 Nubwa Medugu1,2*
Nubwa Medugu1,2* Mabel Kamweli Aworh3,4
Mabel Kamweli Aworh3,4 Kenneth Iregbu2
Kenneth Iregbu2 Philip Nwajiobi-Princewill2
Philip Nwajiobi-Princewill2 Dawn M. Hull4Lyndy Harden4
Dawn M. Hull4Lyndy Harden4 Pallavi Singh5
Pallavi Singh5 Stephen Obaro6
Stephen Obaro6 Abiodun Egwuenu7Faith Adeboye8
Abiodun Egwuenu7Faith Adeboye8 Ruth Egah8
Ruth Egah8 Leonard Uzairue9
Leonard Uzairue9 Yahaya Mohammed10
Yahaya Mohammed10 Nwafia Ifeyinwa11
Nwafia Ifeyinwa11 Siddhartha Thakur4
Siddhartha Thakur4- 1Department of Medical Microbiology and Immunology, Nile University of Nigeria, Abuja, Nigeria
- 2Department of Medical Microbiology and Parasitology, National Hospital Abuja, Abuja, Nigeria
- 3Department of Biological and Forensic Sciences, Fayetteville State University, Fayetteville, NC, United States
- 4Department of Population Health and Pathobiology, College of Veterinary Medicine, North Carolina State University, Raleigh, NC, United States
- 5Department of Biological Sciences, Northwestern Illinois University, DeKalb, IL, United States
- 6Department of Paediatric Infectious Diseases, University of Alabama at Birmingham, Birmingham, AL, United States
- 7Nigeria Centre for Disease Control, Abuja, Nigeria
- 8International Foundation against Infectious Disease in Nigeria, Abuja, Nigeria
- 9Department of Medical Laboratory Science, Federal University, Oye, Nigeria
- 10Department of Microbiology, Usmanu Danfodiyo University Sokoto, Sokoto, Nigeria
- 11Department of Medical Microbiology, University of Nigeria, Nsukka, Nigeria
Background: Multidrug-resistant Escherichia coli poses a critical public health threat in Nigeria, where limited genomic surveillance hinders the understanding of virulence-resistance interplay.
Methods: This cross-sectional study employed whole-genome sequencing to characterize 107 MDR-E isolates from a Nigerian tertiary hospital (2019–2020), analyzing virulence genes, mobile genetic elements (MGEs), phylogroups, sequence types (STs), pathotypes, and antimicrobial resistance (AMR).
Results: We identified 2,021 virulence genes across nine functional categories, dominated by immune evasion (terC, 96.3%), adherence (fimH, 86%), and iron acquisition (fyuA, 63.6%). Strikingly, 81.3% of virulence genes were linked to MGEs, including MITEEc1 (75.7% of isolates) and IS30 (56.1%), with IncFII (17.8%) and Col156 (12.1%) plasmids co-harboring virulence (e.g., traJ/traT, senB) and AMR genes (e.g., blaTEM-1B). Phylogroup B2 (32.7%) dominated, exhibiting high resistance to ampicillin (97.1%) and emerging meropenem resistance (11.4%). Globally disseminated STs (ST131, ST410, ST648) carried significantly more diverse virulence genes than minor clones (p = 0.028) and were strongly associated with double-serine QRDR mutations (gyrA_S83L: 97.6%, parC_S80I: 97.6%), which correlated with more virulence genes (24.2 vs. 22.3 genes) and resistance (MAR index: 0.7 vs. 0.5) compared to minor clones. Notably, 92% (61/66) of high-risk clones harbored these mutations, versus 57% (21/37) of low-risk clones, suggesting a fitness advantage enabling major clones to sustain larger genetic cargoes. Pathotyping revealed 54.2% as extraintestinal pathogenic E. coli (ExPEC), with 72.4% of these being uropathogenic (UPEC) and 5.2% ExPEC/EAEC hybrids, alongside 43.9% atypical ExPEC strains. Hierarchical clustering demonstrated phylogroup B2’s genetic diversity and co-localization of plasmid-borne virulence/AMR genes.
Discussion: These findings underscore Nigeria’s MDR-E crisis, driven by MGE-facilitated gene transfer, hybrid pathotypes, and globally disseminated high-risk clones harboring double-serine QRDR mutations. There is continued need for robust genomic surveillance, stringent infection control measures, enhanced antibiotic stewardship, and exploration of antivirulence strategies (e.g., targeting fimH or yeh) to curb the spread of these highly adaptable pathogens in resource-limited settings and beyond.
1 Introduction
Multidrug-resistant Escherichia coli (MDR-E) poses a significant public health challenge, particularly in low- and middle-income countries (LMICs), where it contributes substantially to morbidity and mortality in severe infections such as sepsis, meningitis, and urinary tract infections (UTIs) (Aiken et al., 2023; Medugu et al., 2022). The global rise of MDR-E is exacerbated by the rapid dissemination of antimicrobial resistance (AMR) genes and virulence factors, often facilitated by mobile genetic elements (MGEs) such as plasmids, transposons, and insertion sequences (Partridge et al., 2024; Schmidt and Hensel, 2004; World Health Organization, 2022).
Pathogenic E. coli strains differ from commensal strains by the presence of virulence genes, which encode factors involved in adherence, colonization, invasion, and immune evasion (Nojoomi and Ghasemian, 2019). Extraintestinal pathogenic E. coli (ExPEC) are particularly concerning due to their extensive repertoire of virulence-associated factors, including adhesins, toxins, iron acquisition systems, and immune modulators, many of which are carried on MGEs (Nojoomi and Ghasemian, 2019; Sora et al., 2021). Despite their critical role in bacterial pathogenicity, the mechanisms by which MGEs contribute to the spread of virulence and AMR genes—such as horizontal gene transfer—remain poorly understood, particularly in resource-limited settings like Nigeria (Iheanacho and Eze, 2022; Morales et al., 2023; Partridge et al., 2024).
Phylogenetic diversity further complicates the epidemiology of E. coli, as different phylogroups exhibit varying degrees of virulence and AMR profiles. For instance, phylogroup B2 is frequently associated with extraintestinal infections, while other phylogroups are more commonly found in commensal gut flora (Clermont et al., 2019; Nojoomi and Ghasemian, 2019). In Nigeria, the burden of MDR-E is exacerbated by limited surveillance, inadequate antimicrobial stewardship, and widespread misuse of antibiotics, driven by factors such as limited healthcare infrastructure and over-the-counter antibiotic availability (Okeke et al., 1999). Although previous studies have characterized AMR trends in E. coli, there remains a gap in understanding the genomic epidemiology of virulence factors, their association with MGEs, sequence types, and pathotypes in this region. This study seeks to address this gap by leveraging whole-genome sequencing (WGS) and in silico analyses to characterize MDR-E isolates from Nigerian patients. Specifically, classify phylogroups, examine the distribution of virulence genes, pathotypes, and identify associated MGEs, while assessing their relationship with AMR patterns and multilocus sequence types (MLST). By providing essential insights into the genomic landscape of MDR-E in Nigeria, this study seeks to inform tailored infection prevention strategies, enhance antimicrobial stewardship programs, and ultimately improve patient outcomes in resource-limited settings.
2 Materials and methods
2.1 Study design, sample collection, transport, and initial processing
This cross-sectional study was undertaken at the National Hospital Abuja (NHA), a 425-bed tertiary care center in Nigeria. A total of 107 unique E. coli isolates, collected from clinical specimens between March 2019 and September 2020, were included. All specimens were obtained according to standard clinical protocols and transported promptly to the microbiology laboratory (Iregbu et al., 2013). Urine specimens were collected as midstream clean-catch. To assess for significant bacteriuria, upon receipt, urine samples were inoculated unto Brilliance UTI Agar (Oxoid, Basingstoke, United Kingdom) using a calibrated 1 μL loop. Plates were incubated at 35.5°C for 18–24 h in ambient air.
Blood specimens were collected using weight-based guidelines, with two sets of 8–10mL drawn for adult patients and age-appropriate volumes for pediatric patients. Samples were inoculated into BacT/Alert culture bottles—Adult Aerobic, Adult Anaerobic, or Pediatric Plus, as appropriate (bioMérieux, Marcy l’Etoile, France)—and incubated in the BacT/Alert automated system. Bottles were continuously monitored for microbial growth for up to 5 days, with positive signals prompting immediate subculture for organism isolation and identification. Bottles flagged as positive were subcultured onto Sheep Blood Agar, Chocolate Agar, and MacConkey Agar. Plates were incubated at 35.5°C for 18–24 h, with Chocolate Agar placed in a 5% CO2 atmosphere to support fastidious organisms. All negative bottles were incubated for the full monitoring 5-day period before being discarded as no growth (Medugu et al., 2023). Other specimen types (e.g., wound swabs, sputum, and body fluids) were inoculated on Sheep Blood Agar, Chocolate Agar, and MacConkey Agar, with incubation conditions as described above. All plates were examined after 18–24 h for colony.
2.2 Identification and antimicrobial susceptibility testing of Escherichia coli isolates
All presumptive Escherichia coli isolates were subjected to species-level identification using the VITEK 2 Compact system (bioMérieux, Marcy l’Etoile, France), employing the GN ID card, which contains a panel of biochemical tests specific for Gram-negative bacteria. Bacterial suspensions were prepared in sterile saline to a 0.5 McFarland turbidity standard using a Densichek turbidity meter and then loaded into the VITEK 2 instrument according to the manufacturer’s instructions. Antimicrobial susceptibility testing was performed on confirmed isolates using the VITEK 2 AST-N280 and AST-N281 cards. Quality control was conducted using E. coli ATCC® 25922. The AST results were automatically generated by the VITEK system and interpreted using the Clinical and Laboratory Standards Institute (CLSI) guidelines, M100, 30th Edition (Clinical and Laboratory Standards Institute, 0000). Interpretations included categorization of each isolate as susceptible, intermediate, or resistant based on minimum inhibitory concentration breakpoints. All results were reviewed and validated by trained clinical microbiologists. Minimum inhibitory concentration results were validated using E. coli ATCC 25922 as a quality control strain. MDR was defined as resistance to three or more antimicrobial classes, consistent with established criteria (Magiorakos et al., 2012). The isolates were shipped to Thakur Molecular Epidemiology laboratory at North Carolina State University (NC State), United States where they underwent microdilution assays with the Gram-negative Sensititre (CMV3AGNF) plate (Trek Diagnostic Systems, OH). The plates were read using Sensititre ARIS and interpreted according to CLSI M100 30th Edition guidelines (Clinical and Laboratory Standards Institute, 0000). E. coli ATCC 25922 was used as a control. All isolates were stored at −80°C in 50% glycerol to preserve viability before further analyses. Relevant sociodemographic and clinical data, including age, sex, diagnosis, and specimen source, were extracted from patient medical records.
2.3 Whole-genome sequencing of Escherichia coli isolates, virulence gene detection, association with mobile genetic elements, and AMR gene detection in study Escherichia coli isolates
WGS was performed on all 107 E. coli isolates to characterize their genomic profiles. Genomic DNA was extracted using the MasterPure Complete DNA and RNA Purification Kit (Epicentre Technologies, Madison, WI), ensuring high-quality DNA for downstream applications. DNA quality was initially assessed using the NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, United States). Purity was evaluated by measuring the absorbance ratio at 260 and 280 nm, with a ratio of approximately 1.8 considered indicative of high-purity DNA suitable for downstream applications. Following this, DNA concentration was quantified using the Qubit 4.0 Fluorometer (Thermo Fisher Scientific) with the dsDNA High Sensitivity (HS) Assay Kit, according to the manufacturer’s instructions. Sequencing libraries were prepared using the Nextera XT DNA Sample Prep Kit (Illumina, San Diego, CA, United States). Sequencing was conducted on the Illumina MiSeq platform (Illumina, San Diego, CA, United States) with a 2 × 250 paired-end configuration, generating high-depth coverage for each isolate. Raw sequencing reads were processed using CLC Genomics Workbench v9.4 (Qiagen, Hilden, Germany) to trim low-quality bases and remove adapter sequences.
The high-quality Illumina paired-end reads generated were assembled de novo into the draft genome sequence for each isolate using SPAdes assembler v.3.13.1 which is optimized for bacterial genomes (Bankevich et al., 2012). The resulting draft genome sequences were subjected to rigorous quality assessment using QUAST (Quality Assessment Tool for Genome Assemblies) (Gurevich et al., 2021). Metrics such as contig number, N50, and genome coverage were evaluated to ensure the reliability and completeness of the assemblies and uploaded to NCBI’s Bio project ID PRJNA293225.
The virulence genes identified in the E. coli isolates were systematically categorized into nine functional categories: adherence, biofilm, and colonization; invasion and intracellular survival; toxin production and secretion; iron acquisition, uptake, transport, and storage; immune evasion and modulation; nutrient acquisition and metabolism; stress response and regulation; motility and chemotaxis; and cell wall and membrane biogenesis. To investigate the genetic context of these virulence factors, we identified MGEs associated with virulence genes using Mobile Element Finder (version 1.0.3, released on 2020-10-09) alongside its database version 1.0.2 (released on 2020-06-09) (Johansson et al., 2021). Each virulence gene was classified according to its location on plasmids, insertion sequences, other MGEs, or categorized as not associated, based on the MGE output. Plasmids were detected employing PlasmidFinder-2.0, with a threshold of at least 95% identity and a minimum coverage of 60%, utilizing draft genome assemblies (Carattoli et al., 2014). MGE detection was conducted with a threshold criterion of 95% sequence similarity and a minimum coverage of 60%, ensuring accurate identification of MGEs linked to virulence genes. Prediction of AMR was conducted by using Mobile Element Finder v1.0.3 (2020-10-09) and selecting Acquired Antimicrobial Resistance genes (ResFinder) (Johansson et al., 2021). Chromosomal point mutation data were retrieved from publicly available whole-genome sequences we had earlier deposited in the NCBI BioProject PRJNA293225 using the NCBI Pathogen Detection Isolates Browser. Mutations in the quinolone resistance-determining regions (QRDRs) of gyrA (Ser83) and parC (Ser80) were identified using the NCBI’s automated AMRFinderPlus tool (v3.10.23) with default parameters (Feldgarden et al., 2021).
2.4 Analysis of virulence gene carriage by sequence type (ST) and QRDR mutation prevalnece
Multilocus sequence typing (MLST) was performed using seven conserved Escherichia coli housekeeping genes (adk, fumC, gyrB, icd, mdh, purA, and recA) as previously described (Wirth et al., 2006) Sequence types (STs) and clonal complexes (CCs) were identified via the PubMLST Achtman scheme with STs assigned to isolates exhibiting 100% allele identity to established MLST databases. Isolates lacking perfect allele matches were classified as “unknown STs”(Larsen et al., 2012). The 35 identified STs were categorized as major or minor clones based on their global epidemiological prevalence and association with AMR outbreaks (Baquero et al., 2013; Manges et al., 2019; Santos et al., 2020b; Shaik et al., 2017).
To investigate the relationship between multidrug resistance and virulence profiles, we analyzed the set of E. coli STs represented by more than one isolate ensure robust statistical estimates, resulting in a final dataset of 20 STs. For each ST, the following variables were evaluated: (1) MAR Index (Multidrug Antibiotic Resistance Index, averaged across isolates), (2) Number of AMR genes (average count), (3) Number of virulence genes (average count), and (4) Virulence Variety (total unique virulence factors per ST). Non-parametric Spearman’s rank correlation analysis was employed to assess pairwise associations between variables, as the data exhibited non-normal distributions. Tied ranks were resolved using averaging, and significance was evaluated via a two-tailed test with an α-level of 0.05. To investigate relationship between major and minor STs with double QRDR mutations, the distinct STs classified as either major or minor clones were evaluated. Each ST was assigned to one of six categories according to the percentage of isolates exhibiting double mutations (0, 0.5, 0.75, 0.97, 0.98, and 1.0). A cross−tabulation of clone type by these double−mutation categories was constructed. Fisher’s exact test was used to assess the statistical significance of the association between clone type and double−mutation category. Statistical computations were performed in R v4.3.1.
2.5 Pathotyping of Escherichia coli
Isolates were screened for ExPEC markers according to Johnson et al. (2003). Briefly, the presence of five established virulence genes were assessed: pap, sfa/foc, afa/dra, iutA, and kps (Supplementary Table 6). An isolate was classified as ExPEC if two or more of these markers were present. Next, ExPEC strains were assessed using the Spurbeck/Hooton–Mobley scheme (Spurbeck et al., 2012), which categorizes isolates as UPEC if they possess fyuA along with at least two of the following three genes: chuA, yfcV, or vat. In the two schema used, each isolate was assessed for the enteroaggregative E. coli (EAEC) regulator gene aggR and when found in an isolate already tagged an ExPEC, was labeled an ExPEC/EAEC hybrid as reported previously (Lara et al., 2017). When an isolate possessed multiple ExPEC-linked genes but did not fulfill either the Johnson criteria or the Spurbeck/HM definition, it was designated “atypical ExPEC,” following the approach described by Sarowska et al. (2019). Lastly, all strains were also screened for hallmark genes of other diarrheagenic E. coli (DEC) pathotypes, including eae, bfp, stx, ipaH, and elt/est, to rule out EPEC, EHEC, EIEC, and ETEC.
2.6 Phylogroups and phylogeny
To determine the O:H serotypes of the E. coli isolates, in silico typing was performed using SerotypeFinder 2.0 (Joensen et al., 2015), with a selected threshold of 90% identity and 60% total serotype gene length. For phylogenetic analysis, single nucleotide polymorphisms (SNPs) were identified using CSI Phylogeny 4.1 (Centre for Genomic Epidemiology) with the E. coli reference sequence NZ_CP028166.1. SNP calling parameters included a minimum depth of coverage ≥ 10 × , SNP quality score ≥ 30, mapping quality score ≥ 25, and Z score ≥ 1.96 (Kaas et al., 2014). The resulting SNP data were used to construct phylogenetic trees, which were visualized and annotated using iTOL version 6 (Interactive Tree of Life).
2.7 Data collection and analysis
Statistical analysis was conducted using STATA software (StataCorp, 2019) on data organized in Microsoft Excel. Descriptive statistics, including frequencies and proportions, were used to summarize the phenotypic and genotypic characteristics of the E. coli isolates. To assess the relationship between phylogroups and sample types, Fisher’s exact test was performed using the Mehta-Patel network algorithm, which is robust for analyzing associations in sparse contingency tables with small, expected cell counts. Differences in the number of virulence genes across phylogroups were evaluated using the non-parametric Kruskal-Wallis test, while variations in resistance percentages for 12 antimicrobials across eight phylogroups were analyzed using Fisher’s exact test. Where applicable, p-values were calculated to determine the statistical significance of observed associations. Statistical significance was set at a threshold of p < 0.05.
2.8 Ethics approval and consent to participate
This study was approved by the Ethics Review Board of the National Hospital Abuja (NHA) under approval number NHA/EC/033/2018. To ensure the protection of patient privacy and confidentiality. A two-stage data encryption process was implemented to ensure that patient information remained strictly limited to the principal investigator and authorized research personnel. All data was de-identified and securely stored in compliance with ethical guidelines.
The bacterial isolates analyzed in this study were obtained from anonymized clinical specimens routinely submitted to the NHA laboratory for diagnostic purposes. All procedures adhered to the ethical guidelines and regulations established by the Ethics Review Board, ensuring the protection of patient rights and confidentiality throughout the research process.
3 Results
3.1 Characteristics of Escherichia coli isolates
A total of 107 E. coli isolates were recovered from clinical specimens obtained from 107 patients. The isolates were predominantly sourced from urine samples (68%), followed by blood (22%) and wound swabs (10%). The patients spanned a broad age range, from 2 months to 84 years, with a mean age of 45 years.
Comprehensive analysis identified 2,639 virulence genes across the isolates (Supplementary Table 1). When variants of the same gene were grouped (e.g., yehA, yehB, yehC, yehD grouped as yeh) the total number of distinct virulence genes was 2,021 (Supplementary Table 2). The number of virulence genes per isolate varied significantly, ranging from 7 to 48 (grouped format) and 7 to 71 (ungrouped format). As illustrated in Figure 1, the distribution of virulence factors (ungrouped format) revealed that 28% of isolates carried 21-25 virulence genes, followed closely by 23% of isolates (25 isolates) with 26-30 virulence genes.
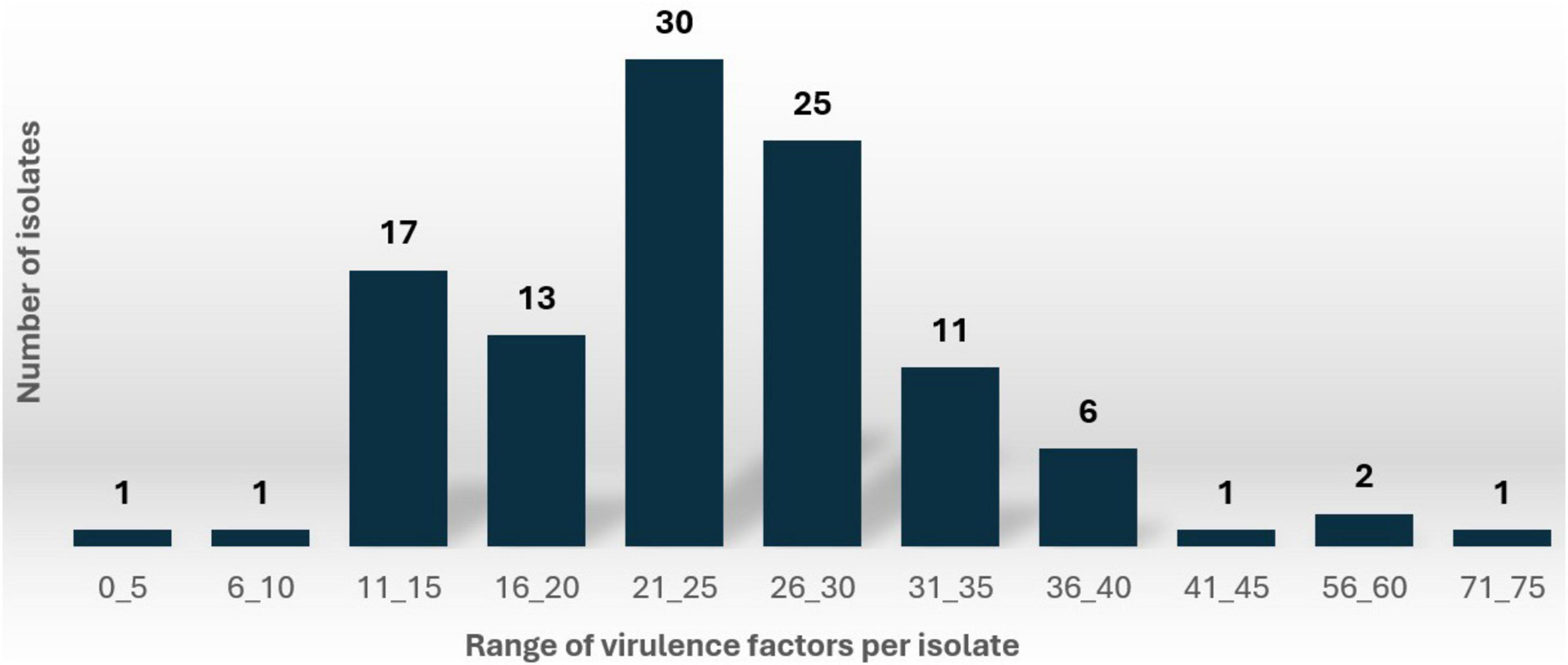
Figure 1. Distribution of isolates by number of virulence per E. coli isolate.
3.2 Functional categorization of virulence genes
The 109 distinct virulence genes identified were categorized into nine functional groups (Table 1). The majority of virulence factors were associated with immune evasion and modulation (1,573 genes), followed by adherence, biofilm formation, and colonization (1,380 genes), and cellular invasion and intracellular survival (821 genes). Notably, some virulence factors, such as the yeh gene complex, exhibited multifunctional properties, contributing to adherence, biofilm formation, and colonization; invasion and intracellular survival; and immune evasion and modulation.
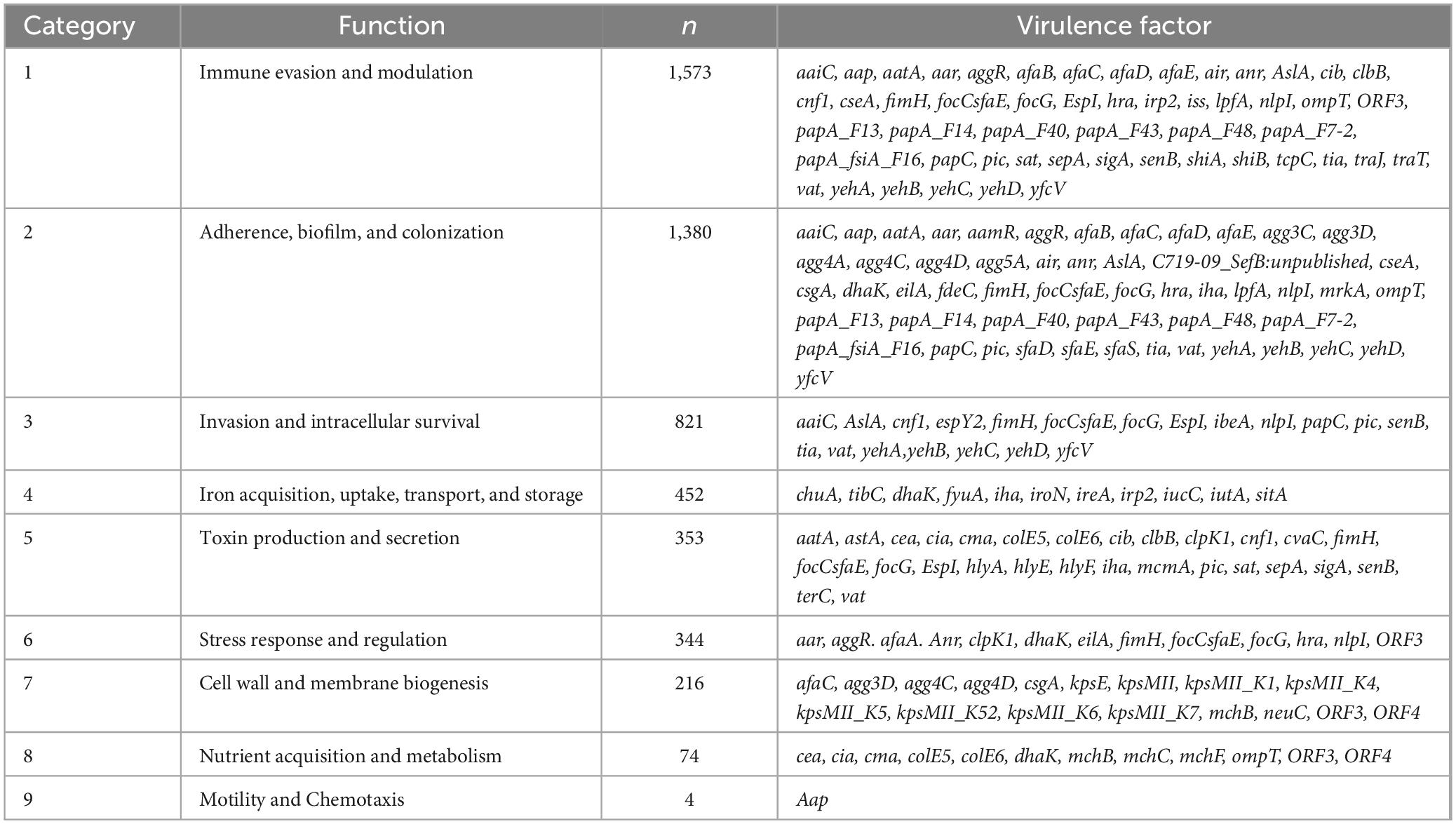
Table 1. Types of Virulence factors detected in E. coli isolates from clinical samples.
3.3 Frequency and types of virulence factors detected in Escherichia coli isolates
The 107 E. coli isolates analyzed in this study displayed a diverse range of virulence genes with a total of 2,021 virulence genes identified or 2,639 when including gene variants separately. The most prevalent was the yeh gene complex, which contributes to adherence, invasion, and immune evasion, and was detected in all isolates (100%). Specific variants of the yeh complex were present at high frequencies, including yehC (97.2%), yehD (96.3%), yehB (93.5%), and yehA (87.9%). The complete distribution of gene alleles is detailed in Supplementary Table 1, with an abridged version provided in Table 2.
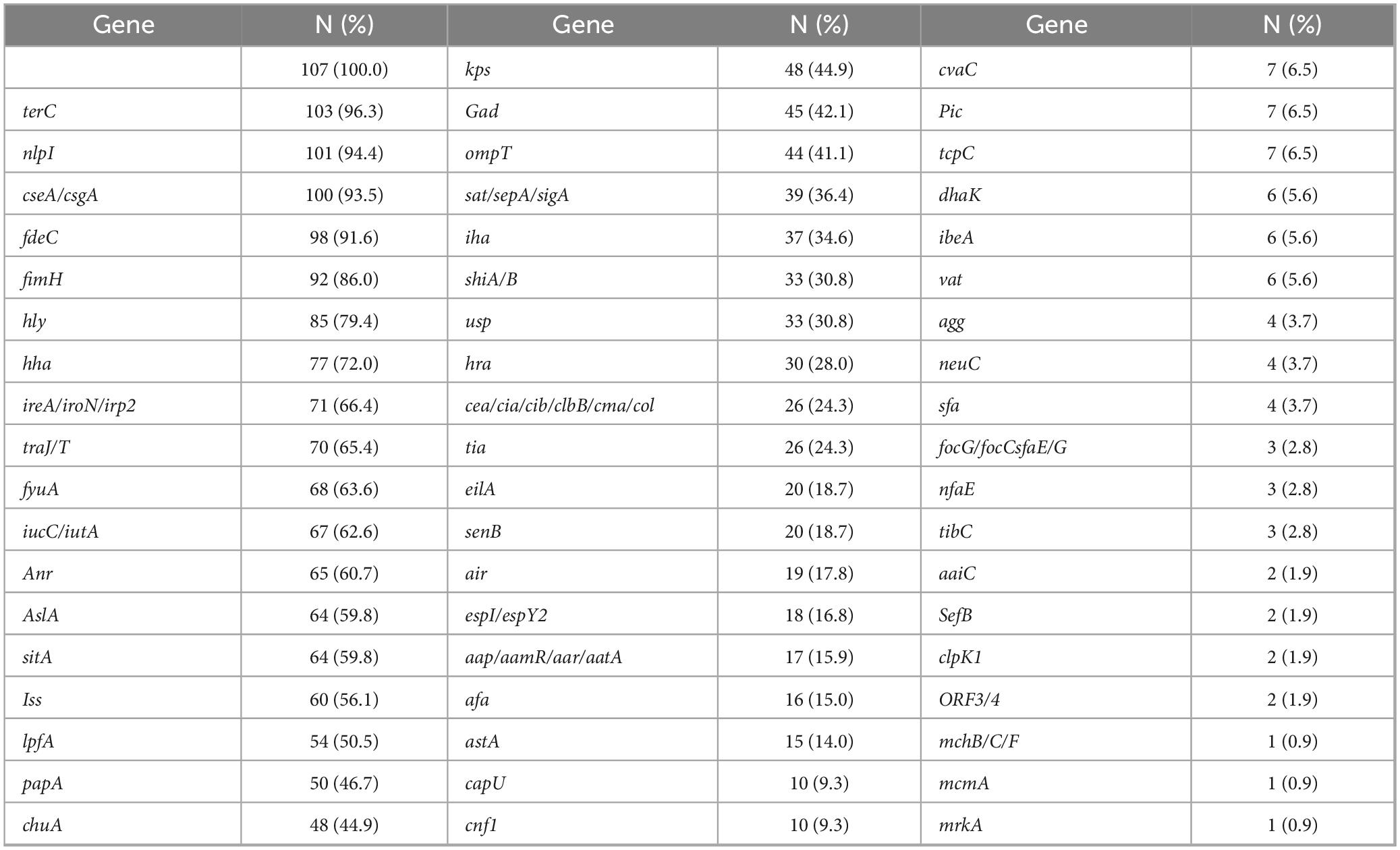
Table 2. Prevalence of virulence genes in 107 E. coli isolates.
Other frequently detected virulence factors were nlpI (immune evasion, adherence, intracellular survival, and stress response, 94.4%), cseA/csgA (immune evasion, adherence, cell wall and membrane biogenesis, 93.5%), terC (toxin production and secretion, 96.3%), and fdeC (adherence, biofilm, and colonization, 91.6%). Notably, the fimH gene, associated with our functional categories 1, 2, 3, 5, and 7, was present in 86.0% of the isolates. Additionally, iron acquisition-related genes from functional category 4, such as fyuA (62.6%), irp2 (59.8%), iucC (62.6%), and iutA (60.7%) were prominent among the virulence factors.
3.4 Distribution and role of virulence genes with mobile genetic elements (MGEs)
In the 107 E. coli isolates analyzed, virulence genes were found located on 48 unique MGEs, including Integrative Conjugative Elements (ICE), miniature inverted-repeat transposable elements (MITEs), insertion sequences, transposons, and plasmid incompatibility types. The classes and most prevalent MGE types are shown in Figure 2 with 2A depicting the classes and 2B depicting the specific classes.
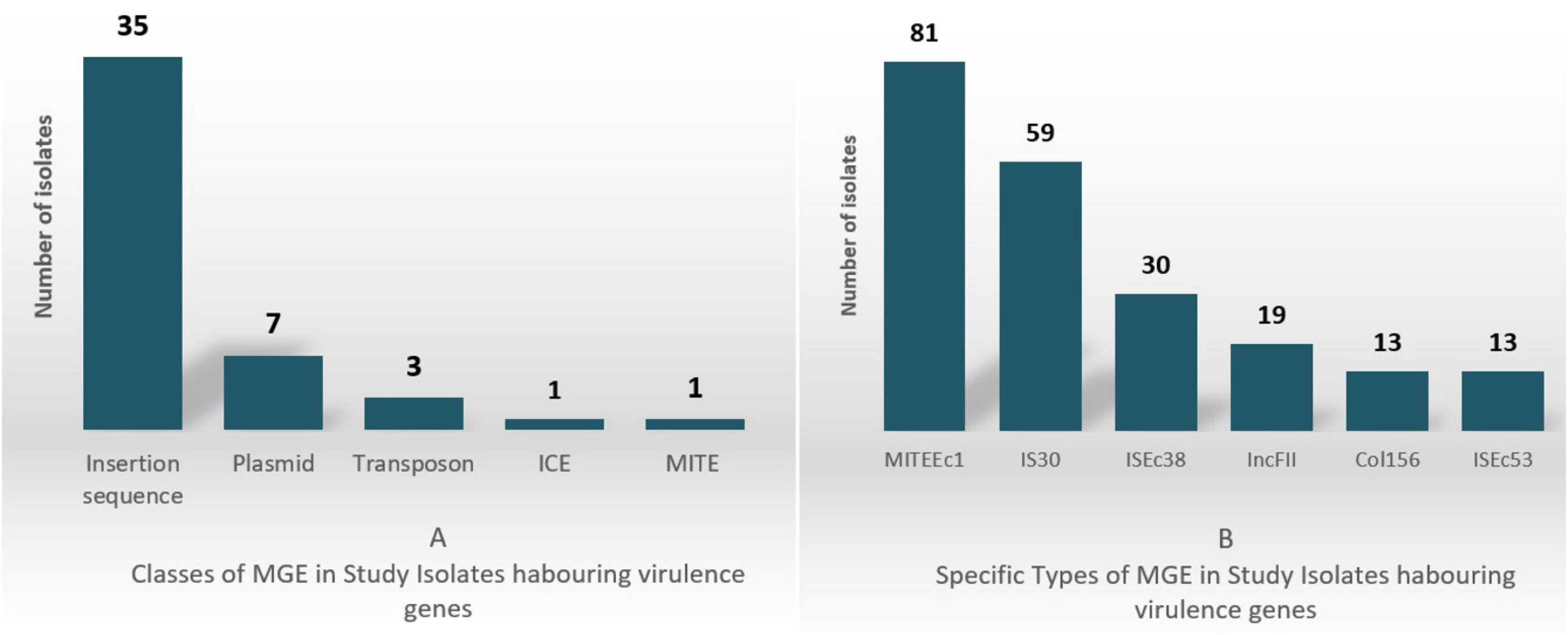
Figure 2. MGEs and virulence gene associations. (A) MGE classes; (B) Specific MGE types.
MITEEc1 and IS30 were the most common, detected in 81 and 59 isolates, respectively. MITEEc1 frequently carried the immune evasion gene terC (46/55 instances), while IS30 was found alongside the SPATE genes sat/sepA/sigA (15/20 instances). ISEc38, identified in 30 isolates, often contained genes like fyuA (13 instances) and irp2 (11 instances), which are involved in iron acquisition and bacterial survival.
Virulence genes involved in immune evasion and intracellular survival (e.g., yeh, yehB, nlpI) were more frequently located on MGEs compared to adherence-related genes like fimH, which were predominantly found on the chromosome. This distribution suggests a potential selective advantage for immune evasion genes in mobile forms, facilitating bacterial survival in host environments.
A total of 27 IncF-type plasmids were identified as carriers of virulence genes. Among these, IncFII plasmids were the most prevalent, found in 19 isolates, with the conjugative transfer genes traJ/traT present in 16 of these. Additionally, Col156 plasmids were detected in 13 isolates, all of which carried the senB gene, a key factor in immune evasion and cellular invasion. Supplementary Table 3 shows in more detail the virulence genes along MGE association. Entries indicate presence (1) or absence (0) of MGE linkage, and MGE type (e.g., ISEc38, ICKp1) if identified.
Some plasmids simultaneously harbored both AMR genes and virulence genes, emphasizing their dual role in pathogenicity and resistance dissemination. Some of which include NHA068 with IncFIC plasmid having both the betalactam resistance gene blaTEM-1B and the virulence gene traT. A representative example from NHA013 shown in the plasmid annotation (Figure 3).
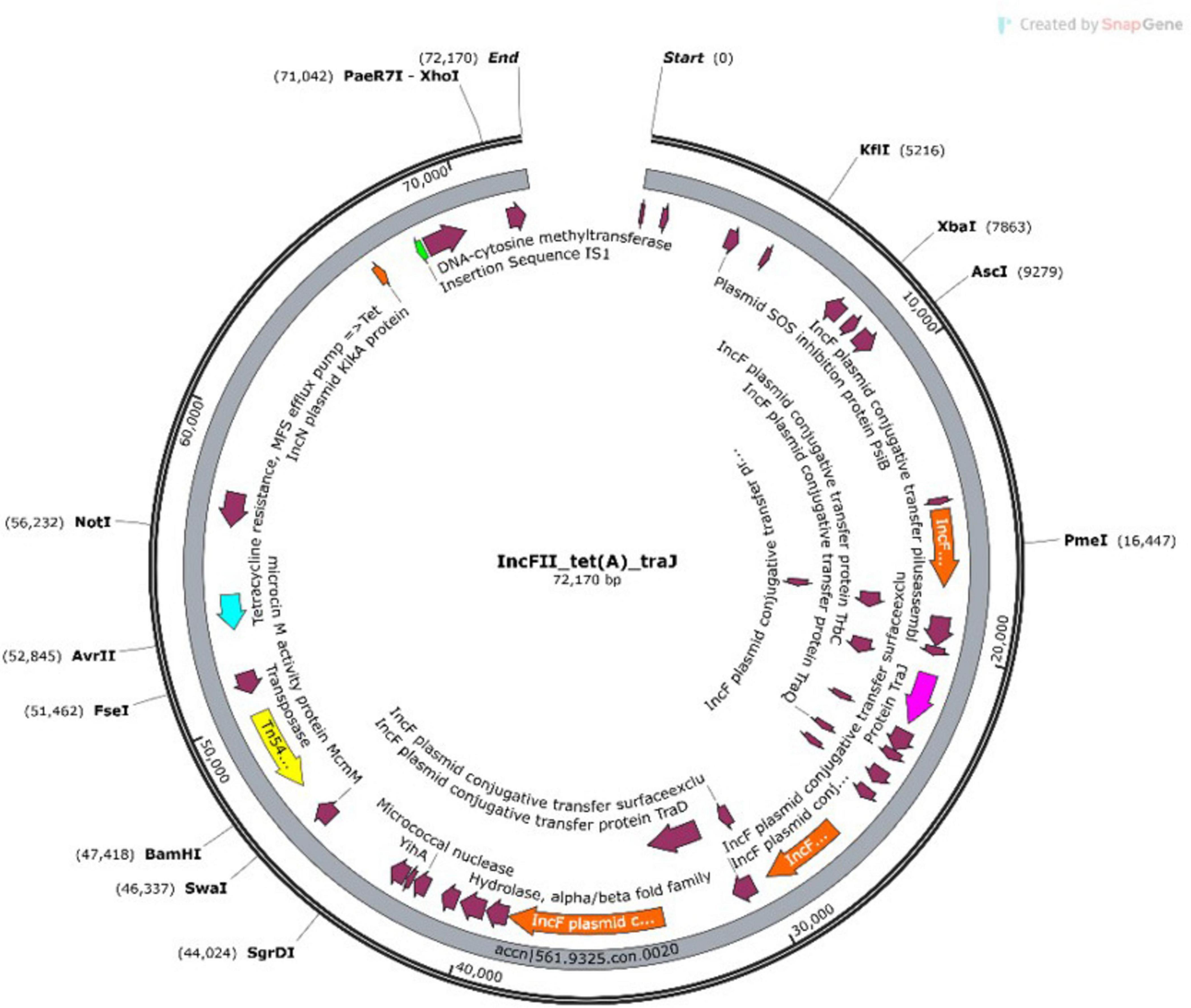
Figure 3. Annotation for NHA013 IncFLL plasmid.
The annotated map highlights an IncFII plasmid carrying a transposon (Tn5403), virulence genes (traJ/traT), and the resistance gene tet(A). These features were identified on contigs 19 and 20. The detailed annotation of contig 20 provides insight into the genetic architecture of the plasmid and its potential role in mediating multidrug resistance and virulence.
3.4.1 Multi-gene clusters on MGEs
Some insertion sequences, such as ISSpu2 and IS5, hosted clusters of virulence genes within single isolates. For example, isolate NHA101 carried ISSpu2 with the genes aatA, agg4A, aggR, and ORF3, as well as an IS5 insertion sequence containing eight virulence genes: ORF4, aatA, agg4A, afaD, aap, agg4C, agg4D, and ORF3.
3.5 Correlation analysis of STs, virulence genes, AMR genes, MAR index, and comparative analysis of major vs. minor clones with DRQR mutations
3.5.1 AMR gene landscape and MAR index
As we previously reported for this dataset (Medugu et al., 2022), 57 distinct AMR genes were found for strains in this study (Supplementary Table 4). The predominant AMR genes detected were β-lactam resistance genes with 13 different variants including the classical ESBL blaCTX–M type (7 types), and AmpC producing blaCMY type (1). Other β-lactam genes found included blaOXA (2) and blaTEM (2). Also reported were blaNDM–1 (1) and blaNDM–5 (4). There were ten genes detected for aminoglycoside resistance including aac(3′)-IIa and aac(3)-Iie found in 30% and 29% of isolates respectfully. We also reported genes conferring erythromycin, macrolide, phenicol, tetracycline, sulfonamide, and trimethoprim resistance (Supplementary Table 4). The multiple antibiotic resistance index (MAR index) ranged from 0.1 to 1 with a mean of 0.6 as detailed in Supplementary Table 4.
3.5.2 Spearman’s correlation analysis
Spearman’s correlation analysis revealed significant associations between multidrug resistance and virulence determinants. A robust positive correlation was observed between the MAR Index and the Number of AMR genes (ρ = 0.813, p < 0.001), indicating that STs with higher multidrug resistance indices harbored greater AMR gene diversity. Similarly, the Number of virulence genes demonstrated a strong positive correlation with Virulence Variety (ρ = 0.741, p < 0.001), underscoring that STs with higher virulence gene counts also exhibited broader functional diversity in virulence factors. Moderate correlations were identified between AMR genes and Virulence Variety (ρ = 0.598, p = 0.006), suggesting a co-occurrence of antibiotic resistance and virulence diversification. The MAR Index exhibited a marginal, non-significant trend with Virulence Variety (ρ = 0.432, p = 0.057), hinting at a potential interplay between multidrug resistance and virulence complexity that warrants further investigation. No significant associations were detected between the MAR Index and Number of virulence genes (ρ = 0.221, p = 0.351) or between AMR genes and virulence gene counts (ρ = 0.128, p = 0.594).
3.5.3 Major vs. minor clones and double QRDR mutations
Analysis of quinolone resistance-determining region (QRDR) mutations revealed a striking predominance of gyrA_S83L and parC_S80I double-serine mutations among high-risk clones. A total of 95.2% (40/42) of major (high-risk) clones harbored both mutations, compared to 68.9% (42/61) of minor (low risk) clones (Table 3; Supplementary Table 7). Notably, 21.3% (13/61) of minor clones lacked QRDR mutations entirely, while only 4.8% (2/42) of major clones exhibited this phenotype. Major clones demonstrated significantly higher antimicrobial resistance (MAR index: 0.7 vs. 0.5) and virulence gene carriage (24.2 vs. 22.3 genes/isolate) compared to minor clones. This near-universal presence of double-serine mutations in major clones may correlate with their enhanced capacity to co-maintain resistance and virulence determinants without apparent fitness trade-offs (p > 0.05).
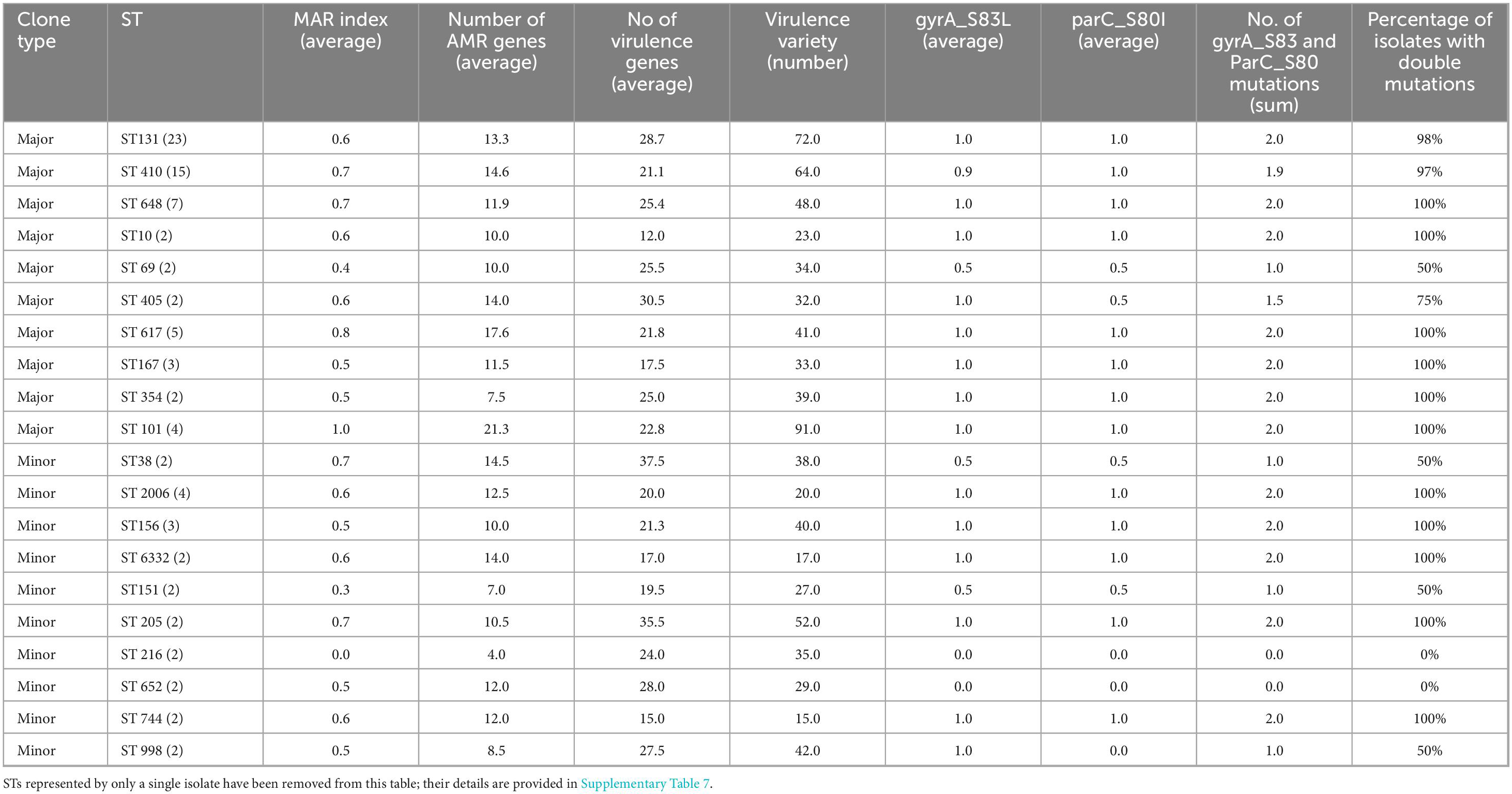
Table 3. Virulence and resistance characteristics of major and minor E. coli sequence types and qRDR double-serine mutations.
A Fisher’s exact test revealed a statistically significant association between clone type and the Single/Double/Nil outcome (p < 0.0001). Data in Table 3 and further detailed in Supplementary Table 4. Specifically, among Major clones, the vast majority displayed double mutation (61/65, 94%), with very few exhibiting the Single (3/65, 5%) or Nil (1/65, 2%) phenotype. In contrast, Minor clones were significantly less likely to present double mutation (21/38, or 55%) and were more frequently associated with the Nil outcome (12/38, or 32%), with a small proportion showing the Single phenotype (5/38, or 13%). Double mutations were found in an average of 88% of major clones and 56% of minor clones and Fisher’s exact test for the presence and absence and absence of double mutations demonstrated a statistically significant association between clone type and the percentage of isolates with double mutations (p = 0.0258). Supplementary Table 7 details the correlations between the major and minor clones alongside AMR genes, Virulence genes, and specific QRDR mutations.
3.6 Pathotypes of Escherichia coli
All 107 E. coli isolates were subjected to Insilco pathotyping. Of these, 58 (54.2%) fulfilled the Johnson criteria for ExPEC. Within this “typical ExPEC” subset, 42 (72.4% of 58) also satisfied the Spurbeck/Hooton–Mobley scheme and were thus classified as UPEC; 13 (22.4%) were deemed ExPEC−only (i.e., meeting Johnson criteria but not the Spurbeck/HM definition); and 3 (5.17%) carried the EAEC regulator gene (aggR), making them ExPEC/EAEC hybrids.
A further 47 isolates (43.93%) were designated “atypical ExPEC” because they possessed various ExPEC−associated genes yet did not meet either the Johnson or the Spurbeck/HM thresholds. One isolate (0.93%) fulfilled only the Spurbeck/HM definition but did not have the required two Johnson markers, so it was labeled “UPEC−only.” Finally, one isolate (0.93%) could not be classified under any of the applied criteria and was reported as unclassified (Supplementary Table 6).
3.7 Distribution of phylogroups in Escherichia coli isolates originating from clinical samples
The distribution of E. coli phylogroups across different clinical sample types is summarized in Table 4. Among the 107 isolates analyzed, phylogroup B2 was the most prevalent, accounting for 32.7% (n = 35) of the isolates, followed by phylogroup B1 at 23.4% (n = 25). The least common phylogroup was G, observed in only 2.8% (n = 3) of the isolates.
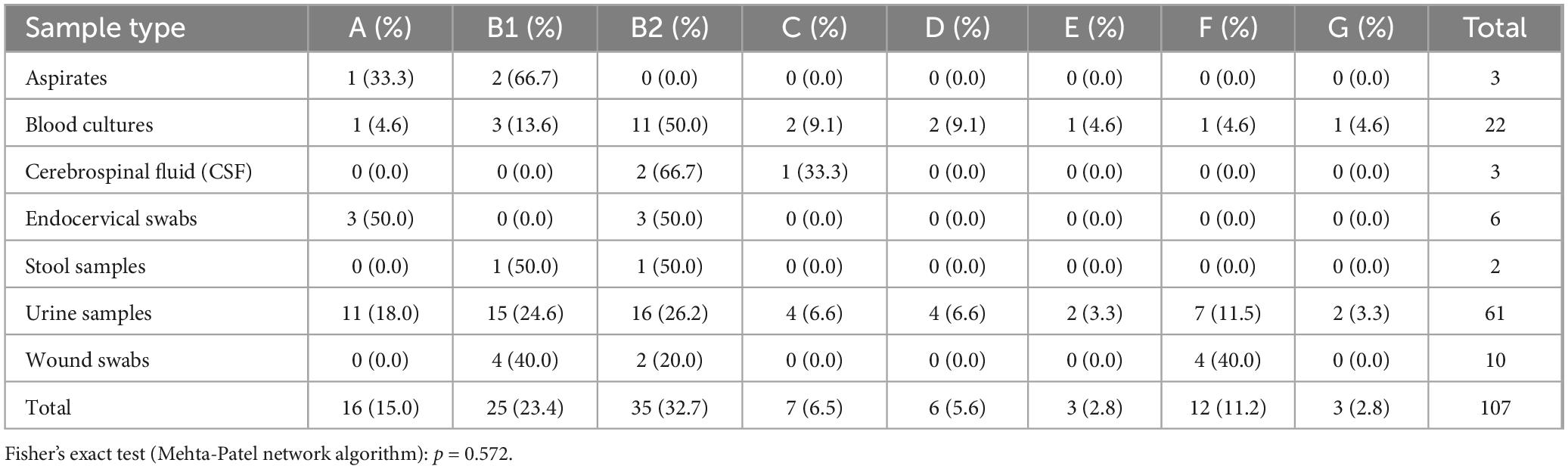
Table 4. Distribution of E. coli phylogroups according to sample type.
When stratified by sample type, phylogroup B2 was most frequently identified in cerebrospinal fluid (CSF) cultures (66.7%, 2/3) and blood cultures (50.0%, 11/22). In contrast, phylogroup A was predominant in endocervical swabs (50%, 3/6), while phylogroup F was most common in wound swabs (40%, 4/10). Urine samples exhibited a more diverse distribution, with phylogroups B2 (26.2%, 16/61) and B1 (24.6%, 15/61) being the most frequently observed.
Statistical analysis using the Chi-square test revealed no significant relationship between phylogroup and sample type (χ2 = 41.68, p = 0.485). This suggests that the distribution of E. coli phylogroups is independent of the clinical sample type.
3.8 Relationship between phylogroups and virulence factors
The number of virulence genes varied across E. coli phylogroups, with phylogroup B2 exhibiting the highest median count. This is shown in detail on Supplementary Table 5. However, the Kruskal-Wallis test revealed no statistically significant differences in the number of virulence genes across phylogroups (H = 7.15, p = 0.413). This suggests that the distribution of virulence gene counts is consistent across the phylogroups analyzed in this study. Figure 4 illustrates the distribution of virulence gene counts for each phylogroup across the 107 E. coli strains.
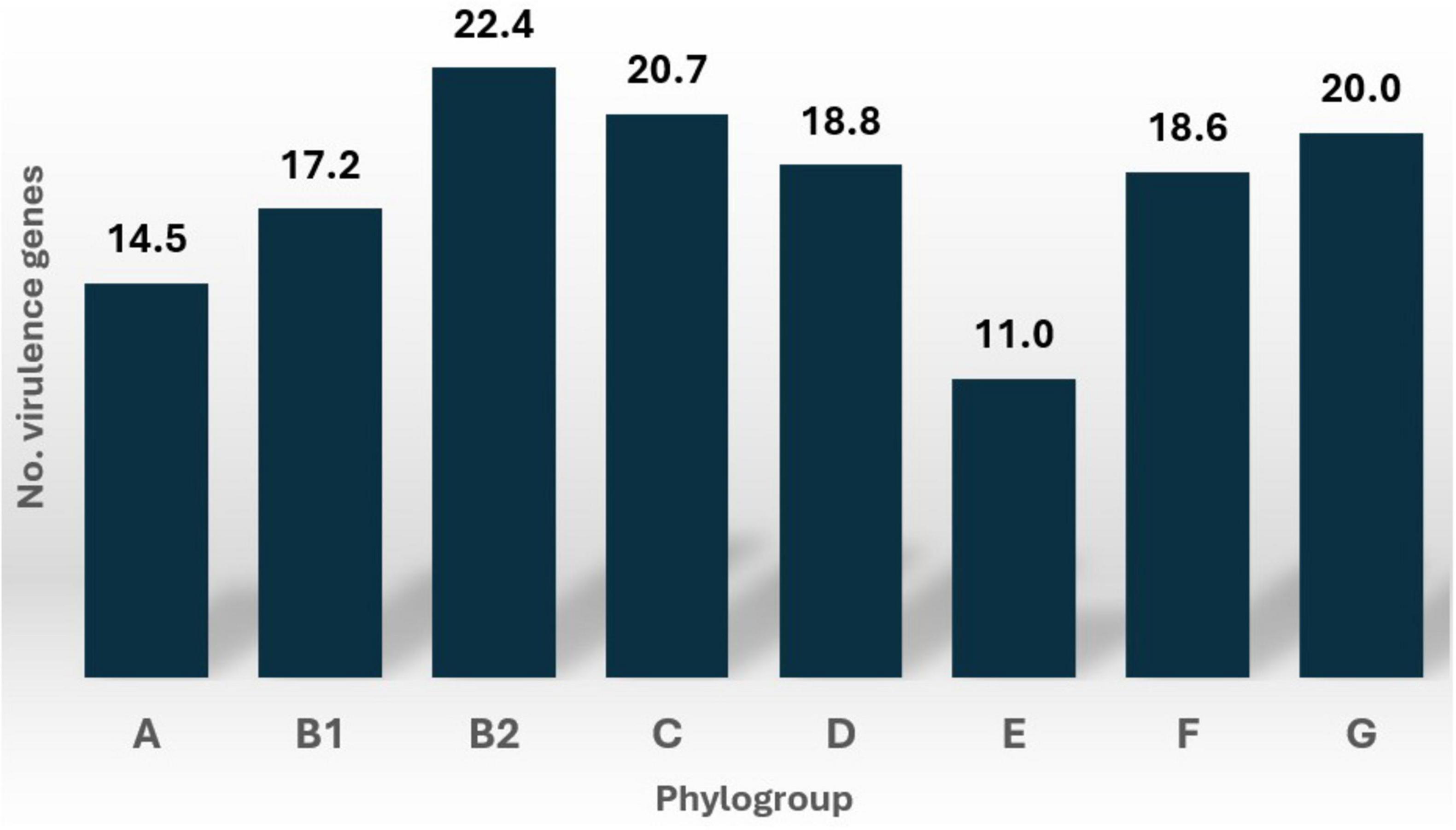
Figure 4. Distribution of virulence genes for each phylogroup across 107 E. coli strains.
Specific virulence factors, such as chuA, yfcV, fimH, and cnf1, were more frequently detected in phylogroup B2. In contrast, other virulence factors, including sfaS, astA, csgA, fdeC, nlpI, terC, and the yeh gene complex, were distributed across various phylogroups with minimal variations. Notably, phylogroups F and G were completely devoid of the csgA gene, which was prevalent in other phylogroups. Detailed information on the distribution of these virulence factors across phylogroups is provided in Supplementary Table 1.
3.9 Phylogroups and antimicrobial resistance
The AMR profiles of E. coli isolates varied significantly across phylogroups, as detailed in Table 5. Phylogroup B2 demonstrated high resistance rates to ampicillin (97.1%, n = 34), ceftriaxone (85.7%, n = 30), and sulfisoxazole (97.1%, n = 34), while phylogroup B1 showed complete resistance to ampicillin (100%, n = 25) and tetracycline (100%, n = 25), along with high resistance to trimethoprim/sulfamethoxazole (96.0%, n = 24).
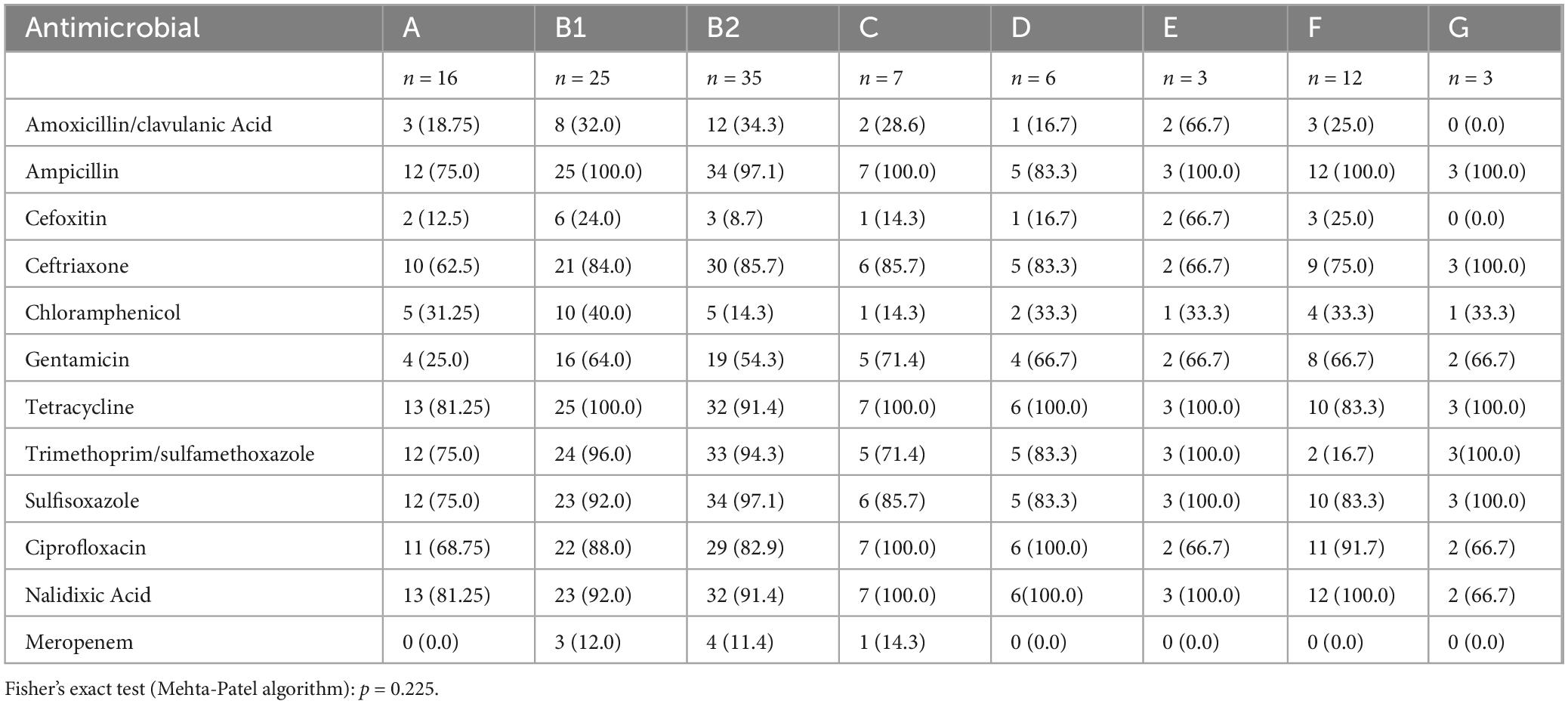
Table 5. Antimicrobial resistance patterns among the phylogroups in E. coli isolates from clinical samples.
Resistance to amoxicillin/clavulanic acid was most prevalent in phylogroup E (66.7%, n = 2), whereas cefoxitin resistance was highest in phylogroup E (66.7%, n = 2) and phylogroup F (25.0%, n = 3). Notably, phylogroup G exhibited complete resistance to ampicillin, ceftriaxone, tetracycline, and sulfisoxazole, but no resistance to meropenem.
Resistance to meropenem was observed only in phylogroups B1 (12.0%, n = 3), B2 (11.4%, n = 4), and C (14.3%, n = 1). Resistance to ciprofloxacin and nalidixic acid was widespread, with phylogroup C showing 100% resistance to both antibiotics.
Kruskal-Wallis analysis of resistance percentages across 12 antibiotics indicated no significant difference among the 8 phylogroups [H (7) ≈ 10.52, p = 0.1625] and Fisher’s exact test (Mehta-Patel algorithm): p = 0.225 (Table 4).
3.10 Hierarchical clustering of virulence genes, plasmid association, and mobile genetic elements
A single nucleotide polymorphism (SNP)-based phylogenetic tree was constructed to examine the genetic relationships among the 107 E. coli isolates. It revealed distinct clustering of isolates according to their phylogroups, with B2 being the most genetically diverse. The presence of virulence genes, sequence types, plasmid associations, and MGEs was mapped onto the tree, illustrating their distribution across different phylogenetic lineages. Notably, virulence genes associated with immune evasion, adherence, and biofilm formation were more prevalent in phylogroup B2, whereas plasmid-encoded virulence genes were commonly found in isolates from multiple phylogroups.
Figure 5 presents the SNP phylogenetic tree, alongside a heatmap indicating the presence (blue) or absence (white) of virulence genes. The association of virulence genes with plasmids is highlighted in dark red, while MGEs linked to virulence factors are represented by pink triangles. Sequence types are shown in a color strip. The bar chart on the right illustrates the number of virulence genes per isolate, and the phylogroups are color-coded.
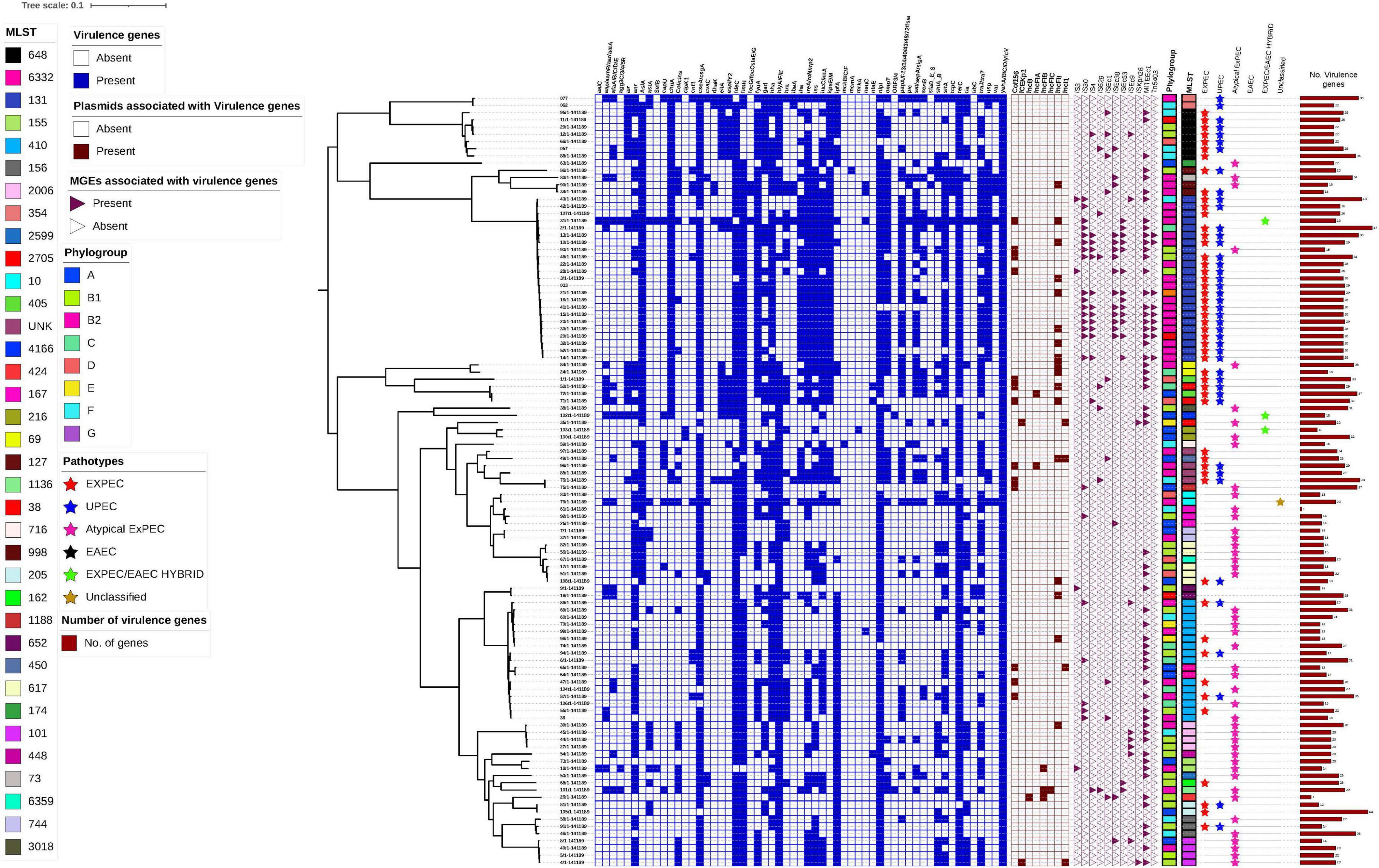
Figure 5. Hierarchical clustering of 107 Escherichia coli isolates based on the presence of virulence genes, plasmid association, and mobile genetic elements (MGEs). The heatmap displays virulence gene presence (blue) and absence (white), while plasmid association with virulence genes is indicated in dark red. MGEs associated with virulence genes are marked with pink triangles (present) or white triangles (absent). The color strips represent Phylogroups and MLSTs with each phylogroup coded in a distinct color. The Pathotypes are represented by different colored triangles. The SNP was based on the reference strain NZ_CP028166.1.
4 Discussion
This study provides a comprehensive genomic analysis of MDR E. coli isolates from a tertiary hospital in Nigeria, offering insights into the relationships between virulence factors, phylogenetic diversity, and AMR profiles. Using WGS, we identified a complex genetic makeup with diverse phylogroups, widespread AMR, and over 100 distinct virulence genes spanning nine functional groups, many of which are located on MGEs. The finding of many of these genes on MGEs—including plasmids like IncFII and Col156—demonstrates how quickly these pathogens could gain and share virulence and resistance traits, potentially complicating clinical management. These findings underscore the urgent need for targeted interventions to curb the spread of MDR E. coli in resource-limited settings like Nigeria.
The identification of 2,021 virulence genes across nine functional categories highlights the pathogenic potential of these MDR-E isolates. The predominance of genes involved in immune evasion (1,573 genes), adherence/biofilm formation (1,380 genes), and iron acquisition (452 genes) aligns with the well-established mechanisms by which E. coli establishes and sustains infections (Croxen and Finlay, 2009; Kaper et al., 2004). The near-universal presence of the yeh gene complex (100%) and the high prevalence of nlpI (94.4%) and fimH (86%) align with previous findings in pathogenic E. coli, particularly ExPEC strains, where these genes are linked to enhanced host adaptability (Croxen and Finlay, 2009; Johnson and Russo, 2002; Kaper et al., 2004; Leimbach et al., 2013). The multifunctional nature of genes like yeh and fimH suggests they have evolved to thrive in diverse niches, from the urinary tract to the bloodstream and wound sites (Yürüyen et al., 2018). The yeh gene complex supports multifunctionality, contributing to adherence, immune evasion, and intracellular survival—traits essential for successful colonization and persistence in host environments (Bien et al., 2012; Ravan and Amandadi, 2015). Similarly, fimH, a type 1 fimbrial adhesin gene, plays a critical role in establishing adherence to host tissues and is also implicated in immune evasion, making it a potential candidate for therapeutic or vaccine-based interventions (Hojati et al., 2015).
Fimbriae play a pivotal role in E. coli pathogenicity by mediating adherence to host cells, a critical step for colonization, biofilm formation, and immune evasion. This study identified key fimbrial adhesins encoded by the afa, sfa, and pap gene clusters, which facilitate attachment to uroepithelial cells, sialic acid receptors, and kidney tissues, respectively, contributing to infections like UTIs (Bergsten et al., 2004; Boisen et al., 2008; Morschhäuser et al., 1993). Biofilm formation, supported by genes like csgA, AslA, and neuC, enhances resistance to antimicrobials and host defenses, with FdeC emerging as a potential vaccine target due to its role in adhesion and biofilm integrity (Vann et al., 2004). Iron acquisition systems, including fyuA, irp2, and iutA, allow E. coli to thrive in iron-limited environments, while immune-modulatory genes like iss and hra promote serum resistance and host tissue persistence, showing the bacterium’s adaptability and resilience in diverse host environments (Johnson et al., 2008; Promite and Saha, 2020).
A particularly striking finding is the finding of virulence genes with MGEs, such as MITEEc1 and IS30. This highlights the role of horizontal gene transfer in the dissemination of virulence determinants (Partridge et al., 2018). The frequent localization of immune evasion genes like terC on MGEs suggests that these genes benefit from being mobile, enabling their rapid spread among bacterial populations (Carattoli, 2013). This is especially concerning in hospital settings, where the exchange of MGEs could lead to the emergence of highly virulent and resistant strains (von Wintersdorff et al., 2025).
Plasmids, such as IncFII and Col156, were frequently found to carry both virulence and AMR genes. For example, IncFII plasmids, detected in 19 isolates, often harbored conjugative transfer genes (traJ/traT) in addition to AMR genes, potentially facilitating the rapid dissemination of these genetic elements (Carattoli, 2009). Col156 plasmids, identified in 13 isolates, consistently carried the senB gene, which is involved in immune evasion and cellular invasion (Smillie et al., 2010). The co-localization of virulence and resistance genes on the same MGEs underscores their potential dual role in driving the spread of both pathogenicity and resistance, posing a significant challenge for clinical management and infection control.
The presence of multi-gene clusters on MGEs, such as ISSpu2 and IS5, further emphasizes their role in the rapid evolution of pathogenic E. coli strains. For instance, isolate NHA101 carried ISSpu2 with genes like aatA, agg4A, and aggR, as well as an IS5 insertion sequence containing eight virulence genes, including afaD and aap (Dobrindt et al., 2004). These findings suggest that MGEs not only facilitate the spread of individual virulence genes but also enable the acquisition of entire virulence gene clusters, potentially leading to the emergence of hypervirulent strains.
Our sequence type (ST) analysis revealed that globally disseminated clones (ST131, ST410, ST648) exhibited greater diversity in virulence genes compared to minor clones (p = 0.028), aligning with prior reports linking these STs to heightened pathogenic potential in extraintestinal infections (Johnson and Stell, 2000; Pitout, 2012). For instance, ST131—a well-documented pandemic clone—carried a median of 15.7 virulence genes, including fimH (98%) and fyuA (89%), consistent with its association with urinary tract and bloodstream infections (Spurbeck et al., 2012).
A key observation in our study is the markedly high prevalence of QRDR double-serine mutations (gyrA_S83L and parC_S80I) in major clones. These mutations, demonstrated to confer a fitness advantage (Fuzi et al., 2020), may facilitate the acquisition and stable retention of a larger gene cargo without imposing significant fitness costs. In our dataset, nearly all major clones harbored these mutations, whereas a lower proportion was observed among minor clones. This near-universal presence of QRDR double-serine mutations in high-risk clones likely underpins their enhanced ability to co-maintain both multidrug resistance and a robust virulence repertoire. While previous studies have noted a potential virulence-resistance trade-off in ST131 (Shaik et al., 2017), our findings suggest that in high-transmission settings like Nigeria—where selective pressures such as antibiotic misuse are intense—the fitness advantage conferred by these QRDR mutations may offset any potential trade-offs. As a result, major clones can thrive even with the added burden of an extensive virulence gene repertoire. This provides a plausible explanation for our observation that MDR strains from major clones exhibit a more diverse virulence gene cargo compared to isolates from minor clones.
Three ExPEC/EAEC hybrids were identified, harboring canonical ExPEC virulence markers (fyuA, chuA, kpsMTII) alongside the enteroaggregative regulator aggR. These hybrids exemplify the genomic plasticity of E. coli, merging the iron acquisition systems and immune evasion strategies of ExPEC with the aggregative adherence and biofilm-forming capacity of EAEC (Lara et al., 2017; Sarowska et al., 2019). For instance, isolate NHA072 carried aggR alongside papG (P fimbriae) and kpsMTII (group 2 capsule synthesis), traits associated with urinary tract colonization and systemic invasion, respectively. Such hybrids are increasingly reported in clinical settings, where their dual virulence repertoires may facilitate colonization of both intestinal and extraintestinal niches, complicating diagnostic categorization and empiric therapy (Santos et al., 2020a). Their detection underscores the need for genomic surveillance to identify emerging pathotypes that evade conventional diagnostic frameworks.
While phenotypic AMR profiles for these isolates were previously published (Medugu et al., 2022), genomic analysis here underscores the role of MGEs in resistance dissemination. IncFII plasmids co-harbored blaTEM-1B and traT in 16 isolates, mirroring findings in other LMICs where such plasmids drive carbapenem and fluoroquinolone resistance (Carattoli, 2013). The strong correlation between AMR gene counts and resistance indices (ρ = + 0.85) highlights the predictive value of WGS for resistance surveillance. However, the persistence of meropenem resistance in phylogroups B1 (12%) and B2 (11%)—despite restricted carbapenem use in Nigeria—may suggest cryptic transmission via mobile elements or clonal expansion, warranting targeted screening.
Phylogroup B2 emerged as the most prevalent (32.7%) and clinically significant group, consistent with its well-documented association with ExPEC (Partridge et al., 2018). The higher diversity of virulence genes and AMR determinants in phylogroup B2 isolates underscores their potential to cause severe infections, such as sepsis, meningitis, and urinary tract infections (UTIs) (Johnson and Stell, 2000). The predominance of phylogroup B2 in cerebrospinal fluid (CSF) and blood cultures further emphasizes its role in invasive infections, necessitating heightened surveillance and targeted interventions (Pitout, 2012).
While the distribution of phylogroups across clinical sample types was not statistically significant, it still provides valuable epidemiological insights. For example, the predominance of phylogroup A in endocervical swabs and phylogroup F in wound swabs suggests niche-specific adaptations that warrant further investigation (Aguirre-Sánchez et al., 2022; Stoppe et al., 2017). The lack of a strong association between phylogroup and sample type, however, indicates that other factors—such as host immune status and antibiotic exposure—may play a more decisive role in infection outcomes (Aguirre-Sánchez et al., 2022; Stoppe et al., 2017; Tenaillon et al., 2010).
5 Limitations
While this study provides valuable insights into the genomic epidemiology of multidrug-resistant Escherichia coli (MDR-E) in Nigeria, several limitations must be acknowledged. The cross-sectional design of this research restricts our ability to establish causal relationships between specific genomic features and clinical outcomes. WGS offers extensive genomic data, this study did not incorporate functional assays to validate the roles of identified virulence genes and their interactions with host factors. Future research should integrate experimental approaches to elucidate the functional significance of these virulence determinants and their impact on infection severity and patient outcomes.
6 Conclusion
This study highlights the genomic diversity and complexity of multidrug-resistant Escherichia coli circulating in Nigeria, characterized by a wide range of sequence types, phylogroups, and virulence–resistance profiles, and a high prevalence of virulence genes frequently associated with MGEs. The high prevalence of ExPEC and UPEC pathotypes, alongside atypical ExPEC and ExPEC/EAEC hybrids, reflects the shifting landscape of E. coli pathogenicity, where classical pathotype boundaries are increasingly blurred. Notably, the universal presence of the yeh gene complex across all isolates highlights a critical virulence mechanism facilitating adherence, invasion, and immune evasion. The co-localization of virulence genes on MGEs, particularly plasmids such as IncFII and Col156, exemplifies the role these elements play in disseminating pathogenicity traits. This genetic linkage poses significant challenges for infection control and clinical management, as it could enable the rapid spread of highly virulent E. coli strains within and potentially beyond healthcare settings.
These findings underscore the urgent need for enhanced genomic surveillance through routine WGS-based monitoring to enable early detection of virulent E. coli strains and timely interventions. Strengthening infection control measures, including strict hygiene and isolation protocols, is crucial to limiting MDR-E transmission in healthcare settings. Additionally, leveraging key virulence factors like the yeh gene complex and fimH for vaccine or antivirulence therapy development could provide novel treatment and prevention strategies. Finally, implementing national policies on AMR, regulating antibiotic sales, and promoting public education on responsible antibiotic use are essential to curbing the spread of MDR-E.
Addressing the spread of highly virulent and adaptable E. coli strains through these measures is imperative for improving patient outcomes and safeguarding public health in resource-limited settings like Nigeria.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
Ethics statement
The studies involving humans were approved by the National Hospital Abuja Ethics Review Board. National Hospital Abuja. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation was not required from the participants or the participants’ legal guardians/next of kin in accordance with the national legislation and institutional requirements.
Author contributions
NM: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. MA: Formal Analysis, Investigation, Methodology, Software, Writing – review & editing. KI: Formal Analysis, Resources, Supervision, Writing – review & editing. PN-P: Investigation, Supervision, Validation, Writing – review & editing. DH: Investigation, Resources, Writing – review & editing. LH: Investigation, Project administration, Software, Writing – review & editing. PS: Methodology, Writing – review & editing. SO: Supervision, Validation, Writing – review & editing. AE: Conceptualization, Investigation, Writing – review & editing. FA: Formal Analysis, Writing – review & editing. RE: Validation, Writing – review & editing. LU: Formal Analysis, Investigation, Validation, Writing – review & editing. YM: Formal Analysis, Writing – review & editing. NI: Validation, Writing – review & editing. ST: Visualization, Writing – review & editing, Conceptualization, Formal Analysis, Investigation, Methodology, Project administration, Resources, Software.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Acknowledgments
We especially appreciate efforts of members of the Thakur Molecular Laboratory, North Carolina State University; toward the success of the research.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2025.1579175/full#supplementary-material
References
Aguirre-Sánchez, J., Valdez-Torres, J., Del Campo, N., Martínez-Urtaza, J., Del Campo, N., Lee, B., et al. (2022). Phylogenetic group and virulence profile classification in Escherichia coli from distinct isolation sources in Mexico. Infect. Genet. Evol. 106:105380. doi: 10.1016/j.meegid.2022.105380
Aiken, A., Rehman, A., de Kraker, M., Madrid, L., Kebede, M., Labi, A., et al. (2023). Mortality associated with third-generation cephalosporin resistance in Enterobacterales bloodstream infections at eight sub-Saharan African hospitals (MBIRA): A prospective cohort study. Lancet Infect. Dis. 23, 1280–1290. doi: 10.1016/S1473-3099(23)00233-5
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A., Dvorkin, M., Kulikov, A., et al. (2012). SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Baquero, F., Tedim, A., and Coque, T. (2013). Antibiotic resistance shaping multi-level population biology of bacteria. Front. Microbiol. 4:15. doi: 10.3389/fmicb.2013.00015
Bergsten, G., Samuelsson, M., Wullt, B., Leijonhufvud, I., Fischer, H., and Svanborg, C. (2004). PapG-dependent adherence breaks mucosal inertia and triggers the innate host response. J. Infect. Dis. 189, 1734–1742. doi: 10.1086/383278
Bien, J., Sokolova, O., and Bozko, P. (2012). Role of uropathogenic Escherichia coli virulence factors in development of urinary tract infection and kidney damage. Int. J. Nephrol. 2012:681473. doi: 10.1155/2012/681473
Boisen, N., Struve, C., Scheutz, F., Krogfelt, K., and Nataro, J. (2008). New adhesin of enteroaggregative Escherichia coli related to the Afa/Dr/AAF family. Infect. Immun. 76, 3281–3292. doi: 10.1128/IAI.01646-07
Carattoli, A. (2009). Resistance plasmid families in Enterobacteriaceae. Antimicrob. Agents Chemother. 53:2227. doi: 10.1128/AAC.01707-08
Carattoli, A. (2013). Plasmids and the spread of resistance. Int. J. Med. Microbiol. 303, 298–304. doi: 10.1016/j.ijmm.2013.02.001
Carattoli, A., Zankari, E., García-Fernández, A., Voldby Larsen, M., Lund, O., Villa, L., et al. (2014). In silico detection and typing of plasmids using PlasmidFinder and plasmid multilocus sequence typing. Antimicrob. Agents Chemother. 58, 3895–3903. doi: 10.1128/AAC.02412-14
Clermont, O., Dixit, O., Vangchhia, B., Condamine, B., Dion, S., Bridier-Nahmias, A., et al. (2019). Characterization and rapid identification of phylogroup G in Escherichia coli, a lineage with high virulence and antibiotic resistance potential. Environ. Microbiol. 21, 3107–3117. doi: 10.1111/1462-2920.14713
Croxen, M., and Finlay, B. (2009). Molecular mechanisms of Escherichia coli pathogenicity. Nat. Rev. Microbiol. 8, 26–38. doi: 10.1038/nrmicro2265
Dobrindt, U., Hochhut, B., Hentschel, U., and Hacker, J. (2004). Genomic islands in pathogenic and environmental microorganisms. Nat. Rev. Microbiol. 2, 414–424. doi: 10.1038/nrmicro884
Feldgarden, M., Brover, V., Gonzalez-Escalona, N., Frye, J., Haendiges, J., Haft, D., et al. (2021). AMRFinderPlus and the reference gene catalog facilitate examination of the genomic links among antimicrobial resistance, stress response, and virulence. Sci. Rep. 11:12728. doi: 10.1038/s41598-021-91456-0
Fuzi, M., Rodriguez Baño, J., and Toth, A. (2020). Global evolution of pathogenic bacteria with extensive use of fluoroquinolone agents. Front. Microbiol. 11:504697. doi: 10.3389/fmicb.2020.00271
Gurevich, A., Saveliev, V., Vyahhi, N., and Tesler, G. (2021). QUAST: Quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. doi: 10.1093/bioinformatics/btt086
Hojati, Z., Zamanzad, B., Hashemzadeh, M., Molaie, R., and Gholipour, A. (2015). The FimH gene in uropathogenic Escherichia coli strains isolated from patients with urinary tract infection. Jundishapur. J. Microbiol. 8:e17520. doi: 10.5812/jjm.17520
Iheanacho, C., and Eze, U. (2022). Antimicrobial resistance in Nigeria: Challenges and charting the way forward. Eur. J. Hosp. Pharm. 29:119. doi: 10.1136/ejhpharm-2021-002762
Iregbu, K., Medugu, N., Abdullahi, N., Aigbe, A., Modibbo, I., Nwajiobi-Princewill, P., et al. (2013). Urine culture contamination: A one-year retrospective study at the national hospital, Abuja. Afr. J. Clin. Experimental Microbiol. 14, 101–104. doi: 10.4314/ajcem.v14i2.10
Joensen, K., Tetzschner, A., Iguchi, A., Aarestrup, F., and Scheutz, F. (2015). Rapid and easy in silico serotyping of Escherichia coli isolates by use of whole-genome sequencing data. J. Clin. Microbiol. 53, 2410–2426. doi: 10.1128/JCM.00008-15
Johansson, M., Bortolaia, V., Tansirichaiya, S., Aarestrup, F., Roberts, A., and Petersen, T. (2021). Detection of mobile genetic elements associated with antibiotic resistance in Salmonella enterica using a newly developed web tool: Mobileelementfinder. J. Antimicrob. Chemother. 76, 101–109. doi: 10.1093/jac/dkaa390
Johnson, J., and Russo, T. (2002). Extraintestinal pathogenic Escherichia coli: “the other bad E coli”. J. Lab. Clin. Med. 139, 155–162. doi: 10.1067/mlc.2002.121550
Johnson, J., and Stell, A. (2000). Extended virulence genotypes of Escherichia coli strains from patients with urosepsis in relation to phylogeny and host compromise. J. Infect. Dis. 181, 261–272. doi: 10.1086/315217
Johnson, J., Murray, A., Gajewski, A., Sullivan, M., Snippes, P., Kuskowski, M., et al. (2003). Isolation and molecular characterization of nalidixic acid-resistant extraintestinal pathogenic Escherichia coli from retail chicken products. Antimicrob. Agents Chemother. 47, 2161–2168. doi: 10.1128/AAC.47.7.2161-2168.2003
Johnson, T., Wannemuehler, Y., and Nolan, L. (2008). Evolution of the iss gene in Escherichia coli. Appl. Environ. Microbiol. 74, 2360–2369. doi: 10.1128/AEM.02634-07
Kaas, R., Leekitcharoenphon, P., Aarestrup, F., and Lund, O. (2014). Solving the problem of comparing whole bacterial genomes across different sequencing platforms. PLoS One 9:e104984. doi: 10.1371/journal.pone.0104984
Kaper, J., Nataro, J., and Mobley, H. (2004). Pathogenic Escherichia coli. Nat. Rev. Microbiol. 2, 123–140. doi: 10.1038/nrmicro818
Lara, F., Nery, D., de Oliveira, P., Araujo, M., Carvalho, F., Messias-Silva, L., et al. (2017). Virulence markers and phylogenetic analysis of Escherichia coli strains with hybrid EAEC/UPEC genotypes recovered from sporadic cases of extraintestinal infections. Front. Microbiol. 8:241651. doi: 10.3389/fmicb.2017.00146
Larsen, M., Cosentino, S., Rasmussen, S., Friis, C., Hasman, H., Marvig, R., et al. (2012). Multilocus sequence typing of total-genome-sequenced bacteria. J. Clin. Microbiol. 50, 1355–1361. doi: 10.1128/JCM.06094-11
Leimbach, A., Hacker, J., and Dobrindt, U. (2013). E. coli as an all-rounder: The thin line between commensalism and pathogenicity. Curr. Top. Microbiol. Immunol. 358, 3–32. doi: 10.1007/82_2012_303
Magiorakos, A., Srinivasan, A., Carey, R., Carmeli, Y., Falagas, M., Giske, C., et al. (2012). Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: An international expert proposal for interim standard definitions for acquired resistance. Clin. Microbiol. Infect. 18, 268–281. doi: 10.1111/j.1469-0691.2011.03570.x
Manges, A., Geum, H., Guo, A., Edens, T., Fibke, C., and Pitout, J. (2019). Global extraintestinal pathogenic Escherichia coli (ExPEC) lineages. Clin. Microbiol. Rev. 32:e00135-18. doi: 10.1128/CMR.00135-18
Medugu, N., Aworh, M., Iregbu, K., Nwajiobi-Princewill, P., Abdulraheem, K., Hull, D., et al. (2022). Molecular characterization of multi drug resistant Escherichia coli isolates at a tertiary hospital in Abuja, Nigeria. Sci. Rep. 12:14822. doi: 10.1038/s41598-022-19289-z
Medugu, N., Tickler, I., Duru, C., Egah, R., James, A., Odili, V., et al. (2023). Phenotypic and molecular characterization of beta-lactam resistant multidrug-resistant enterobacterales isolated from patients attending six hospitals in Northern Nigeria. Sci. Rep. 13:10306. doi: 10.1038/s41598-023-37621-z
Morales, G., Abelson, B., Reasoner, S., Miller, J., Earl, A., Hadjifrangiskou, M., et al. (2023). The role of mobile genetic elements in virulence factor carriage from symptomatic and asymptomatic cases of Escherichia coli bacteriuria. Microbiol. Spectr. 11:e0471022. doi: 10.1128/spectrum.04710-22
Morschhäuser, J., Uhlin, B., and Hacker, J. (1993). Transcriptional analysis and regulation of the sfa determinant coding for S fimbriae of pathogenic Escherichia coli strains. Mol. Gen. Genet. 238, 97–105. doi: 10.1007/BF00279536
Nojoomi, F., and Ghasemian, A. (2019). The relation of phylogroups, serogroups, virulence factors and resistance pattern of Escherichia coli isolated from children with septicemia. New Microbes New Infect. 29:100517. doi: 10.1016/j.nmni.2019.100517
Okeke, I., Lamikanra, A., and Edelman, R. (1999). Socioeconomic and behavioral factors leading to acquired bacterial resistance to antibiotics in developing countries. Emerg. Infect. Dis. 5, 18–27. doi: 10.3201/eid0501.990103
Partridge, S., Kwong, S., Firth, N., and Jensen, S. (2018). Mobile genetic elements associated with antimicrobial resistance. Clin. Microbiol. Rev. 31:e00088-e17. doi: 10.1128/CMR.00088-17
Partridge, S., Kwong, S., Firth, N., and Jensen, S. (2024). Mobile genetic elements associated with antimicrobial resistance. Clin. Microbiol. Rev. 31:e00088-17. doi: 10.1128/CMR.00088-17
Pitout, J. (2012). Extraintestinal pathogenic Escherichia coli: A combination of virulence with antibiotic resistance. Front. Microbiol. 20:9. doi: 10.3389/fmicb.2012.00009
Promite, S., and Saha, S. (2020). Escherichia coli in respiratory tract infections: Evaluating antimicrobial resistance and prevalence of fimA, neuC and iutA virulence genes. Gene Rep. 18:100576. doi: 10.1016/j.genrep.2019.100576
Ravan, H., and Amandadi, M. (2015). Analysis of yeh fimbrial gene cluster in Escherichia coli O157:h7 in order to find a genetic marker for this serotype. Curr. Microbiol. 71, 274–282. doi: 10.1007/s00284-015-0842-6
Santos, A., Santos, F., Silva, R., and Gomes, T. (2020a). Diversity of hybrid- and hetero-pathogenic Escherichia coli and their potential implication in more severe diseases. Front. Cell. Infect. Microbiol. 10:532570. doi: 10.3389/fcimb.2020.00339
Santos, A., Silva, R., Valiatti, T., Santos, F., Santos-Neto, J., Cayô, R., et al. (2020b). Virulence potential of a multidrug-resistant Escherichia coli strain belonging to the emerging clonal group ST101-B1 isolated from bloodstream infection. Microorganisms 8:827. doi: 10.3390/microorganisms8060827
Sarowska, J., Futoma-Koloch, B., Jama-Kmiecik, A., Frej-Madrzak, M., Ksiazczyk, M., Bugla-Ploskonska, G., et al. (2019). Virulence factors, prevalence and potential transmission of extraintestinal pathogenic Escherichia coli isolated from different sources: Recent reports. Gut Pathog. 11:10. doi: 10.1186/s13099-019-0290-0
Schmidt, H., and Hensel, M. (2004). Pathogenicity islands in bacterial pathogenesis. Clin. Microbiol. Rev. 17, 14–56. doi: 10.1128/CMR.17.1.14-56.2004
Shaik, S., Ranjan, A., Tiwari, S., Hussain, A., Nandanwar, N., Kumar, N., et al. (2017). Comparative genomic analysis of globally dominant ST131 clone with other epidemiologically successful extraintestinal pathogenic Escherichia coli (ExPEC) Lineages. mBio 8:e01596-17. doi: 10.1128/mBio.01596-17
Smillie, C., Garcillán-Barcia, M., Francia, M., Rocha, E., and de la Cruz, F. (2010). Mobility of plasmids. Microbiol. Mol. Biol. Rev. 74, 434–452. doi: 10.1128/MMBR.00020-10
Sora, V., Meroni, G., Martino, P., Soggiu, A., Bonizzi, L., and Zecconi, A. (2021). Extraintestinal pathogenic Escherichia coli: Virulence factors and antibiotic resistance. Pathogens 10:1355. doi: 10.3390/pathogens10111355
Spurbeck, R., Dinh, P., Walk, S., Stapleton, A., Hooton, T., Nolan, L., et al. (2012). Escherichia coli isolates that carry vat, fyuA, chuA, and yfcV efficiently colonize the urinary tract. Infect. Immun. 80, 4115–4122. doi: 10.1128/IAI.00752-12
Stoppe, N., Silva, J., Carlos, C., Sato, M., Saraiva, A., Ottoboni, L., et al. (2017). Worldwide phylogenetic group patterns of Escherichia coli from commensal human and wastewater treatment plant isolates. Front Microbiol. 8:289063. doi: 10.3389/fmicb.2017.02512
Tenaillon, O., Skurnik, D., Picard, B., and Denamur, E. (2010). The population genetics of commensal Escherichia coli. Nat. Rev. Microbiol. 8, 207–217. doi: 10.1038/nrmicro2298
Vann, W., Daines, D., Murkin, A., Tanner, M., Chaffin, D., Rubens, C., et al. (2004). The NeuC protein of Escherichia coli K1 is a UDP N-acetylglucosamine 2-epimerase. J. Bacteriol. 186, 706–712. doi: 10.1128/JB.186.3.706-712.2004
von Wintersdorff, C., Penders, J., van Niekerk, J., Mills, N., Majumder, S., van Alphen, L., et al. (2025). Dissemination of antimicrobial resistance in microbial ecosystems through horizontal gene transfer. Front. Microbiol. 7:174871. doi: 10.3389/fmicb.2016.00173
Wirth, T., Falush, D., Lan, R., Colles, F., Mensa, P., Wieler, L., et al. (2006). Sex and virulence in Escherichia coli: An evolutionary perspective. Mol. Microbiol. 60, 1136–1151. doi: 10.1111/j.1365-2958.2006.05172.x
World Health Organization (2022). Global Antimicrobial Resistance and Use Surveillance System (GLASS). Geneva: World Health Organization.
Keywords: multidrug-resistant Escherichia coli, virulence factors, phylogenetic diversity, whole-genome sequencing (WGS), mobile genetic elements (MGEs), IncFII plasmids, antimicrobial resistance (AMR), pathotypes
Citation: Medugu N, Aworh MK, Iregbu K, Nwajiobi-Princewill P, Hull DM, Harden L, Singh P, Obaro S, Egwuenu A, Adeboye F, Egah R, Uzairue L, Mohammed Y, Ifeyinwa N and Thakur S (2025) Whole genome sequencing reveals virulence–mobile element linkages and phylogenetic diversity in multidrug-resistant Escherichia coli from Nigeria. Front. Microbiol. 16:1579175. doi: 10.3389/fmicb.2025.1579175
Received: 18 February 2025; Accepted: 07 April 2025;
Published: 07 May 2025.
Edited by:
Jens Andre Hammerl, Bundesinstitut für Risikobewertung, GermanyReviewed by:
Miklos Fuzi, Independent Researcher, Seattle, WA, United StatesRazib Mazumder, International Centre for Diarrhoeal Disease Research (ICDDR), Bangladesh
Copyright © 2025 Medugu, Aworh, Iregbu, Nwajiobi-Princewill, Hull, Harden, Singh, Obaro, Egwuenu, Adeboye, Egah, Uzairue, Mohammed, Ifeyinwa and Thakur. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nubwa Medugu, bnVid2EubWVkdWd1QGdtYWlsLmNvbQ==