 Lydia M. Mageto1,2*
Lydia M. Mageto1,2* Gabriel Oluga Aboge1
Gabriel Oluga Aboge1 Zelalem H. Mekuria3
Zelalem H. Mekuria3 Peter Gathura1
Peter Gathura1 John Juma4
John Juma4 Michael Mugo5
Michael Mugo5 Collins Kipkorir Kebenei5
Collins Kipkorir Kebenei5 Diana Imoli5
Diana Imoli5 Beatrice Atieno Ongadi5
Beatrice Atieno Ongadi5 Kelvin Kering5
Kelvin Kering5 Cecilia Kathure Mbae5
Cecilia Kathure Mbae5 Samuel Kariuki5
Samuel Kariuki5- 1Department of Public Health, Pharmacology and Toxicology, Faculty of Veterinary Medicine, University of Nairobi, Nairobi, Kenya
- 2Washington State University, Global Health Kenya, Nairobi, Kenya
- 3Department of Veterinary Preventive Medicine, College of Veterinary Medicine, The Ohio State University, Columbus, OH, United States
- 4International Livestock Research Institute (ILRI), Nairobi, Kenya
- 5Centre for Microbiology Research, Kenya Medical Research Institute, Nairobi, Kenya
Background: Cholera remains a public health challenge in Kenya. To better understand its dynamics, we analyzed Vibrio cholerae genomes from clinical and environmental samples collected during the 2022–2023 outbreak. These strains were compared with historical genomes from Kenya, Uganda, Tanzania, and Haiti to inform strategies for cholera prevention, control, and elimination in Kenya.
Methods: Clinical (stool) and environmental (wastewater, drinking water, and household effluent) samples were collected from Nairobi county. Samples were analyzed for V. cholerae using culture and real time PCR. The environmental (n = 17) and clinical (n = 70) isolates were then subjected to phenotypic antimicrobial susceptibility testing using the Kirby-Bauer disk diffusion method. Whole genome sequencing was employed to characterize the genome, detect antimicrobial resistance genes, virulence factors, and mobile genetic elements. Phylogenetic analysis was performed to assess the genetic relationship and diversity of isolates from 2022 to 2023 outbreak, comparing them with isolates from historical outbreaks.
Results: Clinical isolates carried key virulence genes (ctxA, ctxB7, zot, and hlyA) and were 100% resistant to multiple antibiotics, including ampicillin, cefotaxime, ceftriaxone, and cefpodoxime, but remained susceptible to gentamicin and chloramphenicol. In contrast, environmental isolates lacked ctxB gene but harbored toxR, als, and hlyA, showing variable antibiotic resistance (59% to ampicillin, 41% to trimethoprim-sulfamethoxazole, and 47% to nalidixic acid). All clinical isolates from 2022 to 2023 outbreak harbored IncA/C2 plasmids and several antimicrobial resistance genes including blaPER–7. Phylogenetic analysis revealed high genetic diversity in environmental strains, clustering outside the 7th pandemic El Tor lineage, while clinical isolates were highly clonal. Genomes from 2022 to 2023 outbreak were closely related to Kenyan cholera outbreak genomes from 2016 (15 single nucleotide polymorphisms, T13 lineage).
Conclusion: The 2022–2023 outbreak likely resulted from re-emergence of previously circulating strains rather than a new introduction. While the role of environmental reservoirs as a source of human infection remains unclear in our study, environmental isolates possess virulent and antimicrobial resistance genes that may spread via horizontal gene transfer. This highlights the need for continuous genomic surveillance to monitor V. cholerae evolution, track transmission patterns, and mitigate the spread of antimicrobial resistance.
1 Introduction
Vibrio cholerae, the causative agent of cholera is transmitted through consumption of contaminated water or food (Harris et al., 2012). Globally, the burden of cholera in endemic countries is estimated at 1.3–4 million infections with 143,000 mortalities annually (Ali et al., 2015). Out of the 209 different serogroups of V. cholerae, only serogroups O1 and O139 are known to potentially cause epidemics (Chun et al., 2009). Non-O1/non-O139 (NOVC) serogroups are considered environmental strains and part of the normal flora in aquatic ecosystems. However, certain NOVC strains can cause cholera outbreaks characterized by mild diarrhea as reported in a number of countries (Dalsgaard et al., 1999; Chatterjee et al., 2009; Onifade et al., 2011). V. cholerae O1 is categorized into El Tor and classical biotypes based on their phenotypic traits (Tappero and Tauxe, 2011). The classical biotype was associated with the previous six pandemics experienced between 1817 and 1923. These pandemics spread from the Indian subcontinent to other continents (Tappero and Tauxe, 2011). In 1935, EI Tor biotype was found to be the major cause of cholera outbreak in Indonesia. It caused a major pandemic in Asia in 1961 spreading to a number of African countries including Zambia, Zimbabwe, Tanzania, and Uganda, slowly replacing the classical biotype (Banerjee et al., 2014). The two biotypes are further divided into Ogawa, Inaba, and Hikojima serotypes based on antigenic factors (Raychoudhuri et al., 2008). Since 1971 when the first outbreak was reported in Kenya, 16 different cholera outbreaks have been reported up to the year 2015 (Tauxe et al., 1995; Shapiro et al., 1999; Mugoya et al., 2008; Scrascia et al., 2009; Shikanga et al., 2009; Kigen et al., 2020). Its recurrence in 2022 clearly indicates that this disease is a major public health threat in Kenya. According to Kenya’s Ministry of Health, the recent 2022–2023 cholera outbreak affected 27 counties (57%) resulting in 12,123 reported cases and 202 fatalities (IFRC, 2024). The ongoing seventh cholera pandemic El Tor strain (7PET) began in Southeast Asia and so far, three transmission waves (I, II, and III) have been identified globally by phylogenetic analysis of which wave III has the largest number of clusters (Weill et al., 2017).
The ability to express virulence factors is essential for V. cholerae O1 or O139 to cause epidemics. Several genes have been proposed as virulence markers based on in vivo and in vitro studies with cholera toxin and toxin coregulated pilus identified as key pathogenic determinants of V. cholerae (Faruque et al., 2003). Cholera toxin (Ctx) encoded by ctxA and ctxB genes and carried on the CTXφ prophage is the cause of the severe watery diarrhea seen in cholera patients (Davis and Waldor, 2003). The toxin-coregulated pilus (tcp) is responsible for synthesis of fimbriae important for adherence of V. cholerae to the intestinal epithelium of the host (Karaolis et al., 1998). In rare cases, toxin coregulated pili (tcpA) and cholera toxin (ctxA) have been reported in NOVC strains (Singh et al., 2001). Other virulence genes encoding Zonula occludens toxin (Zot), accessory cholera enterotoxin (Ace), hemolysin (hlyA), repeats-in-toxin A toxin (rtxA), and mannose-sensitive hemagglutinin pilus (mshA) have been associated with the endemic disease (Feng et al., 2004; Abana et al., 2019).
Mild to moderate cases of cholera are primarily treated by oral or intravenous hydration (WHO, 2024). Antibiotics such as doxycycline and ciprofloxacin are recommended in patients with severe dehydration and those with underlying conditions. Studies have shown that use of antibiotics in this group of patients decreases the duration of diarrhea, volume of stool and the length of shedding of V. cholerae (Leibovici-Weissman et al., 2014). A major concern that is linked to this is the emergence of antimicrobial resistant (AMR) strains. Furthermore, the acquisition of mobile genetic elements (MGEs) such as plasmids, transposons, integrons, and integrative conjugative elements (ICEs) plays a significant role in spread of antimicrobial resistance genes (Lassalle et al., 2023; Bhandari et al., 2021).
The pathogenesis and virulence patterns of cholera have been studied both globally and regionally (Chaguza et al., 2024; Kimani et al., 2014; Kiiru et al., 2013; Feng et al., 2004; Abana et al., 2019). Even though surveillance of cholera outbreaks has been done in Kenya, the evolutionary trend and mechanisms driving the emergence and spread of virulent AMR strains remain poorly understood. This study aimed at comprehensively analyzing the genomic data of clinical and environmental V. cholerae isolates from the recent 2022–2023 cholera outbreak in Kenya. We performed whole genome sequencing for the isolates in this outbreak and compared them with historical genomes from Kenya, Uganda, Tanzania, and Haiti. We generated data that provides important insights into predicting disease transmission patterns, especially between bordering countries, monitoring the evolution of new variants, and identifying the emergence of virulent and AMR strains. These findings are expected to inform public health strategies for preventing, controlling, and eventually eliminating cholera in Kenya.
2 Materials and methods
2.1 Sample collection
Environmental samples (wastewater, drinking water, and household effluent) were collected from Mukuru informal settlement in Nairobi county (Figure 1). Clinical samples (stool) were collected from Mukuru and other regions in Nairobi county including Kayole, Mathare, Eastleigh, and Dandora. Mukuru informal settlement, the main sampling site has a population of 770,467 people with 97,890 households according to the 2019 national census report. One percent of residents in Mukuru have access to a private toilet and individual water source (Muriithi and Obare, 2017). Poor hygiene, limited access to clean water, inadequate toilets and poor waste disposal system increase the risk for diarrheal diseases including cholera outbreaks in the study area.
Figure 1. Map showing location of Mukuru informal settlement in Nairobi county.
2.2 Sampling procedure
2.2.1 Environmental samples
A total of 121 environmental samples were collected during the epidemic period of January to March 2023 from randomly identified households in the study area. These included 40 drinking water samples, 41 wastewater samples and 40 household effluent samples. Drinking water samples were collected in sterile 100 ml Whirl-Pak bags with or without sodium thiosulfate tablets, following determination of chlorine concentration. Chlorinated water was collected into Whirl-Pak bags with sodium thiosulfate tablets. Household effluents, which majorly included gray water from kitchens, laundry facilities, and baths, were collected using a sterile ladle. Upon ensuring the samples were free of sediments, we carefully transferred the samples into 2-L sterile labeled Whirl-Pak bags. Wastewater containing human waste and household effluent was collected from open drains using the same method as described above for household effluent sampling. All samples in the Whirl-Pak bags were transported to the Centre for Microbiology Research Laboratory at Kenya Medical Research Institute (KEMRI) in cool boxes packed with ice for processing within 6 h of collection.
2.2.2 Clinical samples
Stool samples were collected in clean disposable polypots from suspected cholera patients (patients aged 2 years or more with acute watery diarrhea with or without vomiting) attending hospitals in the study areas in November and December 2022. This was based on Integrated Disease Surveillance and Response Standard case definitions.1 Diarrheal stool samples were transported to the Centre for Microbiology Research Laboratory at KEMRI using Cary Blair transport media (Oxoid, Thermo Fisher Scientific, USA) within 2 h of collection.
2.3 Laboratory analysis
2.3.1 Environmental samples
We measured the Turbidity and PH of each of the samples upon receipt in the laboratory in order to assess sample quality and identify any physicochemical characteristics that could affect V. cholerae recovery. For drinking water, 10 ml of each sample was filtered through a membrane filter of 0.45 μm pore diameter (Millipore, Bedford, MA, United States). The filter paper was transferred into 10 ml Alkaline Peptone Water (APW), well shaken and incubated at 37°C for 18 h. Twenty-five (25) ml of each sample of wastewater and household effluent was added to 25 ml of APW in a conical flask and incubated at 37°C for 18 h. Following enrichment, we streaked the samples onto thiosulfate citrate bile salts sucrose agar (TCBS) plates (Oxoid Ltd.) and incubated at 37°C for 18 h. Characteristic small to large yellow colonies were subcultured on Muller Hinton (MH) agar and incubated for 18 h. Purified isolates were then subjected to oxidase test as described previously by Hounmanou et al. (2016). Those that turned positive were presumed to be V. cholerae isolates. We extracted DNA from pure cultures of suspected V. cholerae colonies using the Zymo Quick-DNA fungal/bacterial Miniprep kit (The Epigenetics Company, CA, United States). Extracted DNA was subjected to Real Time PCR for detection of V. cholerae species-specific gene hlyA based on a highly specific protocol by Huang et al. (2009). Briefly, the 25 μl master-mix comprised 0.4 μM of each hlyA forward and reverse primers, 12.5 μl Bio-Rad iQ Multiplex Powermix, 5 μl molecular water, 0.2 μM hlyA probe, and 5 μl of DNA template. AMPLIRUN V. cholerae DNA control (VIRCELL Microbiologists, MBC118) served as the positive control while nuclease-free water was used as the negative control. Quantitative PCR was run on a Magnetic Induction Cycler (MIC) using the following conditions: activation (at 95°C for 15 min) and 40 cycles of 95°C for 15 s, 55°C for 40 s, and 72°C for 30 s. PCR reactions were duplicated for each sample. We classified cultures with Ct values of 20 or lower and a difference of less than 2 between duplicate Ct values as positive.
2.3.2 Clinical samples
Stool samples were streaked onto TCBS agar plates (Oxoid Ltd.) following enrichment in APW and incubated at 37°C for 18 h. Characteristic small to large yellow colonies were subcultured on MH agar and incubated for 18 h. Purified isolates were then subjected to oxidase test as described by Hounmanou et al. (2016). Those that turned positive were presumed to be V. cholerae isolates. In order to confirm serotype identity, serology was carried out using polyvalent, anti-Ogawa, and anti-Inaba antisera (Denka Seiken, Tokyo, Japan). A similar protocol for DNA extraction and Real Time PCR as for environmental samples was employed for clinical sample analysis.
A total of 100 μl DNA aliquots for 137 positive cultures of clinical (n = 120) and environmental (n = 17) isolates were shipped on dry ice to Ohio State University for library preparations and whole genome sequencing. We stored all confirmed positive isolates at −80°C for further analysis.
2.4 Phenotypic antimicrobial susceptibility testing for clinical and environmental samples
We screened 70 clinical and 17 environmental isolates for antimicrobial susceptibility using the Kirby-Bauer disk diffusion method (Biemer, 1973). The 70 clinical samples screened for AST were randomly selected from the 120 positive isolates with each of the sampling sites represented. The following antimicrobial agents were used; ampicillin (10 μg), gentamicin (30 μg), ciprofloxacin (5 μg), nalidixic acid (30 μg), ceftazidime (30 μg), trimethoprim sulfamethoxazole (25 μg), ceftriaxone (30 μg), cefpodoxime (30 μg), tetracycline (30 μg), azithromycin (15 μg), amoxicillin-clavulanate acid (30 μg), chloramphenicol (30 μg), cefotaxime (30 μg), and Kanamycin (25 μg). Potency of the antibiotic discs and growth of bacteria was tested using Escherichia coli ATCC 25922 as the control.
Briefly, V. cholerae isolates were grown on MH agar plates for 24 h at 37°C. Colonies were emulsified into normal saline to achieve a 0.5 MacFarland suspension. This suspension was evenly spread onto MH agar (Oxoid, Basingstoke, UK) and allowed to air dry. After air drying the antibiotic disks were applied on the agar and incubated at 37°C for 24 h. We measured the diameter of the inhibition ring and determined susceptibility based on 2018 Clinical and Laboratory Standards Institute (CLSI) M45 established guideline for infrequently isolated or fastidious bacteria (CLSI, 2018). We classified isolates showing resistance to at least three categories of antibiotics as multidrug resistant (MDR). Whole genome sequencing analysis was used to detect AMR genes.
2.5 Whole genome sequencing
Genomic DNA of 137 isolates [environmental (n = 17) and clinical (n = 120)] were subjected to whole genome sequencing. We included one clinical isolate from the 2016 cholera outbreak. Library preparation was done using an Illumina paired-end DNA library preparation kit. Briefly, the 4150 Tapestation system was used to determine library size and concentration (Agilent, MA, USA). In order to amplify the tagged DNA and introduce sequencing indexes, the limited-cycle PCR was subsequently employed. We incorporated PhiX Control v3 (Illumina, Inc., San Diego, CA, USA) into each sample prior to library preparation to facilitate a limit of detection assessment for each sample. The prepared libraries were loaded onto a reagent cartridge and subjected to clustering on the NextSeq 2000 System. Subsequently, a paired-end sequencing run with 2 × 150 bp reads was executed using the NextSeq 2000 platform. Raw sequences have been deposited in the National Centre for Biotechnology Information (NCBI) Sequence Read Archive (SRA) BioProject number PRJNA1235657. The SRA accession numbers and other metadata for each sample are provided in Supplementary Table 1.
2.6 Genomic analysis
2.6.1 Serotyping, biotyping, identification of major virulence genes and sequence types, characterization of pathogenicity islands, and assessment of phage susceptibility
Assembly of the raw paired-end reads was done using SPAdes assembler (Bankevich et al., 2012). The assembled sequences were analyzed using CholeraeFinder 1.0 tool in the Centre for Genomic Epidemiology (CGE) platform.2 We confirmed cholera species based on the species-specific ompW gene (Siriphap et al., 2017) with a threshold set at 98% identity. This tool further identified V. cholerae serogroup-specific genes (rfbV-O1 and wbfZ-O139), biotype-specific genes (ctxB, rstR, and tcpA), the gene specific for the 7th pandemic (VC2346), and putative virulence genes. Detection of Vibrio pathogenicity islands mainly VPI-1, VPI-2, VSP-1, VSP-2, and PICI like elements responsible for phage susceptibility (PLE1 and PLE2) was also carried out using CholeraeFinder 1.0. Multilocus sequence typing was done using the bactopia bacterial analysis pipeline available from https://bactopia.github.io/latest/ for short paired-end reads. In the bactopia pipeline, contigs were subjected to the “MLST_MODULE” to determine the sequence type. Briefly, the contigs were queried against a custom Multilocus Sequence Typing (MLST) database (build 2.23.0-20240325) using MLST (version 2.23.0) with automatic detection of scheme, 100% identity, minimum depth of sequence coverage of 10 and minimum alignment score of 50. Additionally, the assembled contigs were annotated using prokka version 1.14.6.
2.6.2 Identification of antimicrobial resistance genes and mobile genetic elements
Antimicrobial resistance genes in the assembled contigs were detected using amrfinderplus (version 3.12.8) against the amrfinderplus-database (build 2024-01-31.1) in the bactopia pipeline. Additional search for MGEs, the different classes of integrons and mutations in gyrA and parC genes (Siriphap et al., 2017) was done by CholeraeFinder on the CGE platform. The PlasmidFinder 1.3 tool of the bactopia pipeline in CGE was used to search for plasmid replicons.
2.6.3 Single nucleotide polymorphism-based phylogenetic analysis
In order to identify the phylogenetic relatedness within and between clinical and environmental V. cholerae isolates, high quality assembled genomes were mapped against a reference genome strain of V. cholerae N16961 (GenBank accession numbers NZ_LT906614 and NZ_LT906615) using Snippy (version 4.6.0).3 Single nucleotide polymorphisms (SNPs) were called with Freebayes (version 1.3.2) using these parameters: minimum mapping quality 60, minimum base quality 13, minimum read coverage 4, and minimum proportion of variant evidence of 75%. With the called SNPs, a core-SNP alignment was then generated. A distance matrix of the SNPs was computed using snp-dists (version 0.8.2) and recombination masked with Gubbins version 3.3.1 (Croucher et al., 2015). A maximum likelihood phylogenetic tree was constructed using IQTREE version 2.2.2.7 (Nguyen et al., 2015) with 1000 ultrafast bootstrap replicates under the HKY model (Posada and Crandall, 2001) and the final tree amended in iTOL.
2.6.4 Global phylogenetic analysis
To understand the evolutionary and temporal dynamics of V. cholerae beyond the 2022–2023 sequenced genomes, we retrieved genomic data of V. cholerae isolated in different countries from pathogen watch.4 The dataset of complete genomes of V. cholerae used in evolutionary and temporal dynamics comprised 606 isolates (study isolates: n = 105; pathogenwatch isolates: n = 501). The 501 included sequences from Kenya (n = 106), Tanzania (n = 69), Uganda (n = 17), and Haiti (n = 308), with the M66 strain used as an outgroup. M66 strain genome was the earliest and ancestral sequence among the seventh pandemic isolates, collected in Indonesia in 1937. We applied the bactopia main module to generate assemblies followed by variant calling with Snippy as described above with inclusion of high-quality genome assemblies. The masked core SNP alignment (n = 322) with 60800 SNPs was used for phylogenetic analysis with M66 strain as an outgroup.
To understand the temporal dynamics of V. cholerae in Kenya, we randomly grouped the genomes by collection time (year), then selected at least 20 genomes per year yielding 37 sequences spanning from 1985 to 2022. For the “global” dataset comprising sequences from the other countries (Kenya, Tanzania, Uganda, and Haiti), we subsampled the genomes by year and selected 12 genomes per sampling time, yielding 94 isolates. For each dataset, maximum phylogenetic inference was performed using IQTREE with generalized time reversible (GTR) applied as the optimal substitution model for evolution (Substitution Models, 2024). To perform phylodynamic analysis, we assessed the temporal signal of the subsampled genome sequences by regressing the genetic distances against collection time (years). Having ascertained sufficient temporal signal, we performed phylodynamic analysis incorporating the sampling locations as traits for phylogeographic reconstructions for dispersal dynamics. For the genomes generated in this study, we inferred the longitude and latitude coordinates using “tidygeocoder” in R using the sampling sites as locations. The associated metadata of the pathogenwatch retrieved genomes were bundled with longitude and latitude information as other relevant annotations. We used Bayesian evolutionary analysis sampling trees (BEAST) version 1.10.4 (Suchard et al., 2018) to perform phylodynamic analysis. For both subsampled datasets, we applied the sampling date (in years) as the tip dates, GTR as the best evolution and Skyline demographic/population models. We used continuous phylogeographic inference by adding the location traits given as longitude and latitude geographical coordinates under a Cauchy distribution model. For the subsampled isolates from Kenya, we run BEAST for 100 million chains, sampling every 10,000th step while the global dataset was run for 300 million generations with sampling at every 30,000th step. We assessed the mixing properties of the relevant estimates for convergence, ensuring effective sample sizes (ESS) > 200 was attained using Tracer version 1.7.2 (Rambaut et al., 2018). Maximum clade credibility trees were obtained using Tree Annotator version 1.10.4 (Rambaut et al., 2018) with a 10% burn-in. Estimates of parameters of interest were reported as median values at 95% highest posterior density or credible interval.
The Phylogenetic tree was constructed using IQ-Tree version 2.0.3 with M66 used as an outgroup and the final tree amended in iTOL (Letunic and Bork, 2021). Accession numbers of strains used in the SNP tree are reported in Supplementary Table 2.
2.7 Ethics statement
Ethical approval for this study was obtained from the Kenyatta National Hospital-University of Nairobi Ethical Review Committee (KNH-UON ERC). The Institutional Review Board reviewed the procedures outlined in this study to ensure the protection of human subjects, the privacy of participants, and the ethical conduct of research (Approval ID: P731/09/2021). Research license was sought from the National Commission for Science, Technology, and Innovation (License No.: NACOSTI/P/22/16171).
3 Results
3.1 Genomic characterization and virulence profiles of clinical and environmental Vibrio cholerae isolates
Out of the 137 sequenced isolates, 105 genomes were included in the final genomic analysis comprising 95 genomes from clinical samples and 10 from environmental samples. The remaining genomes were excluded due to less than 50% coverage when mapped to the N16961 reference strain, thus failing quality control checks. The assembled genomes of Kenyan isolates included in the final analysis are available publicly at the NCBI GenBank (BioProject number: PRJNA1235657), with accession numbers and metadata of each sample provided in Supplementary Tables 3, 4 for clinical and environmental isolates respectively.
Ninety-eight percent (n = 93) of the sequenced strains from the 2016 (n = 1) and 2022 (n = 92) clinical isolates were characterized as serogroup 01, containing both ompW and rfbv-O1 genes. The remaining two isolates were identified as non O1/non-O139. All clinical isolates belonged to the third wave and carried the genetic marker for 7th pandemic V. cholerae strains (VC2346 gene). These isolates were of Ogawa serotype and carried ctxB7 genotype of the ctxB gene. Additionally, V. cholerae O1 isolates contained the rstR and tcpA genes found in El Tor biotype. Multi-locus Sequence Typing revealed that all the clinical strains belonged to sequence type 69 (ST69). There was a similar trend in occurrence of virulence-associated genes and pathogenicity islands across all clinical strains except one isolate that lacked the ctxB gene. This included the key virulence genes such as ctxA, ctxB, zot, ace, hlyA, mshA, als, makA, rtxA, ompU, and toxR. The chxA and stn genes were absent in all strains. Additionally, Vibrio pathogenicity islands VPI-1, VPI-2, VSP-1, and VSP-2 were detected in all clinical strains as shown in Table 1 and Supplementary Table 3.
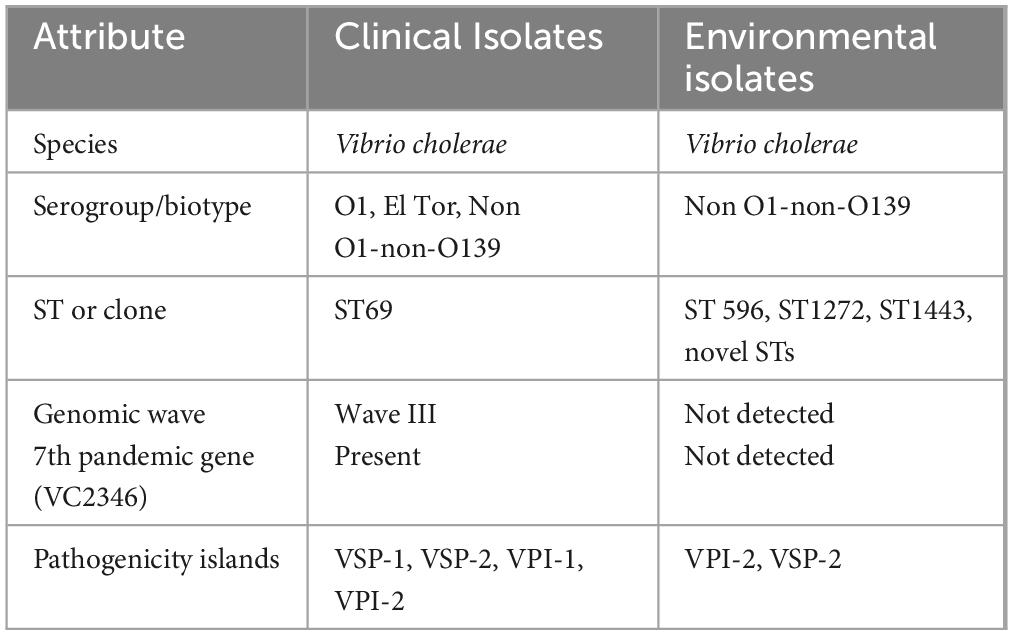
Table 1. Genomic characterization of clinical and environmental isolates from the 2022 to 2023 cholera outbreak in Kenya.
All the 10 environmental isolates neither harbored rfbv-01 nor wbfZ-0139 genes hence were classified as non-O1 and non-O139 V. cholerae (NOVC). Genomes of some of the isolates had virulence factors such as Vibrio pathogenicity islands VPI-2 (n = 1) and VSP-2 (n = 1). Additionally, toxR, als, and hlyA genes were found in all environmental isolates. The rtxA gene was found in 90% of these isolates. In silico MLST revealed that three strains belonged to sequence types 596, 1272, and 1443 with majority (70%) belonging to Novel STs. Both clinical and environmental strains harbored toxR, als, hlyA, and rtxA genes. This is shown in Table 1 and Supplementary Tables 3, 4.
3.2 Phenotypic antibiotic resistance profiles in clinical and environmental Vibrio cholerae isolates
Both clinical and environmental isolates phenotypically expressed antibiotic resistance. The seventy randomly selected clinical isolates had an identical multidrug resistance profile, showing 100% resistance to ampicillin, cefotaxime, ceftriaxone, cefpodoxime, trimethoprim sulfamethoxazole, nalidixic acid, and azithromycin. Susceptibility to gentamicin and chloramphenicol was observed in all clinical strains. The clinical isolates were highly susceptible to tetracycline (99%) while 96% of the isolates exhibited intermediate susceptibility to ciprofloxacin (Table 2). All environmental isolates were susceptible to azithromycin and gentamicin. Phenotypic resistance to ampicillin, trimethoprim sulfamethoxazole, and nalidixic acid was observed in 59%, 41%, and 47% of the environmental isolates respectively as shown in Table 2.
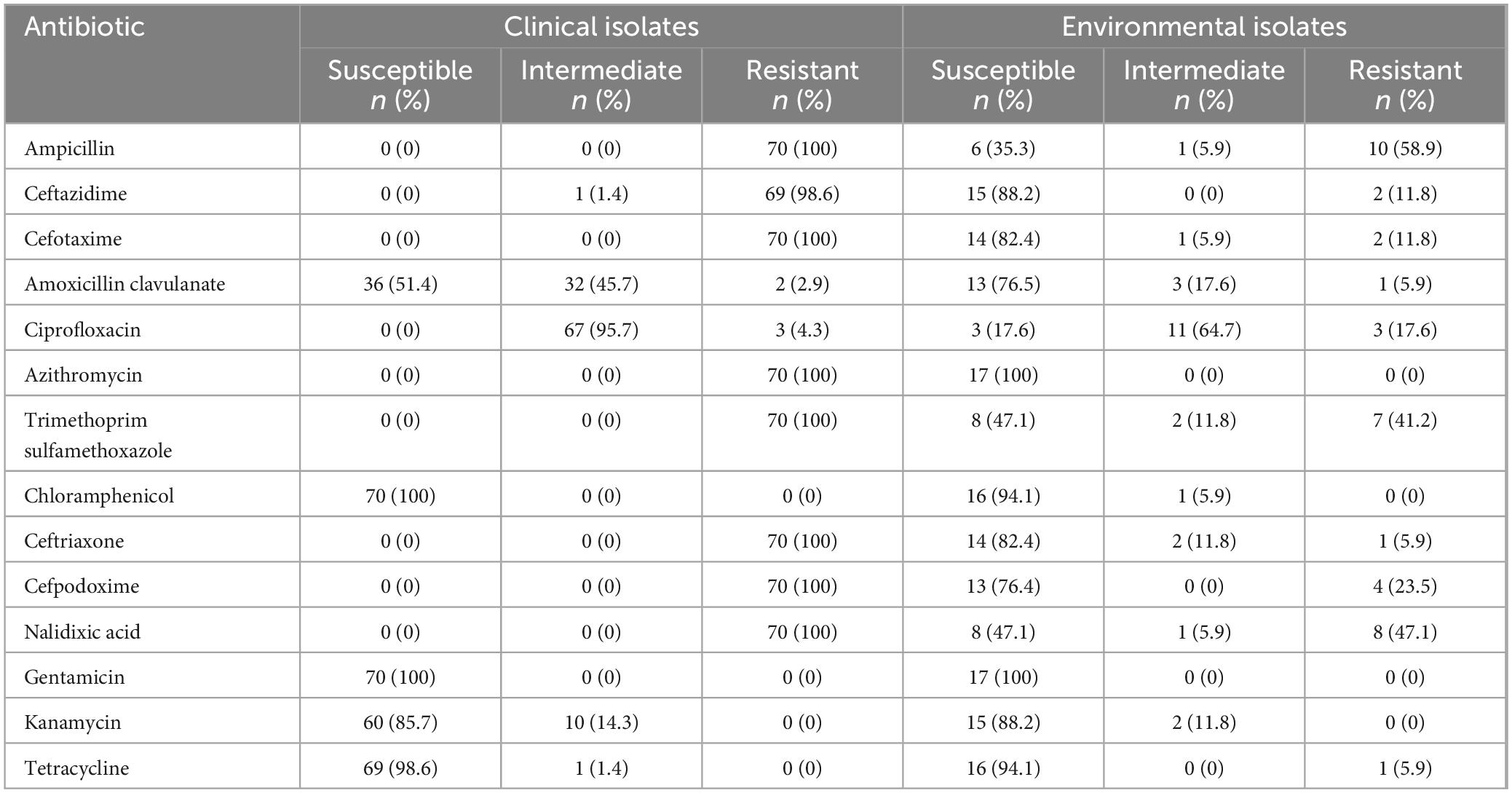
Table 2. Phenotypic antibiotic resistance profile of clinical and environmental isolates.
3.3 Antimicrobial resistance genes and mobile genetic elements in clinical isolates
The dfrA1 gene, conferring resistance to trimethoprim, was detected in all clinical isolates, consistent with their phenotypic resistance to this antibiotic. Furthermore, the sulfonamide resistance gene sul1 was identified in 98% of the isolates. Majority (98%) of the isolates harbored the plasmid-borne extended-spectrum beta-lactamase blaPER-7 gene responsible for third generation cephalosporin resistance. This gene was lacking in the 2016 isolate. Even though all isolates carried the chloramphenicol acetyltransferase gene (catB9), responsible for phenicol resistance, phenotypic resistance to chloramphenicol was not observed. Class 1 integrons, identified by the presence of the intI gene, were detected in 99% isolates. The 2016 isolate lacked intI gene. However, the SXT integron-related resistance determinants dfrA18 and the fluoroquinolone resistance gene qnrVC1 were absent in all samples. Similarly, the SXT-like ICE-borne chloramphenicol resistance gene floR and tetracycline resistance genes were not detected, corresponding to the observed 100% and 98% susceptibility of the clinical isolates to chloramphenicol and tetracycline, respectively (Figure 2 and Supplementary Table 3). Ninety-four (99%) isolates carried the plasmid pVC211, which is associated with high-level resistance to azithromycin, corresponding to the 100% resistance to this antibiotic. The 2016 isolate lacked pVC211. Mutations in gyrA and parC gene were found in all clinical isolates. Additionally, an IncA/C2 plasmid known to carry multiple antimicrobial resistance genes was found in all clinical isolates as shown in Supplementary Table 3.
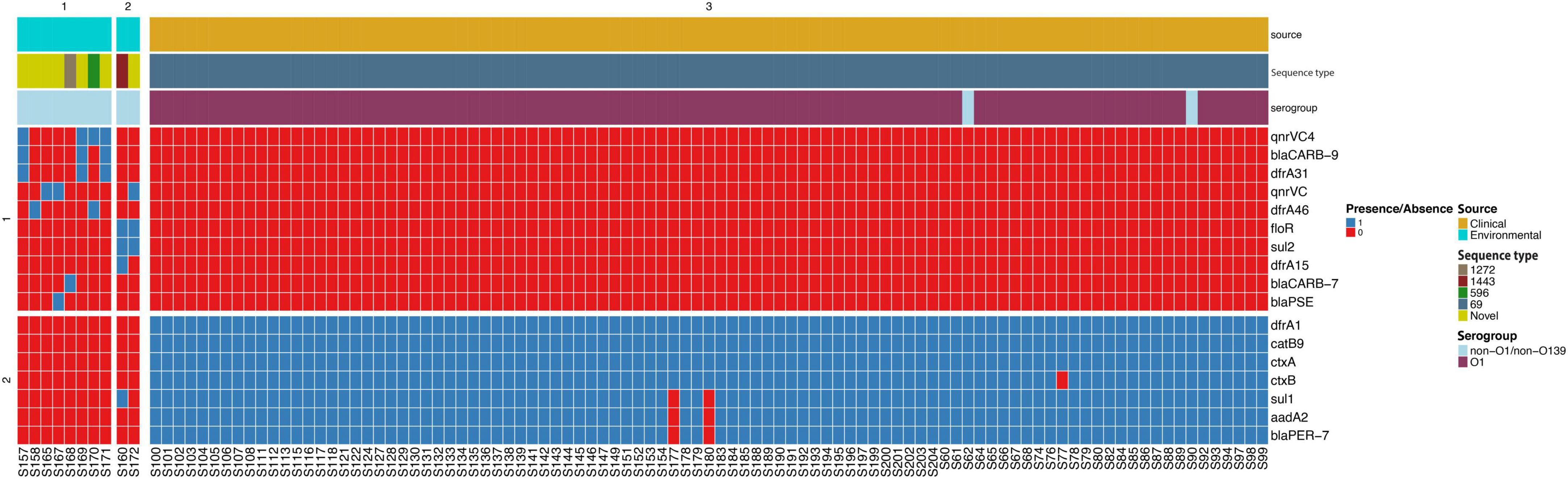
Figure 2. Heat map showing antibiotic resistance genes in clinical and environmental isolates from the 2022 to 2023 cholera outbreak in Kenya. One clinical isolate was collected in 2016. Gene presence is denoted by color blue while red denotes gene absence. The color coded key on the right shows the source of isolates, sequence type, and serogroup.
3.4 Genotypic antibiotic resistance, phage resistance, and mobile genetic elements in environmental isolates
Environmental isolates showed varied resistance profiles. The SXT-like ICE-borne chloramphenicol (CHL) resistance gene floR was detected in 20% of the strains. Two isolates carried the sulfonamide resistance gene sul2 conferring resistance to sulfamethoxazole. The fluoroquinolone resistance gene qnrVC4 was present in 40% of the isolates. Notably, all isolates lacked catB9 gene, a determinant of phenicol resistance. Three isolates carried blaCARB–9 gene, encoding a beta-lactamase enzyme and conferring beta-lactam resistance (Figure 2 and Supplementary Table 4). None of the environmental strains contained the phage susceptibility regions associated with PICI-like elements (PLE1 and PLE2).
3.5 Genetic diversity of environmental and clinical Vibrio cholerae isolates
Phylogenetic analysis of isolates collected during the 2022–2023 cholera outbreak revealed that all the 10 environmental isolates were highly divergent, clustering outside the 7th pandemic El Tor lineage. The difference between these isolates and the reference genome N16961 was between 55,000 and 119,000 SNPs. Multi-locus sequence typing (Jolley et al., 2012) demonstrated significant diversity among the environmental strains with 7 out the 10 isolates classified as novel sequence types that were phylogenetically distinct from previously known types. The other three environmental strains belonged to sequence types (STs) 1272, 1443, and 596 as shown in Figure 3C. This highlights the extensive genetic diversity of V. cholerae in environmental reservoirs. Phylogenetic analysis of the 95 V. cholerae genomes from clinical isolates revealed a high degree of clonality, with fewer than five SNPs differentiating majority of the isolates. However, the 2016 isolate was an exception showing 15 SNP differences. MLST following the methodology described by Jolley et al. (2012) further confirmed the limited genetic diversity, as all the strains were classified under the same rMLST type, ST69 (Figure 3B). It is important to note that all the clinical V. cholerae O1 isolates did not cluster with the 10 environmental isolates (Figure 3A). This further confirms that the clinical strains primarily belonged to the monophyletic 7th pandemic lineage while environmental isolates represent genetically distinct populations.
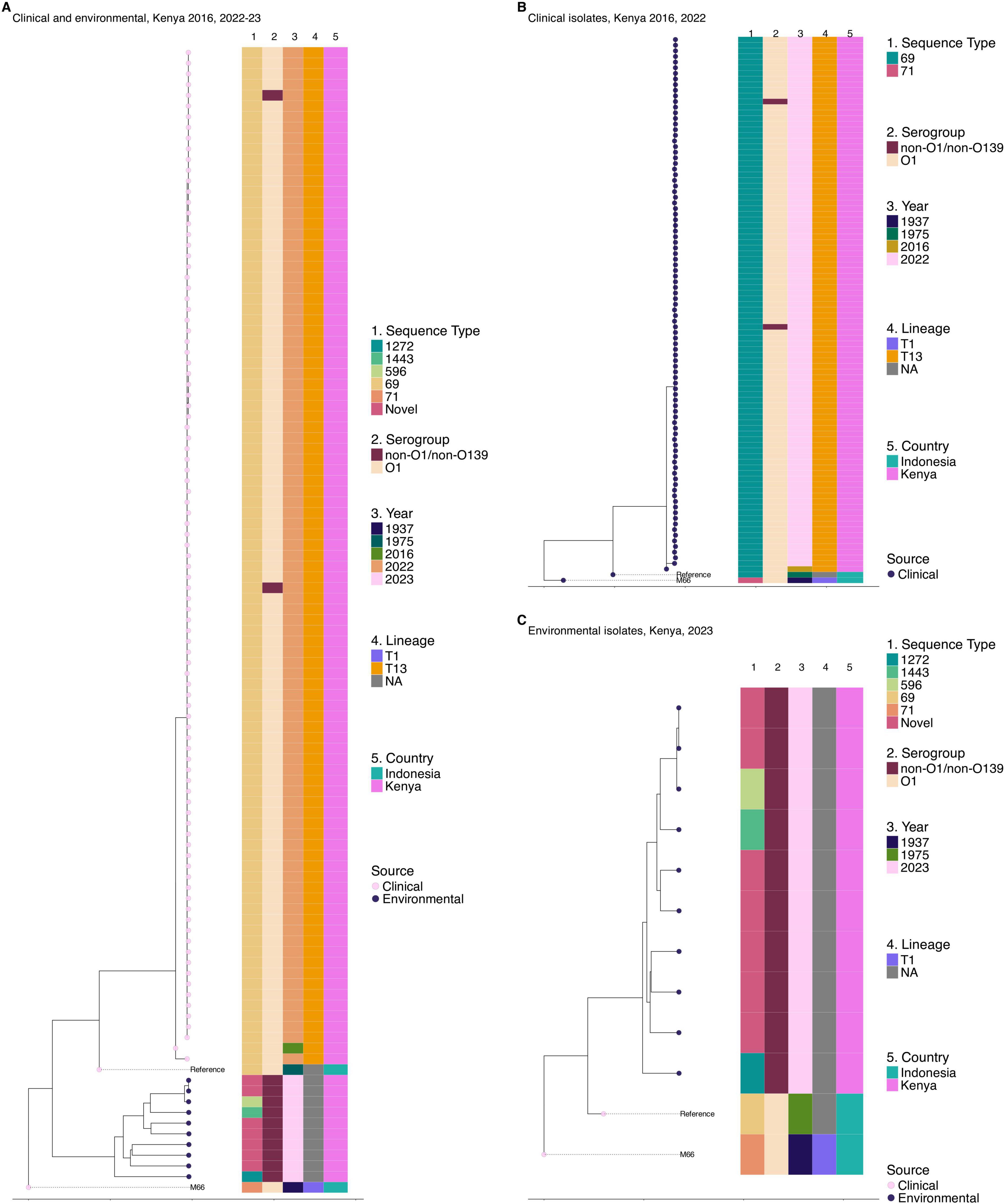
Figure 3. Genetic relatedness of clinical and environmental V. cholerae isolates from 2022 to 2023 outbreak in Kenya. One clinical isolate from 2016 was included. (A) Maximum likelihood phylogenetic tree showing the genetic relatedness of clinical and environmental isolates collected in the 2022–2023 cholera outbreak in Kenya with one clinical isolate from 2016 outbreak included. (B) Maximum likelihood phylogenetic tree illustrating the genetic relatedness of clinical isolates collected in the 2016 (n = 1) and 2022–2023 cholera outbreaks in Kenya. (C) Maximum likelihood phylogenetic tree illustrating the genetic relatedness of environmental isolates collected during the 2022–2023 cholera outbreak in Kenya. The circles at the tip of the phylogeny represent the sample source. Color strips at the tips of each tree represent the sequence type (ST), year of isolation, serogroup, lineage, and country of origin. NA on the lineage for panels (A–C) refers to isolates with no transmission lineage assigned. The phylogeny was constructed based on the reference genome strain of V. cholerae N16961 and rooted based on the pre-seventh pandemic strain M66 used as an outgroup.
Given the high burden of cholera in Haiti and Kenya’s bordering countries, Uganda and Tanzania, further analysis was done to place the 2022–2023 Kenyan isolates within the broader phylogenetic framework of East African and North American 7PET lineages. Comparison of clinical genomes from the 2022 to 2023 Kenyan cholera outbreak revealed clonal relatedness to V. cholerae O1 El Tor isolates from these regions. The maximum likelihood phylogeny showed that the predominant 2022–2023 outbreak-associated 7PET isolates from Kenya formed a single distinct cluster in the phylogeny and belonged to lineage T13 (Figure 4). The historical 7PET isolates from Kenya clustered with sequences from Tanzania and Uganda. Haiti strains were observed to cluster distinctly with clinical and environmental isolates appearing in the same clusters (Figure 4).
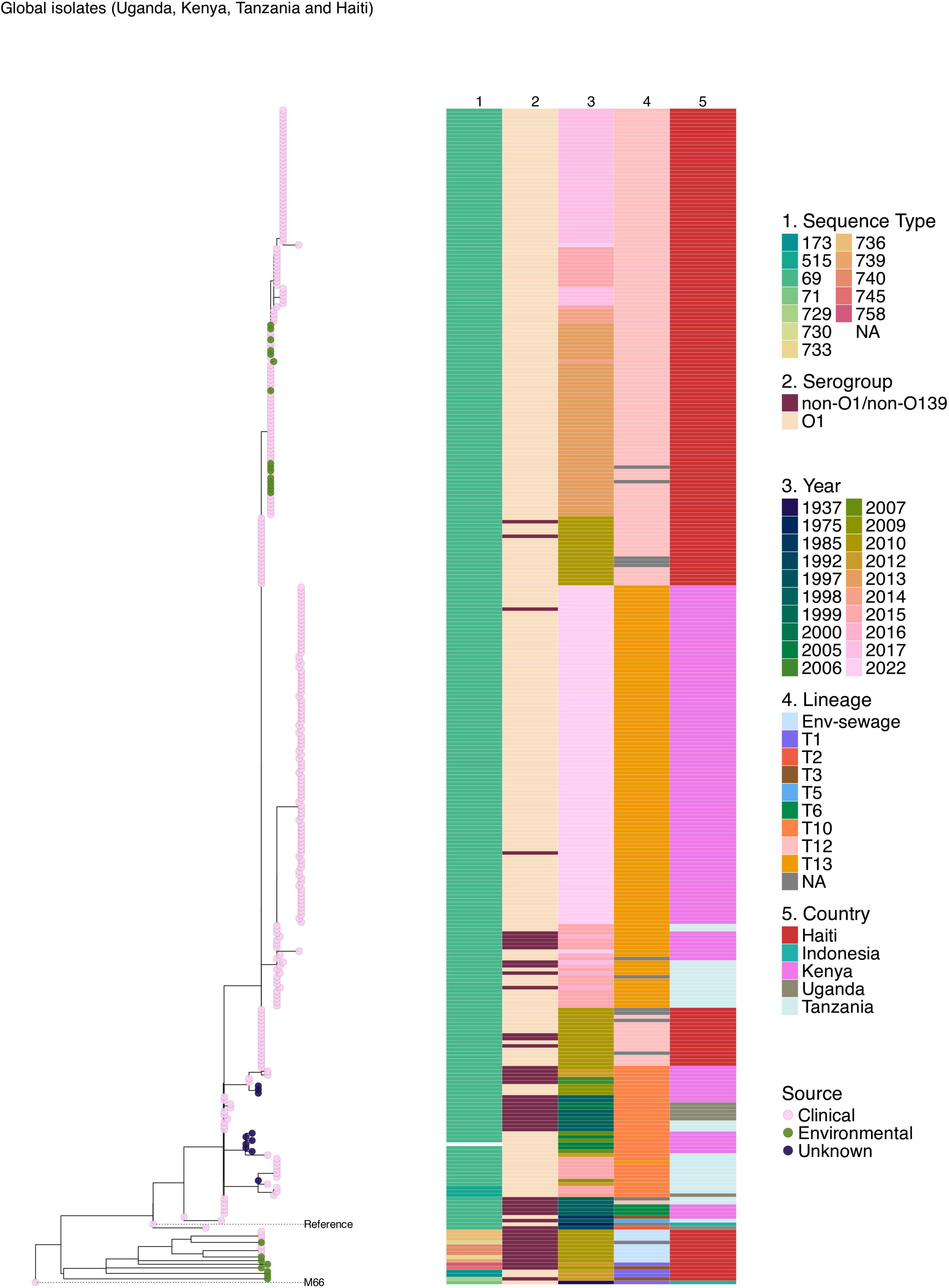
Figure 4. Maximum likelihood phylogenetic tree illustrating the genetic relatedness of clinical and environmental V. cholerae isolates collected during the 2022–2023 cholera outbreak in Kenya and previously published genomes from Kenya, Uganda, Tanzania, and Haiti. The pink, green, and blue circles at the tip of the phylogeny represent the sample type. The phylogeny is annotated by color strips at the tips of each tree representing the sequence type (ST), serogroup (SG), year of isolation, lineage, and country of isolation. The phylogeny was constructed based on the reference genome strain of V. cholerae N16961 and rooted based on the pre-seventh pandemic strain M66 used as an outgroup.
To investigate the potential origin of the 2022–2023 cholera outbreak in Kenya, we conducted a time-scaled phylogenetic analysis incorporating previously published Kenyan genomes alongside clinical genomes from the 2022 to 2023 outbreak (Figure 5). Our analysis revealed that the 2022–2023 isolates showed the highest genetic similarity (15 SNPs, lineage T13) to Kenyan isolates from 2016, suggesting a close evolutionary relationship. We further placed the 2022–2023 clinical isolates within a broader phylogeny including genomes from East Africa and North America, which confirmed a similar pattern of genetic relatedness (Figure 6).
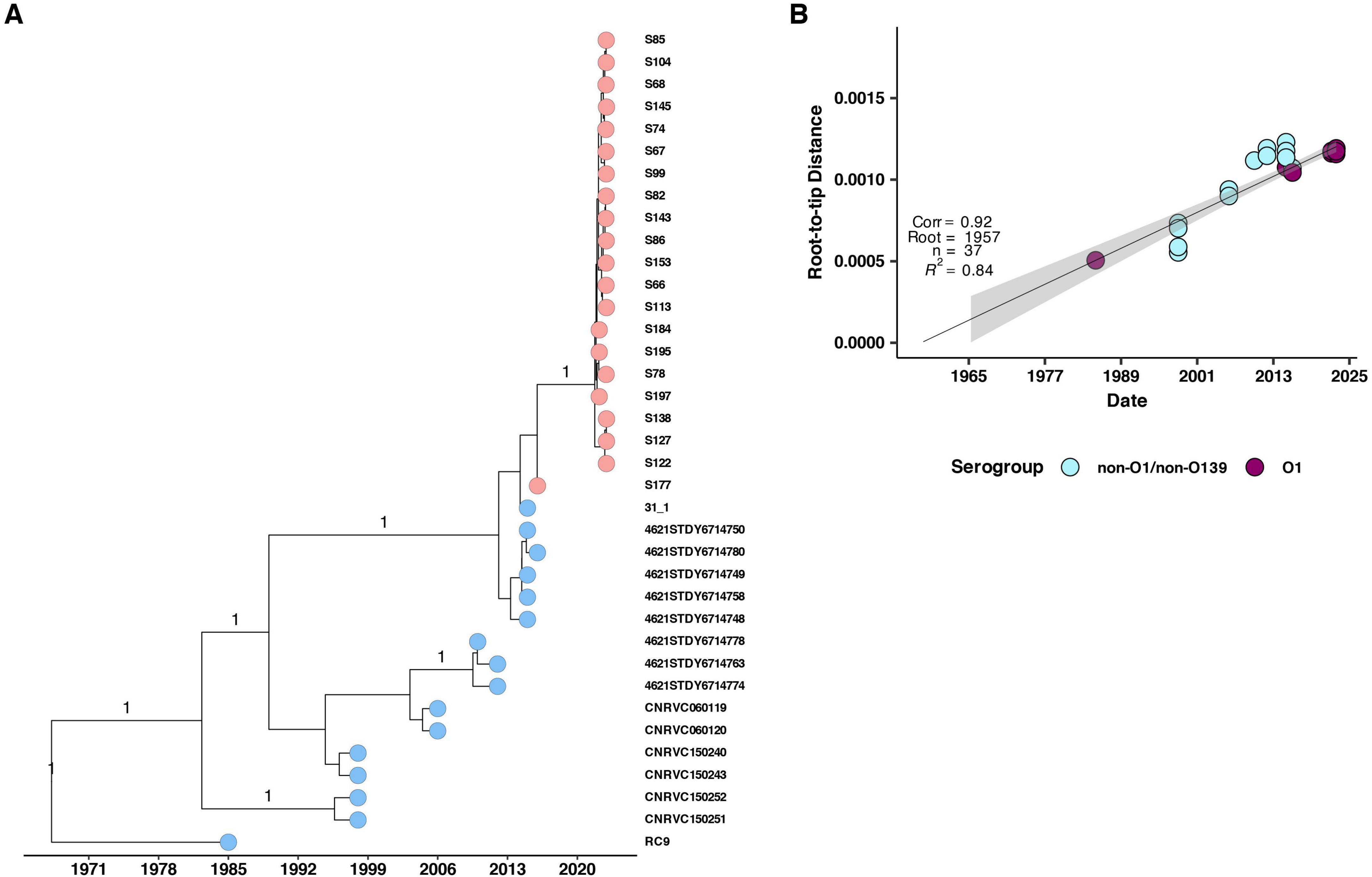
Figure 5. (A) Maximum likelihood phylogenetic tree illustrating the genetic diversity of the 2022 Kenyan clinical V. cholerae isolates and previously published genomes from Kenya. The pink and blue circles at the tip of the phylogeny represent the isolates with historical isolates up to 2015 denoted by blue while 2016 and 2022 isolates are denoted by pink. The horizontal axis indicates the year of isolation. (B) Root to tip distances against collection time (years) on vertical and horizontal axes respectively. We observed a strong correlation (correlation coefficient = 0.92, R2 = 0.84).
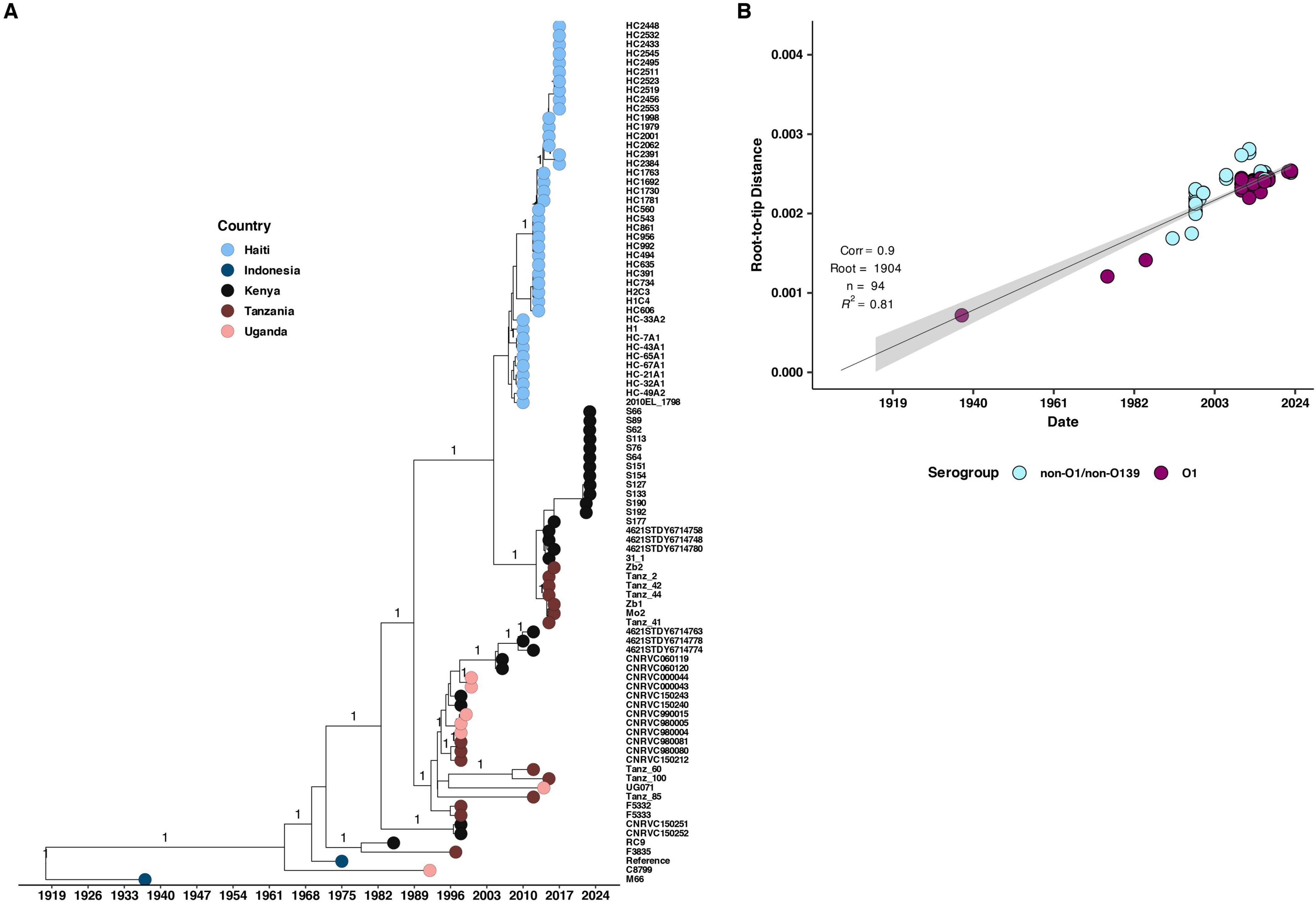
Figure 6. (A) Maximum likelihood phylogenetic tree illustrating the genetic diversity of the 2022 Kenyan clinical V. cholerae isolates and previously published genomes from Kenya, Uganda, Tanzania, and Haiti. The pink, brown, black, and blue circles at the tip of the phylogeny represent the country of isolation while the horizontal axis indicates the year of isolation. (B) Root to tip distances against collection time (years) on vertical and horizontal axes respectively. We observed a strong correlation (correlation coefficient = 0.9, R2 = 0.81).
4 Discussion
Cholera still remains a significant threat to public health since 1971 when the first case was reported in Kenya. Numerous outbreaks have been reported up to the year 2023 highlighting cholera endemic nature in our setting (Tauxe et al., 1995; Shapiro et al., 1999; Mugoya et al., 2008; Scrascia et al., 2009; Shikanga et al., 2009; Mutonga et al., 2013; Kigen et al., 2020). In our study, clinical V. cholerae isolates were primarily identified as serogroup O1 El tor variants and carried the key virulence factors responsible for pathogenicity, including the ctxB7 genotype of the ctxB gene, as well as the rstR and tcpA genes. Earlier studies reported ctxB3 toxin allele in all El tor strains in Kenya (Kiiru et al., 2013). In contrast, none of the environmental isolates tested were positive for these genes. This observation agrees with previous studies, which reported the absence of ctx and tcpA genes in NOVC strains, while all O1 and O139 strains consistently harbored these genes (Faruque et al., 1998; Sharma et al., 1998). However, environmental isolates in our study contained other virulence-associated factors such as VPI-2, VSP-2, and genes including rtxA, als, and hlyA, which have been implicated in diarrheal diseases (Octavia et al., 2013). Among these, the rtxA gene is known to encode a product with cytotoxic activity toward mammalian cells, a key factor driving the virulence of CTX-negative, NOVC strains (Lin et al., 1999). Several studies conducted in tropical regions and globally have linked sporadic cases of gastroenteritis to NOVC strains (Sharma et al., 1998; Schwartz et al., 2019; Siriphap et al., 2017). These findings suggest that although environmental isolates lack the virulence cholera toxin genes, they possess alternative virulence genes that may spread among cholera strains via horizontal gene transfer or MGEs and result in gastrointestinal disease. This highlights the need for continued surveillance and characterization of V. cholerae circulating in our environment.
In our study, a uniform resistance pattern was observed in all clinical isolates highlighting 100% resistance to multiple antibiotics, including ampicillin, cefotaxime, ceftriaxone, cefpodoxime, trimethoprim-sulfamethoxazole, nalidixic acid, and azithromycin. This resistance pattern contrasts with earlier findings from Kenya which documented lower resistance rates to nalidixic acid (64.5%) and ampicillin (3.6%) (Shah et al., 2023) and high susceptibility to ceftriaxone (99%) (Awuor et al., 2020). Additionally, a study by Haque et al. (2023) reported V. cholerae isolates with lower resistance to ceftazidime (27.5%) and cefotaxime (29.4%), findings consistent with reports from other countries (Sahilah et al., 2014; Letchumanan et al., 2015). The mutations observed in gyrA and parC in clinical isolates indicate quinolone resistance, an important evolutionary trait for sub lineages in the 7th cholera pandemic (Opintan et al., 2021; Weill et al., 2017). Majority of the clinical isolates in our study harbored the plasmid-borne extended-spectrum beta-lactamase blaPER-7 gene responsible for third generation cephalosporin resistance. This correlated with observed 100% phenotypic resistance to cefotaxime, ceftriaxone, and cefpodoxime. A study done in Yemen reported similar findings with blaPER-7 gene isolated from multidrug-resistant V. cholerae strains (Lassalle et al., 2023). Clinical strains were all susceptible to chloramphenicol despite presence of catB9 gene possibly due to low gene expression. Similar findings have been reported in a number of studies (Siriphap et al., 2017; Sun et al., 2023) indicating that phenotypic expression is not necessarily related to the presence of the encoding gene. The identification of class 1 integrons in clinical isolates from our study emphasizes their critical role in the acquisition and spread of resistance genes. Notably, these integrons were absent in the sequenced 2016 isolate and in earlier Kenyan studies (Shah et al., 2023) suggesting recent horizontal gene transfer events. The IncA/C2 plasmid found in all clinical isolates in our study is important in horizontal transfer of multiple antimicrobial resistance genes. A previous study in Haiti reported that this plasmid encodes a unique set of resistance determinants and a second copy of the resistance genes hence conferring resistance to multiple antibiotics (Folster et al., 2014). The high number of reported MDR V. cholerae clinical isolates in our study could be attributed to this.
These observations suggest an escalating antibiotic resistance scenario over time. Despite this alarming resistance trend, all clinical strains in our study remained susceptible to gentamicin and chloramphenicol while 99% of the isolates were susceptible to tetracyclines. Tetracyclines (doxycycline) have historically been the drug of choice during cholera outbreaks (Poulin-Laprade et al., 2015). It is critical to monitor usage of these antibiotics to reduce the risk of developing resistance as there are fewer therapeutic options now available.
Environmental isolates showed a more varied resistance profile, with 100% susceptibility to gentamicin and azithromycin. Interestingly, three environmental NOVC isolates carried blaCARB–9 gene conferring beta-lactam resistance. This gene possibly acquired through MGEs has been widely reported in bacteria (De, 2021) with recent studies in Austria and Argentina reporting blaCARB–9 in environmental NOVC strains (Petroni et al., 2004; Lepuschitz et al., 2019). Environmental isolates also carried unique resistance determinants, including the SXT-like ICE-borne floR and qnrVC4 genes, which confer resistance to chloramphenicol and fluoroquinolones, respectively. Furthermore, the lack of phage susceptibility regions associated with PICI-like elements in environmental strains impacts their persistence in the environment. Previously, bacteriophages infective to V. cholerae have been isolated from environmental waters (Maina et al., 2014). There is need to monitor both clinical and environmental strains in order to track the spread of MDR determinants and understand the role of environmental V. cholerae in driving emergence of new MDR strains.
The AMR genes circulating in environmental isolates were different from those found in clinical isolates. For instance, the floR and blaCARB–9 genes were detected in environmental isolates but absent in all clinical samples, whereas the catB9 gene was present in all clinical isolates but absent in environmental isolates. This indicates that the AMR gene profiles in clinical isolates may be evolving differently from those in environmental populations due to a difference in selective pressure.
While majority of clinical isolates clustered together and showed a high degree of genetic relatedness, environmental NOVC isolates were highly divergent with isolates belonging to novel and distinct STs, including ST1272, ST1443, and ST596. This high level of diversity is consistent with previous studies that identified mutation and genetic recombination as key factors driving variation among V. cholerae isolates (Octavia et al., 2013; Salim et al., 2005; Feng et al., 2008). Although these environmental isolates clustered outside the 7th pandemic El Tor lineage, they have the potential to cause mild diarrhea and contribute to spread of AMR determinants hence the need for ongoing surveillance to understand the role of environmental isolates in evolution of clinical V. cholerae strains.
Comparison of clinical genomes from the 2022 to 2023 Kenyan cholera outbreak with previously published genomes from Kenya, Uganda, Tanzania, and Haiti revealed clonal relatedness to V. cholerae O1 El Tor isolates from these regions. This agrees with previous studies (Kiiru et al., 2013; Morita et al., 2020) which showed that V. cholerae O1 El Tor isolates in Kenya and countries in Southeast Asia are clonally related to strains from other regions globally. Similarly, Stine and Morris (2013) reported that isolates from the sixth and seventh pandemics share a single ancestral origin. The historical 7PET isolates from Kenya clustered with sequences from Tanzania and Uganda suggesting cross border spread of cholera. This was in agreement with an earlier study that reported cross border cholera outbreaks as one of the major contributors to the high cholera burden in Sub Saharan Africa (Bwire et al., 2016). Although there is no direct evidence of recent transmission events between Haiti and Kenya, the shared ancestral lineage of isolates from the sixth and seventh pandemics highlights the potential for global dissemination of epidemic V. cholerae strains through international travel and trade. The cholera epidemic that affected Haiti from October 2010 to February 2019 has been attributed to the introduction of V. cholerae by United Nations peacekeepers originating from South Asia (Piarroux et al., 2011). This shows the role of human movement in the transcontinental spread of cholera further highlighting the importance of regional and global surveillance in effectively monitoring and controlling the spread of cholera. Clinical and environmental isolates from Haiti appeared in the same clusters suggesting environmental persistence and possible spill over to human populations.
The 2022–2023 cholera outbreak isolates showed the highest genetic similarity (15 SNPs) to Kenyan isolates from 2016, suggesting a close evolutionary relationship. These results indicate that the 2022–2023 outbreak did not arise from a new introduction but instead resulted from re-emergence of previously circulating strains in Kenya that had persisted since 2016. Sporadic cholera outbreaks were reported in Kenya each year from 2016 to 2022.
While we examined clinical genomes from the 2022 to 2023 cholera outbreak alongside genomes from previous outbreaks in Kenya, Uganda, Tanzania, and Haiti, including more countries would have provided a better global perspective on cholera transmission patterns. However, the selected regions offer important insights into V. cholerae genomics within regional and global contexts. Future genomic surveillance studies incorporating more countries will further enhance our understanding of the spread and genetic diversity of V. cholerae globally.
5 Conclusion
In conclusion, our study shows that the 2022–2023 Kenyan cholera outbreak has been attributed to 7PET O1 ogawa V. cholerae strains carrying IncA/C2 plasmids and multidrug resistant genes and likely resulted from re-emergence of previously circulating strains rather than a new introduction. Kenyan clinical isolates remain susceptible to tetracycline, gentamicin, and chloramphenicol. Although environmental contamination as a source of human infection cannot be clearly elucidated in our study, it is important to note that environmental isolates possess virulent and AMR genes that may spread to clinical and other environmental strains via horizontal gene transfer or MGEs. This highlights the need for continued surveillance in order to track V. cholerae evolution, understand transmission pattern, and limit the development and spread of antimicrobial resistance.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
Ethics statement
The studies involving humans were approved by Kenyatta National Hospital-University of Nairobi Ethical Review Committee (KNH-UON ERC). The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
Author contributions
LM: Conceptualization, Data curation, Formal Analysis, Methodology, Writing – original draft, Writing – review & editing. GA: Conceptualization, Methodology, Supervision, Writing – review & editing. ZM: Data curation, Formal Analysis, Writing – review & editing. PG: Conceptualization, Supervision, Writing – review & editing. JJ: Data curation, Formal Analysis, Writing – review & editing. KK: Conceptualization, Data curation, Methodology, Writing – review & editing. CK: Formal Analysis, Methodology, Writing – review & editing. DI: Formal Analysis, Writing – review & editing. BO: Formal Analysis, Methodology, Writing – review & editing. MM: Formal Analysis, Methodology, Writing – review & editing. CM: Conceptualization, Methodology, Project administration, Supervision, Writing – review & editing. SK: Conceptualization, Data curation, Funding acquisition, Methodology, Supervision, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by a Wellcome and the UK Foreign, Commonwealth and Development Office Grant (215675/Z/19/Z) (SK), and Fogarty International Center and the Institute of Allergy and Infectious Diseases of the National Institutes of Health grant (D43TW011519).
Acknowledgments
We acknowledge the Centre for Microbiology Research Laboratory staff for their invaluable contribution in sample collection and laboratory analysis. Community Health Volunteers’ role during sample collection is appreciated.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Author disclaimer
The content in this study is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2025.1603736/full#supplementary-material
Footnotes
1. ^https://www.nphi.go.ke/sites/default/files/2024-02/IDSR%20Clinicians%20Handbook.pdf
2. ^https://cge.food.dtu.dk/services/CholeraeFinder/
References
Abana, D., Gyamfi, E., Dogbe, M., Opoku, G., Opare, D., Boateng, G., et al. (2019). Investigating the virulence genes and antibiotic susceptibility patterns of Vibrio cholerae O1 in environmental and clinical isolates in Accra, Ghana. BMC Infect. Dis. 19:76. doi: 10.1186/s12879-019-3714-z
Ali, M., Nelson, A. R., Lena Lopez, A., and Sack, D. A. (2015). Updated global burden of cholera in endemic countries. PLoS Negl. Trop. Dis. 9:e0003832. doi: 10.1371/journal.pntd.0003832
Awuor, S. O., Omwenga, E. O., and Daud, I. I. (2020). Geographical distribution and Antibiotic susceptibility patterns of toxigenic Vibrio cholerae isolates from Kisumu County, Kenya. Afr. J. Prim. Health Care Fam. Med. 12, 1–6. doi: 10.4102/PHCFM.V12I1.2264
Banerjee, R., Das, B., Balakrish Nair, G., and Basak, S. (2014). Dynamics in genome evolution of Vibrio cholerae. Infect. Genet. Evol. 23, 32–41. doi: 10.1016/j.meegid.2014.01.006
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Bhandari, M., Jennison, A. V., Rathnayake, I. U., and Huygens, F. (2021). Evolution, distribution and genetics of atypical Vibrio cholerae – A review. Infect. Genet. Evol. 89:104726. doi: 10.1016/j.meegid.2021
Biemer, J. J. (1973). Antimicrobial susceptibility testing by the Kirby-Bauer disc diffusion method. Ann. Clin. Lab. Sci. 3, 135–140.
Bwire, G., Mwesawina, M., Baluku, Y., Kanyanda, S. S. E., and Orach, C. G. (2016). Cross-border cholera outbreaks in Sub-Saharan Africa, the mystery behind the silent illness: What needs to be done? PLoS One 11:e0156674. doi: 10.1371/journal.pone.0156674
Chaguza, C., Chibwe, I., Chaima, D., Musicha, P., Ndeketa, L., Kasambara, W., et al. (2024). Genomic insights into the 2022–2023 Vibrio cholerae outbreak in Malawi. Nat. Commun. 15:6291. doi: 10.1038/s41467-024-50484-w
Chatterjee, S., Ghosh, K., Raychoudhuri, A., Chowdhury, G., Bhattacharya, M. K., Mukhopadhyay, A. K., et al. (2009). Incidence, virulence factors, and clonality among clinical strains of non-O1, non-O139 Vibrio cholerae isolates from hospitalized diarrheal patients in Kolkata, India. J. Clin. Microbiol. 47, 1087–1095. doi: 10.1128/JCM.02026-08
Chun, J., Grim, C. J., Hasan, N. A., Je, H. L., Seon, Y. C., Haley, B. J., et al. (2009). Comparative genomics reveals mechanism for short-term and long-term clonal transitions in pandemic Vibrio cholerae. Proc. Natl. Acad. Sci. U S A. 106, 15442–15447. doi: 10.1073/pnas.0907787106
CLSI (2018). Methods for Antimicrobial Dilution and Disk Susceptibility Testing of Infrequently Isolated or Fastidious Bacteria. Available Online at https://clsi.org/media/1450/m45ed3_sample.pdf (accessed March 31, 2025).
Croucher, N. J., Page, A. J., Connor, T. R., Delaney, A. J., Keane, J. A., Bentley, S. D., et al. (2015). Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucleic Acids Res. 43:e15. doi: 10.1093/nar/gku1196
Dalsgaard, A., Forslund, A., Bodhidatta, L., Serichantalergs, O., Pitarangsi, C., Pang, L., et al. (1999). A high proportion of Vibrio cholerae strains isolated from children with diarrhoea in Bangkok, Thailand are multiple antibiotic resistant and belong to heterogenous non-O1, non-O139 O-serotypes. Epidemiol. Infect. 122, 217–226. doi: 10.1017/S0950268899002137
Davis, B. M., and Waldor, M. K. (2003). Filamentous phages linked to virulence of Vibrio cholerae. Curr. Opin. Microbiol. 6, 35–42. doi: 10.1016/s1369-5274(02)00005-x
De, R. (2021). Mobile genetic elements of Vibrio cholerae and the evolution of its antimicrobial resistance. Front. Trop. Dis. 2:691604. doi: 10.3389/fitd.2021.691604
Faruque, S. M., Asadulghani, Saha, M. N., Alim, A. R., Albert, M. J., Islam, K. M., et al. (1998). Analysis of clinical and environmental strains of non toxigenic Vibrio cholerae for Susceptibility to CTX: Molecular basis for origination of new strains with epidemic potential. Infect. Immun. 66, 5819–5825. doi: 10.1128/IAI.66.12.5819-5825.1998
Faruque, S. M., Kamruzzaman, M., Meraj, I. M., Chowdhury, N., Nair, G. B., Sack, R. B., et al. (2003). Pathogenic potential of environmental Vibrio cholerae strains carrying genetic variants of the toxin-coregulated pilus pathogenicity island. Infect. Immun. 71, 1020–1025. doi: 10.1128/IAI.71.2.1020-1025.2003
Feng, L., Reeves, P. R., Lan, R., Ren, Y., Gao, C., Zhou, Z., et al. (2008). A recalibrated molecular clock and independent origins for the cholera pandemic clones. PLoS One 3:e4053. doi: 10.1371/journal.pone.0004053
Feng, Y., Jadhav, A. P., Rodighiero, C., Fujinaga, Y., Kirchhausen, T., and Lencer, W. I. (2004). Retrograde transport of cholera toxin from the plasma membrane to the endoplasmic reticulum requires the trans-Golgi network but not the Golgi apparatus in Exo2-treated cells. EMBO Rep. 5, 596–601. doi: 10.1038/sj.embor.7400152
Folster, J. P., Katz, L., McCullough, A., Parsons, M. B., Knipe, K., and Sammons, S. A. (2014). Multidrug-resistant IncA/C plasmid in Vibrio cholerae from Haiti. Emerg. Infect. Dis. 20, 1951–1953. doi: 10.3201/eid2011.140889
Haque, Z. F., Islam, M. S., Sabuj, A. A. M., Pondit, A., Sarkar, A. K., Hossain, M. G., et al. (2023). Molecular detection and antibiotic resistance of Vibrio cholerae, Vibrio parahaemolyticus, and Vibrio alginolyticus from Shrimp (Penaeus monodon) and shrimp environments in Bangladesh. Aquac. Res. 1:5436552. doi: 10.1155/2023/5436552
Harris, J. B., LaRocque, R. C., Qadri, F., Ryan, E. T., and Calderwood, S. B. (2012). Cholera. Lancet 379, 2466–2476. doi: 10.1016/S0140-6736(12)60436-X
Hounmanou, Y. M. G., Mdegela, R. H., Dougnon, T. V., Mhongole, O. J., Mayila, E. S., Malakalinga, J., et al. (2016). Toxigenic Vibrio cholerae O1 in vegetables and fish raised in wastewater irrigated fields and stabilization ponds during a non-cholera outbreak period in Morogoro, Tanzania: An environmental health study. BMC Res. Notes 9:466. doi: 10.1186/s13104-016-2283-0
Huang, J., Zhu, Y., Wen, H., Zhang, J., Huang, S., Niu, J., et al. (2009). Quadruplex real-time PCR assay for detection and identification of Vibrio cholerae O1 and O139 strains and determination of their toxigenic potential. Appl. Environ. Microbiol. 75, 6981–6985. doi: 10.1128/AEM.00517-09
IFRC (2024). Kenya: Cholera Outbreak - DREF Final Report (MDRKE054) - Kenya | ReliefWeb. Available online at: https://reliefweb.int/report/kenya/kenya-cholera-outbreak-dref-final-report-mdrke054 (accessed March 29, 2025).
Jolley, K. A., Bliss, C. M., Bennett, J. S., Bratcher, H. B., Brehony, C., Colles, F. M., et al. (2012). Ribosomal multilocus sequence typing: Universal characterization of bacteria from domain to strain. Microbiology 158, 1005–1015. doi: 10.1099/mic.0.055459-0
Karaolis, D. K., Johnson, J. A., Bailey, C. C., Boedeker, E. C., Kaper, J. B., and Reeves, P. R. (1998). A Vibrio cholerae pathogenicity island associated with epidemic and pandemic strains. Proc. Natl. Acad. Sci. U S A. 95, 3134–3139. doi: 10.1073/pnas.95.6.3134
Kigen, H. T., Boru, W., Gura, Z., Githuka, G., Mulembani, R., Rotich, J., et al. (2020). A protracted cholera outbreak among residents in an urban setting, Nairobi County, Kenya, 2015. Pan. Afr. Med. J. 36:127. doi: 10.11604/pamj.2020.36.127.19786
Kiiru, J., Mutreja, A., Mohamed, A. A., Kimani, R. W., Mwituria, J., Sanaya, R. O., et al. (2013). A study on the geophylogeny of clinical and environmental Vibrio cholerae in Kenya. PLoS One 8:e74829. doi: 10.1371/journal.pone.0074829
Kimani, R. W., Muigai, A. W. T., Sang, W., Kiiru, J. N., and Kariuki, S. (2014). Virulence factors in environmental and clinical Vibrio cholerae from endemic areas in Kenya. Afr. J. Lab. Med. 3:41. doi: 10.4102/ajlm.v3i1.41
Lassalle, F., Al-Shalali, S., Al-Hakimi, M., Njamkepo, E., Bashir, I. M., Dorman, M. J., et al. (2023). Genomic epidemiology reveals multidrug resistant plasmid spread between Vibrio cholerae lineages in Yemen. Nat. Microbiol. 8, 1787–1798. doi: 10.1038/s41564-023-01472-1
Leibovici-Weissman, Y., Neuberger, A., Bitterman, R., Sinclair, D., Salam, M. A., and Paul, M. (2014). Antimicrobial drugs for treating cholera. Cochrane Database Syst. Rev. 2014:CD008625. doi: 10.1002/14651858.CD008625.pub2
Lepuschitz, S., Baron, S., Larvor, E., Granier, S. A., Pretzer, C., Mach, R. L., et al. (2019). Phenotypic and genotypic antimicrobial resistance traits of Vibrio cholerae Non-O1/Non-O139 isolated from a large Austrian lake frequently associated with cases of human infection. Front. Microbiol. 10:2600. doi: 10.3389/fmicb.2019.02600
Letchumanan, V., Pusparajah, P., Tan, L. T. H., Yin, W. F., Lee, L. H., and Chan, K. G. (2015). Occurrence and antibiotic resistance of Vibrio parahaemolyticus from Shellfish in Selangor, Malaysia. Front. Microbiol. 6:1417. doi: 10.3389/fmicb.2015.01417
Letunic, I., and Bork, P. (2021). Interactive tree of life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296. doi: 10.1093/nar/gkab301
Lin, W., Fullner, K. J., Clayton, R., Sexton, J. A., Rogers, M. B., Calia, K. E., et al. (1999). Identification of a Vibrio cholerae RTX toxin gene cluster that is tightly linked to the cholera toxin prophage. Proc. Natl. Acad. Sci. U S A. 96, 1071–1076. doi: 10.1073/pnas.96.3.1071
Maina, A. N., Mwaura, F. B., Oyugi, J., Goulding, D., Toribio, A. L., and Kariuki, S. (2014). Characterization of Vibrio cholerae bacteriophages isolated from the environmental waters of the lake victoria region of kenya. Curr. Microbiol. 68, 64–70. doi: 10.1007/s00284-013-0447-x
Morita, M., Okada, K., Yamashiro, T., Sekizuka, T., Roobthaisong, A., Wongboot, W., et al. (2020). Phylogenetic analysis revealed the dissemination of closely related epidemic Vibrio cholerae O1 isolates in laos, Thailand, and Vietnam. Open Forum Infect. Dis. 7:ofaa492. doi: 10.1093/ofid/ofaa492
Mugoya, I., Kariuki, S., Galgalo, T., Njuguna, C., Omollo, J., Njoroge, J., et al. (2008). Rapid spread of Vibrio cholerae O1 Throughout Kenya, 2005. Am. J. Trop. Med. Hyg. 78, 527–533. doi: 10.4269/ajtmh.2008.78.527
Muriithi, E., and Obare, M. (2017). An Assessment of Sanitation situation in Mukuru Kwa Njenga Informal settlement. The Case of Sisal area. BY. ResearchGate. doi: 10.13140/RG.2.2.13672.83204
Mutonga, D., Langat, D., Mwangi, D., Tonui, J., Njeru, M., Abade, A., et al. (2013). National surveillance data on the epidemiology of cholera in Kenya, 1997-2010. J. Infect. Dis. 208, S55–S61. doi: 10.1093/infdis/jit201
Nguyen, L. T., Schmidt, H. A., Von Haeseler, A., and Minh, B. Q. (2015). IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Octavia, S., Salim, A., Kurniawan, J., Lam, C., Leung, Q., Ahsan, S., et al. (2013). Population structure and evolution of Non-O1/Non-O139 Vibrio cholerae by multilocus sequence typing. PLoS One 8:e65342. doi: 10.1371/journal.pone.0065342
Onifade, T. J., Hutchinson, R., Van Zile, K., Bodager, D., Baker, R., and Blackmore, C. (2011). Toxin producing Vibrio cholerae O75 outbreak, United States, March to April 2011. Euro Surveill. 16:19870.
Opintan, J. A., Will, R. C., Kuma, G. K., Osei, M., Akumwena, A., Boateng, G., et al. (2021). Phylogenetic and antimicrobial drug resistance analysis of Vibrio cholerae O1 isolates from Ghana. Microb. Genom. 7:000668. doi: 10.1099/mgen.0.000668
Petroni, A., Melano, R. G., Saka, H. A., Garutti, A., Mange, L., Pasterán, F., et al. (2004). CARB-9, a carbenicillinase encoded in the VCR region of Vibno cholerae non-O1, non-O139 belongs to a family of cassette-encoded β-lactamases. Antimicrob. Agents Chemother. 48, 4042–4046. doi: 10.1128/AAC.48.10.4042-4046.2004
Piarroux, R., Barrais, R., Faucher, B., Haus, R., Piarroux, M., Gaudart, J., et al. (2011). Understanding the cholera epidemic, Haiti. Emerg. Infect. Dis. 17, 1161–1168. doi: 10.3201/eid1707.110059
Posada, D., and Crandall, K. A. (2001). Selecting the best-fit model of nucleotide substitution. Syst. Biol. 50, 580–601. doi: 10.1080/10635150118469
Poulin-Laprade, D., Carraro, N., and Burrus, V. (2015). The extended regulatory networks of SXT/R391 integrative and conjugative elements and IncA/C conjugative plasmids. Front. Microbiol. 6:837. doi: 10.3389/fmicb.2015.00837
Rambaut, A., Drummond, A. J., Xie, D., Baele, G., and Suchard, M. A. (2018). Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 67, 901–904. doi: 10.1093/sysbio/syy032
Raychoudhuri, A., Mukhopadhyay, A., Ramamurthy, T., Nandy, R., Takeda, Y., and Nair, G. (2008). Biotyping of Vibrio cholerae O1: Time to redefine the scheme. Indian J. Med. Res. 128, 695–698.
Sahilah, A. M., Laila, R. A., Sallehuddin, H. M., Osman, H., Aminah, A., and Ahmad Azuhairi, A. (2014). Antibiotic resistance and molecular typing among cockle (Anadara granosa) strains of Vibrio parahaemolyticus by polymerase chain reaction (PCR)-based analysis. World J. Microbiol. Biotechnol. 30, 649–659. doi: 10.1007/s11274-013-1494-y
Salim, A., Lan, R., and Reeves, P. R. (2005). Vibrio cholerae pathogenic clones. Emerg. Infect. Dis. 11, 1758–1760. doi: 10.3201/eid1111.041170
Schwartz, K., Hammerl, J. A., Göllner, C., and Strauch, E. (2019). Environmental and clinical strains of Vibrio cholerae non-O1, non-O139 from Germany possess similar virulence gene profiles. Front. Microbiol. 10:733. doi: 10.3389/fmicb.2019.00733
Scrascia, M., Pugliese, N., Maimone, F., Mohamud, K. A., Grimont, P. A. D., Materu, S. F., et al. (2009). Clonal relationship among Vibrio cholerae O1 El Tor strains isolated in Somalia. Int. J. Med. Microbiol. 299, 203–207. doi: 10.1016/j.ijmm.2008.07.003
Shah, M. M., Bundi, M., Kathiiko, C., Guyo, S., Galata, A., Miringu, G., et al. (2023). Antibiotic-resistant Vibrio cholerae O1 and Its SXT elements associated with two cholera epidemics in Kenya in 2007 to 2010 and 2015 to 2016. Microbiol. Spectr. 11:e0414022. doi: 10.1128/spectrum.04140-22
Shapiro, R. L., Otieno, M. R., Adcock, P. M., Phillips-Howard, P. A., Hawley, W. A., Kumar, L., et al. (1999). Transmission of epidemic Vibrio cholerae O1 in rural western Kenya associated with drinking water from Lake Victoria: An environmental reservoir for cholera? Am. J. Trop. Med. Hyg. 60, 271–276. doi: 10.4269/ajtmh.1999.60.271
Sharma, C., Thungapathra, M., Ghosh, A., Mukhopadhyay, A. K., Basu, A., Mitra, R., et al. (1998). Molecular analysis of non-O1, non-O139 Vibrio cholerae associated with an unusual upsurge in the incidence of cholera-like disease in Calcutta. India. J Clin Microbiol. 36, 756–763. doi: 10.1128/JCM.36.3.756-763.1998
Shikanga, O. T., Mutonga, D., Abade, M., Amwayi, S., Ope, M., Limo, H., et al. (2009). High mortality in a cholera outbreak in Western Kenya after post-election violence in 2008. Am. J. Trop. Med. Hyg. 81, 1085–1090. doi: 10.4269/ajtmh.2009.09-0400
Singh, D. V., Matte, M. H., Matte, G. R., Jiang, S., Sabeena, F., Shukla, B. N., et al. (2001). Molecular analysis of Vibrio cholerae O1, O139, non-O1, and non-O139 strains: Clonal relationships between clinical and environmental isolates. Appl. Environ. Microbiol. 67, 910–921. doi: 10.1128/AEM.67.2.910-921.2001
Siriphap, A., Leekitcharoenphon, P., Kaas, R. S., Theethakaew, C., Aarestrup, F. M., Sutheinkul, O., et al. (2017). Characterization and genetic variation of Vibrio cholerae isolated from clinical and environmental sources in Thailand. PLoS One 12:e0169324. doi: 10.1371/journal.pone.0169324
Stine, O. C., and Morris, J. G. (2013). Circulation and transmission of clones of Vibrio cholerae during cholera outbreaks. Curr Top Microbiol Immunol. 379, 181–193. doi: 10.1007/82_2013_360
Substitution Models (2024). Available online at: http://www.iqtree.org/doc/Substitution-Models (accessed March 20, 2025).
Suchard, M. A., Lemey, P., Baele, G., Ayres, D. L., Drummond, A. J., and Rambaut, A. (2018). Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 4:vey016. doi: 10.1093/ve/vey016
Sun, Y. H., Wu, Y. L., and Liao, B. Y. (2023). Phenotypic heterogeneity in human genetic diseases: Ultrasensitivity-mediated threshold effects as a unifying molecular mechanism. J. Biomed. Sci. 30:58. doi: 10.1186/s12929-023-00959-7
Tappero, J. W., and Tauxe, R. V. (2011). Lessons learned during public health response to cholera epidemic in Haiti and the Dominican Republic. Emerg. Infect. Dis. 17, 2087–2093. doi: 10.3201/eid1711.110827
Tauxe, R. V., Mintz, E. D., and Quick, R. E. (1995). Epidemic cholera in the new world: Translating field epidemiology into new prevention strategies. Emerg. Infect. Dis. 1, 141–146. doi: 10.3201/eid0104.950408
Weill, F. X., Domman, D., Njamkepo, E., Tarr, C., Rauzier, J., Fawal, N., et al. (2017). Genomic history of the seventh pandemic of cholera in Africa. Science 358, 785–789. doi: 10.1126/science.aad5901
WHO (2024). Cholera. Available online at: https://www.who.int/news-room/fact-sheets/detail/cholera (accessed 10 March 2025)
Keywords: cholera, antimicrobial resistance, whole genome sequencing, phylogenetic analysis, virulence
Citation: Mageto LM, Aboge GO, Mekuria ZH, Gathura P, Juma J, Mugo M, Kebenei CK, Imoli D, Ongadi BA, Kering K, Mbae CK and Kariuki S (2025) Genomic characterization of Vibrio cholerae isolated from clinical and environmental sources during the 2022–2023 cholera outbreak in Kenya. Front. Microbiol. 16:1603736. doi: 10.3389/fmicb.2025.1603736
Received: 31 March 2025; Accepted: 30 May 2025;
Published: 07 July 2025.
Edited by:
Manuel Rodriguez-Iglesias, University of Cádiz, SpainReviewed by:
Guillaume Constantin de Magny, Centre for Research on the Ecology and Evolution of Diseases (CREES), FranceJean-Luc Gala, Université catholique de Louvain, Belgium
Copyright © 2025 Mageto, Aboge, Mekuria, Gathura, Juma, Mugo, Kebenei, Imoli, Ongadi, Kering, Mbae and Kariuki. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lydia M. Mageto, bHlkaWFtYWdldG9AZ21haWwuY29t