 Daniel J. Salamango1,2,3*†
Daniel J. Salamango1,2,3*† Reuben S. Harris1,2,3,4*
Reuben S. Harris1,2,3,4*- 1Department of Biochemistry, Molecular Biology and Biophysics, University of Minnesota, Minneapolis, MN, United States
- 2Masonic Cancer Center, University of Minnesota, Minneapolis, MN, United States
- 3Institute for Molecular Virology, University of Minnesota, Minneapolis, MN, United States
- 4Howard Hughes Medical Institute, University of Minnesota, Minneapolis, MN, United States
Accessory proteins are a key feature that distinguishes primate immunodeficiency viruses such as human immunodeficiency virus type I (HIV-1) from other retroviruses. A prime example is the virion infectivity factor, Vif, which hijacks a cellular co-transcription factor (CBF-β) to recruit a ubiquitin ligase complex (CRL5) to bind and degrade antiviral APOBEC3 enzymes including APOBEC3D (A3D), APOBEC3F (A3F), APOBEC3G (A3G), and APOBEC3H (A3H). Although APOBEC3 antagonism is essential for viral pathogenesis, and a more than sufficient functional justification for Vif’s evolution, most viral proteins have evolved multiple functions. Indeed, Vif has long been known to trigger cell cycle arrest and recent studies have shed light on the underlying molecular mechanism. Vif accomplishes this function using the same CBF-β/CRL5 ubiquitin ligase complex to degrade a family of PPP2R5 phospho-regulatory proteins. These advances have helped usher in a new era of accessory protein research and fresh opportunities for drug development.
Introduction
It has been nearly 40 years since the discovery that acquired immune deficiency syndrome (AIDS) is caused by a retrovirus named human immunodeficiency virus type I (HIV-1) (Barré-Sinoussi et al., 1983; Gallo et al., 1984; Popovic et al., 1984). AIDS arises from a profound state of immune suppression brought on by a massive depletion of CD4+ T lymphocytes following HIV-1 infection, which results in patients succumbing to opportunistic infections and rare malignancies (Klatzmann et al., 1984; Masur et al., 1989; Ho et al., 1995). Soon after its discovery, the HIV-1 genome was sequenced and found to contain nine open reading frames that encode over a dozen viral proteins essential for pathogenicity (Emerman and Malim, 1998; Frankel and Young, 1998; Malim and Emerman, 2008).
All retroviruses encode Gag, Pol, and Env polyproteins that are processed by viral and cellular proteases to release individual proteins during nascent particle assembly. Gag encodes structural proteins matrix (MA), capsid (CA), and nucleocapsid (NC), Pol encodes the viral enzymes protease (PR), reverse-transcriptase (RT), and integrase (IN), and Env encodes the surface glycoprotein GP160, which is processed into the receptor-binding GP120 and transmembrane GP41 domains by a cellular furin-like protease (Figure 1A). Lentiviruses encode additional proteins, such as the regulatory proteins Tat and Rev and several accessory proteins (Vif, Vpr, Vpu, and Nef) that perform various functions to enhance HIV-1 pathogenicity (Emerman and Malim, 1998; Malim and Emerman, 2008; Figures 1A,B). In at least three instances, the major function of these accessory proteins is to promote HIV-1 evasion of cell-intrinsic antiviral resistance by either re-localizing antiviral factors from their sites of virus restriction (Vpu-TETHERIN), targeting them for proteasomal degradation (Vif-APOBEC3), or a combination of both activities (Nef-SERINC3/5) (Madani and Kabat, 1998; Simon et al., 1998a; Sheehy et al., 2002; Neil et al., 2008; Van Damme et al., 2008; Rosa et al., 2015; Usami et al., 2015; Figure 1B). In the case of SERINC3/5 counteraction, Nef re-targets these proteins from the plasma membrane to endosomal/lysosomal compartments for degradation (Usami et al., 2015; Rosa et al., 2015; Shi et al., 2018). For Vpr, its role in HIV-1 pathogenesis remains enigmatic, with reported activities ranging from manipulation of the DNA-damage response (Hrecka et al., 2016; Wu et al., 2016) and induction of G2/M cell cycle arrest (Bartz et al., 1996; Sakai et al., 2006), to facilitating nuclear import of the HIV-1 genome (Heinzinger et al., 1994; Jenkins et al., 1998) and enhancing viral gene expression (Thierry et al., 2004; Zhang and Bieniasz, 2020; Figure 1B).
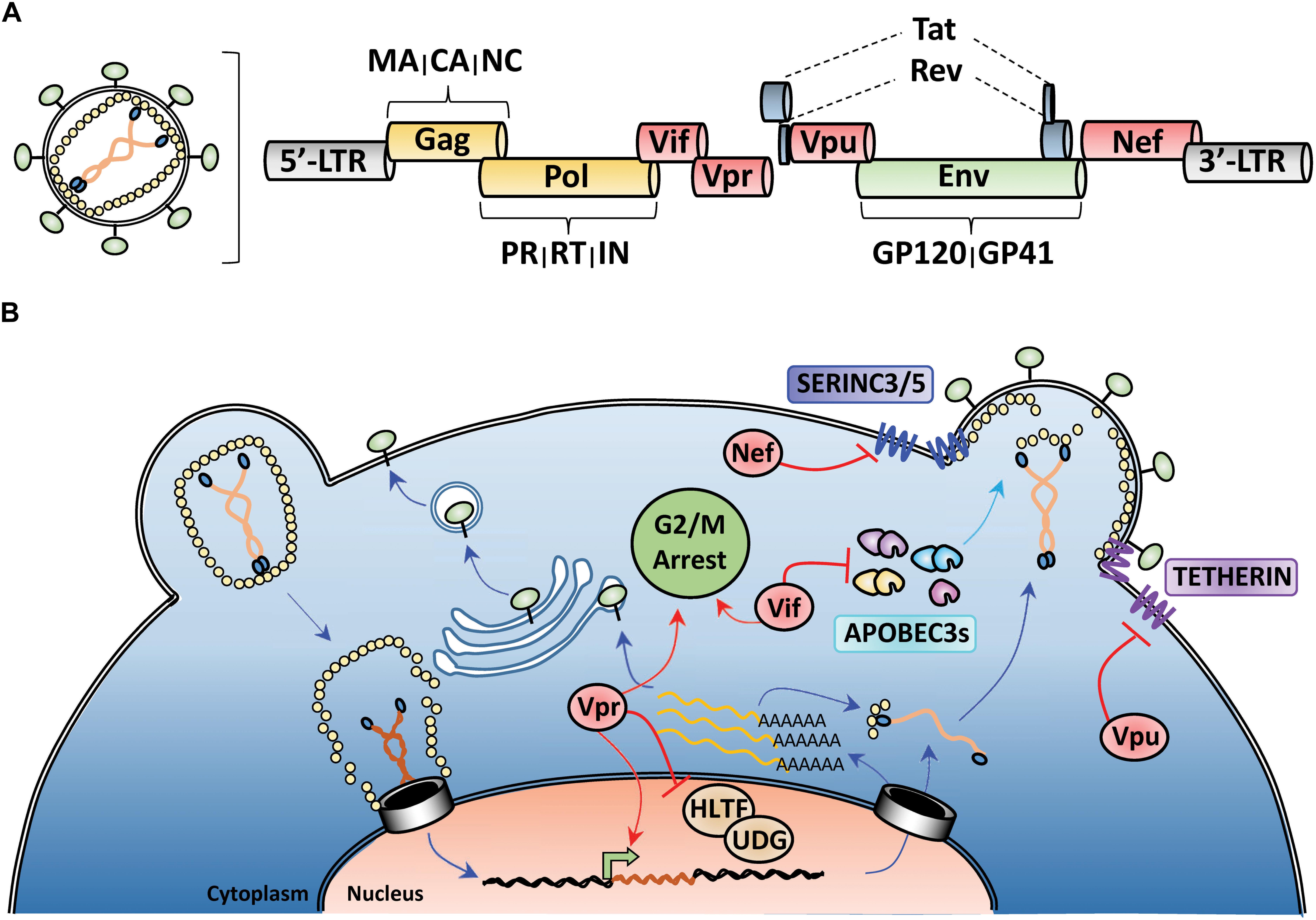
Figure 1. HIV-1 genome organization and counteraction of host-cell restriction factors by viral accessory proteins. (A) Depiction of the genomic organization of HIV-1 with yellow indicating structural proteins and viral enzymes, green indicating the envelope glycoprotein, blue indicating the regulatory proteins, and red highlighting the viral accessory proteins. (B) Simplified schematic of the HIV-1 life-cycle with activities of the viral accessory proteins highlighted in red. Nef and Vpu downmodulate the anti-viral factors TETHERIN and SERINC3/5 from the cell surface. Vif targets cytoplasmic APOBEC3s for proteasomal degradation prior to viron encapsidation. Vpr antagonizes many different cellular processes including DNA-damage response, cell cycle progression, and viral gene expression.
As is true for other viruses (e.g., herpesviruses and poxviruses), immunomodulatory accessory genes are frequently deleted, or dispensable, for viral replication in vitro; however, in vivo they are strongly maintained during natural infections. For example, in vitro, Vif, Vpr, Vpu, and Nef exert functions in a cell-type dependent manner, where these genes are dispensable for producing infectious viral particles in some T cell and myeloid cell lines but not others (i.e., permissive vs. non-permissive cells). However, in physiologically relevant primary CD4+ T cells and monocytes, loss of these genes results in severe impairment of HIV-1 replication (Deacon et al., 1995; Gaddis et al., 2004; Schindler et al., 2006). To further emphasize this point, HIV-1 deficient in the vif gene produces particles that are roughly 1000 times less infectious compared to wild-type virus in primary and non-permissive CD4+ T cells (Fisher et al., 1987; Strebel et al., 1987; Gabuzda et al., 1992).
Discovery of Vif Function in APOBEC3 Degradation
The underlying mechanism of how Vif enhanced HIV-1 pathogenesis remained elusive for many years following its identification. The key to uncovering Vif’s function originated from observations that in some adherent (HeLa and HEK293T) and T cell lines (CEM-SS and SupT1) Vif is dispensable for producing infectious virus. In contrast, loss of Vif in physiologically relevant primary CD4+ T cells and macrophages, and some immortalized T cell lines (CEM and H9), results in the production of almost no infectious virus (Gabuzda et al., 1992; Sakai et al., 1993; Simon et al., 1998b). Comprehensive interrogation of these cell lines using heterokaryon experiments in parallel with subtraction cloning methods indicated the presence of a dominant factor, rather than the lack of a recessive gene product, and led to the identification of the host factor CEM15, now known as APOBEC3G (Madani and Kabat, 1998; Simon et al., 1998a; Sheehy et al., 2002).
APOBEC3G (A3G) belongs to a family of cytosine deaminase enzymes that converts cytosine to uracil (C-to-U) in single-stranded DNA (Harris et al., 2002; Petersen-Mahrt et al., 2002; Figures 2A,B). DNA C-to-U deamination is catalyzed by a zinc-mediated hydrolysis reaction driven by a highly conserved glutamic acid residue located within the substrate-binding pocket. APOBEC3 enzymes exhibit an intrinsic dinucleotide preference with the cytosine base being preceded by either a thymine (TC) or another cytosine (CC) (Carpenter et al., 2010; Kohli et al., 2010; Rathore et al., 2013; Figure 2B). Because of its enzymatic classification, researchers postulated that A3G was able to restrict HIV-1 replication through deamination of the viral RNA during reverse-transcription, when the genome is converted to single-stranded DNA (Harris et al., 2003; Mangeat et al., 2003; Zhang et al., 2003; Di Noia and Neuberger, 2007). In fact, extensive biochemical characterization of A3G and related family members revealed that these enzymes could package into nascent HIV-1 particles and inflict catastrophic levels of C-to-T mutations in the viral genome during its conversion to single-stranded DNA (accounts for G-to-A mutations observed on the genomic plus strand) (Bishop et al., 2004; Desimmie et al., 2014; Feng et al., 2014; Figures 2A,B). In addition, several APOBEC3 enzymes also exert deaminase-independent antiviral activities and hinder reverse transcription, likely through high affinities for RNA and single-stranded DNA viral replication intermediates (Newman et al., 2005; Holmes et al., 2007; Shaban et al., 2018).
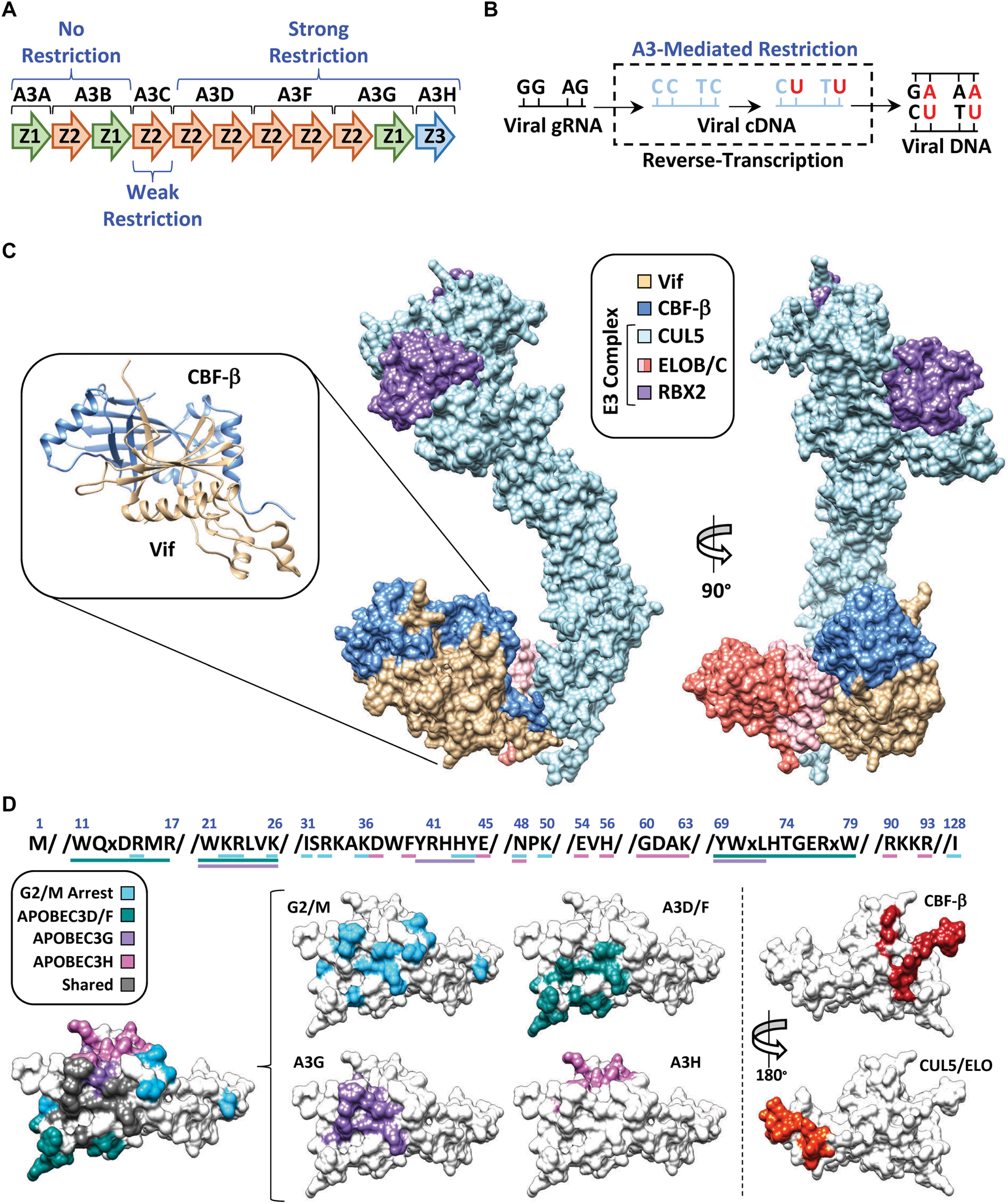
Figure 2. APOBEC3 restriction and counter action by Vif. (A) Schematic of the APOBEC3 locus with anti-HIV-1 activity indicated above or below the respective APOBEC3 enzyme. (B) Depiction of APOBEC3 C-to-T mutations during conversion of the viral genomic RNA (gRNA) to cDNA. Mutations are depicted at either “CC” or “TC” dinucleotide contexts which is reflective of A3G or A3D/F/H target sites, respectively. Fixation of the mutations is depicted by the final double-stranded DNA product (right). (C) Structure of the E3-ubiquitin ligase complex nucleated by Vif (PDB: 4N9F). Full length CUL5 has been modeled in using the partial CUL5 sequence from PDB: 4N9F. RBX2 has been modeled in using RBX1 from PDB: 1LDJ as a template. The inset depicts the Vif/CBF-β hetero-dimer as ribbon diagrams from the crystal structure. (D) Amino acid sequences and surface models of Vif with substrate binding residues highlighted in the indicated color. The shared surface indicated residues that are required for degradation of two or more Vif substrates.
Follow-up studies revealed that Vif counteracts the mutagenic potential of the APOBEC3s by nucleating the formation of an E3-ubiquitin ligase complex that targets A3s for proteasomal degradation prior to virion encapsidation (Marin et al., 2003; Sheehy et al., 2003; Mehle et al., 2004). Proteomic and genetic studies determined that this E3-ubiquitin ligase complex is comprised of Cullin 5 (CUL5), Elongin B and C (ELOB/C), Ring-Box 2 (RBX2), and CBF-β (Yu et al., 2003; Jäger et al., 2011; Zhang et al., 2011; Figure 2C). Interestingly, CBF-β was not part of the original proteomics discovery, but instead, came several years later. Significant efforts were made by many labs to purify Vif from heterologous systems with little success, which led to the hypothesis that Vif may require a cellular co-factor to maintain stability. Quantitative proteomics experiments revealed that this co-factor was the transcription factor CBF-β, which co-precipitated with the E3-ubiquitin ligase complex only in the presence of Vif (Figure 2C, left). Further investigation established that CBF-β is indeed a bona-fide co-factor required for Vif expression and counteraction of APOBEC3 restriction factors in vivo, and that it is an integral part of the complex as evidenced by a high-resolution structure (Guo et al., 2014; Figure 2C).
Dynamic Nature of Vif-Substrate Protein–Protein Interfaces
One of the major questions following the discovery of the Vif-APOBEC3 interaction was how can a relatively small viral protein (∼20 kDa) interact with four restrictive APOBEC3 enzymes (five in total), recruit CBF-β, nucleate the formation of an E3-ubiquitin ligase complex, and induce G2/M cell cycle arrest (discussed in further detail below)? Remarkably, it has become apparent that each Vif-substrate pairwise combination utilizes largely distinct, and genetically separable, interaction surfaces (Figure 2D). Importantly, these biochemical constraints suggest that a single Vif/E3-complex can only degrade one substrate at a time, making Vif’s ability to target so many distinct substrates even more impressive.
Extensive mutagenesis studies have identified several Vif amino acid substitutions that can disrupt individual pair-wise interactions and leave others intact. Distinct clusters of separation-of-function mutants have been identified that disrupt degradation of A3D/F, A3G, or A3H. The Vif surface utilized for A3G recognition is clustered near the N-terminus, encompassing three distinct sets of amino acid residues, with the 40YRHHY44 segment being unique to A3G recognition (Russell and Pathak, 2007; He et al., 2008; Yamashita et al., 2008; Chen et al., 2009; Dang et al., 2009; Pery et al., 2009; Letko et al., 2015; Figure 2D). While the A3D/F interface partially overlaps with A3G, it also extends to residues 11WQxDRMR17 and 74TGERxW79, which imparts specificity (Russell and Pathak, 2007; He et al., 2008; Zhang et al., 2008; Figure 2D). Interestingly, the surface residues used to recognize A3H are clustered at the top of the β-fold and are completely distinct from those used to recognize the other APOBEC3s (Zhen et al., 2012; Ooms et al., 2013, 2017; Nakashima et al., 2017; Wang et al., 2019). Likewise, the same is true for the Vif surfaces that maintain interactions with CBF-β (residues located on β-strand 1 and 6) (Zhou et al., 2012, 2014) and the E3-ubiquitin ligase complex (residues located on α-helix 3 and 4) (Kamura et al., 1998; Yu et al., 2004; Bergeron et al., 2010; Wolfe et al., 2010; Figure 2D).
Previous analyses of available Vif sequences have highlighted evolutionary conservation for some Vif-substrate surfaces but not others (Azimi and Lee, 2020). As one would predict, residues involved in binding CBF-β and the E3-ubiquitin ligase complex are highly conserved; however, the same is not true for residues involved in APOBEC3 recognition. The conservation of interface residues diminishes with A3F > A3G >> A3H, which is consistent with the current model that A3H may only be a threat to the virus in certain regions of the globe (Ebrahimi et al., 2018). This observation is also consistent with the “wobble model” which provides an evolutionary paradigm explaining the biochemical drift between Vif-APOBEC3 interactions (Richards et al., 2015; Harris and Anderson, 2016). This model is predicated on the concept that an ancestral lentivirus was established in a non-primate mammal with a relatively modest APOBEC3 gene-set prior to transmitting into a primate with an expanded APOBEC3 repertoire. In this hypothetical scenario, the strong interaction between Vif and the ancestral APOBEC3 is attenuated following zoonosis. The Vif-APOBEC3 interaction(s) are then restored following compensatory mutations encoding residues at, or near, the periphery of the ancestral Vif-APOBEC3 interaction surface. Importantly, these adaptations would have occurred independently for each novel APOBEC3 restriction factor, giving rise to the distinct interaction surfaces observed on present-day Vif (Figure 2D).
Vif Induces G2/M Cell Cycle Arrest Through Degradation of PPP2R5s
Because counteraction of the APOBEC3s is critical for HIV-1 replication, this was thought to be Vif’s only function for many years. However, in the early 2000s it was discovered that Vif could also induce G2/M cell cycle arrest in a variety of cell lines (Sakai et al., 2006; Wang et al., 2007). Genetic studies established that Vif-induced G2/M arrest requires both CBF-β and the same E3-ubiquitin ligase complex utilized for APOBEC3 degradation (DeHart et al., 2008; Du et al., 2019). Additionally, fine-mapping of the Vif residues required for inducing G2/M arrest revealed that this interface is mostly distinct from those of the APOBEC3s (Izumi et al., 2010; Zhao et al., 2015; Salamango et al., 2019, 2020; Marelli et al., 2020; Nagata et al., 2020; Figure 2D). While these observations collectively pointed toward Vif degrading a cellular factor to induce arrest, the identity of this factor remained elusive for nearly 10 years following the discovery of this activity.
A major breakthrough regarding Vif-induced G2/M arrest came from recent quantitative proteomics studies that revealed Vif-dependent remodeling of the host phosphoproteome (Greenwood et al., 2016; Naamati et al., 2019). These studies discovered that Vif could efficiently deplete multiple members of the PPP2R5 family of protein phosphatase 2A (PP2A) regulators in several cell lines, including primary and immortalized CD4+ T cells (Greenwood et al., 2016; Naamati et al., 2019). PP2As account for a majority of the phosphatase activity in eukaryotic cells and function as heterotrimeric complexes comprised of a phosphatase enzyme (PP2Cα), a scaffolding protein (PPP2R1α or PPP2R1β), and a regulatory subunit (B55, PPP2R5/B56, or PR72/130) (Thompson and Williams, 2018; Nilsson, 2019). The regulatory subunit can be from one of three distinct protein families, which regulate subcellular localization and substrate recognition of PP2A holoenzyme complexes (McCright et al., 1996; Wang J. et al., 2016). Importantly, PP2A/PPP2R5 holoenzymes have been shown to regulate multiple aspects of the G2-to-M phase transition (Moura and Conde, 2019; Nilsson, 2019).
The connection between PP2A/PPP2R5 complexes and regulation of G2/M progression prompted our group, and others, to investigate if these were the factors antagonized by Vif to induce G2/M arrest. Comprehensive mutagenesis using both single amino acid substitutions and large-scale mutagenic libraries revealed that Vif residues required for degradation of PPP2R5 substrates overlap with those required for inducing G2/M arrest (Evans et al., 2018; Salamango et al., 2019, 2020; Marelli et al., 2020; Nagata et al., 2020). These studies also showed that siRNA-mediated knock-down of specific combinations of PPP2R5 transcripts could induce a robust G2/M arrest phenotype in the absence of Vif; however, strong knock-down of single PPP2R5 family members had no impact on cell cycle progression (Salamango et al., 2019; Marelli et al., 2020). While subtle differences were observed for which combinations of PPP2R5 knock-downs were most impactful, the overall conclusions were complementary in that at least two family members need to be simultaneously depleted to induce arrest in the absence of Vif (Salamango et al., 2019; Marelli et al., 2020). Additionally, chemical inhibition of PP2A activity using okadaic acid induced a robust G2/M arrest phenotype, further supporting a direct relationship between Vif-induced PPP2R5 degradation and subsequent G2/M arrest (Salamango et al., 2019; Marelli et al., 2020).
Vif Binds the Same Surface as Physiologic PPP2R5 Substrates
Characterization of Vif separation-of-function mutants suggested that PPP2R5 recognition occurs through electrostatic interactions, as a majority of residues required for degradation are positively charged (8 out of 12 residues) (Salamango et al., 2019, 2020; Marelli et al., 2020; Figure 2D). Using high-resolution crystal structures and homology modeling, it is evident that all Vif-substrate interaction surfaces are largely electrostatic in nature (Kitamura et al., 2012; Li et al., 2012; Bohn et al., 2013; Byeon et al., 2013; Siu et al., 2013; Wang J. et al., 2016; Kouno et al., 2017; Shi et al., 2017; Ito et al., 2018; Shaban et al., 2018; Figure 3A). The PPP2R5 surface recognized by Vif is extremely electronegative and supports a model in which Vif maintains this interaction through a favorable network of electrostatics. Importantly, all of the residues required for Vif recognition are conserved among all five family members, which clarifies how Vif can recognize and degrade five new cellular substrates and explains genetic evidence that loss of at least two family members is required for inducing G2/M arrest.
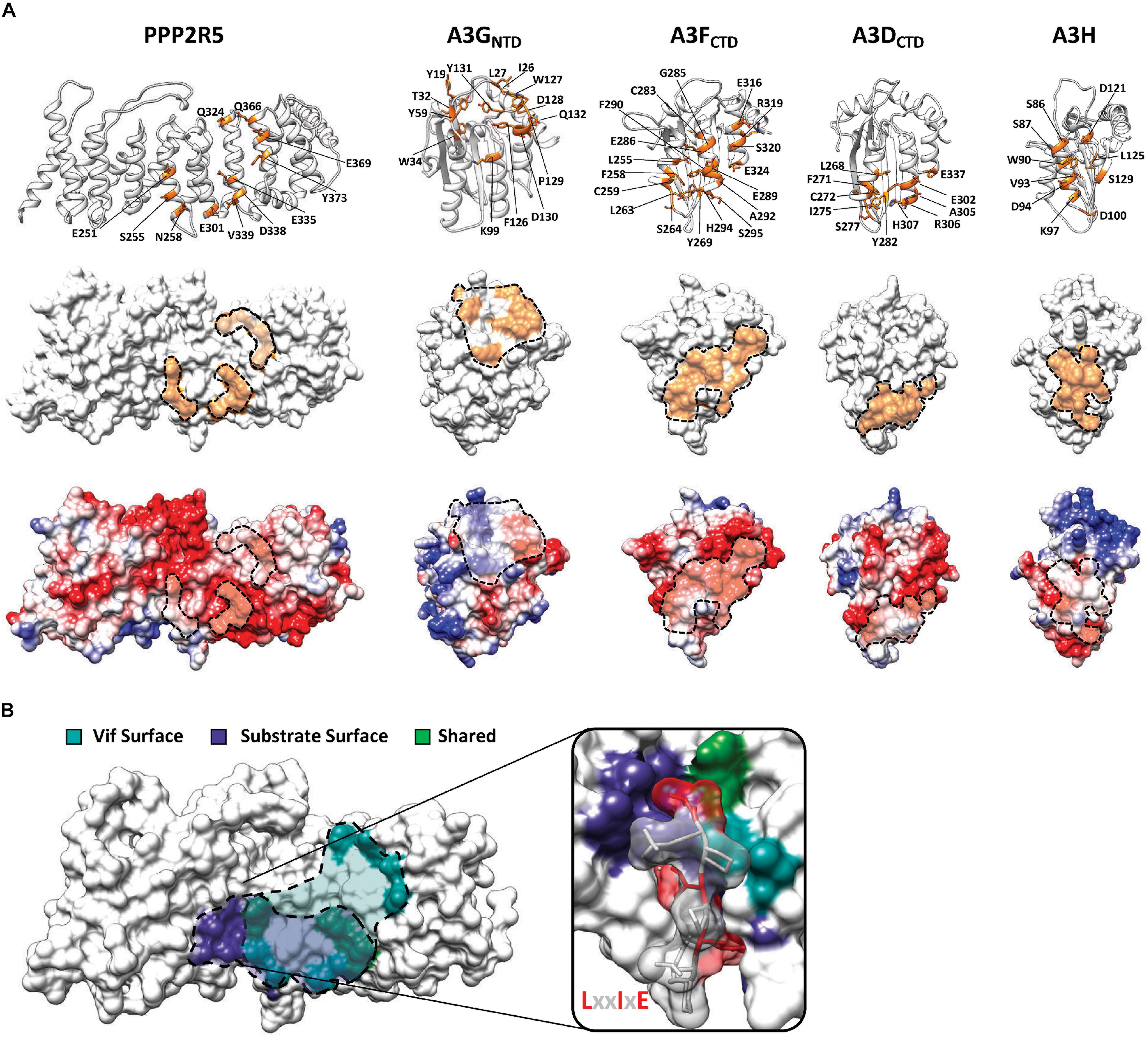
Figure 3. Substrate surfaces recognized by Vif. (A) Amino acid residues on PPP2R5 and APOBEC3 that have been shown to be required for Vif-mediated degradation (top) with substrate surfaces (middle) and electrostatic potential maps (bottom). (B) Depiction of amino acid residues on the surface of PPP2R5 that are recognized by Vif or cellular substrates. The LxxIxE substrate peptide is depicted bound to the PPP2R5 surface to highlight that Vif and cellular substrates compete for an overlapping surface.
Interestingly, the PPP2R5 surface recognized by Vif overlaps with the substrate binding pocket used by PPP2R5s to bind cellular substrates (Hertz et al., 2016; Wang J. et al., 2016; Wang X. et al., 2016; Figure 3B). Crystallographic and proteomics studies have combined to identify the substrate binding motif recognized by PPP2R5 proteins and the mode in which binding occurs. PPP2R5s recognize substrates through a conserved LxxIxE motif that directs the PP2A holoenzyme complex to cellular targets for dephosphorylation (Hertz et al., 2016; Wang J. et al., 2016; Wang X. et al., 2016). The LxxIxE motif is stabilized by two distinct substrate binding pockets on the surface of PPP2R5 proteins. The “Leu pocket,” comprised of residues K208, T184, H187, R188, E251, and I227, and the “Ile pocket,” which contains residues H187, Y215, I227, and I256. In addition, a recent study demonstrated that PPP2R5 substrate recognition is enhanced by electrostatic interactions that occur adjacent to the peptide binding cleft, encompassing residues E335 and D338 (Wang X. et al., 2020). These residues are of interest because E251, E335, and D338 are required for Vif-induced degradation of PPP2R5s (Salamango et al., 2019; Figure 3B). Furthermore, residues S255 and N258, which are located within the “Ile pocket,” are also required for Vif-induced degradation (Figure 3B).
The close proximity and partial overlap between these interfaces raised the question of whether Vif and cellular substrates compete for binding. To test this model, we utilized a well-characterized peptide inhibitor that contains the LxxIxE motif recognized by PPP2R5s to determine if Vif-induced degradation could be blocked. Co-transfection of a plasmid expressing four tandem copies of this LxxIxE peptide caused a dose-dependent inhibition of Vif-mediated degradation of PPP2R5A (Salamango et al., 2020). However, a control plasmid containing an AxxAxA motif had no effect on Vif-induced degradation of either PPP2R5A or APOBEC3G (Salamango et al., 2020). Taken together, these observations support a competitive binding model in which Vif directly interacts with the surface of PPP2R5 proteins and occludes the binding of cellular substrates.
Mechanistic Model for Vif-Induced G2/M Arrest
The discovery of PPP2R5 substrates was a major step forward in understanding Vif-induced cell cycle arrest. However, the regulatory checkpoints altered downstream of PPP2R5 degradation remain unknown. PP2A/PPP2R5 complexes have been shown to regulate entrance and exit of the G2-to-M phase transition at multiple different checkpoints. Below, we discuss these checkpoints in the context of previous observations regarding Vif-induced G2/M arrest and postulate a model in which antagonism of discrete PP2A/PPP2R5 complexes would lead to simultaneous inhibition of multiple checkpoints.
First, arguably the most critical event for mitotic entry is CDC25-mediated activation of CDK1, which allows for nuclear translocation of the CDK1-CyclinB complex (Lee and Kirschner, 1996; Timofeev et al., 2010; Figure 4, top). Activation of CDC25 requires PP2A-mediated dephosphorylation of specific residues to release inhibitory 14-3-3 proteins (Margolis et al., 2006; Figure 4, top). Therefore, Vif-induced antagonism of PP2A/PPP2R5 complexes would render CDC25 inactive and inhibit nuclear translocation of CDK1-CyclinB1 (Figure 4, bottom). In support of this mechanism, a previous study reported that infection of Jurkat T cells with arrest-proficient HIV-1 resulted in constitutive phosphorylation of Tyr15 on CDK1, and subsequent inhibition of CDK1-CyclinB nuclear translocation (Sakai et al., 2011). Additionally, quantitative proteomics and immunoblot analyses revealed elevated levels of CyclinB in CEM T4 cells infected with HIV-1 expressing arrest-proficient Vif (Marelli et al., 2020).
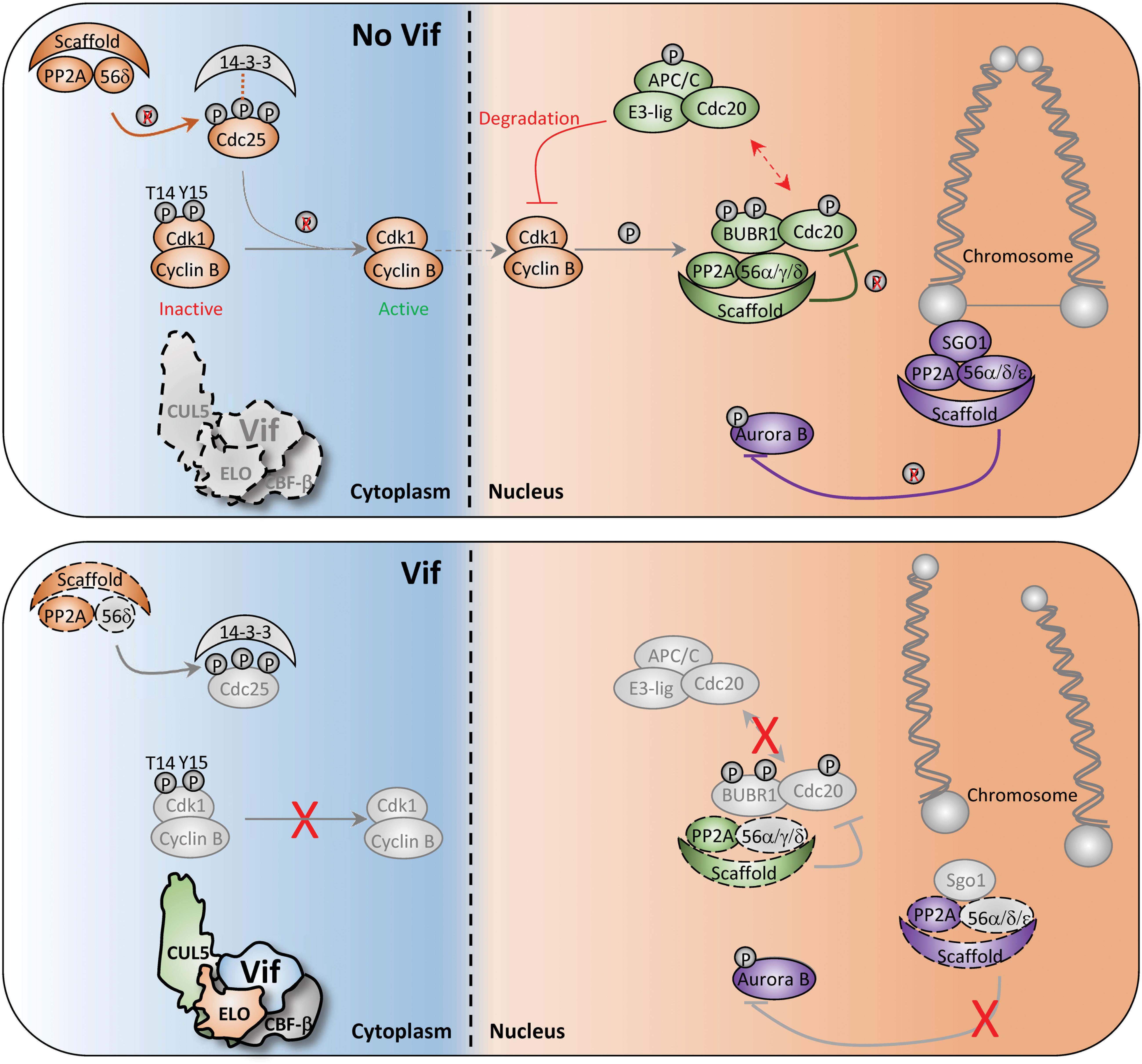
Figure 4. Diagram of potential Vif-induced G2/M arrest mechanisms. Depiction of normal and aberrant regulation of the G2-to-M phase transition in the absence (top) and presence (bottom) of Vif. Key cell cycle checkpoints regulated by PP2A/PPP2R5 complexes are color-coded in the absence or presence of Vif. See text for additional details.
Second, PP2A/PPP2R5 complexes also control G2-to-M progression through spatiotemporal regulation of the Aurora kinases. Aurora kinases are involved in numerous G2-to-M checkpoint controls including spindle assembly, microtubule-kinetochore attachment, and cytokinesis (Fu et al., 2007; Goldenson and Crispino, 2015). To exit mitosis, Aurora kinases are degraded through the APC-ubiquitin-proteasome pathway, which is regulated by PP2A/PPP2R5A, 5C, and/or 5D complexes (Horn et al., 2007; Jeong and Yang, 2013; Lindon et al., 2015; Figure 4, top). Recent studies have demonstrated striking activation of Aurora kinase A and B following infection of CEM T4 cells with HIV-1 expressing arrest-proficient Vif, supporting a model in which Vif simultaneously antagonizes PP2A/PPP2R5 complexes to inactivate the APC system and stall mitotic exit (Figure 4, bottom).
Lastly, PP2A/PPP2R5 holoenzymes have been shown to serve as a scaffold for multiple distinct complexes to initiate exit from mitosis. For example, Shugoshin (SGO1) directly binds to a PP2A/PPP2R5 holoenzyme to form a complex that regulates accurate chromosome segregation during mitosis (Tang et al., 2006; Xu et al., 2009; Figure 4, top). Without the formation of this complex, centromeric cohesion is prematurely cleaved which results in untimely centromere disassociation and chromosome mis-segregation (Tang et al., 2006; Xu et al., 2009; Figure 4, bottom). Additionally, PP2A/PPP2R5 holoenzymes directly bind to BUBR1 and regulate the spindle assembly checkpoint and mitotic progression by counteracting Aurora kinase activity at improperly attached kinetochores (Cheeseman et al., 2006; DeLuca et al., 2006; Kruse et al., 2013). Therefore, disruption of BUBR1 regulatory activity and hyper-activation of Aurora kinases through Vif-mediated antagonism of PP2A/PPP2R5 complexes would result in kinetochore dysfunction and a failure to exit mitosis (Figure 4, bottom).
In addition to manipulation of the PP2A/PPP2R5 axis, it has been suggested that Vif can induce G2/M arrest by blocking MDM2-mediated ubiquitination and nuclear export of TP53 (Izumi et al., 2010). This study observed that Vif binding to TP53 could block MDM2 recognition and subsequent turnover of TP53, which is required for proper cell cycle progression. Although not addressed directly, it is likely that the Vif/TP53 interaction occurs in the nuclear compartment, thus directly shielding TP53 from MDM2 recognition and subsequent nuclear export. If this is indeed the case, it is plausible that there are two pools of Vif that act in concert to stall cell cycle progression. Nuclear localized Vif protects TP53 from MDM2 and antagonizes nuclear PPP2R5C/D substrates, whereas cytoplasmic Vif antagonizes the APOBEC3s and cytoplasmic PPP2R5A/B/E. Additional work will be required to determine if these mechanisms are separate or connected through shared components.
Concluding Remarks
Now, Vif clearly has two distinct sets of cellular substrates, APOBEC3 enzymes and PPP2R5 phospho-regulators (10 proteins total). While APOBEC3 counteraction has been shown to be essential for viral infectivity and pathogenesis in vivo, the importance of PPP2R5 degradation remains to be established. However, at least three key observations have been made that imply that alteration of the cell cycle may be beneficial for HIV-1 pathogenesis.
First, HIV-1 Vpr also potently induces G2/M arrest through a mechanism that is clearly distinct from that of Vif. Vpr activates the ATR DNA damage response pathway by inducing proteasomal degradation of diverse cellular substrates using a Cullin 4 (CUL4), DNA damage-binding protein 1 (DDB1), and DDB1-CUL4-associated factor (DCAF1) E3-ubiquitin ligase complex (Hrecka et al., 2007; Andersen et al., 2008; Greenwood et al., 2019). While Vif degrades a smaller set of cellular proteins, Vpr has been shown to be much more promiscuous. A recent study demonstrated that Vpr induces the degradation of roughly 40 cellular proteins causing a systems-level remodeling of the cellular proteome, which may cumulatively lead to G2/M arrest (Greenwood et al., 2019). Collectively, over half a dozen cellular proteins have been directly implicated as being essential for Vpr-induced arrest [e.g., MCM10, SMN1, CDCA2, ZNF267, MUS81, CCDC137, etc. (Laguette et al., 2014; Romani et al., 2015; Greenwood et al., 2019; Zhang and Bieniasz, 2020)].
At least two phenotypes have been associated with Vpr-induced arrest that may enhance HIV-1 pathogenicity. First, degradation of MUS81 and EME1 by Vpr leads to G2/M arrest and prematurely activates the SLX4 complex, which suppresses spontaneous and HIV-1-mediated induction of type I interferon responses and may contribute to immune evasion (Laguette et al., 2014). Second, depletion of CCDC137 by Vpr has been shown to induce G2/M arrest and enhance HIV-1 gene expression (Zhang and Bieniasz, 2020). It is worth emphasizing that during the G2/M phase of the cell cycle, cellular translation and transcription are significantly diminished (Parsons and Spencer, 1997; Kronja and Orr-Weaver, 2011; Tanenbaum et al., 2015; Strzyz, 2017). Therefore, Vpr’s ability to simultaneously induce arrest and enhance HIV-1 gene expression would lead to an enrichment of viral transcripts during G2/M. Interestingly, PP2A/PPP2R5 complexes have been shown to regulate protein translation kinetics through mTOR, S6 kinase, and 4E-BP1 (Hahn et al., 2010; Gardner et al., 2015). Therefore, it is possible that Vif antagonism of PP2A holoenzymes leads to increased protein translation during G2/M when host cell translation is normally stalled. Taken together, it is possible that Vif and Vpr act in concert to induce G2/M arrest and boost transcription and translation of HIV-1 genes.
Second, arrest-proficient Vif variants are prominent in global populations and in patient derived Vif isolates. Bioinformatic analyses of sequences obtained from the Los Alamos database have drawn correlations between predicted arrest-proficient Vif sequences and the most abundant subtypes in global circulation (Salamango et al., 2019; Marelli et al., 2020). Additionally, a previous study suggested that Vif-induced arrest leads to increased production of HIV-1 particles from both primary and immortalized CD4+ T cells (Izumi et al., 2010). Although this is the only study to-date that demonstrates this phenotype, these findings could explain why arrest-associated Vif variants are prevalent in circulating populations. Furthermore, live cell imaging studies assessing degradation kinetics of APOBEC3G and PPP2R5A in the same cell established that the half-life of PPP2R5A is only ∼2 h slower than that of APOBEC3G following infection, which is striking given that APOBEC3G degradation is essential for HIV-1 infectivity and pathogenesis (Salamango et al., 2020).
Third, subversion of the cell cycle is a mechanism used by diverse viral pathogens to create a favorable environment for replication. Oncogenic viruses, RNA viruses, and DNA viruses have all been shown to hijack the host cell cycle during the course of an infection, many of which arrest cells in the G2/M phase [e.g., human T-lymphotropic virus (HTLV), human polyoma virus, infectious bronchitis virus, simian virus 40, and adenovirus (Zhao and Elder, 2005; Dove et al., 2006; Fan et al., 2018)]. Interestingly, PP2A appears to be a conserved target as well, given that simian virus 40, polyoma virus, HTLV, adenovirus, and HIV-1 affect the enzymatic activity of at least a subset of PP2A complexes (Roopchand et al., 2001; Haoudi et al., 2003; Zhao and Elder, 2005). Importantly, PPP2R5 proteins have also been shown to be hijacked by other viral pathogens. Ebola virus nucleoprotein recruits PPP2R5 proteins through a highly conserved LxxIxE motif to stimulate dephosphorylation of VP30, which initiates transcription of the viral genome and subsequent infection (Kruse et al., 2018). Additionally, HTLV requires PPP2R5C for efficient strand-transfer activity and target integration (Maertens, 2016). HTLV integrase uses a LxxIxE motif to recruit PPP2R5C to the viral integration complex (Bhatt et al., 2020). Taken together, these observations support a model in which PPP2R5 antagonism, and global changes in the cellular phospho-proteome, are likely to be advantageous for the pathogenesis of HIV-1 as well as other prominent viruses. Furthermore, continuing to unravel the complex molecular mechanisms HIV-1 has evolved to subvert cellular processes and enhance pathogenicity may provide major clues for the development of innovative therapeutics that lead to virus eradication.
Author Contributions
DS drafted the manuscript. DS and RH edited the manuscript. Both authors contributed to the article and approved the submitted version.
Funding
This work was supported by NIAID R37-AI064046 (to RH). DS received salary support from an NIAID K99/R00 transition award (K99-AI147811). RH is the Margaret Harvey Schering Land Grant Chair for Cancer Research, a Distinguished University McKnight Professor, and an Investigator of the Howard Hughes Medical Institute.
Conflict of Interest
RH is a co-founder, shareholder, and consultant of ApoGen Biotechnologies Inc.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Andersen, J. L., Le Rouzic, E., and Planelles, V. (2008). HIV-1 Vpr: mechanisms of G2 arrest and apoptosis. Exp. Mol. Pathol. 85, 2–10. doi: 10.1016/j.yexmp.2008.03.015
Azimi, F. C., and Lee, J. E. (2020). Structural perspectives on HIV-1 Vif and APOBEC3 restriction factor interactions. Protein Sci. 29, 391–406. doi: 10.1002/pro.3729
Barré-Sinoussi, F., Chermann, J. C., Rey, F., Nugeyre, M. T., Chamaret, S., Gruest, J., et al. (1983). Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS). Science 220, 868–871. doi: 10.1126/science.6189183
Bartz, S. R., Rogel, M. E., and Emerman, M. (1996). Human immunodeficiency virus type 1 cell cycle control: Vpr is cytostatic and mediates G2 accumulation by a mechanism which differs from DNA damage checkpoint control. J. Virol. 70, 2324–2331. doi: 10.1128/jvi.70.4.2324-2331.1996
Bergeron, J. R., Huthoff, H., Veselkov, D. A., Beavil, R. L., Simpson, P. J., Matthews, S. J., et al. (2010). The SOCS-box of HIV-1 Vif interacts with ELONGINBC by induced-folding to recruit its Cul5-containing ubiquitin ligase complex. PLoS Pathog. 6:e1000925. doi: 10.1371/journal.ppat.1000925
Bhatt, V., Shi, K., Salamango, D. J., Moeller, N. H., Pandey, K., Bera, S., et al. (2020). Structural basis of host protein hijacking in human T-cell leukemia virus integration. Nat. Commun. 11:3121.
Bishop, K. N., Holmes, R. K., Sheehy, A. M., Davidson, N. O., Cho, S. J., and Malim, M. H. (2004). Cytidine deamination of retroviral DNA by diverse APOBEC proteins. Curr. Biol. 14, 1392–1396. doi: 10.1016/j.cub.2004.06.057
Bohn, M. F., Shandilya, S. M., Albin, J. S., Kouno, T., Anderson, B. D., McDougle, R. M., et al. (2013). Crystal structure of the DNA cytosine deaminase APOBEC3F: the catalytically active and HIV-1 Vif-binding domain. Structure 21, 1042–1050. doi: 10.1016/j.str.2013.04.010
Byeon, I. J., Ahn, J., Mitra, M., Byeon, C. H., Hercík, K., Hritz, J., et al. (2013). NMR structure of human restriction factor APOBEC3A reveals substrate binding and enzyme specificity. Nat. Commun. 4:1890. doi: 10.1038/ncomms2883
Carpenter, M. A., Rajagurubandara, E., Wijesinghe, P., and Bhagwat, A. S. (2010). Determinants of sequence-specificity within human AID and APOBEC3G. DNA Repair. 9, 579–587. doi: 10.1016/j.dnarep.2010.02.010
Cheeseman, I. M., Chappie, J. S., Wilson-Kubalek, E. M., and Desai, A. (2006). The conserved KMN network constitutes the core microtubule-binding site of the kinetochore. Cell 127, 983–997. doi: 10.1016/j.cell.2006.09.039
Chen, G., He, Z., Wang, T., Xu, R., and Yu, X. F. (2009). A patch of positively charged amino acids surrounding the human immunodeficiency virus type 1 Vif SLVx4Yx9Y motif influences its interaction with APOBEC3G. J. Virol. 83, 8674–8682. doi: 10.1128/JVI.00653-09
Dang, Y., Wang, X., Zhou, T., York, I. A., and Zheng, Y. H. (2009). Identification of a novel WxSLVK motif in the N terminus of human immunodeficiency virus and simian immunodeficiency virus Vif that is critical for APOBEC3G and APOBEC3F neutralization. J. Virol. 83, 8544–8552. doi: 10.1128/JVI.00651-09
Deacon, N. J., Tsykin, A., Solomon, A., Smith, K., Ludford-Menting, M., Hooker, D. J., et al. (1995). Genomic structure of an attenuated quasi species of HIV-1 from a blood transfusion donor and recipients. Science 270, 988–991. doi: 10.1126/science.270.5238.988
DeHart, J. L., Bosque, A., Harris, R. S., and Planelles, V. (2008). Human immunodeficiency virus type 1 Vif induces cell cycle delay via recruitment of the same E3 ubiquitin ligase complex that targets APOBEC3 proteins for degradation. J. Virol. 82, 9265–9272. doi: 10.1128/JVI.00377-08
DeLuca, J. G., Gall, W. E., Ciferri, C., Cimini, D., Musacchio, A., and Salmon, E. D. (2006). Kinetochore microtubule dynamics and attachment stability are regulated by Hec1. Cell 127, 969–982. doi: 10.1016/j.cell.2006.09.047
Desimmie, B. A., Delviks-Frankenberrry, K. A., Burdick, R. C., Qi, D., Izumi, T., and Pathak, V. K. (2014). Multiple APOBEC3 restriction factors for HIV-1 and one Vif to rule them all. J. Mol. Biol. 426, 1220–1245. doi: 10.1016/j.jmb.2013.10.033
Di Noia, J. M., and Neuberger, M. S. (2007). Molecular mechanisms of antibody somatic hypermutation. Annu. Rev. Biochem. 76, 1–22. doi: 10.1146/annurev.biochem.76.061705.090740
Dove, B., Brooks, G., Bicknell, K., Wurm, T., and Hiscox, J. A. (2006). Cell cycle perturbations induced by infection with the coronavirus infectious bronchitis virus and their effect on virus replication. J. Virol. 80, 4147–4156. doi: 10.1128/JVI.80.8.4147-4156.2006
Du, J., Rui, Y., Zheng, W., Li, P., Kang, J., Zhao, K., et al. (2019). Vif-CBFβ interaction is essential for Vif-induced cell cycle arrest. Biochem. Biophys. Res. Commun. 511, 910–915. doi: 10.1016/j.bbrc.2019.02.136
Ebrahimi, D., Richards, C. M., Carpenter, M. A., Wang, J., Ikeda, T., Cheng, A. Z., et al. (2018). Genetic and mechanistic basis for APOBEC3H alternative splicing, differential retrovirus restriction, and counteraction by HIV-1 protease. Nat. Commun. 9:4137.
Emerman, M., and Malim, M. H. (1998). HIV-1 regulatory/accessory genes: keys to unraveling viral and host cell biology. Science 280, 1880–1884. doi: 10.1126/science.280.5371.1880
Evans, E. L., Becker, J. T., Fricke, S. L., Patel, K., and Sherer, N. M. (2018). HIV-1 Vif’s capacity to manipulate the cell cycle is species specific. J. Virol. 92:e02102-17. doi: 10.1128/JVI.02102-17
Fan, Y., Sanyal, S., and Bruzzone, R. (2018). Breaking bad: how viruses subvert the cell cycle. Front. Cell Infect. Microbiol. 8:396. doi: 10.3389/fcimb.2018.00396
Feng, Y., Baig, T. T., Love, R. P., and Chelico, L. (2014). Suppression of APOBEC3-mediated restriction of HIV-1 by Vif. Front. Microbiol. 5:450. doi: 10.3389/fmicb.2014.00450
Fisher, A. G., Ensoli, B., Ivanoff, L., Chamberlain, M., Petteway, S., Ratner, L., et al. (1987). The sor gene of HIV-1 is required for efficient virus transmission in vitro. Science 237, 888–893. doi: 10.1126/science.3497453
Frankel, A. D., and Young, J. A. (1998). HIV-1: fifteen proteins and an RNA. Annu. Rev. Biochem. 67, 1–25. doi: 10.1146/annurev.biochem.67.1.1
Fu, J., Bian, M., Jiang, Q., and Zhang, C. (2007). Roles of aurora kinases in mitosis and tumorigenesis. Mol. Cancer Res. 5, 1–10. doi: 10.1158/1541-7786.MCR-06-0208
Gabuzda, D. H., Lawrence, K., Langhoff, E., Terwilliger, E., Dorfman, T., Haseltine, W. A., et al. (1992). Role of vif in replication of human immunodeficiency virus type 1 in CD4+ T lymphocytes. J. Virol. 66, 6489–6495. doi: 10.1128/jvi.66.11.6489-6495.1992
Gaddis, N. C., Sheehy, A. M., Ahmad, K. M., Swanson, C. M., Bishop, K. N., Beer, B. E., et al. (2004). Further investigation of simian immunodeficiency virus Vif function in human cells. J. Virol. 78, 12041–12046. doi: 10.1128/JVI.78.21.12041-12046.2004
Gallo, R. C., Salahuddin, S. Z., Popovic, M., Shearer, G. M., Kaplan, M., Haynes, B. F., et al. (1984). Frequent detection and isolation of cytopathic retroviruses (HTLV-III) from patients with AIDS and at risk for AIDS. Science 224, 500–503. doi: 10.1126/science.6200936
Gardner, T. W., Abcouwer, S. F., Losiewicz, M. K., and Fort, P. E. (2015). Phosphatase control of 4E-BP1 phosphorylation state is central for glycolytic regulation of retinal protein synthesis. Am. J. Physiol. Endocrinol. Metab. 309, E546–E556. doi: 10.1152/ajpendo.00180.2015
Goldenson, B., and Crispino, J. D. (2015). The aurora kinases in cell cycle and leukemia. Oncogene 34, 537–545. doi: 10.1038/onc.2014.14
Greenwood, E. J., Matheson, N. J., Wals, K., van den Boomen, D. J., Antrobus, R., Williamson, J. C., et al. (2016). Temporal proteomic analysis of HIV infection reveals remodelling of the host phosphoproteome by lentiviral Vif variants. eLife 5:e018296. doi: 10.7554/eLife.18296
Greenwood, E. J. D., Williamson, J. C., Sienkiewicz, A., Naamati, A., Matheson, N. J., and Lehner, P. J. (2019). Promiscuous targeting of cellular proteins by Vpr drives systems-level proteomic remodeling in HIV-1 infection. Cell Rep. 27, 1579–1596.e7. doi: 10.1016/j.celrep.2019.04.025
Guo, Y., Dong, L., Qiu, X., Wang, Y., Zhang, B., Liu, H., et al. (2014). Structural basis for hijacking CBF-β and CUL5 E3 ligase complex by HIV-1 Vif. Nature 505, 229–233. doi: 10.1038/nature12884
Hahn, K., Miranda, M., Francis, V. A., Vendrell, J., Zorzano, A., and Teleman, A. A. (2010). PP2A regulatory subunit PP2A-B′ counteracts S6K phosphorylation. Cell Metab. 11, 438–444. doi: 10.1016/j.cmet.2010.03.015
Haoudi, A., Daniels, R. C., Wong, E., Kupfer, G., and Semmes, O. J. (2003). Human T-cell leukemia virus-I tax oncoprotein functionally targets a subnuclear complex involved in cellular DNA damage-response. J. Biol. Chem. 278, 37736–37744. doi: 10.1074/jbc.M301649200
Harris, R. S., and Anderson, B. D. (2016). Evolutionary paradigms from ancient and ongoing conflicts between the lentiviral Vif Protein and mammalian APOBEC3 enzymes. PLoS Pathog. 12:e1005958. doi: 10.1371/journal.ppat.1005958
Harris, R. S., Bishop, K. N., Sheehy, A. M., Craig, H. M., Petersen-Mahrt, S. K., Watt, I. N., et al. (2003). DNA deamination mediates innate immunity to retroviral infection. Cell 113, 803–809. doi: 10.1016/s0092-8674(03)00423-9
Harris, R. S., Petersen-Mahrt, S. K., and Neuberger, M. S. (2002). RNA editing enzyme APOBEC1 and some of its homologs can act as DNA mutators. Mol. Cell 10, 1247–1253. doi: 10.1016/s1097-2765(02)00742-6
He, Z., Zhang, W., Chen, G., Xu, R., and Yu, X. F. (2008). Characterization of conserved motifs in HIV-1 Vif required for APOBEC3G and APOBEC3F interaction. J. Mol. Biol. 381, 1000–1011. doi: 10.1016/j.jmb.2008.06.061
Heinzinger, N. K., Bukrinsky, M. I., Haggerty, S. A., Ragland, A. M., Kewalramani, V., Lee, M. A., et al. (1994). The Vpr protein of human immunodeficiency virus type 1 influences nuclear localization of viral nucleic acids in nondividing host cells. Proc. Natl. Acad. Sci. U.S.A. 91, 7311–7315. doi: 10.1073/pnas.91.15.7311
Hertz, E. P. T., Kruse, T., Davey, N. E., López-Méndez, B., Sigurðsson, J. O., Montoya, G., et al. (2016). A conserved motif provides binding specificity to the PP2A-B56 phosphatase. Mol. Cell 63, 686–695. doi: 10.1016/j.molcel.2016.06.024
Ho, D. D., Neumann, A. U., Perelson, A. S., Chen, W., Leonard, J. M., and Markowitz, M. (1995). Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature 373, 123–126. doi: 10.1038/373123a0
Holmes, R. K., Koning, F. A., Bishop, K. N., and Malim, M. H. (2007). APOBEC3F can inhibit the accumulation of HIV-1 reverse transcription products in the absence of hypermutation. Comparisons with APOBEC3G. J. Biol. Chem. 282, 2587–2595. doi: 10.1074/jbc.M607298200
Horn, V., Thélu, J., Garcia, A., Albigès-Rizo, C., Block, M. R., and Viallet, J. (2007). Functional interaction of Aurora-A and PP2A during mitosis. Mol. Biol. Cell 18, 1233–1241. doi: 10.1091/mbc.e06-12-1152
Hrecka, K., Gierszewska, M., Srivastava, S., Kozaczkiewicz, L., Swanson, S. K., Florens, L., et al. (2007). Lentiviral Vpr usurps Cul4-DDB1[VprBP] E3 ubiquitin ligase to modulate cell cycle. Proc. Natl. Acad. Sci. U.S.A. 104, 11778–11783. doi: 10.1073/pnas.0702102104
Hrecka, K., Hao, C., Shun, M. C., Kaur, S., Swanson, S. K., Florens, L., et al. (2016). HIV-1 and HIV-2 exhibit divergent interactions with HLTF and UNG2 DNA repair proteins. Proc. Natl. Acad. Sci. U.S.A. 113, E3921–E3930. doi: 10.1073/pnas.1605023113
Ito, F., Yang, H., Xiao, X., Li, S. X., Wolfe, A., Zirkle, B., et al. (2018). Understanding the structure, multimerization, subcellular localization and mC selectivity of a genomic mutator and Anti-HIV factor APOBEC3H. Sci. Rep. 8:3763. doi: 10.1038/s41598-018-21955-0
Izumi, T., Io, K., Matsui, M., Shirakawa, K., Shinohara, M., Nagai, Y., et al. (2010). HIV-1 viral infectivity factor interacts with TP53 to induce G2 cell cycle arrest and positively regulate viral replication. Proc. Natl. Acad. Sci. U.S.A. 107, 20798–20803. doi: 10.1073/pnas.1008076107
Jäger, S., Kim, D. Y., Hultquist, J. F., Shindo, K., LaRue, R. S., Kwon, E., et al. (2011). Vif hijacks CBF-β to degrade APOBEC3G and promote HIV-1 infection. Nature 481, 371–375. doi: 10.1038/nature10693
Jenkins, Y., McEntee, M., Weis, K., and Greene, W. C. (1998). Characterization of HIV-1 vpr nuclear import: analysis of signals and pathways. J. Cell Biol. 143, 875–885. doi: 10.1083/jcb.143.4.875
Jeong, A. L., and Yang, Y. (2013). PP2A function toward mitotic kinases and substrates during the cell cycle. BMB Rep. 46, 289–294. doi: 10.5483/bmbrep.2013.46.6.041
Kamura, T., Sato, S., Haque, D., Liu, L., Kaelin, W. G., Conaway, R. C., et al. (1998). The Elongin BC complex interacts with the conserved SOCS-box motif present in members of the SOCS, ras, WD-40 repeat, and ankyrin repeat families. Genes Dev. 12, 3872–3881. doi: 10.1101/gad.12.24.3872
Kitamura, S., Ode, H., Nakashima, M., Imahashi, M., Naganawa, Y., Kurosawa, T., et al. (2012). The APOBEC3C crystal structure and the interface for HIV-1 Vif binding. Nat. Struct. Mol. Biol. 19, 1005–1010. doi: 10.1038/nsmb.2378
Klatzmann, D., Barré-Sinoussi, F., Nugeyre, M. T., Danquet, C., Vilmer, E., Griscelli, C., et al. (1984). Selective tropism of lymphadenopathy associated virus (LAV) for helper-inducer T lymphocytes. Science 225, 59–63. doi: 10.1126/science.6328660
Kohli, R. M., Maul, R. W., Guminski, A. F., McClure, R. L., Gajula, K. S., Saribasak, H., et al. (2010). Local sequence targeting in the AID/APOBEC family differentially impacts retroviral restriction and antibody diversification. J. Biol. Chem. 285, 40956–40964. doi: 10.1074/jbc.M110.177402
Kouno, T., Silvas, T. V., Hilbert, B. J., Shandilya, S. M. D., Bohn, M. F., Kelch, B. A., et al. (2017). Crystal structure of APOBEC3A bound to single-stranded DNA reveals structural basis for cytidine deamination and specificity. Nat. Commun. 8:15024. doi: 10.1038/ncomms15024
Kronja, I., and Orr-Weaver, T. L. (2011). Translational regulation of the cell cycle: when, where, how and why? Philos. Trans. R. Soc. Lond. B Biol. Sci. 366, 3638–3652. doi: 10.1098/rstb.2011.0084
Kruse, T., Biedenkopf, N., Hertz, E. P. T., Dietzel, E., Stalmann, G., López-Méndez, B., et al. (2018). The ebola virus nucleoprotein recruits the host PP2A-B56 phosphatase to activate transcriptional support activity of VP30. Mol. Cell 69, 136–145.e6. doi: 10.1016/j.molcel.2017.11.034
Kruse, T., Zhang, G., Larsen, M. S., Lischetti, T., Streicher, W., Kragh Nielsen, T., et al. (2013). Direct binding between BubR1 and B56-PP2A phosphatase complexes regulate mitotic progression. J. Cell Sci. 126(Pt 5), 1086–1092. doi: 10.1242/jcs.122481
Laguette, N., Brégnard, C., Hue, P., Basbous, J., Yatim, A., Larroque, M., et al. (2014). Premature activation of the SLX4 complex by Vpr promotes G2/M arrest and escape from innate immune sensing. Cell 156, 134–145. doi: 10.1016/j.cell.2013.12.011
Lee, T. H., and Kirschner, M. W. (1996). An inhibitor of p34cdc2/cyclin B that regulates the G2/M transition in Xenopus extracts. Proc. Natl. Acad. Sci. U.S.A. 93, 352–356. doi: 10.1073/pnas.93.1.352
Letko, M., Booiman, T., Kootstra, N., Simon, V., and Ooms, M. (2015). Identification of the HIV-1 Vif and human APOBEC3G protein interface. Cell Rep. 13, 1789–1799. doi: 10.1016/j.celrep.2015.10.068
Li, M., Shandilya, S. M., Carpenter, M. A., Rathore, A., Brown, W. L., Perkins, A. L., et al. (2012). First-in-class small molecule inhibitors of the single-strand DNA cytosine deaminase APOBEC3G. ACS Chem. Biol. 7, 506–517. doi: 10.1021/cb200440y
Lindon, C., Grant, R., and Min, M. (2015). Ubiquitin-mediated degradation of aurora kinases. Front. Oncol. 5:307. doi: 10.3389/fonc.2015.00307
Madani, N., and Kabat, D. (1998). An endogenous inhibitor of human immunodeficiency virus in human lymphocytes is overcome by the viral Vif protein. J. Virol. 72, 10251–10255. doi: 10.1128/JVI.72.12.10251-10255.1998
Maertens, G. N. (2016). B′-protein phosphatase 2A is a functional binding partner of delta-retroviral integrase. Nucleic Acids Res. 44, 364–376. doi: 10.1093/nar/gkv1347
Malim, M. H., and Emerman, M. (2008). HIV-1 accessory proteins–ensuring viral survival in a hostile environment. Cell Host Microb. 3, 388–398. doi: 10.1016/j.chom.2008.04.008
Mangeat, B., Turelli, P., Caron, G., Friedli, M., Perrin, L., and Trono, D. (2003). Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature 424, 99–103. doi: 10.1038/nature01709
Marelli, S., Williamson, J. C., Protasio, A. V., Naamati, A., Greenwood, E. J., Deane, J. E., et al. (2020). Antagonism of PP2A is an independent and conserved function of HIV-1 Vif and causes cell cycle arrest. eLife 9:e53036. doi: 10.7554/eLife.53036
Margolis, S. S., Perry, J. A., Forester, C. M., Nutt, L. K., Guo, Y., Jardim, M. J., et al. (2006). Role for the PP2A/B56delta phosphatase in regulating 14-3-3 release from Cdc25 to control mitosis. Cell 127, 759–773. doi: 10.1016/j.cell.2006.10.035
Marin, M., Rose, K. M., Kozak, S. L., and Kabat, D. (2003). HIV-1 Vif protein binds the editing enzyme APOBEC3G and induces its degradation. Nat. Med. 9, 1398–1403. doi: 10.1038/nm946
Masur, H., Ognibene, F. P., Yarchoan, R., Shelhamer, J. H., Baird, B. F., Travis, W., et al. (1989). CD4 counts as predictors of opportunistic pneumonias in human immunodeficiency virus (HIV) infection. Ann. Intern. Med. 111, 223–231. doi: 10.7326/0003-4819-111-3-223
McCright, B., Rivers, A. M., Audlin, S., and Virshup, D. M. (1996). The B56 family of protein phosphatase 2A (PP2A) regulatory subunits encodes differentiation-induced phosphoproteins that target PP2A to both nucleus and cytoplasm. J. Biol. Chem. 271, 22081–22089. doi: 10.1074/jbc.271.36.22081
Mehle, A., Goncalves, J., Santa-Marta, M., McPike, M., and Gabuzda, D. (2004). Phosphorylation of a novel SOCS-box regulates assembly of the HIV-1 Vif-Cul5 complex that promotes APOBEC3G degradation. Genes Dev. 18, 2861–2866. doi: 10.1101/gad.1249904
Moura, M., and Conde, C. (2019). Phosphatases in mitosis: roles and regulation. Biomolecules 9:55. doi: 10.3390/biom9020055
Naamati, A., Williamson, J. C., Greenwood, E. J., Marelli, S., Lehner, P. J., and Matheson, N. J. (2019). Functional proteomic atlas of HIV infection in primary human CD4+ T cells. eLife 8:e041431. doi: 10.7554/eLife.41431
Nagata, K., Shindo, K., Matsui, Y., Shirakawa, K., and Takaori-Kondo, A. (2020). Critical role of PP2A-B56 family protein degradation in HIV-1 Vif mediated G2 cell cycle arrest. Biochem. Biophys. Res. Commun. 527, 257–263. doi: 10.1016/j.bbrc.2020.04.123
Nakashima, M., Tsuzuki, S., Awazu, H., Hamano, A., Okada, A., Ode, H., et al. (2017). Mapping region of human restriction factor APOBEC3H critical for interaction with HIV-1 Vif. J. Mol. Biol. 429, 1262–1276. doi: 10.1016/j.jmb.2017.03.019
Neil, S. J., Zang, T., and Bieniasz, P. D. (2008). Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 451, 425–430. doi: 10.1038/nature06553
Newman, E. N., Holmes, R. K., Craig, H. M., Klein, K. C., Lingappa, J. R., Malim, M. H., et al. (2005). Antiviral function of APOBEC3G can be dissociated from cytidine deaminase activity. Curr. Biol. 15, 166–170. doi: 10.1016/j.cub.2004.12.068
Nilsson, J. (2019). Protein phosphatases in the regulation of mitosis. J. Cell Biol. 218, 395–409. doi: 10.1083/jcb.201809138
Ooms, M., Brayton, B., Letko, M., Maio, S. M., Pilcher, C. D., Hecht, F. M., et al. (2013). HIV-1 Vif adaptation to human APOBEC3H haplotypes. Cell Host Microb. 14, 411–421. doi: 10.1016/j.chom.2013.09.006
Ooms, M., Letko, M., and Simon, V. (2017). The structural interface between HIV-1 Vif and human APOBEC3H. J. Virol. 91:e02289-16. doi: 10.1128/JVI.02289-16
Parsons, G. G., and Spencer, C. A. (1997). Mitotic repression of RNA polymerase II transcription is accompanied by release of transcription elongation complexes. Mol. Cell Biol. 17, 5791–5802. doi: 10.1128/mcb.17.10.5791
Pery, E., Rajendran, K. S., Brazier, A. J., and Gabuzda, D. (2009). Regulation of APOBEC3 proteins by a novel YXXL motif in human immunodeficiency virus type 1 Vif and simian immunodeficiency virus SIVagm Vif. J. Virol. 83, 2374–2381. doi: 10.1128/JVI.01898-08
Petersen-Mahrt, S. K., Harris, R. S., and Neuberger, M. S. (2002). AID mutates E. coli suggesting a DNA deamination mechanism for antibody diversification. Nature 418, 99–103. doi: 10.1038/nature00862
Popovic, M., Sarngadharan, M. G., Read, E., and Gallo, R. C. (1984). Detection, isolation, and continuous production of cytopathic retroviruses (HTLV-III) from patients with AIDS and pre-AIDS. Science 224, 497–500. doi: 10.1126/science.6200935
Rathore, A., Carpenter, M. A., Demir, Ö, Ikeda, T., Li, M., Shaban, N. M., et al. (2013). The local dinucleotide preference of APOBEC3G can be altered from 5′-CC to 5′-TC by a single amino acid substitution. J. Mol. Biol. 425, 4442–4454. doi: 10.1016/j.jmb.2013.07.040
Richards, C., Albin, J. S., Demir, Ö, Shaban, N. M., Luengas, E. M., Land, A. M., et al. (2015). The binding interface between human APOBEC3F and HIV-1 Vif elucidated by genetic and computational approaches. Cell Rep. 13, 1781–1788. doi: 10.1016/j.celrep.2015.10.067
Romani, B., Shaykh Baygloo, N., Aghasadeghi, M. R., and Allahbakhshi, E. (2015). HIV-1 Vpr protein enhances proteasomal degradation of MCM10 DNA replication factor through the Cul4-DDB1[VprBP] E3 Ubiquitin ligase to induce G2/M cell cycle arrest. J. Biol. Chem. 290, 17380–17389. doi: 10.1074/jbc.M115.641522
Roopchand, D. E., Lee, J. M., Shahinian, S., Paquette, D., Bussey, H., and Branton, P. E. (2001). Toxicity of human adenovirus E4orf4 protein in Saccharomyces cerevisiae results from interactions with the Cdc55 regulatory B subunit of PP2A. Oncogene 20, 5279–5290. doi: 10.1038/sj.onc.1204693
Rosa, A., Chande, A., Ziglio, S., De Sanctis, V., Bertorelli, R., Goh, S. L., et al. (2015). HIV-1 Nef promotes infection by excluding SERINC5 from virion incorporation. Nature 526, 212–217. doi: 10.1038/nature15399
Russell, R. A., and Pathak, V. K. (2007). Identification of two distinct human immunodeficiency virus type 1 Vif determinants critical for interactions with human APOBEC3G and APOBEC3F. J. Virol. 81, 8201–8210. doi: 10.1128/JVI.00395-07
Sakai, H., Shibata, R., Sakuragi, J., Sakuragi, S., Kawamura, M., and Adachi, A. (1993). Cell-dependent requirement of human immunodeficiency virus type 1 Vif protein for maturation of virus particles. J. Virol. 67, 1663–1666. doi: 10.1128/JVI.67.3.1663-1666.1993
Sakai, K., Barnitz, R. A., Chaigne-Delalande, B., Bidère, N., and Lenardo, M. J. (2011). Human immunodeficiency virus type 1 Vif causes dysfunction of Cdk1 and CyclinB1: implications for cell cycle arrest. Virol. J. 8:219. doi: 10.1186/1743-422X-8-219
Sakai, K., Dimas, J., and Lenardo, M. J. (2006). The Vif and Vpr accessory proteins independently cause HIV-1-induced T cell cytopathicity and cell cycle arrest. Proc. Natl. Acad. Sci. U.S.A. 103, 3369–3374. doi: 10.1073/pnas.0509417103
Salamango, D., McCann, J. L., Demir, O., Becker, J., Wang, J., Lingappa, J., et al. (2020). Functional and structural insights into a Vif/PPP2R5 complex elucidated using patient HIV-1 isolates and computational modeling. J. Virol. 94:e0631-20.
Salamango, D. J., Ikeda, T., Moghadasi, S. A., Wang, J., McCann, J. L., Serebrenik, A. A., et al. (2019). HIV-1 Vif triggers cell cycle arrest by degrading cellular PPP2R5 Phospho-regulators. Cell Rep. 29, 1057–1065.e4. doi: 10.1016/j.celrep.2019.09.057
Schindler, M., Münch, J., Kutsch, O., Li, H., Santiago, M. L., Bibollet-Ruche, F., et al. (2006). Nef-mediated suppression of T cell activation was lost in a lentiviral lineage that gave rise to HIV-1. Cell 125, 1055–1067. doi: 10.1016/j.cell.2006.04.033
Shaban, N. M., Shi, K., Lauer, K. V., Carpenter, M. A., Richards, C. M., Salamango, D., et al. (2018). The antiviral and cancer genomic DNA deaminase apobec3h is regulated by an RNA-mediated dimerization mechanism. Mol. Cell 69, 75–86.e9. doi: 10.1016/j.molcel.2017.12.010
Sheehy, A. M., Gaddis, N. C., Choi, J. D., and Malim, M. H. (2002). Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 418, 646–650. doi: 10.1038/nature00939
Sheehy, A. M., Gaddis, N. C., and Malim, M. H. (2003). The antiretroviral enzyme APOBEC3G is degraded by the proteasome in response to HIV-1 Vif. Nat. Med. 9, 1404–1407. doi: 10.1038/nm945
Shi, J., Xiong, R., Zhou, T., Su, P., Zhang, X., Qiu, X., et al. (2018). HIV-1 Nef antagonizes SERINC5 restriction by downregulation of SERINC5 via the Endosome/Lysosome system. J. Virol. 92:e00196-18. doi: 10.1128/JVI.00196-18
Shi, K., Carpenter, M. A., Banerjee, S., Shaban, N. M., Kurahashi, K., Salamango, D. J., et al. (2017). Structural basis for targeted DNA cytosine deamination and mutagenesis by APOBEC3A and APOBEC3B. Nat. Struct. Mol. Biol. 24, 131–139. doi: 10.1038/nsmb.3344
Simon, J. H., Gaddis, N. C., Fouchier, R. A., and Malim, M. H. (1998a). Evidence for a newly discovered cellular anti-HIV-1 phenotype. Nat. Med. 4, 1397–1400. doi: 10.1038/3987
Simon, J. H., Miller, D. L., Fouchier, R. A., Soares, M. A., Peden, K. W., and Malim, M. H. (1998b). The regulation of primate immunodeficiency virus infectivity by Vif is cell species restricted: a role for Vif in determining virus host range and cross-species transmission. EMBO J. 17, 1259–1267. doi: 10.1093/emboj/17.5.1259
Siu, K. K., Sultana, A., Azimi, F. C., and Lee, J. E. (2013). Structural determinants of HIV-1 Vif susceptibility and DNA binding in APOBEC3F. Nat. Commun. 4:2593. doi: 10.1038/ncomms3593
Strebel, K., Daugherty, D., Clouse, K., Cohen, D., Folks, T., and Martin, M. A. (1987). The HIV ‘A’ (sor) gene product is essential for virus infectivity. Nature 328, 728–730. doi: 10.1038/328728a0
Strzyz, P. (2017). Transcription: no proper rest in mitosis. Nat. Rev. Mol. Cell Biol. 18, 652–653. doi: 10.1038/nrm.2017.102
Tanenbaum, M. E., Stern-Ginossar, N., Weissman, J. S., and Vale, R. D. (2015). Regulation of mRNA translation during mitosis. eLife 4:e007957. doi: 10.7554/eLife.07957
Tang, Z., Shu, H., Qi, W., Mahmood, N. A., Mumby, M. C., and Yu, H. (2006). PP2A is required for centromeric localization of Sgo1 and proper chromosome segregation. Dev. Cell 10, 575–585. doi: 10.1016/j.devcel.2006.03.010
Thierry, S., Marechal, V., Rosenzwajg, M., Sabbah, M., Redeuilh, G., Nicolas, J. C., et al. (2004). Cell cycle arrest in G2 induces human immunodeficiency virus type 1 transcriptional activation through histone acetylation and recruitment of CBP, NF-kappaB, and c-Jun to the long terminal repeat promoter. J. Virol. 78, 12198–12206. doi: 10.1128/JVI.78.22.12198-12206.2004
Thompson, J. J., and Williams, C. S. (2018). Protein phosphatase 2A in the regulation of wnt signaling, stem cells, and cancer. Genes 9:121. doi: 10.3390/genes9030121
Timofeev, O., Cizmecioglu, O., Settele, F., Kempf, T., and Hoffmann, I. (2010). Cdc25 phosphatases are required for timely assembly of CDK1-cyclin B at the G2/M transition. J. Biol. Chem. 285, 16978–16990. doi: 10.1074/jbc.M109.096552
Usami, Y., Wu, Y., and Göttlinger, H. G. (2015). SERINC3 and SERINC5 restrict HIV-1 infectivity and are counteracted by Nef. Nature 526, 218–223. doi: 10.1038/nature15400
Van Damme, N., Goff, D., Katsura, C., Jorgenson, R. L., Mitchell, R., Johnson, M. C., et al. (2008). The interferon-induced protein BST-2 restricts HIV-1 release and is downregulated from the cell surface by the viral Vpu protein. Cell Host Microb. 3, 245–252. doi: 10.1016/j.chom.2008.03.001
Wang, J., Becker, J. T., Shi, K., Lauer, K. V., Salamango, D. J., Aihara, H., et al. (2019). The role of RNA in HIV-1 Vif-mediated degradation of APOBEC3H. J. Mol. Biol. 431, 5019–5031. doi: 10.1016/j.jmb.2019.09.014
Wang, J., Shackelford, J. M., Casella, C. R., Shivers, D. K., Rapaport, E. L., Liu, B., et al. (2007). The Vif accessory protein alters the cell cycle of human immunodeficiency virus type 1 infected cells. Virology 359, 243–252. doi: 10.1016/j.virol.2006.09.026
Wang, J., Wang, Z., Yu, T., Yang, H., Virshup, D. M., Kops, G. J., et al. (2016). Crystal structure of a PP2A B56-BubR1 complex and its implications for PP2A substrate recruitment and localization. Protein Cell 7, 516–526. doi: 10.1007/s13238-016-0283-4
Wang, X., Bajaj, R., Bollen, M., Peti, W., and Page, R. (2016). Expanding the PP2A interactome by defining a B56-Specific SLiM. Structure 24, 2174–2181. doi: 10.1016/j.str.2016.09.010
Wang, X., Garvanska, D. H., Nasa, I., Ueki, Y., Zhang, G., Kettenbach, A. N., et al. (2020). A dynamic charge-charge interaction modulates PP2A:B56 substrate recruitment. eLife 9:e055966. doi: 10.7554/eLife.55966
Wolfe, L. S., Stanley, B. J., Liu, C., Eliason, W. K., and Xiong, Y. (2010). Dissection of the HIV Vif interaction with human E3 ubiquitin ligase. J. Virol. 84, 7135–7139. doi: 10.1128/JVI.00031-10
Wu, Y., Zhou, X., Barnes, C. O., DeLucia, M., Cohen, A. E., Gronenborn, A. M., et al. (2016). The DDB1-DCAF1-Vpr-UNG2 crystal structure reveals how HIV-1 Vpr steers human UNG2 toward destruction. Nat. Struct. Mol. Biol. 23, 933–940. doi: 10.1038/nsmb.3284
Xu, Z., Cetin, B., Anger, M., Cho, U. S., Helmhart, W., Nasmyth, K., et al. (2009). Structure and function of the PP2A-shugoshin interaction. Mol. Cell 35, 426–441. doi: 10.1016/j.molcel.2009.06.031
Yamashita, T., Kamada, K., Hatcho, K., Adachi, A., and Nomaguchi, M. (2008). Identification of amino acid residues in HIV-1 Vif critical for binding and exclusion of APOBEC3G/F. Microb. Infect. 10, 1142–1149. doi: 10.1016/j.micinf.2008.06.003
Yu, X., Yu, Y., Liu, B., Luo, K., Kong, W., Mao, P., et al. (2003). Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science 302, 1056–1060. doi: 10.1126/science.1089591
Yu, Y., Xiao, Z., Ehrlich, E. S., Yu, X., and Yu, X. F. (2004). Selective assembly of HIV-1 Vif-Cul5-ElonginB-ElonginC E3 ubiquitin ligase complex through a novel SOCS box and upstream cysteines. Genes Dev. 18, 2867–2872. doi: 10.1101/gad.1250204
Zhang, F., and Bieniasz, P. D. (2020). HIV-1 Vpr induces cell cycle arrest and enhances viral gene expression by depleting CCDC137. eLife 9:e055806. doi: 10.7554/eLife.55806
Zhang, H., Yang, B., Pomerantz, R. J., Zhang, C., Arunachalam, S. C., and Gao, L. (2003). The cytidine deaminase CEM15 induces hypermutation in newly synthesized HIV-1 DNA. Nature 424, 94–98. doi: 10.1038/nature01707
Zhang, W., Du, J., Evans, S. L., Yu, Y., and Yu, X. F. (2011). T-cell differentiation factor CBF-β regulates HIV-1 Vif-mediated evasion of host restriction. Nature 481, 376–379. doi: 10.1038/nature10718
Zhang, W., Huang, M., Wang, T., Tan, L., Tian, C., Yu, X., et al. (2008). Conserved and non-conserved features of HIV-1 and SIVagm Vif mediated suppression of APOBEC3 cytidine deaminases. Cell Microbiol. 10, 1662–1675. doi: 10.1111/j.1462-5822.2008.01157.x
Zhao, K., Du, J., Rui, Y., Zheng, W., Kang, J., Hou, J., et al. (2015). Evolutionarily conserved pressure for the existence of distinct G2/M cell cycle arrest and A3H inactivation functions in HIV-1 Vif. Cell Cycle 14, 838–847. doi: 10.1080/15384101.2014.1000212
Zhao, R. Y., and Elder, R. T. (2005). Viral infections and cell cycle G2/M regulation. Cell Res. 15, 143–149. doi: 10.1038/sj.cr.7290279
Zhen, A., Du, J., Zhou, X., Xiong, Y., and Yu, X. F. (2012). Reduced APOBEC3H variant anti-viral activities are associated with altered RNA binding activities. PLoS One 7:e38771. doi: 10.1371/journal.pone.0038771
Zhou, X., Evans, S. L., Han, X., Liu, Y., and Yu, X. F. (2012). Characterization of the interaction of full-length HIV-1 Vif protein with its key regulator CBFβ and CRL5 E3 ubiquitin ligase components. PLoS One 7:e33495. doi: 10.1371/journal.pone.0033495
Keywords: APOBEC3, APOBEC3G, cell cycle arrest, PPP2R5, Vif
Citation: Salamango DJ and Harris RS (2021) Dual Functionality of HIV-1 Vif in APOBEC3 Counteraction and Cell Cycle Arrest. Front. Microbiol. 11:622012. doi: 10.3389/fmicb.2020.622012
Received: 27 October 2020; Accepted: 11 December 2020;
Published: 12 January 2021.
Edited by:
Hirofumi Akari, Kyoto University, JapanReviewed by:
Jean-Christophe Paillart, Université de Strasbourg, FranceYong-Hui Zheng, Michigan State University, United States
Taisuke Izumi, Henry M. Jackson Foundation for the Advancement of Military Medicine (HJF), United States
Copyright © 2021 Salamango and Harris. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Reuben S. Harris, cnNoQHVtbi5lZHU=; Daniel J. Salamango, ZGFuaWVsLnNhbGFtYW5nb0BzdG9ueWJyb29rLmVkdQ==
†Present address: Daniel J. Salamango, Department of Microbiology and Immunology, Stony Brook University, Stony Brook, NY, United States