 Rui Pang1†
Rui Pang1† Shi Wu1†
Shi Wu1† Feng Zhang1Jiahui Huang1
Feng Zhang1Jiahui Huang1 Haoming Wu1
Haoming Wu1 Junhui Zhang2Yanping Li1
Junhui Zhang2Yanping Li1 Yu Ding2
Yu Ding2 Jumei Zhang1
Jumei Zhang1 Moutong Chen1
Moutong Chen1 Xianhu Wei1
Xianhu Wei1 Youxiong Zhang1
Youxiong Zhang1 Qihui Gu1
Qihui Gu1 Zhenwen Zhou3Bingshao Liang3Wenzhi Li4
Zhenwen Zhou3Bingshao Liang3Wenzhi Li4 Qingping Wu1*
Qingping Wu1*- 1State Key Laboratory of Applied Microbiology Southern China, Guangdong Provincial Key Laboratory of Microbial Culture Collection and Application, Guangdong Open Laboratory of Applied Microbiology, Guangdong Institute of Microbiology, Guangdong Academy of Sciences, Guangzhou, China
- 2Department of Food Science and Technology, Jinan University, Guangzhou, China
- 3Clinical Laboratory, Guangzhou Women and Children’s Medical Center, Guangzhou Medical University, Guangzhou, China
- 4Infinitus (China) Company Ltd., Jiangmen, China
Sequence type 59 (ST59) is a predominant clonal lineage of community-acquired, methicillin-resistant Staphylococcus aureus (CA-MRSA) in Asia. Despite its increasing clinical relevance in China, the evolution and geographic expansion of ST59 has been relatively uncared for. Previous study has shown that ST59 was the predominant clone in food-related MRSA in China. This study compared the genomes of 87 clonal complex (CC) 59 S. aureus isolates sourced from food chain and infection cases to reconstruct the molecular evolution and geographical spread of ST59. Accordingly, three major sub-clades of ST59 were identified and these did not correlate with isolation source or location. Phylogenetic analysis estimated that ST59 in mainland China diverged from a most common recent ancestor around 1974, and most of the cases of cross-country transmission occurred between 1987 and 2000. Notably, two recent events of cross-country transmission through the food chain were observed, the isolates from these events diverged within relatively short time intervals. These isolates also showed high similarity in terms of their core genome, accessory genes, and antibiotic resistance patterns. These findings provide a valuable insight into the potential route of ST59 expansion in China and indicate a need for robust food chain surveillance to prevent the spread of this pathogen.
Introduction
Staphylococcus aureus is an opportunistic pathogen that causes serious community- and hospital-acquired disease; it accounts for the majority of skin and soft tissue infections in humans, and is also a causative agent of infective endocarditis, necrotizing pneumonia, septicemia, and toxic shock syndrome (Lowy, 1998; Diep et al., 2006). Particularly concerning is the increasing incidence of methicillin-resistant Staphylococcus aureus (MRSA) infections, which have emerged as a major public health concern. Indeed, MRSA was once considered to be solely healthcare-acquired (HA-MRSA), yet over the past few decades, it has increasingly been identified in community-acquired infections (CA-MRSA) (David and Daum, 2010).
Interestingly, CA-MRSA isolates primarily belong to a subset of clonal lineages and possess specific staphylococcal cassette chromosome mec (SCCmec) elements, which confer methicillin resistance, and the phage-encoded toxin, Panton-Valentine leukocidin (PVL), which contributes to skin and soft tissue infections (Otto, 2013). Currently, five major CA-MRSA clones have been described and are associated with specific geographic areas, sequence type (ST)1, ST8, ST30, ST59, and ST80 (Mediavilla et al., 2012; Chuang and Huang, 2013; Abdulgader et al., 2015). In the Asia-Pacific region including Australia, ST59 predominates, where it accounts for 56% of pediatric CA-MRSA infections in Taiwan (Huang et al., 2008; Chuang and Huang, 2013), and has been associated with an increasing proportion of MRSA infections in mainland China (Li et al., 2018; Liang et al., 2019). In Hong Kong, a recent outbreak of ST59 MRSA has occurred in a neonatal intensive care unit (Cheng et al., 2019). Li et al. (2018) indicated that ST59 possesses a fitness advantage over other MRSA clones and that it poses a critical threat to human health. However, the evolution and expansion of S. aureus ST59 in this region has been relatively understudied.
Previous studies have shown that S. aureus ST59 harbors two major clones: the Taiwan clone (TW), which causes severe infections, and the Asian-Pacific clone (AP), which is typically commensal (Huang et al., 2008). Genomic comparisons have revealed that the TW clone carries a PVL-encoding prophage ΦSa2 (Chen et al., 2013), whereas the AP clone carries a staphylokinase (SAK)-encoding prophage ΦSa3 that enhances the bacterium’s capacity to colonize human hosts (van Wamel et al., 2006; Kwiecinski et al., 2013). Thus, the origins of most ST59 isolates can be determined based on the presence of either PVL- or SAK-related phage. However, a minority of publicly available ST59 genomes indicate the presence of both phages (Feng et al., 2017), which suggests that MRSA ST59 may have multiple origins or that it has experienced recombination events during its evolution and genetic expansion.
Though little is known about the transmission route of the ST59 lineage, a previous study completed by our research group documented the prevalence of this S. aureus clone in various food samples across China (Wu et al., 2019). Indeed, 52.8% of MRSA isolates collected from various food sources were identified as clonal complex (CC)59, which includes ST59, ST338, and ST3355, and suggests that the food chain may serve as a potential transmission vector. However, zoonotic MRSA infections are primarily livestock-associated (LA-MRSA) (Petinaki and Spiliopoulou, 2012), and these are not typically members of CC59. For example, LA-MRSA isolates from Europe and North America primarily belong to CC398, and the vast majority of LA-MRSA isolates from Asia belong to CC9 (Bens et al., 2006; Cui et al., 2009; Smith et al., 2009; Ye et al., 2016). Indeed, CC59 isolates are rarely linked to LA infections but are frequently associated with CA infections. Thus, we hypothesized that the prevalence of S. aureus ST59 in foods is due to human activity that spreads the bacterium through the food chain.
To characterize the potential transmission of S. aureus ST59 in the food chain, we compared the whole-genome sequences of 81 CC59 isolates from geographic areas across China. Using a Bayesian temporal and spatial analysis, we estimated the evolutionary properties of this S. aureus clone during its geographic expansion in China. Two potential food chain transmission events were identified, revealing that the ST59 lineage may have spread across the different regions using food as a vehicle. These findings are critical to understand the evolutionary signatures that correlate with the expansion of this pathogen.
Materials and Methods
Selection of Bacterial Isolates
From July 2011 to June 2016, we collected 4,300 retail food samples from supermarkets, fairs and farmers’ markets covering most of the provincial capitals of China (Supplementary Figure S1). These samples included 604 raw meat (bacon/sausage, poultry, pork, mutton, and beef), 860 aquatic products (freshwater fish, shrimp, and seafood), 601 quick-frozen products (frozen dumplings/steamed stuffed buns and frozen meat), 859 ready-to-eat (RTE) food, 258 pasteurized milk, 419 vegetables, and 699 edible mushrooms. A total of 1,581 S. aureus isolates were obtained from 1,063 positive samples according to the GB 4789.10-2010 food microbiological examination of S. aureus (National Food Safety Standards of China) and the most probable number (MPN) method (Gombas et al., 2003). They were identified by Gram stain, catalase and oxidase tests and API STAPH test strips (BioMerieux, Marcy-1’Etoile, France). According to a multilocus sequence typing (MLST) analysis, 68 typical S. aureus CC59 isolates were further selected (Supplementary Table S1). The presence of the mecA/mecC gene was assessed by PCR for all isolates (Perez-Roth et al., 2001; Stegger et al., 2012). Nineteen additional ST59 isolates that had been previously sequenced were retrieved from the NCBI database for comparative genomic analysis (Supplementary Table S1).
Whole-Genome Sequencing and Assembly
Genomic DNA was extracted from S. aureus isolates using a genomic DNA extraction kit (Magen Biotech, Guangzhou, China) according to the manufacturer’s instructions. Each DNA sample was fragmented into 400-bp fragments by a Covaris M200 sonicator and prepared for sequencing with the Ion Plus Fragment Library Kit (Thermo Fisher Scientific Inc., Waltham, MA, United States). Whole genomes were sequenced on the Life Ion S5 platform with an average coverage of 100×. Clean reads were used for de novo assembly with SPAdes v3.6.2 (Bankevich et al., 2012). Only assemblies that harbored ≥95% of core genome (cg) MLST targets were used for further analysis as previously described (Strauß et al., 2017). If the criteria were not met, the sample was re-sequenced. The presence of PVL- and SAK-coding genes was determined using BLAST.
Single Nucleotide Polymorphisms Calling in Core Genomes and Phylogenetic Inference
The core genome of the ST59 clone was produced using Harvest v1.1.2 using the SA40 genome as a reference (Treangen et al., 2014). After the core-genome alignment was generated, Gubbins was used to conduct recombination analysis and remove the putative recombined regions (Croucher et al., 2015). Single Nucleotide Polymorphisms (SNPs) were then extracted from the recombination-free core-genome alignment using the script available at https://github.com/sanger-pathogens/snp-sites. The ML phylogenetic tree was constructed on the concatenated core SNPs using RAxML v8.2.10 in the GTRGAMMA model (1,000 bootstrap) (Stamatakis, 2014), and was visualized using iTOL (Letunic and Bork, 2016).
Time-Scaled Phylogenetic Analysis
To explore the transmission relationship within ST59 isolates, we inferred the substitution rate, divergence dates, and phylogeographic spread, according to the Bayesian methods implemented in BEAST v1.8.4 package (Drummond et al., 2012). The best nucleotide substitution model was calculated by jModelTest v2.1.7 (Darriba et al., 2012). The core SNP alignment was then used for Bayesian inference with the general time-reversible (GTR) model (inferred best model) of nucleotide substitution, and the symmetric substitution model of discrete trait substitution with the option to perform Bayesian Stochastic Search Variable Selection (BSSVS) selected. Different molecular clock (strict, random, and uncorrelated lognormal) and tree (constant size, exponential growth, Bayesian skyride, and Bayesian skyline) models were run. Each model was run twice for 100,000,000 states, with a sampling frequency of 10,000, to check for convergence in the data set. The convergence of all model combinations was checked using Tracer module in BEAST (threshold: ESS of the tree model over 200), and the maximum clade credibility (MCC) tree was generated with the TreeAnnotator according to the best model (strict molecular clock and constant size model in this study) and visualized with FigTree 1.4.3.
Pan-Genome Analysis
The genomes of all analyzed isolates were annotated using Prokka v1.11 (Seemann, 2014). The output of Prokka was used to construct the pan-genome using Roary v3.11.2, with a BLASTP identity cutoff of 90% (Page et al., 2015). A profile of absence (0) or presence (1) of all genes was generated according to the pan-genome distribution of all samples, and a hierarchically clustered heatmap was created with this matrix by using the Pearson correlation function in R (Graña-Miraglia et al., 2017).
Average Nucleotide Identity Calculation
Pairwise ANIs between all isolates were calculated on whole genome sequences by using the Python module Pyani v0.2.8 with default parameters1. The Average Nucleotide Identity (ANI) result was presented as a heatmap generated according to a matrix of Hadamard product of pairwise alignment coverage, total alignment lengths, similarity errors, and percentage identity; hierarchical clustering in two dimensions was represented by dendrograms constructed by simple linkage of the data (Pritchard et al., 2016).
Assessment of Antibiotic Resistance Patterns
Antimicrobial susceptibility tests were performed using the agar disk diffusion method (Kirby–Bauer) on Mueller–Hinton agar for all of the S. aureus isolates. The antimicrobial agents used for susceptibility testing were amoxicillin/clavulanic acid, ampicillin, cefepime, cefoxitin, penicillin G, ceftazidime, amikacin, gentamicin, kanamycin, streptomycin, chloramphenicol, clindamycin, erythromycin, telithromycin, ciprofloxacin, norfloxacin, tetracycline, linezolid, trimethoprim/sulphamethoxazole (1:19), rifampicin, quinupristin/dalfopristin, teicoplanin, nitrofurantoin, and fusidic acid. MICs for linezolid-resistant isolates by disk diffusion were also confirmed by the agar dilution method on Mueller–Hinton agar. The results were scored and graded as resistant (R) or sensitive (S) according to the guidelines of The Clinical and Laboratory Standards Institute [CLSI] (2015). S. aureus ATCC25923 and Escherichia coli ATCC25922 were included as controls. The pairwise correlation value between each two isolates was estimated using the software available in OmicShare2 based on the Pearson correlation coefficient.
Spatial Phylogenetic Reconstruction
SpreaD3 v0.9.7.1 was used to analyze and visualize phylogeographic relationships resulting from the Bayesian inference of sequences and spatiotemporal diffusion (Bielejec et al., 2016). Here we mapped phylogenies annotated with discrete spatial information on the genomes of ST59 clones generated by the BSSVS procedure. High-dimensional posterior summaries to markup the spatial diffusion through time were identified using a Bayes Factor test (cut-off ≥10).
Data Accessibility
The draft genome sequences of S. aureus generated in this study were submitted to the NCBI database under BioProject PRJNA589244.
Results
Phylogenomic Analysis of the ST59 Lineage
We performed whole genome sequencing on 63 isolates of ST59, four ST338, and one ST3355 isolated from food samples across China (Supplementary Table S1). Of these, 45 isolates were methicillin-resistant, and 23 were methicillin-susceptible S. aureus (MSSA). According to spa typing, most of these isolates were spa type t437 (84.3%), as was previously described by Li et al. (2018). Additionally, 19 publicly available S. aureus ST59 genomes were included in the comparative analysis (Supplementary Table S1). Importantly, these strains were also isolated in Asia.
Phylogenomic analyses were carried out using the combined dataset of 87 genomes, which included isolates from humans (n = 17), aquatic products (n = 24), retail meats (n = 19), quick-frozen products (n = 9), RTE food (n = 7), edible mushrooms (n = 8), and vegetables (n = 3). Only two putative recombination regions were identified, which both occurred on the branch of isolate AR3 (Supplementary Table S2). After the removal of recombination events, these isolates differed in 3,745 core genome SNPs. The concatenated core SNPs were used to construct a maximum-likelihood (ML) phylogenetic tree (Figure 1).
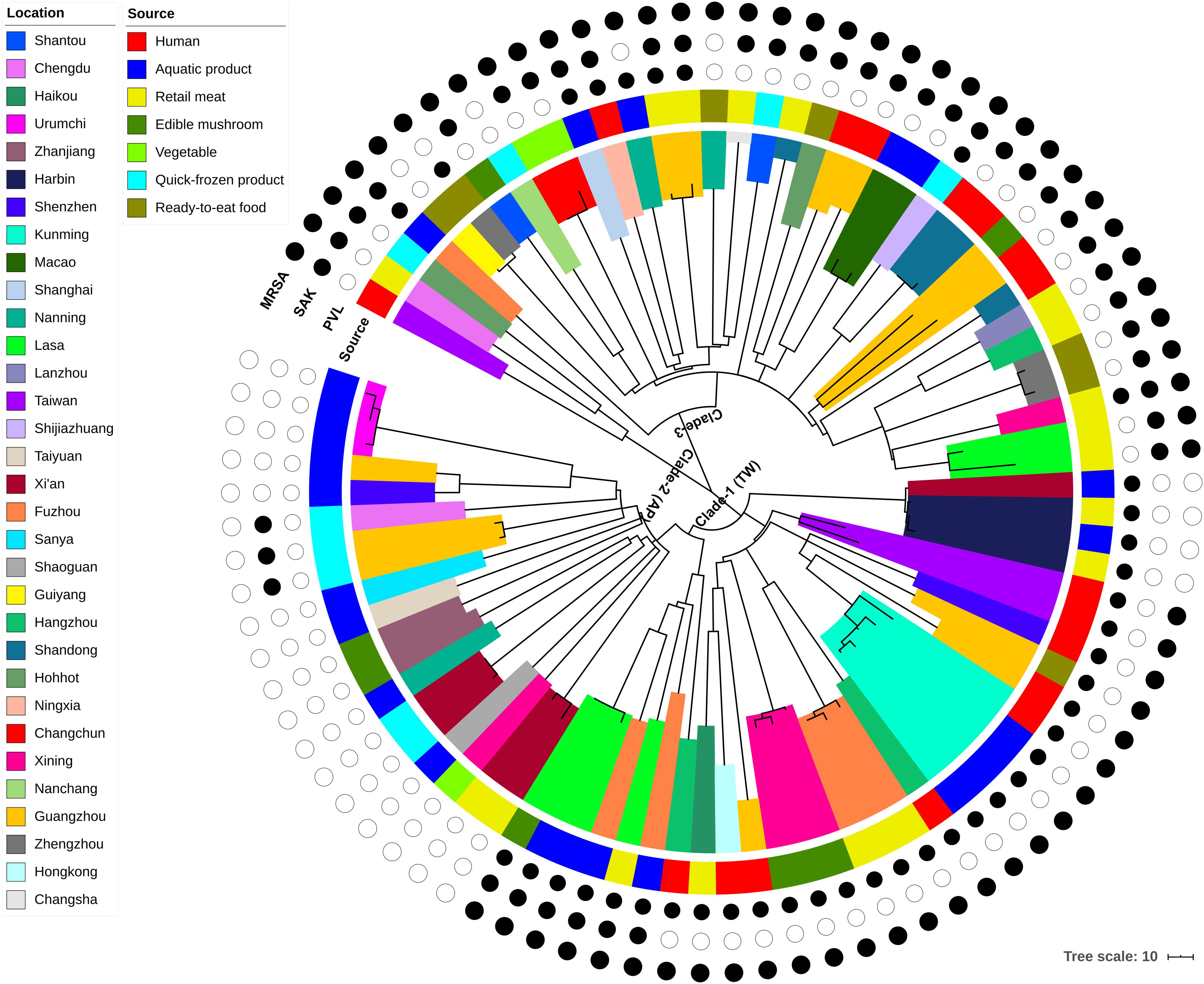
Figure 1. Maximum-likelihood phylogeny of 87 CC59 S. aureus isolates based on 3,745 core genome single nucleotide polymorphisms. The clade colors indicate each isolates region of origin, and the bar color indicates the source of each isolate. Information about the presence (black circle) or absence (white circle) of PVL and SAK is given for each isolate. Scale bar: SNP distance.
According to the node height, the ST59 lineage was divided into three major clades. The clade 1 and clade 2 shared common ancestors with the TW and AP clones respectively, while the clade 3 was genetically distinct from these two widely reported clones. Notably, there was no significant correlation between clades and source or location when the geographic and source origin of isolates were mapped onto the phylogenetic tree. While this revealed the frequent transmission of ST59 clones between geographic areas and food sources, it also suggested that no significant boundary existed between human-infected and food-contaminated isolates. For example, the isolates collected from aquatic products in Kunming were very closely related to another clinical isolate collected in this region (clade marked by cyan in Figure 1). The presence or absence of PVL- and SAK-coding genes was also assessed, as these markers have previously been used to distinguish the origins of ST59 clones (Chen et al., 2013). As such, approximately 78% of isolates carried PVL or SAK, 15% carried both PVL and SAK, and the remainder were negative for PVL and SAK (Figure 1 and Supplementary Table S1). The presence of PVL and SAK in isolates in clade 1 and clade 2 was largely consistent with the previous reports concerning TW (PVL positive and SAK negative) and AP (PVL negative and SAK positive) clones. However, clade 1 has become more diverse as it spread across China, with two potential introductions of SAK. The existence of separate PVL-negative and -positive MSSAs in this clade also indicates that one introduction of PVL and a subsequent loss, and a single introduction of SCCmec may have occurred. For clade 3, repetitive introductions and losses of PVL were observed, indicating that this clade may present more variability in the genomic regions of these genes.
Cross-Country Transmission of the ST59 Lineage
To further reconstruct the evolution and geographic transmission of the ST59 lineage, the core SNPs were used for Bayesian phylogenetic analysis with BEAST. The substitution rate in the core genome of the ST59 lineage was estimated to be 1.31 × 10–6 substitutions per site per year [95% highest posterior density (HPD) interval = 9.89 × 10–7 to 1.62 × 10–6], corresponding to 2.80 SNPs per year (95% HPD = 2.12–3.46). This substitution rate agreed with previously published data for other S. aureus lineages and revealed a relative conservation of the mutation rate across S. aureus lineages (McAdam et al., 2012; Uhlemann et al., 2014, 2017; Moradigaravand et al., 2017).
Using the calculated substitution rate, the date of divergence of the ST59 lineage was estimated according to a MCC tree generated using TreeAnnotator. As such, the estimated time of the most recent common ancestor (MRCA) was approximately 1974 (95% HPD, 1963–1983); the MRCA originated in Guangzhou and diverged into three clades (Figure 2). These clades were then transmitted from Guangzhou to other cities and diverged in different directions. Notably, the ancestors of representative isolates for TW (SA957) and AP (SA40) clones were estimated to be transmitted from Guangzhou to Taiwan around 1988 (95% HPD, 1981–1994) and 1990 (95% HPD, 1983–1995), respectively. Further, an estimated 82% of spread events occurred between 1987 and 2000, which is also the period during which most of the cross-country ST59 transmissions occurred. This indicates that most of the recent food-borne cases of S. aureus ST59 were locally transmitted.
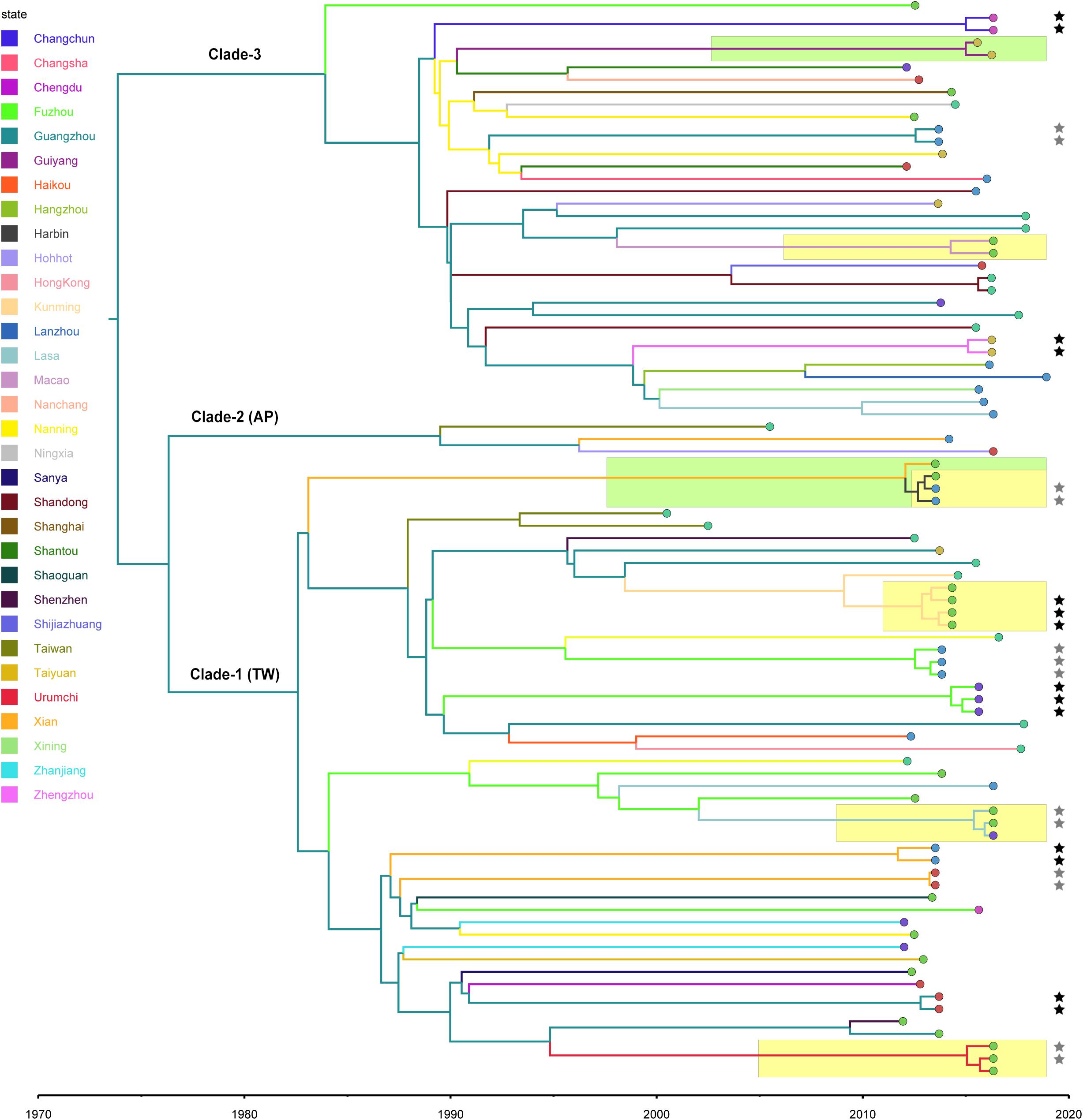
Figure 2. Bayesian maximum clade credibility tree of CC59 isolates. Branch colors represent the region of origin of each sample and their most recent common ancestor, respectively. The color of the circle at the end of each branch indicates the isolate’s source; cyan represents isolation from a human, green from an aquatic source, blue from retail meat, gold from an RTE food, red from a quick-frozen product, purple from an edible mushroom, and orange from a vegetable. Isolates that were source from the same food sample are marked up with colored stars behind the tree tips. Clades that were estimated to have been involved in recent local transmission are highlighted with yellow squares, and those estimated to have been involved in recent cross-country transmission are highlighted with green squares.
Notably, two relatively recent cases of cross-country transmission were observed (marked by green in Figure 2, also see Supplementary Figure S2 for details). Isolates 2939 and fhich were collected from Guiyang in July 2015 and from Zhengzhou in April 2016, respectively, diverged from a common ancestor originating in Zhengzhou in January 2015 (95% HPD, April 2014 to August 2015). These isolates were both identified in RTE foods; thus, RTE foods may have been responsible for the transport of the bacterium from Zhengzhou to Guiyang. Similarly, two aquatic isolates, 1931-0 and 1956-2, which were isolated in Xi’an and Harbin, respectively, in July 2013, shared a recent common ancestor in Xi’an in January 2012 (95% HPD, January 2011 to December 2012). Notably, this sub-clade was also isolated from aquatic products and retail meat in Harbin, which is likely the result of cross-contamination in this region.
To further characterize the putative food chain-transmitted cases, the pairwise SNP distances between isolates were estimated. Excluding those distinct isolates, paired isolates collected from the same sample (direct transmission, marked up with colored stars behind the tree tips) differed by six SNPs (range: 2–10; Supplementary Table S2 and Figure 3). Isolate pairs collected from the same region with shorter divergence time (<2 years, recent local transmission, marked by yellow bar in Figure 2) were also closely related, with an average pairwise SNP distance of six (range: 0–10). In contrast, longitudinal isolates collected from the same region and separated by time interval more than 2 years (independent contamination) harbored a considerably higher average pairwise SNP distance of 169 (range: 32–229). The pairwise SNP distances between isolates 2939 and 1956-2 and their counterparts were both three, indicating that these isolates could be precisely linked to recent transmissions.
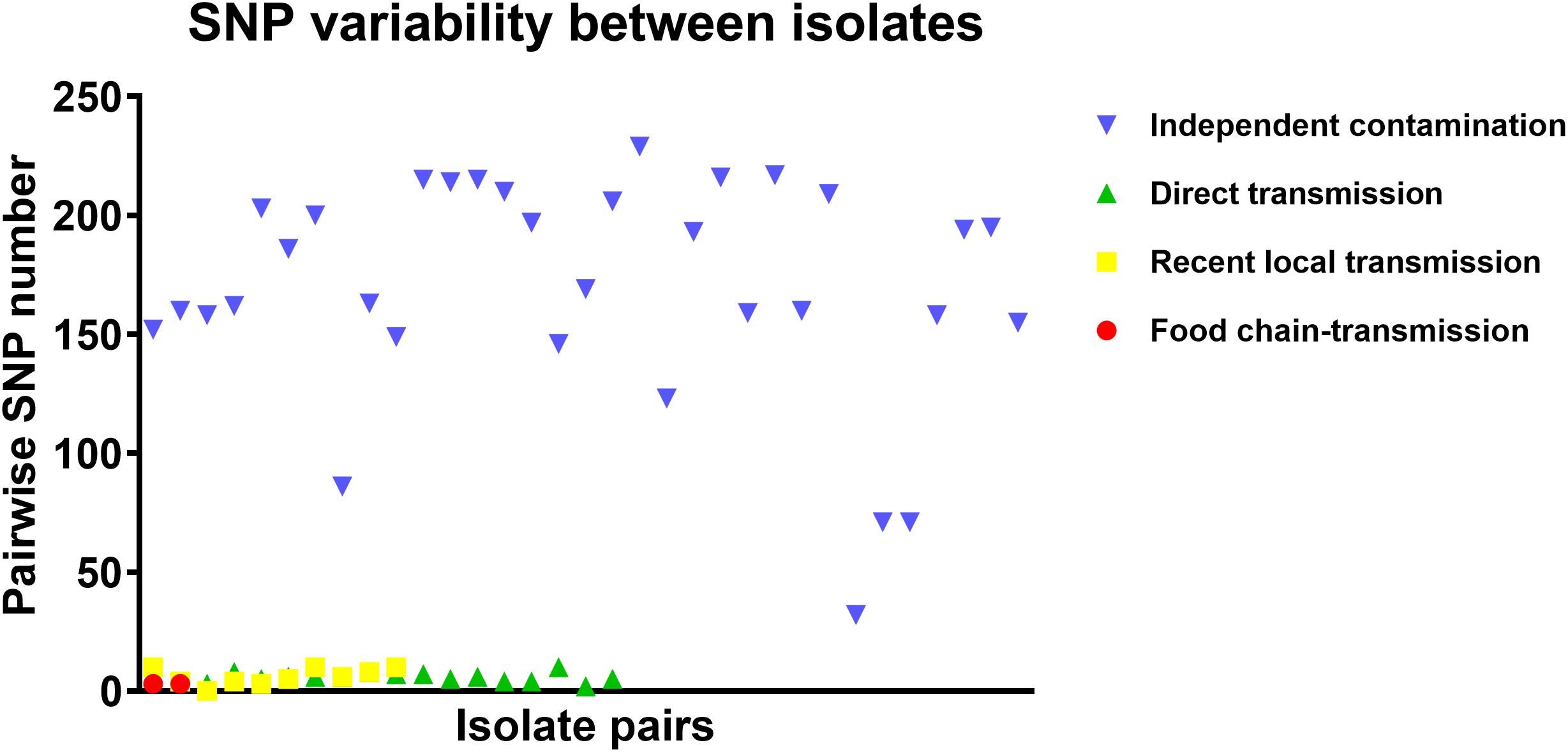
Figure 3. The pairwise SNP comparison between S. aureus isolates from different case types.
Genetic and Phenotypic Evidence for Food Chain Transmission Events
The pan-genome was analyzed to determine whether the putative food chain-transmitted isolates shared similar accessory genome by virtue of their relatively short timescale over which they diversify from a common ancestor. A gene content correlation matrix was constructed according to the profile of accessory gene presence and absence, and was visualized using a heatmap (Figure 4). The dendrogram shown in the heatmap was not fully correlated with the phylogenetic tree inferred from core genome SNPs. However, most of the isolates from direct transmission and recently local transmission still clustered together as was shown in the phylogenetic tree. Further, the clustering of the putative food chain-transmitted isolates is rather similar to the topology of phylogenetic tree. Hence, the similarity between phylogenetic relationships and clustering profiles gathered from gene content variation might due to the short divergence time between these isolates.
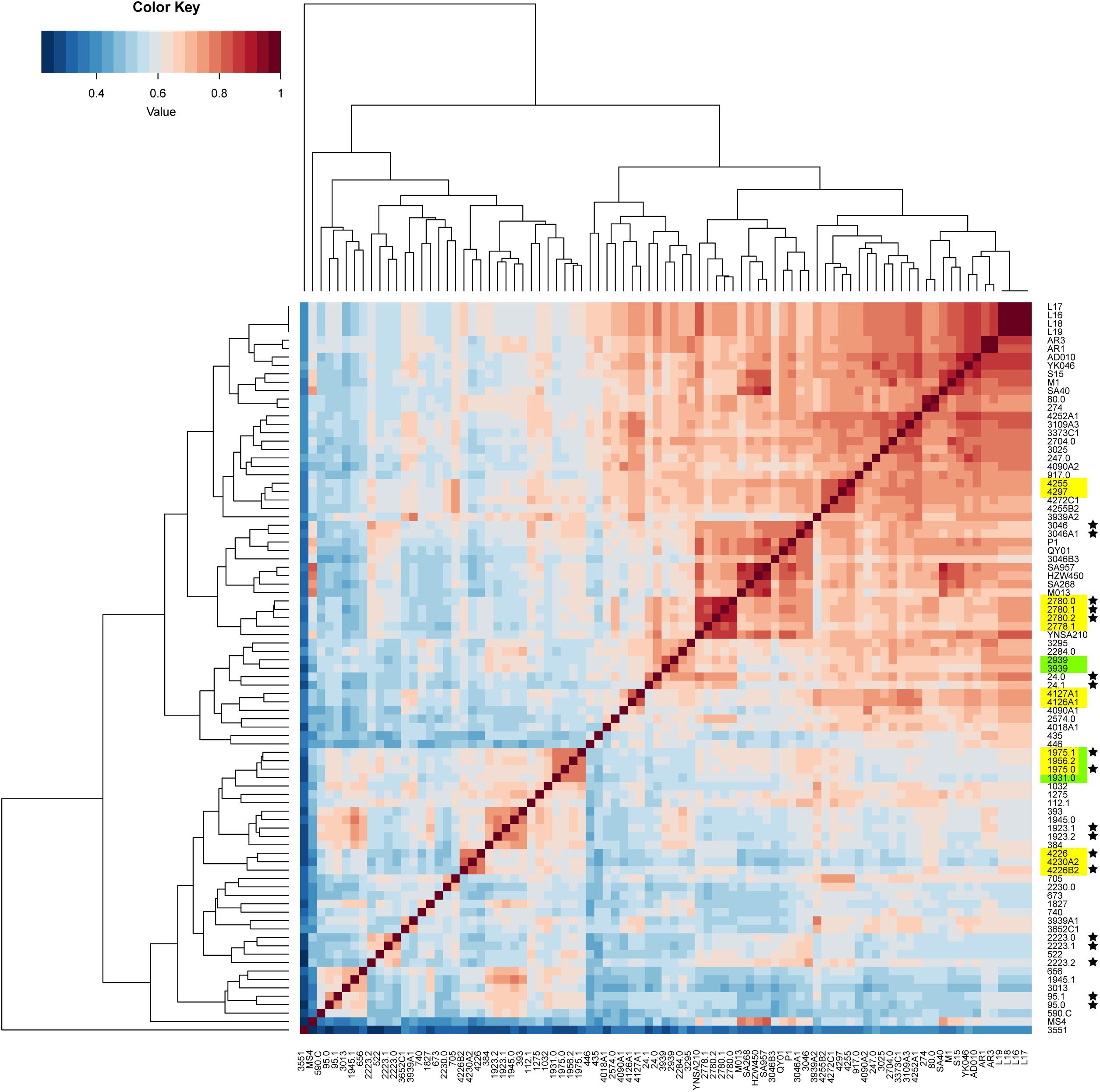
Figure 4. Comparison on the accessory gene profiles among the isolates. The differences in accessory gene contents are presented by a heatmap of the gene content correlation matrix. The dendrograms across the top and side indicate the hierarchical clustering by gene content. Isolates that were source from the same food sample are marked up with colored stars. Isolates estimated to have been involved in recent local transmission are marked with yellow, and those estimated to have been involved in recent cross-country transmission are marked with green.
The pairwise ANI was calculated based on the whole genome sequences of all the isolates, and a dendrogram tree was constructed based on the clustering of the pairwise ANI values (Supplementary Table S3 and Supplementary Figure S3). This analysis takes into account the core and accessory genomes of each isolate. As such, the clustering of the putative food chain-transmitted isolates was highly correlated to the topological patterns inferred from core genome SNPs and accessory gene contents. Thus, the genetic similarities between these isolates and their counterparts are likely the result of relatively recent divergences.
Antibiotic resistance profiles were also considered of putative food chain-transmission, direct transmission, recently local transmission, and independent contaminated case isolates (Supplementary Table S4) and the pairwise correlation was calculated. According to the correlation value, the similarity in antibiotic resistance pattern of paired isolates within direct transmission or recent local transmission was much higher than that of paired isolates collected from independent contamination (Figure 5 and Supplementary Table S5). The resistance profiles were also highly similar between isolates 2939 and 3939 (correlation value 0.9773), and between isolates 1956-2 and 1931-0 (correlation value 0.8856), reinforcing the inference that the divergence time between these isolates and their counterparts was relatively short. Furthermore, these remarkably similarity in phenotypes unveiled a urgent consideration for public health that the antibiotic resistance would probably disseminate through bacterial transmission.
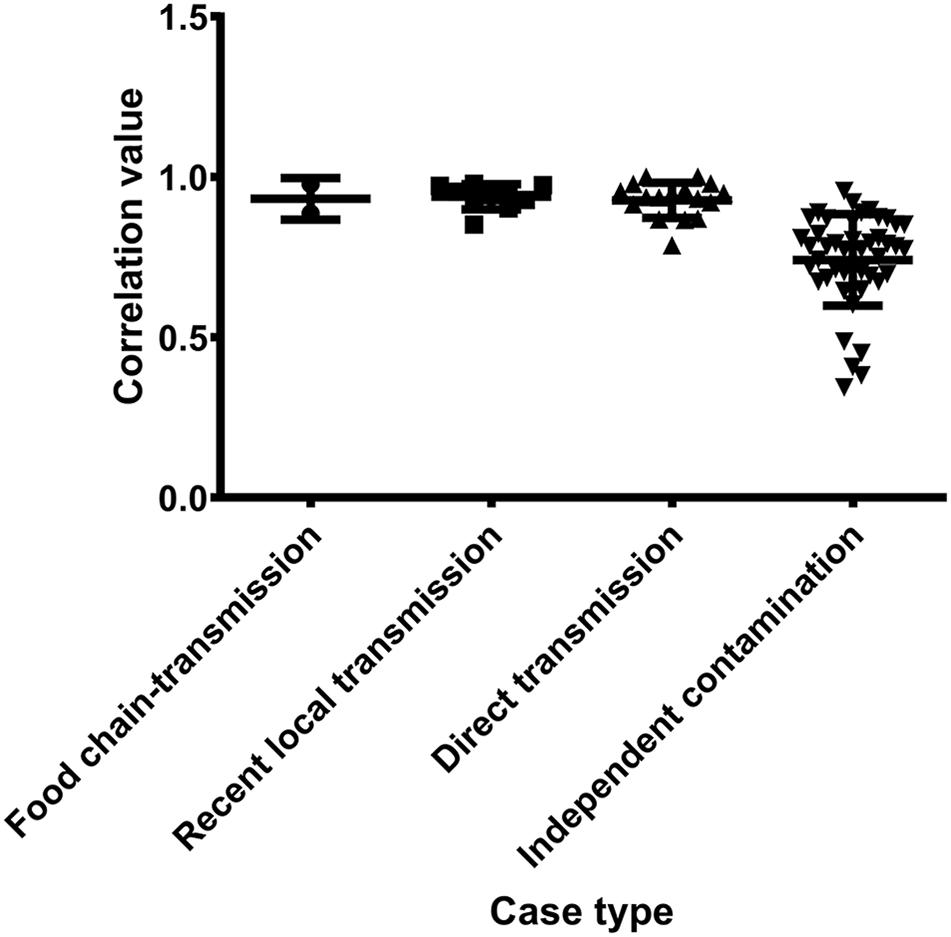
Figure 5. The pairwise correlation of antibiotic resistance patterns in S. aureus isolates between different case types.
Finally, a spatial phylogeographic reconstruction was used to visualize the potential transmission between geographic locations (Figure 6). As such, a Bayes factor cut-off of 10 produced a map that described several waves of ST59 transmission across China. As was estimated above, most transmission events occurred before 2000, and southern China was the most common transmission source of this clone to other regions. Based on the phylogeographic analysis, only two recent transmission events were identified: Zhengzhou to Guiyang in 2015, and Xi’an to Harbin in 2012. Considering that the food sources of contamination at both the departure and destination sites were identical in both events, the food chain may be a considerable reservoir for cross-country transmission of S. aureus ST59 in China.
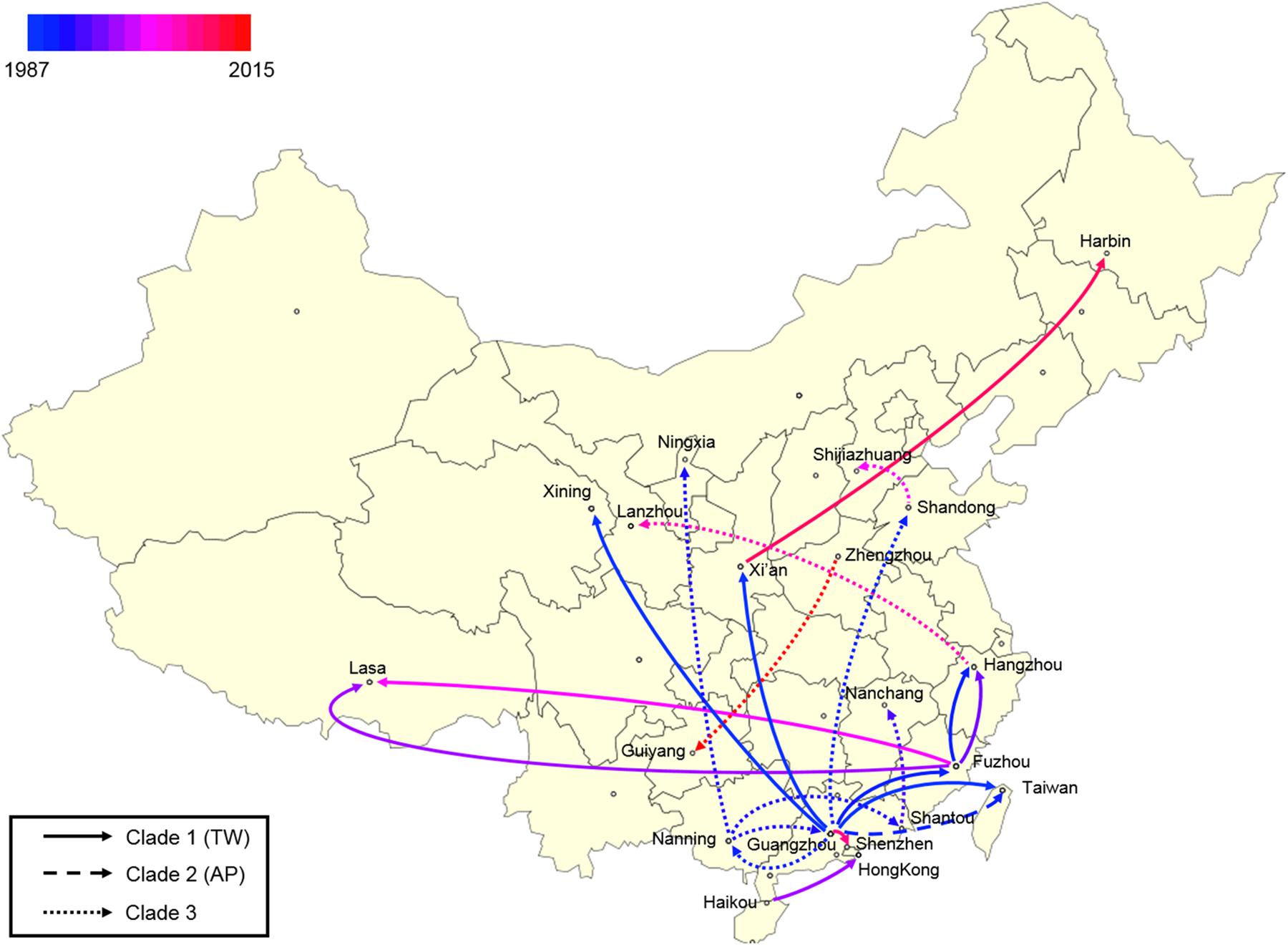
Figure 6. Spatial phylogenetic reconstruction of ST59 transmission dynamics inferred using SpreaD3. A Bayesian phylogeographic reconstruction incorporating discrete spatial spatiotemporal diffusion was used to visualize the transmission between the original regions of ST59 clones and the geographic locations where different lineages were detected throughout the expansion across China. Different linear types indicate that the transmission events fall within different phylogenetic clades.
Discussion
The dissemination of MRSA is a worldwide problem; it is transmitted by humans in both the hospital and community settings, animals in the wild, or by vectors, such as air and water (Diekema et al., 2001; Goodwin et al., 2012; Porrero et al., 2013; Bos et al., 2016). However, most of these transmission pathways lack regularity, making it difficult to determine the origin of the MRSA clone, especially when clones have spread across the country. As a traceable vector, the food chain poses a significant risk for MRSA transmission across geographic regions, as it has been demonstrated by the illegal introduction and commerce of MRSA-contaminated food (Oniciuc et al., 2015; Rodriguez-Lazaro et al., 2017). Thus, the aim of this study was to characterize the evolutionary dynamic and transmission potential of MRSA through the food chain.
This study utilizes ST59 isolates that were isolated from the food chain across China (Wu et al., 2019). Previous studies have demonstrated that ST59 is becoming the predominant MRSA sequence type isolated from hospitalized patients in China (Chuang and Huang, 2013; Yang et al., 2017; Li et al., 2018). There are two distinct clones within the ST59 sequence type, AP, which is PVL-negative/SAK-positive, and TW, which is PVL-positive/SAK-negative (Hung et al., 2016). Both of these clones were identified in our analysis (Figure 1); however, both clones underwent significant genetic changes. In particular, the TW clone acquired the SAK-encoding phage twice, and lost the PVL-encoding phage during its spread between different regions in China. A clone (clade 3) was also identified that was genetically distinct from either the AP or TW clones. This clone showed higher instability in the PVL- and SAK-encoding phages than the other two clones. The variability of these phage genes was also shown in another MRSA linage USA500 (Frisch et al., 2018), unveiling the potential importance of horizontal gene transfer in evolution of pathogenic MRSA.
Several potential direct transmission cases were also identified in the dataset, including local and cross-country transmission. Generally, if two bacterial isolates are involved in a case of direct transmission, they share a low genome-wide SNP distance due to the limited divergence time (Uhlemann et al., 2014). In studies tracing the transmission of CA-MRSA in United States communities, the maximum pairwise distance of isolates within a single household in New York was determined to be 23 SNPs (Uhlemann et al., 2014), and the mean genetic diversities within Los Angeles and Chicago were 17.6 and 12 SNPs per household (Alam et al., 2015). In our study, the maximum pairwise SNP distance between paired isolates collected from the same sample was 10. As such, for isolates involved in putative local transmission, the pairwise SNP distances ranged from 0 to 10, which precisely fell within this range. Both of the putative cross-country transmission cases, isolate 2939 with 3939, and isolate 1956-2 with 1931-0, also harbored pairwise SNP distances lower than 10. The identical sources for these isolates, RTE foods for both 2939 and 3939, and aquatic products for both 1956-2 and 1931-0, reinforced the likelihood of direct transmission between these samples.
Given that the SNP-based analysis was focused on the core genes of these isolates, we also constructed a dendrogram according to the presence and absence of accessory genes. Importantly, the distribution of isolates between the different clades did not fully align with their presentation in the SNP-based phylogenetic tree; although other groups have reported similar discrepancies (Orsi et al., 2011; Anastasi et al., 2016; Graña-Miraglia et al., 2017; Henri et al., 2017). Several reasons may account for this observation, such as natural factors including horizontal gene transfer and recombination, and technical factors including gaps in the draft genomes and inaccurate parameters for the pan-genome estimation (Orsi et al., 2011; Rouli et al., 2015). Despite the discrepancy, most isolates that were estimated to be derived from direct transmissions grouped together, indicating that fewer variations in gene content had occurred between these isolates. The genetic relationships within these isolates was further validated by genome-wide ANI analysis, which considered both core and accessory genes (Pritchard et al., 2016).
Furthermore, this study demonstrated that S. aureus ST59 underwent a series of geographic expansions across China during the 1980s and 1990s (Figure 2) and that southeast coastal cities were the most frequent origins of the spread of this bacterial clone. A previous review has indicated that ST59 lineage mainly spreads along the Asia-Pacific including the southeast coastal of China (Chuang and Huang, 2013), which might exactly explain why S. aureus ST59 in mainland China were originated from this region. Importantly, this also aligns our previous findings, namely that ST59 was the predominant genotype among all MRSAs isolated from aquatic products in China (Wu et al., 2019). Thus, Guangzhou, a city known for consumption of large amounts of aquatic products, had been one of the major distributed cities for ST59 MRSA (Li et al., 2018). In addition, the dominant ST of S. aureus isolated from patients in Guangzhou was exactly belonged to CC59 (Liang et al., 2018). Our analysis also revealed Fuzhou as a hotspot for ST59 transmission. Actually, previous studies has showed the high incidence of ST59 infections in the pediatric population in Taiwan (Huang et al., 2008). The geographical adjacency of Fuzhou to Taiwan suggests that bacterial dissemination may have more frequently occurred through environmental vectors, such as sea water and migratory wildlife (Abulreesh, 2011; Beleneva, 2011; Robb et al., 2013). However, the focus of this study was ST59 food chain isolates, and further studies are necessary to accurately characterize the transmission of ST59 between these two sites.
In summary, the potential for cross-country transmission of S. aureus ST59 through the food chain is supported by this study, which facilitates an increased understanding of the evolution and geographic expansion of CA-MRSA. Given that the food chain is indispensable to human society, the assumption that contaminated food products may serve as vehicles for the spread of pathogens has been widely recognized (Ogata et al., 2012; Schoder et al., 2015; Oniciuc et al., 2017). However, most of the previous studies regarding MRSA in the food chain have only characterized the relationship between multiple bacterial isolates using multi-locus sequence typing (MLST) (Ge et al., 2017; Rodriguez-Lazaro et al., 2017; Rong et al., 2017; Tang et al., 2017; Wu et al., 2019), a method that is dependent on housekeeping genes. Thus, the genetic similarity of housekeeping genes cannot exclude the possibility that these isolates are derived from ancestors separated by earlier divergence events rather than by direct food chain-transmission. This study described a genomic approach to accurately determine the relationship between pathogens isolated from multiple geographic locations, rendering a more accurate system for monitoring and tracking foodborne pathogens.
Data Availability Statement
The datasets generated for this study can be found in the NCBI database under BioProject PRJNA589244.
Author Contributions
RP, SW, and QW conceived and designed the study, and wrote and finalized the manuscript. SW, FZ, JH, YD, JMZ, MC, XW, YZ, QG, ZZ, BL, and WL performed the samples and data collection. RP, JHZ, and YL conducted the genome sequencing. RP, SW, and HW performed the data analysis.
Funding
This work was supported by the National Key Research and Development Program of China (2018YFC1602500), the Science and Technology Planning Project of Guangdong Province (2017B090904004), and GDAS’s Special Project of Science and Technology Development (2017GDASCX-0201).
Conflict of Interest
WL is employed by the company Infinitus (China) Company Ltd., Jiangmen, China.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.00422/full#supplementary-material
Footnotes
References
Abdulgader, S. M., Shittu, A. O., Nicol, M. P., and Kaba, M. (2015). Molecular epidemiology of Methicillin-resistant Staphylococcus aureus in Africa: a systematic review. Front. Microbiol. 6:348. doi: 10.3389/fmicb.2015.00348
Abulreesh, H. H. (2011). “Multidrug-resistant staphylococci in the environment,” in Proceedings of the 2011 International Conference on Biotechnology and Environment Management. Singapore.
Alam, M. T., Read, T. D., Petit, R. A. III, Boyle-Vavra, S., Miller, L. G., Eells, S. J., et al. (2015). Transmission and microevolution of USA300 MRSA in U.S. households: evidence from whole-genome sequencing. mBio 6:e00054-15. doi: 10.1128/mBio.00054-15
Anastasi, E., MacArthur, I., Scortti, M., Alvarez, S., Giguere, S., and Vazquez-Boland, J. A. (2016). Pangenome and phylogenomic analysis of the pathogenic actinobacterium Rhodococcus equi. Genome Biol. Evol. 8, 3140–3148.
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Beleneva, I. A. (2011). Incidence and characteristics of Staphylococcus aureus and Listeria monocytogenes from the Japan and South China seas. Mar. Pollut. Bull. 62, 382–387. doi: 10.1016/j.marpolbul.2010.09.024
Bens, C. C., Voss, A., and Klaassen, C. H. (2006). Presence of a novel DNA methylation enzyme in methicillin-resistant Staphylococcus aureus isolates associated with pig farming leads to uninterpretable results in standard pulsed-field gel electrophoresis analysis. J. Clin. Microbiol. 44, 1875–1876.
Bielejec, F., Baele, G., Vrancken, B., Suchard, M. A., Rambaut, A., and Lemey, P. (2016). SpreaD3: interactive visualization of spatiotemporal history and trait evolutionary processes. Mol. Biol. Evol. 33, 2167–2169. doi: 10.1093/molbev/msw082
Bos, M. E., Verstappen, K. M., van Cleef, B. A., Dohmen, W., Dorado-Garcia, A., Graveland, H., et al. (2016). Transmission through air as a possible route of exposure for MRSA. J. Exp. Sci. Environ. Epidemiol. 26, 263–269. doi: 10.1038/jes.2014.85
Chen, C. J., Unger, C., Hoffmann, W., Lindsay, J. A., Huang, Y. C., and Gotz, F. (2013). Characterization and comparison of 2 distinct epidemic community-associated methicillin-resistant Staphylococcus aureus clones of ST59 lineage. PLoS One 8:e63210. doi: 10.1371/journal.pone.0063210
Cheng, V. C. C., Wong, S. C., Cao, H., Chen, J. H. K., So, S. Y. C., Wong, S. C. Y., et al. (2019). Whole-genome sequencing data-based modeling for the investigation of an outbreak of community-associated methicillin-resistant Staphylococcus aureus in a neonatal intensive care unit in Hong Kong. Eur. J. Clin. Microbiol. Infect. Dis. 38, 563–573. doi: 10.1007/s10096-018-03458-y
Chuang, Y. Y., and Huang, Y. C. (2013). Molecular epidemiology of community-associated meticillin-resistant Staphylococcus aureus in Asia. Lancet Infect. Dis. 13, 698–708.
Croucher, N. J., Page, A. J., Connor, T. R., Delaney, A. J., Keane, J. A., Bentley, S. D., et al. (2015). Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucleic Acids Res. 43, e15. doi: 10.1093/nar/gku1196
Cui, S., Li, J., Hu, C., Jin, S., Li, F., Guo, Y., et al. (2009). Isolation and characterization of methicillin-resistant Staphylococcus aureus from swine and workers in China. J Antimicrob Chemother 64, 680–683.
Darriba, D., Taboada, G. L., Doallo, R., and Posada, D. (2012). jModelTest 2: more models, new heuristics and parallel computing. Nat. Methods 9:772.
David, M. Z., and Daum, R. S. (2010). Community-associated methicillin-resistant Staphylococcus aureus: epidemiology and clinical consequences of an emerging epidemic. Clin. Microbiol. Rev 23, 616–687. doi: 10.1128/CMR.00081-09
Diekema, D. J., Pfaller, M. A., Schmitz, F. J., Smayevsky, J., Bell, J., Jones, R. N., et al. (2001). Survey of infections due to Staphylococcus species: frequency of occurrence and antimicrobial susceptibility of isolates collected in the United States, Canada, Latin America, Europe, and the Western Pacific region for the SENTRY Antimicrobial Surveillance Program, 1997-1999. Clin. Infect. Dis 32, (Suppl. 2), S114–S132.
Diep, B. A., Gill, S. R., Chang, R. F., Phan, T. H., Chen, J. H., Davidson, M. G., et al. (2006). Complete genome sequence of USA300, an epidemic clone of community-acquired methicillin-resistant Staphylococcus aureus. Lancet 367, 731–739.
Drummond, A. J., Suchard, M. A., Xie, D., and Rambaut, A. (2012). Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 29, 1969–1973. doi: 10.1093/molbev/mss075
Feng, Y., Chen, H. L., Chen, C. J., Chen, C. L., and Chiu, C. H. (2017). Genome comparisons of two Taiwanese community-associated methicillin-resistant Staphylococcus aureus ST59 clones support the multi-origin theory of CA-MRSA. Infect. Genet. Evol. 54, 60–65. doi: 10.1016/j.meegid.2017.06.018
Frisch, M. B., Castillo-Ramírez, S., Petit, R. A. I. I. I., Farley, M. M., Ray, S. M., Albrecht, V. S., et al. (2018). Invasive methicillin-resistant Staphylococcus aureus USA500 strains from the U.S. emerging infections program constitute three geographically distinct lineages. mSphere 3:e571-17.
Ge, B., Mukherjee, S., Hsu, C. H., Davis, J. A., Tran, T. T. T., Yang, Q., et al. (2017). MRSA and multidrug-resistant Staphylococcus aureus in U.S. retail meats, 2010-2011. Food Microbiol. 62, 289–297. doi: 10.1016/j.fm.2016.10.029
Gombas, D. E., Chen, Y., Clavero, R. S., and Scott, N. V. (2003). Survey of Listeria monocytogenes in Ready-to-Eat Foods. J. Food Protect. 66, 559–569.
Goodwin, K. D., McNay, M., Cao, Y., Ebentier, D., Madison, M., and Griffith, J. F. (2012). A multi-beach study of Staphylococcus aureus, MRSA, and enterococci in seawater and beach sand. Water Res. 46, 4195–4207. doi: 10.1016/j.watres.2012.04.001
Graña-Miraglia, L., Lozano, L. F., Velázquez, C., Volkow-Fernández, P., Pérez-Oseguera, Á, Cevallos, M. A., et al. (2017). Rapid gene turnover as a significant source of genetic variation in a recently seeded population of a healthcare-associated pathogen. Front. Microbiol. 8:1817. doi: 10.3389/fmicb.2017.01817
Henri, C., Leekitcharoenphon, P., Carleton, H. A., Radomski, N., Kaas, R. S., Mariet, J. F., et al. (2017). An Assessment of different genomic approaches for inferring phylogeny of Listeria monocytogenes. Front Microbiol 8:2351. doi: 10.3389/fmicb.2017.02351
Huang, Y. C., Ho, C. F., Chen, C. J., Su, L. H., and Lin, T. Y. (2008). Comparative molecular analysis of community-associated and healthcare-associated methicillin-resistant Staphylococcus aureus isolates from children in northern Taiwan. Clin. Microbiol. Infect. 14, 1167–1172. doi: 10.1111/j.1469-0691.2008.02115.x
Hung, W. C., Wan, T. W., Kuo, Y. C., Yamamoto, T., Tsai, J. C., Lin, Y. T., et al. (2016). Molecular evolutionary pathways toward two successful community-associated but multidrug-resistant ST59 Methicillin-resistant Staphylococcus aureus Lineages in Taiwan: dynamic modes of mobile genetic element salvages. PLoS One 11:e0162526. doi: 10.1371/journal.pone.0162526
Kwiecinski, J., Jacobsson, G., Karlsson, M., Zhu, X., Wang, W., Bremell, T., et al. (2013). Staphylokinase promotes the establishment of Staphylococcus aureus skin infections while decreasing disease severity. J. Infect. Dis. 208, 990–999. doi: 10.1093/infdis/jit288
Letunic, I., and Bork, P. (2016). Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 44, W242–W245. doi: 10.1093/nar/gkw290
Li, S., Sun, S., Yang, C., Chen, H., Yin, Y., Li, H., et al. (2018). The changing pattern of population structure of Staphylococcus aureus from Bacteremia in China from 2013 to 2016: ST239-030-MRSA replaced by ST59-t437. Front Microbiol 9:332. doi: 10.3389/fmicb.2018.00332
Liang, B., Mai, J., Liu, Y., Huang, Y., Zhong, H., Xie, Y., et al. (2018). Prevalence and characterization of Staphylococcus aureus isolated from women and children in Guangzhou China. Front. Microbiol. 9:2790. doi: 10.3389/fmicb.2018.02790
Liang, Y., Tu, C., Tan, C., El-Sayed Ahmed, M. A. E., Dai, M., Xia, Y., et al. (2019). Antimicrobial resistance, virulence genes profiling and molecular relatedness of methicillin-resistant Staphylococcus aureus strains isolated from hospitalized patients in Guangdong Province China. Infect. Drug Resist. 12, 447–459. doi: 10.2147/IDR.S192611
McAdam, P. R., Templeton, K. E., Edwards, G. F., Holden, M. T., Feil, E. J., Aanensen, D. M., et al. (2012). Molecular tracing of the emergence, adaptation, and transmission of hospital-associated methicillin-resistant Staphylococcus aureus. Proc. Natl. Acad. Sci. U.S.A. 109, 9107–9112. doi: 10.1073/pnas.1202869109
Mediavilla, J. R., Chen, L., Mathema, B., and Kreiswirth, B. N. (2012). Global epidemiology of community-associated methicillin resistant Staphylococcus aureus (CA-MRSA). Curr. Opin. Microbiol. 15, 588–595.
Moradigaravand, D., Jamrozy, D., Mostowy, R., Anderson, A., Nickerson, E. K., Thaipadungpanit, J., et al. (2017). Evolution of the Staphylococcus argenteus ST2250 clone in northeastern thailand is linked with the acquisition of livestock-associated Staphylococcal genes. mBio 8:e00802-17.
Ogata, K., Narimatsu, H., Suzuki, M., Higuchi, W., Yamamoto, T., and Taniguchi, H. (2012). Commercially distributed meat as a potential vehicle for community-acquired methicillin-resistant Staphylococcus aureus. Appl. Environ. Microbiol. 78, 2797–2802. doi: 10.1128/AEM.07470-11
Oniciuc, E. A., Ariza-Miguel, J., Bolocan, A. S., Diez-Valcarce, M., Rovira, J., Hernandez, M., et al. (2015). Foods from black market at EU border as a neglected route of potential methicillin-resistant Staphylococcus aureus transmission. Int. J. Food Microbiol. 209, 34–38. doi: 10.1016/j.ijfoodmicro.2014.11.015
Oniciuc, E.-A., Nicolau, A. I., Hernández, M., and Rodríguez-Lázaro, D. (2017). Presence of methicillin-resistant Staphylococcus aureus in the food chain. Trends Food Sci. Technol. 61, 49–59.
Orsi, R. H., den Bakker, H. C., and Wiedmann, M. (2011). Listeria monocytogenes lineages: genomics, evolution, ecology, and phenotypic characteristics. Int. J. Med. Microbiol. 301, 79–96. doi: 10.1016/j.ijmm.2010.05.002
Otto, M. (2013). Community-associated MRSA: what makes them special? Int. J. Med. Microbiol. 303, 324–330. doi: 10.1016/j.ijmm.2013.02.007
Page, A. J., Cummins, C. A., Hunt, M., Wong, V. K., Reuter, S., Holden, M. T., et al. (2015). Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693. doi: 10.1093/bioinformatics/btv421
Perez-Roth, E., Claverie-Martın, F., Villar, J., and Mendez-Alvarez, S. (2001). Multiplex PCR for simultaneous identification of Staphylococcus aureus and detection of methicillin and mupirocin resistance. J. Clin. Microbiol. 39, 4037–4041.
Petinaki, E., and Spiliopoulou, I. (2012). Methicillin-resistant Staphylococcus aureus among companion and food-chain animals: impact of human contacts. Clin. Microbiol. Infect. 18, 626–634. doi: 10.1111/j.1469-0691.2012.03881.x
Porrero, M. C., Mentaberre, G., Sanchez, S., Fernandez-Llario, P., Gomez-Barrero, S., Navarro-Gonzalez, N., et al. (2013). Methicillin resistant Staphylococcus aureus (MRSA) carriage in different free-living wild animal species in Spain. Vet. J. 198, 127–130. doi: 10.1016/j.tvjl.2013.06.004
Pritchard, L., Glover, R. H., Humphris, S., Elphinstone, J. G., and Toth, I. K. (2016). Genomics and taxonomy in diagnostics for food security: soft-rotting enterobacterial plant pathogens. Anal. Methods 8, 12–24.
Robb, A., Pennycott, T., Duncan, G., and Foster, G. (2013). Staphylococcus aureus carrying divergent mecA homologue (mecA LGA251) isolated from a free-ranging wild bird. Vet. Microbiol. 162, 300–301.
Rodriguez-Lazaro, D., Oniciuc, E. A., Garcia, P. G., Gallego, D., Fernandez-Natal, I., Dominguez-Gil, M., et al. (2017). Detection and characterization of Staphylococcus aureus and methicillin-resistant S. aureus in Foods Confiscated in EU Borders. Front. Microbiol. 8:1344. doi: 10.3389/fmicb.2017.01344
Rong, D., Wu, Q., Xu, M., Zhang, J., and Yu, S. (2017). Prevalence, virulence genes, antimicrobial susceptibility, and genetic diversity of Staphylococcus aureus from retail aquatic products in China. Front. Microbiol. 8:714. doi: 10.3389/fmicb.2017.00714
Rouli, L., Merhej, V., Fournier, P. E., and Raoult, D. (2015). The bacterial pangenome as a new tool for analysing pathogenic bacteria. New Microbes New Infect. 7, 72–85. doi: 10.1016/j.nmni.2015.06.005
Schoder, D., Strauss, A., Szakmary-Brandle, K., Stessl, B., Schlager, S., and Wagner, M. (2015). Prevalence of major foodborne pathogens in food confiscated from air passenger luggage. Int. J. Food Microbiol. 209, 3–12. doi: 10.1016/j.ijfoodmicro.2014.08.010
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Smith, T. C., Male, M. J., Harper, A. L., Kroeger, J. S., Tinkler, G. P., Moritz, E. D., et al. (2009). Methicillin-resistant Staphylococcus aureus (MRSA) strain ST398 is present in midwestern U.S. swine and swine workers. PLoS One 4:e4258. doi: 10.1371/journal.pone.0004258
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Stegger, M., Andersen, P. S., Kearns, A., Pichon, B., Holmes, M. A., Edwards, G., et al. (2012). Rapid detection, differentiation and typing of methicillin-resistant Staphylococcus aureus harbouring either mecA or the new mecA homologue mecA LGA251. Clin. Microbiol. Infect. 18, 395–400. doi: 10.1111/j.1469-0691.2011.03715.x
Strauß, L., Stegger, M., Akpaka, P. E., Alabi, A., Breurec, S., Coombs, G., et al. (2017). Origin, evolution, and global transmission of community-acquired Staphylococcus aureus ST8. Proc. Natl. Acad. Sci. U.S.A. 114, E10596–E10604. doi: 10.1073/pnas.1702472114
Tang, Y., Larsen, J., Kjeldgaard, J., Andersen, P. S., Skov, R., and Ingmer, H. (2017). Methicillin-resistant and -susceptible Staphylococcus aureus from retail meat in Denmark. Int. J. Food Microbiol. 249, 72–76.
The Clinical and Laboratory Standards Institute [CLSI], (2015). Performance Standards for Antimicrobial Susceptibility Testing; Twenty-Fifth Informational Supplement. Approved Standard-M02-A12. Wayne, PA: The Clinical and Laboratory Standards Institute.
Treangen, T. J., Ondov, B. D., Koren, S., and Phillippy, A. M. (2014). The Harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes. Genome Biol. 15:524.
Uhlemann, A. C., Dordel, J., Knox, J. R., Raven, K. E., Parkhill, J., Holden, M. T., et al. (2014). Molecular tracing of the emergence, diversification, and transmission of S. aureus sequence type 8 in a New York community. Proc. Natl. Acad. Sci. U.S.A. 111, 6738–6743. doi: 10.1073/pnas.1401006111
Uhlemann, A. C., McAdam, P. R., Sullivan, S. B., Knox, J. R., Khiabanian, H., Rabadan, R., et al. (2017). Evolutionary dynamics of pandemic methicillin-sensitive Staphylococcus aureus ST398 and its international spread via routes of human migration. mBio 8:e01375-16.
van Wamel, W. J., Rooijakkers, S. H., Ruyken, M., van Kessel, K. P., and van Strijp, J. A. (2006). The innate immune modulators staphylococcal complement inhibitor and chemotaxis inhibitory protein of Staphylococcus aureus are located on beta-hemolysin-converting bacteriophages. J. Bacteriol. 188, 1310–1315.
Wu, S., Huang, J., Zhang, F., Wu, Q., Zhang, J., Pang, R., et al. (2019). Prevalence and characterization of food-related Methicillin-Resistant Staphylococcus aureus (MRSA) in China. Front. Microbiol. 10:304.
Yang, X., Qian, S., Yao, K., Wang, L., Liu, Y., Dong, F., et al. (2017). Multiresistant ST59-SCCmec IV-t437 clone with strong biofilm-forming capacity was identified predominantly in MRSA isolated from Chinese children. BMC Infect. Dis. 17:733. doi: 10.1186/s12879-017-2833-7
Keywords: Staphylococcus aureus, molecular evolution, ST59 lineage, transmission, food chain
Citation: Pang R, Wu S, Zhang F, Huang J, Wu H, Zhang J, Li Y, Ding Y, Zhang J, Chen M, Wei X, Zhang Y, Gu Q, Zhou Z, Liang B, Li W and Wu Q (2020) The Genomic Context for the Evolution and Transmission of Community-Associated Staphylococcus aureus ST59 Through the Food Chain. Front. Microbiol. 11:422. doi: 10.3389/fmicb.2020.00422
Received: 14 November 2019; Accepted: 27 February 2020;
Published: 17 March 2020.
Edited by:
Haitao Lu, Shanghai Jiao Tong University, ChinaReviewed by:
Santiago Castillo Ramírez, National Autonomous University of Mexico, MexicoPhil Giffard, Charles Darwin University, Australia
Copyright © 2020 Pang, Wu, Zhang, Huang, Wu, Zhang, Li, Ding, Zhang, Chen, Wei, Zhang, Gu, Zhou, Liang, Li and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qingping Wu, d3VxcDIwM0AxNjMuY29t
†These authors have contributed equally to this work