 Eriko Ohsaki
Eriko Ohsaki Keiji Ueda*
Keiji Ueda*- Division of Virology, Department of Microbiology and Immunology, Osaka University Graduate School of Medicine, Suita, Osaka, Japan
Kaposi’s sarcoma-associated herpesvirus (KSHV) is thought to be an oncogenic member of the γ-herpesvirus subfamily. The virus usually establishes latency upon infection as a default infection pattern. The viral genome replicates according to the host cell cycle by recruiting the host cellular replication machinery. Among the latently expressing viral factors, LANA plays pivotal roles in viral genome replication, partitioning, and maintenance. LANA binds with two LANA-binding sites (LBS1/2) within a terminal repeat (TR) sequence and is indispensable for viral genome replication in latency. The nuclear matrix region seems to be important as a replication site, since LANA as well as cellular replication factors accumulate there and recruit the viral replication origin in latency (ori-P) by its binding activity to LBS. KSHV ori-P consists of LBS followed by a 32-bp GC-rich segment (32GC). Although it has been reported that LANA recruits cellular pre-replication complexes (pre-RC) such as origin recognition complexes (ORCs) to the ori-P through its interaction with ORCs, this mechanism does not account completely for the requirement of the 32GC. On the other hand, there are few reports about the partitioning and maintenance of the viral genome. LANA interacts with many kinds of chromosomal proteins, including Brd2/RING3, core histones, such as H2A/H2B and histone H1, and so on. The detailed molecular mechanisms by which LANA enables KSHV genome partitioning and maintenance still remain obscure. By integrating the findings reported thus far on KSHV genome replication, partitioning, and maintenance in latency, we will summarize what we know now, discuss what questions remain to be answered, and determine what needs to be done next to understand the mechanisms underlying viral replication, partitioning, and maintenance strategy.
Introduction
Kaposi’s sarcoma (KS)-associated herpesvirus (KSHV) is a gamma-2 herpesvirus discovered from KS specimens in 1994 (Chang et al., 1994). KSHV is closely associated with KS and several non-Hodgkin lymphomas, including primary effusion lymphoma (PEL) and multicentric Castleman’s disease (MCD; Cesarman et al., 1995, 1996; Soulier et al., 1995). While KS is the most common cancer in acquired immune deficiency syndrome patients (Potthoff et al., 2010), KSHV is detected in about 95% of all types of KS lesions by PCR analysis (Dupin et al., 1995; Huang et al., 1995; Moore and Chang, 1995). PEL is a rare B cell lymphoma originated from preterminal B cells, and PEL in AIDS patients is often associated with KSHV as well as EBV. Several KSHV-infected PEL cell lines have been established, and EBV is frequently lost in the course of establishment (Arvanitakis et al., 1996; Gaidano et al., 1996; Renne et al., 1996; Said et al., 1996; Carbone et al., 1997, 1998; Katano et al., 1999). MCD is a plasmacytic lymphadenopathy with polyclonal hyper-immunoglobulinemia and high levels of serum IL-6 (Frizzera et al., 1983; Yoshizaki et al., 1989).
Like all herpesviruses, KSHV has two life cycles: latent and lytic replication phases (for review, see Boshoff and Chang, 2001). Whereas KSHV is usually in latency when it infects KS and PEL cells, in MCD some cells express lytic genes (Katano et al., 2000; Parravicini et al., 2000). On the other hand, it has been reported that KSHV infection itself and/or viral lytic proteins promote cell proliferation and angiogenesis as well as lymphatic reprogramming (Ciufo et al., 2001; Gao et al., 2003; Carroll et al., 2004; Hong et al., 2004; Naranatt et al., 2004; Pan et al., 2004; Wang et al., 2004; Sharma-Walia et al., 2006; Qian et al., 2007, 2008; Sadagopan et al., 2007; Ye et al., 2007).
In latency, the KSHV genome is present as an episome, which is capable of autonomously replicating during S phase of the host cell cycle without integration into host chromosomes, and only limited genes are expressed during latency. Therefore, there is no generation of progeny virions. It is very important to elucidate and learn the virus’s survival strategy in order to control infection and to formulate treatment for KSHV-related diseases.
In this review, we would like to focus on studies on the mechanisms underlying viral DNA replication, genome segregation and maintenance, and gene expression regulation in latency, and to discuss these topics in the light of studies on cellular mechanisms.
Gene Expression Control in KSHV Latency
The KSHV genome is a double-stranded linear DNA in the virion. It is circularized upon infection and is maintained as an episome in the infected nucleus. The complete genome is about 160–170 kbp, including a 40 ∼ 50 times repeated sequence called a terminal repeat (TR), which is 801 bp as a unit, at each end of the genome (Figure 1). The viral genome encodes approximately 90 ORFs in the unique region (for reviews, see Moore and Chang, 2001; Dourmishev et al., 2003). In latency, the viral genome is maintained in a cell cycle-dependent manner, and extremely limited viral genes, such as latent gene clusters, are expressed. Although the establishment of latent infection could be this virus’s default infection mode, it is not good for the virus to continue latent infection for a long time, since it will vanish from the earth without progeny virus production. Thus, latency is a state in which the virus waits for an opportunity for the explosive production of progeny virions. Nevertheless, it is very interesting how KSHV establishes latency and is maintained in infected host cells without losing the genomes.
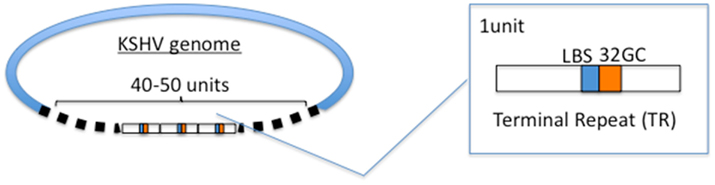
Figure 1. Terminal repeat (TR) sequences. The KSHV genome has 40–50 units of TR sequences. TR contains LANA-binding site (LBS) and GC-rich sequences termed 32GC. It is thought that one of the LBS-32GC sequence is selected as DNA replication origin (ori-P).
As mentioned above, the limited region within the KSHV genome is transcriptionally active in latency, and this region contains only several genes, including the latency-associated nuclear antigen (Wang and Frappier, 2009), viral cyclin (v-CYC), viral FLICE-inhibitory protein (v-FLIP), kaposin, 17 microRNAs (miRNAs), and viral interferon regulatory factor 3 (v-IRF-3; Chang et al., 1996; Thome et al., 1997; Muralidhar et al., 1998; Lubyova and Pitha, 2000; Gomez-Roman et al., 2001; Rivas et al., 2001; Staudt and Dittmer, 2003; Cai et al., 2005; Pearce et al., 2005; Pfeffer et al., 2005; Samols et al., 2005). Such genes, except for v-IRF-3, are in that limited region, and LANA, v-CYC, and v-FLIP are in one of the unit’s genes. This region forms an active locus for expression including miRNAs and kaposin. It is unclear why this region is active for the expression of genes and is insulated from inactive lytic genes, such as ORF69 and K14 just downstream and upstream, respectively.
Recently, it was reported that CTCF, which is the only insulator protein found in vertebrates, coupled with SMC3, endows this gene insulation. More interestingly, the binding sites are in the genes, i.e., downstream from the LANA transcription start sites, not at the boundary regions (Stedman et al., 2008; Kang and Lieberman, 2009). Transcriptional analysis using the KSHV-BAC system demonstrated that mutations of CTCF binding sites abolished latency-regulated transcription such as K14 and ORF74 during latency (Kang and Lieberman, 2009). CTCF usually binds at the boundary regions between active and inactive loci in mammalian genomes, forming locus control regions (LCRs; Tanimoto et al., 2003). A typical example is an LCR seen in the beta-globin locus. CTCF binds to several DNase I hypersensitivity sites (HS), called HS4 and HS5, and forms boundaries to insulate this locus from the outside locus (Tanimoto et al., 2003; Hou et al., 2008). Thus, latent gene expression in KSHV-infected cells might be regulated differently from the mechanism observed in the beta-globin locus.
Inversely, it is interesting how the viral lytic genes are tightly inactivated in latency. Epigenetic regulation seems to be essential for inactivation as well as activation of latent genes. LANA recruits heterochromatin components to the TR by the interaction between LANA and SUV39H1, which is a key factor that methylates histone H3, which in turn recruits heterochromatin protein 1 (HP1; Sakakibara et al., 2004). Because this mechanism contributes to the propagation and maintenance of heterochromatin, it appears that heterochromatin could spread over the KSHV genome during latency. The propagation of heterochromatin into the active latent gene zone might be blocked by the boundary effect and by the enhancer-blocking activity of an insulator, CTCF which has multiple functions such as gene activation or inactivation, X-chromosome inactivation, and gene imprinting (for review, see Zlatanova and Caiafa, 2009).
Thus, it is thought that not the overall lytic genes region, except for the latent gene clusters, forms heterochromatin during latency, because recent genome-wide analysis using ChIP-on-chip showed that not only latent gene clusters but also several regions of lytic genes are enriched in activating histone marks (acetylated H3 and H3K4me3). However, H3K27me3, which is a bivalent histone marker, is widely distributed through the KSHV genome (Toth et al., 2010), meaning that the genome is poised for reactivation. Furthermore, the treatment of specific histone demethylases of H3K27me3 such as JMJD3 and UTX could induce the lytic reactivation. Immunoprecipitation of methylated DNA assay showed that the KSHV genome was methylated during latency (Gunther and Grundhoff, 2010). Gunther and Grundhoff (2010) suggested that the CpG methylation process could take a long time to prevail over the genome, and thus could not control early latency. There are several reports that DNA methylation of viral genomes is related to the regulation of the gene expression of gammaherpesviruses such as EBV and herpesvirus saimiri (HVS; Minarovits, 2006). Heterochromatin formation on the viral genomes, however, seems to be inconvenient for the rapid induction of lytic replication. Further investigations are needed to clarify how viruses are ready for lytic induction if heterochromatin and/or DNA methylation was formed on the genome.
Viral factors play key roles in maintaining gene expression profiles in latency. Otherwise, modulation by viral and cellular factors maintains viral latency. In addition to the recruitment of heterochromatin factors on to the genome, LANA itself tends to repress viral lytic gene expression. LANA physically associates with recombination signal sequence-binding protein Jκ (RBP-Jκ) and represses the replication and transcription activator (RTA) promoter through the RBP-Jκ binding site existing within its promoter (Lan et al., 2005a).
Viral FLICE-inhibitory protein, also known as K13, interacts with several NF-κB-related signaling proteins and activates the NF-κB pathways, thus enhancing cell survival (Chaudhary et al., 1999; Field et al., 2003; Matta et al., 2003, 2007; Matta and Chaudhary, 2004). It should be noted that the effect of NF-κB signaling on reactivation depends on cellular context (Grossmann and Ganem, 2008) and seems to be regulated by an intricate balance within the cellular environment. Previous studies, however, demonstrated that v-FLIP repressed the RTA promoter by activating NF-κB binding to the cognate sequence, but in that case it deregulates vIL-6 and hIL-6 expression (Zhao et al., 2007). The reactivation is initiated by RTA, which is a lytic switch protein and a homolog of EBV BRLF1 (Liang et al., 2002). The RTA promoter region is highly responsive to 12-O-tetradecanoylphorbol 13-acetate (TPA) or phorbol 12-myristate 13-acetate (PMA), sodium butylate (NaB), and trichostatin A (TSA), and is associated with several histone deacetylase proteins such as HDAC, which leads to chromatin remodeling of a nucleosome and then regulates KSHV reactivation from latency (Lu et al., 2003). RTA activates various viral genes through direct binding with RTA-responsive elements existing within the K8 and ORF57 promoters (Byun et al., 2002) and also through indirect mechanisms on RTA itself and vIRF-1 (Nishimura et al., 2001; Sakakibara et al., 2001; Ueda et al., 2002). Although RTA is a strong transactivator and inducer of lytic replication, it also enhances LANA expression and then is involved in the establishment of latency in the early infection phase (Lan et al., 2005b). This feedback mechanism explains the low efficiency of lytic replication and the generation of complete viral particles in KSHV-infected cell lines.
MicroRNAs (miRNAs) are single-stranded and 20- to 23-nucleotide RNA molecules that are involved in gene expression (Bartel, 2004; Bartel and Chen, 2004). Recent studies have highlighted the critical role of viral microRNAs (miRNAs) in the maintenance of KSHV latency (for review, see Ganem and Ziegelbauer, 2008; Boss et al., 2009; Lei et al., 2010a). The KSHV genome contains 17 miRNAs that are clustered and located in the intragenic region between kaposin and v-FLIP (Cai et al., 2005; Pearce et al., 2005; Samols et al., 2005; Cai and Cullen, 2006). Surprisingly, a new proteomic approach suggests that a single miRNA can directly lead to the suppression of the synthesis of hundreds of proteins at both mRNA and translation levels, although the level of suppression is mild (Baek et al., 2008; Selbach et al., 2008).
Kaposi’s sarcoma-associated herpesvirus miRNAs are reported to regulate, directly or indirectly, various factors including lytic genes (Murphy et al., 2008; Bellare and Ganem, 2009) and cellular factors such as NF-κB and IκBα, the latter of which is directly regulated by KSHV miR-K1 (Lei et al., 2010b) and Bcl-2 associated factor (BCLAF1) as a target of miR-K5 (Ziegelbauer et al., 2009). miR-K12-7 (Lin et al., 2011) and miR-K9 (Bellare and Ganem, 2009) directly target RTA and contribute to the maintenance of latency. miR-K12-11 shows remarkable homology to cellular miR-155;it inhibits a BACH-1 3′UTR-containing reporter and downregulates the expression of BACH-1, which is a broadly expressed transcriptional repressor that regulates genes involved in the hypoxia response (Gottwein et al., 2007; Skalsky et al., 2007). Thrombospondin 1 (THBS1), an inhibitor of angiogenesis, is targeted by multiple KSHV miRNAs, such as miR-K12-1, miR-K12-3-3p, miR-K12-6-3p, and miR-K12-11(Samols et al., 2007). miR-K1 represses the expression of p21 via the 3′UTR and attenuates p21-mediated cell cycle arrest during KSHV latency (Gottwein and Cullen, 2010).
Cellular DNA Licensing and Viral DNA Replication in Latency
Because eukaryotic DNA replication is strictly regulated by a licensing mechanism, the genome is replicated only once per cell cycle. DNA replication starts at multiple sites on a chromosome; these sites are called the replication origin, whose number is predicted to be 30,000 ∼ 50,000 (Huberman and Riggs, 1966). The genome size of eukaryotes is about 107 to 1011 bp (fungi to mammals), and the entire DNA must be replicated within a limited time (Wyrick et al., 2001). To achieve this, many proteins participate in DNA replication licensing, including origin recognition complex (ORC), Cdc6, Cdt1, and mini-chromosomal maintenance (MCM) helicase, and so on. First, ORC recognizes and binds to the origins and then recruits a Cdc6 followed by the association of another replication protein, called Cdt1. Finally, the MCM helicase is loaded onto the complex to establish a complete pre-replication complex (pre-RC) (reviews in Nishitani and Lygerou, 2002; DePamphilis, 2003, 2005).
KSHV Origin of Replication in Latency
The features of DNA replication origins have been reported. Although there are no consensus sequences for the replication origin, recent studies showed that CpG islands, promoter regions, DNA topology, and nucleosome positioning are involved in origin selection (Mechali, 2010). Saccharomyces cerevisiae (S. cerevisiae) has autonomous replication sequence (ARS) elements that are specific 12 bp consensus sequences and has origin activity (Stinchcomb et al., 1979; Bell and Stillman, 1992). S. pombe ARS also has been identified, but it does not share a consensus sequences as in S. cerevisiae (Segurado et al., 2003; Dai et al., 2005; Heichinger et al., 2006). Substitution experiments showed that the ARS region could be replaced with a 40-bp poly (dA/dT) fragment (Okuno et al., 1999).
In higher eukaryotes, no consensus sequences are identified, though known origin sequences have been reported (for review, see Aladjem, 2004). It is not yet known why there is no consensus sequence among ORC binding sites of higher eukaryotes, or how they are selected. Replication origins should be determined by different mechanisms, and recent genome-wide analyses show that the origin sequences are closely related with transcriptional regulatory elements and CpG islands but not sequence motifs (Cadoret et al., 2008; Sequeira-Mendes et al., 2009).
The KSHV genome appears to replicate once per cell cycle during latency, as cellular DNA replication. The number of the genome copies is supposed to be 50–100 per KSHV-infected PEL cell and the copy number is kept at the same number, at least appears to be kept at the same copy number (Cesarman et al., 1995; Ballestas et al., 1999; Ueda et al., 2006). This observation suggests that the KSHV genome uses cellular replication machinery so that viral DNA replication synchronizes with the cell cycle. The KSHV origin of replication in latency, called ori-P, consists of two LANA-binding sites (LBS), in which LBS1 has a higher affinity with LANA (Garber et al., 2002), and the following 32 bp GC-rich segment (termed 32GC in this manuscript) and the ori-P is in the TR region of the KSHV episome (Hu and Renne, 2005; Figure 1). LANA directly binds to LBS and supports viral DNA replication (Garber et al., 2002; Hu et al., 2002; Fejer et al., 2003; Grundhoff and Ganem, 2003). The components of pre-RC, such as ORC, Cdc6, and MCM were recruited to the TR sequences in a LANA-dependent manner (Lim et al., 2002; Ohsaki et al., 2004; Stedman et al., 2004; Verma et al., 2006).
In the case of EBV ori-P, the dyad symmetry (DS) and family of repeats (FR) are essential for the ori-P activity (Reisman et al., 1985), though FR rather works for viral genome maintenance. The DS element contains two EBNA1 binding sites and is the functional replicator in the presence of EBNA1 (Wysokenski and Yates, 1989; Harrison et al., 1994). The FR element contains 20 copies of a 30-bp repeat sequence and has an essential role in the long-term maintenance of ori-P-containing plasmid (Krysan et al., 1989; Marechal et al., 1999). It is suggested that cellular replication factors bind to the sequences adjacent to EBNA1 binding sites through the interaction with EBNA1 (Yates et al., 2000; Koons et al., 2001). A chromatin immunoprecipitation assay suggested that the ORC complex and EBNA1 bound to chromatin and ori-P in G0-arrested cells (Ritzi et al., 2003). Nucleosome assembly proteins, such as NAP1 and TAF-I, interact with EBNA1 and are recruited to the ori-P regions. These proteins contribute to the activation of transcription, although TAF-I negatively regulates DNA replication (Wang and Frappier, 2009). The EBNA1 LR1 and LR2 domains are critical for the interaction with ORC and for disrupting this association by binding with G-rich RNA (Norseen et al., 2009). Thus, the latent replication of KSHV and EBV totally depends on cellular DNA replication machinery with the only exception of the requirement of LANA and EBNA1, respectively.
How LANA Works
LANA is a nuclear protein with 1162 amino acids. It shows a functional homology to EBNA1 of EBV and, in part, to E1/E2 of human papillomavirus and to SV40 large T antigen. Especially, the C-terminus of LANA and that of EBNA1 conserve secondary and tertiary structures (Han et al., 2010). The N-terminus of LANA contains a chromosome binding site (CBS) and a nuclear localization signal (NLS), and the C-terminus contains a DNA binding domain and a dimerization domain, called DBD. In the DBD, there appears to be another NLS, but this NLS is rather cryptic and weak, since an N-terminal deleted mutant, which contains 108–1162 aa, is localized in the cytoplasm (Ohsaki et al., 2009). The central region is composed of a proline-rich region, an aspartate (D)- and glutamate (E)-rich repetitious region, and a glutamine-rich domain (Garber et al., 2001; Piolot et al., 2001; Figure 2). DBD (923–1162 aa) is necessary and partially sufficient to support ori-P replication compared to the full-length LANA (Hu et al., 2002; Ohsaki et al., 2009), and when considering their expression levels in in vitro study.
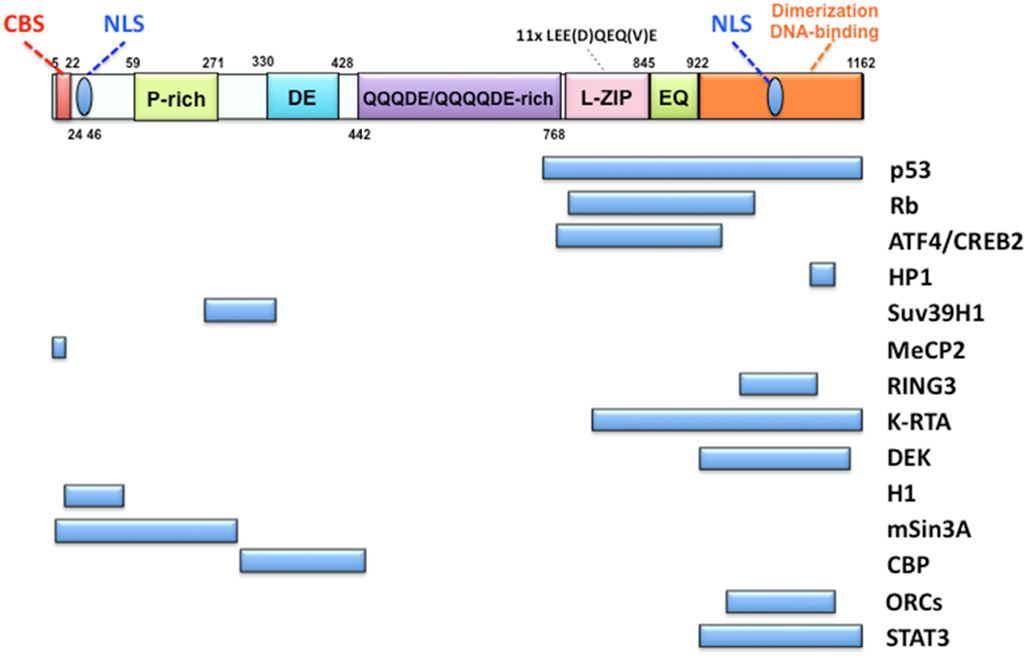
Figure 2. Organization of LANA and its interacting proteins. The top Figure shows the organization of the LANA protein. LANA has two nuclear localization signals (NLS): one at the N-terminal region and the other at the C-terminal region. The former is a primary NLS and the latter is cryptic. A chromosome binding site (CBS) was reported to be present at the N-terminal end as a histone H1 binding region. The mid-part of LANA consists of a very peculiar amino acid composition called a proline-rich region (P-rich), an extremely aspartate (D)- and glutamate (E)-rich region (DE) and a glutamine (Q)-aspartate (D)-glutamate (E)-rich region (QQQDE/QQQQDE). LEE(D)QEQ(V)E looks like a leucine zipper but has not been recognized as a functional determinant. The C-terminal region has roles in both homo-dimerization and DNA binding to LBS. The main interacting proteins with LANA are listed below. The C-terminal region seems to be a region that interacts with many proteins.
Many studies show that LANA binds to LBS and recruits ORC to the origin as described above. What is the mechanism by which ORC is recruited to ori-P? One possible mechanism is that LANA directly interacts with ORC so that ORC is loaded to the origin (Lim et al., 2002; Stedman et al., 2004; Verma et al., 2006). If LANA could directly interact with ORC and recruit to the origin, we are confronted with this question: why is 32GC required for ori-P activity despite LANA’s ability to bind to the ori-P? It could be that the 32GC is required to load ORC and the other pre-RC components to the region next to LBS, although the underlying molecular mechanism is not yet known. Further study is needed to resolve this question.
LANA is highly expressed in KSHV-related malignancies, and plays an essential in episomal maintenance. It interacts with multiple cellular proteins, including tumor suppressors such as p53 (Friborg et al., 1999) and Rb (Radkov et al., 2000), as well as transcription factors such as ATF4/CREB2 (Lim et al., 2000) and STAT3 (Muromoto et al., 2006), chromatin-associated proteins such as HP1 (Lim et al., 2003), histone H2A/B (Barbera et al., 2006b), MeCP2 (Krithivas et al., 2002; Matsumura et al., 2010), and Brd4 (Ottinger et al., 2006), in addition to signal transducers such as GSK-3b (Fujimuro and Hayward, 2003) and so on (Figure 2). Almost all of these proteins interact with the C-terminal domain of LANA, implying the functional importance of this domain. However, careful analysis of protein–protein interaction is required, because only DBD sometimes shows non-specific binding with other proteins and may exhibit different properties from the full-length LANA (our personal observation).
Regulation of KSHV ori-P Activation
The origin number varies from species to species. In mammals, it, 30,000–50,000 origins are thought to exist at each cell cycle (Huberman and Riggs, 1966). However, not all of these origins start DNA synthesis at the same time. Some origins are activated early in the S phase, whereas others are activated in the late S phase; that is, the DNA replication timing is controlled (Dimitrova and Gilbert, 1999; Cimbora et al., 2000). How is the timing of DNA replication determined? Some groups have provided possible answers to this question. It was reported that heterochromatin could change the timing of DNA replication by transgene insertion into a mammalian genome (Lin et al., 2003). Sir proteins, which are silencer proteins, can delay replication and correlate with transcriptional silencing (Zappulla et al., 2002). Thus, various studies suggest that heterochromatin modulates replication timing (Goren et al., 2008; Klochkov et al., 2009; Schwaiger et al., 2010) and EBV replication in latency occurs in mid-late S phase (Zhou et al., 2009). In the case of KSHV, the replication timing of the viral genome is not yet known. Considering that ori-P is present in the proximity of heterochromatin because of LANA-dependent accumulation of heterochromatin, DNA replication of the KSHV genome may start at the middle or late S phase.
The frequency of origin usage also differs from origin to origin; some origins are used in every cell cycle, whereas others are used rarely. Various studies including in Drosophila, Xenopus, and mammals demonstrate how specific origins are selected, but several questions about the decision mechanism of origins remain unanswered. Considering the genome size of this virus, one origin is enough to complete replication within the S phase; and because of the existence of “origin interference” by the ATR and ATM pathways (Shechter et al., 2004), a single origin must be chosen.
The micrococcal nuclease digestion pattern at TR in G1-arrested cells leads to change, suggesting that the chromatin structure became more accessible to enzymatic digestion (Stedman et al., 2004). Therefore, the chromatin structure may be changed by the recruitment of the replication machinery during the late G1 phase.
A recent study shows that the cellular deubiquitylating enzyme USP7 stimulates EBNA1 binding to its recognition sites so that histone modification at the EBV ori-P is changed by EBNA1 mediating the recruitment of USP7 (Sarkari et al., 2009).
The Roles of Nuclear Architectures
Nuclear Matrix as a Scaffold for DNA Replication and Transcription
The nucleus consists of a well-organized structure and is highly complex. The structures of nuclear matrix proteins such as lamins, nuclear mitotic apparatus (NuMA), hnRNP, and so on, are important for the organization of chromatin, DNA replication, and transcription (Dechat et al., 2008). The nuclear matrix, isolated by Berezney and Coffey (1974), is believed to support the spatial distribution of several nuclear factors, such as DNA replication machinery and transcription factors. The nuclear matrix fraction contains DNase I-resistant and high salt-resistant proteins. Because the nuclear matrix can be visualized only after chromatin extraction, there has been a debate that such a nuclear matrix is an essential component of in vivo nuclear architectures. In previous studies, replication origins have come to the nuclear matrix (van der Velden et al., 1984; Amati and Gasser, 1990; Adom et al., 1992; Brylawski et al., 1993; Fallaux et al., 1996). Eukaryotic DNA is organized into DNA loops generated by the attachment of chromatin to the nuclear matrix via specific regions, referred to as scaffold/matrix attachment regions (Pardoll et al., 1980; Vogelstein et al., 1980; Laemmli et al., 1992; Roberge and Gasser, 1992). DNA loop formation is essential for DNA replication, transcription, and chromosomal packaging (Gasser and Laemmli, 1987; Berezney et al., 1995; Bode et al., 1995; Nickerson et al., 1995; Razin et al., 1995; Jackson, 1997; Volpi et al., 2000; Mahy et al., 2002; Sumer et al., 2003; Marenduzzo et al., 2007).
In EBV, both the latent ori-P and the lytic ori-Lyt are bound to the nuclear matrix (Jankelevich et al., 1992; Mattia et al., 1999). EBV nuclear antigen leader protein (EBNA-LP) is a nuclear matrix-associated protein, and its nuclear matrix-targeting signal is a 10-aa segment, which also functions as a NLS (Yokoyama et al., 2001). However, this is not the case for LANA, because it was reported that C-terminal truncation up to 1128 aa could not be localized in a high salt-resistant fraction (nuclear matrix fraction), but the C-terminal region (1129–1143 aa) is high salt extractable (Viejo-Borbolla et al., 2003). We previously showed that the N-terminal region up to 107 aa is localized in nucleo-cytoplasmic and chromatin fractions (Ohsaki et al., 2009). The localization to the nuclear matrix fraction of LANA might depend on the conformation of LANA or post-translational modifications.
On the other hand, a cell fractionation assay has shown that cellular pre-RC components, such as ORC2, Cdc6, and Cdt1, preferentially localize in the nuclear matrix fraction in a cell cycle-dependent manner, and LANA itself also can localize in the nuclear matrix fraction. Accordingly, TR accumulates in the nuclear matrix fraction during the late G1 phase, suggesting that LANA recruits the ori-P to the nuclear matrix, so that cellular replication machinery is abundant and available for viral DNA replication during latency (Ohsaki et al., 2009; Figure 3).
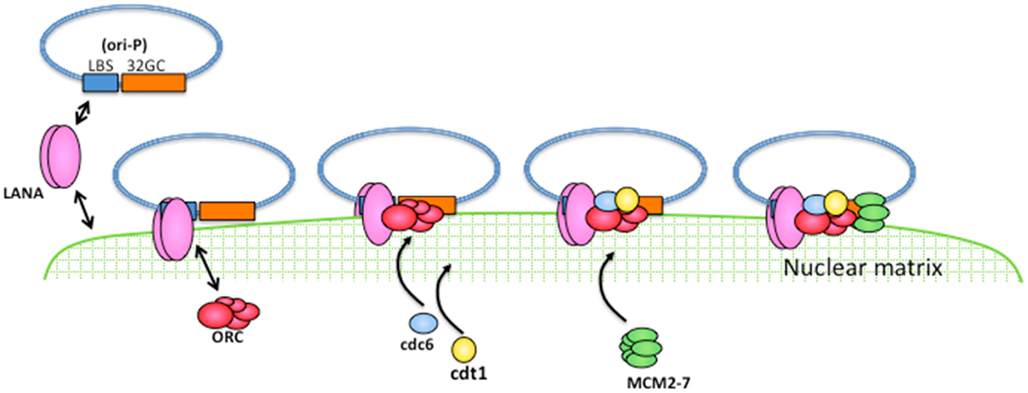
Figure 3. A model for latent DNA replication of KSHV. LANA can associate with the nuclear matrix and can directly bind to the ori-P so that LANA can recruit the ori-P to the nuclear matrix. Cellular DNA replication machinery assembles to the nuclear matrix in a cell cycle-dependent manner, and is therefore available for viral DNA replication.
Genome Segregation Mechanisms of KSHV in Latency
The same copy number of KSHV genomes appears to be maintained in daughter cells after every cell division (Ballestas et al., 1999; Ueda et al., 2006), indicating that a strict genome maintenance mechanism is working. In the KSHV-infected PEL cell lines, LANA associates with a condensed mitotic chromatin (Ballestas et al., 1999; Cotter and Robertson, 1999; Tetsuka et al., 2004). It was reported that MeCP2, a methyl CpG-binding protein, interacts with the N-terminal of LANA and that DEK protein interacts with the C-terminal of LANA. These two independent interactions are involved in the tethering of LANA to chromosomes (Krithivas et al., 2002). Various studies have reported the interaction between LANA and multiple cellular proteins associated with chromatin.
Brd4, which is a member of the BET family that carries two bromodomains and associates with mitotic chromosomes, interacts with LANA on mitotic chromosomes (You et al., 2006). It has also been reported that core histones such as H2A and H2B are essential for LANA N-terminal chromosome binding (Barbera et al., 2006a,b). Furthermore, LANA interacts with Brd2/Ring3, which is a member of the BET family of double bromodomain-containing genes and contains two tandem bromodomains (Viejo-Borbolla et al., 2005).
Xiao et al. (2010) reported that LANA is associated with centromeres via the formation of complexes with Cenp-F and Bub1, which are kinetochore-associated proteins. This suggests that LANA preferentially interacts with kinetochore-associated proteins and that its association is critical for segregation into daughter cells. Though the interaction of LANA with kinetochore factors might interfere with correct spindle formation, it suggests that LANA should support viral genome segregation along with condensed chromatin.
A NuMA plays a critical role in the nuclear architecture in the interphase. After nuclear envelope breakdown in mitosis, NuMA is hyperphosphorylated by p34cdc2 and is distributed at spindle poles, where it remains until the anaphase and plays an essential role in tethering spindle microtubules to each pole (Merdes et al., 1996; Gehmlich et al., 2004). Although NuMA drastically alters the localization and functions in the interphase and M phase, biochemical fractionation analysis shows that NuMA is localized in the insoluble nuclear matrix fraction in both phases. A recent study demonstrated that the C-terminus of LANA was co-localized with NuMA during the interphase and that the knockdown of NuMA expression caused the disruption of genome segregation and TR-containing plasmid maintenance (Bhaumik et al., 2008) and thus, KSHV genome segregation is disrupted in the absence of NuMA. Through siRNA and knockdown strategies in mice, NuMA has been shown to be an essential protein for early embryogenesis and cellular proliferation (Harborth et al., 2001; Silk et al., 2009), and it is thus unclear how the interaction with LANA actually works for viral genome segregation.
In either case, LANA has the capability of associating with various cellular proteins so that KSHV can maintain the genome stably if the cells are divided into two daughter cells. Nuclear matrix proteins function as a scaffold of DNA replication, transcription, and repair during the interphase, and also play an essential role in the segregation of condensed chromosomes in mitosis. Condensed chromosomes include, for example, NuMA, which can behave as a component of a spindle pole during mitosis, or Cenp-F, which is a nuclear matrix protein during the interphase and is distributed to kinetochores in mitosis. Taken together, the previous and present results suggest that nuclear architectures such as the nuclear matrix have essential roles not only in DNA replication and transcription but also in genome segregation during mitosis (Figure 4).
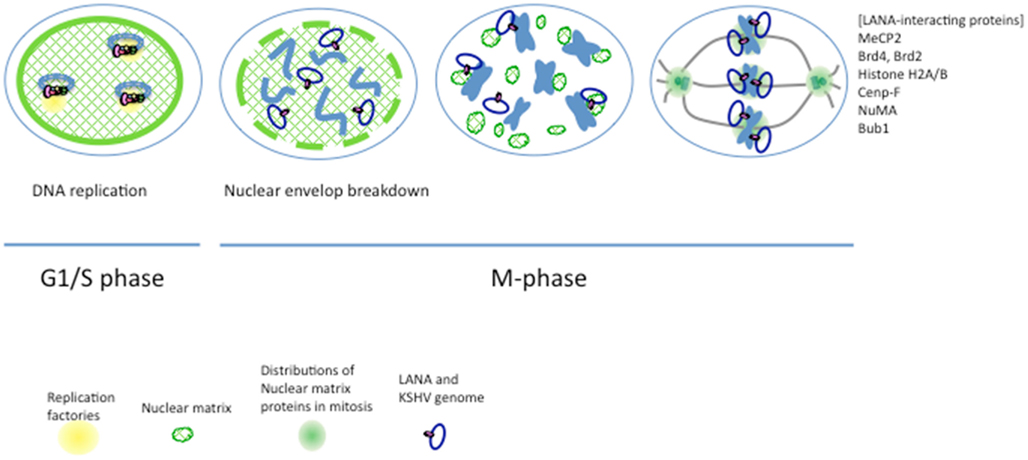
Figure 4. A model for KSHV genome segregation. The KSHV genome associates with the nuclear matrix through LANA, which exhibits nuclear matrix localization activity. DNA replication factories are supposed to anchor to the nuclear matrix, and DNA replication of the KSHV genome occurs on the nuclear matrix at the G1/S phase. Nuclear architectures, including the nuclear matrix, are disrupted at the prophase (nuclear envelope breakdown), and nuclear matrix proteins drastically change these functions and distributions; such as spindle pole or kinetochore proteins. It has been reported that KSHV LANA interacts with various proteins such as MeCP2, Brd2, Brd4, Histone H2A/B, Cenp-F, NuMA, Bub1, and so on. These interactions can associate a sister-chromatid with LANA, and thus KSHV genomes can separate into two daughter cells.
Conclusion and Perspectives for Future Analysis
The importance of nuclear architecture is increasingly recognized as important in various nuclear events, such as DNA replication, transcription, and DNA repair. It is well accepted that the chromosomes are organized into distinct territories in the interphase. These distributions of chromosomes are closely related to the place for active or inactive transcription, the presence of DNA replication machinery, and the formation of higher-order structures of chromatin loops. DNA looping appears to be mediated by attachment to the nuclear matrix and thus achieves transcriptional control (Ostermeier et al., 2003). Other studies have suggested that the gene-rich chromosomes are frequently located in the nuclear interior. On the other hand, gene-poor chromosomes are located in the nuclear periphery (Tanabe et al., 2002; Reddy et al., 2008). A recent study shows that the transcriptional silencing might be accomplished by the binding of a specific promoter region to lamin type A (Lee et al., 2009). It is reported that replication foci at the middle to late S phase are also preferentially located toward the nuclear periphery, whereas early replication foci are located in the nuclear interior (Grasser et al., 2008) or throughout the nucleus (Izumi et al., 2004).
The spatial and temporal analyses using live-cell imaging revealed that replication forks are generated at the same origin and are closely associated during replication (Kitamura et al., 2006). Interestingly, two replication loci, located at the same distance from the origin, were in closer proximity when DNA replication took place at these loci, after which they moved apart from each other after replication. It is speculated that the replication factory anchors some region and that replicated DNA can move away from a replication factory immediately after DNA synthesis. This anchored region may be a nuclear scaffold such as the nuclear matrix.
The cellular environment is critical for viral survival, for which viruses make full use of the cellular machinery. The analysis of insoluble proteins, including nuclear matrix proteins, is difficult for the investigation of protein–protein and protein–DNA interactions in vitro, because the conditions of in vitro experiments always include soluble fractions. To overcome these problems, recent new approaches, such as genome-wide analysis using chromatin immunoprecipitation or live-cell imaging that reflects the in vivo environment, may be more powerful and accurate.
As for KSHV genome replication and maintenance, it would be first necessary to clarify how LANA is involved in both. Considering the real and actual cellular environments, we need to develop more powerful tools to know what the virus does in cells.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Adom, J. N., Gouilleux, F., and Richard-Foy, H. (1992). Interaction with the nuclear matrix of a chimeric construct containing a replication origin and a transcription unit. Biochim. Biophys. Acta 1171, 187–197.
Aladjem, M. I. (2004). The mammalian beta globin origin of DNA replication. Front. Biosci. 9, 2540–2547.
Amati, B., and Gasser, S. M. (1990). Drosophila scaffold-attached regions bind nuclear scaffolds and can function as ARS elements in both budding and fission yeasts. Mol. Cell. Biol. 10, 5442–5454.
Arvanitakis, L., Mesri, E. A., Nador, R. G., Said, J. W., Asch, A. S., Knowles, D. M., and Cesarman, E. (1996). Establishment and characterization of a primary effusion (body cavity-based) lymphoma cell line (BC-3) harboring Kaposi’s sarcoma-associated herpesvirus (KSHV/HHV-8) in the absence of Epstein-Barr virus. Blood 88, 2648–2654.
Baek, D., Villen, J., Shin, C., Camargo, F. D., Gygi, S. P., and Bartel, D. P. (2008). The impact of microRNAs on protein output. Nature 455, 64–71.
Ballestas, M. E., Chatis, P. A., and Kaye, K. M. (1999). Efficient persistence of extrachromosomal KSHV DNA mediated by latency-associated nuclear antigen. Science 284, 641–644.
Barbera, A. J., Chodaparambil, J. V., Kelley-Clarke, B., Joukov, V., Walter, J. C., Luger, K., and Kaye, K. M. (2006a). The nucleosomal surface as a docking station for Kaposi’s sarcoma herpesvirus LANA. Science 311, 856–861.
Barbera, A. J., Chodaparambil, J. V., Kelley-Clarke, B., Luger, K., and Kaye, K. M. (2006b). Kaposi’s sarcoma-associated herpesvirus LANA hitches a ride on the chromosome. Cell Cycle 5, 1048–1052.
Bartel, D. P., and Chen, C. Z. (2004). Micromanagers of gene expression: the potentially widespread influence of metazoan microRNAs. Nat. Rev. Genet. 5, 396–400.
Bell, S. P., and Stillman, B. (1992). ATP-dependent recognition of eukaryotic origins of DNA replication by a multiprotein complex. Nature 357, 128–134.
Bellare, P., and Ganem, D. (2009). Regulation of KSHV lytic switch protein expression by a virus-encoded microRNA: an evolutionary adaptation that fine-tunes lytic reactivation. Cell Host Microbe 6, 570–575.
Berezney, R., and Coffey, D. S. (1974). Identification of a nuclear protein matrix. Biochem. Biophys. Res. Commun. 60, 1410–1417.
Berezney, R., Mortillaro, M. J., Ma, H., Wei, X., and Samarabandu, J. (1995). The nuclear matrix: a structural milieu for genomic function. Int. Rev. Cytol. 162A, 1–65.
Bhaumik, D., Scott, G. K., Schokrpur, S., Patil, C. K., Campisi, J., and Benz, C. C. (2008). Expression of microRNA-146 suppresses NF-kappaB activity with reduction of metastatic potential in breast cancer cells. Oncogene 27, 5643–5647.
Bode, J., Schlake, T., Rios-Ramirez, M., Mielke, C., Stengert, M., Kay, V., and Klehr-Wirth, D. (1995). Scaffold/matrix-attached regions: structural properties creating transcriptionally active loci. Int. Rev. Cytol. 162A, 389–454.
Boshoff, C., and Chang, Y. (2001). Kaposi’s sarcoma-associated herpesvirus: a new DNA tumor virus. Annu. Rev. Med. 52, 453–470.
Boss, I. W., Plaisance, K. B., and Renne, R. (2009). Role of virus-encoded microRNAs in herpesvirus biology. Trends Microbiol. 17, 544–553.
Brylawski, B. P., Tsongalis, G. J., Cordeiro-Stone, M., May, W. T., Comeau, L. D., and Kaufman, D. G. (1993). Association of putative origins of replication with the nuclear matrix in normal human fibroblasts. Cancer Res. 53, 3865–3868.
Byun, H., Gwack, Y., Hwang, S., and Choe, J. (2002). Kaposi’s sarcoma-associated herpesvirus open reading frame (ORF) 50 transactivates K8 and ORF57 promoters via heterogeneous response elements. Mol. Cells 14, 185–191.
Cadoret, J. C., Meisch, F., Hassan-Zadeh, V., Luyten, I., Guillet, C., Duret, L., Quesneville, H., and Prioleau, M. N. (2008). Genome-wide studies highlight indirect links between human replication origins and gene regulation. Proc. Natl. Acad. Sci. U.S.A. 105, 15837–15842.
Cai, X., and Cullen, B. R. (2006). Transcriptional origin of Kaposi’s sarcoma-associated herpesvirus microRNAs. J. Virol. 80, 2234–2242.
Cai, X., Lu, S., Zhang, Z., Gonzalez, C. M., Damania, B., and Cullen, B. R. (2005). Kaposi’s sarcoma-associated herpesvirus expresses an array of viral microRNAs in latently infected cells. Proc. Natl. Acad. Sci. U.S.A. 102, 5570–5575.
Carbone, A., Cilia, A. M., Gloghini, A., Canzonieri, V., Pastore, C., Todesco, M., Cozzi, M., Perin, T., Volpe, R., Pinto, A., and Gaidano, G. (1997). Establishment of HHV-8-positive and HHV-8-negative lymphoma cell lines from primary lymphomatous effusions. Int. J. Cancer 73, 562–569.
Carbone, A., Cilia, A. M., Gloghini, A., Capello, D., Todesco, M., Quattrone, S., Volpe, R., and Gaidano, G. (1998). Establishment and characterization of EBV-positive and EBV-negative primary effusion lymphoma cell lines harbouring human herpesvirus type-8. Br. J. Haematol. 102, 1081–1089.
Carroll, P. A., Brazeau, E., and Lagunoff, M. (2004). Kaposi’s sarcoma-associated herpesvirus infection of blood endothelial cells induces lymphatic differentiation. Virology 328, 7–18.
Cesarman, E., Chang, Y., Moore, P. S., Said, J. W., and Knowles, D. M. (1995). Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N. Engl. J. Med. 332, 1186–1191.
Cesarman, E., Nador, R. G., Aozasa, K., Delsol, G., Said, J. W., and Knowles, D. M. (1996). Kaposi’s sarcoma-associated herpesvirus in non-AIDS related lymphomas occurring in body cavities. Am. J. Pathol. 149, 53–57.
Chang, Y., Cesarman, E., Pessin, M. S., Lee, F., Culpepper, J., Knowles, D. M., and Moore, P. S. (1994). Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science 266, 1865–1869.
Chang, Y., Moore, P. S., Talbot, S. J., Boshoff, C. H., Zarkowska, T., Godden, K., Paterson, H., Weiss, R. A., and Mittnacht, S. (1996). Cyclin encoded by KS herpesvirus. Nature 382, 410.
Chaudhary, P. M., Jasmin, A., Eby, M. T., and Hood, L. (1999). Modulation of the NF-kappa B pathway by virally encoded death effector domains-containing proteins. Oncogene 18, 5738–5746.
Cimbora, D. M., Schubeler, D., Reik, A., Hamilton, J., Francastel, C., Epner, E. M., and Groudine, M. (2000). Long-distance control of origin choice and replication timing in the human beta-globin locus are independent of the locus control region. Mol. Cell. Biol. 20, 5581–5591.
Ciufo, D. M., Cannon, J. S., Poole, L. J., Wu, F. Y., Murray, P., Ambinder, R. F., and Hayward, G. S. (2001). Spindle cell conversion by Kaposi’s sarcoma-associated herpesvirus: formation of colonies and plaques with mixed lytic and latent gene expression in infected primary dermal microvascular endothelial cell cultures. J. Virol. 75, 5614–5626.
Cotter, M. A. II, and Robertson, E. S. (1999). The latency-associated nuclear antigen tethers the Kaposi’s sarcoma-associated herpesvirus genome to host chromosomes in body cavity-based lymphoma cells. Virology 264, 254–264.
Dai, J., Chuang, R. Y., and Kelly, T. J. (2005). DNA replication origins in the Schizosaccharomyces pombe genome. Proc. Natl. Acad. Sci. U.S.A. 102, 337–342.
Dechat, T., Pfleghaar, K., Sengupta, K., Shimi, T., Shumaker, D. K., Solimando, L., and Goldman, R. D. (2008). Nuclear lamins: major factors in the structural organization and function of the nucleus and chromatin. Genes Dev. 22, 832–853.
DePamphilis, M. L. (2003). The ‘ORC cycle’: a novel pathway for regulating eukaryotic DNA replication. Gene 310, 1–15.
DePamphilis, M. L. (2005). Cell cycle dependent regulation of the origin recognition complex. Cell Cycle 4, 70–79.
Dimitrova, D. S., and Gilbert, D. M. (1999). The spatial position and replication timing of chromosomal domains are both established in early G1 phase. Mol. Cell 4, 983–993.
Dourmishev, L. A., Dourmishev, A. L., Palmeri, D., Schwartz, R. A., and Lukac, D. M. (2003). Molecular genetics of Kaposi’s sarcoma-associated herpesvirus (human herpesvirus-8) epidemiology and pathogenesis. Microbiol. Mol. Biol. Rev. 67, 175–212.
Dupin, N., Gorin, I., Deleuze, J., Agut, H., Huraux, J. M., and Escande, J. P. (1995). Herpes-like DNA sequences, AIDS-related tumors, and Castleman’s disease. N. Engl. J. Med. 333, 798.
Fallaux, F. J., Hoeben, R. C., Cramer, S. J., Van Den Wollenberg, D. J., Briet, E., Van Ormondt, H., and Van Der Eb, A. J. (1996). The human clotting factor VIII cDNA contains an autonomously replicating sequence consensus- and matrix attachment region-like sequence that binds a nuclear factor, represses heterologous gene expression, and mediates the transcriptional effects of sodium butyrate. Mol. Cell. Biol. 16, 4264–4272.
Fejer, G., Medveczky, M. M., Horvath, E., Lane, B., Chang, Y., and Medveczky, P. G. (2003). The latency-associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus interacts preferentially with the terminal repeats of the genome in vivo and this complex is sufficient for episomal DNA replication. J. Gen. Virol. 84, 1451–1462.
Field, N., Low, W., Daniels, M., Howell, S., Daviet, L., Boshoff, C., and Collins, M. (2003). KSHV vFLIP binds to IKK-gamma to activate IKK. J. Cell. Sci. 116, 3721–3728.
Friborg, J. Jr., Kong, W., Hottiger, M. O., and Nabel, G. J. (1999). p53 Inhibition by the LANA protein of KSHV protects against cell death. Nature 402, 889–894.
Frizzera, G., Banks, P. M., Massarelli, G., and Rosai, J. (1983). A systemic lymphoproliferative disorder with morphologic features of Castleman’s disease. Pathological findings in 15 patients. Am. J. Surg. Pathol. 7, 211–231.
Fujimuro, M., and Hayward, S. D. (2003). The latency-associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus manipulates the activity of glycogen synthase kinase-3beta. J. Virol. 77, 8019–8030.
Gaidano, G., Cechova, K., Chang, Y., Moore, P. S., Knowles, D. M., and Dalla-Favera, R. (1996). Establishment of AIDS-related lymphoma cell lines from lymphomatous effusions. Leukemia 10, 1237–1240.
Ganem, D., and Ziegelbauer, J. (2008). MicroRNAs of Kaposi’s sarcoma-associated herpes virus. Semin. Cancer Biol. 18, 437–440.
Gao, S. J., Deng, J. H., and Zhou, F. C. (2003). Productive lytic replication of a recombinant Kaposi’s sarcoma-associated herpesvirus in efficient primary infection of primary human endothelial cells. J. Virol. 77, 9738–9749.
Garber, A. C., Hu, J., and Renne, R. (2002). Latency-associated nuclear antigen (LANA) cooperatively binds to two sites within the terminal repeat, and both sites contribute to the ability of LANA to suppress transcription and to facilitate DNA replication. J. Biol. Chem. 277, 27401–27411.
Garber, A. C., Shu, M. A., Hu, J., and Renne, R. (2001). DNA binding and modulation of gene expression by the latency-associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 75, 7882–7892.
Gasser, S. M., and Laemmli, U. K. (1987). Improved methods for the isolation of individual and clustered mitotic chromosomes. Exp. Cell Res. 173, 85–98.
Gehmlich, K., Haren, L., and Merdes, A. (2004). Cyclin B degradation leads to NuMA release from dynein/dynactin and from spindle poles. EMBO Rep. 5, 97–103.
Gomez-Roman, J. J., Sanchez-Velasco, P., Ocejo-Vinyals, G., Hernandez-Nieto, E., Leyva-Cobian, F., and Val-Bernal, J. F. (2001). Human herpesvirus-8 genes are expressed in pulmonary inflammatory myofibroblastic tumor (inflammatory pseudotumor). Am. J. Surg. Pathol. 25, 624–629.
Goren, A., Tabib, A., Hecht, M., and Cedar, H. (2008). DNA replication timing of the human beta-globin domain is controlled by histone modification at the origin. Genes Dev. 22, 1319–1324.
Gottwein, E., and Cullen, B. R. (2010). A human herpesvirus microRNA inhibits p21 expression and attenuates p21-mediated cell cycle arrest. J. Virol. 84, 5229–5237.
Gottwein, E., Mukherjee, N., Sachse, C., Frenzel, C., Majoros, W. H., Chi, J. T., Braich, R., Manoharan, M., Soutschek, J., Ohler, U., and Cullen, B. R. (2007). A viral microRNA functions as an orthologue of cellular miR-155. Nature 450, 1096–1099.
Grasser, F., Neusser, M., Fiegler, H., Thormeyer, T., Cremer, M., Carter, N. P., Cremer, T., and Muller, S. (2008). Replication-timing-correlated spatial chromatin arrangements in cancer and in primate interphase nuclei. J. Cell. Sci. 121, 1876–1886.
Grossmann, C., and Ganem, D. (2008). Effects of NFkappaB activation on KSHV latency and lytic reactivation are complex and context-dependent. Virology 375, 94–102.
Grundhoff, A., and Ganem, D. (2003). The latency-associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus permits replication of terminal repeat-containing plasmids. J. Virol. 77, 2779–2783.
Gunther, T., and Grundhoff, A. (2010). The epigenetic landscape of latent Kaposi sarcoma-associated herpesvirus genomes. PLoS Pathog. 6, e1000935. doi:10.1371/journal.ppat.1000935
Han, S. J., Hu, J., Pierce, B., Weng, Z., and Renne, R. (2010). Mutational analysis of the latency-associated nuclear antigen DNA-binding domain of Kaposi’s sarcoma-associated herpesvirus reveals structural conservation among gammaherpesvirus origin-binding proteins. J. Gen. Virol. 91, 2203–2215.
Harborth, J., Elbashir, S. M., Bechert, K., Tuschl, T., and Weber, K. (2001). Identification of essential genes in cultured mammalian cells using small interfering RNAs. J. Cell. Sci. 114, 4557–4565.
Harrison, S., Fisenne, K., and Hearing, J. (1994). Sequence requirements of the Epstein-Barr virus latent origin of DNA replication. J. Virol. 68, 1913–1925.
Heichinger, C., Penkett, C. J., Bahler, J., and Nurse, P. (2006). Genome-wide characterization of fission yeast DNA replication origins. EMBO J. 25, 5171–5179.
Hong, Y. K., Foreman, K., Shin, J. W., Hirakawa, S., Curry, C. L., Sage, D. R., Libermann, T., Dezube, B. J., Fingeroth, J. D., and Detmar, M. (2004). Lymphatic reprogramming of blood vascular endothelium by Kaposi sarcoma-associated herpesvirus. Nat. Genet. 36, 683–685.
Hou, C., Zhao, H., Tanimoto, K., and Dean, A. (2008). CTCF-dependent enhancer-blocking by alternative chromatin loop formation. Proc. Natl. Acad. Sci. U.S.A. 105, 20398–20403.
Hu, J., Garber, A. C., and Renne, R. (2002). The latency-associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus supports latent DNA replication in dividing cells. J. Virol. 76, 11677–11687.
Hu, J., and Renne, R. (2005). Characterization of the minimal replicator of Kaposi’s sarcoma-associated herpesvirus latent origin. J. Virol. 79, 2637–2642.
Huang, Y. Q., Li, J. J., Kaplan, M. H., Poiesz, B., Katabira, E., Zhang, W. C., Feiner, D., and Friedman-Kien, A. E. (1995). Human herpesvirus-like nucleic acid in various forms of Kaposi’s sarcoma. Lancet 345, 759–761.
Huberman, J. A., and Riggs, A. D. (1966). Autoradiography of chromosomal DNA fibers from Chinese hamster cells. Proc. Natl. Acad. Sci. U.S.A. 55, 599–606.
Izumi, M., Yatagai, F., and Hanaoka, F. (2004). Localization of human Mcm10 is spatially and temporally regulated during the S phase. J. Biol. Chem. 279, 32569–32577.
Jackson, D. A. (1997). Chromatin domains and nuclear compartments: establishing sites of gene expression in eukaryotic nuclei. Mol. Biol. Rep. 24, 209–220.
Jankelevich, S., Kolman, J. L., Bodnar, J. W., and Miller, G. (1992). A nuclear matrix attachment region organizes the Epstein-Barr viral plasmid in Raji cells into a single DNA domain. EMBO J. 11, 1165–1176.
Kang, H., and Lieberman, P. M. (2009). Cell cycle control of Kaposi’s sarcoma-associated herpesvirus latency transcription by CTCF-cohesin interactions. J. Virol. 83, 6199–6210.
Katano, H., Hoshino, Y., Morishita, Y., Nakamura, T., Satoh, H., Iwamoto, A., Herndier, B., and Mori, S. (1999). Establishing and characterizing a CD30-positive cell line harboring HHV-8 from a primary effusion lymphoma. J. Med. Virol. 58, 394–401.
Katano, H., Sato, Y., Kurata, T., Mori, S., and Sata, T. (2000). Expression and localization of human herpesvirus 8-encoded proteins in primary effusion lymphoma, Kaposi’s sarcoma, and multicentric Castleman’s disease. Virology 269, 335–344.
Kitamura, E., Blow, J. J., and Tanaka, T. U. (2006). Live-cell imaging reveals replication of individual replicons in eukaryotic replication factories. Cell 125, 1297–1308.
Klochkov, D. B., Gavrilov, A. A., Vassetzky, Y. S., and Razin, S. V. (2009). Early replication timing of the chicken alpha-globin gene domain correlates with its open chromatin state in cells of different lineages. Genomics 93, 481–486.
Koons, M. D., Van Scoy, S., and Hearing, J. (2001). The replicator of the Epstein-Barr virus latent cycle origin of DNA replication, oriP, is composed of multiple functional elements. J. Virol. 75, 10582–10592.
Krithivas, A., Fujimuro, M., Weidner, M., Young, D. B., and Hayward, S. D. (2002). Protein interactions targeting the latency-associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus to cell chromosomes. J. Virol. 76, 11596–11604.
Krysan, P. J., Haase, S. B., and Calos, M. P. (1989). Isolation of human sequences that replicate autonomously in human cells. Mol. Cell. Biol. 9, 1026–1033.
Laemmli, U. K., Kas, E., Poljak, L., and Adachi, Y. (1992). Scaffold-associated regions: cis-acting determinants of chromatin structural loops and functional domains. Curr. Opin. Genet. Dev. 2, 275–285.
Lan, K., Kuppers, D. A., and Robertson, E. S. (2005a). Kaposi’s sarcoma-associated herpesvirus reactivation is regulated by interaction of latency-associated nuclear antigen with recombination signal sequence-binding protein Jkappa, the major downstream effector of the Notch signaling pathway. J. Virol. 79, 3468–3478.
Lan, K., Kuppers, D. A., Verma, S. C., Sharma, N., Murakami, M., and Robertson, E. S. (2005b). Induction of Kaposi’s sarcoma-associated herpesvirus latency-associated nuclear antigen by the lytic transactivator RTA: a novel mechanism for establishment of latency. J. Virol. 79, 7453–7465.
Lee, D. C., Welton, K. L., Smith, E. D., and Kennedy, B. K. (2009). A-type nuclear lamins act as transcriptional repressors when targeted to promoters. Exp. Cell Res. 315, 996–1007.
Lei, X., Bai, Z., Ye, F., Huang, Y., and Gao, S. J. (2010a). Regulation of herpesvirus lifecycle by viral microRNAs. Virulence 1, 433–435.
Lei, X., Bai, Z., Ye, F., Xie, J., Kim, C. G., Huang, Y., and Gao, S. J. (2010b). Regulation of NF-kappaB inhibitor IkappaBalpha and viral replication by a KSHV microRNA. Nat. Cell Biol. 12, 193–199.
Liang, Y., Chang, J., Lynch, S. J., Lukac, D. M., and Ganem, D. (2002). The lytic switch protein of KSHV activates gene expression via functional interaction with RBP-Jkappa (CSL), the target of the Notch signaling pathway. Genes Dev. 16, 1977–1989.
Lim, C., Lee, D., Seo, T., Choi, C., and Choe, J. (2003). Latency-associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus functionally interacts with heterochromatin protein 1. J. Biol. Chem. 278, 7397–7405.
Lim, C., Sohn, H., Gwack, Y., and Choe, J. (2000). Latency-associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus (human herpesvirus-8) binds ATF4/CREB2 and inhibits its transcriptional activation activity. J. Gen. Virol. 81, 2645–2652.
Lim, C., Sohn, H., Lee, D., Gwack, Y., and Choe, J. (2002). Functional dissection of latency-associated nuclear antigen 1 of Kaposi’s sarcoma-associated herpesvirus involved in latent DNA replication and transcription of terminal repeats of the viral genome. J. Virol. 76, 10320–10331.
Lin, C. M., Fu, H., Martinovsky, M., Bouhassira, E., and Aladjem, M. I. (2003). Dynamic alterations of replication timing in mammalian cells. Curr. Biol. 13, 1019–1028.
Lin, X., Liang, D., He, Z., Deng, Q., Robertson, E. S., and Lan, K. (2011). miR-K12-7-5p encoded by Kaposi’s sarcoma-associated herpesvirus stabilizes the latent state by targeting viral ORF50/RTA. PLoS ONE 6, e16224. doi:10.1371/journal.pone.0016224
Lu, F., Zhou, J., Wiedmer, A., Madden, K., Yuan, Y., and Lieberman, P. M. (2003). Chromatin remodeling of the Kaposi’s sarcoma-associated herpesvirus ORF50 promoter correlates with reactivation from latency. J. Virol. 77, 11425–11435.
Lubyova, B., and Pitha, P. M. (2000). Characterization of a novel human herpesvirus 8-encoded protein, vIRF-3, that shows homology to viral and cellular interferon regulatory factors. J. Virol. 74, 8194–8201.
Mahy, N. L., Perry, P. E., Gilchrist, S., Baldock, R. A., and Bickmore, W. A. (2002). Spatial organization of active and inactive genes and noncoding DNA within chromosome territories. J. Cell Biol. 157, 579–589.
Marechal, V., Dehee, A., Chikhi-Brachet, R., Piolot, T., Coppey-Moisan, M., and Nicolas, J. C. (1999). Mapping EBNA-1 domains involved in binding to metaphase chromosomes. J. Virol. 73, 4385–4392.
Marenduzzo, D., Faro-Trindade, I., and Cook, P. R. (2007). What are the molecular ties that maintain genomic loops? Trends Genet. 23, 126–133.
Matsumura, S., Persson, L. M., Wong, L., and Wilson, A. C. (2010). The latency-associated nuclear antigen interacts with MeCP2 and nucleosomes through separate domains. J. Virol. 84, 2318–2330.
Matta, H., and Chaudhary, P. M. (2004). Activation of alternative NF-kappa B pathway by human herpes virus 8-encoded Fas-associated death domain-like IL-1 beta-converting enzyme inhibitory protein (vFLIP). Proc. Natl. Acad. Sci. U.S.A. 101, 9399–9404.
Matta, H., Mazzacurati, L., Schamus, S., Yang, T., Sun, Q., and Chaudhary, P. M. (2007). Kaposi’s sarcoma-associated herpesvirus (KSHV) oncoprotein K13 bypasses TRAFs and directly interacts with the IkappaB kinase complex to selectively activate NF-kappaB without JNK activation. J. Biol. Chem. 282, 24858–24865.
Matta, H., Sun, Q., Moses, G., and Chaudhary, P. M. (2003). Molecular genetic analysis of human herpes virus 8-encoded viral FLICE inhibitory protein-induced NF-kappaB activation. J. Biol. Chem. 278, 52406–52411.
Mattia, E., Ceridono, M., Chichiarelli, S., and D’erme, M. (1999). Interactions of Epstein-Barr virus origins of replication with nuclear matrix in the latent and in the lytic phases of viral infection. Virology 262, 9–17.
Mechali, M. (2010). Eukaryotic DNA replication origins: many choices for appropriate answers. Nat. Rev. Mol. Cell Biol. 11, 728–738.
Merdes, A., Ramyar, K., Vechio, J. D., and Cleveland, D. W. (1996). A complex of NuMA and cytoplasmic dynein is essential for mitotic spindle assembly. Cell 87, 447–458.
Minarovits, J. (2006). Epigenotypes of latent herpesvirus genomes. Curr. Top. Microbiol. Immunol. 310, 61–80.
Moore, P. S., and Chang, Y. (1995). Detection of herpesvirus-like DNA sequences in Kaposi’s sarcoma in patients with and without HIV infection. N. Engl. J. Med. 332, 1181–1185.
Moore, P. S., and Chang, Y. (2001). Molecular virology of Kaposi’s sarcoma-associated herpesvirus. Philos. Trans. R. Soc. Lond. B Biol. Sci. 356, 499–516.
Muralidhar, S., Pumfery, A. M., Hassani, M., Sadaie, M. R., Kishishita, M., Brady, J. N., Doniger, J., Medveczky, P., and Rosenthal, L. J. (1998). Identification of kaposin (open reading frame K12) as a human herpesvirus 8 (Kaposi’s sarcoma-associated herpesvirus) transforming gene. J. Virol. 72, 4980–4988.
Muromoto, R., Okabe, K., Fujimuro, M., Sugiyama, K., Yokosawa, H., Seya, T., and Matsuda, T. (2006). Physical and functional interactions between STAT3 and Kaposi’s sarcoma-associated herpesvirus-encoded LANA. FEBS Lett. 580, 93–98.
Murphy, E., Vanicek, J., Robins, H., Shenk, T., and Levine, A. J. (2008). Suppression of immediate-early viral gene expression by herpesvirus-coded microRNAs: implications for latency. Proc. Natl. Acad. Sci. U.S.A. 105, 5453–5458.
Naranatt, P. P., Krishnan, H. H., Svojanovsky, S. R., Bloomer, C., Mathur, S., and Chandran, B. (2004). Host gene induction and transcriptional reprogramming in Kaposi’s sarcoma-associated herpesvirus (KSHV/HHV-8)-infected endothelial, fibroblast, and B cells: insights into modulation events early during infection. Cancer Res. 64, 72–84.
Nickerson, J. A., Blencowe, B. J., and Penman, S. (1995). The architectural organization of nuclear metabolism. Int. Rev. Cytol. 162A, 67–123.
Nishimura, K., Ueda, K., Sakakibara, S., Ishikawa, K., Chen, J., Okuno, T., and Yamanishi, K. (2001). Functional analysis of Kaposi’s sarcoma-associated herpesvirus RTA in an RTA-depressed cell line. J. Hum. Virol. 4, 296–305.
Nishitani, H., and Lygerou, Z. (2002). Control of DNA replication licensing in a cell cycle. Genes Cells 7, 523–534.
Norseen, J., Johnson, F. B., and Lieberman, P. M. (2009). Role for G-quadruplex RNA binding by Epstein-Barr virus nuclear antigen 1 in DNA replication and metaphase chromosome attachment. J. Virol. 83, 10336–10346.
Ohsaki, E., Suzuki, T., Karayama, M., and Ueda, K. (2009). Accumulation of LANA at nuclear matrix fraction is important for Kaposi’s sarcoma-associated herpesvirus replication in latency. Virus Res. 139, 74–84.
Ohsaki, E., Ueda, K., Sakakibara, S., Do, E., Yada, K., and Yamanishi, K. (2004). Poly(ADP-ribose) polymerase 1 binds to Kaposi’s sarcoma-associated herpesvirus (KSHV) terminal repeat sequence and modulates KSHV replication in latency. J. Virol. 78, 9936–9946.
Okuno, Y., Satoh, H., Sekiguchi, M., and Masukata, H. (1999). Clustered adenine/thymine stretches are essential for function of a fission yeast replication origin. Mol. Cell. Biol. 19, 6699–6709.
Ostermeier, G. C., Liu, Z., Martins, R. P., Bharadwaj, R. R., Ellis, J., Draghici, S., and Krawetz, S. A. (2003). Nuclear matrix association of the human beta-globin locus utilizing a novel approach to quantitative real-time PCR. Nucleic Acids Res. 31, 3257–3266.
Ottinger, M., Christalla, T., Nathan, K., Brinkmann, M. M., Viejo-Borbolla, A., and Schulz, T. F. (2006). Kaposi’s sarcoma-associated herpesvirus LANA-1 interacts with the short variant of BRD4 and releases cells from a BRD4- and BRD2/RING3-induced G1 cell cycle arrest. J. Virol. 80, 10772–10786.
Pan, H., Zhou, F., and Gao, S. J. (2004). Kaposi’s sarcoma-associated herpesvirus induction of chromosome instability in primary human endothelial cells. Cancer Res. 64, 4064–4068.
Pardoll, D. M., Vogelstein, B., and Coffey, D. S. (1980). A fixed site of DNA replication in eucaryotic cells. Cell 19, 527–536.
Parravicini, C., Chandran, B., Corbellino, M., Berti, E., Paulli, M., Moore, P. S., and Chang, Y. (2000). Differential viral protein expression in Kaposi’s sarcoma-associated herpesvirus-infected diseases: Kaposi’s sarcoma, primary effusion lymphoma, and multicentric Castleman’s disease. Am. J. Pathol. 156, 743–749.
Pearce, M., Matsumura, S., and Wilson, A. C. (2005). Transcripts encoding K12, v-FLIP, v-cyclin, and the microRNA cluster of Kaposi’s sarcoma-associated herpesvirus originate from a common promoter. J. Virol. 79, 14457–14464.
Pfeffer, S., Sewer, A., Lagos-Quintana, M., Sheridan, R., Sander, C., Grasser, F. A., Van Dyk, L. F., Ho, C. K., Shuman, S., Chien, M., Russo, J. J., Ju, J., Randall, G., Lindenbach, B. D., Rice, C. M., Simon, V., Ho, D. D., Zavolan, M., and Tuschl, T. (2005). Identification of microRNAs of the herpesvirus family. Nat. Methods 2, 269–276.
Piolot, T., Tramier, M., Coppey, M., Nicolas, J. C., and Marechal, V. (2001). Close but distinct regions of human herpesvirus 8 latency-associated nuclear antigen 1 are responsible for nuclear targeting and binding to human mitotic chromosomes. J. Virol. 75, 3948–3959.
Potthoff, A., Brockmeyer, N. H., Stucker, M., Wieland, U., Kreuter, A., and Competence Network, H. A. (2010). Kaposi sarcoma in a HIV uninfected man who has sex with men. Eur. J. Med. Res. 15, 79–80.
Qian, L. W., Greene, W., Ye, F., and Gao, S. J. (2008). Kaposi’s sarcoma-associated herpesvirus disrupts adherens junctions and increases endothelial permeability by inducing degradation of VE-cadherin. J. Virol. 82, 11902–11912.
Qian, L. W., Xie, J., Ye, F., and Gao, S. J. (2007). Kaposi’s sarcoma-associated herpesvirus infection promotes invasion of primary human umbilical vein endothelial cells by inducing matrix metalloproteinases. J. Virol. 81, 7001–7010.
Radkov, S. A., Kellam, P., and Boshoff, C. (2000). The latent nuclear antigen of Kaposi sarcoma-associated herpesvirus targets the retinoblastoma-E2F pathway and with the oncogene Hras transforms primary rat cells. Nat. Med. 6, 1121–1127.
Razin, S. V., Gromova, I. I., and Iarovaia, O. V. (1995). Specificity and functional significance of DNA interaction with the nuclear matrix: new approaches to clarify the old questions. Int. Rev. Cytol. 162B, 405–448.
Reddy, K. L., Zullo, J. M., Bertolino, E., and Singh, H. (2008). Transcriptional repression mediated by repositioning of genes to the nuclear lamina. Nature 452, 243–247.
Reisman, D., Yates, J., and Sugden, B. (1985). A putative origin of replication of plasmids derived from Epstein-Barr virus is composed of two cis-acting components. Mol. Cell. Biol. 5, 1822–1832.
Renne, R., Zhong, W., Herndier, B., Mcgrath, M., Abbey, N., Kedes, D., and Ganem, D. (1996). Lytic growth of Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) in culture. Nat. Med. 2, 342–346.
Ritzi, M., Tillack, K., Gerhardt, J., Ott, E., Humme, S., Kremmer, E., Hammerschmidt, W., and Schepers, A. (2003). Complex protein-DNA dynamics at the latent origin of DNA replication of Epstein-Barr virus. J. Cell. Sci. 116, 3971–3984.
Rivas, C., Thlick, A. E., Parravicini, C., Moore, P. S., and Chang, Y. (2001). Kaposi’s sarcoma-associated herpesvirus LANA2 is a B-cell-specific latent viral protein that inhibits p53. J. Virol. 75, 429–438.
Roberge, M., and Gasser, S. M. (1992). DNA loops: structural and functional properties of scaffold-attached regions. Mol. Microbiol. 6, 419–423.
Sadagopan, S., Sharma-Walia, N., Veettil, M. V., Raghu, H., Sivakumar, R., Bottero, V., and Chandran, B. (2007). Kaposi’s sarcoma-associated herpesvirus induces sustained NF-kappaB activation during de novo infection of primary human dermal microvascular endothelial cells that is essential for viral gene expression. J. Virol. 81, 3949–3968.
Said, J. W., Tasaka, T., Takeuchi, S., Asou, H., De Vos, S., Cesarman, E., Knowles, D. M., and Koeffler, H. P. (1996). Primary effusion lymphoma in women: report of two cases of Kaposi’s sarcoma herpes virus-associated effusion-based lymphoma in human immunodeficiency virus-negative women. Blood 88, 3124–3128.
Sakakibara, S., Ueda, K., Chen, J., Okuno, T., and Yamanishi, K. (2001). Octamer-binding sequence is a key element for the autoregulation of Kaposi’s sarcoma-associated herpesvirus ORF50/Lyta gene expression. J. Virol. 75, 6894–6900.
Sakakibara, S., Ueda, K., Nishimura, K., Do, E., Ohsaki, E., Okuno, T., and Yamanishi, K. (2004). Accumulation of heterochromatin components on the terminal repeat sequence of Kaposi’s sarcoma-associated herpesvirus mediated by the latency-associated nuclear antigen. J. Virol. 78, 7299–7310.
Samols, M. A., Hu, J., Skalsky, R. L., and Renne, R. (2005). Cloning and identification of a microRNA cluster within the latency-associated region of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 79, 9301–9305.
Samols, M. A., Skalsky, R. L., Maldonado, A. M., Riva, A., Lopez, M. C., Baker, H. V., and Renne, R. (2007). Identification of cellular genes targeted by KSHV-encoded microRNAs. PLoS Pathog. 3, e65. doi:10.1371/journal.ppat.0030065
Sarkari, F., Sanchez-Alcaraz, T., Wang, S., Holowaty, M. N., Sheng, Y., and Frappier, L. (2009). EBNA1-mediated recruitment of a histone H2B deubiquitylating complex to the Epstein-Barr virus latent origin of DNA replication. PLoS Pathog. 5, e1000624. doi:10.1371/journal.ppat.1000624
Schwaiger, M., Kohler, H., Oakeley, E. J., Stadler, M. B., and Schubeler, D. (2010). Heterochromatin protein 1 (HP1) modulates replication timing of the Drosophila genome. Genome Res. 20, 771–780.
Segurado, M., De Luis, A., and Antequera, F. (2003). Genome-wide distribution of DNA replication origins at A+T-rich islands in Schizosaccharomyces pombe. EMBO Rep. 4, 1048–1053.
Selbach, M., Schwanhausser, B., Thierfelder, N., Fang, Z., Khanin, R., and Rajewsky, N. (2008). Widespread changes in protein synthesis induced by microRNAs. Nature 455, 58–63.
Sequeira-Mendes, J., Diaz-Uriarte, R., Apedaile, A., Huntley, D., Brockdorff, N., and Gomez, M. (2009). Transcription initiation activity sets replication origin efficiency in mammalian cells. PLoS Genet. 5, e1000446. doi:10.1371/journal.pgen.1000446
Sharma-Walia, N., Raghu, H., Sadagopan, S., Sivakumar, R., Veettil, M. V., Naranatt, P. P., Smith, M. M., and Chandran, B. (2006). Cyclooxygenase 2 induced by Kaposi’s sarcoma-associated herpesvirus early during in vitro infection of target cells plays a role in the maintenance of latent viral gene expression. J. Virol. 80, 6534–6552.
Shechter, D., Costanzo, V., and Gautier, J. (2004). ATR and ATM regulate the timing of DNA replication origin firing. Nat. Cell Biol. 6, 648–655.
Silk, A. D., Holland, A. J., and Cleveland, D. W. (2009). Requirements for NuMA in maintenance and establishment of mammalian spindle poles. J. Cell Biol. 184, 677–690.
Skalsky, R. L., Samols, M. A., Plaisance, K. B., Boss, I. W., Riva, A., Lopez, M. C., Baker, H. V., and Renne, R. (2007). Kaposi’s sarcoma-associated herpesvirus encodes an ortholog of miR-155. J. Virol. 81, 12836–12845.
Soulier, J., Grollet, L., Oksenhendler, E., Cacoub, P., Cazals-Hatem, D., Babinet, P., d’Agay, M. F., Clauvel, J. P., Raphael, M., Degos, L., and Sigaux, F. (1995). Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman’s disease. Blood 86, 1276–1280.
Staudt, M. R., and Dittmer, D. P. (2003). Viral latent proteins as targets for Kaposi’s sarcoma and Kaposi’s sarcoma-associated herpesvirus (KSHV/HHV-8) induced lymphoma. Curr. Drug Targets Infect. Disord. 3, 129–135.
Stedman, W., Deng, Z., Lu, F., and Lieberman, P. M. (2004). ORC, MCM, and histone hyperacetylation at the Kaposi’s sarcoma-associated herpesvirus latent replication origin. J. Virol. 78, 12566–12575.
Stedman, W., Kang, H., Lin, S., Kissil, J. L., Bartolomei, M. S., and Lieberman, P. M. (2008). Cohesins localize with CTCF at the KSHV latency control region and at cellular c-myc and H19/Igf2 insulators. EMBO J. 27, 654–666.
Stinchcomb, D. T., Struhl, K., and Davis, R. W. (1979). Isolation and characterisation of a yeast chromosomal replicator. Nature 282, 39–43.
Sumer, H., Craig, J. M., Sibson, M., and Choo, K. H. (2003). A rapid method of genomic array analysis of scaffold/matrix attachment regions (S/MARs) identifies a 2.5-Mb region of enhanced scaffold/matrix attachment at a human neocentromere. Genome Res. 13, 1737–1743.
Tanabe, H., Habermann, F. A., Solovei, I., Cremer, M., and Cremer, T. (2002). Non-random radial arrangements of interphase chromosome territories: evolutionary considerations and functional implications. Mutat. Res. 504, 37–45.
Tanimoto, K., Sugiura, A., Omori, A., Felsenfeld, G., Engel, J. D., and Fukamizu, A. (2003). Human beta-globin locus control region HS5 contains CTCF- and developmental stage-dependent enhancer-blocking activity in erythroid cells. Mol. Cell. Biol. 23, 8946–8952.
Tetsuka, T., Higuchi, M., Fukushi, M., Watanabe, A., Takizawa, S., Oie, M., Gejyo, F., and Fujii, M. (2004). Visualization of a functional KSHV episome-maintenance protein LANA in living cells. Virus Genes 29, 175–182.
Thome, M., Schneider, P., Hofmann, K., Fickenscher, H., Meinl, E., Neipel, F., Mattmann, C., Burns, K., Bodmer, J. L., Schroter, M., Scaffidi, C., Krammer, P. H., Peter, M. E., and Tschopp, J. (1997). Viral FLICE-inhibitory proteins (FLIPs) prevent apoptosis induced by death receptors. Nature 386, 517–521.
Toth, Z., Maglinte, D. T., Lee, S. H., Lee, H. R., Wong, L. Y., Brulois, K. F., Lee, S., Buckley, J. D., Laird, P. W., Marquez, V. E., and Jung, J. U. (2010). Epigenetic analysis of KSHV latent and lytic genomes. PLoS Pathog. 6, e1001013. doi:10.1371/journal.ppat.1001013
Ueda, K., Ishikawa, K., Nishimura, K., Sakakibara, S., Do, E., and Yamanishi, K. (2002). Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) replication and transcription factor activates the K9 (vIRF) gene through two distinct cis elements by a non-DNA-binding mechanism. J. Virol. 76, 12044–12054.
Ueda, K., Sakakibara, S., Ohsaki, E., and Yada, K. (2006). Lack of a mechanism for faithful partition and maintenance of the KSHV genome. Virus Res. 122, 85–94.
van der Velden, H. M., Van Willigen, G., Wetzels, R. H., and Wanka, F. (1984). Attachment of origins of replication to the nuclear matrix and the chromosomal scaffold. FEBS Lett. 171, 13–16.
Verma, S. C., Choudhuri, T., Kaul, R., and Robertson, E. S. (2006). Latency-associated nuclear antigen (LANA) of Kaposi’s sarcoma-associated herpesvirus interacts with origin recognition complexes at the LANA binding sequence within the terminal repeats. J. Virol. 80, 2243–2256.
Viejo-Borbolla, A., Kati, E., Sheldon, J. A., Nathan, K., Mattsson, K., Szekely, L., and Schulz, T. F. (2003). A Domain in the C-terminal region of latency-associated nuclear antigen 1 of Kaposi’s sarcoma-associated Herpesvirus affects transcriptional activation and binding to nuclear heterochromatin. J. Virol. 77, 7093–7100.
Viejo-Borbolla, A., Ottinger, M., Bruning, E., Burger, A., Konig, R., Kati, E., Sheldon, J. A., and Schulz, T. F. (2005). Brd2/RING3 interacts with a chromatin-binding domain in the Kaposi’s sarcoma-associated herpesvirus latency-associated nuclear antigen 1 (LANA-1) that is required for multiple functions of LANA-1. J. Virol. 79, 13618–13629.
Vogelstein, B., Pardoll, D. M., and Coffey, D. S. (1980). Supercoiled loops and eucaryotic DNA replicaton. Cell 22, 79–85.
Volpi, E. V., Chevret, E., Jones, T., Vatcheva, R., Williamson, J., Beck, S., Campbell, R. D., Goldsworthy, M., Powis, S. H., Ragoussis, J., Trowsdale, J., and Sheer, D. (2000). Large-scale chromatin organization of the major histocompatibility complex and other regions of human chromosome 6 and its response to interferon in interphase nuclei. J. Cell. Sci. 113(Pt 9), 1565–1576.
Wang, H. W., Trotter, M. W., Lagos, D., Bourboulia, D., Henderson, S., Makinen, T., Elliman, S., Flanagan, A. M., Alitalo, K., and Boshoff, C. (2004). Kaposi sarcoma herpesvirus-induced cellular reprogramming contributes to the lymphatic endothelial gene expression in Kaposi sarcoma. Nat. Genet. 36, 687–693.
Wang, S., and Frappier, L. (2009). Nucleosome assembly proteins bind to Epstein-Barr virus nuclear antigen 1 and affect its functions in DNA replication and transcriptional activation. J. Virol. 83, 11704–11714.
Wyrick, J. J., Aparicio, J. G., Chen, T., Barnett, J. D., Jennings, E. G., Young, R. A., Bell, S. P., and Aparicio, O. M. (2001). Genome-wide distribution of ORC and MCM proteins in S. cerevisiae: high-resolution mapping of replication origins. Science 294, 2357–2360.
Wysokenski, D. A., and Yates, J. L. (1989). Multiple EBNA1-binding sites are required to form an EBNA1-dependent enhancer and to activate a minimal replicative origin within oriP of Epstein-Barr virus. J. Virol. 63, 2657–2666.
Xiao, B., Verma, S. C., Cai, Q., Kaul, R., Lu, J., Saha, A., and Robertson, E. S. (2010). Bub1 and CENP-F can contribute to Kaposi’s sarcoma-associated herpesvirus genome persistence by targeting LANA to kinetochores. J. Virol. 84, 9718–9732.
Yates, J. L., Camiolo, S. M., and Bashaw, J. M. (2000). The minimal replicator of Epstein-Barr virus oriP. J. Virol. 74, 4512–4522.
Ye, F. C., Blackbourn, D. J., Mengel, M., Xie, J. P., Qian, L. W., Greene, W., Yeh, I. T., Graham, D., and Gao, S. J. (2007). Kaposi’s sarcoma-associated herpesvirus promotes angiogenesis by inducing angiopoietin-2 expression via AP-1 and Ets1. J. Virol. 81, 3980–3991.
Yokoyama, A., Kawaguchi, Y., Kitabayashi, I., Ohki, M., and Hirai, K. (2001). The conserved domain CR2 of Epstein-Barr virus nuclear antigen leader protein is responsible not only for nuclear matrix association but also for nuclear localization. Virology 279, 401–413.
Yoshizaki, K., Matsuda, T., Nishimoto, N., Kuritani, T., Taeho, L., Aozasa, K., Nakahata, T., Kawai, H., Tagoh, H., and Komori, T. (1989). Pathogenic significance of interleukin-6 (IL-6/BSF-2) in Castleman’s disease. Blood 74, 1360–1367.
You, J., Srinivasan, V., Denis, G. V., Harrington, W. J. Jr., Ballestas, M. E., Kaye, K. M., and Howley, P. M. (2006). Kaposi’s sarcoma-associated herpesvirus latency-associated nuclear antigen interacts with bromodomain protein Brd4 on host mitotic chromosomes. J. Virol. 80, 8909–8919.
Zappulla, D. C., Sternglanz, R., and Leatherwood, J. (2002). Control of replication timing by a transcriptional silencer. Curr. Biol. 12, 869–875.
Zhao, J., Punj, V., Matta, H., Mazzacurati, L., Schamus, S., Yang, Y., Yang, T., Hong, Y., and Chaudhary, P. M. (2007). K13 blocks KSHV lytic replication and deregulates vIL6 and hIL6 expression: a model of lytic replication induced clonal selection in viral oncogenesis. PLoS ONE 2, e1067. doi:10.1371/journal.pone.0001067
Zhou, J., Snyder, A. R., and Lieberman, P. M. (2009). Epstein-Barr virus episome stability is coupled to a delay in replication timing. J. Virol. 83, 2154–2162.
Ziegelbauer, J. M., Sullivan, C. S., and Ganem, D. (2009). Tandem array-based expression screens identify host mRNA targets of virus-encoded microRNAs. Nat. Genet. 41, 130–134.
Keywords: Kaposi’s sarcoma-associated herpesvirus, human herpesvirus 8, latency-associated nuclear antigen, ori-P, DNA replication, genome maintenance, pre-replication complex, nuclear matrix
Citation: Ohsaki E and Ueda K (2012) Kaposi’s sarcoma-associated herpesvirus genome replication, partitioning, and maintenance in latency. Front. Microbio. 3:7. doi: 10.3389/fmicb.2012.00007
Received: 30 November 2011;
Accepted: 05 January 2012;
Published online: 24 January 2012.
Edited by:
Tatsuya Tsurumi, Aichi Cancer Center, JapanReviewed by:
Masahiro Fujimuro, Kyoto Pharmaceutical University, JapanTeru Kanda, Aichi cancer Center Research Institute, Japan
Copyright: © 2012 Ohsaki and Ueda. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Keiji Ueda, Division of Virology, Department of Microbiology and Immunology, Osaka University Graduate School of Medicine, 2-2 Yamadaoka, Suita, Osaka 565-0871, Japan. e-mail:a3VlZGFAdmlydXMubWVkLm9zYWthLXUuYWMuanA=