 Anjney Sharma1,2
Anjney Sharma1,2 Ram Nageena Singh3
Ram Nageena Singh3 Xiu-Peng Song2
Xiu-Peng Song2 Rajesh Kumar Singh1,2
Rajesh Kumar Singh1,2 Dao-Jun Guo1,2,4
Dao-Jun Guo1,2,4 Pratiksha Singh1,2
Pratiksha Singh1,2 Krishan K. Verma1,2
Krishan K. Verma1,2 Yang-Rui Li1,2,4*
Yang-Rui Li1,2,4*- 1Key Laboratory of Sugarcane Biotechnology and Genetic Improvement, Ministry of Agriculture, Sugarcane Research Center, Chinese Academy of Agricultural Sciences, Guangxi Academy of Agricultural Sciences (GXXAS), Nanning, Guangxi, China
- 2Guangxi Key Laboratory of Sugarcane Genetic Improvement, Sugarcane Research Institute, Guangxi Academy of Agricultural Sciences, Nanning, Guangxi, China
- 3Department of Chemical and Biological Engineering, South Dakota School of Mines and Technology, Rapid City, SD, United States
- 4State Key Laboratory of Conservation and Utilization of Subtropical, College of Agriculture, Agro-Bioresources, Guangxi University, Nanning, Guangxi, China
Globally, due to widespread dispersion, intraspecific diversity, and crucial ecological components of halophilic ecosystems, halophilic bacteria is considered one of the key models for ecological, adaptative, and biotechnological applications research in saline environments. With this aim, the present study was to enlighten the plant growth-promoting features and investigate the systematic genome of a halophilic bacteria, Virgibacillus halodenitrificans ASH15, through single-molecule real-time (SMRT) sequencing technology. Results showed that strain ASH15 could survive in high salinity up to 25% (w/v) NaCl concentration and express plant growth-promoting traits such as nitrogen fixation, plant growth hormones, and hydrolytic enzymes, which sustain salt stress. The results of pot experiment revealed that strain ASH15 significantly enhanced sugarcane plant growth (root shoot length and weight) under salt stress conditions. Moreover, the sequencing analysis of the strain ASH15 genome exhibited that this strain contained a circular chromosome of 3,832,903 bp with an average G+C content of 37.54%: 3721 predicted protein-coding sequences (CDSs), 24 rRNA genes, and 62 tRNA genes. Genome analysis revealed that the genes related to the synthesis and transport of compatible solutes (glycine, betaine, ectoine, hydroxyectoine, and glutamate) confirm salt stress as well as heavy metal resistance. Furthermore, functional annotation showed that the strain ASH15 encodes genes for root colonization, biofilm formation, phytohormone IAA production, nitrogen fixation, phosphate metabolism, and siderophore production, which are beneficial for plant growth promotion. Strain ASH15 also has a gene resistance to antibiotics and pathogens. In addition, analysis also revealed that the genome strain ASH15 has insertion sequences and CRISPRs, which suggest its ability to acquire new genes through horizontal gene transfer and acquire immunity to the attack of viruses. This work provides knowledge of the mechanism through which V. halodenitrificans ASH15 tolerates salt stress. Deep genome analysis, identified MVA pathway involved in biosynthesis of isoprenoids, more precisely “Squalene.” Squalene has various applications, such as an antioxidant, anti-cancer agent, anti-aging agent, hemopreventive agent, anti-bacterial agent, adjuvant for vaccines and drug carriers, and detoxifier. Our findings indicated that strain ASH15 has enormous potential in industries such as in agriculture, pharmaceuticals, cosmetics, and food.
Introduction
With the ever-changing climate and global warming, rising sea level are a growing concern which increase soil salinity across the coastal areas, thereby globally imposing a detrimental effect on soil quality, decreasing land areas, and reducing agricultural crop production, resulting in an instable national economy (Corwin, 2021; Godde et al., 2021; Ullah et al., 2021). Salinity stress causes changes in various physiological and metabolic processes of the plant, ultimately inhibiting the quality and productivity of agriculturally important crops (Sharma et al., 2021). In the current scenario, with limited cultivated land resources, growing crops in saline soil may be a feasible opportunity (Xiaoqin et al., 2021). In light of the rising global need for food and agricultural production, scientists are looking for new, more eco-friendly, greener, and more sustainable alternatives pesticides and chemical fertilizers (Ullah et al., 2021). In this framework, the unusual halophilic bacteria thriving in saline environments have been a subject of study for the last few years due to their interesting physiological and metabolic adaptation properties to their extreme environmental conditions (Dutta and Bandopadhyay, 2022). These incredibly resilient microorganisms can thrive at 0% to saturation salt concentrations. The basic mechanisms of these extremophiles microbes for halo-adaptation and surviving in saline habitats are based on two strategies to regulate intracellular osmolarity and evade water loss (Zhou et al., 2017). The first is the “salt-in” approach, where KCl inorganic salt is stored to provide the cellular osmotic pressure and balance the external osmotic pressure. The other is known as the “salt-out” (compatible solutes) strategy, in which low-molecular-weight, highly soluble organic molecules are cumulated to ensure cellular osmotic balance without hindering crucial cellular processes even when they are occurring at high levels (Sharma et al., 2016). Extremely halophilic and anaerobic moderately halophilic bacteria adopted the “salt-in” strategy, while the majority of bacteria used the “salt-out” (compatible solutes) approach (Zhou et al., 2017). There are numerous reports of new halophilic bacteria and archaea species available from diverse hypersaline locations in various nations, primarily from USA, Australia, Korea, China, India, Thailand, Taiwan, Russia, France, Austria, Spain, Japan, Egypt, Iran, Indonesia, Mexico, the Philippines, Poland, and Romania (Naghoni et al., 2017; Corral et al., 2019; Reang et al., 2022).
The exploitation of new halophilic microorganisms is always of special importance and interest in the current era for the evaluation and development of new biomolecules with potential applications in agriculture and several industries. Halophilic microorganisms help in the improvement of soil structure, plant salt tolerance, and growth through various mechanisms such as phytohormones production (IAA and ABA), solubilizing the essential nutrients (P, K, and Zn, etc.), regulating the ethylene level, and inducing systemic resistance (ISR) against the harmful plant pathogens through the production of secondary metabolite/antimicrobial peptides (Arora et al., 2020; Masmoudi et al., 2023). Additionally, halophilic microorganisms mitigate the salt stress in plants by maintaining high K+/Na+ and Ca2+ and scavenging ROS by regulating the expression of antioxidant enzymes and stress-responsive genes (Sharma et al., 2021). In addition, low nutritional requirements, genetic pieces of machinery, and great metabolic versatility for adaptation to harsh environmental environments make halophilic microorganisms a promising candidate and a hope for new sources of enzymes, drug discovery, and other biological materials with applications in various human welfare fields (Vaidya et al., 2018). Several extremely halophilic and halotolerant bacteria, such as Bacillus, Haloferax, Micrococcus, Salinibacter Halobacterium, Halobacillus, Virgibacillus, and Haloarcula, were reported from various saline environments (Gupta et al., 2015; Satari et al., 2021). Also, the plant growth-promoting effectiveness of so many halophilic bacteria was investigated in seed germination and growth promotion of several agriculturally important crops such as rice (Abbas et al., 2019; Suriani et al., 2020), tomato, cotton, maize (Anbumalar and Ashokumar, 2016), and sugarcane (Sharma et al., 2021).
Among the various bacterial genera inhabiting various extreme environments, the morphologically, biochemically, and genetically diverse genus Virgibacillus is widely recognized as an important model group for agriculture and industrial applications (Sánchez-Porro et al., 2014; Fayez et al., 2022). A wide range of different species of Virgibacillus have been reported globally from various saline environments, such as seawater, marine sediments, lakes, soil, and fermented seafoods (Montriwong et al., 2012; Amziane et al., 2013; Xu et al., 2018; Mechri et al., 2019; Bhatt and Singh, 2022). To date, 39 validated and 381 non-validated species in the Virgibacillus genus have been published in the NCBI database (Chen et al., 2018), whereas six complete genomic sequences of Virgibacillus species are available in the NCBI database (2017) (Chen et al., 2018). Virgibacillus halodenitrificans is one of such bacteria whose potential is less explored (Lee et al., 2012; Kumaunang et al., 2019; Fayez et al., 2022). In 1989, the first report of a halophilic denitrifier, Bacillus halodenitrificans, from a solar saltern was reported by Denariaz et al. (1989). The isolates grew and survived well in various NaCl (0.35 to 4.25 M NaCl) supplement mediums, with optimum growth was considered between 0.5 and 1.35 M (3 to 8%) NaCl. Later on, in 2004, Bacillus halodenitrificans was transferred into the genus Virgibacillus as Virgibacillus halodenitrificans (Yoon et al., 2004). Although numerous halophiles have been thoroughly defined to date, V. halodenitrificans is still one of the least explored organisms in terms of the number of published research studies, strain characterizations, and whole genome sequence analysis (Lee et al., 2012). Thus, in order to explore its capabilities to be commercialized, it is required to understand its genetic structure and metabolic mechanisms involved in the biosynthesis of beneficial biomolecules.
With the advancement of powerful tools and “omics” approaches such as whole-genome sequencing analysis, deciphering new insights into halophilic microorganisms (Durán-Viseras et al., 2021; Lam et al., 2021). Also, in response to the extreme environments, the concomitant advances in genomics are disclosing uncountable encoding genes for understanding the adaptation strategies, physiological attributes, and metabolic features of the halophilic bacteria (Corral et al., 2019). This resulted in a plethora of genomic information that was mined for potential agricultural and industrial applications (Ziemert et al., 2016; Othoum et al., 2019; Passari et al., 2019). Draft genomes with inaccurate or incomplete genomic data and low completeness are not fully reliable for phylogenomics, genome structure, genome synteny, and pan-genomic investigations (Denton et al., 2014). Thus, to know more about physiological, metabolic, and functional mechanisms, there is a need to generate high-quality whole genome sequences of halophilic microorganisms on a large scale to further understand the complete role of genes and their proteins in various extreme environments. The information from whole genome sequence analysis will constitute an exciting period for microbiology and the allied sector in the near future, which is generally not fully explored in the draft genome sequencing analysis. Since the first report of Virgibacillus halodenitrificans (Denariaz et al., 1989; Yoon et al., 2004), only one draft genome (Lee et al., 2012) and one complete genome (Zhou et al., 2017) have been published.
Therefore, in this study, we isolated and characterized a halophilic bacterium Virgibacillus halodenitrificans strain ASH15, from sugarcane-grown field in the coastal regions of Beihai, China and sequenced its entire genome. In addition, the plant growth-promoting efficiency of V. halodenitrificans ASH15 was evaluated for sugarcane plant growth under greenhouse conditions. Furthermore, systematic analysis of whole genome sequence data and the identification of genes will aid our understanding of the molecular mechanisms of osmoadaptation and the metabolic activities of the strain. Moreover, the obtained genome information will help to develop the strain ASH15 as an eco-friendly model for industrially important biomolecules and sustainable agriculture production.
Materials and methods
Sampling and isolation of halophilic bacteria
The soil sample was collected from the sugarcane-growing field of the sea city of Beihai, China (Latitude 21.4811° N, Longitude 109.1201° E). For the isolation of specific halophilic bacteria, the collected soil sample was enriched in a nutrient broth (NB) medium containing 10% NaCl at 37°C for 72 h. After the enrichment, the enriched soil sample was heat-treated at 80°C for 15 minutes to kill vegetative cells (Sharma et al., 2015). Spore-forming halophilic bacteria were isolated through the standard serial dilution method by spreading the diluted soil sample (10−4) over a nutrient agar (NA) growth medium plate supplemented with 10% NaCl. Plates were incubated at 37°C for 72 h. After the incubation, a dominant bacterial isolate designated as ASH15 was recovered and purified by sub-culturing on the same NaCl-amended growth medium. The purified culture was preserved in 50% glycerol stock at −80°C for further study.
Growth kinetics studies under different levels of NaCl stress
Bacterial growth kinetics under different NaCl concentrations was spectroscopically determined in a 96-well microplate at 37°C. In brief, 1% of pure bacterial culture was transferred to an individual well containing 200 μl of NB broth with different NaCl concentrations, viz., 0, 5, 10, 15, 20, and 25%, and incubated to grow at 37°C under shaking condition at 150 rpm. The growth was spectroscopically monitored in a microplate reader by taking absorbance at 600 nm at every 12 h time interval.
Scanning electron microscopic analysis
A pure single bacterial colony was inoculated in NB and incubated for 48 h at 37°C under shaking conditions. After incubation, the cell pellet of bacterial culture was collected via centrifugation at 10,000 rpm at room temperature for 10 min. The collected pellet was washed 2–3 times with 100 mM phosphate buffer and then fixed with a 2.5% glutaraldehyde solution and incubated at 4°C for 10–12 h. After fixation, treated cells were further washed with phosphate buffer and with a gradient concentration of ethanol (10 to 100%) every 10 min. Following by, sample was dried in desiccators, mounted onto SEM stubs, and coated thinly with gold and palladium (60:40), and then sample was examined using the SEM machine.
Metabolic characterization through BIOLOG
The metabolic potentiality of the strain ASH15 was tested based on the carbon (C) utilization pattern in the BIOLOG Micro-Array TM GENIII plate (Biolog Inc., Hayward, CA) which contained 95 different carbon sources. Briefly, a single pure colony of strain ASH15 was streaked on NA plates and incubated at 30°C for 24 h. After the incubation, the bacterial culture mass was scraped from the surface of the plate and transferred into a sterile 2-ml centrifuge tube. Collected cells were washed with phosphate buffer and suspended in 20 ml of inoculation fluid (IF) (Biolog Inc., Hayward, CA) to reach a transmittance of 81–85% as per the manufacturer's instructions. A 100 μl of suspension was inoculated in each well of the GNIII plate, which contained 95 different C sources. After incubation to record the results, the plate was read in an automated BIOLOG(R) Micro-Station Reader according to the manufacturer's instructions.
Biofilm formation and motility assay
The biofilm formation capacity of the strain ASH15 was assayed according to the method of Qurashi and Sabri (2012). 100 μl of pure bacterial culture suspension (108 CFU/ml) was transferred into a well of a microtiter plate which contained 200 μl NB of different NaCl concentrations. The plate was airtight packed and incubated at 37°C for 72 h. The medium from each well of the plate was thrown out, and the well was washed 2–3 times using distilled water. A 0.01% solution of crystal violet dye was added to each dried well. After 10 min of incubation, the dye was drained, and the plate was rinsed 2–3 times with sterile distilled water (D/W). Following that, in a rinsed and dried plate, 100 μL of acetic acid (30%) was added to solubilize the cell's bound remaining dye. Absorbance at 570 nm was taken to quantify the biofilm formation.
Swarming motility was detected by adopting the method of Connelly et al. (2004). Freshly grown bacterial cultures was point-inoculated on swarm plates consisted of 0.5% Bacto-agar (w/v) and 8 g l−1 of NB supplemented with 5 g l−1 of dextrose. After 24 h of incubation at 37°C, positive results as a swarming zone was recorded.
Characterization of different plant growth-promoting traits
The indole acetic acid (IAA) production ability of the test strain ASH15 was determined by following the standard protocol of Brick et al. (1991) using the Salkowski reagent. The phosphate solubilizing ability of the strain was analyzed by spot inoculation of the bacterial culture on the National Botanical Research Institute's Phosphate Medium agar plate (NBRIP) (Mehta and Nautiyal, 2001). The in-vitro zinc solubilization ability of the strain was carried out by employing plate assay of Sharma et al. (2012). The siderophore production ability of the test strain was assayed on a chrome azurol S (CAS) agar plate by adopting the standard method of Schwyn and Neilands (1987). The qualitative nitrogen (N) fixation capacity of the strain ASH15 was tested on an Ashby's Mannitol agar medium (Ashby, 1907). Exopolysaccharide (EPS) production was determined according to the method of Kumari et al. (2015). All the abovementioned plant growth-promoting traits were assayed under normal and different NaCl concentrations.
Effect of strain ASH15 on sugarcane growth under greenhouse condition
To evaluate the plant growth-promoting activity of the strain ASH15, a greenhouse experiment with sugarcane under salt stress (NaCl) and non-stressed conditions was performed at the Sugarcane Research Institute, Guangxi Academy of Agricultural Sciences, Nanning, China. The bacterial inoculum was prepared by centrifugation of freshly grown bacterial cultures. The collected bacterial cell pellet was washed 2–3 times with 0.1 M phosphate buffer and resuspended in the same buffer to make the bacterial suspension. Sugarcane seedlings GT42 (15 days old), were washed 3–4 times with sterilized distilled water (D/W) and treated with bacterial suspension (CFU 108) for 3 h. For control treatment, D/W was used in place of bacterial suspension. All the treated and non-treated sugarcane plants were planted in plastic pots that contained sterilized soil: sand mixture (3:1). A salt stress treatment of 200 mM NaCl was given after 10 days of plant establishment in the pot. The following treatments (T) were applied in the greenhouse experiment: T-1 (control: un-inoculated, no stress), T-2 (bacterial treatment), T-3 (200 mM salt stress treatment), and T-4 (200 mM salt stress + bacterial treatment). The experiment was conducted in a completely randomized manner in triplicate under a 16/8 h light/dark cycle with 80% field water capacity (FWC) moisture at 28 ± 2°C temperature. After 30 days of stress treatment, the plants from all treatments were uprooted, cleaned, and vegetative growth parameters, such as shoot length (SL), root length (RL), shoot fresh weight (SFW), and shoot dry weight (SDW), root fresh weight (RFW), and root dry weight (RDW), were taken.
DNA extraction, library construction and whole genome sequencing analysis
For complete genome sequencing, genomic DNA was extracted from a full-grown culture of strain ASH15 using the DNA extraction kit (CWBIO, Beijing, China) according to the manufacturer's instructions. DNA quality and quantity were assessed using the TBS-380 fluorometer (Turner BioSystems Inc., Sunnyvale, CA, United States), and high-quality genomic DNA (OD260/280 = 1.8–2.0, >20 μg) was used for further processing. The genome sequencing was performed using single-molecule real-time (SMRT) Oxford-Nanopore and Pacbio (third generation) sequencing technology. Moreover, 15 μg of high-quality DNA was processed for fragmentation using Covaris G-TUBE (Covaris, MA, United States) for 60 s at 6,000 rpm. Genomic DNA fragments were purified, end-repaired, and ligated via SMRTbell sequencing adapters according to the manufacturer's protocol (Pacific Biosciences, CA, United States) and purified using AMPureXP beads (Beckman Coulter Genomics, MA, United States). Further, approximately 10 kb insert library was sequenced on one SMRT cell by standard procedures. For Nanopore sequencing, high-quality genomic DNA with a large fraction was selected using Blue Pippin (Sage Science, USA), followed by end-repair/dA tailing. End-repaired DNA fragments are processed for adapter ligation using a ligation sequencing kit (NBD103 and NBD114, Oxford Nanopore Technologies USA). Finally, the DNA library was quantified through Qubit 3.0 (Thermo Fisher Technologies USA). Afterward, 11 μL of DNA library was loaded into a 1 flow cell and sequenced on a PromethION sequencer (Oxford Nanopore Technologies USA).
Genome assembly, gene prediction, and functional annotation
The raw sequence data was processed for quality checks by employing Majorbio Cloud Platform1 (Shanghai Majorbio Co., Ltd.). Quality-passed raw sequence data reads were then assembled into contigs using the hierarchical genome assembly method (HGAP) (Chin et al., 2013). The final genome assembly was finished using Pilon. The assembled genome was further processed for gene prediction and annotations. Prediction of coding sequence (CDS) was conducted with Glimmer version 3.02, followed by annotation using multiple databases, i.e., Pfam, Swiss-Prot, NR, Clusters of Orthologous Groups (COG), Kyoto Encyclopedia of Genes and Genomes (KEGG, http://www.genome.jp/kegg/), and Gene Ontology (GO) (Delcher et al., 2007) with Basic Local Alignment Search Tool (BLAST), DIAMOND sequence alignment and HMMER, and other tools. tRNAs and rRNAs were predicted using tRNA-scan-SE (v1.2.1) (Borodovsky and Mcininch, 1993) and Barrnap. A circular genome map of strain ASH15 was constructed using genome annotation files on the CGviewer server (Grant and Stothard, 2008).
Taxonomic identification and phylogenetic analysis
The PCR 16S rRNA gene amplification was carried out using the universal primer pairs pA_F and pH_R (Sharma et al., 2022). The purified PCR product was sequenced using Sanger dideoxy-chain termination chemistry. The obtained sequence was assembled to make a consensus sequence by converting one strand to a reverse complement. For strain ASH15 identification, the assembled consensus 16S rRNA gene sequence was used for a BLAST (BLASTn) search against the available bacterial 16S rRNA gene sequences in the NCBI GenBank database. Further, a neighbor-joining (NJ)-based phylogenetic tree of 16S rRNA gene sequence of the strain ASH15 with 11 reference 16S rRNA gene sequences was constructed through MEGA-X. The bootstrap analysis was conducted using 1,000 replications by the Felsenstein method (Felsenstein, 1985). The evolutionary distances were calculated by the Jukes–Cantor coefficient procedure (Tamura et al., 2004).
Identification of biosynthetic gene clusters and metabolic system analysis
The genome sequence of strain ASH15 was analyzed using antiSMASH (Blin et al., 2019) software for predicting biosynthetic gene clusters (BCGs), such as non-ribosomal peptide synthetases (NRPSs), polyketide synthases (PKSs), post-translationally modified peptides (RiPPs), hybrid lipopeptides (NRPS-PKS), and bacteriocins. Less than 70% of the amino acid identity shared by the biosynthetic Gene Clusters compared to the known clusters was considered novel. The Carbohydrate Active Enzyme Database (CAZy, http://www.cazy.org/) is a professional database for enzymes synthesizing or decomposing complex carbohydrates and sugar complexes. Carbohydrate activity enzymes derived from different species are divided into glycoside hydrolases (GHs), polysaccharide lyases (PLs), glycosyltransferases (GTs), carbohydrate esterases (CEs), auxiliary activities (AAs), carbohydrate-binding modules (CBMs), and other six major protein families.
Additional genome analysis (sRNA prediction, repeat sequence prediction, tandem repeat prediction, and scattered repetitive sequence prediction)
Bacterial sRNA is a type of non-coding RNA with a length of 50 to 500 nt. They are located in the intergenic region of genomes, and some are in the 5′and 3′UTR regions of coding genes. We used Infernal software (http://eddylab.org/infernal/) and the Rfam database (https://rfam.xfam.org/) to predict and annotate the sRNA from the genome of strain ASH15. Tandem Repeats Finder software (Benson, 1999) was used to predict the tandem repeat sequences. Interspersed repeat, also known as a transposon element, includes DNA transposons and retrotransposons transposed by DNA-DNA. RepeatMasker software (Tarailo-Graovac and Chen, 2009) was used to identify these sequences as similar to known repetitive sequences and classify them. IslandViewer (Bertelli et al., 2017) was used to identify genomic islands in strain ASH15.
Statistical analysis
The experimental data of the present study were subjected to analysis of variance (ANOVA) followed by DMRT (Duncan's multiple range test) with a significance level of p of ≤0.05 (Duncan, 1955). Bioinformatics analysis of the strain ASH15 genome was carried out through Majorbio I-Sanger (www.i-sanger.com).
Results and discussion
Isolation and characterization of halophilic bacteria
This study undertook the isolation, characterization and systematic genome analysis of a halophilic bacterial strain ASH15 and identification of their key genes that contribute in osmoadaptation (stress tolerance), plant growth-promoting (PGP) traits and other industrially important biomolecules production. A Gram-positive, spore-forming, rod-shaped, and motile halophilic bacteria strain ASH15 was isolated from the collected soil sample (Figure 1). Strain ASH15 showed medium-sized, round colonies with a ceramic-white, opaque appearance and smooth margins. Further, the carbon source utilization on GNIII MicroPlate™ (Biolog Inc., Hayward, CA), was used to extricate the metabolic sensitivity of the isolated halophilic bacterial strain ASH15 (Zhao et al., 2019). Results showed that strain ASH15 was able to metabolize a wide range of carbon sources. Strain ASH15 was positive for 24 sugars and chemically sensitive to 16 substrates, 2 hexose-PO4, 8 amino acids, 8 hexose acids, and 13 carboxylic acids, esters, and fatty acids (Supplementary Table 1). The utilization of different carbon sources by the bacterial cell assists as components of the metabolic network, whereby they are broken down to facilitate the source of amino acids and other building blocks to make up a cell (Wang et al., 2019). These metabolic properties of halophilic bacteria might lead to their response and adaptation in specific extreme environments (Sharma et al., 2021).
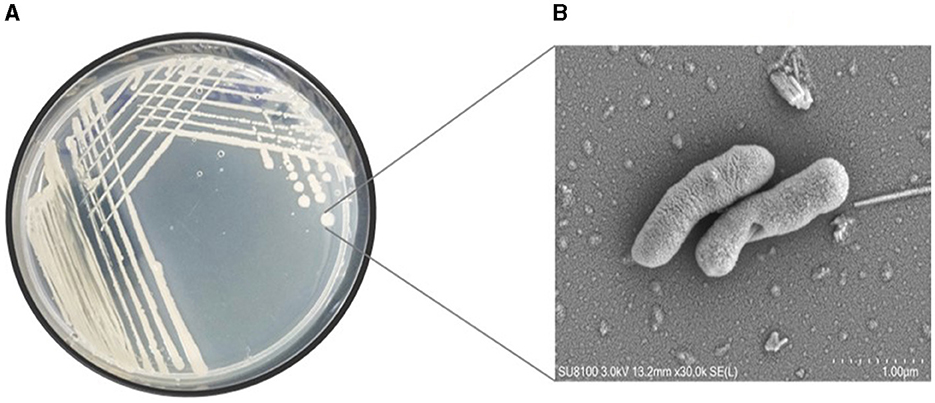
Figure 1. (A) Pure culture and (B) SEM micrograph of the halophilic strain Virgibacillus halodenitrificans ASH15 cell.
When testing the growth of the strain ASH15 under various NaCl concentrations, we observed that supplementing nutrient broth with 0.5–25% NaCl had a positive effect on bacterial growth. The maximum growth (OD at 600 nm) of the strain ASH15 was observed at 15% as compared to NB with 0.5% NaCl. Further results showed that the growth of ASH15 was in contrast, slightly delayed when NaCl concentration was increased up to 25% (Figure 2). These findings demonstrate that strain ASH15 is moderately halophilic and is able to withstand up to 25% NaCl concentrations. In accordance with our findings recently, Srivastava et al. (2022) reported that Chromohalobacter salexigens ANJ207 was able to grow up to 30% NaCl concentration. The capacity to withstand moderate salt stress might help ASH15 survive as a free-living bacterium in saline soils.
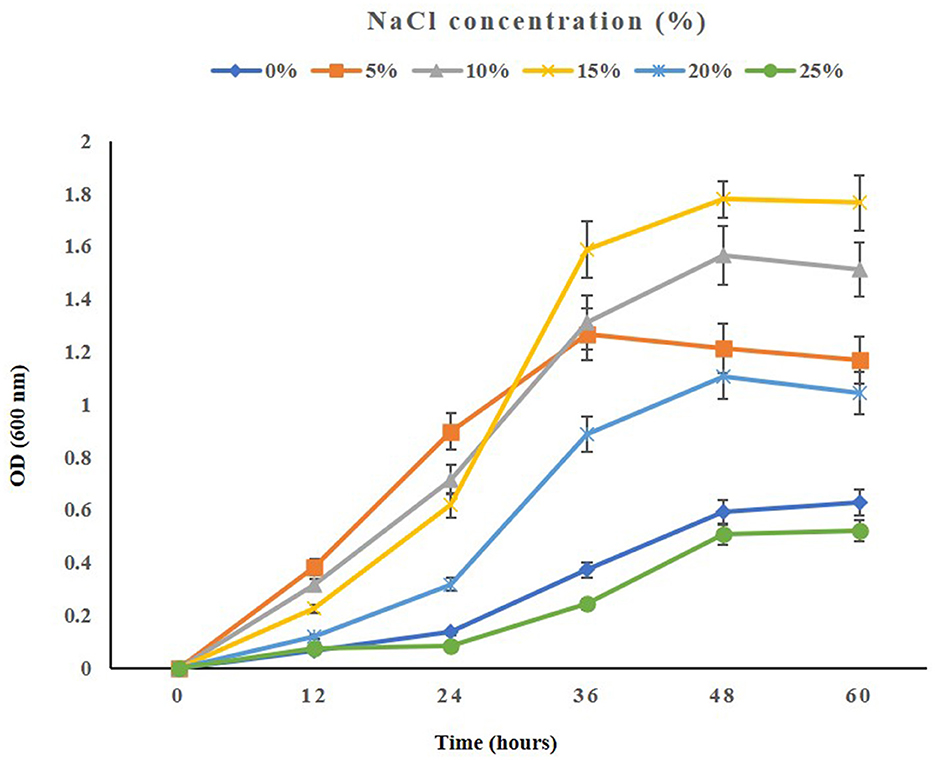
Figure 2. The growth pattern of the halophilic strain Virgibacillus halodenitrificans ASH15 at different NaCl concentrations.
The qualitative evaluation of plant growth-promoting attributes revealed that strain ASH15 was able to fix nitrogen, solubilize phosphate and zinc, and produce siderophore and ammonia. In addition, quantitative estimation of IAA revealed that strain ASH15 produced 54.7, 76.1, 82.8, and 80.3 μg/ml IAA at 0, 5, 10, and 15% NaCl concentrations respectively. Interestingly, strain ASH15 has all the PGP activity up to higher NaCl concentrations. Furthermore, strain ASH15 produces EPS at all the tested NaCl concentrations (Table 1). Results of the biofilm formation assay showed that strain ASH15 was able to produced biofilm in all the tested NaCl concentrations with maximum at 15% NaCl. These results showed the possible strategies that the strain ASH15 might employ on its host plant for salt stress alleviation and plant growth promotion. These findings are consistent with previous research studies, where halophilic bacteria, such as Bacillus halophilus, Marinococcus halophilus, Halobacillus litoralis, Saliiococcus hispanicus, and Halobacillus halophilus, were recognized to grow optimally between 10 and 15% NaCl concentrations (Sarwar et al., 2015; Reang et al., 2022; Srivastava et al., 2022).

Table 1. Plant growth promoting attributes of the strain Virgibacillus halodenitrificans ASH15.
Phylogeny of strain ASH15
Based on the 16S rRNA gene sequencing analysis and BLASTn search, strain ASH15 showed 100% similarity with Virgibacillus halodenitrificans of the NCBI database. A neighbor-joining (NJ) phylogenetic tree was constructed with the similar bacterial sequences of the NCBI GenBank database (Figure 3). The phylogenetic analysis provides an important depiction of the evolutionary relationship between different strains (Srivastava et al., 2022).
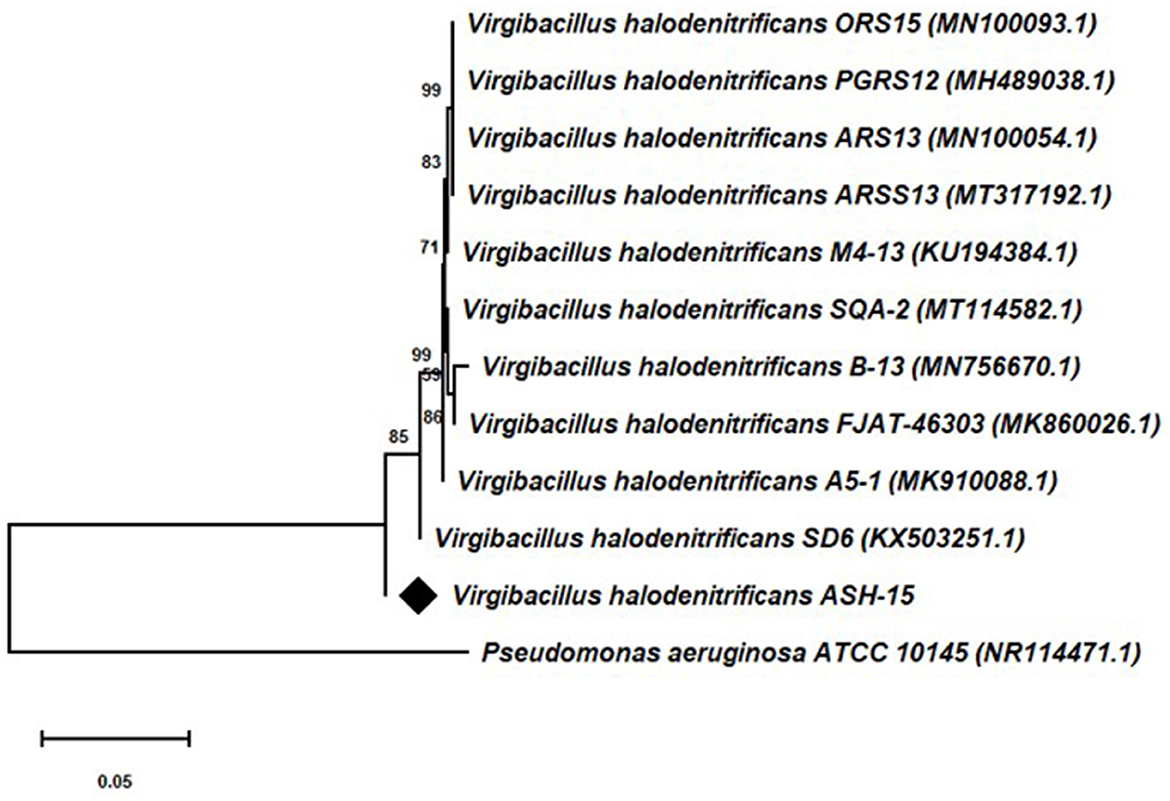
Figure 3. 16S rRNA gene sequencing-based phylogenetic tree of the Virgibacillus halodenitrificans ASH15 with its nearest member of the NCBI GenBank database.
Effect of strain ASH15 on sugarcane growth under greenhouse conditions
In the present study, the positive effect of V. halodenitrificans strain ASH15 on sugarcane growth under salt stress conditions was assessed under normal and salt stress (200 mM NaCl) conditions (Figure 4). Results of the greenhouse study showed that salinity stress imposes adverse effects on sugarcane vegetative growth. However, strain ASH15 application significantly (p < 0.05) enhanced the growth of sugarcane plants under normal as well as NaCl stress conditions (Figure 4). The results of greenhouse pot experiments showed that salt stress treatment (T-3, 200 mM NaCl) decreased root length (RL) and shoot length (SL) growth by 32.3% and 25.8%, respectively, as compared to uninoculated non-stressed (T1). Whereas, application of strain ASH15 (T-4) treatment remarkably (p < 0.05) enhanced the root length and shoot length by 71.2% and 64.4%, respectively, over un-inoculated NaCl-stressed plants (Figure 4). Similarly, salt stress (T-3) treatment decreases root fresh weight (RFW) and shoot fresh weight (SFW) by 42.1 % and 30.9%, respectively, over uninoculated non-stress plants (T-1). In contrast, strain ASH15 (T-4) boost up the RFW and SFW by 53.5% and 72.0%, respectively, as compared to uninoculated salt-stressed plants (T-3). Moreover, similar trends were observed in the case of root dry weight (RDW) and shoot dry weight (SDW), where salt stress (T-3) reduced the RDW and SDW by 45.8% and 57.6%, respectively, over their uninoculated control (T-1). However, strain ASH15 (T-4) increased the RDW and SDW by 54.1% and 109.1%, respectively, compared to the uninoculated salt-stressed control (T-3) (Figure 4). The results of the pot experiment demonstrated that the sugarcane plant's overall health, growth, and development were reliant on the presence of strain ASH15, which regulated an adequate amount of multiple plant nutrient levels (Alishahi et al., 2020; Khumairah et al., 2022). Therefore, in this study, we explored the strain ASH15 genome and mined the gene codes for almost all PGP traits like IAA, nitrogen fixation, phosphate solubilization, and siderophore production. Sultana et al. (2020) recently reported that salt-tolerant bacteria significantly increased rice plant growth. The results are also in accordance with those reported by Bhattacharyya et al. (2017), Asaf et al. (2018), and Abdullahi et al. (2021), where they analyzed the presence of multiple genes encoding for PGP mechanisms in plant growth-promoting bacteria.
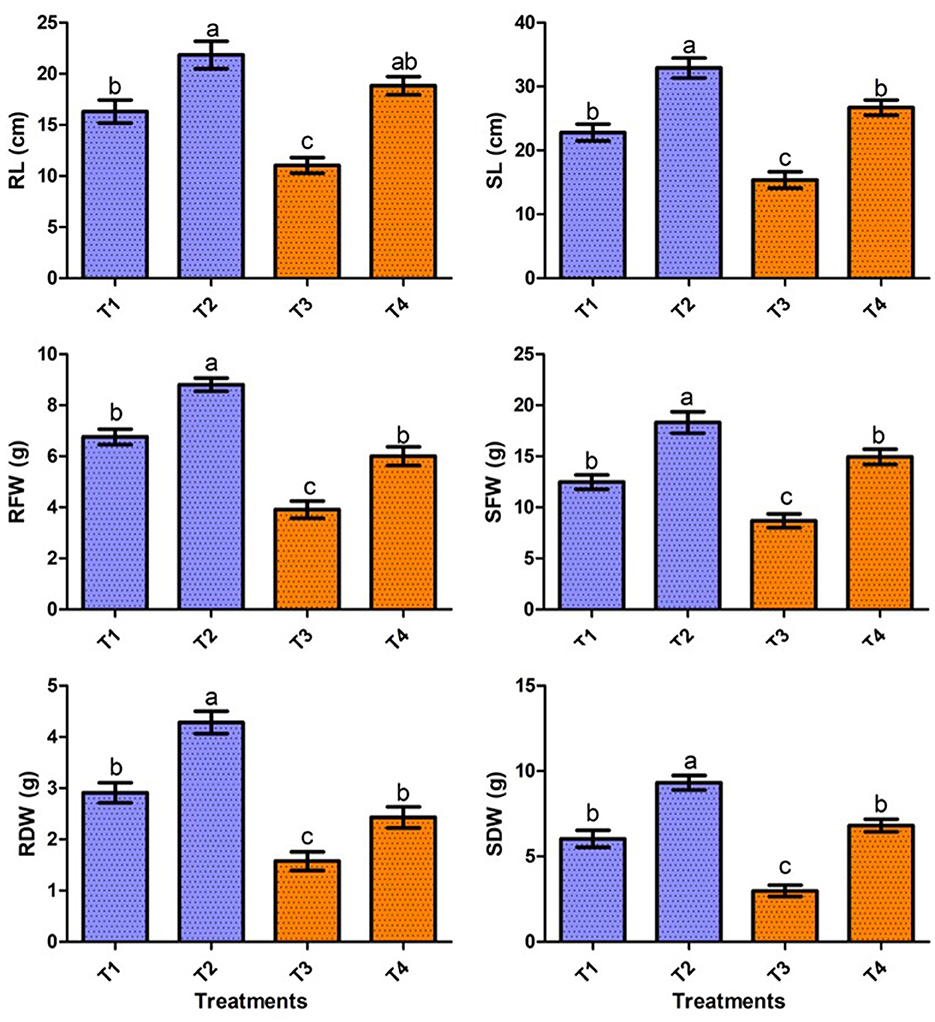
Figure 4. Effect of strain ASH15 on sugarcane growth under normal and NaCl (200 mM) stressed conditions. (RL, root length; RFW, root fresh weight; RDW, root dry weight; SL, shoot length; SFW, shoot fresh weight; and SDW, shoot dry weight). The figures represent the mean values ± standard error of three independent experiments. Bars indicate ± SE (standard error of mean). The letters on the bars indicate DMRT at a p-value of <0.05.
Genome analysis of V. halodenitrificans ASH15
The genome assembly details of the strain ASH15 are given in Table 2. The high-quality raw sequence data was assembled with a hybrid genome assembly, and a single scaffold was achieved. The genome of V. halodenitrificans strain ASH15 is composed of a circular chromosome (Figure 5) of 3,832,903 base pairs with an average G+C content of 37.54%. There was no plasmid identified in the genome assembly. The genome was processed for gene prediction, and the total predicted genes were 3,807, which included ~3,721 protein-coding genes (CDS), 62 tRNAs, and 24 rRNA genes (Table 2). CDS constitute 3,207,696 bases (83.69%) of the genome, with an average gene length of 862.05 bases. Approximately 10.78% (803483 bases) of the genome was found to be intergenic. Furthermore, predicted genes against various databases were characterized. The number of COG genes, Gene Ontology (GO), KEGG, NR (Non-redundant Protein Database), and SwissProt were 3,268, 2,575, 2,008, 3,644, and 2,683 respectively (Table 2 and Figures 6A–C). The complete genome sequence of the strain V. halodenitrificans ASH15 has been deposited at the NCBI/GeneBank with accession number CP090006.
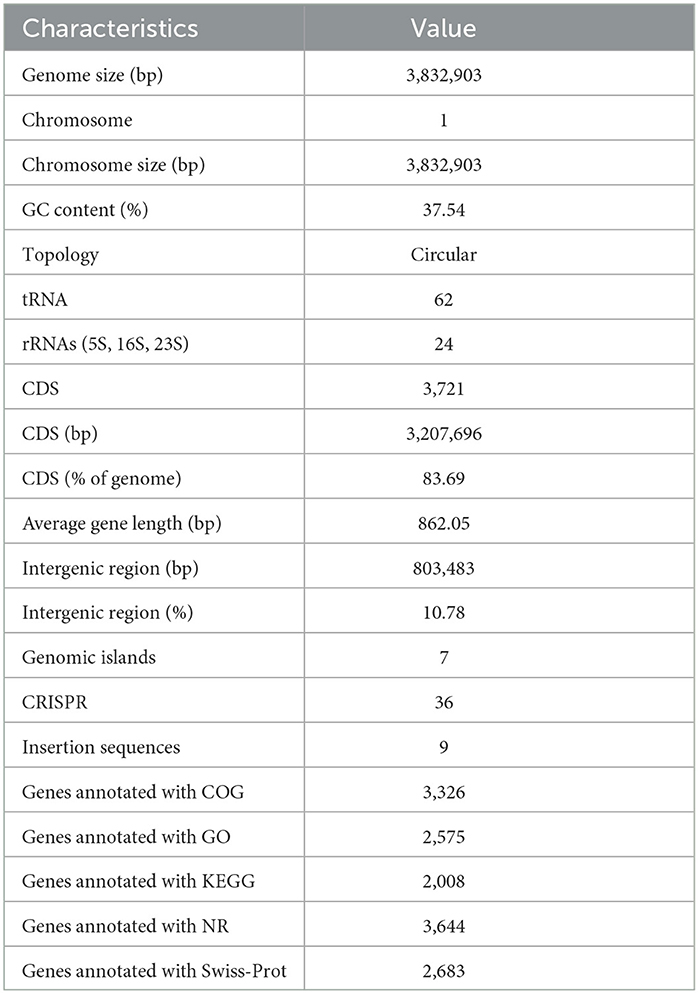
Table 2. Genome characteristics of Virgibacillus halodenitrificans strain ASH15.
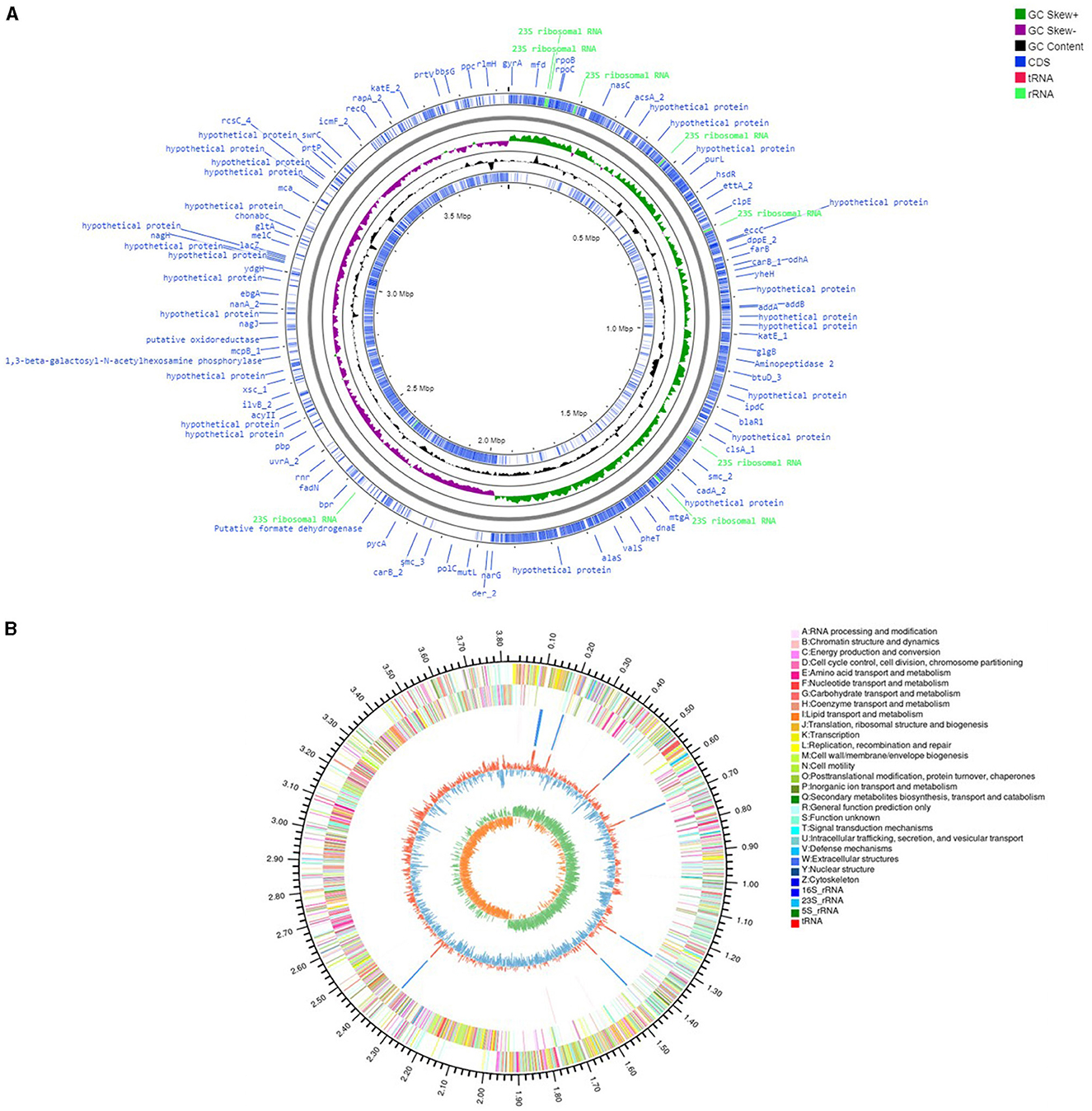
Figure 5. (A) Circular representation of the chromosome/genome of Virgibacillus halodenitrificans strain ASH15. The Outermost and innermost rings represent two strands of the genome. The outermost ring displays the functional annotation of genes (CDS), tRNA, and rRNA distribution in the genome. The third ring (black in color) shows the GC content of the genome, while the fourth ring (green in color) represents the GC skewness throughout the genome. (B) A–Z, the outer two circles, show the functional classification of the CDS genes in the chromosome with the colors of the COG. The inner circle shows rRNAs and tRNAs.
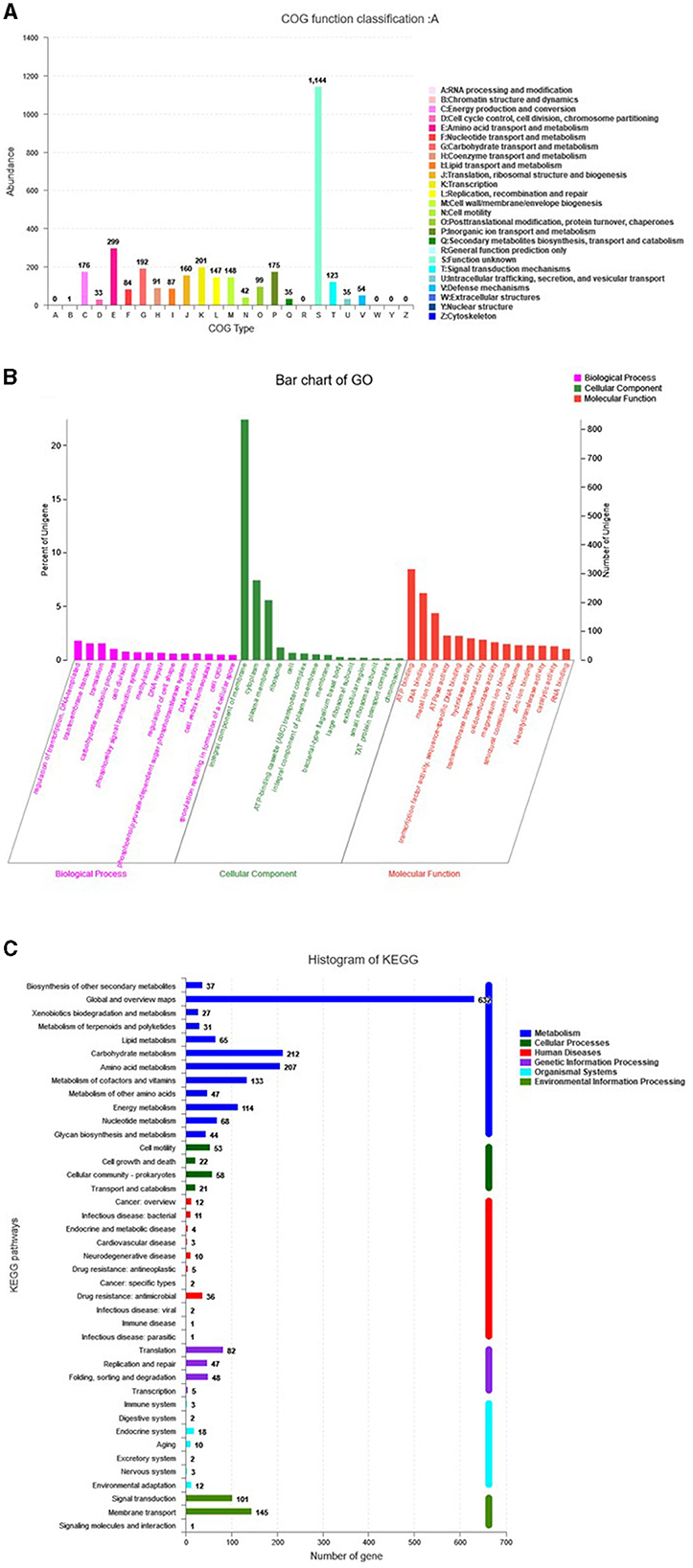
Figure 6. Predicted genes of the Virgibacillus halodenitrificans strain ASH15 genome through (A) COG, (B) GO, and (C) KEGG databases.
Genetic potential of various stress tolerance in the ASH15 genome
Halophilic bacteria have unique inherent osmoadaptation mechanisms for stress adaptation, which could be used for agriculture, food, and fermentation industries (Gunde-Cimerman et al., 2018; Vaidya et al., 2018). Genome analysis of the strain ASH15 confirmed the presence of several key genes responsible for different abiotic stress tolerances, mainly osmotic stress (proVWXSBA, fadANM, betBA, trkAH, opuBDCA, opcR, putP, yrgG, kch, and nhaC), ectoine biosynthesis (ectCBAD), and oxidative stress (hmp, pfpI, Usp, katE, and osmC) (Table 3). These osmolytes, or compatible solutes, provide osmotic balance to the bacteria without disturbing their cell functions (Roberts, 2005; León et al., 2018).
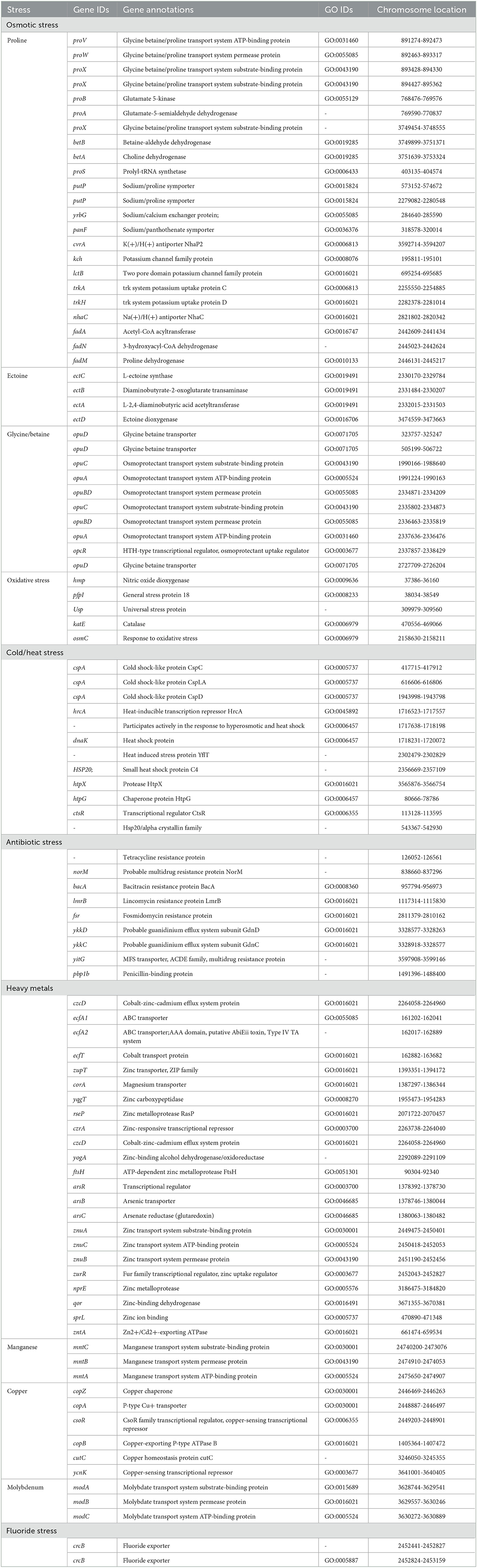
Table 3. Genes associated with abiotic stress responses in Virgibacillus halodenitrificans strain ASH15.
In addition, further analysis revealed that the strain ASH15 genome has various other abiotic stress tolerance genes such as cold-shock protein (cspA), heat shock proteins (hrcA, dnaK, hsp20, htpX, htpG, and ctsR), heavy metals such as arsenic (arsRBC), cobalt (czcD, ecfT, ecfA1, ecfA2), zinc (zupT, yqgT, rseP, czrA, znuACB, zurR, nprE, qor, sprL), cadmium (zntA), magnesium (corA), molybdenum (modABC), copper (copZA, csoR, copB, cutC, ycnK), and manganese (mntCBA), antibiotics (norM, bacA, lmrB, fsr, pbp1b, ykkDC, and yitG), and fluoride resistance (crcB) (Table 3). These groups of genes provide stress-tolerant capabilities to strain ASH15 and enable it to survive in extreme conditions. Fluoride exporter genes, crcB, were involved in multilevel stress responses (Calero et al., 2022). Several PGPR genera have been described to succeed in heavy metal stresses to improve plant tolerance, especially abiotic stresses, and crop yields (Tiwari and Lata, 2018). These proteins are linked to tolerance to cobalt, zinc, copper, arsenic and cadmium (Kang et al., 2020).
Genes related to plant growth promotion in the strain ASH15
Sugarcane is a long-term economically important crop, and for its growth, various types of plant nutrients such as N, P, K, and phytohormones are required. Thus, to decrease the application of chemical fertilizers in the current era, PGPR with various PGP attributes promises an alternative approach to plant nutrient requirements (Sharma et al., 2021). Therefore, in the present study, we revealed the plant growth promotion potential of V. halodenitrificans ASH15. The genome of the strain covers so many genes that encode various PGP traits, such as phytohormone IAA production, nitrogen fixation, phosphate solubilization, ammonia assimilation, and siderophore production (Table 4, Guo et al., 2020).
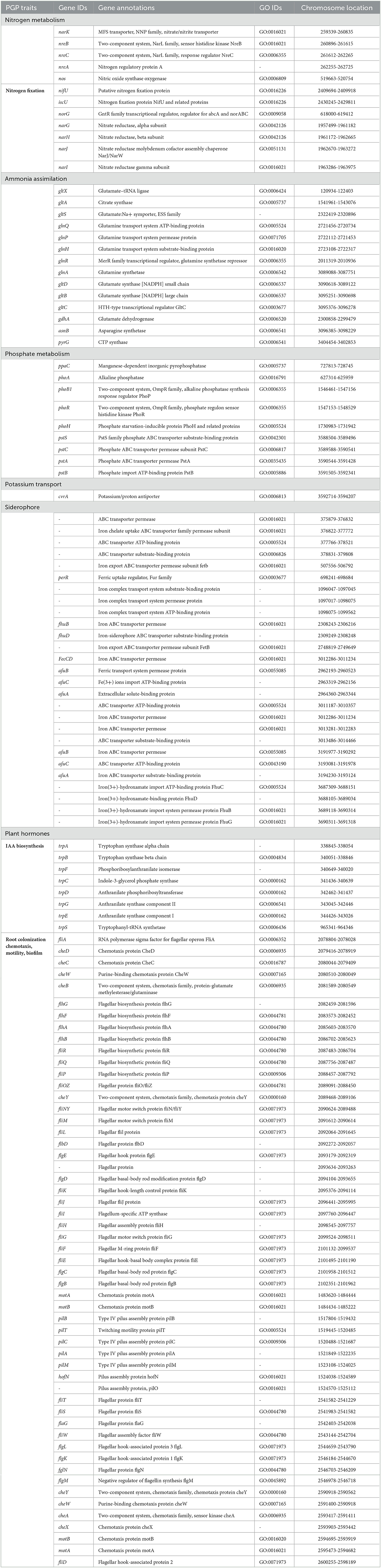
Table 4. Genes associated with PGP traits in Virgibacillus halodenitrificans strain ASH15.
Indole-3-acetic acid (IAA) is an important phytohormone involved in various physiological processes, including cell enlargement and division, tissue differentiation, and responses to light and gravity. The ability to synthesize IAA is a well-characterized trait in halophilic PGPR (Pérez-Inocencio et al., 2022). Bacterial IAA is involved in overcoming stress, serving as a C/N source, and playing a role in plant–microbe interactions (Defez et al., 2019). In the current study, we observed that strain ASH15 was capable of synthesizing IAA, and its genome consists of trpA, trpB, trpC, trpD, trpE, trpF, trpG, and trpS genes, which code for enzymes of the IAA biosynthesis pathway (Table 4). Similar to our findings, tryptophan biosynthesis genes (trpABD) are involved in IAA production in Sphingomonas sp. LK11 (Asaf et al., 2018).
Another strategy of PGPR to enhance plant growth is to fix atmospheric nitrogen. Nitrogen is an essential nutrient element for soil fertility, sugarcane plant growth and development, physiological and metabolic activities, and sustainable sugarcane crop production (Singh et al., 2022). PGPR catalyzes nitrogen fixation through the nif (nitrogenase complex) gene-coded nitrogenase enzyme. In this study, the strain ASH15 genome lacks genes (nifDHK) coding the nitrogenase enzyme, but contains genes related to dissimilatory nitrate reduction (Table 4). These include narGHIJ, a nitrate/nitrite ABC transporter (narK), a putative nitrogen fixation protein (nifU), and various other genes associated with nitrogen metabolism and transport (iscU, norG, nreBCA, and nos). Strain ASH15 also has genes coding for ammonia assimilation, such as gltXASDBC, glnQPHRA, gdhA, asnB, and pyrG (Table 4). These results showed that strain ASH15 is able to incorporate nitrate and nitrite for assimilation into ammonia and can incorporate ammonia directly.
Together with N, phosphorus (P) is also an important nutrient required for plant growth (Bergkemper et al., 2016). PGPR plays a key role in plant growth by facilitating the conversion of the available insoluble inorganic phosphate to the soluble PO (Bergkemper et al., 2016). In PGPR, a mineral's phosphate-dissolving ability has been directly related to the presence of various genes responsible for producing organic acids. In this study, the genome of ASH15 contains genes coding for inorganic pyrophosphatase (ppaC) and alkaline phosphatase (phoA). The two-component system CS PhoB1/PhoR is involved in the alkaline phosphatase, phosphate starvation response (phoH), and an ABC transporter for phosphate uptake (pstSCAB), which are responsible for solubilizing the inorganic phosphate (Table 4). Moreover, the presence of an effective system in the PGPR for absorbing iron can help to protect the host plant from pathogen infestations (Herlihy et al., 2020; Lahlali et al., 2022). In the strain ASH15 genome, we also detected the presence of several siderophore-related genes in the ASH15 genome, including several iron ABC transporters (fhuBD, afuABC, and FecCD), a ferric uptake regulator (perR), an iron export ABC transporter permease (fetB), and a ferric transport system and ions import (fhuBCG) (Table 4). Our findings are in line with the fact that PGPR with salt-tolerant properties provides a range of benefits such as phytohormones, nitrogen fixation, P solubilization, ammonia production, and siderophore production for plant's stress tolerance and growth promotion (Egamberdieva et al., 2019; Arora et al., 2020; Khumairah et al., 2022).
In addition, genes like antimicrobial peptides and hydrolase genes, such as GTP cyclohydrolase (ribBA), α-amylase (treC), α-glucosidase (malZ), and glutamate dehydrogenase are also involved in plant immune responses. Moreover, oxidoreductase genes such as glutathione hydrolase proenzyme (ggt), superoxide dismutase (SOD), glutathione transport system (gsiDCB), and peroxiredoxin (DOT5, tpx) have been categorized. Strain ASH15′s genome predicted some key genes of volatile substances such as keto-acid metabolism (ilvABCDEH), 2,3-butanediol catabolism (acuABC), and Isopentenyl-diphosphate delta-isomerase (idi), which may be related to the biocontrol mechanism of strain ASH15.
Biofilm-related genes in strain ASH15
The motility of bacteria is another important feature that enables them to move, colonize, and systematically spread in plants (Palma et al., 2022). The motility ability of strain ASH15 allows it to move through the soil matrix and into the plant, as confirmed by the genes involved in the flagella biosynthesis and assembly such as flhA, flhB, flhF, flhG; fliAROPLJIHFTSW, flbD; flagellar proteins fliO/fliZ and flbD; flagellar motor switch proteins, fliN, fliY, fliM, and fliG; and flagellar hook associated protein, fliDEK and flgBCDEMN, and two sets of genes coding for the flagellar motor proteins (motA and motB) (Table 4). Genome analysis of the strain ASH15 showed that two genes hofN and pilBCAM are involved in the biosynthesis and assembly of the type IV pilus system (T4PS): (Table 4).
Secretion systems of strain ASH15
Bacteria have a set of different protein secretion systems that are essential for their growth and plant interaction. Bacteria secrete secondary metabolites, peptides, antibiotics, enzymes, and toxins to compete with nearby microbes or interact with host plants (Netzker et al., 2015; Köhl et al., 2019). Among the bacterial secretion systems, to transport proteins across the plasma membrane, the twin-arginine translocation (Tat) and general secretion (Sec) pathways are most commonly in use (Natale et al., 2008). The Tat pathway is mostly used to secrete folded proteins, while the Sec pathway primarily secretes unfolded proteins (Natale et al., 2008; Green and Mecsas, 2016). In this study, the strain ASH15 genome demonstrated five types of secretion systems: Type II/Type IV, Type III, Type VII (yukDC; ESX secretion system), and twin-arginine translocase (tatAEC). The operon acuABC encodes proteins to utilize acetoin and butanediol as carbon sources for energy requirements (Thanh et al., 2010). The presence of operon acuABC confirms that strain ASH15 has the ability to metabolize complex cyclic organic compounds and could be utilized for bioremediation.
Genome mining for biosynthetic gene clusters and metabolic system analysis
The PGPR secretes bioactive secondary metabolites in the soil (rhizosphere-niche), which are related to plant-microbe interaction and root colonization as well as play a significant role in the plant immune response (Backer et al., 2018; Sharma et al., 2019; Bukhat et al., 2020; Jamali et al., 2020). The antimicrobial potential of strain ASH15 to produce hydrolytic enzymes and siderophores was confirmed by genome analysis (Tables 4, 5). The biosynthesis potential of the halophilic PGPR was assessed using antiSMASH 6.0.0 to predict both known and unidentified functional secondary metabolites in order to better understand its antagonistic action. Results showed (Figure 7) that five biosynthetic gene clusters were present in the genome: the first cluster of T3PKS (45 genes), the second cluster of terpenes (23 genes), the third cluster of T3PKS (48 genes), the fourth cluster of ectoine (9 genes), and the fifth cluster of terpenes (17 genes) (Figure 7).
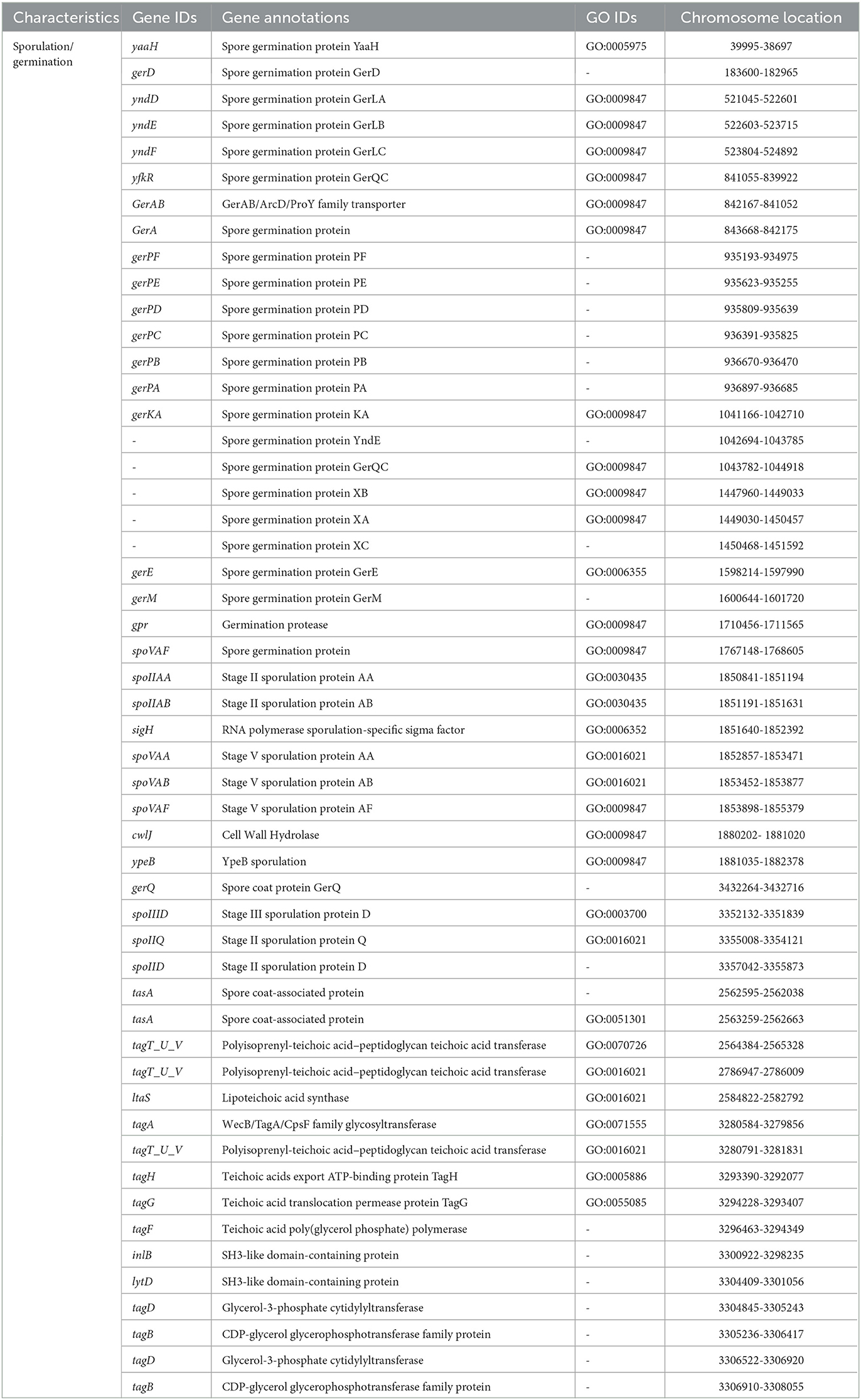
Table 5. Sporulation/germination genes in Virgibacillus halodenitrificans strain ASH15.

Figure 7. Biosynthesis gene clusters identified in the genome of Virgibacillus halodenitrificans strain ASH15. (A) T3PKS cluster with 45 genes; (B) terpene cluster with 23 genes (C) T3PKS cluster with 48 genes (D) ectoine cluster with 9 genes, and (E) terpene cluster with 17 genes.
Carbohydrates play several important roles in different biological functions. Carbohydrate activity enzymes derived from various species are divided into glycoside hydrolases (GHs), polysaccharide lyases (Ls), glycosyltransferases (GTs), auxiliary activities (AAs), carbohydrate-binding modules (CBMs), carbohydrate esterases (CEs), and other six major protein families. CAZyme analysis resulted in the identification of 80 genes in the genome ASH15 and annotated them (Figure 6, Supplementary Table 4) as 9 genes for AAs, 22 genes for CEs, 25 genes for GHs, and 24 genes for GTs.
Other important genes identified in the genome
V. halodenitrificans strain ASH15 is an endospore-forming halophilic bacterium. Table 5 shows the predicted and identified genes (gene IDs, annotations, GO IDs, and chromosome locations) related to spore formation and spore germination in the genome of strain ASH15.
Additional findings in the genome
Small RNAs play a significant role in metabolic pathway regulation and are thus very important. In the present investigation genome-wide analysis predicted a total of 120 sRNAs, which constitute 16,299 bases and 0.4252% of the strain ASH15 genome (Supplementary Table 2). We also identified tandem repeat sequences in the genome of strain ASH15. A total of 220 repeats were identified, which constitute 55,602 bases and 1.73% of the genome. Interspersed repeats, also known as transposable elements (transposon), includes DNA transposons and retrotransposons transposed by DNA-RNA. Common retrotransposons (also called Class I transposable elements or transposons via RNA intermediates) are LTR, LINE, and SINE. Genome analysis found that the strain has 6-SINE, 16-LINE, and 5 transposons, constituting 1,733 bases and 0.05% of the genome. Bacterial sRNA is a type of non-coding RNA with a length of 50 to 500 nt. They are mostly found in the intergenic region, while some are found in the coding genes' 5′ and 3′UTR sections. Bacterial sRNA mainly performs a variety of biological functions by binding to target mRNA or target protein. For example, bacterial sRNA plays an important role in regulating outer membrane protein expression, iron ion balance, community sense, and bacterial pathogenicity. Tandem repeats refer to the occurrence of two or more repeats in adjacent positions of the genome.
Analysis of movable components of the genome
In the long evolutionary process, to adapt to changes in the environment or improve their own survival competitiveness, bacterial genomes often take in some foreign gene fragments and integrate them into their own genomes. These fragments generally contain some genes encoding specific functions, such as virulence genes, drug resistance genes, and metabolic genes, etc., which can change the phenotype of bacteria and help bacteria get through “difficulties” or occupy a dominant niche. These exogenous genome fragments are collectively called mobile elements; this phenomenon is called horizontal gene transfer (HGT). In the present genome analysis, three types of movable elements were predicted in strain ASH15: Genome Island, Prophage, and Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR). Prophage is a bacteriophage genome integrated into the circular genome of the host bacteria (Piligrimova et al., 2021). As a ubiquitous mobile genetic element, prophage plays a key role in bacterial genetics, evaluation, and increasing survival and virulence potential through multiple mechanisms (Ramisetty and Sudhakari, 2019).
Conversely, the CRISPR system provides inherited and acquired sequence-specific adaptive immunity against the phage and other horizontally acquired elements like plasmid. CRISPR-associated (Cas) proteins constitute an RNA-guided adaptive immune system found in several prokaryotes. It is not only a type of bacteriophage defense system but also a regulator of bacterial physiology (Newsom et al., 2021). In recent times, to fulfill the demand for the next generation of industrial biotechnology (NGIB), the CRISPR/Cas system has been evaluated as a potential editing tool for customizing and reprogramming the genome of many extremophilic bacterial species for the production of natural compounds (Cress et al., 2016; Qin et al., 2018; Singh et al., 2021).
Insertion sequences
The insertion sequence is a transposon encoding the enzyme required for transposition, and it is flanked by short, inverted terminal repeats. A total of 9 IS were identified in the genome of ASH15 using ISEScan software (Supplementary Figure 1).
Genome island analysis
Genome island (GI) is one of the most important forms of horizontal transfer elements. It contains genes related to a variety of biological functions. According to the different genes, genome islands can generally be divided into virulence, drug-resistant, metabolic, symbiotic islands, among others. Genome islands are usually large, ranging from 10 to 200 kb. A total of 7 GIs were predicted in the genome, which constituted 162,069 bases, where the smallest island was 8,201 bases and the largest was 82,517 bases. The island genome has coded for 167 CDS, mainly belonging to proton transporters, iron transport, sugar transporters, chaperons, and many hypothetical proteins (Supplementary Figure 2 and Supplementary Table 3).
Two-component systems identified in the genome of strain ASH15
Deep genome analysis revealed that in the strain ASH15 cell several two-component systems (nreBC, cssSR, liaSR, phoBR, resDE, cheAY, degUS, citTS, yesMN, desRKA, walRK, and vicKR) working, which play an important role in the modulation and regulation of the expression of critical proteins under stressful conditions. Specifically, nreBC is involved in nitrogen metabolisms, cssSR is a sensory system, phoBR is associated with the phosphate metabolism mechanism, and histidine kinase sensor response regulators (resDE, yesMN, and desRK) are responsible for various cellular processes. Additionally, vicKR is a regulator for cell wall metabolism.
LiaSR two-component system
Strain ASH15 has a LiaIFRS operon in the genome. The LiaSR is a two-component system widely found in Gram (+) bacteria. The LiaSR system is most studied in Bacillus subtilis as a part of the LiaIHGFSR operon. A striking characteristic of the LiaSR system is that its expression is induced upon exposure to antibiotics that target the cell envelope (Jordan et al., 2006; Suntharalingam et al., 2009; Shankar et al., 2015).
CheAY, two-component system
CheAY two-component system plays a regulatory role in the signal transduction of chemotaxis (Zschiedrich et al., 2016). The CheY and CheA system are well studied in B. subtilis and E. coli (Rao et al., 2004; Minato et al., 2017). In B. subtilis, the autophosphorylating activity of CheA increases through the binding of attractants to transmembrane receptors (Karatan et al., 2001). Phosphorylated CheA donates the phosphate to the response regulator CheY (Wang et al., 2014). The CheY that has been phosphorylated interacts with the flagellar motor switch complex to insist the flagella rotate counterclockwise (CCW), which induces a smooth swimming motion (Minamino et al., 2019; Mukherjee et al., 2019).
DegUS two-component system
DegUS two component system are identified in strain ASH15; it controls degradative enzyme synthesis. DegUS is involved in the complex network that mediates the regulation of transition state-specific processes. It contributes to controlling the development of natural competence for DNA uptake, motility, and degradative enzyme synthesis (Meliawati et al., 2022).
CitST two-component system
The CitST system [carbon catabolite repression (CCR)] is involved in citrate fermentation metabolism and citrate/succinate transport. This two component system has also been studied in other bacterial genomes (Repizo et al., 2006).
A unique finding in the genome of strain ASH15
Isoprenoid (squalene/phytoene) biosynthesis pathway
Genome analysis revealed that strain ASH15 has an MVA pathway to synthesize isoprenoids. The MVA pathway starts with the condensation of two acetyl-CoA molecules followed by a series of reduction (six) steps that produce IPP involving the expression (Figure 7) of genes: acsA (gene number 0346), HMG-CoA Synthase (gene number 00029; mvaS), HMG-CoA reductase (gene number 0215; mvaE), mevalonate kinase (gene number 0216; mvaK1), phosphomevalonate kinase (gene number 0218; mvaK2), and diphosphomevalonate decarboxylase (gene number 0217; mvaD) (Figure 7). IPP and DMAPP are isomers, and inter-conversion is catalyzed by the enzyme isopentenyl-diphosphate Delta-isomerase (IdI). idI (gene number 0398) is the key regulatory gene that maintains the IPP to DMAPP ratio and has a significant impact on isoprenoid biosynthesis. The gene (gene number 1929), which codes for geranylgeranyl diphosphate synthase (GGPPS), converts farnesyl diphosphate (FPP) to geranylgeranyl diphosphate (GGPP). Farnesyl diphosphate (FPP) is an intermediate molecule that could be converted to many different terpenoids (Rinaldi et al., 2022), while GGPP could be further converted to diterpenoids and tetraterpenoids. Strain ASH15 has genes (SQS_PSY; 0327 and SQS_PSY; 2893) to produce triterpenoids and tetraterpenoids, Squalene (C30) and Phytoene (C40). Squalene synthase (gene number 0327) converts two molecules of FPP to squalene, and phytoene synthase (gene number 2893) could convert GGPP to Phytoene.
Squalene has been known for its various applications, such as an anti-cancer agent, an anti-oxidant agent, an anti-bacterial agent, a chemopreventive agent, an anti-aging agent, a detoxifier, and an adjuvant for drug carriers and vaccines (Kim and Karadeniz, 2012; Paramasivan and Mutturi, 2022). Therefore, it has enormous potential in the food, cosmetics, and pharmaceutical industries (Huang et al., 2009; Gohil et al., 2019). The global squalene market demand in 2014 was approximately 2.67 kilotons, and by 2022, it is estimated to reach a value of USD$ 241.9 million, with the majority of sales coming from personal care and cosmetic items (Rosales-Garcia et al., 2017). There is a pressing need to generate squalene in a renewable and sustainable way to meet this ever-increasing demand. The development of microbial cell factories could be a solution to fulfill this demand, and strain ASH15 is a natural bacterium that has all the genes for the biosynthesis of squalene.
Conclusion
According to the findings of the present study, the availability of V. halodenitrificans ASH15's entire genome will shed additional light on complex biological systems, which showed that strain ASH15 has open a number of opportunities to study this efficient plant growth-promoting bacterium. These results indicate that strain ASH15 may be used as a possible eco-friendly bioresource alternative for chemical fertilizers to promote plant growth in salt stressed agriculture area. However, the usability of V. halodenitrificans ASH15 under field trials is required for establishing it as a potential plant growth promoter for utilizing in sustainable agriculture under saline conditions.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/nuccore/CP090006.1/.
Author contributions
AS, X-PS, and Y-RL conceived the idea and designed the experiments. AS performed the experiments and wrote the original draft of the manuscript. RNS assisted in analysis and software support. RS, PS, and D-JG assisted in the experiments. X-PS contributed to resource management. KV and Y-RL critically revised the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by Guangxi Innovation Teams of Modern Agriculture Technology (nycytxgxcxtd-2021-03), National Natural Science Foundation of China (31760415), Youth Program of the National Natural Science Foundation of China (31901594), and Fund of the Guangxi Academy of Agricultural Sciences (2021YT011).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1229955/full#supplementary-material
References
Abbas, R., Rasul, S., Aslam, K., Baber, M., Shahid, M., Mubeen, F., et al. (2019). Halotolerant PGPR: a hope for the cultivation of saline soils. J. King Saud Univ. Sci. 31, 1195–1201. doi: 10.1016/j.jksus.02019
Abdullahi, S., Haris, H., Zarkasi, K. Z., and Amir, H. G. (2021). Complete genome sequence of plant growth-promoting and heavy metal-tolerant Enterobacter tabaci 4M9 (CCB-MBL 5004). J. Basic Microbiol. 61, 293–304. doi: 10.1002/jobm.202000695
Alishahi, F., Alikhani, H. A., Khoshkholgh-Sima, N. A., and Etesami, H. (2020). Mining the roots of various species of the halophyte Suaeda for halotolerant nitrogen-fixing endophytic bacteria with the potential for promoting plant growth. Int. Microbiol. 23, 415–427. doi: 10.1007/s10123-019-00115-y
Amziane, M., Metiaz, F., Darenfed-Bouanane, A., Djenane, Z., Selama, O., Abderrahmani, A., et al. (2013). Virgibacillus natechei sp. nov., a moderately halophilic bacterium isolated from sediment of a saline lake in southwest of Algeria. Curr. Microbiol. 66, 462–466. doi: 10.1007/s00284-012-0300-7
Anbumalar, S., and Ashokumar, P. (2016). Effect of Halobacterium in promoting the plant growth. Int. J. Sci. Res. 5, 934938.
Arora, N. K., Fatima, T., Mishra, J., Mishra, I., Verma, S., Verma, R., et al. (2020). Halo-tolerant plant growth promoting rhizobacteria for improving productivity and remediation of saline soils. J. Adv. Res. 26, 69–82. doi: 10.1016/j.jare.07003
Asaf, S., Khan, A. L., Khan, M. A., Al-Harrasi, A., and Lee, I. J. (2018). Complete genome sequencing and analysis of endophytic Sphingomonas sp. LK11 and its potential in plant growth. Biotech. 8, 1–14. doi: 10.1007/s13205-018-1403-z
Ashby, S. F. (1907). Some observations on the assimilation of atmospheric nitrogen by a free living soil organism, Azotobacter chroococcum of Beijerinck. J. Agric. Sci. 2, 35–51. doi: 10.1017/S0021859600000988
Backer, R., Rokem, J. S., Ilangumaran, G., Lamont, J., Praslickova, D., Ricci, E., et al. (2018). Plant growth-promoting rhizobacteria: context, mechanisms of action, and roadmap to commercialization of biostimulants for sustainable agriculture. Front. Plant Sci. 9, 1473. doi: 10.3389/fpls.2018.01473
Benson, G. (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580. doi: 10.1093/nar/27.2.573
Bergkemper, F., Schöler, A., Engel, M., Lang, F., Krüger, J., Schloter, M., et al. (2016). Phosphorus depletion in forest soils shapes bacterial communities toward phosphorus recycling systems. Environ. Microbiol. 18, 1988–2000. doi: 10.1111/1462-2920.13188
Bertelli, C., Laird, M. R., Williams, K. P., Lau, B. Y., Hoad, G., et al. (2017). Island Viewer 4: expanded prediction of genomic islands for larger-scale datasets. Nuc. Acids Res. 45, W30–W35. doi: 10.1093/nar/gkx343
Bhatt, H. B., and Singh, S. P. (2022). Diversity of cultivable bacteria in a saline desert of Little Rann of Kutch, India: a phylogenetic perspective. Front. Mar. Sci. 9, 472. doi: 10.3389/fmars.2022.769043
Bhattacharyya, C., Bakshi, U., Mallick, I., Mukherji, S., Bera, B., Ghosh, A., et al. (2017). Genome-guided insights into the plant growth promotion capabilities of the physiologically versatile Bacillus aryabhattai strain AB211. Front. Microbiol. 8, 411. doi: 10.3389/fmicb.2017.00411
Blin, K., Shaw, S., Steinke, K., Villebro, R., Ziemert, N., Lee, S. Y., et al. (2019). antiSMASH 5.0: updates to the secondary metabolite genome mining pipeline. Nucl. Acids Res. 47, W81–W87. doi: 10.1093/nar/gkz310
Borodovsky, M., and Mcininch, J. (1993). Gene mark, parallel gene recognition for both DNA strands. Comput. Chem. 17, 123–133. doi: 10.1016/0097-8485(93)85004-V
Brick, J. M., Bostock, R. M., and Silverstone, S. E. (1991). Rapid in situ assay for indole acetic acid production by bacteria immobilized on nitrocellulose membrane. Appl. Environ. Microbiol. 57, 535–538. doi: 10.1128/aem.57.2.535-538.1991
Bukhat, S., Imran, A., Javaid, S., Shahid, M., Majeed, A., Naqqash, T., et al. (2020). Communication of plants with microbial world: exploring the regulatory networks for PGPR mediated defense signaling. Microbiol. Res. 238, 126486. doi: 10.1016/j.micres.2020.126486
Calero, P., Gurdo, N., and Nikel, P. I. (2022). Role of the CrcB transporter of Pseudomonas putida in the multilevel stress response elicited by mineral fluoride. Environ. Microbiol. 24, 5082–5104. doi: 10.1111/1462-2920.16110
Chen, Y. H., Shyu, Y. T., and Lin, S. S. (2018). Characterization of candidate genes involved in halotolerance using high-throughput omics in the halotolerant bacterium Virgibacillus chiguensis. PLoS ONE. 13, e0201346. doi: 10.1371/journal.pone.0201346
Chin, C. S., Alexander, D. H., Marks, P., Klammer, A. A., Drake, J., Heiner, C., et al. (2013). Non-hybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat Methods 10, 563–569. doi: 10.1038/nmeth.2474
Connelly, M. B., Young, G. M., and Sloma, A. (2004). Extracellular proteolytic activity plays a central role in swarming motility in Bacillus subtilis. J Bacteriol. 186, 4159–4167. doi: 10.1128/JB.186.13.4159-4167.2004
Corral, P., Amoozegar, M. A., and Ventosa, A. (2019). Halophiles and their biomolecules: recent advances and future applications in biomedicine. Marine Drugs 18, 33. doi: 10.3390/md18010033
Corwin, D. L. (2021). Climate change impacts on soil salinity in agricultural areas. Eur. J. Soil Sci. 72, 842–862. doi: 10.1111/ejss.13010
Cress, B. F., Jones, J. A., Kim, D. C., Leitz, Q. D., Englaender, J. A., Collins, S. M., et al. (2016). Rapid generation of CRISPR/dCas9-regulated, orthogonally repressible hybrid T7-lac promoters for modular, tuneable control of metabolic pathway fluxes in Escherichia coli. Nucl. Acids Res. 44, 4472–4485. doi: 10.1093/nar/gkw231
Defez, R., Andreozzi, A., Romano, S., Pocsfalvi, G., Fiume, I., Esposito, R., et al. (2019). Bacterial IAA-delivery into medicago root nodules triggers a balanced stimulation of C and N metabolism leading to a biomass increase. Microorganisms. 7, 403. doi: 10.3390/microorganisms7100403
Delcher, A. L., Bratke, K. A., Powers, E. C., and Salzberg, S. L. (2007). Identifying bacterial genes and endosymbiont DNA with GLIMMER. Bioinformatics. 23, 673–679. doi: 10.1093/bioinformatics/btm009
Denariaz, G., Payne, W. J., and Le Gall, J. (1989). A halophilic denitrifier, Bacillus halodenitrificans sp. nov. Int. J. Syst. E39, 145–151. doi: 10.1099/00207713-39-2-145
Denton, J. F., Lugo-Martinez, J., Tucker, A. E., Schrider, D. R., Warren, W. C., Hahn, M. W., et al. (2014). Extensive error in the number of genes inferred from draft genome assemblies. PLoS Comput. Biol. 10, e1003998. doi: 10.1371/journal.pcbi.1003998
Duncan, D. B. (1955). Multiple range and multiple F tests. Biometrics 11, 1–42. doi: 10.2307/3001478
Durán-Viseras, A., Sánchez-Porro, C., and Ventosa, A. (2021). Genomic insights into new species of the genus Halomicroarcula reveals potential for new osmoadaptative strategies in halophilic archaea. Front. Microbiol. 3336. doi: 10.3389./fmicb.2021.751746
Dutta, B., and Bandopadhyay, R. (2022). Biotechnological potentials of halophilic microorganisms and their impact on mankind. Beni-Suef Univ. J. Basic Appl. Sci. 11, 75. doi: 10.1186/s43088-022-00252-w
Egamberdieva, D., Wirth, S., Bellingrath-Kimura, S. D., Mishra, J., and Arora, N. K. (2019). Salt-tolerant plant growth promoting rhizobacteria for enhancing crop productivity of saline soils. Front. Microbiol. 10, 2791. doi: 10.3389/fmicb.2019.02791
Fayez, D., Youssif, A., Sabry, S., Ghozlan, H., and Eltarahony, M. (2022). Carotegenic Virgibacillus halodenitrificans from Wadi El-Natrun Salt Lakes: isolation, optimization, characterization and biological activities of carotenoids. Biology 11, 1407. doi: 10.3390/biology11101407
Felsenstein, J. (1985). Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39, 783–791. doi: 10.1111/j.1558-5646.1985.tb00420.x
Godde, C. M., Mason-D'Croz, D., Mayberry, D. E., Thornton, P. K., and Herrero, M. (2021). Impacts of climate change on the livestock food supply chain; a review of the evidence. Global Food Secur. 28, 100488. doi: 10.1016/j.gfs.2020.100488
Gohil, N., Bhattacharjee, G., Khambhati, K., Braddick, D., and Singh, V. (2019). Engineering strategies in microorganisms for the enhanced production of squalene: advances, challenges and opportunities. Front. Bioeng. Biotechnol. 7, 50. doi: 10.3389/fbioe.2019.00050
Grant, J. R., and Stothard, P. (2008). The CGView Server: a comparative genomics tool for circular genomes. Nucleic Acids Res. 36, W181–W184. doi: 10.1093/nar/gkn179
Green, E. R., and Mecsas, J. (2016). Bacterial secretion systems: an overview. Microbiol. Spectr. 4. doi: 10.1128/microbiolspec.VMBF-0012-2015
Gunde-Cimerman, N., Plemenitaš, A., and Oren, A. (2018). Strategies of adaptation of microorganisms of the three domains of life to high salt concentrations. FEMS Microbiol. Rev. 42, 353–375. doi: 10.1093/femsre/fuy009
Guo, D. J., Singh, R. K., Singh, P., Li, D. P., Sharma, A., Xing, Y. X., et al. (2020). Complete genome sequence of Enterobacter roggenkampii ED5, a nitrogen fixing plant growth promoting endophytic bacterium with biocontrol and stress tolerance properties, isolated from sugarcane root. Front. Microbiol. 11, 580081. doi: 10.3389/fmicb.2020.580081
Gupta, S., Sharma, P., Dev, K., Srivastava, M., and Sourirajan, A. (2015). A diverse group of halophilic bacteria exist in Lunsu, a natural salt water body of Himachal Pradesh, India. SpringerPlus. 4, 1–9. doi: 10.1186/s40064-015-1028-1
Herlihy, J. H., Long, T. A., and McDowell, J. M. (2020). Iron homeostasis and plant immune responses: recent insights and translational implications. J. Biol. Chem. 295, 13444–13457. doi: 10.1074/jbc.REV120.010856
Huang, Z. R., Lin, Y. K., and Fang, J. Y. (2009). Biological and pharmacological activities of squalene and related compounds: potential uses in cosmetic dermatology. Molecules 14, 540–554. doi: 10.3390/molecules14010540
Jamali, H., Sharma, A., and Srivastava, A. K. (2020). Biocontrol potential of Bacillus subtilis RH5 against sheath blight of rice caused by Rhizoctonia solani. J. Basic Microbiol. 60, 268–280. doi: 10.1002/jobm.201900347
Jordan, S., Junker, A., Helmann, J. D., and Mascher, T. (2006). Regulation of LiaRS-dependent gene expression in Bacillus subtilis: identification of inhibitor proteins, regulator binding sites, and target genes of a conserved cell envelope stress-sensing two-component system. J. Bacteriol. 188, 5153–5166. doi: 10.1128/JB.00310-06
Kang, S. M., Asaf, S., Khan, A. L., Lubna Khan, A., Mun, B. G., et al. (2020). Complete genome sequence of Pseudomonas psychrotolerans CS51, a plant growth-promoting bacterium, under heavy metal stress conditions. Microorganisms 8, 382. doi: 10.3390/microorganisms8030382
Karatan, E., Saulmon, M. M., Bunn, M. W., and Ordal, G. W. (2001). Phosphorylation of the response regulator CheV is required for adaptation to attractants during Bacillus subtilischemotaxis. J. Biol. Chem. 276, 43618–43626. doi: 10.1074/jbc.M104955200
Khumairah, F. H., Setiawati, M. R., Fitriatin, B. N., Simarmata, T., Alfaraj, S., Ansari, M. J., et al. (2022). Halotolerant Plant growth-promoting rhizobacteria isolated from saline soil improve nitrogen fixation and alleviate salt stress in rice plants. Front. Microbiol. 13, 1607. doi: 10.3389/fmicb.2022.905210
Kim, S. K., and Karadeniz, F. (2012). Biological importance and applications of squalene and squalane. Adv. Food Nutr. Res. 65, 223–233. doi: 10.1016/B978-0-12-416003-3.00014-7
Köhl, J., Kolnaar, R., and Ravensberg, W. J. (2019). Mode of action of microbial biological control agents against plant diseases: relevance beyond efficacy. Front. Plant Sci. 845. doi: 10.3389./fpls.2019.00845
Kumari, S., Vaishnav, A., Jain, S., Varma, A., and Choudhary, D. K. (2015). Bacterial-mediated induction of systemic tolerance to salinity with expression of stress alleviating enzymes in soybean Glycine max L. Merrill. J. Plant Growth Regul. 343, 558–573. doi: 10.1007/s00344-015-9490-0
Kumaunang, M., Sanchart, C., Suyotha, W., and Maneerat, S. (2019). Virgibacillus halodenitrificans MSK-10P, a potential protease-producing starter culture for fermented shrimp paste (kapi) production. J. Aquat. Food Prod. Technol. 28, 877–890. doi: 10.1080/10498850.2019.1652874
Lahlali, R., Ezrari, S., Radouane, N., Kenfaoui, J., Esmaeel, Q., El Hamss, H., et al. (2022). Biological control of plant pathogens: a global perspective. Microorganisms 10, 596. doi: 10.3390/microorganisms10030596
Lam, M. Q., Chen, S. J., Goh, K. M., Abd Manan, F., Yahya, A., Shamsir, M. S., et al. (2021). Genome sequence of an uncharted halophilic bacterium Robertkochia marina with deciphering its phosphate-solubilizing ability. Braz. J. Microbiol. 52, 251–256. doi: 10.1007/s42770-020-00401-2
Lee, S. J., Lee, Y. J., Jeong, H., Lee, S. J., Lee, H. S., Pan, J. G., et al. (2012). Draft genome sequence of Virgibacillus halodenitrificans 1806, 6332–6333. doi: 10.1128/JB.01280-12
León, M. J., Hoffmann, T., Sánchez-Porro, C., Heider, J., Ventosa, A., Bremer, E., et al. (2018). Compatible solute synthesis and import by the moderate halophile Spiribacter salinus: physiology and genomics. Front. Microbiol. 9, 108. doi: 10.3389/fmicb.2018.00108
Masmoudi, F., Alsafran, M., Jabri, H. A., Hosseini, H., Trigui, M., Sayadi, S., et al. (2023). Halobacteria-based biofertilizers: a promising alternative for enhancing soil fertility and crop productivity under biotic and abiotic stresses—A review. Microorganisms 11, 1248. doi: 10.3390/microorganisms11051248
Mechri, S., Bouacem, K., Amziane, M., Dab, A., Nateche, F., Jaouadi, B., et al. (2019). Identification of a new serine alkaline peptidase from the moderately halophilic Virgibacillus natechei sp. nov., strain FarD T and its application as bioadditive for peptide synthesis and laundry detergent formulations. Biomed Res. Int. 2019. doi: 10.1155./2019/6470897
Mehta, S., and Nautiyal, C. S. (2001). An efficient method for qualitative screening of phosphate-solubilizing bacteria. Curr. Microbiol. 43, 51–56. doi: 10.1007/s002840010259
Meliawati, M., May, T., Eckerlin, J., Heinrich, D., Herold, A., Schmid, J., et al. (2022). Insights in the complex DegU, DegS, and Spo0A regulation system of Paenibacillus polymyxa by CRISPR-Cas9-based targeted point mutations. Appl. Environ. Microbiol. 88, e00164–22. doi: 10.1128/aem.00164-22
Minamino, T., Kinoshita, M., and Namba, K. (2019). Directional switching mechanism of the bacterial flagellar motor. Comput. Struct. Biotechnol. J. 17, 1075-1081. doi: 10.1016/j.csbj.07020
Minato, Y., Ueda, T., Machiyama, A., Iwaï, H., and Shimada, I. (2017). Dynamic domain arrangement of CheA-CheY complex regulates bacterial thermotaxis, as revealed by NMR. Sci. Rep. 7, 16462. doi: 10.1038/s41598-017-16755-x
Montriwong, A., Kaewphuak, S., Rodtong, S., Roytrakul, S., and Yongsawatdigul, J. (2012). Novel fibrinolytic enzymes from Virgibacillus halodenitrificans SK1-3-7 isolated from fish sauce fermentation. Process Biochem. 47, 2379–2387. doi: 10.1016/j.procbio.09020
Mukherjee, T., Elmas, M., Vo, L., Alexiades, V., Hong, T., Alexandre, G., et al. (2019). Multiple CheY homologs control swimming reversals and transient pauses in Azospirillum brasilense. Biophys. J. 116, 1527–1537. doi: 10.1016/j.bpj.03006
Naghoni, A., Emtiazi, G., Amoozegar, M. A., Cretoiu, M. S., Stal, L. J., Etemadifar, Z., et al. (2017). Microbial diversity in the hypersaline Lake Meyghan, Iran. Sci. Rep. 7, 11522. doi: 10.1038/s41598-017-11585-3
Natale, P., Brüser, T., and Driessen, A. J. (2008). Sec-and Tat-mediated protein secretion across the bacterial cytoplasmic membrane—distinct translocases and mechanisms. Biochim. Biophys. Acta - Biomembr. 1778, 1735–1756. doi: 10.1016/j.bbamem.07015
Netzker, T., Fischer, J., Weber, J., Mattern, D. J., König, C. C., Valiante, V., et al. (2015). Microbial communication leading to the activation of silent fungal secondary metabolite gene clusters. Front. Microbiol. 6, 299. doi: 10.3389/fmicb.2015.00299
Newsom, S., Parameshwaran, H. P., Martin, L., and Rajan, R. (2021). The CRISPR-Cas mechanism for adaptive immunity and alternate bacterial functions fuels diverse biotechnologies. Front. Cell. Infect. Microbiol. 10, 619763. doi: 10.3389/fcimb.2020.619763
Othoum, G., Bougouffa, S., Bokhari, A., Lafi, F. F., Gojobori, T., Hirt, H., et al. (2019). Mining biosynthetic gene clusters in Virgibacillus genomes. BMC Genom. 20, 1–10. doi: 10.1186/s12864-019-6065-7
Palma, V., Gutiérrez, M. S., Vargas, O., Parthasarathy, R., and Navarrete, P. (2022). Methods to evaluate bacterial motility and its role in bacterial–host interactions. Microorganisms. 10, 563. doi: 10.3390/microorganisms10030563
Paramasivan, K., and Mutturi, S. (2022). Recent advances in the microbial production of squalene. World J. Microbiol. Biotechnol. 38, 91. doi: 10.1007/s11274-022-03273-w
Passari, A. K., Rajput, V., Priya, L. P., Dharne, M., Dastager, S., and Singh, B. P. (2019). Draft genome sequence of plant growth-promoting endophytic Microbacterium hydrothermale BPSAC84, isolated from the medicinal plant Mirabilis jalapa. Microbiol. Resour. Announc. 8, 10–1128. doi: 10.1128/MRA.00406-19
Pérez-Inocencio, J., Iturriaga, G., Aguirre-Mancilla, C. L., Ramírez-Pimentel, J. G., Vásquez-Murrieta, M. S., Álvarez-Bernal, D., et al. (2022). Identification of halophilic and halotolerant bacteria from the root soil of the halophyte Sesuvium verrucosum Raf. Plants 11, 3355. doi: 10.3390/plants11233355
Piligrimova, E. G., Kazantseva, O. A., Kazantsev, A. N., Nikulin, N. A., Skorynina, A. V., Koposova, O. N., et al. (2021). Putative plasmid prophages of Bacillus cereus sensu lato may hold the key to undiscovered phage diversity. Sci. Rep. 11, 7611. doi: 10.1038/s41598-021-87111-3
Qin, Q., Ling, C., Zhao, Y., Yang, T., Yin, J., Guo, Y., et al. (2018). CRISPR/Cas9 editing genome of extremophile Halomonas spp. Metab. Eng. 47, 219–229. doi: 10.1016/j.ymben.03018
Qurashi, A. W., and Sabri, A. N. (2012). Bacterial exopolysaccharide and biofilm formation stimulate chickpea growth and soil aggregation under salt stress. Braz. J. Microbiol. 43, 1183–1191. doi: 10.1590/S1517-83822012000300046
Ramisetty, B. C. M., and Sudhakari, P. A. (2019). Bacterial “grounded” prophages: hotspots for genetic renovation and innovation. Front. Gen. 10, 65. doi: 10.3389/fgene.2019.00065
Rao, C. V., Kirby, J. R., and Arkin, A. P. (2004). Design and diversity in bacterial chemotaxis: a comparative study in Escherichia coli and Bacillus subtilis. PLoS Biol. 2, e49. doi: 10.1371/journal.pbio.0020049
Reang, L., Bhatt, S., Tomar, R. S., Joshi, K., Padhiyar, S., Vyas, U. M., et al. (2022). Plant growth promoting characteristics of halophilic and halotolerant bacteria isolated from coastal regions of Saurashtra Gujarat. Sci. Rep. 12, 4699. doi: 10.1038/s41598-022-08151-x
Repizo, G. D., Blancato, V. S., Sender, P. D., Lolkema, J., and Magni, C. (2006). Catabolite repression of the citST two-component system in Bacillus subtilis. FEMS Microbiol. Lett. 260, 224–231. doi: 10.1111/j.1574-6968.2006.00318.x
Rinaldi, M. A., Ferraz, C. A., and Scrutton, N. S. (2022). Alternative metabolic pathways and strategies to high-titre terpenoid production in Escherichia coli. Nat. Product Rep. 39, 90118. doi: 10.1039/d1np00025j
Roberts, M. F. (2005). Organic compatible solutes of halotolerant and halophilic microorganisms. Saline Sys. 1, 1–30. doi: 10.1186/1746-1448-1-5
Rosales-Garcia, T., Jimenez-Martinez, C., and Dávila-Ortiz, G. (2017). Squalene extraction: biological sources and extraction methods. Int. J. Environ. Agric. Biotechnol. 2, 1662–1670. doi: 10.22161/ijeab/2.4.26
Sánchez-Porro, C., de la Haba, R. R., and Ventosa, A. (2014). The genus Virgibacillus. Prokar. Firm. Teneri. 455-465. doi: 10.1007./978-3-642-30120-9_353
Sarwar, M. K., Azam, I., and Iqbal, T. (2015). Biology and applications of halophilic bacteria and archaea. A. Electron. J. Biotechnol. 11, 98–103.
Satari, L., Guillén, A., Latorre-Pérez, A., and Porcar, M. (2021). Beyond archaea: the table salt bacteriome. Front. Microbiol. 3157. doi: 10.3389./fmicb.2021.714110
Schwyn, B., and Neilands, J. B. (1987). Universal chemical assay for the detection and determination of siderophores. Anal. Biochem. 160, 47–56. doi: 10.1016/0003-2697(87)90612-9
Shankar, M., Mohapatra, S. S., Biswas, S., and Biswas, I. (2015). Gene regulation by the LiaSR two-component system in Streptococcus mutans. PLoS ONE. 10, e0128083. doi: 10.1371/journal.pone.0128083
Sharma, A., Kashyap, P. L., Srivastava, A. K., Bansal, Y. K., and Kaushik, R. (2019). Isolation and characterization of halotolerant bacilli from chickpea (Cicer arietinum L.) rhizosphere for plant growth promotion and biocontrol traits. Eur J Plant Pathol. 153, 787–800. doi: 10.1007/s10658-018-1592-7
Sharma, A., Singh, P., Kumar, S., Kashyap, P. L., Srivastava, A. K., Chakdar, H., et al. (2015). Deciphering diversity of salt-tolerant bacilli from saline soils of eastern Indo-gangetic plains of India. Geomicrobiol. J. 32, 170–180. doi: 10.1080/01490451.2014.938205
Sharma, A., Singh, R. K., Singh, P., Vaishnav, A., Guo, D. J., Verma, K. K., et al. (2021). Insights into the bacterial and nitric oxide-induced salt tolerance in sugarcane and their growth-promoting abilities. Microorganisms 9, 2203. doi: 10.3390/microorganisms9112203
Sharma, A., Song, X. P., Singh, R. K., Vaishnav, A., Gupta, S., Singh, P., et al. (2022). Impact of carbendazim on cellular growth, defence system and plant growth promoting traits of Priestia megaterium ANCB-12 isolated from sugarcane rhizosphere. Front. Microbiol. 13. doi: 10.3389./fmicb.2022.1005942
Sharma, A., Vaishnav, A., Jamali, H., Srivastava, A. K., Saxena, A. K., Srivastava, A. K., et al. (2016). Halophilic bacteria: potential bioinoculants for sustainable agriculture and environment management under salt stress. Plant-Microbe Int. App. Sustain. Agricult. 4, 297–325. doi: 10.1007./978-981-10-2854-0_14
Sharma, S. K., Sharma, M. P., and Aketi, R., and Joshi, O. P. (2012). Characterization of zinc-solubilizing Bacillus isolates and their potential to influence zinc assimilation in soybean seeds. J. Microbiol. Biotechnol. 22, 352–359. doi: 10.4014/jmb.1106.05063
Singh, R. K., Singh, P., Sharma, A., Guo, D. J., Upadhyay, S. K., Song, Q. Q., et al. (2022). Unraveling nitrogen fixing potential of endophytic diazotrophs of different saccharum species for sustainable sugarcane growth. Int. J. Mol. Sci. 23, 6242. doi: 10.3390/ijms23116242
Singh, T. A., Passari, A. K., Jajoo, A., Bhasin, S., Gupta, V. K., Hashem, A., et al. (2021). Tapping into actinobacterial genomes for natural product discovery. Front. Microbiol. 12, 655620. doi: 10.3389/fmicb.2021.655620
Srivastava, A. K., Srivastava, R., Sharma, A., Bharati, A. P., Yadav, J., Singh, A. K., et al. (2022). Transcriptome analysis to understand salt stress regulation mechanism of Chromohalobacter salexigens ANJ207. Front. Microbiol. 13, 9276. doi: 10.3389./fmicb.2022.909276
Sultana, S., Paul, S. C., Parveen, S., Alam, S., Rahman, N., Jannat, B., et al. (2020). Isolation and identification of salt-tolerant plant-growth-promoting rhizobacteria and their application for rice cultivation under salt stress. Can. J. Microbiol. 66, 144–160. doi: 10.1139/cjm-2019-0323
Suntharalingam, P., Senadheera, M. D., Mair, R. W., Levesque, C. M., and Cvitkovitch, D. G. (2009). The LiaFSR system regulates the cell envelope stress response in Streptococcus mutans. J. Bacteriol. 191, 2973–2984. doi: 10.1128/JB.01563-08
Suriani, N. L., Suprapta, D., Novizar, N., Parwanayoni, N., Darmadi, A., Dewi, D., et al. (2020). A mixture of piper leaves extracts and rhizobacteria for sustainable plant growth promotion and biocontrol of blast pathogen of organic bali rice. Sustainability 12, 8490. doi: 10.3390/su12208490
Tamura, K., Nei, M., and Kumar, S. (2004). Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc. Natl. Acad. Sci. USA. 101, 11030–11035. doi: 10.1073/pnas.0404206101
Tarailo-Graovac, M., and Chen, N. (2009). Using Repeat Masker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinform. 5, 4–10. doi: 10.1002/0471250953.bi0410s25
Thanh, T. N., Jürgen, B., Bauch, M., Liebeke, M., Lalk, M., Ehrenreich, A., et al. (2010). Regulation of acetoin and 2, 3.-butanediol utilization in Bacillus licheniformis. Appl. Microbiol. Biotechnol. 87, 2227–2235. doi: 10.1007/s00253-010-2681-5
Tiwari, S., and Lata, C. (2018). Heavy metal stress, signaling, and tolerance due to plant-associated microbes: an overview. Front. Plant Sci. 9, 452. doi: 10.3389/fpls.2018.00452
Ullah, A., Bano, A., and Khan, N. (2021). Climate change and salinity effects on crops and chemical communication between plants and plant growth-promoting microorganisms under stress. Front. Sustain. Food Syst. 5, 618092. doi: 10.3389/fsufs.2021.618092
Vaidya, S., Dev, K., and Sourirajan, A. (2018). Distinct osmoadaptation strategies in the strict halophilic and halotolerant bacteria isolated from Lunsu salt water body of North West Himalayas. Curr. Microbiol. 75, 888–895. doi: 10.1007/s00284-018-1462-8
Wang, X., Vallurupalli, P., Vu, A., Lee, K., Sun, S., Bai, W. J., et al. (2014). The linker between the dimerization and catalytic domains of the CheA histidine kinase propagates changes in structure and dynamics that are important for enzymatic activity. Biochem. 53, 855–861. doi: 10.1021/bi4012379
Wang, X., Xia, K., Yang, X., and Tang, C. (2019). Growth strategy of microbes on mixed carbon sources. Nat. Commun. 10, 1279. doi: 10.1038/s41467-019-09261-3
Xiaoqin, S., Dongli, S., Yuanhang, F., Hongde, W., and Lei, G. (2021). Three dimensional fractal characteristics of soil pore structure and their relationships with hydraulic parameters in biochar-amended saline soil. Soil Tillage Res. 205, 104809. doi: 10.1016/j.still.2020.104809
Xu, B., Hu, B., Wang, J., Lan, Y., Zhu, Y., Dai, X., et al. (2018). Virgibacillus indicus sp. nov. and Virgibacillus profundi sp. nov, two moderately halophilic bacteria isolated from marine sediment by using microfluidic streak plates. Int. J. Syst. Evol. Microbiol. 68, 2015–2023. doi: 10.1099/ijsem.0.002782
Yoon, J. H., Oh, T. K., and Park, Y. H. (2004). Transfer of Bacillus halodenitrificans Denariaz 1989 to the genus Virgibacillus as Virgibacillus halodenitrificans comb. nov. Int. J. Syst. Evol. Microbiol. 54, 2163–2167. doi: 10.1099/ijs.0.63196-0
Zhao, M., Yin, C., Tao, Y., Li, C., and Fang, S. (2019). Diversity of soil microbial community identified by Biolog method and the associated soil characteristics on reclaimed Scirpus mariqueter wetlands. SN Appl. Sci. 1, 1–8. doi: 10.1007/s42452-019-1443-y
Zhou, Y., Sun, Z. Y., Li, H., Qian, C. J., Wu, X., Tang, H., et al. (2017). Investigation of compatible solutes synthesis and transport of Virgibacillus halodenitrificans PDB-F2 with complete genome analysis. Int. Biodeterior. Biodegrad. 122, 165–172. doi: 10.1016/j.ibiod.05005
Ziemert, N., Alanjary, M., and Weber, T. (2016). The evolution of genome mining in microbes–a review. Nat. Prod. Rep. 33, 988–1005. doi: 10.1039/C6NP00025H
Keywords: Virgibacillus halodenitrificans, whole genome, salt-tolerant, plant growth promoting traits, CRISPRs, isoprenoids, squalene
Citation: Sharma A, Singh RN, Song X-P, Singh RK, Guo D-J, Singh P, Verma KK and Li Y-R (2023) Genome analysis of a halophilic Virgibacillus halodenitrificans ASH15 revealed salt adaptation, plant growth promotion, and isoprenoid biosynthetic machinery. Front. Microbiol. 14:1229955. doi: 10.3389/fmicb.2023.1229955
Received: 27 May 2023; Accepted: 31 July 2023;
Published: 22 September 2023.
Edited by:
D. K. Choudhary, Amity University, IndiaReviewed by:
Ajit Kumar Passari, Scotland's Rural College, United KingdomPawan Kumar Jayaswal, Indian Council of Agricultural Research, India
Copyright © 2023 Sharma, Singh, Song, Singh, Guo, Singh, Verma and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yang-Rui Li, bGl5ckBneGFhcy5uZXQ=