 Andrés Julián Gutiérrez-Escobar1*
Andrés Julián Gutiérrez-Escobar1* Billie Velapatiño2,3
Billie Velapatiño2,3 Victor Borda4
Victor Borda4 Charles S. Rabkin1Eduardo Tarazona-Santos3,5Lilia Cabrera6Jaime Cok3Catherine C. Hooper3Helena Jahuira-Arias3Phabiola Herrera3Mehwish Noureen7,8Difei Wang1Judith Romero-Gallo9Bao Tran10Richard M. Peek Jr.9Douglas E. Berg11Robert H. Gilman12†M. Constanza Camargo1†
Charles S. Rabkin1Eduardo Tarazona-Santos3,5Lilia Cabrera6Jaime Cok3Catherine C. Hooper3Helena Jahuira-Arias3Phabiola Herrera3Mehwish Noureen7,8Difei Wang1Judith Romero-Gallo9Bao Tran10Richard M. Peek Jr.9Douglas E. Berg11Robert H. Gilman12†M. Constanza Camargo1†- 1Division of Cancer Epidemiology and Genetics, National Cancer Institute, Rockville, MD, United States
- 2Department of Pathology and Laboratory Medicine, Faculty of Medicine, The University of British Columbia, Vancouver, BC, Canada
- 3Universidad Peruana Cayetano Heredia, Lima, Peru
- 4Laboratório de Bioinformática, Laboratório Nacional de Computação Científica (LNCC/MCTIC), Petrópolis, Brazil
- 5Department of Genetics, Ecology and Evolution, Institute of Biological Sciences, Federal University of Minas Gerais, Belo Horizonte, Brazil
- 6Asociación Benéfica PRISMA, Lima, Peru
- 7National Institute of Genetics, Mishima, Japan
- 8Department of Genetics, Graduate School of Life Sciences, The Graduate University for Advanced Studies (SOKENDAI), Mishima, Japan
- 9Division of Gastroenterology, Hepatology and Nutrition, Department of Medicine, Vanderbilt University Medical Center, Nashville, TN, United States
- 10Frederick National Laboratory for Cancer Research, National Cancer Institute, Frederick, MD, United States
- 11Department of Molecular Microbiology, Washington University School of Medicine in St. Louis, St. Louis, MO, United States
- 12Department of International Health, Bloomberg School of Public Health, Johns Hopkins University, Baltimore, MD, United States
Region-specific Helicobacter pylori subpopulations have been identified. It is proposed that the hspAmerind subpopulation is being displaced from the Americans by an hpEurope population following the conquest. Our study aimed to describe the genomes and methylomes of H. pylori isolates from distinct Peruvian communities: 23 strains collected from three groups of Native Americans (Asháninkas [ASHA, n = 9], Shimaas [SHIM, n = 5] from Amazonas, and Punos from the Andean highlands [PUNO, n = 9]) and 9 modern mestizos from Lima (LIM). Closed genomes and DNA modification calls were obtained using SMRT/PacBio sequencing. We performed evolutionary analyses and evaluated genomic/epigenomic differences among strain groups. We also evaluated human genome-wide data from 74 individuals from the selected Native communities (including the 23 H. pylori strains donors) to compare host and bacterial backgrounds. There were varying degrees of hspAmerind ancestry in all strains, ranging from 7% in LIM to 99% in SHIM. We identified three H. pylori subpopulations corresponding to each of the Native groups and a novel hspEuropePeru which evolved in the modern mestizos. The divergence of the indigenous H. pylori strains recapitulated the genetic structure of Native Americans. Phylogenetic profiling showed that Orthogroups in the indigenous strains seem to have evolved differentially toward epigenomic regulation and chromosome maintenance, whereas OGs in the modern mestizo (LIM) seem to have evolved toward virulence and adherence. The prevalence of cagA+/vacA s1i1m1 genotype was similar across populations (p = 0.32): 89% in ASHA, 67% in PUNO, 56% in LIM and 40% in SHIM. Both cagA and vacA sequences showed that LIM strains were genetically differentiated (p < 0.001) as compared to indigenous strains. We identified 642 R-M systems with 39% of the associated genes located in the core genome. We found 692 methylation motifs, including 254 population-specific sequences not previously described. In Peru, hspAmerind is not extinct, with traces found even in a heavily admixed mestizo population. Notably, our study identified three new hspAmerind subpopulations, one per Native group; and a new subpopulation among mestizos that we named hspEuropePeru. This subpopulation seems to have more virulence-related elements than hspAmerind. Purifying selection driven by variable host immune response may have shaped the evolution of Peruvian subpopulations, potentially impacting disease outcomes.
Introduction
Helicobacter pylori is an ancestral member of the gastric microbiota and remains as a common cause of stomach diseases, including cancer (Khalifa et al., 2010). H. pylori has accompanied humans in their migrations and mirrored their biogeographic distributions (Falush et al., 2003; Linz et al., 2007; Yamaoka, 2009). Native Americans diverged from East Asians ∼23,000 years ago (ya) and settled in Beringia (Moreno-Mayar et al., 2018a,b). They later migrated to the Americas ∼16,000 ya via the Bering Strait, and rapidly dispersed through this vast territory (Gravel et al., 2013; Waters, 2019), arriving in South America initially in the Amazonas, and then progressively moving to Andes and Pacific coastal regions ∼12,000–15,000 ya (Gravel et al., 2013; Mendes et al., 2020).
In Peru, the Native population was exceedingly affected by Inca rules that forced the population to migrate and admix in the Andes (O’Fallon and Fehren-Schmitz, 2011; Harris et al., 2018). Immigration tended to occur toward the Amazon and coast from the Andes (Harris et al., 2018) due to high-altitude (Bigham, 2016), although gene flow in the reverse direction was also observed (Rodriguez-Delfin et al., 2000; Sandoval et al., 2013). When the Spanish conquerors invaded the territory, they imposed the assimilation rule that hindered even more other population movements (Mumford, 2012). After Peruvian independence, a considerable proportion of the Native population admixed with the Spanish to generate the modern mestizo population (Lovell, 1992; Harris et al., 2018) that still preserve a strong Amerindian ancestry, unlike other Latin American mestizos (Pereira et al., 2012; Homburger et al., 2015). A recent report found a clear differentiation between central Andean and Amazonian Native populations (Borda et al., 2020).
Phylogenetically, modern H. pylori strains are divided in six major populations according to Multilocus Sequence Typing (MLST) analysis (based on seven housekeeping genes), including hspEAsia and hpEurope; hspEAsia is further subdivided into hspMaori and hspAmerind (Falush et al., 2003; Linz et al., 2007; Yamaoka, 2009). A seminal work by Kersulyte et al. (2010) found that Shimaa strains of H. pylori (hspAmerind) from the Amazonas are derived from hspEAsia, while Lima strains intermingled with hpEurope. It is proposed that hspAmerind have been progressively displaced by hpEurope due to selection for more fit genotypes (Dominguez-Bello et al., 2008; Maldonado-Contreras et al., 2013). More recently, Thorell et al. (2017) identified several H. pylori subpopulations that rapidly evolved in the Americas during the last 500 years, but their study had a limited number of strains from the central Andes and Amazonas, and did not include samples from Peruvian modern mestizos.
Our study aimed to describe the evolution and genetic structure of genomes and methylomes of H. pylori isolates from four distinct Peruvian populations, residing in the Andes, Amazonas and urban regions. We addressed two research questions: i) Do H. pylori strains isolated from modern mestizos have an Amerindian component? and ii) Does the genetic diversity of Peruvian H. pylori populations recapitulate the genetic structure of their host human populations?
Materials and Methods
Samples
Bacterial Samples
The genomes of 32 H. pylori strains from four geographically and culturally distinct regions were fully sequenced: 9 from Amazonian Asháninkas (ASHA), 5 from Amazonian Shimaas (SHIM), 9 from Andean Puno (PUNO), and 9 from modern mestizos in Lima (LIM). The strains were isolated from gastric material collected by swallowed string (ASHA and SHIM) or tissue biopsies (PUNO and LIM). LIM strains were isolated from patients with histologically confirmed non-atrophic gastritis. All individuals provided informed consent, and the study was approved by the Human Studies Committees of Johns Hopkins University (Baltimore, MD, United States), of AB Prisma and of Universidad Peruana Cayetano Heredia (Lima, Peru).
Human Samples
We used genotyping data from 74 Native American individuals from the same study populations (Borda et al., 2020). This set included the 23 individuals from whom the H. pylori strains were isolated. Briefly, these human populations correspond to an Aymara-speaking group (n = 16) collected near the Titicaca lake shore in Puno region, and two Amazonian groups that inhabit the Amazon Yunga area and belong to the Arawakan linguistic family (Asháninkas [n = 35] and Shimaa [n = 23]). DNA samples were genotyped using the Illumina Human Omni array 2.5M. A total of 1,927,769 autosomal single nucleotide polymorphisms (SNPs) passed quality control and were combined with 1000 Genomes populations (n = 250), resulting in a dataset with 324 individuals (Supplementary Table S1).
SMRT/PacBio Sequencing, Genome Assembly, Annotation and Methylation Calls
Bacterial genomic DNA was extracted from the 32 H. pylori strains using the QIAamp DNA Minikit (QIAGEN, Hilden, Germany) and purified with QIAGEN Genomic-tip 100/G columns. The genomic DNA was sequenced using PacBio RSII at the NCI’s Frederick National Laboratory for Cancer Research following the manufacturer’s protocol to obtain complete and circular genome sequences. The de novo assembly of each genome was performed following the instructions of the hierarchical genome assembly process (HGAP), version 2.0 (Chin et al., 2013); a complete closed contig was obtained for each bacterial genome. The genomes were annotated in Prokka v1.12 software (Seemann, 2014). DNA methylation detection was performed using kinetic data from the sequencing process, and base modification detection was conducted using the protocol “RS_Modification_and_Motif_Analysis.1” from PacBio using SMRT software analysis (version 1.4.0). Each motif was analyzed on REBASE1 to find the associated restriction-modification (R-M) systems against the H. pylori gold standard DNA methylation motifs. Only methylation sites with a Phred-like quality value score of ≥50 were used for subsequent analysis. Plasmids were assembled independently into extrachromosomal elements.
Bioinformatic Analyses
We employed 95 NCBI genomes from different H. pylori populations as references (Supplementary Table S2), including the Canadian hspAmerind strains Aklavik86 and Aklavik117 that were sequenced using 454 technology (Kersulyte et al., 2010). To characterize and compare the circularized bacterial chromosomes, the following analyses were conducted: (i) phylogenomics and population structure with and without external NCBI reference sequences; (ii) population genetics analyses of the two major virulence factors vacA and cagA; (iii) methylation motif frequencies and densities. In addition, we performed a phylogenetic analysis of plasmid sequences.
Phylogenomics Analysis
Average nucleotide identity using blast (ANIb) among genomes was calculated in Pyani v0.2.7 (Richter and Rossello-Mora, 2009) and the corresponding identity scores were hierarchical clustered using Morpheus2. Phylogenomics analysis was performed by applying two approaches. First, to understand the global distribution of the Peruvian strains, we reconstructed a SNP core phylogeny using KSNP v3.0 (Gardner et al., 2015; van Vliet and Kusters, 2015). Second, the study genomes were annotated with Prokka, and then the gff files were used to inferred the pan-genome with Roary v3.7.0 (Page et al., 2015) using a blast identity of 80% and the -s option (van Vliet, 2017). Then, the gene presence/absence matrix and the core genome alignment from the Roary outputs were used as template to obtain a local phylogenomics tree among strains using Fasttree 2 (Price et al., 2009). A root-to-tip analysis using the local phylogenomics tree was determined in TempEst (Rambaut et al., 2016). The divergence times were estimated using the LSD software (To et al., 2016), applying the bacterial mutation rates reported by Moodley et al. (2012), and the human ancestral divergence times reported by the Peruvian Human Genome Project (Harris et al., 2018). In addition, we built a phylogenomics tree to compare the hspEuropePeru with hpEuropeColombia and hpEuropeNicaragua. The phylogenies were visualized using iTOL v3 software (Letunic and Bork, 2016).
Bacterial and Human Ancestry Analyses
To determine the bacterial population structure, we obtained a genome-wide co-ancestry matrix extracted from the study genomes using in silico chromosome painting. Subsequently, we used the co-ancestry matrix as input to run fineSTRUCTURE v4 for 100,000 iterations to perform model-based clustering using a Markov chain Monte Carlo as previously described (Lawson et al., 2012; Yahara et al., 2013).
The human population structure was inferred using genetic clustering analysis on ∼1.9M SNPs. We also included data from 1000 Genomes populations (Genomes Project et al., 2015) representing the subcontinental groups: West African (GWD), West Central African (YRI), East African (LWK), South European (IBS), North European (CEU), South Asian (ITU), East Asian (CDX and JPT) and two admixed Latin American populations (CLM and PEL). We performed a linkage disequilibrium pruning with PLINK (Purcell et al., 2007) and running the genetic clustering with ADMIXTURE (Alexander et al., 2009). We used a cross-validation approach to identify the best K value for the clustering. We ran admixture for K values ranging from 4 to 8 ancestral clusters.
Bacterial Genome Consensus
To identify differences in the bacterial genome architecture, we applied the approach proposed by Tada et al. (2017). Briefly, orthologs gene clusters were obtained by the bidirectional best hit method and used to create a consensus genome template. The template was aligned against each complete genome from the study population and clustered based on blast similarity scores.
Bacterial Orthogroups Determination and Population Genetics of Major Virulence Factors
Orthogroups (OGs) among H. pylori strains were identified using OrthoFinder v2.2.3 (Emms and Kelly, 2015). We used the generated gene count matrix and the local phylogenomics tree to evaluate the gain and loss patterns of OGs across all the study strains. Briefly, we applied the gain-loss-duplication model with Poisson distribution and four discrete gamma categories using Count software (Csuros, 2010). We screened all OGs to identify differential gene families either gained or lost among the groups. We defined four categories: OGs lost in all hspEuropePeru strains, OGs gain in all hspEuropePeru strains, OGs lost in most hspAmerind strains, and OGs gain in most hspAmerind strains. Functional classification of the identified families was performed by BLAST + software against the conserved domain database (Marchler-Bauer et al., 2015).
We calculated the prevalence of cagA and vacA alleles and determined the number of haplotypes (H), haplotype diversity (Hd) and nucleotide diversity (Pi) for both genes using DnaSP v6 software (Rozas et al., 2017). Then, we estimated the genetic differentiation for cagA and vacA alleles among study strains by using the nearest neighbor statistic (Snn) test with gene flow (Nm) under 1000 iterations in DnaSP v6 software (Rozas et al., 2017). Neutrality deviations were calculated by the z-tests in Kumar et al. (2016). Natural selection intensification or relaxation of cagA alleles were obtained by using the RELAX algorithm (Wertheim et al., 2015). EPIYA and CRPIA motifs were detected according to the approach by Suzuki (Suzuki et al., 2011).
Finally, using point mutations (A2142G, A2143G, and A2142C) in 23S ribosomal RNA gene, we identified resistance to clarithromycin, a core antibiotic in H. pylori eradication therapy.
Phylogenetic Analysis of Plasmids
We compared our H. pylori plasmids with 44 NCBI complete plasmid sequences (Supplementary Table S3) with lengths ranging from 5 to 25 kilobases (Kb). Phylogenetic analysis of all plasmids was performed using KSNP v3.0 (Gardner et al., 2015; van Vliet and Kusters, 2015). The phylogeny was visualized using iTOL v3 software (Letunic and Bork, 2016).
Bacterial Methylation Analysis
The presence of R-M systems: R (I, II, and III), S and M genes in the core and accessory genomes were determined using Spine, AGEnt and ClustAGE (Ozer et al., 2014; Ozer, 2018). Densities per 1 kb for total N6-methyladenine (m6A), N4-methylcytosine (m4C) and other methylation types were estimated using an in-house customized bash script (available upon request). The frequencies of motifs with at least 80% methylation fractions (n = 621) in the sequenced H. pylori genomes were visualized as a Venn diagram using a web tool3. Subsequently, shared and novel methylation motifs were identified using REBASE database against the H. pylori gold standard (Roberts et al., 2015).
Results
Overall, the genome lengths and coding sequences ranged from 1.63 to 1.65 megabases (Mb) and from 1.59 to 1.62 Mb, respectively, similar to H. pylori genomes from other populations. The ANIb analysis revealed two major groups of strains: indigenous (ASHA, SHIM, and PUNO) and modern mestizo (LIM) (Figure 1). Although the two groups presented a high sequence similarity (ANIb ≥ 91–100%), the genomes from modern mestizo were more similar to those of hpEurope. We also identified that strains from the indigenous group (ASHA-003, PUNO-003, PUNO-009, and PUNO-010) shared components with the modern mestizo group, and one strain from the modern mestizo group (LIM-007) clustered with the indigenous (Figure 1). These five genomes represent heavily admixed strains.
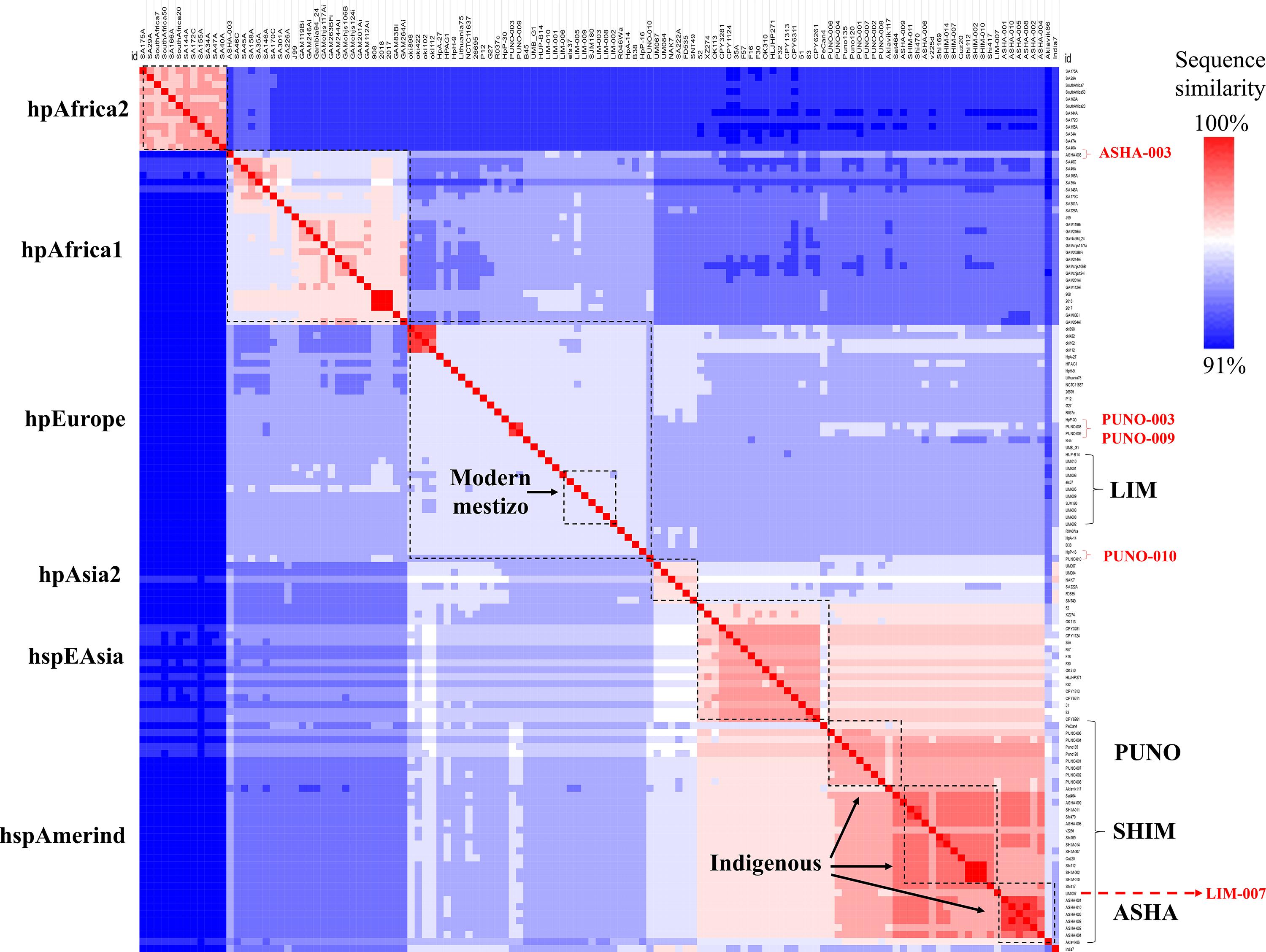
Figure 1. Hierarchical clustering analysis of ANIb values. Each line represents the similarity score of each H. pylori genome (32 study and 95 references). Left, ancestral H. pylori populations. Right, H. pylori study populations. Heavily admixed genomes (ASHA-003, PUNO-003, PUNO-009, PUNO-010, and LIM-007) are shown in red font.
The core phylogenomic tree constructed from a total of 930,403 SNPs with a K value of 29 showed that all indigenous strains were grouped into independent clusters that define the hspAmerind subpopulation located next to the hspEAsia population. The modern mestizo strains were located near to the hpEurope population (Figure 2A). The divergence time estimates for all indigenous strains (ASHA, SHIM, and PUNO) was ∼13,512–9,000 ya. The ancestry analysis confirmed that the indigenous group was composed by three hspAmerind-like subclades. The hspAmerind ancestry component varied by population: ASHA ranged from 13 to 86%, SHIM from 90 to 99%, and PUNO from 15 to 68%. Except for LIM-007, with 64% of hspAmerind ancestry, all other LIM strains had less than 15% hspAmerind ancestry and were classified as hspEuropePeru. This new subpopulation is different from hpEuropeNicaragua and hpEuropeColombia (Supplementary Figure S1). Interestingly, we observed that ASHA-003 had a 50% of hpAfrica1 ancestry, while PUNO-003 and PUNO-009, 46 and 47% of hpEurope ancestry, respectively (Supplementary Table S4). The human ancestry analysis also showed that the Central Andean population (Puno) is differentiated from the Amazon populations (Shimaa and Asháninkas) (Figures 2B,C). Supplementary Figure S2 shows the evolution of the human clusters; K = 4 identified the four continental populations (Africa, Europe, Asian, and Native American), while K = 8 further discriminate the three African populations as Native American groups have higher genetic drift levels than Africans. A cross-validation approach identified K = 6 as the best value for the clustering.
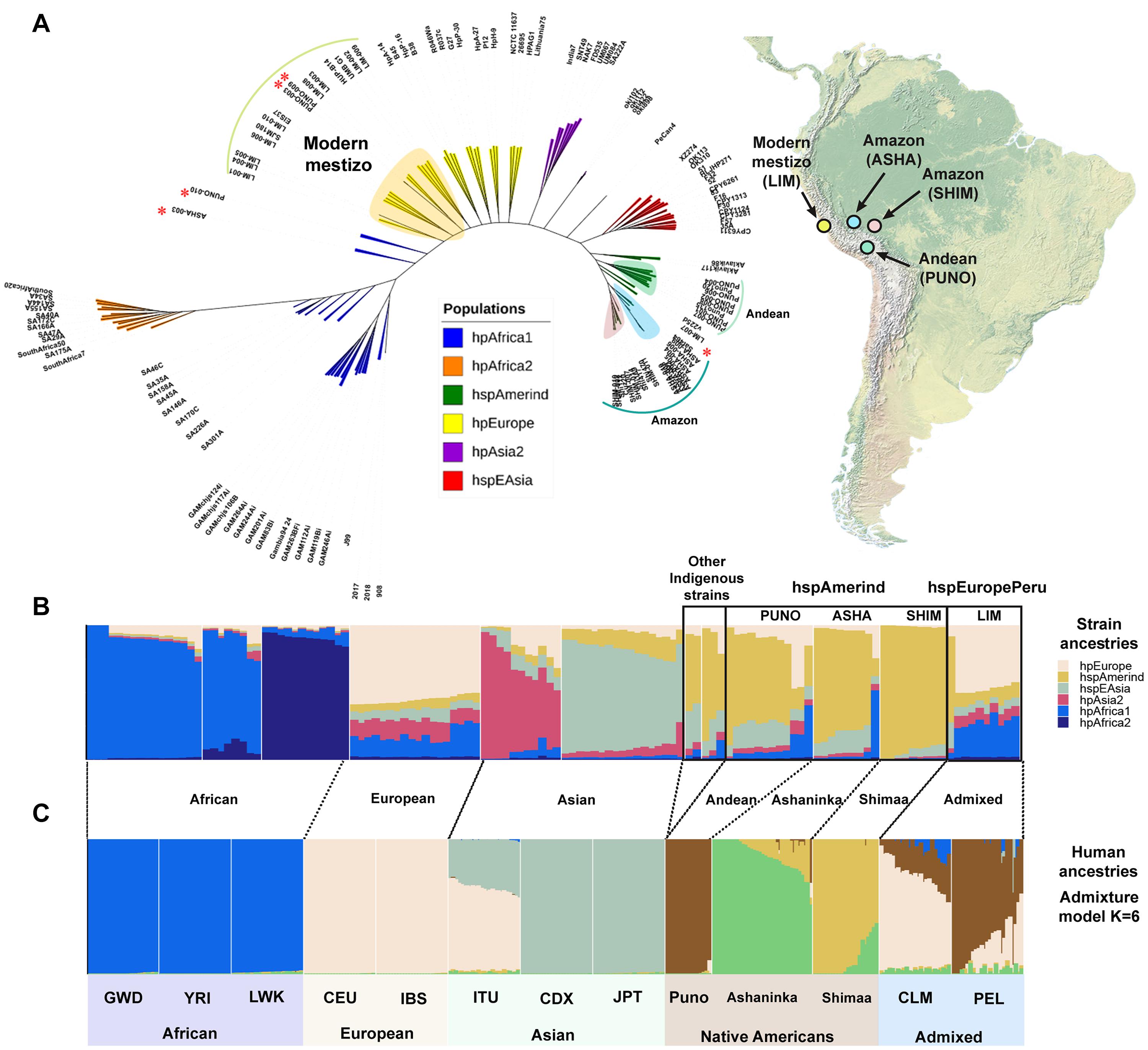
Figure 2. Phylogenomic relationships and population structures of H. pylori and human populations. (A) Global phylogenomic tree for 127 H. pylori strains. The tree was constructed from a total of 930,403 SNPs with a K value of 29 using KSNPV3.0. The shading blue (ASHA) and red (SHIM) are the strains obtained from the Amazon, the light green (PUNO) are the strains isolated from Puno in the Andes, and the light yellow (LIM) represent the strains isolated from Lima. The Peruvian map shows the regions where the samples were collected. The asterisks represent the heavily admixed strains (ASHA-003, PUNO-003, PUNO-009, PUNO-010, and LIM-007). (B) Ancestry profiles inferred using Chromopainter v2/fineSTRUCTURE for 127 H. pylori strains (32 study and 95 references; Supplementary Table S4). (C) ADMIXTURE results for 10 human populations from 1000 Genomes (n = 250) and Borda et al. (2020) (n = 74; Supplementary Table S1). We plot admixture results for the K value with the lowest cross validation error (K = 6; Supplementary Figure S2).
The bacterial consensus analysis using complete genomes showed that the indigenous group (ASHA, SHIM, and PUNO) have a homogeneous genomic architecture except for a few small insertions, transpositions and deletions. Likewise, although the indigenous group and modern mestizo strains share some rearrangements, the latter appeared to be more similar to the hpEurope strains (Figure 3).
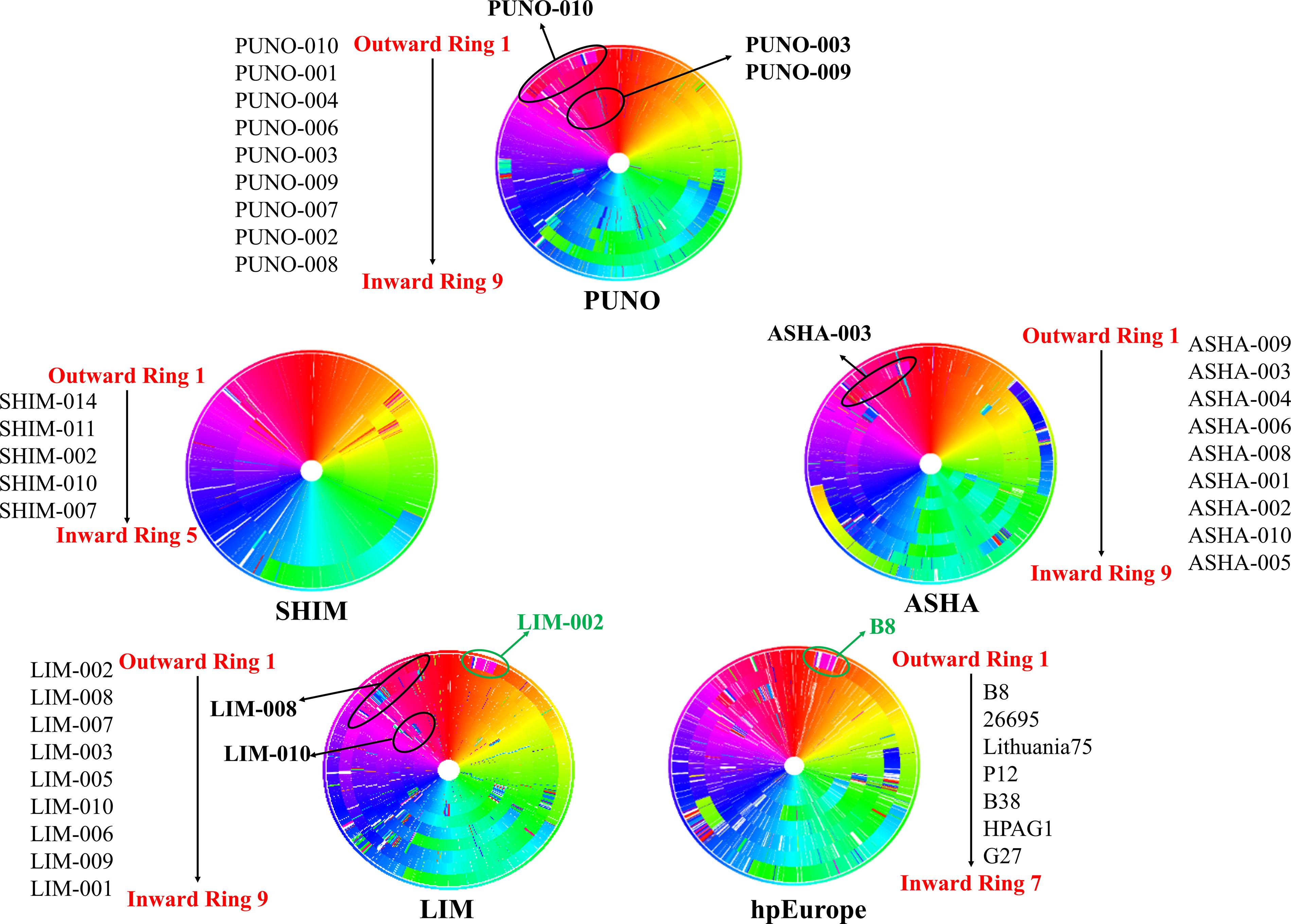
Figure 3. Genomic consensus of the study populations and hpEurope references. These circular views were obtained using the method developed by Tada et al. (2017), which creates a consensus genome that is used as a template for alignment. Each ring represents one complete genome and each block in the ring represents a genomic region. Different colors represent the genes according to their genomic position in the consensus. The shifts in the color represent the rearrangements. The outermost ring is the distant genome from the consensus. The names alongside each circle indicate the genomes going from outward to inward direction. The indicated areas (black circles) in the ASHA (ring 2) and PUNO (rings 1, 5, and 6) genomes show the regions in which these genomes are similar to the LIM (rings 2 and 6). The green circles indicate the similarities between the LIM genomes and the hpEurope references. The genomes ASHA-003, PUNO-003, PUNO-009, and PUNO-010 had an inversion from 10 o’clock to 11 o’clock which was not observed in SHIM genomes.
The pangenome analysis indicates the core (i.e., >99% sequence similarity) genome contained 1,238 genes. Likewise, we also identified soft (i.e., 95 to 99%), shell (i.e., 15 to 94%), and cloud (i.e., <15%) genes (18, 375, and 346, respectively) that integrated a 1,996 pangenome. The genes were further clustered into 1,819 OGs that accounted for 99.4% of the genes. Table 1 shows the OGs by the evolutionary patterns of gene gain and loss based on a phylogenetic profiling. In general, OGs in the indigenous strains seem to have evolved differentially toward epigenomic regulation and chromosome maintenance, whereas OGs in the modern mestizo (LIM) seem to have evolved toward virulence, adherence, and phage protection. Supplementary Figure S3 shows examples of the gain and loss of OGs among study strains.

Table 1. Gain and loss patterns of orthogroup in hspAmerind and hspEuropePeru strains.
The prevalence of the combination cagA+/vacA s1i1m1 genotype was similar across populations (p = 0.32; Fischer’s exact): 89% in ASHA, 67% in PUNO, and 56% in LIM and 40% in SHIM (Supplementary Table S5). For both cagA+ and vacA s1i1m1 genes, Pi was considerably low, with overall values of 0.083 for cagA and 0.089 for vacA. Both genes also showed high Hd, with population averages of 0.994 for cagA and 0.987 for vacA. For both cagA and vacA, the snn tests showed that LIM was genetically differentiated (p < 0.001) as compared to the indigenous strains. Gene flow was also low (Nm 0.70 for cagA and 1.74 for vacA) and indicated a limited genetic exchange among populations. The z-tests showed signals of balancing and purifying selection for both virulence factors (Table 2). We found that the test RELAX showed significant results for selection intensification (k = 1.15, p = 0.037, and LR = 4.37). As expected, all study strains contain the EPIYA-ABC motif (Supplementary Figure S4). We found the AM-CRPIA motif in 70% (16/23) of indigenous strains and the W-CRPIA motif in all mestizo strains.

Table 2. Population and evolutionary statistics for cagA and vacA. Gdiv, genetic diversity estimators; n, number of sequences; S, number of segregating sites; h, number of haplotypes; Hd, haplotype diversity; K, average number of differences; Pi, Nucleotide diversity.
Regarding the cag pathogenicity island (cagpai), the average number of genes among the 27 H. pylori genomes with this genomic region was 33 for all indigenous populations combined and 38 for mestizos. DNA alignments of cagpai sequence showed that SHIM strains were very similar with a small difference in length, with no insertions, inversions, transpositions or deletions. ASHA and PUNO shared similar patterns of inversions and transpositions, but ASHA showed more variability in sequence length than PUNO. On the other hand, LIM showed the highest inter and intra sequence complexity with many insertions, deletions and transpositions (Supplementary Figure S5).
We determined that one (LIM-003; A2142G mutation) of the 9 LIM strains could be classified as clarithromycin resistant, while no mutations were found in the 23 indigenous H. pylori strains.
We identified a total of 642 R-M systems among the four populations with 39% of them located in the core genome and 61% in the accessory. The average number of R-M genes by population was 174, the lowest found in SHIM strains with 108, and the highest in LIM strains with 198. Type I M, type I S and type III R genes were present in both the core and accessory genomes. The average number of core genes of type I M, type I S, and type III R genes were 39, 15, and 8 in the core genome and 37, 12, and 7 in the accessory genome, respectively (Figure 4A). In contrast, we found that the type I R, type II R, and DNMT1 genes were only present in the accessory genome; including duplications, the 32 strains had averages of 14, 13, and 17 genes of these types, respectively.
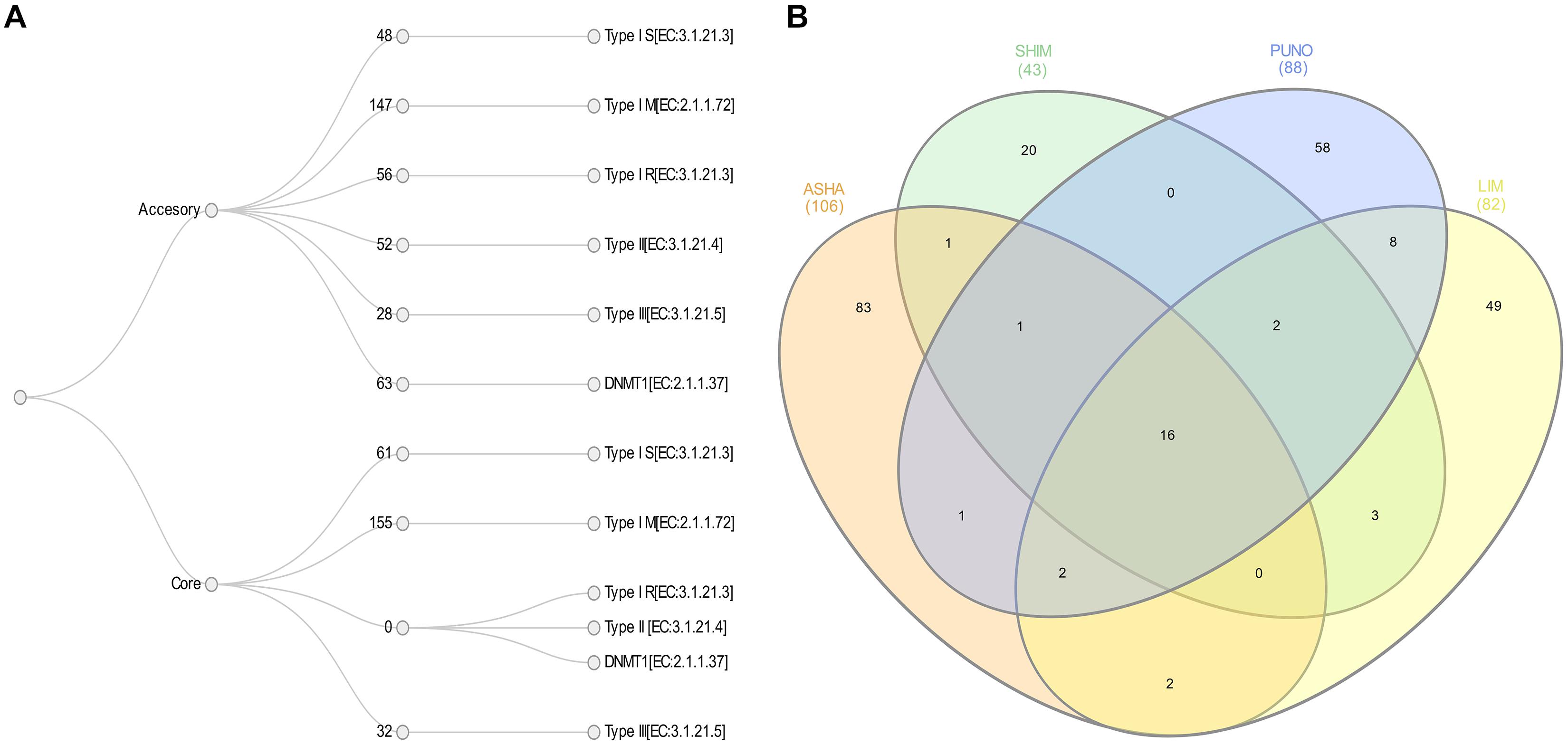
Figure 4. (A) Restriction-modification systems content in core and accessory H. pylori genomes from indigenous (9 ASHA, 5 SHIM, and 9 PUNO) and mestizos (9 LIM). (B) Venn diagram of methylation motifs with at least 80% methylation fractions.
The average methylation densities for m6A motifs per kb were 42 for ASHA and 39 for the other SHIM, PUNO and LIM; whereas for m4C motifs were 6 for ASHA, 8 for PUNO and LIM and 10 for SHIM. We found a total of 692 methylation motifs (with ≥80% of methylated sites) in the 32 genomes, including 254 novel motif sequences. Only 16 motifs were present in all four populations (at least one strain). There were no significant differences in the average motif number by population (22 for ASHA, 17 for SHIM, 21 for PUNO, and 22 for LIM) or in the average number of unique motifs (9 in ASHA, 4 in SHIM, 6 in PUNO, and 5 in LIM) (Figures 4A,B).
Among 32 genomes, we identified five (15.7%) plasmids, ASHA-003, ASHA-006, LIM-002, LIM-003, and LIM-005. Considering study and NCBI (n = 44) plasmids, lengths and GC content ranged from 5 to 25 Kb and from 31.7 to 37.5%, respectively. All plasmid sequences shared 2,916 common SNPs, 561 of which were homoplastic. The SNP phylogeny revealed five major clades showing a mixture of H. pylori populations and subpopulations as follows: (i) hpAsia2/hspEAsia, (ii) hpAsia2, (iii) hspEAsia/hspAmerind, (iv) hpEurope/hpAsia2/hpAfrica2, and (v) hspAfrica1/hpEurope/HpAsia2 (Supplementary Table S4 and Supplementary Figure S6).
Discussion
Based on MLST analysis, it is proposed that the hspAmerind subpopulation has been progressively displaced by the hpEurope population. We used a cutting-edge sequencing technology to describe the genomic and epigenomic microevolution of H. pylori isolates from Peruvian populations. We found that hspAmerind is present in Native Americans and even traces are observed in modern mestizos.
Our findings suggest that hspAmerind-like populations in Peru may have evolved by a founder effect, following the divergence between the human Central-Southern Andean and Amazon populations (Borda et al., 2020). We found that the H. pylori divergence estimates dates follow along with the human divergence timing dates (Rodriguez-Delfin et al., 2000; Rothhammer and Dillehay, 2009; Gravel et al., 2013; Sandoval et al., 2013; Harris et al., 2018), and that the three hspAmerind-like populations had followed the genetic structure of their corresponding Amerindian human populations. It is of interest to determine the minimum rate of evolution at which new H. pylori subpopulations emerge and its determinants.
The genome consensus and ancestry analyses showed that indigenous and modern mestizo strains shared not only some rearrangements but also ∼15% of common ancestry, suggesting that modern mestizo strains still retain a significant hspAmerind component. However, modern mestizo strains have transitioned toward an hpEurope-like subpopulation and have been subject to a more aggressive genome erosion than the indigenous strains. Our results confirmed that the indigenous group is composed by a set of three well-differentiated hspAmerind-like subpopulations (SHIM, ASHA, and PUNO) that support the idea that hspAmerind-like subpopulations are present even in urbanized cities (Puno) that were affected by the Spanish conquerors. Complementary, modern mestizo strains were assigned to the hpEurope population, suggesting that their demography was recently shaped due to the introduction of new genetic material after the conquest in early 1,500s (Dufour and Piperata, 2004; Homburger et al., 2015; Adhikari et al., 2016; Mendes et al., 2020). Thus, following the nomenclature convention proposed by Thorell et al. (2017), we named this new subpopulation hspEuropePeru.
Our data suggest that hspEuropePeru and hspAmerind-like subpopulations seem to have evolved different gene content repertoires with potential phenotypic consequences. The following examples illustrate previous supporting evidence for the importance of some of the identified OGs. Kojima and Kobayashi (2015) found that in hspAmerind strains, the pab1 restriction endonuclease gene was replaced by the hrgC (encoding a potential toxin) before its divergence from the hspEastAsia. In agreement, we found that the hspAmerind-like subpopulations (including LIM-007) have a copy of the hrgC, while the hspEuropePeru subpopulation have a copy of the pab1. Remarkably, our data suggest that the pab1 was recently acquired by the hspEuropePeru subpopulation as a result of the human admixture with the conquerors. We also observed that hspEuropePeru contains the AbiEii system that is involved in phage-infected cell abortion (Dy et al., 2014), the fecA2 that is associated with iron metabolism (van Vliet, 2017), and the tonB nickel transporter gene that is important for the stomach colonization (Schauer et al., 2007). In contrast, we found that most hspAmerind have lost sabA that encodes a sialic acid-binding adhesion protein with an important function on H. pylori infection chronicity (Mahdavi et al., 2002), contributing to its virulence. Future studies are warranted to replicate our findings and further characterize and understand the potential selective advantage of the modern mestizo H. pylori strains.
Related to major virulence factors, we found that the hspEuropePeru had a Western CagA type, whereas the hspAmerind-like carried a less virulent Amerindian type. As an expansion of our previous work (Kersulyte et al., 2010), we showed that cagA has diversified into a set of well-differentiated alleles that may represent a response against host immune challenge. It seems that the hspAmerind-like subpopulations optimized their co-evolutionary balance with the indigenous host. On the other hand, hspEuropePeru is still under the evolutionary arms race with its host following a red queen pattern (Morran et al., 2011; Defraine et al., 2018) that may have induced the evolution of a more aggressive bacterial phenotype.
Helicobacter pylori has a massive R-M system repertoire (Krebes et al., 2014) that continues to be revealed by technological advances. Our results suggest that ∼10% of the genome encodes R-M systems. Notably, the type I and II R-M systems were located exclusively in the accessory genome, supporting the hypothesis that restriction enzymes may be part of a bacterial defensive network that contribute to lineage homogenization (Sneppen et al., 2015; Oliveira et al., 2016). Likewise, we found that overall, 1/3 of methylation motifs were population-specific with no previous report in REBASE. There is not a universal motif across our 32 methylomes; only 2.4% (15 motifs) were present in all four population at least in one strain. The high diversity observed in population-specific methylation motifs suggests a reduction of gene transfer among populations with different motifs set, but also points toward the existence of specific gene fluxes among populations with the same motif repertoire (Oliveira et al., 2016). This diversity implies that each population was subject to a very intense diversifying population-specific selection that shaped its methylomes contributing to the geographic differentiation observed among the bacterial subpopulations in Peru (Xu et al., 2000; Kobayashi, 2001; Vale et al., 2009). Functional validation of identified R-M systems is critical as some methylation motifs may be spurious.
Plasmids are key extrachromosomal elements that not only provide novel functions to bacterial cells (i.e., antibiotic resistance), but also, they can increase the mutation rate and fitness (Hulter et al., 2017). Unlike the bacterial core-genome, the phylogenetic tree of plasmids is characterized by the presence of clades with mixed populations. Although, we identified a set of core SNPs shared by all plasmids suggesting a common ancestor, the lack of phylogeographic discrimination might be a consequence of the limited number of homoplastic traits that emerged independently in the mixed clades by convergent evolution. Plasmid diversity may reflect deep roots of evolutionary history of H. pylori. However, for a full characterization of plasmid diversity and evolution of H. pylori, large-scale studies in diverse populations are needed.
In conclusion, our study describes the evolution of hspAmerind and hspEuropePeru subpopulations from a larger ecological perspective, sampling individuals from different isolated communities. Both hspAmerind-like and hspEuropePeru subpopulations shared a significant common ancestry. We identified three hspAmerind-like subpopulations in Peru, one of them identified in Puno, a colonial city heavily impacted by the Spanish conquest. Also, we found that hspEuropePeru locally evolved in the modern mestizo. All subpopulations presented a very diverse methylome characterized by its population-specific motif repertoire. While our study adds to the understanding of the H. pylori admixture, further studies should address this phenomenon in other human communities with complex and recent migration patterns. We speculate that immune selection and lineage homogenization due to the bacterial R-M defensive system may be the force forging the evolution of H. pylori subpopulations not only in Peru but also in the Americas, and might help explaining the variable clinical outcomes associated with chronic H. pylori infection.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author Contributions
AJG-E, ET-S, DEB, RHG, and MCC: study concept and design. BV, VB, ET-S, LC, JC, CCH, HJ-A, PH, JR-G, BT, RP, and DEB: acquisition of data. AJG-E, VB, CSR, ET-S, MN, DW, DEB, RG, and MCC: analysis and interpretation of data. AJG-E, MH, DW, and VB: statistics and bioinformatics. AJG-E, CSR, DW, and MCC: drafting of the manuscript. CSR, RHG, and MCC: obtained funding. RHG and MCC: study supervision. All authors: critical revision of the manuscript for important intellectual content. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the Extramural (Grants numbers, R01 DK 58587, R01 CA 77955, P01 CA 116087, and P30 DK 058404) and Intramural Research Program of the U.S. National Cancer Institute. ET-S was supported by the Brazilian National Council for Scientific and Technological Development (CNPq) and the Department of Science and Technology of the Brazilian Ministry of Health (MS-DECIT).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We gratefully acknowledge laboratory assistance from Castle Raley, Xiongfong Chen, Bailey Kessing, and Yongmei Zhao from the National Cancer Institute’s Center for Cancer Research at the Frederick National Laboratory for Cancer Research. We also acknowledge expert advice on the ancestry analysis provided by Roberto Torres from the Mexican Institute of Social Security.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.601839/full#supplementary-material
Footnotes
- ^ http://rebase.neb.com/rebase/rebase.html
- ^ https://software.broadinstitute.org/morpheus/
- ^ http://bioinformatics.psb.ugent.be/webtools/Venn
References
Adhikari, K., Mendoza-Revilla, J., Chacon-Duque, J. C., Fuentes-Guajardo, M., and Ruiz-Linares, A. (2016). Admixture in Latin America. Curr. Opin. Genet. Dev. 41, 106–114. doi: 10.1016/j.gde.2016.09.003
Alexander, D. H., Novembre, J., and Lange, K. (2009). Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664. doi: 10.1101/gr.094052.109
Bigham, A. W. (2016). Genetics of human origin and evolution: high-altitude adaptations. Curr. Opin. Genet. Dev. 41, 8–13. doi: 10.1016/j.gde.2016.06.018
Borda, V., Alvim, I., Aquino, M. M., Silva, C., Soares-Souza, G. B., Leal, T. P., et al. (2020). The genetic structure and adaptation of Andean highlanders and Amazonian dwellers is influenced by the interplay between geography and culture. bioRxiv [Preprint]. doi: 10.1101/2020.01.30.916270
Chin, C. S., Alexander, D. H., Marks, P., Klammer, A. A., Drake, J., Heiner, C., et al. (2013). Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 10, 563–569. doi: 10.1038/nmeth.2474
Csuros, M. (2010). Count: evolutionary analysis of phylogenetic profiles with parsimony and likelihood. Bioinformatics 26, 1910–1912. doi: 10.1093/bioinformatics/btq315
Defraine, V., Fauvart, M., and Michiels, J. (2018). Fighting bacterial persistence: current and emerging anti-persister strategies and therapeutics. Drug Resist. Updat. 38, 12–26. doi: 10.1016/j.drup.2018.03.002
Dominguez-Bello, M. G., Perez, M. E., Bortolini, M. C., Salzano, F. M., Pericchi, L. R., Zambrano-Guzman, O., et al. (2008). Amerindian Helicobacter pylori strains go extinct, as european strains expand their host range. PLoS One 3:e3307. doi: 10.1371/journal.pone.0003307
Dufour, D. L., and Piperata, B. A. (2004). Rural-to-urban migration in Latin America: an update and thoughts on the model. Am. J. Hum. Biol. 16, 395–404. doi: 10.1002/ajhb.20043
Dy, R. L., Przybilski, R., Semeijn, K., Salmond, G. P. C., and Fineran, P. C. (2014). A widespread bacteriophage abortive infection system functions through a Type IV toxin-antitoxin mechanism. Nucleic Acids Res. 42, 4590–4605. doi: 10.1093/nar/gkt1419
Emms, D. M., and Kelly, S. (2015). OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 16:157.
Falush, D., Wirth, T., Linz, B., Pritchard, J. K., Stephens, M., Kidd, M., et al. (2003). Traces of human migrations in Helicobacter pylori populations. Science 299, 1582–1585. doi: 10.1126/science.1080857
Gardner, S. N., Slezak, T., and Hall, B. G. (2015). kSNP3.0: SNP detection and phylogenetic analysis of genomes without genome alignment or reference genome. Bioinformatics 31, 2877–2878. doi: 10.1093/bioinformatics/btv271
Genomes Project, C., Auton, A., Brooks, L. D., Durbin, R. M., Garrison, E. P., Kang, H. M., et al. (2015). A global reference for human genetic variation. Nature 526, 68–74. doi: 10.1038/nature15393
Gravel, S., Zakharia, F., Moreno-Estrada, A., Byrnes, J. K., Muzzio, M., Rodriguez-Flores, J. L., et al. (2013). Reconstructing native American migrations from whole-genome and whole-exome data. PLoS Genet. 9:e1004023. doi: 10.1371/journal.pgen.1004023
Harris, D. N., Song, W., Shetty, A. C., Levano, K. S., Caceres, O., Padilla, C., et al. (2018). Evolutionary genomic dynamics of Peruvians before, during, and after the Inca Empire. Proc. Natl. Acad. Sci. U.S.A. 115, E6526–E6535. doi: 10.1073/pnas.1720798115
Homburger, J. R., Moreno-Estrada, A., Gignoux, C. R., Nelson, D., Sanchez, E., Ortiz-Tello, P., et al. (2015). Genomic insights into the ancestry and demographic history of South America. PLoS Genet. 11:e1005602. doi: 10.1371/journal.pgen.1005602
Hulter, N., Ilhan, J., Wein, T., Kadibalban, A. S., Hammerschmidt, K., and Dagan, T. (2017). An evolutionary perspective on plasmid lifestyle modes. Curr. Opin. Microbiol. 38, 74–80. doi: 10.1016/j.mib.2017.05.001
Kersulyte, D., Kalia, A., Gilman, R. H., Mendez, M., Herrera, P., Cabrera, L., et al. (2010). Helicobacter pylori from Peruvian Amerindians: traces of human migrations in strains from remote amazon, and genome sequence of an Amerind strain. PLoS One 5:e15076. doi: 10.1371/journal.pone.0015076
Khalifa, M. M., Sharaf, R. R., and Aziz, R. K. (2010). Helicobacter pylori: a poor man’s gut pathogen? Gut Pathog. 2:2. doi: 10.1186/1757-4749-2-2
Kobayashi, I. (2001). Behavior of restriction-modification systems as selfish mobile elements and their impact on genome evolution. Nucleic Acids Res. 29, 3742–3756. doi: 10.1093/nar/29.18.3742
Kojima, K. K., and Kobayashi, I. (2015). Transmission of the PabI family of restriction DNA glycosylase genes: mobility and long-term inheritance. BMC Genomics 16:817. doi: 10.1186/s12864-015-2021-3
Krebes, J., Morgan, R. D., Bunk, B., Sproer, C., Luong, K., Parusel, R., et al. (2014). The complex methylome of the human gastric pathogen Helicobacter pylori. Nucleic Acids Res. 42, 2415–2432. doi: 10.1093/nar/gkt1201
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for Bigger Datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Lawson, D. J., Hellenthal, G., Myers, S., and Falush, D. (2012). Inference of population structure using dense haplotype data. PLoS Genet. 8:e1002453. doi: 10.1371/journal.pgen.1002453
Letunic, I., and Bork, P. (2016). Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 44, W242–W245. doi: 10.1093/nar/gkw290
Linz, B., Balloux, F., Moodley, Y., Manica, A., Liu, H., Roumagnac, P., et al. (2007). An African origin for the intimate association between humans and Helicobacter pylori. Nature 445, 915–918. doi: 10.1038/nature05562
Lovell, W. G. (1992). Heavy shadows and black night - disease and depopulation in colonial Spanish-America. Ann. Assoc. Am. Geogr. 82, 426–443. doi: 10.1111/j.1467-8306.1992.tb01968.x
Mahdavi, J., Sonden, B., Hurtig, M., Olfat, F. O., Forsberg, L., Roche, N., et al. (2002). Helicobacter pylori SabA adhesin in persistent infection and chronic inflammation. Science 297, 573–578. doi: 10.1126/science.1069076
Maldonado-Contreras, A., Mane, S. P., Zhang, X. S., Pericchi, L., Alarcon, T., Contreras, M., et al. (2013). Phylogeographic evidence of cognate recognition site patterns and transformation efficiency differences in H. pylori: theory of strain dominance. BMC Microbiol 13:211. doi: 10.1186/1471-2180-13-211
Marchler-Bauer, A., Derbyshire, M. K., Gonzales, N. R., Lu, S., Chitsaz, F., Geer, L. Y., et al. (2015). CDD: NCBI’s conserved domain database. Nucleic Acids Res. 43, D222–D226. doi: 10.1093/nar/gku1221
Mendes, M., Alvim, I., Borda, V., and Tarazona-Santos, E. (2020). The history behind the mosaic of the Americas. Curr. Opin. Genet. Dev. 62, 72–77. doi: 10.1016/j.gde.2020.06.007
Moodley, Y., Linz, B., Bond, R. P., Nieuwoudt, M., Soodyall, H., Schlebusch, C. M., et al. (2012). Age of the association between Helicobacter pylori and man. PLoS Pathog. 8:e1002693. doi: 10.1371/journal.ppat.1002693
Moreno-Mayar, J. V., Potter, B. A., Vinner, L., Steinrucken, M., Rasmussen, S., Terhorst, J., et al. (2018a). Terminal Pleistocene Alaskan genome reveals first founding population of Native Americans. Nature 553, 203–207. doi: 10.1038/nature25173
Moreno-Mayar, J. V., Vinner, L., Damgaard, P. D., de la Fuente, C., Chan, J., Spence, J. P., et al. (2018b). Early human dispersals within the Americas. Science 362:eaav2621. doi: 10.1126/science.aav2621
Morran, L. T., Schmidt, O. G., Gelarden, I. A., Parrish, R. C. II, and Lively, C. M. (2011). Running with the Red Queen: host-parasite coevolution selects for biparental sex. Science 333, 216–218. doi: 10.1126/science.1206360
Mumford, J. (2012). Vertical Empire: The General Resettlement of Indians in the Colonial Andes. Durham, StateNC: Duke University Press Books.
O’Fallon, B. D., and Fehren-Schmitz, L. (2011). Native Americans experienced a strong population bottleneck coincident with European contact. Proc. Natl. Acad. Sci. U.S.A. 108, 20444–20448. doi: 10.1073/pnas.1112563108
Oliveira, P. H., Touchon, M., and Rocha, E. P. (2016). Regulation of genetic flux between bacteria by restriction-modification systems. Proc. Natl. Acad. Sci. U.S.A. 113, 5658–5663. doi: 10.1073/pnas.1603257113
Ozer, E. A. (2018). ClustAGE: a tool for clustering and distribution analysis of bacterial accessory genomic elements. BMC Bioinformatics 19:150. doi: 10.1186/s12859-018-2154-x
Ozer, E. A., Allen, J. P., and Hauser, A. R. (2014). Characterization of the core and accessory genomes of Pseudomonas aeruginosa using bioinformatic tools Spine and AGEnt. BMC Genomics 15:737. doi: 10.1186/1471-2164-15-737
Page, A. J., Cummins, C. A., Hunt, M., Wong, V. K., Reuter, S., Holden, M. T., et al. (2015). Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693. doi: 10.1093/bioinformatics/btv421
Pereira, L., Zamudio, R., Soares-Souza, G., Herrera, P., Cabrera, L., Hooper, C. C., et al. (2012). Socioeconomic and nutritional factors account for the association of gastric cancer with Amerindian ancestry in a Latin American admixed population. PLoS One 7:e41200. doi: 10.1371/journal.pone.0041200
Price, M. N., Dehal, P. S., and Arkin, A. P. (2009). FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 26, 1641–1650. doi: 10.1093/molbev/msp077
Purcell, S., Neale, B., Todd-Brown, K., Thomas, L., Ferreira, M. A., Bender, D., et al. (2007). PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575. doi: 10.1086/519795
Rambaut, A., Lam, T. T., Carvalho, L. M., and Pybus, O. G. (2016). Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2:vew007. doi: 10.1093/ve/vew007
Richter, M., and Rossello-Mora, R. (2009). Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. U.S.A. 106, 19126–19131. doi: 10.1073/pnas.0906412106
Roberts, R. J., Vincze, T., Posfai, J., and Macelis, D. (2015). REBASE-a database for DNA restriction and modification: enzymes, genes and genomes. Nucleic Acids Res. 43, D298–D299. doi: 10.1093/nar/gku1046
Rodriguez-Delfin, L. A., Rubin-de-Celis, V. E., and Zago, M. A. (2000). Genetic diversity in an Andean population from country-regionPeru and regional migration patterns of Amerindians in South America: data from Y chromosome and mitochondrial DNA. Hum. Hered. 51, 97–106. doi: 10.1159/000022964
Rothhammer, F., and Dillehay, T. D. (2009). The late pleistocene colonization of South America: an interdisciplinary perspective. Ann. Hum. Genet. 73, 540–549. doi: 10.1111/j.1469-1809.2009.00537.x
Rozas, J., Ferrer-Mata, A., Sanchez-DelBarrio, J. C., Guirao-Rico, S., Librado, P., Ramos-Onsins, S. E., et al. (2017). DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 34, 3299–3302. doi: 10.1093/molbev/msx248
Sandoval, J. R., Salazar-Granara, A., Acosta, O., Castillo-Herrera, W., Fujita, R., Pena, S. D. J., et al. (2013). Tracing the genomic ancestry of Peruvians reveals a major legacy of pre-Columbian ancestors. J. Hum. Genet. 58, 627–634. doi: 10.1038/jhg.2013.73
Schauer, K., Gouget, B., Carriere, M., Labigne, A., and de Reuse, H. (2007). Novel nickel transport mechanism across the bacterial outer membrane energized by the TonB/ExbB/ExbD machinery. Mol. Microbiol. 63, 1054–1068. doi: 10.1111/j.1365-2958.2006.05578.x
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Sneppen, K., Semsey, S., Seshasayee, A. S., and Krishna, S. (2015). Restriction modification systems as engines of diversity. Front. Microbiol. 6:528. doi: 10.3389/fmicb.2015.00528
Suzuki, M., Kiga, K., Kersulyte, D., Cok, J., Hooper, C. C., Mimuro, H., et al. (2011). Attenuated CagA oncoprotein in Helicobacter pylori from Amerindians in Peruvian Amazon. J. Biol. Chem. 286, 29964–29972. doi: 10.1074/jbc.M111.263715
Tada, I., Tanizawa, Y., and Arita, M. (2017). Visualization of consensus genome structure without using a reference genome. BMC Genomics 18:208. doi: 10.1186/s12864-017-3499-7
Thorell, K., Yahara, K., Berthenet, E., Lawson, D. J., Mikhail, J., Kato, I., et al. (2017). Rapid evolution of distinct Helicobacter pylori subpopulations in the Americas. PLoS Genet. 13:e1006546. doi: 10.1371/journal.pgen.1006546
To, T. H., Jung, M., Lycett, S., and Gascuel, O. (2016). Fast dating using least-squares criteria and algorithms. Syst. Biol. 65, 82–97. doi: 10.1093/sysbio/syv068
Vale, F. F., Megraud, F., and Vitor, J. M. B. (2009). Geographic distribution of methyltransferases of Helicobacter pylori: evidence of human host population isolation and migration. BMC Microbiol. 9:193. doi: 10.1186/1471-2180-9-193
van Vliet, A. H. (2017). Use of pan-genome analysis for the identification of lineage-specific genes of Helicobacter pylori. FEMS Microbiol. Lett. 364:fnw296. doi: 10.1093/femsle/fnw296
van Vliet, A. H. M., and Kusters, J. G. (2015). Use of alignment-free phylogenetics for rapid genome sequence-based typing of Helicobacter pylori virulence markers and antibiotic susceptibility. J. Clin. Microbiol. 53, 2877–2888. doi: 10.1128/jcm.01357-15
Waters, M. R. (2019). Late Pleistocene exploration and settlement of the Americas by modern humans. Science 365:eaat5447. doi: 10.1126/science.aat5447
Wertheim, J. O., Murrell, B., Smith, M. D., Kosakovsky Pond, S. L., and Scheffler, K. (2015). RELAX: detecting relaxed selection in a phylogenetic framework. Mol. Biol. Evol. 32, 820–832. doi: 10.1093/molbev/msu400
Xu, Q., Morgan, R. D., Roberts, R. J., and Blaser, M. J. (2000). Identification of type II restriction and modification systems in Helicobacter pylori reveals their substantial diversity among strains. Proc. Natl. Acad. Sci. U.S.A. 97, 9671–9676. doi: 10.1073/pnas.97.17.9671
Yahara, K., Furuta, Y., Oshima, K., Yoshida, M., Azuma, T., Hattori, M., et al. (2013). Chromosome painting in silico in a bacterial species reveals fine population structure. Mol. Biol. Evol. 30, 1454–1464. doi: 10.1093/molbev/mst055
Keywords: Amerindians, ancestry, indigenous, hspAmerind, mestizo, Peru
Citation: Gutiérrez-Escobar AJ, Velapatiño B, Borda V, Rabkin CS, Tarazona-Santos E, Cabrera L, Cok J, Hooper CC, Jahuira-Arias H, Herrera P, Noureen M, Wang D, Romero-Gallo J, Tran B, Peek RM Jr, Berg DE, Gilman RH and Camargo MC (2020) Identification of New Helicobacter pylori Subpopulations in Native Americans and Mestizos From Peru. Front. Microbiol. 11:601839. doi: 10.3389/fmicb.2020.601839
Received: 01 September 2020; Accepted: 16 November 2020;
Published: 14 December 2020.
Edited by:
Frank T. Robb, University of Maryland, Baltimore, United StatesReviewed by:
Bodo Linz, Friedrich Alexander University Erlangen-Nuremberg, GermanyNagendran Tharmalingam, Rhode Island Hospital, United States
Copyright © 2020 Gutiérrez-Escobar, Velapatiño, Borda, Rabkin, Tarazona-Santos, Cabrera, Cok, Hooper, Jahuira-Arias, Herrera, Noureen, Wang, Romero-Gallo, Tran, Peek, Berg, Gilman and Camargo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andrés Julián Gutiérrez-Escobar, YW5kcmVzanVsaWFuLmd1dGllcnJlemVzY29iYXJAbmloLmdvdg==
†These authors share last authorship