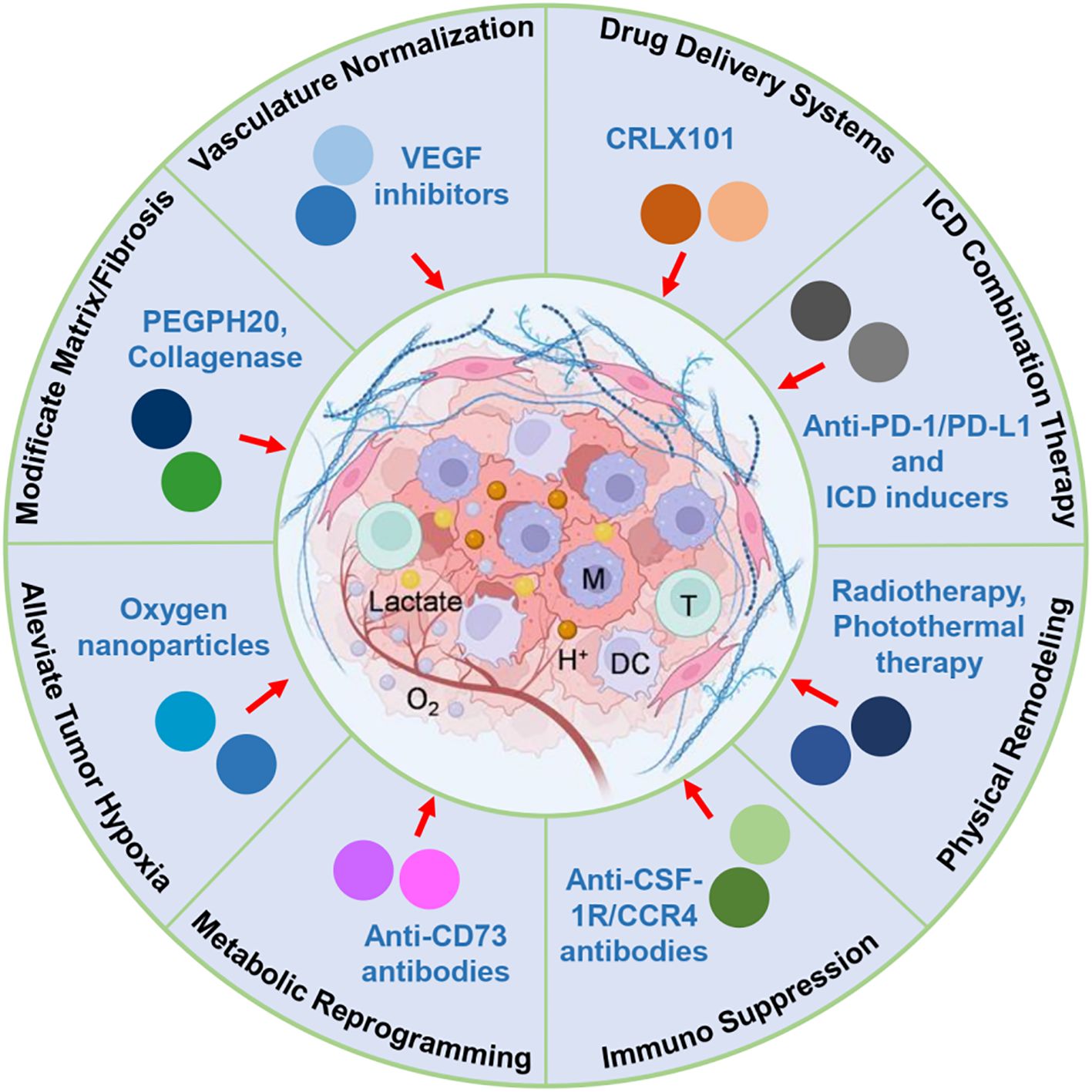
Figure 1. Schematic illustration of key tumor microenvironmental barriers limiting ICD efficacy and corresponding therapeutic strategies. This figure illustrates the major immunological, physical, and metabolic barriers within the solid tumor microenvironment that hinder the effectiveness of ICD-based therapies. Key immunosuppressive components include Tregs, MDSCs, and TAMs, which inhibit antigen presentation and effector T-cell responses. Physical barriers, such as dense ECM, fibrotic stroma, and abnormal vasculature, obstruct immune cell infiltration and drug penetration. Metabolic stressors, including hypoxia, glucose depletion, and accumulation of immunosuppressive metabolites (e.g., lactate, adenosine), further compromise immune function. Therapeutic strategies to overcome these obstacles are highlighted, including vascular normalization, ECM remodeling, metabolic reprogramming, immunosuppressive cell targeting, and nanomedicine-based delivery. These approaches aim to enhance immune accessibility, sustain cytotoxic activity, and improve the therapeutic outcome of ICD in solid tumors.
3 Current strategies to overcome microenvironmental barriers
To effectively translate ICD into clinically successful cancer immunotherapy, numerous therapeutic strategies have been developed to specifically target and overcome the complex TME barriers. These approaches are primarily designed to reduce immunosuppression, enhance immune cell infiltration, and reverse unfavorable metabolic conditions within the solid tumor environment. To orient the reader, Sections 3.1 – 3.7 map each major barrier (vascular, stromal/ECM, hypoxic–metabolic, immunosuppressive cells, physical/ablative, and delivery constraints) to the corresponding intervention, and Section 4 synthesizes representative clinical studies that evaluate these approaches in patients.
3.1 Normalization of tumor vasculature
Aberrant angiogenesis creates dysfunctional tumor blood vessels, significantly limiting immune cell infiltration and therapeutic drug delivery. Therapeutic strategies, such as VEGF inhibitors (e.g., bevacizumab), have shown promise in normalizing tumor vasculature by pruning immature vessels, stabilizing vessel structures, and improving tumor perfusion (26, 27). Clinically, combining anti-VEGF therapy with immune checkpoint inhibitors enhances immune infiltration and efficacy, with early evidence from trials such as NCT02366143 (advanced renal cell carcinoma) (28, 29).
3.2 Modification of extracellular matrix and fibrosis
ECM deposition and tumor-associated fibrosis restrict immune cell infiltration and drug diffusion (30). Agents targeting ECM components, including collagenase and hyaluronidase enzymes, have demonstrated preclinical efficacy by enhancing immune infiltration and drug penetration (31). Notably, recombinant hyaluronidase (PEGPH20) in combination with chemotherapy or immunotherapy has entered clinical trials (NCT01839487, NCT03481920), aiming to alleviate ECM density and thus promote immune cell access and therapeutic efficacy in pancreatic and other solid tumors (32, 33). However, the phase III HALO 301 study in HA-high metastatic pancreatic ductal adenocarcinoma showed no overall survival (OS) or progression-free survival (PFS) benefit with PEGPH20 plus nab-paclitaxel/gemcitabine versus placebo plus nab-paclitaxel/gemcitabine: median OS 11.2 vs 11.5 months (HR 1.00; 95% CI 0.80 – 1.27; P = 0.97) and median PFS 7.1 vs 7.1 months (HR 0.97; 95% CI 0.75 – 1.26). Although ORR was higher (47% vs 36%), this did not translate into survival improvement, and grade ≥3 AEs (e.g., fatigue, muscle spasm, hyponatremia) occurred more frequently in the PEGPH20 arm (34).
The failure of HALO 301 to achieve survival benefit may be related to the heterogeneity of hyaluronan expression among patients, incomplete stromal remodeling, and treatment-limiting toxicities, which may have offset the potential pharmacokinetic gains from ECM targeting. This interpretation is supported by both clinical and mechanistic evidence. Subgroup analyses from earlier PEGPH20 trials demonstrated that clinical benefit was largely restricted to patients with uniformly high hyaluronan (HA) expression, whereas HALO - 301 enrolled an HA-high cohort with substantial intra- and inter-tumoral variability, potentially leading to patient misclassification and dilution of effect (32). Mechanistic studies further reveal that enzymatic depletion of HA can transiently decompress tumor vessels and enhance drug delivery, but the degree and durability of stromal remodeling in patients may be insufficient to translate into consistent survival gains (7). Additionally, higher rates of grade ≥3 adverse events in the PEGPH20 arm, including thromboembolic events, likely reduced treatment adherence and dose intensity, offsetting pharmacokinetic advantages (34).
3.3 Alleviation of tumor hypoxia
Hypoxia-driven metabolic adaptations profoundly suppress immune function within tumors. Therapeutics aimed at targeting hypoxia-inducible factors (HIF - 1α inhibitors) and innovative oxygen-releasing nanoparticles have been explored to mitigate hypoxia (35). Small molecules like evofosfamide (TH - 302), designed to target hypoxic regions selectively, have shown promise in preclinical and clinical studies (e.g., NCT01497444), improving immune cell functionality and tumor sensitivity to immunotherapy when combined with immune checkpoint inhibitors (36). Nevertheless, the randomized phase III SARC021 trial in soft-tissue sarcoma found that adding evofosfamide to doxorubicin did not improve OS versus doxorubicin alone; therefore, this combination cannot be recommended as first-line therapy in that setting (37). Similarly, the lack of efficacy in SARC021 may be explained by insufficient hypoxia selectivity, heterogeneous tumor oxygenation profiles, and possible limitations in drug penetration to hypoxic niches.
3.4 Metabolic reprogramming of the immunosuppressive microenvironment
Tumor metabolism profoundly shapes immunological responses through accumulation of immunosuppressive metabolites such as lactate, adenosine, and kynurenine. Therapeutics such as adenosine pathway inhibitors (e.g., anti-CD73, anti-A2A receptor antibodies) are actively being investigated in clinical trials (NCT02503774, NCT03367819) to neutralize adenosine-driven immune suppression (38, 39). Similarly, lactate transport inhibitors targeting monocarboxylate transporters (MCTs) have entered preclinical testing, aiming to restore immune effector cell functionality within tumors.
3.5 Targeting immunosuppressive cell populations
Various approaches target immunosuppressive cells such as Tregs, MDSCs, and TAMs. Anti-CSF-1R antibodies (e.g., cabiralizumab), aimed at reprogramming TAM polarization from an immunosuppressive M2 phenotype toward a pro-inflammatory M1 phenotype, are being clinically evaluated (NCT02526017, NCT02880371) (40, 41). Similarly, anti-CCR4 antibody (mogamulizumab) targeting Tregs has shown potential for enhancing immune activation in solid tumors (NCT02946671) (42).
3.6 Physical and ablative strategies to remodel TME
Localized physical approaches, including radiotherapy, photodynamic therapy (PDT), and photothermal therapy (PTT), have been utilized to physically disrupt the tumor architecture and induce ICD (43). Clinical evidence from radiotherapy combined with immune checkpoint inhibitors (e.g., PEMBRO-RT, NCT02492568) supports further evaluation by promoting immune cell infiltration and reversing local immune suppression through induced inflammatory responses and DAMP release (44).
3.7 Nanotechnology-based drug delivery systems:
Advanced nanoparticle-based delivery systems are being employed to specifically modulate the TME, enhance drug delivery, and amplify ICD efficacy. For instance, camptothecin-based nanoparticle formulations (CRLX101, NCT02769962) enhance tumor-specific ICD induction by promoting controlled intratumoral release, reducing systemic toxicity, and overcoming TME barriers through improved bioavailability and therapeutic targeting (45, 46).
These multifaceted strategies to overcome immunological, physical, and metabolic barriers demonstrate substantial potential to amplify ICD-induced antitumor immunity. Continued clinical development, optimization of combination regimens, and identification of predictive biomarkers will further improve the therapeutic efficacy of ICD in solid tumors, ultimately enhancing patient outcomes. A comprehensive overview of these therapeutic strategies, representative agents, mechanisms of action, and corresponding clinical trials is provided in Table 1. In the next section, we transition from mechanistic rationale to clinical translation, explicitly pairing each strategy with outcomes from representative trials.
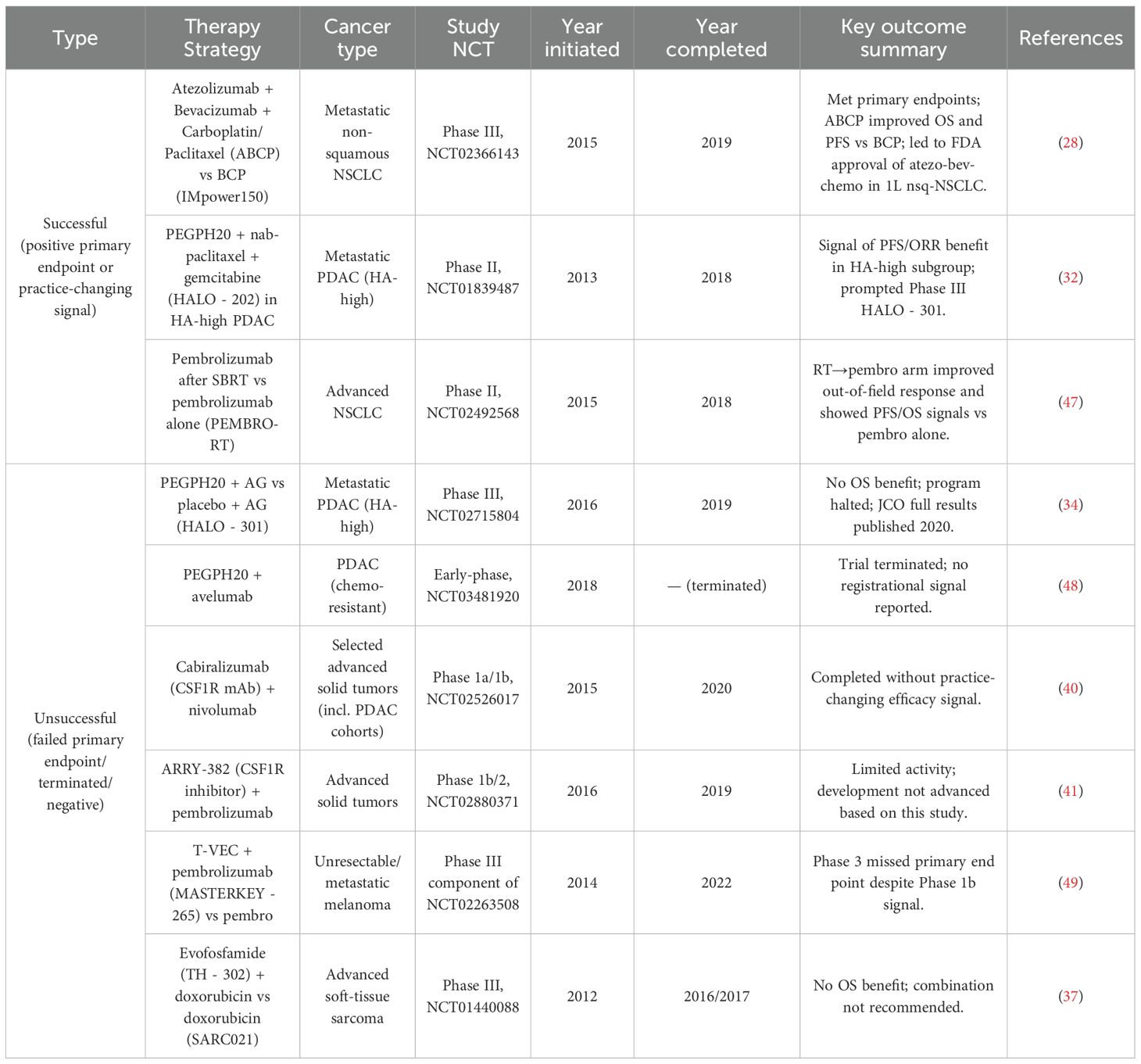
Table 1. Clinical studies of stroma/ECM or TME-modulating strategies (split by outcome) with cancer type and timeline.
4 Clinical translation and current evidence
Clinical translation of strategies targeting TME barriers to enhance ICD represents a rapidly evolving area in oncology, underscored by a growing number of clinical trials and promising therapeutic outcomes (43). Various innovative combinations designed to reshape the immunosuppressive, metabolic, and physical landscape of solid tumors have advanced into clinical evaluation, highlighting both the feasibility and therapeutic potential of TME-targeted ICD enhancement. Building on the mechanistic rationale outlined in Section 3, we connect each intervention to the specific barrier it addresses and summarize key endpoints (PFS, OS, ORR) from representative studies to clarify the strength and limitations of the evidence.
4.1 Combining ICD-inducing chemotherapy with microenvironmental modulators
Emerging clinical evidence suggests potential benefit from combining conventional ICD-inducing chemotherapies, such as oxaliplatin, cyclophosphamide, and paclitaxel, with TME-targeted therapies in selected populations. For example, oxaliplatin-based chemotherapy combined with immune checkpoint inhibitors and antiangiogenic agents (e.g., bevacizumab) has been investigated in metastatic colorectal cancer (NCT02375672) and pancreatic cancer (NCT04181645) (50, 51). These studies demonstrated improved immune cell infiltration, reduced immunosuppression, and enhanced overall response rates compared to chemotherapy alone, reflecting synergistic effects from simultaneous ICD induction and microenvironmental modulation. For instance, in the phase 3 IMpower150 study, atezolizumab + bevacizumab + carboplatin/paclitaxel achieved a median PFS of 8.3 vs 6.8 months and 6-month PFS rates of 71.7% vs 57.0% compared with bevacizumab + chemotherapy; subsequent analyses confirmed an overall survival advantage for the combination, albeit with effect sizes on the order of months and varying across subgroups (52). In part, the clinical success of IMpower150 may be attributed to complementary mechanisms, including VEGF inhibition–mediated vascular normalization, relief of VEGF-driven immunosuppression, and chemotherapy-induced immunogenic cell death, which together facilitate enhanced T-cell infiltration and activation.
4.2 Radiotherapy-induced ICD combined with immune checkpoint blockade
Radiotherapy, a potent ICD inducer, has shown preliminary or modest benefit in combination with immune checkpoint inhibitors by simultaneously alleviating physical barriers, triggering immunogenic cell death, and reducing TME-associated immune suppression. The PEMBRO-RT trial (NCT02492568) combined pembrolizumab with focal radiotherapy in metastatic non-small-cell lung cancer (NSCLC), showing increased intratumoral cytotoxic T-cell infiltration, elevated immune activation biomarkers, and improved progression-free survival (47). In PEMBRO-RT, the ORR at 12 weeks was 36% vs 18% (P = 0.07), median PFS 6.6 vs 1.9 months (HR 0.71; P = 0.19), and median OS 15.9 vs 7.6 months (HR 0.66; P = 0.16) for pembrolizumab + SBRT versus pembrolizumab alone; although numerical improvements were observed, differences did not reach statistical significance, with the largest signal seen in PD-L1-negative tumors (47). Similarly, the RADVAX study (NCT01497808) involving prostate cancer patients combined radiotherapy and ipilimumab, revealing significant immune modulation and encouraging clinical responses, thereby providing strong clinical evidence supporting this combinational approach (53).
4.3 Nanoparticle-based delivery systems for enhanced ICD and TME modulation
Clinical trials investigating nanoparticle formulations designed for targeted ICD induction and TME modulation represent another promising translational avenue. CRLX101, a camptothecin-based nanoparticle formulation, demonstrated enhanced ICD induction via potent DNA damage and ER stress, and has been clinically tested in combination with pembrolizumab for advanced solid tumors including ovarian and colorectal cancers (NCT02769962) (54). Preliminary results indicate improved intratumoral drug retention, enhanced immune cell recruitment, and promising safety profiles, affirming the clinical feasibility of nanoparticle-based ICD enhancement strategies. However, efficacy signals remain preliminary and no randomized phase III data are currently available for most nanoformulations; thus, any clinical benefit should be regarded as investigational pending definitive trials.
4.4 Oncolytic viruses and peptide-based therapies for TME remodeling
Localized ICD induction approaches such as oncolytic viruses and intratumoral peptide therapies are gaining clinical traction. Talimogene laherparepvec (T-VEC), an FDA-approved oncolytic virus, induces robust ICD through direct tumor lysis and immune activation, remodeling the local TME. Clinical trials combining T-VEC with immune checkpoint inhibitors (e.g., KEYNOTE - 034, NCT02263508) have reported substantial improvement in overall response rates and prolonged patient survival compared to historical monotherapy outcomes (55, 56). By contrast, in the randomized phase 3 MASTERKEY - 265 trial, T-VEC + pembrolizumab did not significantly improve PFS (HR 0.86; 95% CI 0.71 – 1.04; P = 0.13) or OS (HR 0.96; 95% CI 0.76 – 1.22; P = 0.74) compared with pembrolizumab plus placebo; ORR was 48.6% vs 41.3% (CR 17.9% vs 11.6%), and grade ≥3 treatment-related AEs occurred in 20.7% vs 19.5% (49). For MASTERKEY - 265, the absence of benefit despite robust intratumoral viral replication suggests that oncolytic virus–mediated immune priming alone may be insufficient in poorly immunogenic tumors, highlighting the need for improved patient selection and combination sequencing. Likewise, LTX - 315, an oncolytic peptide designed to trigger ICD and local TME modulation, demonstrated encouraging immunological responses and tumor regression in early-phase clinical studies (NCT01986426), further validating the clinical potential of localized ICD approaches (57).
4.5 Clinical management of toxicities and safety considerations
Although promising, these combinational strategies necessitate careful clinical management due to potential additive toxicities, particularly immune-related adverse events (irAEs). Clinical experiences from trials such as PEMBRO-RT in advanced non-small cell lung cancer (NSCLC) (NCT02492568), RADVAX in metastatic melanoma (NCT01497808), and nanoparticle-based therapies in triple-negative breast cancer (TNBC) (NCT02769962) highlight the importance of meticulous patient monitoring, precise dosing strategies, and supportive management protocols to ensure optimal safety and therapeutic effectiveness (47, 58, 59). As an example, in MASTERKEY - 265, grade ≥3 treatment-related adverse events occurred in approximately one-fifth of patients in both arms (20.7% vs 19.5%), underscoring the need for careful monitoring and patient selection when deploying TME-modulating combinations (49).
4.6 Summary of clinical insights
Taken together, current clinical studies suggest a potential for enhancing ICD through targeted modulation of the TME; however, the magnitude of benefit is generally modest (often measured in months), heterogeneous across tumor types and biomarkers, and not consistently accompanied by overall survival gains. Positive signals (e.g., IMpower150) coexist with neutral phase III results (e.g., HALO 301 for PEGPH20; SARC021 for evofosfamide; MASTERKEY - 265 for T-VEC + pembrolizumab), highlighting unresolved questions around patient selection, optimal sequencing/combination, and toxicity management. Consequently, biomarker-guided, randomized trials with rigorous time-to-event endpoints remain essential to define where these strategies deliver clinically meaningful benefit (25).
5 Challenges and concluding perspectives
Although multiple TME-targeted approaches can potentiate ICD in principle, most improvements observed clinically are incremental and become evident only over several months of follow-up. Moreover, not all mechanistically compelling strategies translate into survival benefits in randomized settings (e.g., PEGPH20 in HALO 301, evofosfamide in SARC021, and T-VEC + pembrolizumab in MASTERKEY - 265), emphasizing the need for stringent patient stratification and robust phase III validation before broad adoption (34, 49). These examples illustrate that despite strong mechanistic rationale, limitations such as inadequate biomarker-driven patient selection, treatment-related toxicities, and challenges in trial design often contribute to the lack of survival benefit, highlighting the importance of refining future clinical approaches.
Despite significant clinical progress in targeting TME barriers to enhance ICD, several critical challenges remain. Tumor heterogeneity continues to complicate therapeutic strategies, as variable patient responses demand personalized approaches guided by robust biomarkers (60). Additionally, the complexity of combinatorial regimens increases the risk of immune-related adverse events (irAEs), emphasizing the need for refined clinical management protocols (61). Translating promising preclinical findings to clinical success requires development of advanced preclinical models that accurately reflect human tumor biology and immune interactions.
Nevertheless, integrating ICD induction with precise TME modulation offers substantial promise. For clarity, this section has been revised to more clearly articulate how the preceding discussion of barriers is directly connected to the rationale for these integrated strategies. Future directions should focus on personalized therapeutic regimens based on individual tumor characteristics, innovative delivery platforms (e.g., nanoparticle technologies), and improved clinical trial designs featuring adaptive strategies and immune-specific endpoints. Interdisciplinary collaboration among clinicians, immunologists, bioengineers, and pharmaceutical scientists remains essential to addressing these challenges. Equally important, these integrated strategies—particularly the combination of ICD induction with microenvironment-targeted therapies—hold substantial clinical promise. However, their translation into routine practice will critically depend on rigorous validation in well-designed clinical trials. Ultimately, continued refinement of these integrated approaches is poised to significantly expand patient populations benefiting from ICD-based therapies, reshaping the landscape of cancer immunotherapy and substantially improving clinical outcomes.
Author contributions
ML: Writing – original draft, Writing – review & editing. WH: Writing – original draft, Data curation. MZ: Writing – review & editing, Data curation. JZ: Data curation, Writing – original draft, Writing – review & editing. YY: Data curation, Supervision, Writing – original draft.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be constructed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Lv J, Cai J, Yuan K, Wang J, Xu Y, Huang X, et al. Immunogenic cell death inducers:Current advances and future perspectives. Coordination Chem Rev. (2025) 542:216846. doi: 10.1016/j.ccr.2025.216846
2. Arimoto K-I, Miyauchi S, Liu M, and Zhang D-E. Emerging role of immunogenic cell death in cancer immunotherapy. Front Immunol. (2024) 15-2024:1390263. doi: 10.3389/fimmu.2024.1390263
3. Galluzzi L, Guilbaud E, Schmidt D, Kroemer G, and Marincola FM. Targeting immunogenic cell stress and death for cancer therapy. Nat Rev Drug Discov. (2024) 23:445–60. doi: 10.1038/s41573-024-00920-9
4. Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini J-L, et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med. (2007) 13:54–61. doi: 10.1038/nm1523
5. Fucikova J, Kralikova P, Fialova A, Brtnicky T, Rob L, Bartunkova J, et al. Human tumor cells killed by anthracyclines induce a tumor-specific immune response. Cancer Res. (2011) 71:4821–33. doi: 10.1158/0008-5472.CAN-11-0950
6. Jiang J, Yan Y, Yang C, and Cai H. Immunogenic cell death and metabolic reprogramming in cancer: mechanisms, synergies, and innovative therapeutic strategies. Biomedicines. (2025) 13. doi: 10.3390/biomedicines13040950
7. Provenzano Paolo P, Cuevas C, Chang Amy E, Goel Vikas K, Von Hoff Daniel D, and Hingorani Sunil R. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell. (2012) 21:418–29. doi: 10.1016/j.ccr.2012.01.007
8. Jacobetz MA, Chan DS, Neesse A, Bapiro TE, Cook N, Frese KK, et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut. (2013) 62:112. doi: 10.1136/gutjnl-2012-302529
9. Lunardi S, Jamieson NB, Lim SY, Griffiths KL, Carvalho-Gaspar M, Al-Assar O, et al. IP-10/CXCL10 induction in human pancreatic cancer stroma influences lymphocytes recruitment and correlates with poor survival. Oncotarget. (2014) 5. doi: 10.18632/oncotarget.v5i22
10. Tschumperlin DJ and Lagares D. Mechano-therapeutics: targeting mechanical signaling in fibrosis and tumor stroma. Pharmacol Ther. (2020) 212:107575. doi: 10.1016/j.pharmthera.2020.107575
11. Zheng Y, Xu R, Chen X, Lu Y, Zheng J, Lin Y, et al. Metabolic gatekeepers: harnessing tumor-derived metabolites to optimize T cell-based immunotherapy efficacy in the tumor microenvironment. Cell Death Dis. (2024) 15:775. doi: 10.1038/s41419-024-07122-6
12. Alvarado-Ortiz E and Sarabia-SáNchez MA. Hypoxic link between cancer cells and the immune system: The role of adenosine and lactate. Oncol Res. (2025) 33. doi: 10.32604/or.2025.065953
13. Meng X, Che C, Yi Y, and Qu X. Reprogramming the tumor-immune landscape via nanomaterial-induced immunogenic cell death: a mini review. Front Bioengineering Biotechnol. (2025) 13-2025:1635747. doi: 10.3389/fbioe.2025.1635747
14. Brand A, Singer K, Koehl Gudrun E, Kolitzus M, Schoenhammer G, Thiel A, et al. LDHA-associated lactic acid production blunts tumor immunosurveillance by T and NK cells. Cell Metab. (2016) 24:657–71. doi: 10.1016/j.cmet.2016.08.011
15. Weiss SA, Djureinovic D, Jessel S, Krykbaeva I, Zhang L, Jilaveanu L, et al. A phase I study of APX005M and cabiralizumab with or without nivolumab in patients with melanoma, kidney cancer, or non–small cell lung cancer resistant to anti-PD-1/PD-L1. Clin Cancer Res. (2021) 27:4757–67. doi: 10.1158/1078-0432.CCR-21-0903
16. Tie Y, Tang F, Wei Y-Q, and Wei X-W. Immunosuppressive cells in cancer: mechanisms and potential therapeutic targets. J Hematol Oncol. (2022) 15:61. doi: 10.1186/s13045-022-01282-8
17. Park K, Veena MS, and Shin DS. Key players of the immunosuppressive tumor microenvironment and emerging therapeutic strategies. Front Cell Dev Biol. (2022) 10-2022:830208. doi: 10.3389/fcell.2022.830208
18. Spranger S, Bao R, and Gajewski TF. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature. (2015) 523:231–5. doi: 10.1038/nature14404
19. Chen T-T, Li X, Zhang Y, Kang X-J, Zhang S-F, Zhang T, et al. Breaking down physical barriers: strategies to improve lymphocyte infiltration for effective neoantigen-based therapies. Front Immunol. (2025) 16-2025:1614228. doi: 10.3389/fimmu.2025.1614228
20. Zhang M and Zhang B. Extracellular matrix stiffness: mechanisms in tumor progression and therapeutic potential in cancer. Exp Hematol Oncol. (2025) 14:54. doi: 10.1186/s40164-025-00647-2
21. Chauhan Vikash P, Boucher Y, Ferrone Cristina R, Roberge S, Martin John D, Stylianopoulos T, et al. Compression of pancreatic tumor blood vessels by hyaluronan is caused by solid stress and not interstitial fluid pressure. Cancer Cell. (2014) 26:14–5. doi: 10.1016/j.ccr.2014.06.003
22. Jensen C, Nissen NI, Von Arenstorff CS, Karsdal MA, and Willumsen N. Serological assessment of collagen fragments and tumor fibrosis may guide immune checkpoint inhibitor therapy. J Exp Clin Cancer Res. (2021) 40:326. doi: 10.1186/s13046-021-02133-z
23. Ohta A, Gorelik E, Prasad SJ, Ronchese F, Lukashev D, Wong MKK, et al. A2A adenosine receptor protects tumors from antitumor T cells. Proc Natl Acad Sci. (2006) 103:13132–7. doi: 10.1073/pnas.0605251103
24. Renner K, Bruss C, Schnell A, Koehl G, Becker HM, Fante M, et al. Restricting glycolysis preserves T cell effector functions and augments checkpoint therapy. Cell Rep. (2019) 29:135–150.e139. doi: 10.1016/j.celrep.2019.08.068
25. Socinski Mark A, Jotte Robert M, Cappuzzo F, Orlandi F, Stroyakovskiy D, Nogami N, et al. Atezolizumab for first-line treatment of metastatic nonsquamous NSCLC. New Engl J Med. (2018) 378:2288–301. doi: 10.1056/NEJMoa1716948
26. Chen K, Jiang B, Yan H, Yang L, and Chen Z. Case report: Bevacizumab-induced cerebrovascular events: a case series report and literature review. Front Oncol. (2025) 15-2025:1395129. doi: 10.3389/fonc.2025.1395129
27. Zhang J, Yu J, Yang D, Jiang L, Dong X, Liu Z, et al. Bevacizumab reduces cerebral radiation necrosis due to stereotactic radiotherapy in non-small cell lung cancer patients with brain metastases: an inverse probability of treatment weighting analysis. Front Immunol. (2024) 15-2024:1399613. doi: 10.3389/fimmu.2024.1399613
28. Nogami N, Barlesi F, Socinski MA, Reck M, Thomas CA, Cappuzzo F, et al. IMpower150 final exploratory analyses for atezolizumab plus bevacizumab and chemotherapy in key NSCLC patient subgroups with EGFR mutations or metastases in the liver or brain. J Thorac Oncol. (2022) 17:309–23. doi: 10.1016/j.jtho.2021.09.014
29. Socinski MA, Mok TS, Nishio M, Jotte RM, Cappuzzo F, Orlandi F, et al. Abstract CT216: IMpower150 final analysis: Efficacy of atezolizumab (atezo) + bevacizumab (bev) and chemotherapy in first-line (1L) metastatic nonsquamous (nsq) non-small cell lung cancer (NSCLC) across key subgroups. Cancer Res. (2020) 80:CT216–6. doi: 10.1158/1538-7445.AM2020-CT216
30. Li L, Fu H, and Liu Y. The fibrogenic niche in kidney fibrosis: components and mechanisms. Nat Rev Nephrol. (2022) 18:545–57. doi: 10.1038/s41581-022-00590-z
31. Jiratchayamaethasakul C, Ding Y, Hwang O, Im S-T, Jang Y, Myung S-W, et al. In vitro screening of elastase, collagenase, hyaluronidase, and tyrosinase inhibitory and antioxidant activities of 22 halophyte plant extracts for novel cosmeceuticals. Fisheries Aquat Sci. (2020) 23:6. doi: 10.1186/s41240-020-00149-8
32. Hingorani SR, Zheng L, Bullock AJ, Seery TE, Harris WP, Sigal DS, et al. HALO 202: randomized phase II study of PEGPH20 plus nab-paclitaxel/gemcitabine versus nab-paclitaxel/gemcitabine in patients with untreated, metastatic pancreatic ductal adenocarcinoma. J Clin Oncol. (2017) 36:359–66. doi: 10.1200/JCO.2017.74.9564
33. Morosi L, Meroni M, Ubezio P, Fuso Nerini I, Minoli L, Porcu L, et al. PEGylated recombinant human hyaluronidase (PEGPH20) pre-treatment improves intra-tumour distribution and efficacy of paclitaxel in preclinical models. J Exp Clin Cancer Res. (2021) 40:286. doi: 10.1186/s13046-021-02070-x
34. Van Cutsem E, Tempero MA, Sigal D, Oh D-Y, Fazio N, Macarulla T, et al. Randomized phase III trial of pegvorhyaluronidase alfa with nab-paclitaxel plus gemcitabine for patients with hyaluronan-high metastatic pancreatic adenocarcinoma. J Clin Oncol. (2020) 38:3185–94. doi: 10.1200/JCO.20.00590
35. Chawla SP, Cranmer LD, Van Tine BA, Reed DR, Okuno SH, Butrynski JE, et al. Phase II study of the safety and antitumor activity of the hypoxia-activated prodrug TH-302 in combination with doxorubicin in patients with advanced soft tissue sarcoma. J Clin Oncol. (2014) 32:3299–306. doi: 10.1200/JCO.2013.54.3660
36. Tran NH, Foster NR, Mahipal A, Byrne T, Hubbard J, Silva A, et al. Phase IB study of sorafenib and evofosfamide in patients with advanced hepatocellular and renal cell carcinomas (NCCTG N1135, Alliance). Investigational New Drugs. (2021) 39:1072–80. doi: 10.1007/s10637-021-01090-w
37. Tap WD, Papai Z, Van Tine BA, Attia S, Ganjoo KN, Jones RL, et al. Doxorubicin plus evofosfamide versus doxorubicin alone in locally advanced, unresectable or metastatic soft-tissue sarcoma (TH CR-406/SARC021): an international, multicentre, open-label, randomised phase 3 trial. Lancet Oncol. (2017) 18:1089–103. doi: 10.1016/S1470-2045(17)30381-9
38. Bendell J, LoRusso P, Overman M, Noonan AM, Kim D-W, Strickler JH, et al. First-in-human study of oleclumab, a potent, selective anti-CD73 monoclonal antibody, alone or in combination with durvalumab in patients with advanced solid tumors. Cancer Immunology Immunotherapy. (2023) 72:2443–58. doi: 10.1007/s00262-023-03430-6
39. Paolo Andrea Z, Chia-Chi L, Bradley CC, Todd MB, Marcello T, Antoine I, et al. Targeting CD38 and PD-1 with isatuximab plus cemiplimab in patients with advanced solid Malignancies: results from a phase I/II open-label, multicenter study. J ImmunoTherapy Cancer. (2022) 10:e003697.
40. A phase 1a/1b study of cabiralizumab in combination with nivolumab in patients with selected advanced cancers. (2015).
41. Johnson M, Dudek AZ, Sukari A, Call J, Kunk PR, Lewis K, et al. ARRY-382 in combination with pembrolizumab in patients with advanced solid tumors: results from a phase 1b/2 study. Clin Cancer Res. (2022) 28:2517–26. doi: 10.1158/1078-0432.CCR-21-3009
42. Phase I study of pre-operative combination therapy with mogamulizumab (Anti-CCR4) and nivolumab (Anti-PD-1) against solid cancer patients. (2016).
43. Gao J, Wang W-Q, Pei Q, Lord MS, and Yu H-J. Engineering nanomedicines through boosting immunogenic cell death for improved cancer immunotherapy. Acta Pharmacologica Sin. (2020) 41:986–94. doi: 10.1038/s41401-020-0400-z
44. Randomized phase II, 2-arm study of pembrolizumab after high dose radiation (SBRT) versus pembrolizumab alone in patients with advanced non-small cell lung cancer. (2015).
45. Efficacy, safety and immunogenicity study of GSK biologicals' Candidate malaria vaccine (SB257049) evaluating schedules with or with-out fractional doses, early dose 4 and yearly doses, in children 5 – 17 months of age. (2017).
46. Serrano-Martínez A, Victoria-Montesinos D, García-Muñoz AM, Hernández-Sánchez P, Lucas-Abellán C, and González-Louzao R. A systematic review of clinical trials on the efficacy and safety of CRLX101 cyclodextrin-based nanomedicine for cancer treatment. Pharmaceutics. (2023) 15. doi: 10.3390/pharmaceutics15071824
47. Theelen WSME, Peulen HMU, Lalezari F, van der Noort V, de Vries JF, Aerts JGJV, et al. Effect of pembrolizumab after stereotactic body radiotherapy vs pembrolizumab alone on tumor response in patients with advanced non–small cell lung cancer: results of the PEMBRO-RT phase 2 randomized clinical trial. JAMA Oncol. (2019) 5:1276–82. doi: 10.1001/jamaoncol.2019.1478
48. Rodriguez LM, Guillén-Ponce C, Feliu J, and Hidalgo M. A pilot trial of PEGPH20 (Pegvorhyaluronidase alfa) in combination with avelumab (anti-PD-L1 MSB0010718C) in chemotherapy resistant pancreatic cancer (PDAC)-Trial in progress. Ann Oncol. (2018) 29:v51. doi: 10.1093/annonc/mdy151.182
49. Chesney JA, Ribas A, Long GV, Kirkwood JM, Dummer R, Puzanov I, et al. Randomized, double-blind, placebo-controlled, global phase III trial of talimogene laherparepvec combined with pembrolizumab for advanced melanoma. J Clin Oncol. (2022) 41:528–40. doi: 10.1200/JCO.22.00343
50. Shahda S, Noonan AM, Bekaii-Saab TS, O'Neil BH, Sehdev A, Shaib WL, et al. A phase II study of pembrolizumab in combination with mFOLFOX6 for patients with advanced colorectal cancer. J Clin Oncol. (2017) 35:3541–1.
51. Cui J, Yang H, Hu J, Yao J, Wang Y, Liang Y, et al. Anti-PD-1 antibody combined with albumin-bound paclitaxel and gemcitabine (AG) as first-line therapy and Anti-PD-1 monotherapy as maintenance in metastatic pancreatic ductal adenocarcinoma (PDAC). J Clin Oncol. (2021) 39:e16218–8.
52. Socinski MA, Nishio M, Jotte RM, Cappuzzo F, Orlandi F, Stroyakovskiy D, et al. IMpower150 final overall survival analyses for atezolizumab plus bevacizumab and chemotherapy in first-line metastatic nonsquamous NSCLC. J Thorac Oncol. (2021) 16:1909–24. doi: 10.1016/j.jtho.2021.07.009
53. Maity A, Mick R, Rengan R, Mitchell TC, Amaravadi RK, Schuchter LM, et al. A stratified phase I dose escalation trial of hypofractionated radiotherapy followed by ipilimumab in metastatic melanoma: long-term follow-up and final outcomes. OncoImmunology. (2021) 10:1863631. doi: 10.1080/2162402X.2020.1863631
54. Schmidt KT, Huitema ADR, Dorlo TPC, Peer CJ, Cordes LM, Sciuto L, et al. Population pharmacokinetic analysis of nanoparticle-bound and free camptothecin after administration of NLG207 in adults with advanced solid tumors. Cancer Chemotherapy Pharmacol. (2020) 86:475–86. doi: 10.1007/s00280-020-04134-9
55. Kjeldsen JW, Lorentzen CL, Martinenaite E, Ellebaek E, Donia M, Holmstroem RB, et al. A phase 1/2 trial of an immune-modulatory vaccine against IDO/PD-L1 in combination with nivolumab in metastatic melanoma. Nat Med. (2021) 27:2212–23. doi: 10.1038/s41591-021-01544-x
56. A phase 1b/3, multicenter, trial of talimogene laherparepvec in combination with pembrolizumab (MK-3475) for treatment of unresectable stage IIIB to IVM1c melanoma (MASTERKEY-265). (2014).
57. A phase I, open-label, multi-arm, multi-centre, multi-dose, dose escalation study of LTX-315 as monotherapy or in combination with either ipilimumab or pembrolizumab in patients with transdermally accessible tumours. (2013).
58. Radvax™: A stratified phase I/ii dose escalation trial of hypofractionated radiotherapy followed by ipilimumab in metastatic melanoma. (2011).
59. A phase I/II trial of EP0057, a nanoparticle camptothecin with olaparib in patients with relapsed/refractory small cell lung, bladder and prostate cancers. (2016).
60. Proietto M, Crippa M, Damiani C, Pasquale V, Sacco E, Vanoni M, et al. Tumor heterogeneity: preclinical models, emerging technologies, and future applications. Front Oncol. (2023) 13-2023:1164535. doi: 10.3389/fonc.2023.1164535
Keywords: immunogenic cell death (ICD), tumor microenvironment (TME), immune suppression, tumor microenvironmental barriers, metabolic reprogramming
Citation: Zhang J, Han W, Zhang M, Yi Y and Long M (2025) Targeting tumor microenvironmental barriers to enhance immunogenic cell death in solid tumors. Front. Immunol. 16:1672601. doi: 10.3389/fimmu.2025.1672601
Received: 24 July 2025; Accepted: 27 August 2025;
Published: 11 September 2025.
Edited by:
Rahul Shivahare, The Ohio State University, United StatesReviewed by:
Zhengrui Li, Shanghai Jiao Tong University, ChinaRavi Thakur, CHRIST (Deemed to be University), India
Copyright © 2025 Zhang, Han, Zhang, Yi and Long. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yingjie Yi, eWl5aW5namllMjAxM0AxNjMuY29t; Mei Long, MTg1NjAyOTA1MzJAMTYzLmNvbQ==
†These authors share first authorship