 Brook E. Santangelo1*
Brook E. Santangelo1* Madison Apgar1
Madison Apgar1 Angela Sofia Burkhart Colorado1Casey G. Martin1
Angela Sofia Burkhart Colorado1Casey G. Martin1 John Sterrett2Elena Wall1
John Sterrett2Elena Wall1 Marcin P. Joachimiak3
Marcin P. Joachimiak3 Lawrence E. Hunter1Catherine A. Lozupone1
Lawrence E. Hunter1Catherine A. Lozupone1- 1Department of Biomedical Informatics, University of Colorado School of Medicine, Aurora, CO, United States
- 2Department of Integrative Physiology, University of Colorado, Boulder, CO, United States
- 3Lawrence Berkeley National Laboratory, Environmental Genomics and Systems Biology Division, Biosystems Data Science Department, Berkeley, CA, United States
Advances in high-throughput technologies have enhanced our ability to describe microbial communities as they relate to human health and disease. Alongside the growth in sequencing data has come an influx of resources that synthesize knowledge surrounding microbial traits, functions, and metabolic potential with knowledge of how they may impact host pathways to influence disease phenotypes. These knowledge bases can enable the development of mechanistic explanations that may underlie correlations detected between microbial communities and disease. In this review, we survey existing resources and methodologies for the computational integration of broad classes of microbial and host knowledge. We evaluate these knowledge bases in their access methods, content, and source characteristics. We discuss challenges of the creation and utilization of knowledge bases including inconsistency of nomenclature assignment of taxa and metabolites across sources, whether the biological entities represented are rooted in ontologies or taxonomies, and how the structure and accessibility limit the diversity of applications and user types. We make this information available in a code and data repository at: https://github.com/lozuponelab/knowledge-source-mappings. Addressing these challenges will allow for the development of more effective tools for drawing from abundant knowledge to find new insights into microbial mechanisms in disease by fostering a systematic and unbiased exploration of existing information.
1 Introduction
The structure and function of the human microbiome can be both a driver and consequence of various disease states (Falony et al., 2019; King et al., 2019). Microbiome signatures are associated with a range of conditions including auto-immune and gastrointestinal disease, cancer, and neurological disease (Berg et al., 2020). Understanding interactions between the gut microbiome and the host at a mechanistic level requires a sophisticated synthesis of individual microbial functions, such as metabolic output, and how these functions interact with host processes that influence human physiology.
Mechanistic prediction of microbe-host interactions often involves in-depth analyses of multi-omic datasets. For example, in one study that related immune markers, microbiome composition, metabolomic data, diet, and demographic measures to markers of metabolic health in people living with human immunodeficiency virus (HIV), they found that butyrate production and mucolytic activity of particular gut microbes play a potential role in intestinal barrier dysfunction, suggesting more targeted followup studies (Armstrong et al., 2021). In another study that used metagenomic, metatranscriptomic, metaproteomic, and metabolic data to explore the functional attributes of the microbiome that influence Parkinson’s disease (PD) pathogenesis, a preliminary result found the metabolite 2-hydroxypyridine (2-HP) and the microbe Methanobrevibacter smithii to be enriched in PD, prompting several experiments which verified their effects on alpha synuclein aggregation (Wilmes et al., 2022). Large scale metagenomics studies can hone in on these interactions at a species or strain level, and bioinformatics analyses can further hypothesize how the microbial community contributes to health or disease (Armour et al., 2019; Wallen et al., 2022). Such studies provide rich information in the scientific literature on associations between microbes, host pathways and diseases, which brings us closer toward a mechanistic understanding of the microbiome. Despite advances in bioinformatics techniques to evaluate multi-omic datasets and laboratory methods to further explore promising results, there is no efficient and reliable solution for using existing knowledge to identify the most promising potential mechanisms involving the microbiome in human disease for further experimental validation.
Public resources that organize microbial knowledge serve an important purpose in mechanistic inference. These can be summarized into six categories, with relevant concepts defined in Box 1: (1) Ontologies and taxonomies which provide a standardized nomenclature and hierarchical ranking of biological entities such as microorganisms, proteins or metabolites, (2) Annotated databases that have some information about the given concept that is linked to an experimental result, (3) Mechanistic curated knowledge bases that contain knowledge drawn from multiple data sources and render known explanations about biological interactions (4) Integrated knowledge bases which aggregate relationships and identifiers represented across many different sources (5) Correlative curated knowledge bases which include associations found between two unique concept types, e.g., a microbe and a disease, and (6) Inference-ready knowledge bases that enable mechanistic inference (Figure 1A). Synthesizing microbial and host information is critical to achieve a systems level perspective of the microbiome. Such integrated resources elucidate relationships between microbes and other biological concepts, allowing researchers to access the increasing amount of information to draw new conclusions.
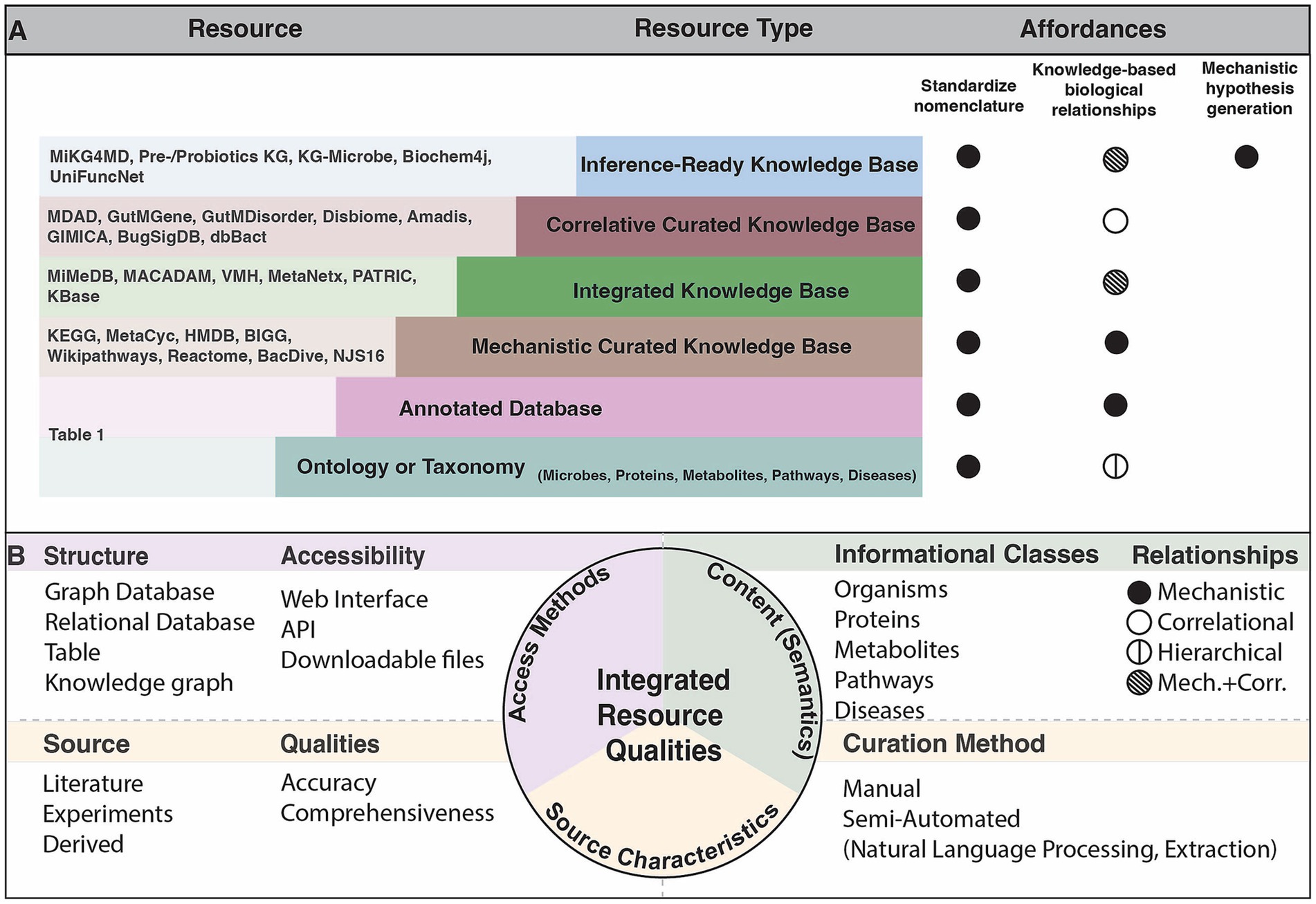
Figure 1. Characterization of known resources relevant to microbiome research. (A) Schematic of the types of resources that exist and the purpose that they serve in microbiome research. Note that resource characterization is based on the prominent qualities, though many resources span these types. Affordances represent the primary purpose of the given resource type. The standardized nomenclature affordance indicates that the resource introduces new identifiers to uniquify concepts. The knowledge-based biological relationships affordance implies that the resource describes interactions among the concepts by the indicated relationship type in (B). The mechanistic hypothesis inference affordance indicates that the resource is uniquely suited to provide a mechanistic explanation when given specific queries. (B) The evaluations performed over existing resources mentioned in the Resources column of (A) within this review.
BOX 1 Key terms
• Data repository: an archive of any data formats to enable public sharing
• Primary knowledge source: a source of knowledge that is used as a nomenclature standard for a knowledge base, for example an annotated database or ontology
• Ontology or Taxonomy: a system that is used as a semantic standard with a hierarchical classification scheme approved by groups of experts (Carpendale et al., 2014)
• Annotated database: database that stores experimentally derived content, such as sequences or structures from an instrument, with data labels potentially from manual curation
• Knowledge Base: structured repository describing the relationships between categories and the standardizing mappings of such categories*
• Mechanistic knowledge: an assertion of causal relationships between two categories
• Correlative knowledge: an assertion of statistical associations between two categories
• Mechanistic curated knowledge base: knowledge base derived from manual curation over multiple knowledge sources
• Correlative curated knowledge base: repository of correlative knowledge derived from manual curation over multiple knowledge sources
• Integrated knowledge base: knowledge base that incorporates content from multiple knowledge sources, most often cross-linking identifiers over such resources
• Inference-ready knowledge base: knowledge base that represents relationships in a logically consistent and semantically well-defined manner
*A category here signifies a class of knowledge based on empirical evidence, often referred to as a concept or entity. The three terms are used interchangeably throughout this review.
We evaluate the accessibility of the many resources that fall within these categories, which alludes to the structure that the content is made available in, and the interfaces that users are able to access the resource. The various ways that knowledge bases are made available, whether through downloadable files, programmatically, or via a user interface, influences how useful they are among scientists. We identify the content of each resource including the types of information classes that are represented and the types of relationships between concepts. Lastly, we critique the source characteristics of each resource by assessing the source of knowledge, the curation method, and the qualities such as accuracy that result from those curation methods. We assess comprehensiveness and accuracy by examining how each resource was constructed and how automated processes can lack the specific or validated evidence provided by manual curation. The most effective integrated resources are those which link all categories of knowledge, and align the concepts represented to identifiers of ontologies or primary knowledge bases (Figure 1B). These resources thereby allow for sophisticated computational analyses and inference to understand microbial mechanisms. In this review, we assess how integrated resources and tools can be used to address mechanistic questions in microbiome research.
2 Efforts to standardize microbiome studies
Understanding the host-associated microbiome is particularly challenging given the need to incorporate both microbial and host processes into analyses. Due to the interdisciplinary nature of microbiome research, there have been many efforts to develop broad standardization of experimental design, metadata, and reported results of observational and genetic studies in the field. The Genomic Standards Consortium (GSC) introduced two important standards: minimum information about any (x) sequence (MIxS) and minimum information about marker gene sequence (MIMARKS), and a checklist for microbiome study reporting and manuscript preparation (Yilmaz et al., 2011; Mirzayi and Renson, 2021). Platforms such as Qiita, which allows users to perform microbiome analyses for one or more studies, require the metadata to be entered according to MIxS standards (Gonzalez et al., 2018). These standards ensure consistency in reporting metadata of new, published experimental results and support integration of data across studies seamlessly.
In addition to standardizing metadata, it is important to unify the representation of concepts for multi-omic studies. Integrated resources harmonize biological content by mapping entities to standardized ontologies or other primary databases. The nomenclature of microbes, proteins, and metabolites that are involved in a microbiome study may vary, and aligning these terms is important for aggregation. It is most useful if the concepts represented are mapped to universally accepted identifiers such as ontologies or taxonomies (Box 1). Many domain specific ontologies exist in the Open Biological and Biomedical Ontology Foundry (OBO) that are widely relevant to biomedicine, such as the Gene Ontology (GO) that provides a directed acyclic graph (DAG) structure to the biological processes, cellular components, and molecular functions that result from gene products (The Gene Ontology Consortium, 2019; Jackson et al., 2021). Structured databases that consolidate external annotations, align nomenclature, and provide frequent updates can also be the main source of identifiers. PubChem is one such resource of chemical information including molecular formula, structure, and physical properties, while DrugBank expands on this information to include drug target sequences and pharmacological properties (Wishart et al., 2018; Kim et al., 2023). This process of standardization thus enables the contextualization of specific experimental results to a broader class of concepts (chemicals, proteins, organisms, etc.). The primary databases relevant for microbiome research are described in Table 1. These efforts for standardization of both metadata and microbial concepts have supported the development of integrated resources that combine functional and metabolic concepts of microbes and the host.
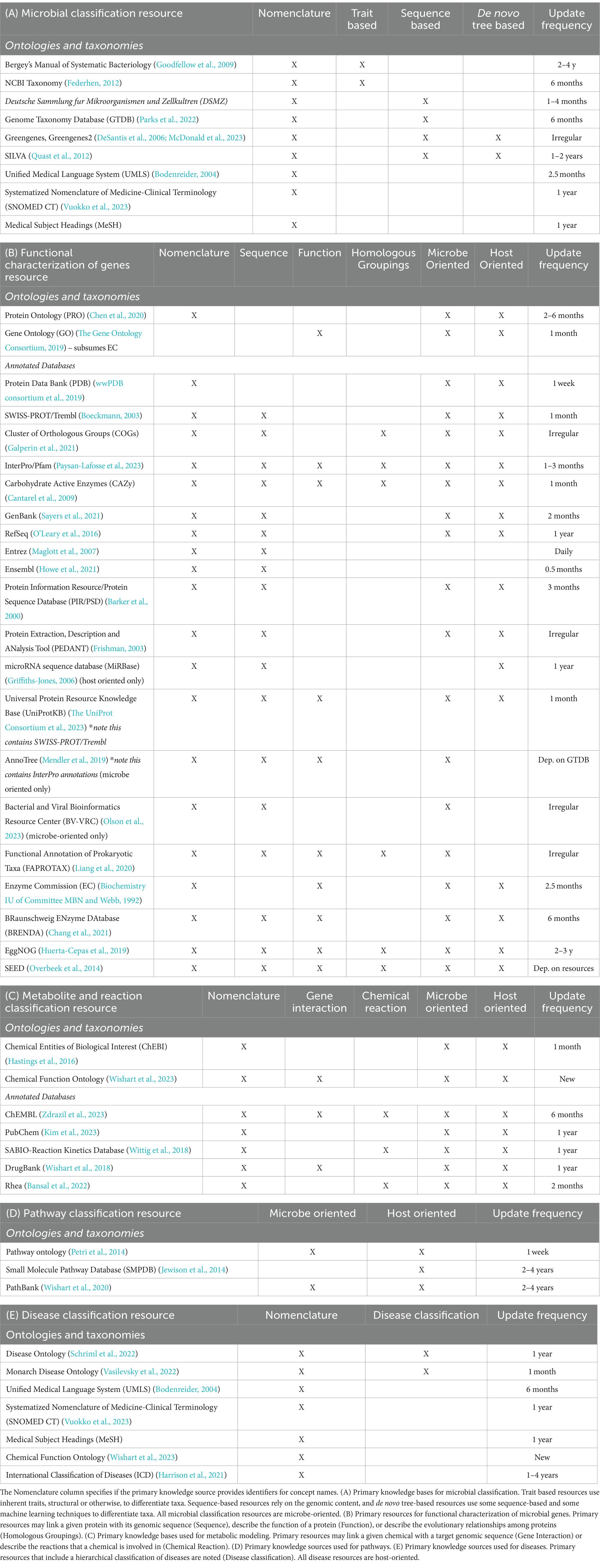
Table 1. Primary knowledge sources for the standardization of all entity types.
3 Microbiome-relevant knowledge bases and their applications
Our understanding of the microbiome is improved through knowledge of the relationships between individual microbial taxa, the functional characterization of their genes and how genomic content contributes to their metabolic outputs, and other information on microbial traits and functions determined through experimentation. Relating microbial taxonomic and functional information to host pathways, physiology or disease can provide mechanistic detail that informs our understanding of microbe-host interactions. This knowledge is made available through methods to collect and curate knowledge of microbial functions from the literature using natural language processing or manual annotation and representing the information in the form of integrated resources. We assess the relationships among integrated knowledge bases and their mappings to primary databases in Figure 2. The varying categories of integrated resources are highly applicable to three primary use cases: effectively accessing systems level microbiome information, contextualizing new findings with existing findings, and inferring new relationships to better understand how microbes influence disease. More detail of these resource qualities is described in Supplementary Table 1.
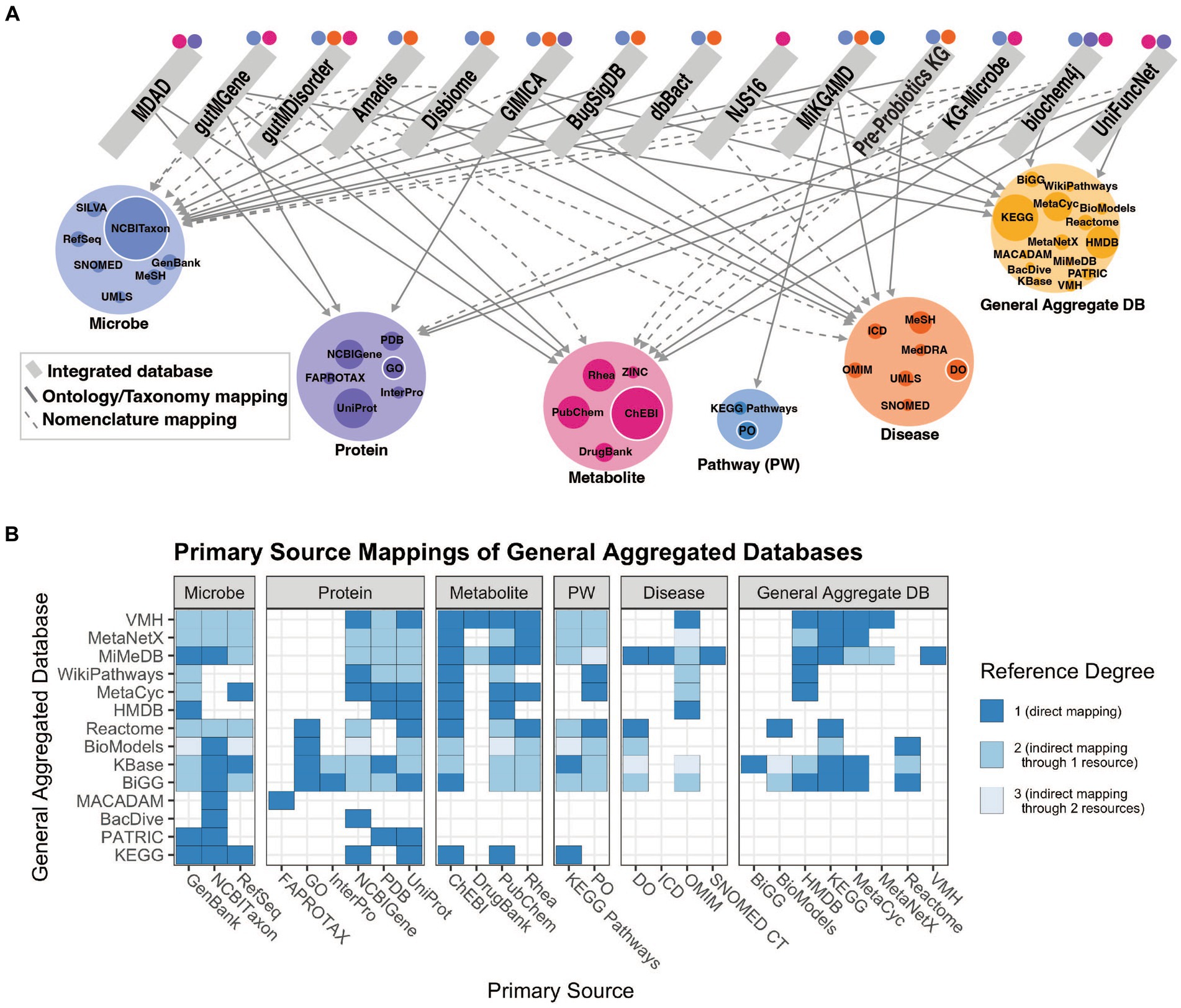
Figure 2. (A) Network of relationships included in integrated resources. Edges between an integrated resource and some type of primary knowledge source (microbe, protein, metabolite, pathway, or disease) represent either a categorization of the concept via an ontology or taxonomy (solid lines), or a nomenclature mapping to some identifier (dashed lines). Node size of the primary knowledge source (colored) represents the in-degree from integrated databases, where the largest nodes are those most often used for standardization. Colored points above integrated resources specify to which concept type the integrated resource maps, including indirect mappings through a general aggregate database. (B) Relationships among all general aggregate databases and primary databases, separated by category. Reference degree shows the degree to which a primary database may be referenced, indicating those most often used for standardization. E.g., If database i references another database j, a general aggregate database that in turn references database k, then i and k have a reference degree of 2. That primary database is only referenced if shown in (A). This figure was generated using code and data available in github repository: https://github.com/lozuponelab/knowledge-source-mappings.
3.1 Knowledge bases that streamline access
An increasing number of knowledge bases have been developed that synthesize microbial and host content for systems biology research, including mechanistic curated knowledge bases and integrated knowledge bases. The relationships represented capture biological processes in a causal way, and are rooted in human curation of specific, validating experiments. In most cases, these resources introduce new unique identifiers for informational classes, which, in combination with other primary databases discussed previously, supports standardization and integration of correlative and inference-ready knowledge bases. In order to connect microbial sequences from experimental studies to these resources, sequence search tools such as Basic Local Alignment Search Tool (BLAST) or functional annotation tools such as InterPro and EggNog Mapper are used, alongside additional web applications such as MolEvolvR facilitating protein characterization across phylogenetic contexts (McGinnis and Madden, 2004; Jones et al., 2014; Cantalapiedra et al., 2021; Krol et al., 2022).
3.1.1 Resources that include microbial and host genes, reactions, pathways, and metabolites
The Kyoto Encyclopedia of Genes and Genomes (KEGG) and MetaCyc are examples of mechanistic curated knowledge bases that represent relationships among microbial and host genes, reactions/pathways, and metabolites (Kanehisa et al., 2017; Caspi et al., 2020). KEGG and MetaCyc provide direct taxonomic mappings to NCBI Taxonomy, RefSeq, or GenBank, as well as direct mappings to multiple primary sources of metabolites and proteins (Figure 2). Both knowledge bases integrate knowledge of metabolic pathways for many organisms (Mendoza et al., 2019; Caspi et al., 2020). These knowledge bases organize pathway and molecular content in unique ways, often making comparison difficult, however a primary difference is in the supported tools surrounding the resource. KEGG introduces several tools including the BlastKOALA (short for KEGG Orthology And Links Annotation), a protein annotation web service, KEGG Mapper, a genome annotation service, and Pathogen Checker, a service supporting search of antimicrobial resistance genes (Kanehisa et al., 2017). KEGG also introduced drug and disease links to the pathways represented in 2005 and 2008, respectively. MetaCyc introduces Pathway Tools consisting of key components such as PathoLogic, a method to predict metabolic pathways of a given organism, and MetaFlux to generate genome-scale metabolic networks (GSMNs or GEMs) using flux based analysis (Caspi et al., 2020). Each of these platforms support extensive web-based interfaces for exploring the content represented, and both have moved to a subscription model. The large diversity of life represented among MetaCyc and KEGG render them broadly relevant to understanding microbiome-related results.
3.1.2 Resources that host GSMNs
A method that systematically evaluates microbial phenotypes relevant for microbe:host interactions is GSMNs. GSMNs are in silico models and can be used to infer metabolic phenotypes. GSMNs use the annotated genes of an organism, which describe the associated biochemical reactions that the enzymatic products of such genes are capable of affecting. These gene annotations are found using annotation tools such as GapMind or aggregated from publicly available curated databases (Price et al., 2020). GSMNs serve two purposes: they synthesize knowledge of that organism’s metabolism, and they are a mathematical model which can be used to simulate metabolic phenotypes in environments of interest (Moretti et al., 2021). Moreover, GSMNs from multiple organisms can be aggregated in order to simulate entire microbial communities with tools such as MICOM (Swainston et al., 2017). Recently, MICOM was used to predict the risk of Clostridium difficile infection, the leading cause of antibiotic associated diarrhea, based on the metabolic strategies of C. difficile in different host microbiome and diet contexts (Carr et al., 2023). GSMNs can therefore serve as a blueprint for the suite of metabolic transformations possible and facilitate the understanding of the metabolic potential of a given microbial community (Mendoza et al., 2019; Esvap and Ulgen, 2021; Passi et al., 2021).
GSMNs are highly dependent on knowledge sources used in their construction. Because many different methods to generate GSMNs exist and it is often a manual process, there are often inconsistencies in the resulting models (Heinken et al., 2023). These differences are influenced by the reconstruction approach and attributed to the chosen database or annotation tools from which the information is gathered (Magnúsdóttir et al., 2017; Machado et al., 2018; Hsieh et al., 2023). The consistent mapping of the biological concepts represented in public databases and knowledge bases is a critical aspect of their broad utility. This standardization challenge expands beyond GSMNs to all resources that combine unique forms of knowledge based on prior studies, and remains a major limitation in the causal mechanism generating task.
There are several key resources hosting GSMNs, including the Biochemical, Genetic and Genomic knowledge base (BiGG), MetaNetX, BioModels, the Department of Energy Systems Biology Knowledgebase (KBase), and the Virtual Metabolic Human (VMH) (Arkin et al., 2018; Malik-Sheriff et al., 2019; Noronha et al., 2019; Norsigian et al., 2019; Moretti et al., 2021). BiGG enables an efficient search over multiple GSMNs by integrating published models of different organisms with standardized nomenclature of all components, with models of 108 organisms included as of 2019(Norsigian et al., 2019). BiGG and BioModels make high-quality GSMNs available to the academic community. Over the years, these have been updated to introduce features such as including genome annotations, standardizing reactions and metabolites to primary sources, and a greater taxonomic diversity of models (Malik-Sheriff et al., 2019; Norsigian et al., 2019). The VMH connects human metabolism, genetics, and disease with microbial metabolism and diet. The VMH cross-references over 57 resources to combine GSMNs of humans and microbes drawn from existing metabolic maps, experimental data from literature, and other integrated resources including BiGG (Noronha et al., 2019). VMH is a useful resource for studies seeking available knowledge of metabolite profiles. For example, the VMH was used in an evaluation of the influence of the Mediterranean diet on aging and the gut microbiome (Ghosh et al., 2020). MetaNetX is an integrated knowledge base that provides a mapping between major GSMN databases for more standardized representation of metabolic processes (Moretti et al., 2021). The goal of this resource is to reconcile the biochemical and metabolic content represented in key public databases. MetaNetX both provides cross-links and merges equivalent biochemical reactions and metabolites into a single identifier, such that entities are merged based on reaction context or chemical formula (Moretti et al., 2021). MetaNetX provides a straightforward way to access the relationships among metabolites through many GSMN sources. KBase is a resource funded by the US Department of Energy that integrates external repositories with data generated on the system, e.g., for public access to genomes and their corresponding metabolic models, including KEGG, BiGG, and MetaCyc, thus including metabolic models for 773 gut microbes as of 2018 and potentially more GSMNs that are currently private (Arkin et al., 2018). KBase also supports a suite of tools that allow for the construction of these metabolic models and workflows supporting the assembly of genomes all the way through to metabolic reconstruction, as well as many other computational tools for omic analyses. These user generated tools can generate data linkages to Functional Annotation of Prokaryotic Taxa (FAPROTAX) and InterPro, and all database entries and mappings are inherited from ModelSEED (Arkin et al., 2018; Seaver et al., 2021).
3.1.3 Resources that host microbe and host metabolic content
There are several curated and integrated knowledge bases focused on centralizing known metabolic traits and output in host and microbial environments. The Human Microbial Metabolome Database (MiMeDB) connects microbial and human metabolism among many resources, including the VMH, as well as genome or proteome information with a focus on how the human microbiome influences health (Wishart et al., 2023). While MiMeDB represents fewer diseases than KEGG, they are constrained to those that are understood to be affected by microbial metabolism. MiMeDB furthermore supports specific constraints on the search of all entity types within the web-interface (e.g., co-metabolite, microbial, or human metabolite type). The MetAboliC pAthways DAtabase for Microbial taxonomic groups (MACADAM) is focused on functional annotations and integrates pathway genome databases (PGDBs) from MetaCyc, MicroCyc, FAPROTAX, and International Journal of Systematic and Evolutionary Microbiology (IJSEM) with genomes from RefSeq (Le Boulch et al., 2019). The Human Metabolome Database (HMDB) has accelerated the standardization of metabolic output and originally provided a uniquely centralized resource of broadly relevant human metabolomic data (Wishart et al., 2022). In 2021 microbial or gut-derived metabolites were added to the HMDB, supporting disease-focused investigation of microbial pathways. With the comprehensive array of metabolites documented, the known metabolite-disease associations in HMDB were used in a deep learning method intended to predict novel disease associated metabolites (Sun et al., 2022). The HMDB ecosystem also introduces tools such as DeepMet, a deep generative model for identifying new metabolites and potential hypotheses surrounding them (Wishart et al., 2022). Other deep learning based methods for understanding microbe-metabolite relationships include MMVec, BiomNED, and MiMeNet (Morton et al., 2019; Le et al., 2020; Reiman et al., 2021).
3.1.4 Resources that include microbial trait and genomic content
Curated databases that incorporate microbial trait information or genomic content can illuminate functional qualities of microbes. These include the Bacterial Diversity Metadatabase (BacDive) and the Pathosystems Resource Integration Center (PATRIC) (Gillespie et al., 2011; Söhngen et al., 2014). PATRIC integrates genomic, transcriptomic, protein–protein interaction, protein structure, and other diverse data types for 22 genera of prokaryotic bacteria, mainly pathogens (Gillespie et al., 2011). This integrated knowledge base, which also includes some correlative results of host-pathogen-disease associations, compiles this information from publicly available datasets for users to easily view and analyze such results. BacDive is the largest standardized resource of prokaryotic information, consisting of strain level details of phenotypes, morphology, growth patterns, metabolism, and sequences for over 70,000 strains (Söhngen et al., 2014).
3.1.5 Resources that include microbial or host pathway content
Several graph relational databases exist that support more complex queries based on their structure. These resources incorporate semantically defined relationships between concepts at a much greater depth than those represented in KEGG or MetaCyc. The Reactome Knowledgebase (Reactome) is a graph database that synthesizes human molecular processes in a standardized way such that all concepts are rooted in ontologies or primary databases (Fabregat et al., 2018). With over 10,000 human genes and their function incorporated, Reactome provides a high-level metabolic map for the interaction between the genome, the proteome, and the metabolome in humans. Reactome is not as broadly relevant to the specific microbe-human interactions that exist elsewhere as only pathogenic bacteria and infectious diseases are included. WikiPathways is another graph database of biological pathway models for all species, though mostly focused on human biology (Martens et al., 2021). Reactome and WikiPathways are community driven, which results in content that reflects the current consensus and supports more frequent updates. Reactome and WikiPathways provide interactive network visualizations of curated processes and pathways for the user to browse the concepts represented.
3.2 Contextualizing experimental findings
Whereas the previous section described curated and integrated knowledge bases that allow scientists to effectively access systems-level microbiome information, a second category of knowledge bases represent literature findings. Correlative knowledge bases allow researchers to contextualize new findings with existing findings in the literature, such as previous studies that have detected a relationship between a microbe and a disease, pathway, or other entity through laboratory or population level studies. These resources organize previously found associations between microbes and other entities, making knowledge computationally accessible.
In response to the growing number of drug resistant bacteria, the Microbe-Drug Association Database (MDAD) was built through manual curation of literature describing microbe-drug relationships based on PubMed keywords (Sun et al., 2018). The studies represented are either microbe-drug relationships identified through lab experiments or those found effective in clinical trials. GutMGene is another database created after manually searching PubMed articles for evidence of associations between microbes and metabolites produced or consumed, and microbial influence on human gene expression (Cheng et al., 2022). GutMDisorder similarly synthesizes associations between microbes and human diseases or phenotypes found in the literature (Cheng et al., 2020). Disbiome contains microbe-disease associations found from population level studies that identified significant differences in abundance between a control and disease state (Janssens et al., 2018). Amadis similarly provides evidence of associations between diseases and microbes, with a similar number of disease entries as Disbiome (Amadis includes relationships between 221 human diseases and 774 microbes, while Disbiome includes 190 human diseases and 800 microbes) (Janssens et al., 2018; Li et al., 2021). The Host Genetic and Immune Factors Shaping Human Microbiota (GIMICA) is another database representative of multiple human body sites and the immune, environmental, and genetic factors that they interact with (Tang et al., 2021). Several other link-based aggregate databases introduced more stringent manual curation techniques to adequately represent the variable aspects of studies, such as experimental setting or sequencing technique. BugSigDB is a community-supported effort of over 2,500 curated microbial signatures cited in over 600 scientific articles. With over 1,400 unique taxa represented, BugSigDB is rich in metadata, experimental conditions, and design of each experiment and is well standardized to a range of ontologies. Another knowledge base, dbBact, includes over 900 experiments and supports similar use cases aligning results across many studies (Amir et al., 2023). These literature-based databases support easy access to information in a context dependent manner. The provenance of such associations is also easily made available within these resources by PubMed Identifier (PMID). NJS16 is an integrative network that incorporates manually curated knowledge from literature of gut microbes and how they interact via metabolite transport (Sung et al., 2017). NJS16 uses a metagenomic analysis of a cohort of Type 2 Diabetes individuals to showcase a framework that can predict microbe-metabolite interactions that influence host physiology in other contexts. Such manually curated resources play a critical role in allowing researchers to contextualize their results by easily accessing literature that describes correlative microbial findings.
Findings of a specific experimental result can be related to a more complete mechanistic path by using relationships summarized in correlative databases. These databases have been used for corroboration of the findings of targeted experiments. For example, gutMGene has been used to corroborate the hypothesis that the gut microbial community plays an important role in cardiovascular disease through short chain fatty acid production by citing searchable microbe-metabolite relationships in the form of a network. Additionally, gutMDisorder has been used to validate polysaccharides identified to have a regulatory effect in disease through microbe-disease relationships in the form of a network (Hu et al., 2022; Wei et al., 2023). BugSigDB demonstrates the value in having a heterogeneous resource to explore patterns of microbial composition across studies, examines the commonly co-occurring or mutually exclusive individual or groups of microbes, and evaluates differences in microbial communities across body sites (Geistlinger et al., 2022). However, despite these highly useful applications of manually curated, correlative knowledge bases, there are key challenges that contribute to their limited use. A primary limitation of these databases is the small number of relationship types represented (designated as path length in Figure 3). Furthermore, it is difficult to align experimental results to such databases when concepts are not mapped to common primary knowledge sources, discussed more in challenges and future perspectives.
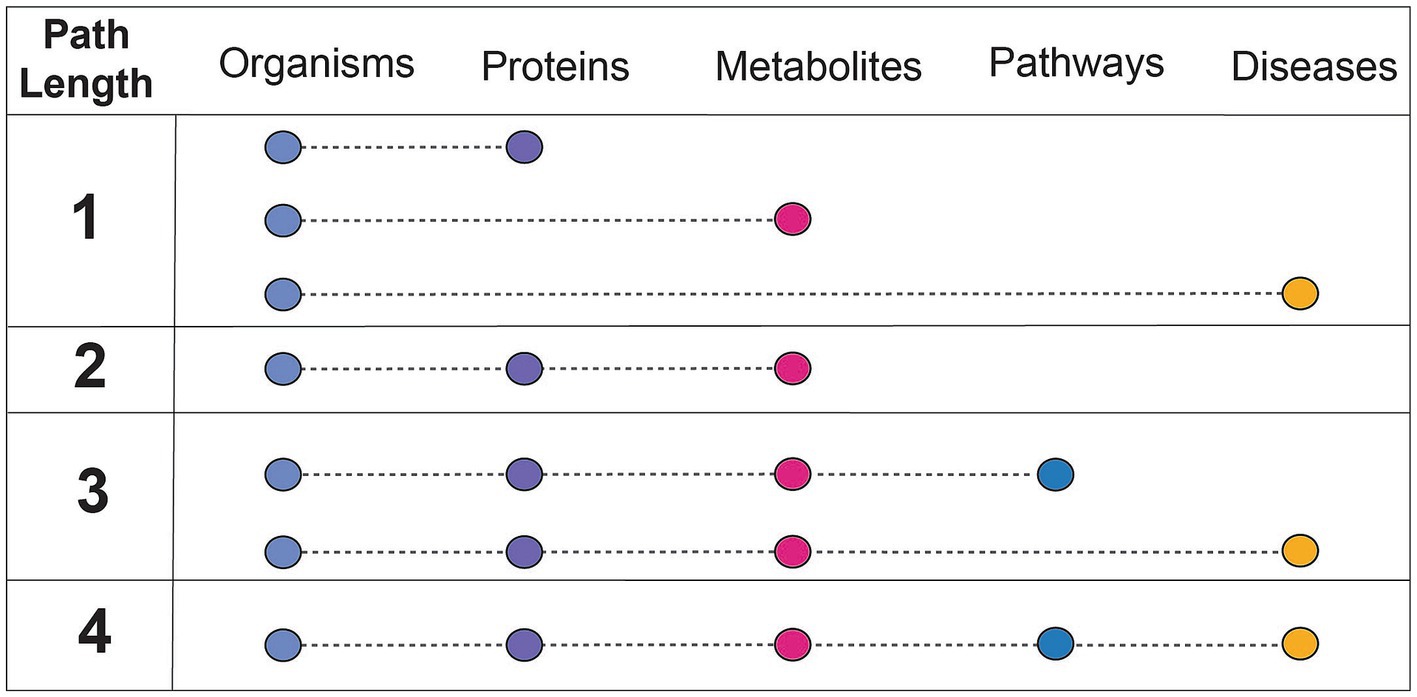
Figure 3. Understanding the connectedness of integrated databases based on path length. Path length refers to the number of relationships between unique concepts, or feature types, that are included within a resource. The feature types discussed in this context are microbes, proteins (or genes, human or microbial), metabolites (human or microbial), pathways (human or microbial), and diseases (human). The concept of path length is used to assess how comprehensively a resource can be used for mechanistic inference, or which relationships are needed from other databases to do so.
3.3 Mechanistic hypothesis generation
Hypothesis generation in microbiome research requires a diverse range of knowledge. To date, no resource or methodology supports the task of hypothesizing mechanisms of microbial processes that influence disease by including all categories of data described in Figure 3. However, some resources represent data in a way that supports inference, linking multiple complex relationships into a derived explanation. Structured, microbiome-relevant resources can support this automated inference. Knowledge graphs (KGs) are commonly used for this purpose due to their logical representation conducive to automated inference. KGs are simplified representations of related concepts through nodes (concepts) and edges (relationships between those concepts). The KG construction process involves the aggregation of content and harmonization to ontologies, most often through ingests that extract, transform, and load such information into a semantically consistent format. Graph-based models, the basis of KGs, enable complex queries and reasoning, which is especially useful for understanding the intricate interactions between microbes and the host. The following resources have varying levels of specificity to a particular disease, solely focus on microbial trait data, or lack the wider context necessary for disease-based inference.
MiKG4MD is one resource that represents how microbes are involved with mental disorders in the form of a knowledge graph (Liu et al., 2021). MiKG4MD was used to form specific queries that identify several sources describing the relationship between Bifidobacterium dentium and anxiety or depression via the neurotransmitter gamma-aminobutyric acid (GABA) (Liu et al., 2021). MiKG4MD has not been applied beyond the case studies that demonstrate its purpose, though these queries exemplify the hypothesis generating potential. The Pre-/Probiotics Knowledge Graph (PPKG) represents over 29,000 articles describing prebiotics and probiotics, combined with three other primary public databases, MeSH, UMLS and SNOMED CT (Table 1) (Liu et al., 2022). Similar to MiKG4MD, a specific query of PPKG showed 114 direct relationships identified between Bifidobacterium bifidum and disease, suggesting an influence on blood lipids, gut microbiome profiles, brain connectivity, and gene expression (Liu et al., 2022). KG-microbe is a resource that more broadly represents how microbes interact with their environment (Joachimiak et al., n.d.). KG-Microbe is useful to understand microbial traits and environments, such as soil or water as well as human anatomical sites, though it does not yet include information which connects microbes to disease. Several relevant ontologies that play an important role in the representation of the complex knowledge associated with the microbiome also exist. The ontology of host–microbe interactions (OHMI) is the only known OBO ontology resource that introduces a structured representation of microbe-host interactions (He et al., 2019). This resource makes a critical step in developing standards for how to represent host–microbe interactions through flexible and interoperable representations. Furthermore, OHMI aligns to several OBO ontologies including NCBI Taxonomy, the Environmental Ontology (ENVO), and the Uber-anatomy Ontology (UBERON). Importantly, OHMI does not include the mechanistic detail of proteins and metabolites necessary for inference, however the logical representation introduced can provide a framework for mechanistic inference (He et al., 2019). OHMI has not been updated since the original publication. OHMI introduces over 1,000 terms including microbes, host–microbe interactions, and study details (He et al., 2019).
There are also frameworks that synthesize multi-omic content in a graph database or network representation. BioChem4j is one such framework that automatically ingests content from multiple ontologies and represents microbes and their functional traits using the UniProt API (Swainston et al., 2017). BioChem4j is therefore an extensible resource from which researchers can gather the enzymes and metabolites involved in microbial biochemical reactions that may occur in any environment. BioChem4j has been applied toward a pipeline for the discovery and optimization of biosynthetic pathways, specifically for understanding a range of industrial microorganisms. The pipeline examined flavonoid production pathways and an alkaloid pathway in Escherichia coli for the purposes of microbial engineering for chemical production (Carbonell et al., 2018). The Unified Functional Network (UniFuncNet) is another framework that integrates multiple resources necessary for the construction of GSMNs (Queirós et al., 2022). The UniFuncNet framework can take as input a list of entities from different databases (e.g., proteins, genes, metabolites, etc) and output a network representation of all associations among such entities. UniFuncNet’s applications are demonstrated through two workflows which, for example, expanded existing GSMNs of Akkermansia muciniphila to include the biosynthesis and metabolism of glycans, or to relate compounds identified in a metabolomics dataset to relevant pathways and organisms (Queirós et al., 2022).
4 Challenges associated with the construction and applications of knowledge bases
4.1 Inconsistent taxonomy and metabolite nomenclature assignment
A major challenge arising from the availability of multiple taxonomic databases as well as multiple versions of the same taxonomic database are the resultant inconsistencies in the labeling of a microbe. The classification method of microbes curated from the literature is often overlooked, and in many cases a microbe may be assigned the wrong identifier (e.g., a microbe originally labeled via SILVA is assigned an NCBI Taxonomy identifier). Methods of taxonomic assignment in sequence-based studies of microbial population differ depending on whether small subunit (SSU) ribosomal RNA (rRNA) is targeted, also known as 16S sequencing for bacteria and archaea, or shotgun metagenomic sequencing is performed. Inconsistent classification, whether varying labels is due to lack of information or poor quality of sequencing reads, can impede the ability to relate findings about a given microbe across studies to each other and to their functional attributes, which is important for ultimately trying to understand microbe-host interactions at the mechanistic level. Additional challenges arise when microbial nomenclature is revised based on a better resolution of evolutionary relationships from sequencing data or phenotypic information, resulting in the same taxa having different names depending on the date of publication.
SILVA and Greengenes, which are built using sequences from the European Nucleotide Archive (ENA) and GenBank, respectively, are the most used taxonomic databases for 16S sequencing (Pruesse et al., 2007; Ceccarani and Severgnini, 2023; McDonald et al., 2023). SILVA uses a Bergey’s seed alignment (Garrity et al., 2004), then partially manually builds upon that classification to construct a phylogenetic tree which is used as a guide. In order to classify sequences, SILVA uses the SILVA Incremental Aligner (SINA) reference-based alignment tool for multiple sequence alignment, and assigns organism names according to the Deutsche Sammlung fur Mikroorganismen und Zellkultren (DSMZ) (Pruesse et al., 2012). In contrast to SILVA, which uses a pre-constructed tree, Greengenes constructs a de novo tree for taxonomic classification (DeSantis et al., 2006). Greengenes2 made significant updates by linking a substantial number of whole genome sequences from the International Nucleotide Sequence Database Collaboration (INSDC) (Arita et al., 2021), amplicons from the Living Tree Project (Yilmaz et al., 2014) and other resources, to create the largest tree with the broadest phylogenetic coverage to date (McDonald et al., 2023). A new version of the SILVA database is released semi-annually, whereas Greengenes2 only recently was released, 9 years after the prior version (Pruesse et al., 2007; McDonald et al., 2023). Although it is well established that use of different taxonomic databases and their versions can greatly impact taxonomic assignments made, there are limited solutions for dealing with this ambiguity when creating integrated resources.
Similar problems arise for the nomenclature of metabolites that are represented in manually curated databases. Main technologies used for metabolomics include mass spectrometry (MS)-based or nuclear magnetic resonance (NMR)-based approaches. Metabolomics can be approached with untargeted techniques (for hypothesis generation) or targeted techniques (for hypothesis testing) (Johnson et al., 2016). The naming and mapping of these metabolites therefore can introduce some uncertainty and similar discontinuity as microbial taxonomy. Metabolite identification is done by comparing the spectra obtained experimentally with that included in the curated knowledge bases or primary knowledge sources described above, such as ChEBI or ChEMBL (Hastings et al., 2016; Zdrazil et al., 2023). The mismatch of metabolite names and identifiers across these standardized resources, presents challenges for researchers to contextualize their findings and formulate hypotheses regarding their data using integrated resources (Merlet et al., 2016; Shaffer et al., 2017). Resources also exist that facilitate the classification relating their spectra to those of known metabolites to improve direct mapping such as the Global Natural Products Social molecular networking (GNPS) (Overbeek et al., 2014). The challenge of mismatching metabolite labels is especially prominent in the construction and alignment of GSMNs, which draw from these standardized databases. HMDB is one of the most comprehensive resources of known host and microbiome associated metabolites, still only representing a fraction of the metabolome, that cross-links many of standard chemical databases and identifiers to make this process more straightforward (Wishart et al., 2022). MetaNetX further facilitates mapping experimental results to representations in GSMNs to contextualize metabolomics findings (Moretti et al., 2021). An important direction of understanding microbiome-host relationships is evaluating how the microbiome and the metabolome interact with exogenous factors, such as diet, collectively called the exposome (Shaffer et al., 2017). The VMH is an important resource for this, as it introduces known relationships between the exposome and the metabolome (Noronha et al., 2019). Nomenclature challenges also are confronted in constructing correlative knowledge bases, such as gutMGene, in that chemical names that are manually curated, or text mined potentially, cannot be mapped to an identifier in a primary knowledge source (Cheng et al., 2022). As such, what exists in these resources may not accurately represent what was found in the corresponding study. The increased utility and standardization of naming of integrated knowledge bases is critical for addressing the challenges described, as integrated resources provide expansive knowledge that will support mechanistic exploration.
4.2 Semantic standardization
Another limitation of these resources is the extent to which entities are mapped to existing primary knowledge sources (e.g., ChEBI). Without mappings to a semantic standard, it is impossible to combine a resource with others as the concepts represented are not identical. Mechanistic curated knowledge bases such as KEGG, which introduce new identifiers due to their broadly represented and cross-linked information, are particularly useful resources to map to because of their scale and connectivity to other resources. Integrated knowledge bases play an important role in enabling one to search a broader field of knowledge, or relationships between more concept types (a higher path length as defined in Figure 3). The benefits of standardizing to ontologies are two-fold; first, ontologies offer a full hierarchy of relationships in a machine-readable format. Ontologies are curated by experts in both data engineering and the scientific area represented and provide a logical interpretation of knowledge categories. This makes it possible to abstract or concretize a concept depending on the mechanistic detail desired. Experimental results can be mapped to ontological concepts as an exact term (e.g., AKT-interacting protein isoform 2) or more broadly characterize the concept to a parent term (e.g., AKT-interacting protein). Second, KGs built of the logical representation of concepts in ontologies can be used to contextualize scientific results and infer mechanistic explanations. More comprehensive KGs can be constructed when all the knowledge represented in these resources is correctly mapped to useful ontologies.
Microbiome relevant knowledge bases primarily lack standardization in microbial and disease categories. The few databases that incorporate human diseases are limited in their degree of standardization. Ontologies such as the Monarch Disease Ontology (MONDO) and the Human Phenotype Ontology (HPO) have been developed as part of the Monarch initiative and provide logically coherent hierarchical representations of concepts (Köhler et al., 2021; Vasilevsky et al., 2022). MONDO, which includes resources such as Online Mendelian Inheritance in Man (OMIM) and Orphanet, is updated monthly, and introduces thousands of diseases and disorders. Mappings to resources such as MONDO support the applications of aggregate databases toward understanding microbial mechanisms in human disease. Microbes in gutMGene, gutMDisorder, Disbiome, Amadis, and GIMICA are mapped to NCBI Taxonomy, however those in MDAD and NJS16 are not (Figure 2). MDAD includes protein mappings to UniProt and metabolite mappings to DrugBank, while NJS16 only includes metabolite mappings to KEGG (Goodfellow et al., 2009; wwPDB consortium et al., 2019). The absence of mappings to NCBI Taxonomy or any structured phylogenetic database limits usability due to the inconsistencies in naming and taxonomic classification strategies. It is important that new resources map to the primary sources most often referenced by current integrated resources, as shown in Figure 2A by the colored node size, to ensure that concepts can be consistently identified. These standardization challenges limit the capacity to integrate sources of knowledge and make mechanistic claims using such knowledge.
4.3 Access methods and source characteristics of resources
The source characteristics of integrated resources can influence both their comprehensiveness and accuracy. Manually curated resources can have increased accuracy as content is provided through curation by experts directly from literature. While manual curation is nearly always at play due to the requirement of specific expertise in understanding microbes, the field is clearly approaching a new era of automated content extraction. Text mining approaches make this task more efficient, allowing for more content to be easily accessed by scientists with a wide variety of research interests.
Knowledge bases can be accessed in many ways depending on the type of users that they serve (Figure 1B). Wet-lab focused researchers interested in accessing the broad store of knowledge offered by these resources are primarily interested in interactive web interfaces. Curated knowledge bases KEGG and MetaCyc each offer interactive visual interfaces and useful pathway diagrams. Integrated knowledge bases such as MiMeDB and MACADAM also offer an interface to easily query the desired content, though not the same support in pathway diagrams as Wikipathways and Reactome. Reactome is even more uniquely suited to show interactive cartoon diagrams which can greatly increase accessibility to all users. Other programmatic ways of accessing these resources are important for the analyses that bioinformaticians do using complex datasets. The Simple Protocol And Resource Description Framework Query Language (SPARQL) is a query language for the Resource Description Framework (RDF), a framework that supports relationship-based data made available on the web (Candan et al., 2001). When SPARQL queries are supported, whether programmatically or via an API, computational users can easily access information through highly specialized queries. API support also enables this functionality.
Many mechanistic curated and integrated knowledge bases are offered as relational databases or tables, which supports fast access to a range of knowledge. Graph databases require traversal across a wider domain of information, and therefore are not quick in retrieval. However, graph databases can host information to a greater level of detail. For example, the “glycolysis” pathway in KEGG and MetaCyc host fewer than 30 metabolites or gene/gene products, whereas Reactome includes over 40 metabolites and 100 proteins. In relational databases such as KEGG, the detail comes in the nodes (genes, metabolites, organisms) and the relationships represent some interaction or input/output more generally. In graph databases such as Reactome, the edges provide a hierarchical set of content in themselves with much more detail, for example the edge “ADPGK:Mg2+ phosphorylates Glc to G6P” connects alpha-D-Glucose to alpha-D-glucose 6-phosphate. All reactions are rooted in literature evidence, providing a detailed account of biological interactions.
The formal representation of knowledge introduced by KGs can include heterogeneous biological content that is flexible and interoperable. The network structure of a KG supports inference based on both the semantic representation of knowledge and the structure of the graph, allowing one to infer new edges (hypothesized relationships between distinct concepts) or classify biological concepts. An important consideration for KGs is the model used to represent such complex knowledge. A logical semantic representation is critical for inference, and this can be difficult with such complex concepts as microbe-host interactions. It is generally useful to follow a predefined schema for interoperability and introducing new information, such as the Web Ontology Language (OWL) or the Biolink model (Bechhofer, 2009; Unni et al., 2022). These models allow harmonization of data sources across all knowledge types, which is especially important in the multi-omic nature of microbiome science. The types of edges within MiKG4MD are arbitrary and do not align with previously existing repositories, such as the Biolink Model or the Relationships Ontology (RO), both of which provide some standardized structure to the organization of a KG (Smith et al., 2005; Liu et al., 2021; Unni et al., 2022). However KG-microbe does align to the Biolink schema, which ingests microbial trait databases and combines them with ontologies such as ChEBI and GO (Joachimiak et al., n.d.). This was done using automated graph construction libraries that are a part of KG-Hub. Through some manual curation, KG-Microbe includes specific microbial traits to be represented in a way that aligns with the Biolink schema (Joachimiak et al., n.d.). It is important for the chosen schema to support interoperability between KGs, incorporation of any ontology or primary knowledge source, and correctly represent the heterogeneous data types necessary within a microbiome-relevant KG.
5 Future perspective
By indexing and linking multi-omic knowledge, integrated resources can contextualize results at the systems-level, corroborating findings from experimental observations, and provide promise toward uncovering novel hypotheses. We evaluate key categories of microbiome-relevant knowledge including microbes, host and microbial proteins, host and microbial metabolites, host and microbial pathways, and host diseases and argue that the extent to which these categories are covered by such integrated resources influences their ability to be adopted for mechanistic inquiry. It is important for users to evaluate the resource based on the six categories present here and their affordances (Figure 1A); ontologies and taxonomies, annotated databases, mechanistic curated knowledge bases, integrated knowledge bases, correlative curated knowledge bases, and inference-ready knowledge bases, in order to derive the best applications of such resources. We have also evaluated the primary traits of these resources including access methods, content, and source characteristics (summarized in Supplementary Table 1). The access points of the knowledge contained in these resources, whether programmatically or via a user-friendly web interface, can greatly affect the adoption by the intended user. Ensuring they support downloadable flat files or APIs translates to more readily available content for automatic hypothesis generation. Mapping the concepts represented in each resource is an important factor to consider in utilizing these resources, as it can limit the capacity for connecting it with other resources. Many correlative knowledge bases, for example, lack the level of nomenclature standardization to commonly used primary knowledge sources that is essential for wide adoption and integration of such resources. Future resources should always keep the nomenclature limitations in mind during construction and ensure that the level of standardization supports the intended use case.
Inference-ready knowledge bases such as KGs serve an important purpose in the microbiome field in supporting mechanistic hypothesis generation using existing knowledge. As shown, there are few resources that adequately map all categories of knowledge mentioned to enable explanations for microbe-disease associations to be understood. A focus on this connectedness, highly dependent on the level of standardization discussed previously, will drive the microbiome field toward a deeper understanding of microbe-host interactions via automated inference (Figure 2). Furthermore, it is important that these KGs use a data model that is highly interoperable and flexible to integrate heterogeneous data types. Applying these resources to mechanistic inference can help assess health outcomes and derive new understandings of multi-omic data sets through many methodologies such as linear modeling or machine learning based approaches. While these methodologies are not addressed in great detail, it is important to recognize their complexities.
Through this review of resources, we have provided evidence of the efforts to consolidate the rapidly increasing number of experimental findings surrounding the microbiome. We have published data resource mappings in a git-hub repository to ensure reproducibility and to support updates.1 We recognize that this review does not capture all possible resources, therefore encourage contributions to this repository in hopes of maintaining a useful source of information for researchers to select the most appropriate knowledge sources. We argue that the adoption of these resources and contributions to the field will be maximized with further standardization and connectedness. The application of these resources to understanding microbe-host-disease related questions holds promise for advancing biomedical understanding.
Data availability statement
The repository used for developing the figures can be found in our git-hub repository https://github.com/lozuponelab/knowledge-source-mappings.
Author contributions
BS: Conceptualization, Investigation, Visualization, Writing – original draft, Writing – review & editing. MA: Conceptualization, Investigation, Writing – review & editing. AC: Conceptualization, Investigation, Writing – review & editing. CM: Conceptualization, Investigation, Software, Visualization, Writing – review & editing. JS: Conceptualization, Investigation, Writing – review & editing. EW: Conceptualization, Investigation, Writing – review & editing. MPJ: Conceptualization, Investigation, Writing – review & editing. LH: Conceptualization, Investigation, Writing – review & editing. CL: Conceptualization, Investigation, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The authors gratefully acknowledge the support of NIH grants R01LM13400 and OT2TR003422 to LH, and NSF GRFP which supported EW. This work is supported as part of the Genomic Sciences Program. The DOE Systems Biology Knowledgebase (KBase) is funded by the U.S. Department of Energy, Office of Science, Office of Biological and Environmental Research under Award Numbers DEAC02- 05CH11231, DE-AC02-06CH11357, DEAC05-00OR22725, and DE-AC02-98CH10886.
Acknowledgments
The authors would like to acknowledge Alan Morris for their consultations on manuscript figures and general comments.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2024.1351678/full#supplementary-material
Footnotes
References
Amir, A., Ozel, E., Haberman, Y., and Shental, N. (2023). Achieving pan-microbiome biological insights via the dbBact knowledge base. Nucleic Acids Res. 51, 6593–6608. doi: 10.1093/nar/gkad527
Arita, M., Karsch-Mizrachi, I., and Cochrane, G. (2021). The international nucleotide sequence database collaboration. Nucleic Acids Res. 49, D121–D124. doi: 10.1093/nar/gkaa967
Arkin, A. P., Cottingham, R. W., Henry, C. S., Harris, N. L., Stevens, R. L., Maslov, S., et al. (2018). KBase: the United States Department of Energy Systems Biology Knowledgebase. Nat. Biotechnol. 36, 566–569. doi: 10.1038/nbt.4163
Armour, C. R., Nayfach, S., Pollard, K. S., and Sharpton, T. J. (2019). A metagenomic Meta-analysis reveals functional signatures of health and disease in the human gut microbiome. mSystems 4:e00332. doi: 10.1128/mSystems.00332-18
Armstrong, A. J. S., Quinn, K., Fouquier, J., Li, S. X., Schneider, J. M., Nusbacher, N. M., et al. (2021). Systems analysis of gut microbiome influence on metabolic disease in HIV-positive and high-risk populations. mSystems 6:e01178-20. doi: 10.1128/mSystems.01178-20
Bansal, P., Morgat, A., Axelsen, K. B., Muthukrishnan, V., Coudert, E., Aimo, L., et al. (2022). Rhea, the reaction knowledgebase in 2022. Nucleic Acids Res. 50, D693–D700. doi: 10.1093/nar/gkab1016
Barker, W. C., Garavelli, J. S., Huang, H., McGarvey, P. B., Orcutt, B. C., Srinivasarao, G. Y., et al. (2000). The protein information resource (PIR). Nucleic Acids Res. 28, 41–44. doi: 10.1093/nar/28.1.41
Bechhofer, S. O. W. L. (2009). “Web ontology language” in Encyclopedia of database systems. eds. L. Liu and M. T. Özsu (Boston, MA: Springer US)
Berg, G., Rybakova, D., Fischer, D., Cernava, T., Vergès, M. C. C., Charles, T., et al. (2020). Microbiome definition re-visited: old concepts and new challenges. Microbiome 8:103. doi: 10.1186/s40168-020-00875-0
Biochemistry IU of Committee MBNWebb, EC . (1992). Enzyme Nomenclature: recommendations of the Nomenclature Committee of the International Union of Biochemistry and Molecular Biology on the Nomenclature and Classification of Enzymes. International Union of Biochemistry and Molecular Biology. Available at: https://books.google.com/books?id=353mzgEACAAJ
Bodenreider, O. (2004). The unified medical language system (UMLS): integrating biomedical terminology. Nucleic Acids Res. 32, 267D–270D. doi: 10.1093/nar/gkh061
Boeckmann, B. (2003). The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 31, 365–370. doi: 10.1093/nar/gkg095
Candan, K. S., Liu, H., and Suvarna, R. (2001). Resource description framework: metadata and its applications. ACM SIGKDD Explor Newsl. 3, 6–19. doi: 10.1145/507533.507536
Cantalapiedra, C. P., Hernández-Plaza, A., Letunic, I., Bork, P., and Huerta-Cepas, J. (2021). eggNOG-mapper v2: functional annotation, Orthology assignments, and domain prediction at the metagenomic scale. Tamura K, editor. Mol. Biol. Evol. 38, 5825–5829. doi: 10.1093/molbev/msab293
Cantarel, B. L., Coutinho, P. M., Rancurel, C., Bernard, T., Lombard, V., and Henrissat, B. (2009). The carbohydrate-active EnZymes database (CAZy): an expert resource for Glycogenomics. Nucleic Acids Res. 37, D233–D238. doi: 10.1093/nar/gkn663
Carbonell, P., Jervis, A. J., Robinson, C. J., Yan, C., Dunstan, M., Swainston, N., et al. (2018). An automated design-build-test-learn pipeline for enhanced microbial production of fine chemicals. Commun Biol. 1:66. doi: 10.1038/s42003-018-0076-9
Carpendale, S., Chen, M., Evanko, D., Gehlenborg, N., Gorg, C., Hunter, L., et al. (2014). Ontologies in Biological Data Visualization. IEEE Comput. Graph. Appl. 34, 8–15. doi: 10.1109/MCG.2014.33
Carr, A., Baliga, N. S., Diener, C., and Gibbons, S. M. (2023). Personalized Clostridioides difficile engraftment risk prediction and probiotic therapy assessment in the human gut. bioRxiv. doi: 10.1101/2023.04.28.538771
Caspi, R., Billington, R., Keseler, I. M., Kothari, A., Krummenacker, M., Midford, P. E., et al. (2020). The MetaCyc database of metabolic pathways and enzymes - a 2019 update. Nucleic Acids Res. 48, D445–D453. doi: 10.1093/nar/gkz862
Ceccarani, C., and Severgnini, M. (2023). A comparison between Greengenes, SILVA, RDP, and NCBI reference databases in four published microbiota datasets. bioRxiv. doi: 10.1101/2023.04.12.535864v1
Chang, A., Jeske, L., Ulbrich, S., Hofmann, J., Koblitz, J., Schomburg, I., et al. (2021). BRENDA, the ELIXIR core data resource in 2021: new developments and updates. Nucleic Acids Res. 49, D498–D508. doi: 10.1093/nar/gkaa1025
Chen, C., Huang, H., Ross, K. E., Cowart, J. E., Arighi, C. N., Wu, C. H., et al. (2020). Protein ontology on the semantic web for knowledge discovery. Sci. Data 7:337. doi: 10.1038/s41597-020-00679-9
Cheng, L., Qi, C., Yang, H., Lu, M., Cai, Y., Fu, T., et al. (2022). gutMGene: a comprehensive database for target genes of gut microbes and microbial metabolites. Nucleic Acids Res. 50, D795–D800. doi: 10.1093/nar/gkab786
Cheng, L., Qi, C., Zhuang, H., Fu, T., and Zhang, X. (2020). gutMDisorder: a comprehensive database for dysbiosis of the gut microbiota in disorders and interventions. Nucleic Acids Res. 48, D554–D560. doi: 10.1093/nar/gkz843
DeSantis, T. Z., Hugenholtz, P., Larsen, N., Rojas, M., Brodie, E. L., Keller, K., et al. (2006). Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 72, 5069–5072. doi: 10.1128/AEM.03006-05
Esvap, E., and Ulgen, K. O. (2021). Advances in genome-scale metabolic modeling toward microbial community analysis of the human microbiome. ACS Synth. Biol. 10, 2121–2137. doi: 10.1021/acssynbio.1c00140
Fabregat, A., Jupe, S., Matthews, L., Sidiropoulos, K., Gillespie, M., Garapati, P., et al. (2018). The Reactome pathway knowledgebase. Nucleic Acids Res. 46, D649–D655. doi: 10.1093/nar/gkx1132
Falony, G., Vandeputte, D., Caenepeel, C., Vieira-Silva, S., Daryoush, T., Vermeire, S., et al. (2019). The human microbiome in health and disease: hype or hope. Acta Clin. Belg. 74, 53–64. doi: 10.1080/17843286.2019.1583782
Federhen, S. (2012). The NCBI taxonomy database. Nucleic Acids Res. 40, D136–D143. doi: 10.1093/nar/gkr1178
Frishman, D. (2003). The PEDANT genome database. Nucleic Acids Res. 31, 207–211. doi: 10.1093/nar/gkg005
Galperin, M. Y., Wolf, Y. I., Makarova, K. S., Vera Alvarez, R., Landsman, D., and Koonin, E. V. (2021). COG database update: focus on microbial diversity, model organisms, and widespread pathogens. Nucleic Acids Res. 49, D274–D281. doi: 10.1093/nar/gkaa1018
Garrity, G. M., Bell, J. A., and Lilburn, T. G. (2004). Taxonomic outline of the prokaryotes release 5.0 Bergey’s manual of systematic bacteriology. 2nd Edn. New York: Springer-Verlag.
Geistlinger, L., Mirzayi, C., Zohra, F., Azhar, R., Elsafoury, S., Grieve, C., et al. (2022). BugSigDB captures patterns of differential abundance across a broad range of host-associated microbial signatures. Nat. Biotechnol. Advance online publication. doi: 10.1038/s41587-023-01872-y
Ghosh, T. S., Rampelli, S., Jeffery, I. B., Santoro, A., Neto, M., Capri, M., et al. (2020). Mediterranean diet intervention alters the gut microbiome in older people reducing frailty and improving health status: the NU-AGE 1-year dietary intervention across five European countries. Gut 69, 1218–1228. doi: 10.1136/gutjnl-2019-319654
Gillespie, J. J., Wattam, A. R., Cammer, S. A., Gabbard, J. L., Shukla, M. P., Dalay, O., et al. (2011). PATRIC: the comprehensive bacterial bioinformatics resource with a focus on human pathogenic species. Maurelli AT, editor. Infect. Immun. 79, 4286–4298. doi: 10.1128/IAI.00207-11
Gonzalez, A., Navas-Molina, J. A., Kosciolek, T., McDonald, D., Vázquez-Baeza, Y., Ackermann, G., et al. (2018). Qiita: rapid, web-enabled microbiome meta-analysis. Nat. Methods 15, 796–798. doi: 10.1038/s41592-018-0141-9
Goodfellow, M, Kampfer, P, Busse, HJ, Trujillo, ME, Suzuki, K, Ludwig, W., et al. (2009). Bergey’s Manual of Systematic Bacteriology. Vol. 5: The Actinobacteria.
Griffiths-Jones, S. (2006). miRBase: the microRNA sequence database. Methods Mol Biol Clifton NJ. 342, 129–138. doi: 10.1385/1-59745-123-1:129
Harrison, J. E., Weber, S., Jakob, R., and Chute, C. G. (2021). ICD-11: an international classification of diseases for the twenty-first century. BMC Med. Inform. Decis. Mak. 21:206. doi: 10.1186/s12911-021-01534-6
Hastings, J., Owen, G., Dekker, A., Ennis, M., Kale, N., Muthukrishnan, V., et al. (2016). ChEBI in 2016: improved services and an expanding collection of metabolites. Nucleic Acids Res. 44, D1214–D1219. doi: 10.1093/nar/gkv1031
He, Y., Wang, H., Zheng, J., Beiting, D. P., Masci, A. M., Yu, H., et al. (2019). OHMI: the ontology of host-microbiome interactions. J. Biomed. Semant. 10:25. doi: 10.1186/s13326-019-0217-1
Heinken, A., Hertel, J., Acharya, G., Ravcheev, D. A., Nyga, M., Okpala, O. E., et al. (2023). Genome-scale metabolic reconstruction of 7,302 human microorganisms for personalized medicine. Nat. Biotechnol. 41, 1320–1331. doi: 10.1038/s41587-022-01628-0
Howe, K. L., Achuthan, P., Allen, J., Allen, J., Alvarez-Jarreta, J., Amode, M. R., et al. (2021). Ensembl 2021. Nucleic Acids Res. 49, D884–D891. doi: 10.1093/nar/gkaa942
Hsieh, Y. E., Tandon, K., Verbruggen, H., and Nikoloski, Z. (2023). Comparative analysis of metabolic models of microbial communities reconstructed from automated tools and consensus approaches. bioRxiv. doi: 10.1101/2023.09.13.557568
Hu, T., Wu, Q., Yao, Q., Jiang, K., Yu, J., and Tang, Q. (2022). Short-chain fatty acid metabolism and multiple effects on cardiovascular diseases. Ageing Res. Rev. 81:101706. doi: 10.1016/j.arr.2022.101706
Huerta-Cepas, J., Szklarczyk, D., Heller, D., Hernández-Plaza, A., Forslund, S. K., Cook, H., et al. (2019). eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 47, D309–D314. doi: 10.1093/nar/gky1085
Jackson, R., Matentzoglu, N., Overton, J. A., Vita, R., Balhoff, J. P., Buttigieg, P. L., et al. (2021). OBO foundry in 2021: operationalizing open data principles to evaluate ontologies. Database 2021:baab069. doi: 10.1093/database/baab069
Janssens, Y., Nielandt, J., Bronselaer, A., Debunne, N., Verbeke, F., Wynendaele, E., et al. (2018). Disbiome database: linking the microbiome to disease. BMC Microbiol. 18:50. doi: 10.1186/s12866-018-1197-5
Jewison, T., Su, Y., Disfany, F. M., Liang, Y., Knox, C., Maciejewski, A., et al. (2014). SMPDB 2.0: big improvements to the small molecule pathway database. Nucleic Acids Res. 42, D478–D484. doi: 10.1093/nar/gkt1067
Joachimiak, M. P., Hegde, H., Duncan, W. D., Reese, J. T., Cappelletti, L., Mungall, C. J., et al. (2021). “International Conference on Biomedical Ontologies,” KG-Microbe: a reference knowledge-graph and platform for harmonized microbial information.
Johnson, C. H., Ivanisevic, J., and Siuzdak, G. (2016). Metabolomics: beyond biomarkers and towards mechanisms. Nat. Rev. Mol. Cell Biol. 17, 451–459. doi: 10.1038/nrm.2016.25
Jones, P., Binns, D., Chang, H. Y., Fraser, M., Li, W., McAnulla, C., et al. (2014). InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240. doi: 10.1093/bioinformatics/btu031
Kanehisa, M., Furumichi, M., Tanabe, M., Sato, Y., and Morishima, K. (2017). KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 45, D353–D361. doi: 10.1093/nar/gkw1092
Kim, S., Chen, J., Cheng, T., Gindulyte, A., He, J., He, S., et al. (2023). PubChem 2023 update. Nucleic Acids Res. 51, D1373–D1380. doi: 10.1093/nar/gkac956
King, C. H., Desai, H., Sylvetsky, A. C., LoTempio, J., Ayanyan, S., Carrie, J., et al. (2019). Baseline human gut microbiota profile in healthy people and standard reporting template. PLoS One 14:e0206484. doi: 10.1371/journal.pone.0206484
Köhler, S., Gargano, M., Matentzoglu, N., Carmody, L. C., Lewis-Smith, D., Vasilevsky, N. A., et al. (2021). The human phenotype ontology in 2021. Nucleic Acids Res. 49, D1207–D1217. doi: 10.1093/nar/gkaa1043
Krol, J. D., Burke, J. T., Chen, S. Z., Sosinski, L., Alquaddoomi, F. S., Brenner, E. P., et al. (2022). MolEvolvR: A web-app for characterizing proteins using molecular evolution and phylogeny. bioRxiv. doi: 10.1101/2022.02.18.461833
Le Boulch, M., Déhais, P., Combes, S., and Pascal, G. (2019). The MACADAM database: a MetAboliC pAthways DAtabase for microbial taxonomic groups for mining potential metabolic capacities of archaeal and bacterial taxonomic groups. Database J. Biol. Databases Cur. 2019:baz049
Le, V., Quinn, T. P., Tran, T., and Venkatesh, S. (2020). Deep in the bowel: highly interpretable neural encoder-decoder networks predict gut metabolites from gut microbiome. BMC Genomics 21:256. doi: 10.1186/s12864-020-6652-7
Li, L., Jing, Q., Yan, S., Liu, X., Sun, Y., Zhu, D., et al. (2021). Amadis: A comprehensive database for association between microbiota and disease. Front. Physiol. 12:697059. doi: 10.3389/fphys.2021.697059
Liang, S., Deng, J., Jiang, Y., Wu, S., Zhou, Y., and Zhu, W. (2020). Functional distribution of bacterial community under different Land use patterns based on FaProTax function prediction. Pol. J. Environ. Stud. 29, 1245–1261. doi: 10.15244/pjoes/108510
Liu, T., Lan, G., Feenstra, K. A., Huang, Z., and Heringa, J. (2022). Towards a knowledge graph for pre−/probiotics and microbiota–gut–brain axis diseases. Sci. Rep. 12:18977. doi: 10.1038/s41598-022-21735-x
Liu, T., Pan, X., Wang, X., Feenstra, K. A., Heringa, J., and Huang, Z. (2021). Predicting the relationships between gut microbiota and mental disorders with knowledge graphs. Health Inf. Sci. Syst. 9:3. doi: 10.1007/s13755-020-00128-2
Machado, D., Andrejev, S., Tramontano, M., and Patil, K. R. (2018). Fast automated reconstruction of genome-scale metabolic models for microbial species and communities. Nucleic Acids Res. 46, 7542–7553. doi: 10.1093/nar/gky537
Maglott, D., Ostell, J., Pruitt, K. D., and Tatusova, T. (2007). Entrez gene: gene-centered information at NCBI. Nucleic Acids Res. 35, D26–D31. doi: 10.1093/nar/gkl993
Magnúsdóttir, S., Heinken, A., Kutt, L., Ravcheev, D. A., Bauer, E., Noronha, A., et al. (2017). Generation of genome-scale metabolic reconstructions for 773 members of the human gut microbiota. Nat. Biotechnol. 35, 81–89. doi: 10.1038/nbt.3703
Malik-Sheriff, R. S., Glont, M., Nguyen, T. V. N., Tiwari, K., Roberts, M. G., Xavier, A., et al. (2019). BioModels—15 years of sharing computational models in life science. Nucleic Acids Res. 48, D407–D415. doi: 10.1093/nar/gkz1055
Martens, M., Ammar, A., Riutta, A., Waagmeester, A., Slenter, D. N., Hanspers, K., et al. (2021). WikiPathways: connecting communities. Nucleic Acids Res. 49, D613–D621. doi: 10.1093/nar/gkaa1024
McDonald, D., Jiang, Y., Balaban, M., Cantrell, K., Zhu, Q., Gonzalez, A., et al. (2023). Greengenes2 enables a shared data universe for microbiome studies. Nat. Biotechnol. doi: 10.1038/s41587-023-01845-1
McGinnis, S., and Madden, T. L. (2004). BLAST: at the core of a powerful and diverse set of sequence analysis tools. Nucleic Acids Res. 32, W20–W25. doi: 10.1093/nar/gkh435
Mendler, K., Chen, H., Parks, D. H., Lobb, B., Hug, L. A., and Doxey, A. C. (2019). AnnoTree: visualization and exploration of a functionally annotated microbial tree of life. Nucleic Acids Res. 47, 4442–4448. doi: 10.1093/nar/gkz246
Mendoza, S. N., Olivier, B. G., Molenaar, D., and Teusink, B. (2019). A systematic assessment of current genome-scale metabolic reconstruction tools. Genome Biol. 20:158. doi: 10.1186/s13059-019-1769-1
Merlet, B., Paulhe, N., Vinson, F., Frainay, C., Chazalviel, M., Poupin, N., et al. (2016). A computational solution to automatically map metabolite libraries in the context of genome scale metabolic networks. Front. Mol. Biosci. 3:e002. doi: 10.3389/fmolb.2016.00002/abstract
Mirzayi, C., and Renson, A. (2021). Genomic standards consortium, massive analysis and quality control society, Furlanello C, Sansone SA, et al. reporting guidelines for human microbiome research: the STORMS checklist. Nat. Med. 27, 1885–1892. doi: 10.1038/s41591-021-01552-x
Moretti, S., Tran, V. D. T., Mehl, F., Ibberson, M., and Pagni, M. (2021). MetaNetX/MNXref: unified namespace for metabolites and biochemical reactions in the context of metabolic models. Nucleic Acids Res. 49, D570–D574. doi: 10.1093/nar/gkaa992
Morton, J. T., Aksenov, A. A., Nothias, L. F., Foulds, J. R., Quinn, R. A., Badri, M. H., et al. (2019). Learning representations of microbe–metabolite interactions. Nat. Methods 16, 1306–1314. doi: 10.1038/s41592-019-0616-3
Noronha, A., Modamio, J., Jarosz, Y., Guerard, E., Sompairac, N., Preciat, G., et al. (2019). The virtual metabolic human database: integrating human and gut microbiome metabolism with nutrition and disease. Nucleic Acids Res. 47, D614–D624. doi: 10.1093/nar/gky992
Norsigian, C. J., Pusarla, N., McConn, J. L., Yurkovich, J. T., Dräger, A., Palsson, B. O., et al. (2019). BiGG models 2020: multi-strain genome-scale models and expansion across the phylogenetic tree. Nucleic Acids Res. 48, D402–D406. doi: 10.1093/nar/gkz1054
O’Leary, N. A., Wright, M. W., Brister, J. R., Ciufo, S., Haddad, D., McVeigh, R., et al. (2016). Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 44, D733–D745. doi: 10.1093/nar/gkv1189
Olson, R. D., Assaf, R., Brettin, T., Conrad, N., Cucinell, C., Davis, J. J., et al. (2023). Introducing the bacterial and viral bioinformatics resource center (BV-BRC): a resource combining PATRIC, IRD and ViPR. Nucleic Acids Res. 51, D678–D689. doi: 10.1093/nar/gkac1003
Overbeek, R., Olson, R., Pusch, G. D., Olsen, G. J., Davis, J. J., Disz, T., et al. (2014). The SEED and the rapid annotation of microbial genomes using subsystems technology (RAST). Nucleic Acids Res. 42, D206–D214. doi: 10.1093/nar/gkt1226
Parks, D. H., Chuvochina, M., Rinke, C., Mussig, A. J., Chaumeil, P. A., and Hugenholtz, P. (2022). GTDB: an ongoing census of bacterial and archaeal diversity through a phylogenetically consistent, rank normalized and complete genome-based taxonomy. Nucleic Acids Res. 50, D785–D794. doi: 10.1093/nar/gkab776
Passi, A., Tibocha-Bonilla, J. D., Kumar, M., Tec-Campos, D., Zengler, K., and Zuniga, C. (2021). Genome-scale metabolic modeling enables in-depth understanding of big data. Meta 12:14. doi: 10.3390/metabo12010014
Paysan-Lafosse, T., Blum, M., Chuguransky, S., Grego, T., Pinto, B. L., Salazar, G. A., et al. (2023). InterPro in 2022. Nucleic Acids Res. 51, D418–D427. doi: 10.1093/nar/gkac993
Petri, V., Jayaraman, P., Tutaj, M., Hayman, G., Smith, J. R., De Pons, J., et al. (2014). The pathway ontology – updates and applications. J. Biomed. Semant. 5:7. doi: 10.1186/2041-1480-5-7
Price, M. N., Deutschbauer, A. M., and Arkin, A. P. (2020). GapMind: automated annotation of amino acid biosynthesis. mSystems 5:e00291. doi: 10.1128/mSystems.00291-20
Pruesse, E., Peplies, J., and Glöckner, F. O. (2012). SINA: accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinform. Oxf. Engl. 28, 1823–1829. doi: 10.1093/bioinformatics/bts252
Pruesse, E., Quast, C., Knittel, K., Fuchs, B. M., Ludwig, W., Peplies, J., et al. (2007). SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 35, 7188–7196. doi: 10.1093/nar/gkm864
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2012). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596. doi: 10.1093/nar/gks1219
Queirós, P., Hickl, O., Arbas, S. M., Wilmes, P., and May, P. (2022). UniFuncNet: a flexible network annotation framework. bioRxiv. doi: 10.1101/2022.03.15.484380
Reiman, D., Layden, B. T., and Dai, Y. (2021). MiMeNet: exploring microbiome-metabolome relationships using neural networks. PLoS Comput. Biol. 17:e1009021. doi: 10.1371/journal.pcbi.1009021
Sayers, E. W., Cavanaugh, M., Clark, K., Pruitt, K. D., Schoch, C. L., Sherry, S. T., et al. (2021). GenBank. Nucleic Acids Res. 49, D92–D96. doi: 10.1093/nar/gkaa1023
Schriml, L. M., Munro, J. B., Schor, M., Olley, D., McCracken, C., Felix, V., et al. (2022). The human disease ontology 2022 update. Nucleic Acids Res. 50, D1255–D1261. doi: 10.1093/nar/gkab1063
Seaver, S. M. D., Liu, F., Zhang, Q., Jeffryes, J., Faria, J. P., Edirisinghe, J. N., et al. (2021). The ModelSEED Biochemistry database for the integration of metabolic annotations and the reconstruction, comparison and analysis of metabolic models for plants, fungi and microbes. Nucleic Acids Res. 49, D575–D588. doi: 10.1093/nar/gkaa746
Shaffer, M., Armstrong, A. J. S., Phelan, V. V., Reisdorph, N., and Lozupone, C. A. (2017). Microbiome and metabolome data integration provides insight into health and disease. Transl. Res. 189, 51–64. doi: 10.1016/j.trsl.2017.07.001
Smith, B., Ceusters, W., Klagges, B., Köhler, J., Kumar, A., Lomax, J., et al. (2005). Relations in biomedical ontologies. Genome Biol. 6:R46. doi: 10.1186/gb-2005-6-5-r46
Söhngen, C., Bunk, B., Podstawka, A., Gleim, D., and Overmann, J. (2014). BacDive—the bacterial diversity Metadatabase. Nucleic Acids Res. 42, D592–D599. doi: 10.1093/nar/gkt1058
Sun, F., Sun, J., and Zhao, Q. (2022). A deep learning method for predicting metabolite–disease associations via graph neural network. Brief. Bioinform. 23:bbac266. doi: 10.1093/bib/bbac266
Sun, Y. Z., Zhang, D. H., Cai, S. B., Ming, Z., Li, J. Q., and Chen, X. (2018). MDAD: A special resource for microbe-drug associations. Front. Cell. Infect. Microbiol. 8:424. doi: 10.3389/fcimb.2018.00424
Sung, J., Kim, S., Cabatbat, J. J. T., Jang, S., Jin, Y. S., Jung, G. Y., et al. (2017). Global metabolic interaction network of the human gut microbiota for context-specific community-scale analysis. Nat. Commun. 8:15393. doi: 10.1038/ncomms15393
Swainston, N., Batista-Navarro, R., Carbonell, P., Dobson, P. D., Dunstan, M., Jervis, A. J., et al. (2017). biochem4j: integrated and extensible biochemical knowledge through graph databases. PLoS One 12:e0179130. doi: 10.1371/journal.pone.0179130
Tang, J., Wu, X., Mou, M., Wang, C., Wang, L., Li, F., et al. (2021). GIMICA: host genetic and immune factors shaping human microbiota. Nucleic Acids Res. 49, D715–D722. doi: 10.1093/nar/gkaa851
The Gene Ontology Consortium (2019). The gene ontology resource: 20 years and still GOing strong. Nucleic Acids Res. 47, D330–D338. doi: 10.1093/nar/gky1055
The UniProt ConsortiumBateman, A., Martin, M. J., Orchard, S., Magrane, M., Ahmad, S., et al. (2023). UniProt: the universal protein knowledgebase in 2023. Nucleic Acids Res. 51, D523–D531. doi: 10.1093/nar/gkac1052
Unni, D. R., Moxon, S. A. T., Bada, M., Brush, M., Bruskiewich, R., Caufield, J. H., et al. (2022). Biolink model: a universal schema for knowledge graphs in clinical, biomedical, and translational science. Clin. Transl. Sci. 15, 1848–1855. doi: 10.1111/cts.13302
Vasilevsky, N. A., Matentzoglu, N. A., Toro, S., Flack, J. E., Hegde, H., Unni, D. R., et al. Mondo: unifying diseases for the world, by the world. medRxiv. doi: 10.1101/2022.04.13.22273750
Vuokko, R., Vakkuri, A., and Palojoki, S. (2023). Systematized nomenclature of medicine–clinical terminology (SNOMED CT) clinical use cases in the context of electronic health record systems: systematic literature review. JMIR Med. Inform. 11:e43750. doi: 10.2196/43750
Wallen, Z. D., Demirkan, A., Twa, G., Cohen, G., Dean, M. N., Standaert, D. G., et al. (2022). Metagenomics of Parkinson’s disease implicates the gut microbiome in multiple disease mechanisms. Nat. Commun. 13:6958. doi: 10.1038/s41467-022-34667-x
Wei, W., Li, Z., Li, S., Wu, S., Zhang, D., An, Y., et al. (2023). Fingerprint profiling and gut microbiota regulation of polysaccharides from Fritillaria species. Int. J. Biol. Macromol. 237:123844. doi: 10.1016/j.ijbiomac.2023.123844
Wilmes, P., Trezzi, J.P., Aho, V., Jäger, C., Schade, S., Janzen, A., et al. An archaeal compound as a driver of Parkinson’s disease pathogenesis. In Review (2022).
Wishart, D. S., Feunang, Y. D., Guo, A. C., Lo, E. J., Marcu, A., Grant, J. R., et al. (2018). DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 46, D1074–D1082. doi: 10.1093/nar/gkx1037
Wishart, D. S., Girod, S., Peters, H., Oler, E., Jovel, J., Budinski, Z., et al. (2023). ChemFOnt: the chemical functional ontology resource. Nucleic Acids Res. 51, D1220–D1229. doi: 10.1093/nar/gkac919
Wishart, D. S., Guo, A., Oler, E., Wang, F., Anjum, A., Peters, H., et al. (2022). HMDB 5.0: the human metabolome database for 2022. Nucleic Acids Res. 50, D622–D631. doi: 10.1093/nar/gkab1062
Wishart, D. S., Li, C., Marcu, A., Badran, H., Pon, A., Budinski, Z., et al. (2020). PathBank: a comprehensive pathway database for model organisms. Nucleic Acids Res. 48, D470–D478. doi: 10.1093/nar/gkz861
Wishart, D. S., Oler, E., Peters, H., Guo, A., Girod, S., Han, S., et al. (2023). MiMeDB: the human microbial metabolome database. Nucleic Acids Res. 51, D611–D620. doi: 10.1093/nar/gkac868
Wittig, U., Rey, M., Weidemann, A., Kania, R., and Müller, W. (2018). SABIO-RK: an updated resource for manually curated biochemical reaction kinetics. Nucleic Acids Res. 46, D656–D660. doi: 10.1093/nar/gkx1065
wwPDB consortiumBurley, S. K., Berman, H. M., Bhikadiya, C., Bi, C., Chen, L., et al. (2019). Protein data Bank: the single global archive for 3D macromolecular structure data. Nucleic Acids Res. 47, D520–D528. doi: 10.1093/nar/gky949
Yilmaz, P., Kottmann, R., Field, D., Knight, R., Cole, J. R., Amaral-Zettler, L., et al. (2011). Minimum information about a marker gene sequence (MIMARKS) and minimum information about any (x) sequence (MIxS) specifications. Nat. Biotechnol. 29, 415–420. doi: 10.1038/nbt.1823
Yilmaz, P., Parfrey, L. W., Yarza, P., Gerken, J., Pruesse, E., Quast, C., et al. (2014). The SILVA and “all-species living tree project (LTP)” taxonomic frameworks. Nucleic Acids Res. 42, D643–D648. doi: 10.1093/nar/gkt1209
Keywords: microbiome, databases, ontologies, inference, microbiology, computational biology
Citation: Santangelo BE, Apgar M, Colorado ASB, Martin CG, Sterrett J, Wall E, Joachimiak MP, Hunter LE and Lozupone CA (2024) Integrating biological knowledge for mechanistic inference in the host-associated microbiome. Front. Microbiol. 15:1351678. doi: 10.3389/fmicb.2024.1351678
Edited by:
Jana Seifert, University of Hohenheim, GermanyReviewed by:
Joon-Yong Lee, PrognomiQ Inc., United StatesChristopher L. Hemme, University of Rhode Island, United States
Copyright © 2024 Santangelo, Apgar, Colorado, Martin, Sterrett, Wall, Joachimiak, Hunter and Lozupone. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Brook E. Santangelo, YnJvb2suc2FudGFuZ2Vsb0BjdWFuc2NodXR6LmVkdQ==