 Letícia Fernandes da Rocha1†
Letícia Fernandes da Rocha1† Grazielle Motta Rodrigues2†
Grazielle Motta Rodrigues2† Gabriela Simões de Oliveira3,4
Gabriela Simões de Oliveira3,4 Aymê Duarte Echevarria5Priscila Wink5
Aymê Duarte Echevarria5Priscila Wink5 Fabiana Volpato5Mayana Berdichevski3,5
Fabiana Volpato5Mayana Berdichevski3,5 Larissa Lutz1
Larissa Lutz1 Dariane Castro Pereira1,5
Dariane Castro Pereira1,5 Afonso Luís Barth2,3,5*
Afonso Luís Barth2,3,5* Andreza Francisco Martins2,3,4,5,6
Andreza Francisco Martins2,3,4,5,6- 1Unidade de Microbiologia e Biologia Molecular, Hospital de Clínicas de Porto Alegre, Porto Alegre, Brazil
- 2PPGCM - Programa de Pós-Graduação em Ciências Médicas, Universidade Federal do Rio Grande do Sul, Porto Alegre, Brazil
- 3PPGCF - Programa de Pós-Graduação em Ciências Farmacêuticas, Universidade Federal do Rio Grande do Sul, Porto Alegre, Brazil
- 4Departamento de Microbiologia, Imunologia e Parasitologia, Instituto de Ciências Básicas, Universidade Federal do Rio Grande do Sul, Porto Alegre, Brazil
- 5LABRESIS - Laboratório de Pesquisa em Resistência Bacteriana, Hospital de Clínicas de Porto Alegre, Porto Alegre, Brazil
- 6Departamento de Microbiologia, Imunologia e Parasitologia, Instituto de Ciências Básicas, Universidade Federal do Rio Grande do Sul, Porto Alegre, Brazil
Introduction: Serratia marcescens is a significant causative agent of hospital-acquired infections (HAIs), particularly in intensive care units (ICUs). Carbapenem resistance represents a major concern in HAI management, as carbapenem-resistant bacteria can trigger outbreaks in hospital settings. While molecular evaluation of outbreaks typically relies on pulse field gel electrophoresis (PFGE) or core genome multilocus sequence typing (cgMLST) methods, alternative rapid, reliable, and cost-effective methods for assessing clonal relatedness are needed.
Methods: This study aimed to characterize a carbapenem-resistant S. marcescens outbreak that occurred during the COVID-19 pandemic in a tertiary care hospital, using the flagellin gene as a single-locus sequence typing (SLST) method. In addition, we evaluated the genetic context of carbapenemase genes through whole-genome sequencing (WGS).
Results: Among the 170 carbapenem-resistant Serratia marcescens isolates recovered, high resistance to gentamicin, ciprofloxacin, and cefepime was observed. The predominant carbapenemase gene detected by qPCR-HRM was blaKPC (92.2%). Phylogenetic analysis of the flagellin gene grouped the sequences into two distinct clades, with all outbreak-related blaKPC-positive S. marcescens isolates clustering within clade B. The blaKPC gene was carried on an IncP6 plasmid.
Discussion: Our findings indicate that the flagellin gene serves as an effective marker for characterizing carbapenem-resistant S. marcescens carrying blaKPC, confirming that the outbreak was caused by the clonal expansion of isolates harboring blaKPC on an IncP6 plasmid.
1 Introduction
Serratia marcescens is a ubiquitous, fermentative, rod-shaped Gram-negative bacteria belonging to the Enterobacterales order. This organism typically exhibits multiple resistance mechanisms, including intrinsic resistance to polymyxins, which significantly limits therapeutic options (Iguchi et al., 2014). As an opportunistic pathogen, S. marcescens has been associated with high mortality rates, particularly among immunocompromised patients, during hospital outbreaks (Šiširak, 2013; Iguchi et al., 2014).
Carbapenems are the primary antibiotics used to treat infections caused by Enterobacterales, including strains of S. marcescens that are resistant to other antimicrobials (da Silva et al., 2021). However, there has been a significant increase in carbapenem-resistant Enterobacterales (CRE) worldwide, particularly in recent years. This rise has been especially noted during the COVID-19 pandemic period, when an overall increase in CRE incidence was documented (Hamers et al., 2022; Pintado et al., 2022).
The first report of a plasmid-encoded carbapenem-hydrolyzing enzyme (KPC-2) in S. marcescens was documented in Hangzhou, China. The three isolates obtained from patients at a hospital in China exhibited identical plasmid profiles, indicating that the same plasmid had been transmitted among these S. marcescens isolates (Zhang et al., 2007). Currently, nosocomial infections caused by carbapenem-resistant Serratia spp. have become increasingly common worldwide, including in Brazil, and are typically attributed to carbapenemase production (Cayô et al., 2017; Barberino et al., 2018; Streling et al., 2018; NOTA TÉCNICA No. 74/2022-CGLAB/DAEVS/SVS/MS-Agência Nacional de Vigilância Sanitária-Anvisa, 2025).
Prompt and accurate identification of sources and transmission routes is crucial for implementing infection control measures and preventing the further nosocomial spread of bacteria. DNA-based typing methods, such as multi-locus sequence typing (MLST), have been developed for key human pathogens. For Serratia marcescens, an established MLST scheme is available on PubMLST, which currently includes 1832 sequence types (STs).1 This scheme has proven to be valuable for the molecular characterization of S. marcescens strains and serves as an important tool for epidemiological surveillance (Martineau et al., 2018). Currently, whole-genome sequencing (WGS)-based typing is employed for the majority of bacterial species, including S. marcescens (Zingg et al., 2017; Muyldermans et al., 2021). However, both MLST and WGS are considered time-consuming, labor-intensive, and expensive methods. In contrast, techniques utilizing single- or double-locus sequence typing have been successfully employed for the rapid assignment of clonal lineages in various bacterial species (Weissman et al., 2012; Pournaras et al., 2014; Fernández-Huerta et al., 2020; Magalhães et al., 2020).
During the 2-year period of the COVID-19 pandemic, we observed an increase in infections caused by carbapenem-resistant S. marcescens (outbreak) at our institution, which is a tertiary care hospital in southern Brazil. Therefore, we evaluated a novel approach using the flagellin gene as a single-locus sequence typing (SLST) method for the molecular characterization of S. marcescens isolates. In addition, we investigated the genetic environment of the carbapenemase genes present in the outbreak isolates.
2 Materials and methods
2.1 Isolate collection and identification
The study was conducted at Hospital de Clínicas de Porto Alegre, Porto Alegre, Brazil, which is an 860-bed tertiary care university hospital. During a surveillance study focused on carbapenem-resistant Enterobacterales, a total of 170 S. marcescens isolates non-susceptible to meropenem—according to the European Committee on Antimicrobial Susceptibility Testing (EUCAST) criteria, (Eucast: MIC Determination, 2025)—were obtained from January 2020 to January 2022. The incidence rates of meropenem-non-susceptible S. marcescens (MNSSm) per 1,000 patient-days were evaluated for each month to monitor the increase in the case numbers.
Only one isolate from each patient was included. The isolates were identified by mass spectrometry using the VITEK® MALDI-TOF MS system (bioMérieux, France) and MYLA® (version 3.0) for clinical use.
2.2 Antimicrobial susceptibility profile
Antimicrobial susceptibility was evaluated for all isolates using the disc diffusion method following the EUCAST guidelines (EUCAST, 2024). The antibiotics tested included amikacin, cefepime, ciprofloxacin, norfloxacin, ceftazidime, gentamicin, meropenem, piperacillin/tazobactam, and sulfamethoxazole/trimethoprim. The susceptibility profile of tigecycline was determined through broth microdilution following the EUCAST guidelines (EUCAST, 2024), and quality control of this test was performed in parallel using E. coli ATCC 25922.
Minimum inhibitory concentrations (MICs) of meropenem, ceftazidime-avibactam, and meropenem-vaborbactam were determined for a subset of 69 isolates using concentration gradient strips (MTS, Liofilchem, Inc., Waltham, MA) according to the EUCAST guidelines. The isolates were selected based on recovery data (during the outbreak period). One isolate per patient was included, sourced from different care units, with at least one isolate collected each month.
2.3 Molecular detection of carbapenemase genes
Total genomic DNA was extracted from the isolates by thermal lysis (Dashti et al., 2009), and DNA concentration and purity were evaluated using a NanoDrop 2000 spectrophotometer (Thermo Scientific, Wilmington, DE, United States), with DNA concentrations ranging from 20 to 50 ng/μL. For the qPCR reactions, 1 μL of DNA template was used. The presence of carbapenemase genes was detected using multiplex high-resolution melting real-time PCR (qPCR-HRM) with primers previously described by Monteiro et al. (2012) for blaIMP, blaVIM, blaNDM-1, blaKPC, blaGES, and blaOXA-48-like.
2.4 Single-locus sequence typing
Reference sequences of the flagellin (fliC) gene from S. marcescens (Jimenez et al., 2020; Moradigaravand et al., 2016; Iguchi et al., 2014; Nodari et al., 2017; Supplementary Table S1) were extracted, aligned, and trimmed to identify the polymorphic region. A phylogenetic tree was reconstructed to compare the relationship between these sequences, and the best region was selected to design the primers using Geneious 9.0 (Kearse et al., 2012). The primers fliC_F (5′-CGCTTCTCAGTCCCGTATCC-3′) and fliC_R (5′-AATAGCC CGATTCCCCCG-3′) were designed to be complementary to the positions 701–1,150 of the fliC gene, resulting in a product length of 450 bp.
Total genomic DNA from the 69 isolates was extracted and evaluated according to the protocol cited above (Dashti et al., 2009). In addition, we also sequenced a meropenem-susceptible isolate using Sanger sequencing to serve as an outgroup in the phylogenetic tree. PCR amplification of the fliC gene was carried out using 10 ng of DNA template and Platinum® Taq DNA Polymerase (Invitrogen Corporation, United States). The PCR conditions were as follows: 94°C for 5 min, followed by 35 cycles of 94°C for 30 s, 64°C for 45 s, and 72°C for 30 s, with a final extension at 72°C for 5 min. The amplified products were analyzed using 1.5% agarose gel electrophoresis (40 min at 110 v) and purified using ExoSAP-IT PCR Product Cleanup (Afymetrix, Santa Clara, CA, United States).
For Sanger sequencing, the PCR products were labeled using the BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, United States) and purified using the BigDye XTerminator Purification Kit (Applied Biosystems, Foster City, California, United States). The samples were sequenced in both forward and reverse directions using the ABI 3500 Genetic Analyzer (Applied Biosystems, Foster City, CA, United States).
Phylogeny was reconstructed using IQTree (Nguyen et al., 2015) from consensus sequences generated by aligning a fliC gene fragment with MAFFT v7.475 (Katoh and Standley, 2013), using 20 reference sequences (Supplementary Table S1). This fragment was created by systematically removing nucleotides from both ends to identify a DNA sequence that can resolve all phylogenetic clades, aligning with the previously published phylogeny inference (Jimenez et al., 2020). Subsequently, the sequences of this fragment obtained from the isolates in this study were aligned (using MAFFT v7.475) and subjected to maximum likelihood (ML) analysis under the K80 nucleotide substitution model, as selected by the ModelFinder application (Kalyaanamoorthy et al., 2017). Branch support was assessed using the approximate likelihood-ratio test based on the Shimodaira–Hasegawa procedure (SHaLRT) with 1,000 replicates. The phylogenetic tree was visualized using MEGA X (v.10.2.3) (Kumar et al., 2018).
2.5 Sequencing and plasmid characterization
One isolate recovered during the outbreak was sequenced using both Illumina MiSeq (2 × 250 bp; average coverage ∼100×) and MinION (R9.4 flow cell) for plasmid characterization. Genomic DNA was extracted from colonies grown in BHI broth (KASVI®) using the QIAamp DNA Mini Extraction Kit (QIAGEN®). DNA concentration was determined using the Qubit dsDNA HS Assay Kit with a Qubit 4 fluorometer (Thermo Fisher Scientific), and fragment lengths were assessed using TapeStation 2,200 (Agilent, United Kingdom). The quality of the DNA was determined using NanoDrop™, and the 260/280 ratio was considered.
The paired-end library was constructed using the Nextera XT DNA Library Prep Kit (Illumina), while for long reads (MinION; fast model base-calling; Q ≥8; Guppy v6.3.9; MinKNOW 22.10.10), the library was prepared using the Rapid Barcoding Sequencing Kit (SQK-RBK004; Oxford Nanopore), following the manufacturer’s protocols.
Raw short reads were quality-trimmed (Q > 30) and assembled using CLC Genomics Workbench 23. Antimicrobial resistance genes were identified (contigs >200 bp; >10x average coverage) in silico using the QIAGEN Microbial Insight-Antimicrobial Resistance database (QMI-AR). Plasmid replicon typing and IS typing were performed using the PlasmidFinder (2.0.1) and MobileElementFinder (v1.0.3) databases, respectively.
CLC Genomics Workbench (v. 23.0) was used to extract reads from base-called MinION sequencing data and to generate de novo assemblies, which were error-corrected using short-read Illumina data and the assembly polisher tool. Alignments of the fully reconstructed plasmid sequences were visualized and annotated using Geneious Prime (v. 2023.0.4).
For plasmid characterization, a hybrid assembly was generated using QIAGEN CLC Genomics Workbench (version 23.0). Comparison analyses were performed using Geneious Prime (v. 2023.0.4) and BLAST Ring Generator (BRIG v. 0.95) to compare the circularized plasmids from this study with similar plasmids deposited in the NCBI database. Prokka (v. 1.14.6) and reference sequences were used for preliminary annotation, and the coding sequences (CDS) were manually curated.
3 Results
During the 2-year study period (January 2020 to January 2022), the incidence rates of MNSSm ranged from 0 to 1.39 cases/1,000 patient-days, with a median of 1.14 cases/1,000 patient-days. The highest rates were observed in December 2020, January 2021, February 2021, and March 2021 with 0.24, 0.19, 0.35, and 1.39 cases/1000 patient-days, respectively. The incidence curve (Supplementary Figure S1) revealed that the outbreak began in December 2020 and concluded in November 2021. Clinical data indicated that 77.65% (132/170) of the isolates were recovered from COVID-19-positive patients. Among these patients, the majority (83%; 109/132) were admitted to the intensive care unit (ICU).
The MNSSm isolates were predominantly obtained from tracheal aspirate samples (77%; 131/170). High resistance rates were observed for cefepime (100%), ceftazidime (98.2%), gentamicin (94.4%), ciprofloxacin (93.6%), sulfamethoxazole-trimethoprim (73.8%), and tigecycline (73.8%) (Supplementary Table S2). Susceptibility to amikacin was observed in 51.2% of the isolates. The MICs for meropenem (4.0–250.0 μg/mL), ceftazidime-avibactam (0.5–256 μg/mL), and meropenem-vaborbactam (0.06–8 μg/mL) are presented in Table 1. The MIC₅₀/MIC₉₀ values for meropenem, ceftazidime-avibactam, and meropenem-vaborbactam were 8.0/256, 0.5/8, and 0.125/4 μg/mL, respectively. We successfully recovered 166 out of 170 MNSSm isolates for carbapenemase gene detection. The blaKPC gene was the most prevalent carbapenemase gene (92.2%, 153/166), followed by blaNDM-1 (3.6%; 6/166).
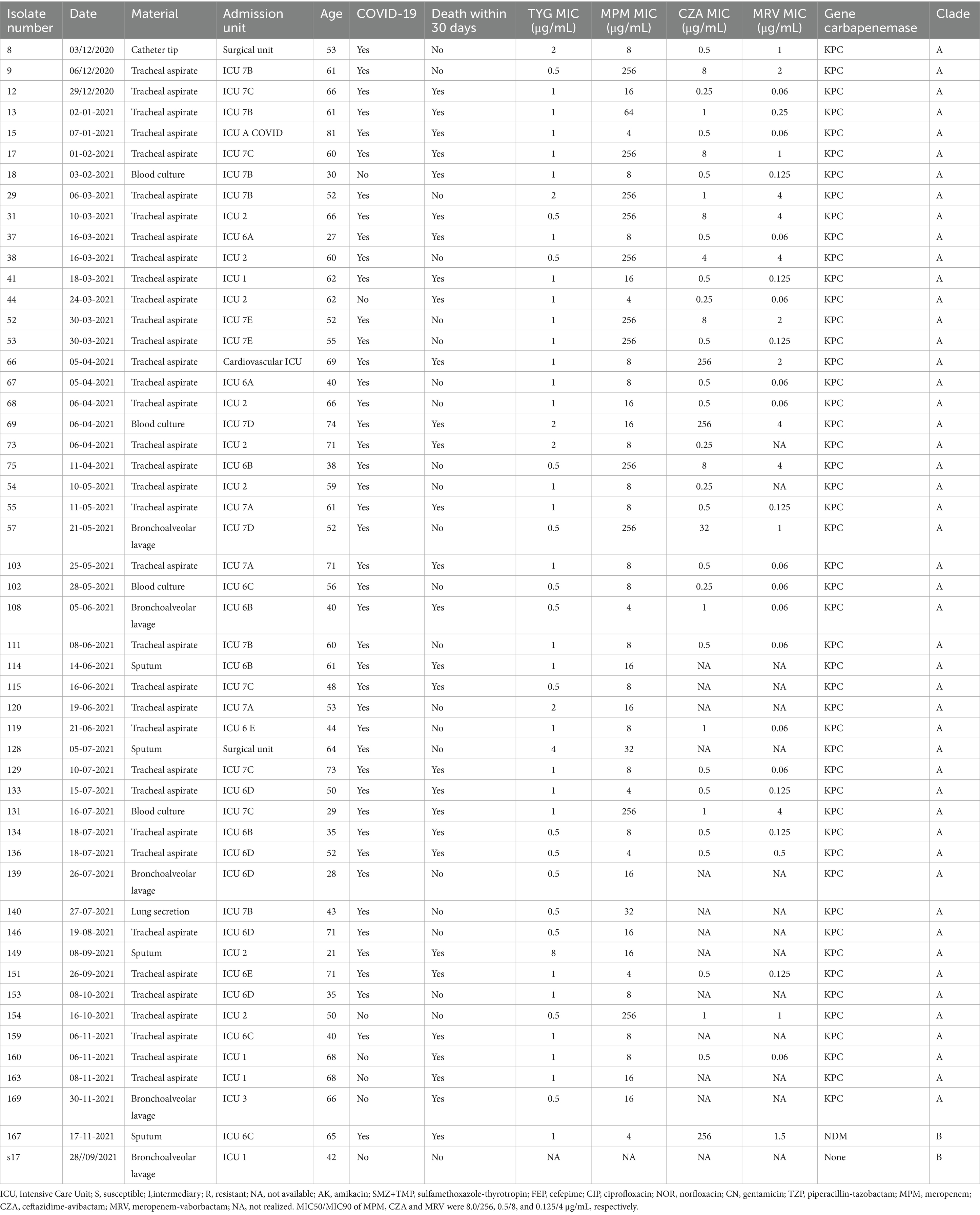
Table 1. Clinical characteristics of the isolates for phylogenetic analysis.
For the SLST method using the flagellin gene, eight polymorphic sites were identified in the reference sequences, and a 353 bp DNA sequence was sufficient to resolve all previously reported phylogenetic clades (Table 1). This sequence was designated as the fliC gene typing region. Of the 69 isolates amplified by PCR for Sanger sequencing, high-quality sequence data were obtained for 50 isolates (Supplementary Figure S2). Phylogenetic analysis grouped these isolates into two distinct clades: Clade B comprised all blaKPC-2-positive isolates (49/50), while Clade A contained the single meropenem-susceptible isolate, which was closely related to a blaNDM-1-positive isolate (Figure 1). WGS revealed that the Serratia marcescens isolate GSMA0007 belongs to sequence type 807 (ST807).
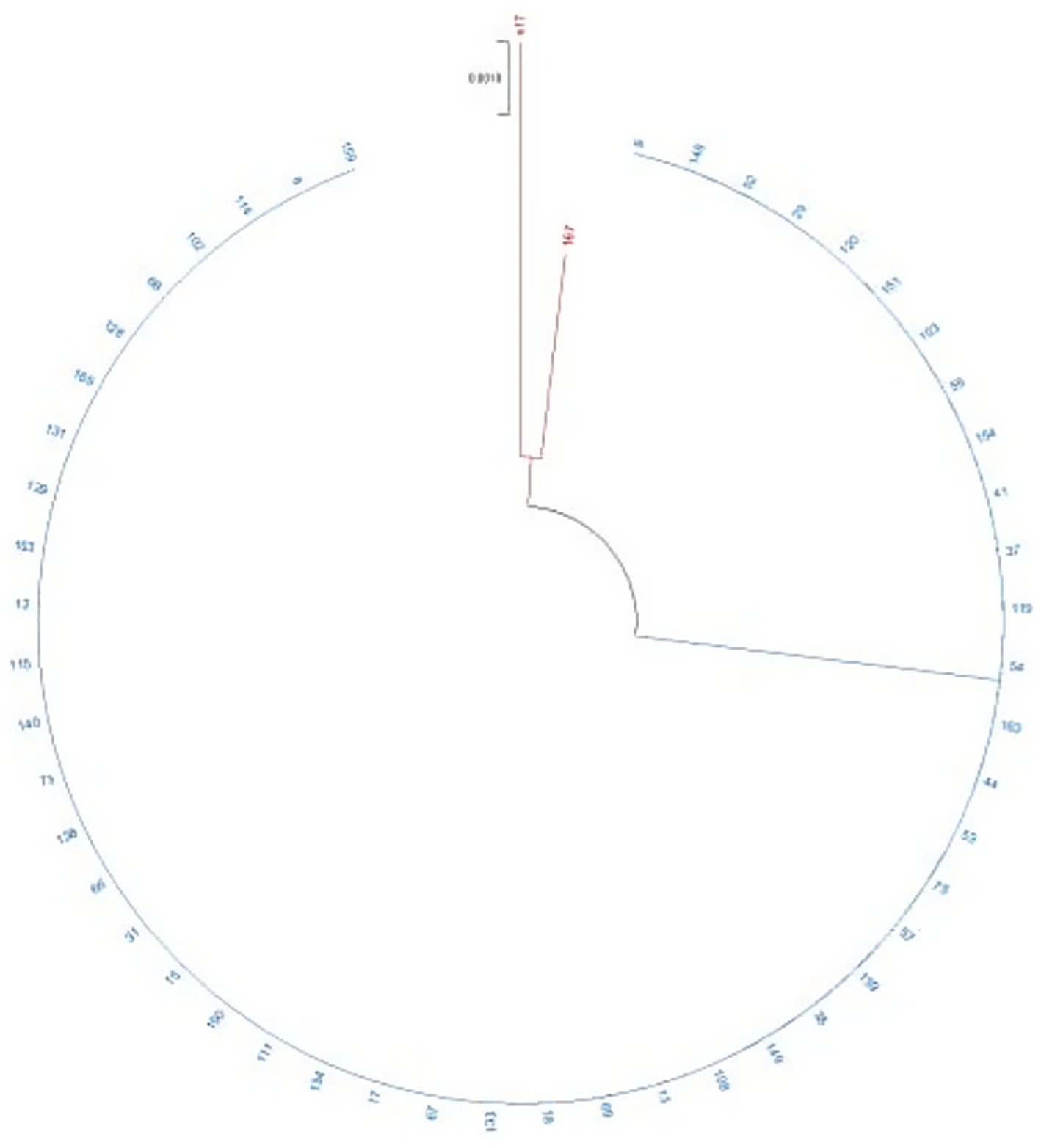
Figure 1. Phylogenetic analysis of the Serratia marcescens isolates. The phylogenetic tree was inferred using the maximum likelihood (ML) method. The bootstrap test was based on 1,000 replicates. Branches in red represent clade A, and branches in blue represent clade B. Branch lengths are shown above the branches in black. S17 is the meropenem-susceptible isolate obtained during the outbreak period.
The blaKPC-2 gene was located on a plasmid with 99.93% identity and 83% coverage to pWP8-S18-CRE-01_2 (GenBank accession number AP022243.1). PlasmidFinder identified the incompatibility group as IncP6, with 99.8% identity and 100% coverage. The complete circularized IncP6 plasmid exhibited a GC content of 58.3% and measured 51,220 kb in size; it was designated pLB_GSMA0007 (accession number CP130614 and CP130615). A graphical comparison of the IncP6 plasmids harboring blaKPC-2 is presented in Figure 2. The blaKPC-2 gene was inserted within a classical Tn3-family transposon alongside other antibiotic resistance genes, including blaTEM-1, mph(A), qacE, sul1, and aac(6′)-lb-cr, which confer resistance to cephalosporins, macrolides, chlorhexidine and benzalkonium chloride, sulfamethoxazole, fluoroquinolones, and aminoglycosides, respectively. Complete information regarding the whole genome analysis is provided in Supplementary Table S3.
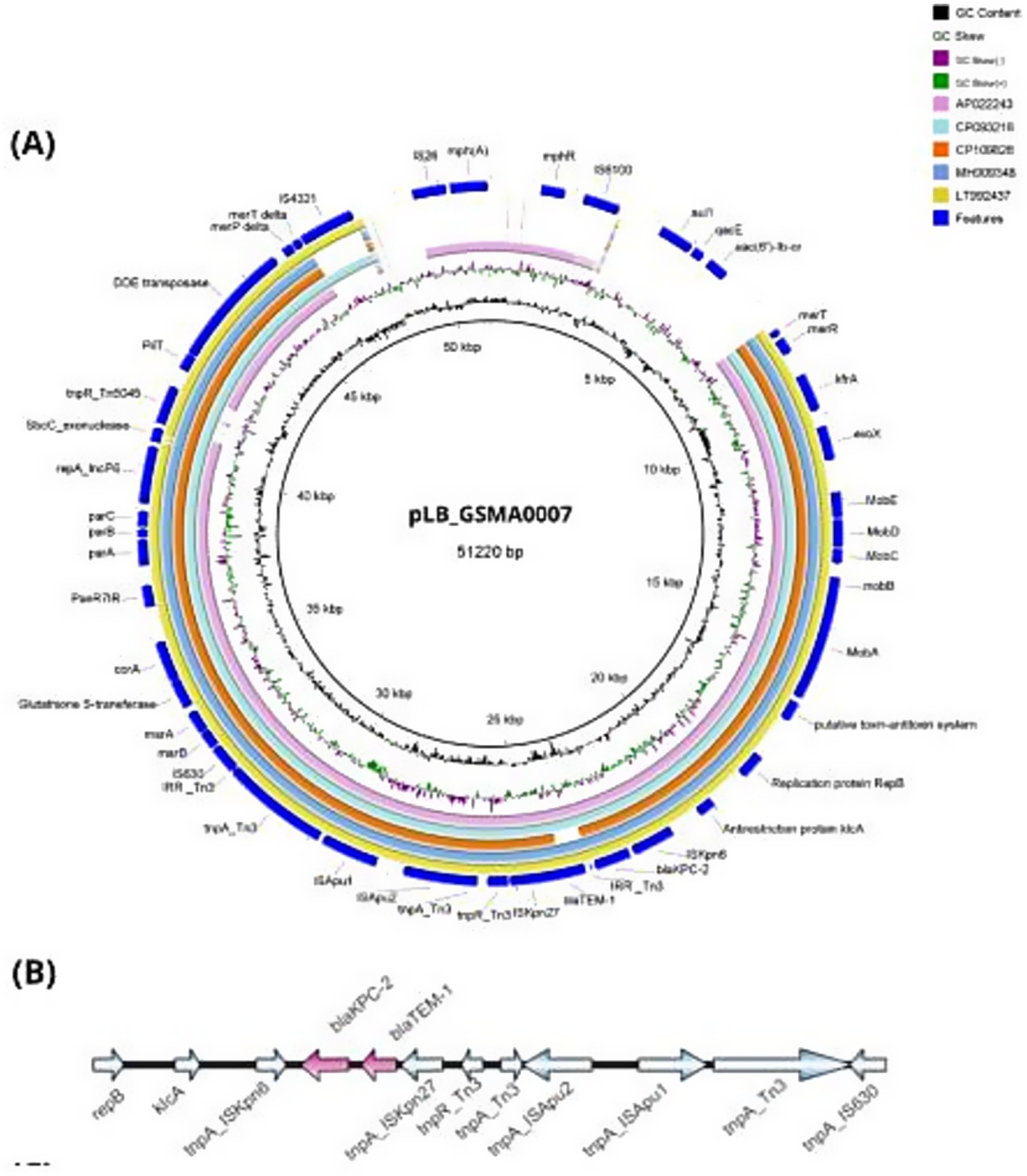
Figure 2. Comparisons among the plasmids belonging to the IncP6 group harboring blaKPC-2. (A) Circular alignment of pLB_GSMA0007 (51,220 kb) with other sequences previously deposited in Genbank (AP022243, CP093216, CP109826, MH909348, LT992437), generated using BLAST Ring Image Generator (BRIG). The inner circles represent the assembly of pLB_GSMA0007, GC content (black), and GC skew (dark green and purple). (B) Genetic environment of blaKPC-2 in pLB_GSMA0007, generated using IBS 2.0. Resistance genes are represented in pink (for details regarding the isolates, see Supplementary Table S3).
4 Discussion
The COVID-19 pandemic significantly disrupted hospital settings worldwide, increasing the demand for ICU beds, medical supplies, and healthcare workers. This surge severely impacted hospital healthcare systems. The prolonged and complex course of SARS-CoV-2 infections weakened surveillance measures for multi-drug resistant (MDR) organisms, creating favorable conditions for hospital-acquired infections (HAIs) (Falcone et al., 2022; Kozłowski et al., 2022). Extensive antimicrobial exposure, prolonged hospitalization, use of invasive devices, and compromised host immunity are considered the primary factors associated with antimicrobial resistance development (De Waele et al., 2020). According to data from our institution, the highest number of hospitalized COVID-19 patients was recorded between 19 February 2021 and 17 March 2021 (Martins et al., 2021) Notably, the highest incidence density of MNSSm was observed in March 2021. During the outbreak in our institution, meropenem was the fourth most commonly used antimicrobial among COVID-19 patients (Silva et al., 2020). Its consumption, measured in days of therapy (DOT) per 1,000 patient-days, was higher in 2021 than in 2020 (101.4 vs. 90.9, respectively) (data not shown).
S. marcescens has long been recognized as a cause of nosocomial outbreaks. During the pandemic, several hospital outbreaks of carbapenem-resistant S. marcescens were linked to COVID-19 dedicated units (Vera-Leiva et al., 2017; World Health Organization, 2017; Nedeljković et al., 2021). These outbreaks were due to S. marcescens carrying the blaKPC-2 gene, primarily located on plasmid groups IncA/C and IncN (Prado et al., 2022). The transmission of S. marcescens in healthcare settings is often associated with direct patient contact, contaminated medical equipment, and healthcare personnel. In our outbreak, the predominance of cases among ICU inpatients suggests a likely role of invasive procedures, such as mechanical ventilation and central venous catheters, as potential facilitators of bacterial spread. In addition, environmental reservoirs, including sinks and disinfectant solutions, have been previously implicated in S. marcescens outbreaks. Upon identifying the outbreak, immediate infection control measures were implemented, including cohorting of infected patients, enhanced hand hygiene reinforcement among healthcare workers, and decontamination of high-touch surfaces.
Treatment of infections caused by carbapenem-resistant S. marcescens is challenging due to this bacterium’s intrinsic resistance to polymyxins. Newer beta-lactam/beta-lactamase inhibitor combinations may be effective against carbapenem-resistant S. marcescens but only when resistance is mediated by serine carbapenemases rather than metallo-carbapenemases. Therefore, identification of bacterial resistance mechanisms plays a crucial role in determining appropriate clinical treatment for patients with carbapenem-resistant infections. Our findings demonstrated that the S. marcescens isolates carrying blaKPC were susceptible to ceftazidime-avibactam and meropenem-vaborbactam, consistent with previous reports (Prado et al., 2022).
Evaluating clonal relatedness of isolates during an outbreak is essential, with PFGE and cgMLST schemes being the most common typing methods. However, developing faster, reliable, and cost-effective methods remains necessary. Recently, various typing approaches using single- or double-locus sequence typing have been proposed (Weissman et al., 2012; Pournaras et al., 2014; Fernández-Huerta et al., 2020; Magalhães et al., 2020) to enable rapid evaluation of outbreak isolates. In this study, we evaluated a rapid approach to characterize an S. marcescens outbreak using a 353 bp region of the fliC gene. This gene encodes flagellin, the primary protein constituting the flagellar structure in various bacterial species. The flagellin sequence contains highly conserved regions across species, as well as a hypervariable central region (Nedeljković et al., 2021), making the fliC gene an interesting molecular marker for typing. Using this gene, our phylogenetic analysis clustered all blaKPC-2-positive isolates into the same clade while distinguishing both blaNDM-1-positive and meropenem-susceptible isolates. Although this molecular marker produced promising results, it is important to emphasize that confirmation of isolate clonality should utilize more robust methods.
In this study, the blaKPC-2 gene was carried on an IncP6 incompatibility plasmid of 51,220 kb (pLB_GSMA0007). The genetic environment of the carbapenemase gene harbored a Tn3 transposon formed by ISKpn6/blaKPC-2/ΔblaTEM-1/ISKpn27, identical to the structure previously reported by Yao et al. (2017). While the genetic context of blaKPC-2 varies across different plasmids, the most common transposon in Brazil is Tn4401, which has been responsible for the widespread dissemination of this gene in the country (Vera-Leiva et al., 2017). IncP6 plasmids carrying blaKPC-2 have rarely been reported, and to the best of our knowledge, this is the first report of an IncP6 plasmid from a clinical isolate in Brazil.
Our findings demonstrate that an outbreak of clonal-related carbapenem-resistant S. marcescens occurred during the COVID-19 pandemic, primarily affecting ICU inpatients. The spread of the resistance gene was facilitated by an IncP6 plasmid containing blaKPC-2, reported here for the first time from a clinical isolate in Brazil. In addition, our approach using the fliC gene for SLST successfully enabled molecular characterization of the S. marcescens outbreak. This method is particularly valuable given that whole-genome sequencing, while considered the gold standard, is not always feasible. The SLST method represents a promising tool for genomic surveillance due to its lower cost and faster turnaround time.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/supplementary material.
Ethics statement
The studies involving humans were approved by Comitê de Ética em Pesquisa do Hospital de Clínicas de Porto Alegre. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation was not required from the participants or the participants’ legal guardians/next of kin because This project involved minimal risks, as it used biological samples, not direct participation of patients. The data obtained for the study were accessed through patient records via the AGHuse institutional system. The privacy of patients, as well as their respective associated information contained in the medical records was preserved.
Author contributions
LR: Data curation, Investigation, Methodology, Writing – original draft, Writing – review & editing. GR: Investigation, Methodology, Writing – original draft, Writing – review & editing. GO: Data curation, Formal analysis, Investigation, Software, Writing – review & editing. AE: Methodology, Writing – review & editing. PW: Visualization, Writing – review & editing. FV: Data curation, Formal analysis, Investigation, Methodology, Writing – review & editing. MB: Methodology, Writing – review & editing. LL: Data curation, Methodology, Supervision, Writing – original draft. DP: Conceptualization, Formal analysis, Investigation, Software, Supervision, Writing – original draft, Writing – review & editing. AB: Funding acquisition, Project administration, Resources, Supervision, Visualization, Writing – original draft, Writing – review & editing. AM: Conceptualization, Investigation, Methodology, Project administration, Resources, Software, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq 407953/2022–1), the Instituto de Pesquisa em Resistência aos Antimicrobianos (INPRA)-INCT CNPq 465718/2014–0, the Fundo de Amparo à Pesquisa doEstado do Rio Grande do Sul (INPRA-INCT FAPERGS 17/2551–0000514-7), and the Fundo de Incentivo à Pesquisa e Eventos do Hospital de Clínicas de Porto Alegre (FIPE/HCPA).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2025.1525543/full#supplementary-material
SUPPLEMENTARY FIGURE 1 | Incidence rate of the 170 MNSSm isolates from January 2020 to January 2022. Incidence rate was calculated as MNSSm isolates per 1000 Patient Days.
SUPPLEMENTARY FIGURE 2 | Alignment of fliC gene (350 bp fragment). This MAFFT alignment shows the identity of the fliC gene fragment among outbreak isolates and an outgroup isolate, used to build the phylogenetic tree (Figure 1). The colours yellow (G), green (T), red (A) and blue (C) represent the SNPs in each position.
Footnotes
References
Barberino, M. G., Cruvinel, S. d. A., Faria, C., Salvino, M. A., and Silva, M. d. O. (2018). Isolation of blaNDM-producing Enterobacteriaceae in a public hospital in Salvador, Bahia, Brazil. Braz. J. Infect. Dis. 22, 47–50. doi: 10.1016/j.bjid.2017.10.002
Cayô, R., Leme, R. C. P., Streling, A. P., Matos, A. P., Nodari, C. S., Chaves, J. R. E., et al. (2017). Serratia marcescens harboring SME-4 in Brazil: a silent threat. Diagn. Microbiol. Infect. Dis. 87, 357–358. doi: 10.1016/j.diagmicrobio.2017.01.008
da Silva, K. E., Rossato, L., Jorge, S., de Oliveira, N. R., Kremer, F. S., Campos, V. F., et al. (2021). Three challenging cases of infections by multidrug-resistant Serratia marcescens in patients admitted to intensive care units. Braz. J. Microbiol. 52, 1341–1345. doi: 10.1007/s42770-021-00477-4
Dashti, A. A., Jadaon, M. M., Abdulsamad, A. M., and Dashti, H. M. (2009). Heat treatment of bacteria: a simple method of DNA extraction for molecular techniques. Kuw. Med. J. 41, 117–122.
De Waele, J. J., Boelens, J., and Leroux-Roels, I. (2020). Multidrug-resistant bacteria in ICU: fact or myth. Curr. Opin. Anaesthesiol. 33, 156–161. doi: 10.1097/ACO.0000000000000830
EUCAST Breakpoint tables for interpretation of MICs and zone diameters, version 14.0. EUCAST (2024). Available online at: https://www.eucast.org/clinical_breakpoints/
Eucast: MIC Determination (2025). Available at: https://www.eucast.org/ast_of_bacteria/mic_determination (Accessed October 24, 2024).
Falcone, M., Suardi, L. R., Tiseo, G., Galfo, V., Occhineri, S., Verdenelli, S., et al. (2022). Superinfections caused by carbapenem-resistant Enterobacterales in hospitalized patients with COVID-19: a multicentre observational study from Italy (CREVID study). JAC Antimicrob. Resist. 4:dlac064. doi: 10.1093/jacamr/dlac064
Fernández-Huerta, M., Serra-Pladevall, J., Esperalba, J., Moreno-Mingorance, A., Fernández-Naval, C., Barberá, M.-J., et al. (2020). Single-locus-sequence-based typing of the mgpB gene reveals transmission dynamics in Mycoplasma genitalium. J. Clin. Microbiol. 58:e01886. doi: 10.1128/JCM.01886-19
Hamers, R. L., Cassini, A., Asadinia, K. S., and Bertagnolio, S. (2022). Developing a priority global research agenda for antimicrobial resistance in the human health sector: protocol for a scoping review. BMJ Open 12:e060553. doi: 10.1136/bmjopen-2021-060553
Iguchi, A., Nagaya, Y., Pradel, E., Ooka, T., Ogura, Y., Katsura, K., et al. (2014). Genome evolution and plasticity of Serratia marcescens, an important multidrug-resistant nosocomial pathogen. Genome Biol. Evol. 6, 2096–2110. doi: 10.1093/gbe/evu160
Jimenez, A., Abbo, L. M., Martinez, O., Shukla, B., Sposato, K., Iovleva, A., et al. (2020). KPC-3–producing Serratia marcescens outbreak between acute and long-term care facilities, Florida, USA. Emerg. Infect. Dis. 26, 2746–2750. doi: 10.3201/eid2611.202203
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., von Haeseler, A., and Jermiin, L. S. (2017). ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589. doi: 10.1038/nmeth.4285
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., et al. (2012). Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. doi: 10.1093/bioinformatics/bts199
Kozłowski, B., Kubiak-Pulkowska, J., Pałka, J., Bożiłow, D., Zając, M., and Deptuła, A. (2022). Healthcare-associated infections in COVID-19 ICU patients - two-Centre study. Cent. Eur. J. Public Health 30, 196–200. doi: 10.21101/cejph.a7135
Kumar, S., Stecher, G., Li, M., Knyaz, C., and Tamura, K. (2018). MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Martins, A., Zavascki, A., Wink, P., Volpato, F., Monteiro, F., Rosset, C., et al. (2021). Detection of SARS-CoV-2 lineage P.1 in patients from a region with exponentially increasing hospitalisation rate, February 2021, Rio Grande do Sul, Southern Brazil. Euro Surveill. 26. doi: 10.2807/1560-7917.ES.2021.26.12.2100276
Magalhães, B., Valot, B., Abdelbary, M. M. H., Prod’hom, G., Greub, G., Senn, L., et al. (2020). Combining standard molecular typing and whole genome sequencing to investigate Pseudomonas aeruginosa epidemiology in intensive care units. Front. Public Health 8:3. doi: 10.3389/fpubh.2020.00003
Martineau, C., Li, X., Lalancette, C., Perreault, T., Fournier, E., Tremblay, J., et al. (2018). Serratia marcescens outbreak in a neonatal intensive care unit: new insights from next-generation sequencing applications. J. Clin. Microbiol. 56:e00235. doi: 10.1128/JCM.00235-18
Monteiro, J., Widen, R. H., Pignatari, A. C. C., Kubasek, C., and Silbert, S. (2012). Rapid detection of carbapenemase genes by multiplex real-time PCR. J. Antimicrob. Chemother. 67, 906–909. doi: 10.1093/jac/dkr563
Moradigaravand, D., Boinett, C. J., Martin, V., Peacock, S. J., and Parkhill, J. (2016). Recent independent emergence of multiple multidrug-resistant Serratia marcescens clones within the United Kingdom and Ireland. Genome Res. 26, 1101–9. doi: 10.1101/gr.205245.116
Muyldermans, A., Crombé, F., Bosmans, P., Cools, F., Piérard, D., and Wybo, I. (2021). Serratia marcescens outbreak in a neonatal intensive care unit and the potential of whole-genome sequencing. J. Hosp. Infect. 111, 148–154. doi: 10.1016/j.jhin.2021.02.006
Nedeljković, M., Sastre, D. E., and Sundberg, E. J. (2021). Bacterial flagellar filament: a supramolecular multifunctional nanostructure. Int. J. Mol. Sci. 22:7521. doi: 10.3390/ijms22147521
Nguyen, L.-T., Schmidt, H. A., Von Haeseler, A., and Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Nodari, C. S., Siebert, M., Matte, U. D. S., and Luís Barth, A. (2017). Draft genome sequence of a GES-5-producing Serratia marcescens isolated in southern Brazil. Braz J Microbiol. 48, 191–192. doi: 10.1016/j.bjm.2016.08.002
NOTA TÉCNICA No. 74/2022-CGLAB/DAEVS/SVS/MS-Agência Nacional de Vigilância Sanitária-Anvisa (2025). Available at: https://www.gov.br/anvisa/pt-br/centraisdeconteudo/publicacoes/servicosdesaude/notas-tecnicas/notas-tecnicas-vigentes/nota-tecnica-no-74-2022-cglab-daevs-svs-ms/view (Accessed October 24, 2024).
Pintado, V., Ruiz-Garbajosa, P., Escudero-Sanchez, R., Gioia, F., Herrera, S., Vizcarra, P., et al. (2022). Carbapenemase-producing Enterobacterales infections in COVID-19 patients. Infect. Dis. 54, 36–45. doi: 10.1080/23744235.2021.1963471
Pournaras, S., Gogou, V., Giannouli, M., Dimitroulia, E., Dafopoulou, K., Tsakris, A., et al. (2014). Single-locus-sequence-based typing of blaOXA-51-like genes for rapid assignment of Acinetobacter baumannii clinical isolates to international clonal lineages. J. Clin. Microbiol. 52, 1653–1657. doi: 10.1128/JCM.03565-13
Prado, G., Mendes, E. T., Martins, R. C. R., Perdigão-Neto, L. V., Freire, M. P., Marchi, A. P., et al. (2022). Phenotypic and genotypic characteristics of a carbapenem-resistant Serratia marcescens cohort and outbreak: describing an opportunistic pathogen. Int. J. Antimicrob. Agents 59:106463. doi: 10.1016/j.ijantimicag.2021.106463
Šiširak, M. (2013). An outbreak of multidrug-resistant Serratia marcescens: the importance of continuous monitoring of nosocomial infections. Acta Med. Acad. 42, 25–31. doi: 10.5644/ama2006-124.67
Silva, C. F. da, Deutschendorf, C., Nagel, F. M., Dalmora, C. H., Santos, R. P. dos, and Lisboa, T. C. (2020). Impact of the pandemic on antimicrobial consumption patterns. Infect. Control Hosp. Epidemiol. 1, 1170–1172. doi: 10.1017/ice.2020.1227
Streling, A. P., Barbosa, P. P., Marcondes, M. F., Nicoletti, A. G., Picão, R. C., Pinto, E. C., et al. (2018). Genetic and biochemical characterization of GES-16, a new GES-type β-lactamase with carbapenemase activity in Serratia marcescens. Diagn. Microbiol. Infect. Dis. 92, 147–151. doi: 10.1016/j.diagmicrobio.2018.05.003
Vera-Leiva, A., Barría-Loaiza, C., Carrasco-Anabalón, S., Lima, C., Aguayo-Reyes, A., Domínguez, M., et al. (2017). KPC: Klebsiella pneumoniae carbapenemasa, principal carbapenemasa en enterobacterias. Rev. Chil. Infectol. 34, 476–484. doi: 10.4067/S0716-10182017000500476
Weissman, S. J., Johnson, J. R., Tchesnokova, V., Billig, M., Dykhuizen, D., Riddell, K., et al. (2012). High-resolution two-locus clonal typing of Extraintestinal pathogenic Escherichia coli. Appl. Environ. Microbiol. 78, 1353–1360. doi: 10.1128/AEM.06663-11
World Health Organization. (2017). Guidelines for the prevention and control of carbapenem-resistant Enterobacteriaceae, Acinetobacter baumannii and Pseudomonas aeruginosa in health care facilities. Geneva: World Health Organization. Available online at: https://apps.who.int/iris/handle/10665/259462 (Accessed September 27, 2022).
Yao, Y., Lazaro-Perona, F., Falgenhauer, L., Valverde, A., Imirzalioglu, C., Dominguez, L., et al. (2017). Insights into a novel blaKPC-2 -encoding IncP-6 plasmid reveal carbapenem-resistance circulation in several Enterobacteriaceae species from wastewater and a hospital source in Spain. Front. Microbiol. 8:1143. doi: 10.3389/fmicb.2017.01143
Zhang, R., Zhou, H. W., Cai, J. C., and Chen, G.-X. (2007). Plasmid-mediated carbapenem-hydrolysing beta-lactamase KPC-2 in carbapenem-resistant Serratia marcescens isolates from Hangzhou, China. J. Antimicrob. Chemother. 59, 574–576. doi: 10.1093/jac/dkl541
Zingg, W., Soulake, I., Baud, D., Huttner, B., Pfister, R., Renzi, G., et al. (2017). Management and investigation of a Serratia marcescens outbreak in a neonatal unit in Switzerland – the role of hand hygiene and whole genome sequencing – R1, ARIC-D-17-00143. Antimicrob. Resist. Infect. Control 6:125. doi: 10.1186s13756-017-0285-x
Keywords: Serratia marcescens, blaKPC, single-locus sequence typing, IncP6 plasmid, fliC
Citation: da Rocha LF, Rodrigues GM, de Oliveira GS, Echevarria AD, Wink P, Volpato F, Berdichevski M, Lutz L, Pereira DC, Barth AL and Martins AF (2025) Molecular epidemiology of a carbapenem-resistant Serratia marcescens outbreak during the COVID-19 pandemic. Front. Microbiol. 16:1525543. doi: 10.3389/fmicb.2025.1525543
Edited by:
Maria Jorge Campos, Polytechnic Institute of Leiria, PortugalReviewed by:
Jorge Aníbal Reyes, Universidad San Francisco de Quito, EcuadorIvan Ivanov, National Center of Infectious and Parasitic Diseases (NCIPD), Bulgaria
Copyright © 2025 da Rocha, Rodrigues, de Oliveira, Echevarria, Wink, Volpato, Berdichevski, Lutz, Pereira, Barth and Martins. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Afonso Luís Barth, YWxiYXJ0aEBoY3BhLmVkdS5icg==
†These authors have contributed equally to this work and share first authorship