 Laura de Boni1
Laura de Boni1 Ullrich Wüllner2*
Ullrich Wüllner2*- 1Dementia Research Institute, University College London, London, United Kingdom
- 2Department of Neurology, University Hospital Bonn, German Center for Neurologic Diseases, Bonn, Germany
Parkinson’s disease (PD) is the second most common neurodegenerative disorder next to Alzheimer’s disease. Most PD cases are considered to be sporadic and despite considerable scientific effort, the underlying cause(s) still remain(s) enigmatic. In particular, it is unknown to which extent epigenetic alterations contribute to the pathophysiology of this devastating disorder. This is partly due to the fact that appropriate PD models are not yet available. Moreover, epigenetic patterns and mechanisms are species specific and murine systems reflect only a few of the idiosyncrasies of human neurons. For several years now, patient-specific stem cell-derived neural and non-neural cells have been employed to overcome this limitation allowing the analysis and establishment of humanized disease models for PD. Thus, several studies tried to dissect epigenetic alterations such as aberrant DNA methylation or microRNA patterns using lund human mesencephalic cell lines or neurons derived from (patient-specific) induced pluripotent stem cells. These studies demonstrate that human neurons have the potential to be used as model systems for the study of epigenetic modifications in PD such as characterizing epigenetic changes, correlating epigenetic changes to gene expression alterations and hopefully using these insights for the development of novel therapeutics. However, more research is required to define the epigenetic (age-associated) landscape of human in vitro neurons and compare these to native neurons before they can be established as suitable models for epigenetic studies in PD. In this review, we summarize the knowledge about epigenetic studies performed on human neuronal PD models, and we discuss advantages and current limitations of these (stem cell-derived) neuronal models for the study of epigenetic alterations in PD.
Introduction
Parkinson’s disease (PD) is the second most common neurodegenerative disorder affecting 1–2% of the population aged >65 years (Tanner and Goldman, 1996). The vast majority (>90%) of PD cases are non-familial and are considered to be sporadic (sPD) (Deng et al., 2018). Despite considerable scientific effort in the last decades, the underlying cause for sPD still remains enigmatic. Not a single cause, but rather several mechanisms appear to underlie the individual susceptibility of sPD (Marras and Lang, 2013). Indeed, several cluster analysis of clinical features identified subtypes in PD pointing to different clinic phenotypes which may correspond to different gene-environment interactions and individual susceptibility (Erro et al., 2013).
Epigenetic alterations of genes could contribute to the pathophysiology of this individual susceptibility (Landgrave-Gomez et al., 2015). Epigenetic modifications comprise DNA methylation, modification of histones, non-coding RNA regulation, RNA editing and nucleosome remodeling/positioning (Figure 1A) (Portela and Esteller, 2010; Wen et al., 2016; Qureshi and Mehler, 2018). These epigenetic modifications regulate gene expression without impact on the DNA sequence itself (Portela and Esteller, 2010; Wen et al., 2016; Qureshi and Mehler, 2018). Epigenetic modifications regulate fundamental cellular processes (e.g., gene transcription, X-chromosome inactivation) and fill the gap between environmental and age-associated gene expression (Wen et al., 2016; Qureshi and Mehler, 2018). They are dynamic, cell specific, display interindividual variability and can also occur in non-dividing cells such as neurons (Fraga et al., 2005; Feng et al., 2007; Fraga and Esteller, 2007). DNA methylation in particular modulates gene expression at promoter or intragenic loci and importantly also via modification of (regulatory), non-coding DNA-sequences (Wullner et al., 2016). Epigenetic patterns change in cancer tissue (Figure 1B) and upon aging (Figure 1B) in various organs and the analysis of DNA methylation levels can provide an independent indicator of aging (i.e., the “epigenetic clock”) (Horvath, 2013). Mutations of epigenetic regulatory genes in neurological diseases include DNA methyltransferases [DNMT1, hereditary sensory and autonomic neuropathy type 1 (Klein et al., 2011)], histone transferases [CREB binding protein, Rubinstein-Taybi syndrome (Petrif et al., 1995)], or Methyl-CpG-binding proteins [MeCP2, Rett syndrome (Amir et al., 1999)]. In addition, mutations in genes can lead to secondary epigenetic modifications. For example, CGG expansion mutations in the fragile X mental retardation 1 (FMR1) gene result in hypermethylation of the 5′ UTR of FMR1 in patients, leading to decreased FMR1 protein levels (Liu et al., 2018). Furthermore, different groups of cortical genes undergo profound and characteristic age-dependent DNA methylation alterations (Siegmund et al., 2007). In general, aging is associated with a global decrease in DNA methylation levels and aged monozygotic twins display significant epigenetic differences, a process referred to as epigenetic drift (Fraga et al., 2005; Teschendorff et al., 2013). Profound cortical DNA methylation differences have been reported in monozygotic twins discordant for Alzheimer’s disease (Mastroeni et al., 2009).
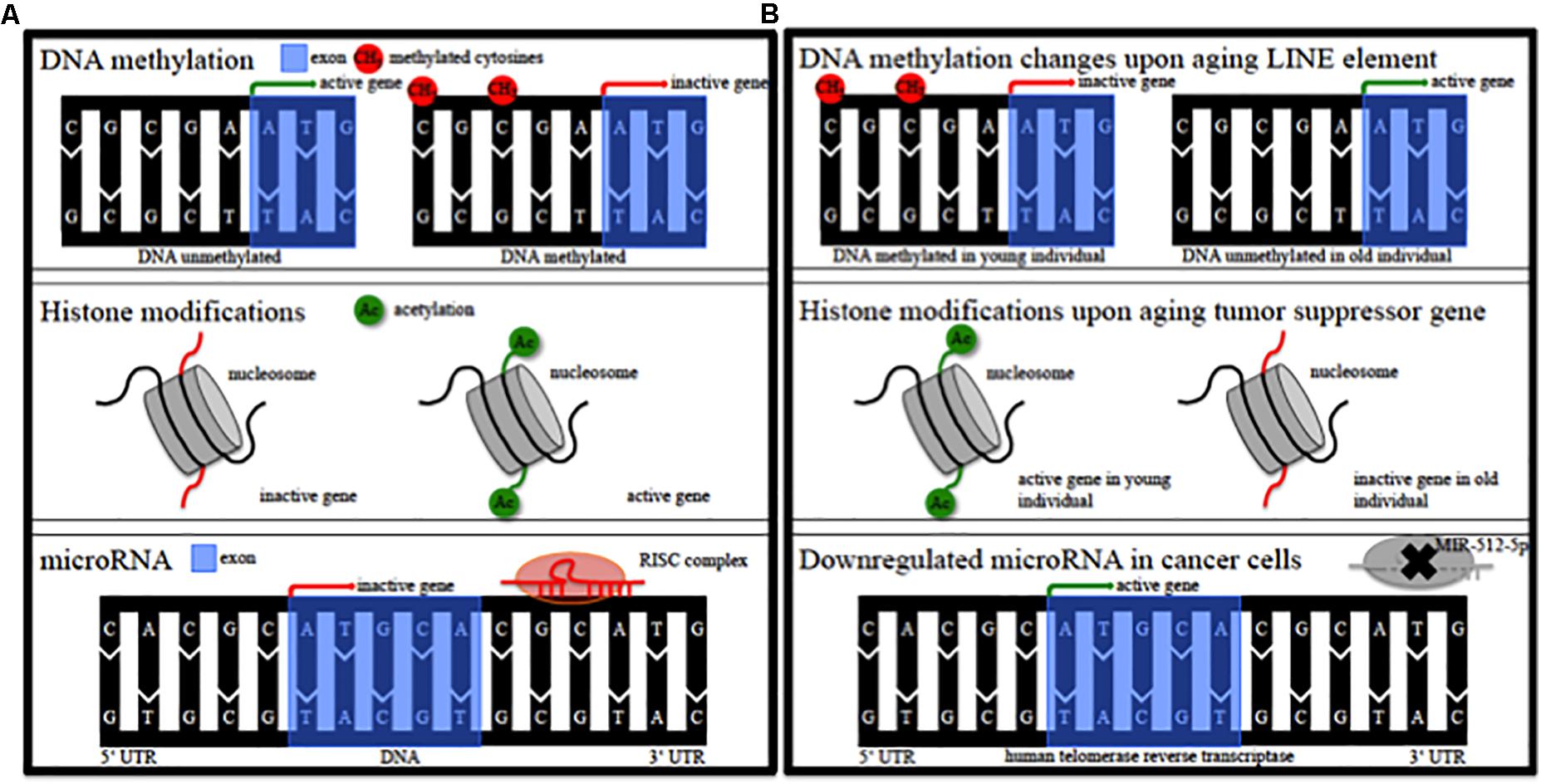
Figure 1. Epigenetic modifications. (A) Schematic of epigenetic modifications comprising DNA methylation, histone modifications, and microRNA. RISC, RNA-induced silencing complex. (B) Schematic of possible epigenetic modifications during aging and in cancer tissue. LINE, long interspersed nuclear element; MIR, microRNA.
Elevated expression of α-synuclein, an important key player in PD, has been linked to the development of PD (Singleton et al., 2003; Ibáñez et al., 2004). Thus, studies tried to identify DNA methylation alterations of the α-synuclein gene (SNCA) (Spillantini et al., 1997; Jowaed et al., 2010; Matsumoto et al., 2010; de Boni et al., 2011). However, these findings remain controversial as some studies identified DNA methylation alterations of SNCA in post-mortem brain tissue from PD patients (Jowaed et al., 2010; Matsumoto et al., 2010; Desplats et al., 2011), whereas others could not replicate these findings (de Boni et al., 2011; Guhathakurta et al., 2017).
The in-depth analysis of presumed epigenetic alterations in sPD is hampered by the lack of appropriate sPD models. Several phenomena have to be considered:
(I) Neurodegenerative diseases (NDDs) do not have any equivalents in the animal kingdom and are specific to humans except for some rare examples in aged dolphins or baboons which are not dispensable for research (Lowenstine et al., 2016; Gunn-Moore et al., 2018).
(II) Epigenetics both in terms of structure (DNA methylation), histone code and functional interplay of the various enzymes are species specific (Mendizabal et al., 2014; Lowdon et al., 2016). Thus, murine (or other) model systems are not representative for human conditions.
(III) NDD occur as non-familial diseases in the vast majority of cases. A limited number of autosomal (and recessive) mutations have been identified. Exploring these inherited forms in transgenic cell culture and animal models to transfer the results to the sporadic cases has been the mainstream research strategy for the past 30 years. However, at least in PD, the discovery and exploitation has not resulted in a decisive clue to the etiology of the non-familial PD cases. Clinical trials of compounds identified in several transgenic and toxin models have failed (Olanow et al., 2008; Löhle and Reichmann, 2010).
(IV) More appropriate disease models, i.e., living human neurons have become available only recently.
Challenges Choosing the Right Model to Study Epigenetic Alterations in sPd
To date, there is no decisive clue to the aetiology of sPD and consequently no causal treatment. This lack of knowledge represents a tremendous burden for the patients, their families and caregivers. PD treatment relies on symptomatic therapies, neither slowing nor halting the process of neurodegeneration. Further, a biomarker identifying PD patients before the onset of motor symptoms, i.e., before pronounced neurodegeneration, is lacking. Thus, the development of novel therapeutics is challenged and current clinical trials (e.g., Isradipine, phase III) try to focus on early PD patients not requiring a symptomatic therapy (Biglan et al., 2017). Moreover, current animal models do not fully reflect the underlying pathology and hallmarks of PD and despite huge efforts, could not elucidate the underlying causal pathology towards sPD. The analyses of native brain tissue is thus necessary, but limited to post-mortem tissue, which is not suited for mechanistic studies. Studies on epigenetic alterations using native bulk post-mortem brain tissue generated conflicting results (Jowaed et al., 2010; de Boni et al., 2011).
Common cellular human models comprise the neuroblastoma cell line SK-N-SH and its subclone SH-SY5Y. Indeed, these cancer cells helped to identify important pathways and mechanisms in PD, e.g., mitochondrial dysfunction or impaired autophagy (Zeng et al., 2018). Upon treatment of SK-N-SH cells with the DNMT inhibitor 5-Azacytidine, DNA demethylation of the SNCA intron 1 was associated with an increase in SNCA mRNA and α-synuclein protein expression (Jowaed et al., 2010). To date, these findings could not be replicated in human neurons due to the inaccessibility of living disease-affected cells. It remains to be determined whether alteration in cis or yet undetermined exogenous factors underly epigenetic modifications of the SNCA gene.
Epigenetic variation plays an important role in evolution (Richards, 2008; Mendizabal et al., 2014). Many animal genomes (mostly vertebrates) are depleted of CpG dinucleotides and genes associated with neurological functions exhibit human-specific patterns of DNA methylation (Mendizabal et al., 2014). Whole-genome DNA methylation analysis of prefrontal cortical tissue of humans and chimpanzees revealed extensive species-specific variation associated with strong gene expression changes (Zeng et al., 2012). Moreover, it has already been demonstrated on a single-cell level that mouse and human embryos are characterized by distinct differences in DNA methylation and histone modification patterns (Chavez et al., 2014).
To date, there is no decisive clue to the etiology of sPD and consequently no causal treatment. This lack of knowledge represents a tremendous burden for the patients, their families and caregivers. PD treatment relies on symptomatic therapies, neither slowing nor halting the process of neurodegeneration. Further, the lack of a biomarker which would identify PD patients before the onset of motor symptoms, i.e., before pronounced neurodegeneration has occurred. Current clinical trials (e.g., Isradipine, phase III) try to focus on early PD patients not requiring a symptomatic therapy (Biglan et al., 2017). Isradipine was identified in epidemiological studies and has shown “rejuvenating” effects in vitro and in vivo models. It is supposed that Isradipine blocks L-type Ca(v)1.3 Ca2+ channels maintaining their rhythmic pacemaking which in turn results in decreased vulnerability of the neuron to cell stressors (Chan et al., 2007). It remains to be determined whether the current clinical trial, in contrast to the previous trials of compounds identified in transgenic and toxin models will be successful (Olanow et al., 2008; Löhle and Reichmann, 2010).
More appropriate disease models, i.e., living human neurons have become available only recently and could be used to study epigenetic modifications in PD. Two immortalized human male fetal neural precursor lines were developed from cortex (CX) and the ventral mesencephalon (VM) carrying the c-myc or v-myc oncogene, respectively. These so-called ReNcell CX and VM express typical markers for neural precursor cells and upon differentiation, ReNcell VM showed positive staining for tyrosine hydroxylase (Donato et al., 2007). Another mesencephalic human cell line was derived from 8-week-old female human ventral mesencephalic tissue and a subclone, the lund human mesencephalic (LUHMES) cell line, can be differentiated into morphologically and biochemically mature dopamine-like neurons (Lotharius et al., 2005). The advantage of these cell lines is that they consist of a quite homogenous population, have the ability to be cultured and expanded long-term, have the capacity to be differentiated into more mature post-mitotic dopaminergic phenotypes and are suitable for to novel genetic editing tools such as the Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)/Cas system.
Human stem cell-derived neurons can be generated from embryonic stem cells (ESC), induced pluripotent stem cells (iPSCs) or tissue-specific (stem) cells. ESC are obtained from the inner cell mass of the blastocyst and are pluripotent (Thomson et al., 1998; Dhara and Stice, 2008). Diverse protocols were utilized to directly differentiate ESC into neural (and glial) cells under defined culture conditions using different factors, signaling molecules, supplements, and additives (Dhara and Stice, 2008). Differentiated cells such as fibroblasts can be reprogrammed by defined factors to iPSC resetting an embryonic state (Nakagawa et al., 2008; Eguchi and Kuboki, 2016). IPSC can be derived from patients and controls allowing the study of disease- and non-affected defined neuronal populations and other cellular subtypes. Tissue specific cells such as dermal (Joannides et al., 2004) or mesenchymal stem cells (Singh et al., 2017) can also be differentiated into neural cells. Direct conversion – omitting a pluripotent intermediate state – of somatic cells into so-called induced neurons (iN) exhibiting typical neuronal and differentiation properties is achieved by defined transcription factors (Ladewig et al., 2012; Prasad et al., 2016; Xu et al., 2017). Regarding PD, most attempts have focused on generating in vitro DAn to study disease-related mechanisms as cardinal motor manifestations of PD refer to the progressive loss of DAn. Stem cell-derived DAn were generated by many research groups using quite different approaches and culture conditions. Most protocols are based on activation of the important Sonic Hedgehog and WNT signaling pathway in combination with floor plate based and feeder cell protocols or other small molecules and together with transcription factors required for proper DAn specification such as FOXA2, LMX1A, or Oct4 (Chen et al., 2009; Cooper et al., 2010; Deleidi et al., 2011; Kriks et al., 2011; Kirkeby et al., 2012; Arenas et al., 2015; Fernandez-Santiago et al., 2015; Fedele et al., 2017; Qiu et al., 2017). Other protocols used retinoic acid (Tsai et al., 2015), Vitamin C (Wulansari et al., 2017), supplementation with steroids (Agbay et al., 2018), FGF20 (Correia et al., 2007), or plating in Lam-111 (Kirkeby et al., 2017) to increase the yield of DAn. Indeed, the attempt to study a defined and especially vulnerable neuronal population is imperative. However, it should be noted that the marker expression and efficiency generating DAn vary highly depending on the applied protocol (Xu et al., 2017). Thus, observed PD related phenotypes (i.e., mitochondrial dysfunction, α-synuclein accumulation, synaptic dysfunction) might not be solely linked to DAn. PD related phenotypes have nevertheless been detected in iPSC-derived DAn and neurons from PARK2/Parkin (Imaizumi et al., 2012; Ren et al., 2015; Chung et al., 2016), PINK1 patients (Chung et al., 2016), PD patients carrying glucocerebrosidase (GBA) mutations (Schondorf et al., 2014; Woodard et al., 2014; Aflaki et al., 2016; Fernandes et al., 2016; Yang et al., 2017), patients harboring the SNCA triplication mutation (Byers et al., 2011; Flierl et al., 2014; Oliveira et al., 2015), and in iPSC-derived DAn and sensory neurons from patients carrying leucine-rich repeat kinase 2 (LRRK2) mutations (Nguyen et al., 2011; Bahnassawy et al., 2013; Schwab and Ebert, 2015; Hsieh et al., 2016; Lopez de Maturana et al., 2016; Sandor et al., 2017; Schwab et al., 2017). Several studies have already tried to decipher mechanistic processes and tested drugs using iPSC-derived DAn: DAn from SNCA triplication carriers secreted high levels of α-synuclein which was taken up by neighboring neurons (Reyes et al., 2015). FACS-sorted post-mortem DAn from LRRK2-G2019S carriers displayed a distinct transcriptome compared to post-mortem DAn from non-affected individuals and these DAn also expressed a distinct set of genes compared to control DAn (Sandor et al., 2017). Clioquinol, a compound known to alleviate dopamine neuron loss, rescued these pathogenic expression changes (Sandor et al., 2017). The macrolide compound rapamycine and the PGC1α-transcription factor inducing disaccharide trehalose protected against mitochondrial neurotoxicity in iPSC-derived DAn exhibiting the Parkin Q311X mutation (Siddiqui et al., 2015). High-throughput screening identified the compounds NCGC607 and NCGC00188758 and the chaperone ambroxol, which restore glucocerebrosidase activity and rescue PD-related phenotypes in DAn from GBA carriers (Aflaki et al., 2016; Mazzulli et al., 2016; Yang et al., 2017). The effect of NCGC00188758 was confirmed in iPSC-derived DAn of SNCA triplication, SNCA A53T, sPD and PARK9 patients (Mazzulli et al., 2016). These aforementioned studies however bear only little impact regarding the pathophysiological processes of sPD. It remains to be determined whether human (patient-specific) neurons might also be used to generate useful models to identify novel pathophysiological processes to such as epigenetic modifications in PD.
Epigenetic Studies in Human Neurons
Epigenetic Studies Using Immortalized Human Fetal-Derived Neurons
HDACi Rescue WT α-Synuclein-Induced DNA Damage in LUHMES Cells
Paiva et al. (2017) demonstrated transcriptional deregulation (e.g., downregulation of the dopamine receptor; DAT) in α-synuclein wild type (WT) and A30P lentiviral transfected LUHMES cells differentiated for 8 days (Table 1). RNAseq demonstrated a downregulation of DNA repair genes due to the overexpression of WT and A30P α-synuclein. The expression of WT α-synuclein was associated with a significant reduction in the levels of acetylated Histone 3 and treatment of cells using the HDACi sodium butyrate could ameliorate the observed phenotype. These results were confirmed in LUHMES cells treated with 1-methyl-4-phenylpyridinium (MPP+).
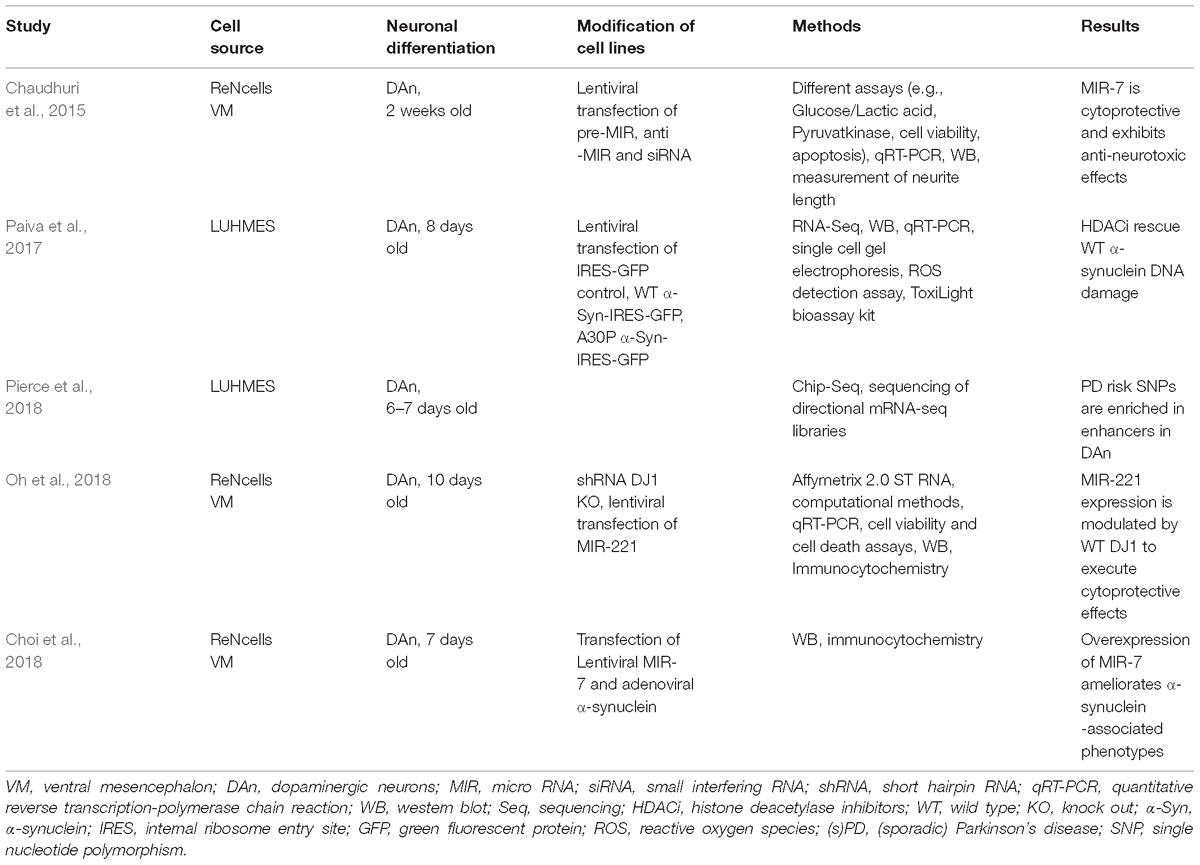
Table 1. Epigenetic studies in immortalized human fetal-derived neurons.
PD-Risk SNPs Are Enriched in Enhancers in DAn From LUHMES Cells
Pierce et al. (2018) evaluated genome-wide histone H3K27 acetylation, CCCTC-binding factor (CTCF) occupancy, and RNA expression in differentiated (6–7 days old) and undifferentiated LUHMES cells (Table 1). The differentiation into DAn was accompanied by distinct changes in gene expression. 57,905 genes were mapped in total and 6,147 genes were significantly downregulated whereas 7,621 genes were significantly up-regulated. Chip-Seq revealed 25,000 active enhancers, 12,000 active promoters, and 40,000 occupied CTCF sites between differentiated and undifferentiated LUHMES cells. PD-risk SNPs were enriched in enhancers for synapse and vesicle trafficking categories in DAn from LUHMES. However, the fold-change to enhancer location explained only 10% of variability in their data set. The authors suggested that enhancers do not only control the expression of single genes, but many genes are affected by particular loci.
MIR-7 Is Cytoprotective and Exhibits Anti-Neurotoxic Effects in ReNcells
MIR are small, highly conserved, sequence-specific non-coding RNA molecules regulating post-transcriptional gene expression (Sato et al., 2011) (Table 1). Concerning MIR alterations, the group of E. Junn showed that lentiviral overexpressed MIR-7 promoted glycolysis via downregulation of RelA in 2 weeks old ReNcell (human ventral mesencephalic neural progenitor)-derived DAn (Chaudhuri et al., 2015). MIR-7 is highly expressed in the brain, including DAn from the Substantia nigra. In their cell model, MIR-7 also increased the expression of the Glucose Transporter Glut3 and finally resulted in cytoprotection and exhibition of anti-neurotoxic effect after treatment with MPP+. The authors pointed out that an intact glycolytic pathway is necessary for neuronal survival and promoting effects of MIR-7 could be beneficial in PD. In a subsequent study, Choi et al. (2018) demonstrated that overexpression of MIR-7 in ReNcells resulted in a decrease of monomeric and high molecular weight forms of α-synuclein (Table 1). Interestingly, α-synuclein aggregates due to transfection of preformed α-synuclein fibrils were also diminished. The authors could show that an increased autophagic reflux promoting α-synuclein clearance was responsible for the observed effects.
MIR-221 Expression Is Modulated by WT DJ1 to Execute Cytoprotective Effects
DJ1 (PARK7) mutations lead to fPD (Bonifati et al., 2003). Thus, Oh et al. (2018) used an array-based screen to identify RNA transcripts regulated by DJ1 (Table 1). Using SH-SY5Y cells, they demonstrated that MIR-221 exhibited a 55% reduction of expression in DJ1 knock out (KO) cells and confirmed these findings via qRT-PCR as well as using a DJ1 KO mouse model. Solely the WT DJ1 protein seems to influence MIR-221 expression levels through the MAPK/ERK pathway. They further consolidated these findings in ReNcell VM showing that lentiviral transfection of MIR-221 was cytoprotective in DJ1 KO cells.
Overall, these studies demonstrated various epigenetic PD-associated changes in fetal human neurons. Their impact on PD pathogenesis requires further studies, especially using patient-specific neurons derived from aged individuals.
Epigenetic Studies in iPSC-Derived Neurons From PD Patients
Aberrant DNA Methylation Profiles in iPSC-Derived DAn From PD Patients
Fernandez-Santiago et al. (2015) demonstrated that iPSC-derived DAn from PD patients exhibit an aberrant DNA methylation pattern compared to controls (Table 2). They differentiated fibroblast-derived iPSC into 30 days old dopaminergic neurons of the A9 subtype. In total, they generated DAn from four PD patients carrying LRRK2 mutations (G2019S, L2PD), six sPD patients and four controls. The expression of DAn markers ranged from 44 to 67%. Applying array-based analysis for DNA methylation (450K, Illumina) and gene expression (Genechip Human Exon 1.0 ST, Affymetrix) this study demonstrated that L2PD and sPD exhibit similar DNA methylation and gene expression profiles. DAn from L2PD and sPD patients showed an aberrant DNA methylation and gene expression profile compared to fibroblasts, iPSC and non-dopaminergic neurons from PD patients and controls. DNA methylation alterations in DAn were enriched in enhancer elements, transcription factor (TF) binding sites and were partially correlated with gene expression alterations validated via qRT-PCR. Upregulated TFs and proteins included e.g., OTX2/PAX6 or SNCA/DCC. The authors noted that DAn from PD patients might have undergone incomplete epigenomic remodeling compared to controls. The study showed an aberrant DNA methylation profile in DAn only – not other non-dopaminergic phenotypes - and very similar DNA methylations differences in DAn derived from the fPD and sPD cases. It is quite remarkable that familial and idiopathic PD cases show common epigenetic modifications in the most vulnerable neuronal subtype affected in PD.
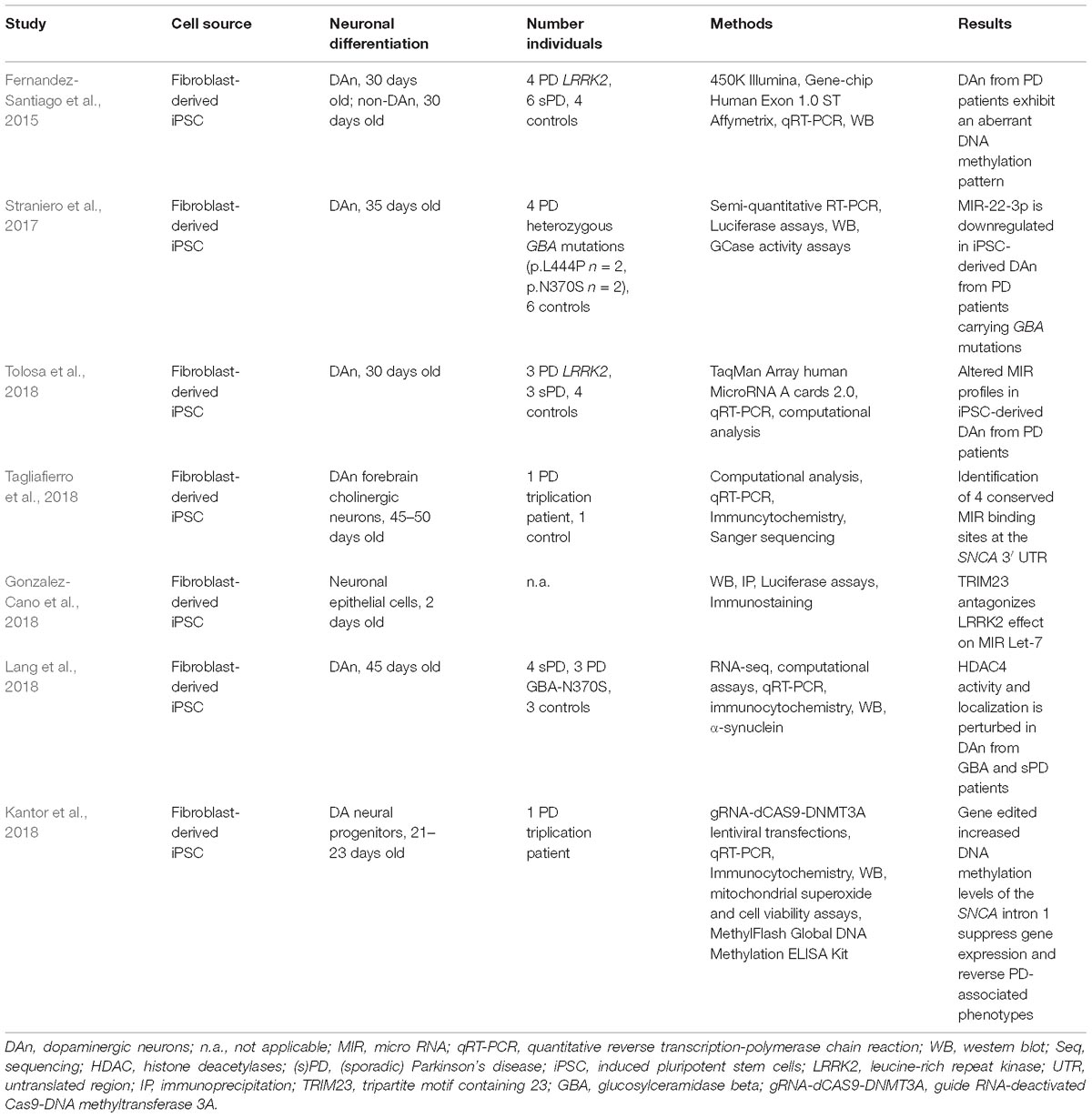
Table 2. Epigenetic studies in human iPSC-derived neurons.
Altered MIR Profiles in iPSC-Derived DAn From PD Patients
Tolosa et al. identified MIR alterations in DAn from sPD and L2PD patients compared to controls (Tolosa et al., 2018) (Table 2). They used the very same samples as in the study of Fernandez Santiago et al. (Fernandez-Santiago et al., 2015). They first performed array-based and qPCR analyses to identify differentially expressed MIR. Different computational approaches were then applied to perform enrichment analysis and to compare the results with global gene expression data from the previous study. The functional analysis revealed two main clusters associated with proper neuronal (cluster 1) and diverse homeostatic (cluster 2) functions. Interestingly, the alterations were again identified in DAn from sPD and L2PD patients to a very similar extent. MIR alterations involved up- and downregulated MIR. These expression differences seem to display early molecular changes preceeding neuropathological cellular PD phenotypes such as inclusion formation.
TRIM23 Reverses Impaired LRRK2-Mediated MIR Let-7a Activity in Neuronal Epithelial Cells
Tripartite Motif Containing 23 (TRIM23) has been described as an activator of MIR-associated proteins and is supposed to regulate SNCA (Schwamborn et al., 2009; Pavlou et al., 2017). Gonzalez-Cano et al. (2018) showed that LRRK2, TRIM23, argonaute protein 2 and the MIR Let-7a interact in vitro and in vivo (Table 2). Vector-mediated expression of GFP-tagged TRIM23 and/or fly-tagged LRRK2-RR141H in neuronal epithelial cells (differentiated for 48 h) suggested that LRRK2 inhibits MIR Let-7a resulting in impaired neuronal differentiation. These effects could be antagonized by TRIM23 expression to some extent.
The SNCA 3′ UTR Contains Conserved MIR Binding Sites in iPSC-Derived Cholinergic Neurons
Tagliafierro et al. (2018) tried to identify conserved MIR binding sites at the 3′ UTR of SNCA (Table 2). They carried out a computational approach and further validated the MIR expression in iPSC-derived forebrain cholinergic neurons (45–50 days old) from a SNCA triplication patient and 1 control cell line. The SNCA 3′ UTR exhibited four conserved binding sites for MIR-7-5p, MIR-140-3p, MIR-153-3p, and MIR-223-3p. It has been previously shown that MIR-7 and MIR-153 regulate SNCA mRNA expression levels (Junn et al., 2009; Doxakis, 2010) and MIR-223 is involved in the differentiation of mature neurons (Harraz et al., 2014). However, the expression levels of all 4 MIR varied between iPSC, neural precursor cells and iPSC-derived neurons. Moreover, an association between genetic variants of the 3′ UTR and the conserved MIR binding sites could not be established.
MIR-22-3p Is Downregulated in iPSC-Derived DAn From PD Patients Carrying GBA Mutations
Straniero et al. (2017) focused on GBA expression regulation evaluating the effect of the MIR-22-3p and MIR-132 (Table 2). Preliminary analysis in HEK293 and HeLa cells demonstrated, that MIR-22-3p targets GBA and the GBA pseudogene GBAP1. These findings were validated in 35 days old iPSC-derived DAn from six controls and four PD patients carrying GBA mutations according to the differentiation protocol of Kriks et al. (2011). Upon neuronal differentiation, GBA and GBAP1 were up-regulated and MIR-22-3p was downregulated in control DAn compared to the neuronal precursors. GBA transcripts were downregulated in iPSC-derived DAn from PD patients accompanied by a slight up-regulation of MIR-22-3p. Though a connection between the RNA-based network and PD pathology has not been firmly established, the results point toward a possible link between GBA/GBAP1and MIR-22-3p.
HDAC4 Mislocalization Leads to Protein Dyshomeostasis in iPSC-Derived DAn From GBA-N370S Patients
Another link between GBA mutations and epigenetic alterations was proposed by Lang et al. (2018) (Table 2). They observed an increase in nuclear localization of HDAC4 in iPSC-derived DAn from PD GBA-N370S patients compared to controls. Non-dopaminergic neurons did not exhibit this phenotype. The mislocalization of HDAC4 was associated with a downregulation of HDAC4-controlled genes. It is exciting that four compounds – used in other diseases such as prostate cancer – targeted HDAC4 activity, reversed the impaired expression of the HDAC4-controlled genes and subsequently ameliorated pertubations in autophagy and lysosomal pathways. Furthermore, these findings were also confirmed in four sPD cases.
DNA Methylation Editing of the SNCA Intron 1 Results in Gene Downregulation and Improvement of PD Phenotypes in iPSC-Derived Neuronal DA Progenitor Cell Lines
As already mentioned, the SNCA intron 1 might be a putative promoter region regulating SNCA expression (Jowaed et al., 2010). Thus, Kantor et al. (2018) generated guide RNA (gRNA)-deactivated Cas9 (dCas9)-DNMT3A lentiviral vectors and increased the DNA methylation levels at the SNCA intron 1 in neuronal DA progenitors from a patient with a SNCA triplication mutation (Table 2). The targeted DNA methylation editing resulted in decreased SNCA expression, reduced mRNA/protein levels and improved pathological phenotypes (superoxide production, cell viability) in the iPSC-derived neuronal DA progenitors. The transgenes increased the methylation levels across several CpGs of the SNCA intron 1 with minimal effects on global DNA methylation levels or expression of certain selected genes. CpG 6 and 7 of the SNCA intron 1 seemed to be major target sites. To the best of our knowledge, this is the first study showing mechanistic epigenetic analysis and their direct impact on gene expression being not only relevant for fPD but also sPD. We would like to point out that such studies indeed require human neurons and cannot be performed using post-mortem brain tissue.
Perspectives
Human neuronal cells bear promising potential to study epigenetic alterations. Several studies already identified characteristic features with regard to DNA methylation, MIR and histone modifications in human neuronal in vitro PD models. These findings are quite encouraging and point toward possible epigenetic pathophysiological processes in PD. This will hopefully lead to the development of novel therapeutic strategies – however a definite confirmation of such findings in vivo and of re-juvenation awaits clarification.
Human iPSC-derived neurons offer several advantages over other cellular and in vivo models. They
• are patient-derived
• can be obtained in an unlimited amount
• offer the possibility to generate highly pure neuronal populations
• are available for co-culture studies or generating 3D brain (cerebral organoids) better recapitulating the native cerebral environment, as astrocytes and microglia are considered to be implicated in the pathogenesis of PD (Fellner et al., 2011; Booth et al., 2017; Subramaniam and Federoff, 2017)
• allow the direct investigation of the presumed effects of epigenetic alterations on gene expression
• allow high-throughput screening
• are accessible for gene editing
However, it is under intense scrutiny whether human neuronal cellular models are appropriate to study epigenetic modifications. Particularly, fetal neurons might not be suitable to study age-associated epigenetic patterns related to NDD. Concerning iPSC-derived neurons, three major caveats have to be addressed:
(1) The process of directed reprogramming itself is based on epigenetic modifications (Rando and Chang, 2012).
(2) Reprogramming resets the epigenetic pattern almost to zero (“epigenetic rejuvenation”), impairing studies of age-associated alterations (Rando and Chang, 2012).
(3) The epigenome of iPSC-derived neurons might differ substantially from native neurons.
These three issues have yet to be resolved. It has already been demonstrated, using isogenic human neural stem cell systems, that the reprogramming process itself does not lead to an aberrant DNA methylation or gene expression pattern (Choi et al., 2015; de Boni et al., 2018). To overcome the limitation of epigenetic rejuvenation, fibroblast-derived neurons were generated via direct conversion omitting a pluripotent state (Mertens et al., 2015). However, preservation of age-associated signatures was not replicated in a current study generating directly converted neurons without the use of Oct4 (Sheng et al., 2018). Another approach was proposed by introducing progerin, a truncated version of lamin A protein involved in Hutchinson–Gilford progeria syndrome (Miller et al., 2013). The authors suggested that the lack of a distinct neurodegenerative phenotype, especially neuronal protein inclusions, could be due to the age-reset during reprogramming (Miller et al., 2013). Indeed, the analysis of DAn grafts exhibiting progerin overexpression in mice demonstrated large multilamellar inclusions which are considered to be precursors of Lewy bodies (Miller et al., 2013).
However, a dysregulation of progerin expression has not been observed in PD. Moreover, it has been shown that the genetic background and differentiation protocols have a high impact on the epigenetic landscape in iPSC-derived neurons (de Boni et al., 2018). Comparing iPSC-derived forebrain neurons to native, post-mortem temporal lobe bulk brain tissue, it has been shown that upon neuronal differentiation, differences in actively transcribed genes, long intergenic non-coding RNAs (lincRNA), and DNA methylation patterns were diminished (Hjelm et al., 2013). IPSC-derived neurons differentiated for 140 days exhibited <25% gene expression, <16% lincRNA, and <30% DNA methylation differences compared to native brain tissue (Hjelm et al., 2013). Single gene analysis demonstrated variable gene expression patterns for neuronal, astrocytic, or oligodendroglial markers: some markers are remarkably similar to native brain tissue [e.g., microtubule-associated protein 2 (MAP2) or the serine/threonine protein kinase 6 (PAK6)], some increased [e.g., Tubulin Beta 3 Class III (TUBB3) or neurotrophic growth factor (NGF)], whereas astrocytic and oligodendroglial markers remained downregulated [e.g., oligodendrocyte transcription factor 2 (Olig2) or glial fibrillary acidic protein (GFAP)], possibly due to the optimization of in vitro cell culture conditions for neuronal differentiation (Hjelm et al., 2013). Moreover, iPSC-derived DAn exhibited substantially different gene expression patterns and an incorrect anterior–posterior axis patterning compared to native midbrain bulk tissue (Xia et al., 2016). Genes associated with immature DAn were highly expressed in iPSC-derived DAn (Xia et al., 2016). In this context, a comparison of iPSC-derived neurons to, e.g., FACS-sorted native neurons is required. Using native laser-capture microdissected or FACS-sorted neuronal populations (e.g., DAn and glutamatergic neurons), recent studies have begun to demonstrate profound epigenetic and transcriptomic differences between iPSC-derived neurons and native neurons (Montaño et al., 2013; Kozlenkov et al., 2014, 2015; Dong et al., 2018). However, the precise epigenetic and transcriptomic landscape of native neurons has yet to be determined and compared to iPSC-derived neurons.
Conclusion
Human in vitro (patient-specific) neurons bear the potential to generate useful tools to study epigenetic modifications in PD. However, major efforts are still required to define the epigenetic landscape of these neurons compared to native neurons and determine age-associated epigenetic patterns. First of all, native (single cell) epigenomic genome-wide analyses, from, e.g., FACS-sorted brain tissue, have to be performed to define the variation of the normal epigenetic landscape of different neuronal and glial populations. Including cells from aged individuals, such analyses could be used to create a reference epigenome for further epigenetic evaluations using human (patient-specific) in vitro neurons. The epigenetic comparison of native and in vitro human neurons could then help to improve current differentiation protocols and might reveal whether observed epigenetic changes in vitro indeed correspond to a PD or other phenotypes.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
The study was supported by the BONFOR program of the University Clinic of Bonn (LdB), the Thiemann Foundation (LdB), the German Center for Neurodegenerative Diseases [Deutsches Zentrum für Neurodegenerative Erkrankungen (DZNE)]; and by the BMBF/ANR through the EpiPD (Epigenomics of Parkinson’s disease) project, under the auspices of the bilateral Epigenomics of Common and Age-related Diseases Programme (Grant No. 01KU1403B; UW), by the EU/EFPIA Innovative Medicines Initiative Joint Undertaking Aetionomy (Grant No. 115568; UW), the ParkinsonFonds Deutschland gGmbH (UW) and the Deutsche Parkinson Vereinigung (dPV).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Adam Cantlon for critical reading of the manuscript.
References
Aflaki, E., Borger, D. K., Moaven, N., Stubblefield, B. K., Rogers, S. A., Patnaik, S., et al. (2016). A new glucocerebrosidase chaperone reduces alpha-synuclein and glycolipid levels in iPSC-derived dopaminergic neurons from patients with gaucher disease and parkinsonism. J. Neurosci. 36, 7441–7452. doi: 10.1523/JNEUROSCI.0636-16.2016
Agbay, A., De La Vega, L., Nixon, G., and Willerth, S. (2018). Guggulsterone-releasing microspheres direct the differentiation of human induced pluripotent stem cells into neural phenotypes. Biomed. Mater. 13:034104. doi: 10.1088/1748-605X/aaaa77
Amir, R. E., Van Den Veyver, I. B., Wan, M., Tran, C. Q., Francke, U., and Zoghbi, H. Y. (1999). Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl- CpG-binding protein 2. Nat. Genet. 23, 185–188. doi: 10.1038/13810
Arenas, E., Denham, M., and Villaescusa, J. C. (2015). How to make a midbrain dopaminergic neuron. Development 142, 1918–1936. doi: 10.1242/dev.097394
Bahnassawy, L., Nicklas, S., Palm, T., Menzl, I., Birzele, F., Gillardon, F., et al. (2013). The parkinson’s disease-associated LRRK2 mutation R1441G inhibits neuronal differentiation of neural stem cells. Stem Cells Dev. 22, 2487–2496. doi: 10.1089/scd.2013.0163
Biglan, K. M., Oakes, D., Lang, A. E., Hauser, R. A., Hodgeman, K., Greco, B., et al. (2017). A novel design of a Phase III trial of isradipine in early Parkinson disease (STEADY-PD III). Ann. Clin. Transl. Neurol. 4, 360–368. doi: 10.1002/acn3.412
Bonifati, V., Rizzu, P., van Baren, M., Schaap, O., Breedveld, G. J., Krieger, E., et al. (2003). Mutations in the DJ1 gene associated with autosomal recessive early-onset parkinsonism. Science 299, 256–259. doi: 10.1126/science.299.5604.256
Booth, H. D. E., Hirst, W. D., and Wade-Martins, R. (2017). The role of astrocyte dysfunction in Parkinson’s Disease Pathogenesis. Trends Neurosci. 40, 358–370. doi: 10.1016/j.tins.2017.04.001
Byers, B., Cord, B., Nguyen, H. N., Schule, B., Fenno, L., Lee, P. C., et al. (2011). SNCA triplication Parkinson’s patient’s iPSC-derived DA neurons accumulate alpha-synuclein and are susceptible to oxidative stress. PLoS One 6:e26159. doi: 10.1371/journal.pone.0026159
Chan, C. S., Guzman, J. N., Ilijic, E., Mercer, J. N., Rick, C., Tkatch, T., et al. (2007). Rejuvenation’ protects neurons in mouse models of Parkinson’s disease. Nature 447, 1081–1086. doi: 10.1038/nature05865
Chaudhuri, A. D., Kabaria, S., Choi, D. C., Mouradian, M. M., and Junn, E. (2015). MicroRNA-7 promotes glycolysis to protect against 1-methyl-4-phenylpyridinium-induced cell death. J. Biol. Chem. 290, 12425–12434. doi: 10.1074/jbc.M114.625962
Chavez, S. L., McElroy, S. L., Bossert, N. L., De Jonge, C. J., Rodriguez, M. V., Leong, D. E., et al. (2014). Comparison of epigenetic mediator expression and function in mouse and human embryonic blastomeres. Hum. Mol. Genet. 23, 4970–4984. doi: 10.1093/hmg/ddu212
Chen, J., Sai, S.-Y. T., Vazin, T., Coggiano, M., and Freed, W. J. (2009). Human embryonic stem cells which express hrGFP in the undifferentiated state and during dopaminergic differentiation. Restor. Neurol. Neurosci. 27, 359–370. doi: 10.3233/RNN-2009-0521
Choi, D. C., Yoo, M., Kabaria, S., and Junn, E. (2018). MicroRNA-7 facilitates the degradation of alpha-synuclein and its aggregates by promoting autophagy. Neurosci. Lett. 678, 118–123. doi: 10.1016/j.neulet.2018.05.009
Choi, J., Lee, S., Mallard, W., Clement, K., Tagliazucchi, G. M., Lim, H., et al. (2015). A comparison of genetically matched cell lines reveals the equivalence of human iPSCs and ESCs. Nat. Biotechnol. 33, 1173–1181. doi: 10.1038/nbt.3388
Chung, S. Y., Kishinevsky, S., Mazzulli, J. R., Graziotto, J., Mrejeru, A., Mosharov, E. V., et al. (2016). Parkin and PINK1 Patient iPSC-derived midbrain dopamine neurons exhibit mitochondrial dysfunction and alpha-synuclein accumulation. Stem Cell Rep. 7, 664–677. doi: 10.1016/j.stemcr.2016.08.012
Cooper, O., Hargus, G., Deleidi, M., Blak, A., Osborn, T., Marlow, E., et al. (2010). Differentiation of human ES and Parkinson’s disease iPS cells into ventral midbrain dopaminergic neurons requires a high activity form of SHH, FGF8a and specific regionalization by retinoic acid. Mol. Cell. Neurosci. 45, 258–266. doi: 10.1016/j.mcn.2010.06.017
Correia, A. S., Anisimov, S. V., Roybon, L., Li, J.-Y., and Brundin, P. (2007). Fibroblast growth factor-20 increases the yield of midbrain dopaminergic neurons derived from human embryonic stem cells. Front. Neuroanat. 1:4. doi: 10.3389/neuro.05.004.2007
de Boni, L., Gasparoni, G., Haubenreich, C., Tierling, S., Schmitt, I., Peitz, M., et al. (2018). DNA methylation alterations in iPSC- and hESC-derived neurons: potential implications for neurological disease modeling. Clin. Epigenetics 10:13. doi: 10.1186/s13148-018-0440-0
de Boni, L., Tierling, S., Roeber, S., Walter, J., Giese, A., and Kretzschmar, H. A. (2011). Next-generation sequencing reveals regional differences of the α-synuclein methylation state independent of Lewy body disease. Neuromolecular Med. 13, 310–320. doi: 10.1007/s12017-011-8163-9
Deleidi, M., Cooper, O., Hargus, G., Levy, A., and Isacson, O. (2011). Oct4-induced reprogramming is required for adult brain neural stem cell differentiation into midbrain dopaminergic neurons. PLoS One 6:e19926. doi: 10.1371/journal.pone.0019926
Deng, H., Wang, P., and Jankovic, J. (2018). The genetics of Parkinson disease. Ageing Res. Rev. 42, 72–85. doi: 10.1016/j.arr.2017.12.007
Desplats, P., Spencer, B., Coffee, E., Patel, P., Michael, S., Patrick, C., et al. (2011). Alpha-synuclein sequesters Dnmt1 from the nucleus: a novel mechanism for epigenetic alterations in Lewy body diseases. J. Biol. Chem. 286, 9031–9037. doi: 10.1074/jbc.C110.212589
Dhara, S. K., and Stice, S. L. (2008). Neural differentiation of human embryonic stem cells. J. Cell. Biochem. 105, 633–640. doi: 10.1002/jcb.21891
Donato, R., Miljan, E. A., Hines, S. J., Aouabdi, S., Pollock, K., Patel, S., et al. (2007). Differential development of neuronal physiological responsiveness in two human neural stem cell lines. BMC Neurosci. 8:36. doi: 10.1186/1471-2202-8-36
Dong, X., Liao, Z., Gritsch, D., Hadzhiev, Y., Bai, Y., Locascio, J. J., et al. (2018). Enhancers active in dopamine neurons are a primary link between genetic variation and neuropsychiatric disease. Nat. Neurosci. 21, 1482–1492. doi: 10.1038/s41593-018-0223-0
Doxakis, E. (2010). Post-transcriptional regulation of α-synuclein expression by mir-7 and mir-153. J. Biol. Chem. 285, 12726–12734. doi: 10.1074/jbc.M109.086827
Eguchi, T., and Kuboki, T. (2016). Cellular reprogramming using defined factors and MicroRNAs. Stem Cells Int. 637, 201–206. doi: 10.1155/2016/7530942
Erro, R., Vitale, C., Amboni, M., Picillo, M., Moccia, M., Longo, K., et al. (2013). The heterogeneity of early Parkinson’s disease: a cluster analysis on newly diagnosed untreated patients. PLoS One 8:e70244. doi: 10.1371/journal.pone.0070244
Fedele, S., Collo, G., Behr, K., Bischofberger, J., Muller, S., Kunath, T., et al. (2017). Expansion of human midbrain floor plate progenitors from induced pluripotent stem cells increases dopaminergic neuron differentiation potential. Sci. Rep. 7:6036. doi: 10.1038/s41598-017-05633-1
Fellner, L., Jellinger, K. A., Wenning, G. K., and Stefanova, N. (2011). Glial dysfunction in the pathogenesis of α-synucleinopathies: emerging concepts. Acta Neuropathol. 121, 675–693. doi: 10.1007/s00401-011-0833-z
Feng, J., Fouse, S., and Fan, G. (2007). Epigenetic regulation of neural gene expression and neuronal function. Pediatr. Res. 61, 58R–63R. doi: 10.1203/pdr.0b013e3180457635
Fernandes, H. J. R., Hartfield, E. M., Christian, H. C., Emmanoulidou, E., Zheng, Y., Booth, H., et al. (2016). ER stress and autophagic perturbations lead to elevated extracellular alpha-synuclein in GBA-N370S Parkinson’s iPSC-derived dopamine neurons. Stem Cell Rep. 6, 342–356. doi: 10.1016/j.stemcr.2016.01.013
Fernandez-Santiago, R., Carballo-Carbajal, I., Castellano, G., Torrent, R., Richaud, Y., Sanchez-Danes, A., et al. (2015). Aberrant epigenome in iPSC-derived dopaminergic neurons from Parkinson’s disease patients. EMBO Mol. Med. 7, 1529–1546. doi: 10.15252/emmm.201505439
Flierl, A., Oliveira, L. M. A., Falomir-Lockhart, L. J., Mak, S. K., Hesley, J., Soldner, F., et al. (2014). Higher vulnerability and stress sensitivity of neuronal precursor cells carrying an alpha-synuclein gene triplication. PLoS One 9:e112413. doi: 10.1371/journal.pone.0112413
Fraga, M. F., Ballestar, E., Paz, M. F., Ropero, S., Setien, F., Ballestar, M. L., et al. (2005). Epigenetic differences arise during the lifetime of monozygotic twins. Proc. Natl. Acad. Sci. U.S.A. 102, 10604–10609. doi: 10.1073/pnas.0500398102
Fraga, M. F., and Esteller, M. (2007). Epigenetics and aging: the targets and the marks. Trends Genet. 23, 413–418. doi: 10.1016/j.tig.2007.05.008
Gonzalez-Cano, L., Menzl, I., Tisserand, J., Nicklas, S., and Schwamborn, J. C. (2018). Parkinson’s Disease-Associated Mutant LRRK2-Mediated Inhibition of miRNA Activity is Antagonized by TRIM32. Mol. Neurobiol. 55, 3490–3498. doi: 10.1007/s12035-017-0570-y
Guhathakurta, S., Evangelista, B. A., Ghosh, S., Basu, S., and Kim, Y. S. (2017). Hypomethylation of intron1 of α-synuclein gene does not correlate with Parkinson’s disease. Mol. Brain 10, 1–6. doi: 10.1186/s13041-017-0285-z
Gunn-Moore, D., Kaidanovich-Beilin, O., Gallego Iradi, M. C., Gunn-Moore, F., and Lovestone, S. (2018). Alzheimer’s disease in humans and other animals: a consequence of postreproductive life span and longevity rather than aging. Alzheimers Dement. J. Alzheimers Assoc. 14, 195–204. doi: 10.1016/j.jalz.2017.08.014
Harraz, M. M., Xu, J.-C., Guiberson, N., Dawson, T. M., and Dawson, V. L. (2014). MiR-223 regulates the differentiation of immature neurons. Mol. Cell. Ther. 2:18. doi: 10.1186/2052-8426-2-18
Hjelm, B. E., Salhia, B., Kurdoglu, A., Szelinger, S., Reiman, R. A., Sue, L. I., et al. (2013). In vitro-differentiated neural cell cultures progress towards donor-identical brain tissue. Hum. Mol. Genet. 22, 3534–3546. doi: 10.1093/hmg/ddt208
Horvath, S. (2013). DNA methylation age of human tissues and cell types DNA methylation age of human tissues and cell types. Proc. Natl. Acad. Sci. U.S.A. 111, 15538–15543. doi: 10.1073/pnas.1412759111
Hsieh, C.-H., Shaltouki, A., Gonzalez, A. E., Bettencourt da Cruz, A., Burbulla, L. F., St Lawrence, E., et al. (2016). Functional impairment in miro degradation and mitophagy is a shared feature in familial and sporadic Parkinson’s Disease. Cell Stem Cell 19, 709–724. doi: 10.1016/j.stem.2016.08.002
Ibáñez, P., Bonnet, A.-M., Débarges, B., Lohmann, E., Tison, F., Pollak, P., et al. (2004). Causal relation between alpha-synuclein gene duplication and familial Parkinson’s disease. Lancet 364, 1169–1171. doi: 10.1016/S0140-6736(04)17104-3
Imaizumi, Y., Okada, Y., Akamatsu, W., Koike, M., Kuzumaki, N., Hayakawa, H., et al. (2012). Mitochondrial dysfunction associated with increased oxidative stress and α-synuclein accumulation in PARK2 iPSC-derived neurons and postmortem brain tissue. Mol. Brain 5:35. doi: 10.1186/1756-6606-5-35
Joannides, A., Gaughwin, P., Schwiening, C., Majed, H., Sterling, J., Compston, A., et al. (2004). Efficient generation of neural precursors from adult human skin: astrocytes promote neurogenesis from skin-derived stem cells. Lancet 364, 172–178. doi: 10.1016/S0140-6736(04)16630-0
Jowaed, A., Schmitt, I., Kaut, O., and Wüllner, U. (2010). Methylation regulates alpha-synuclein expression and is decreased in Parkinson’s disease patients’ brains. J. Neurosci. 30, 6355–6359. doi: 10.1523/JNEUROSCI.6119-09.2010
Junn, E., Lee, K.-W., Jeong, B. S., Chan, T. W., Im, J.-Y., and Mouradian, M. M. (2009). Repression of alpha-synuclein expression and toxicity by microRNA-7. Proc. Natl. Acad. Sci. U.S.A. 106, 13052–13057. doi: 10.1073/pnas.0906277106
Kantor, B., Tagliafierro, L., Gu, J., Zamora, M. E., Ilich, E., Grenier, C., et al. (2018). Downregulation of SNCA expression by targeted editing of dna methylation: a potential strategy for precision therapy in PD. Mol. Ther. 26, 2638–2649. doi: 10.1016/j.ymthe.2018.08.019
Kirkeby, A., Grealish, S., Wolf, D. A., Nelander, J., Wood, J., Lundblad, M., et al. (2012). Generation of regionally specified neural progenitors and functional neurons from human embryonic stem cells under defined conditions. Cell Rep. 1, 703–714. doi: 10.1016/j.celrep.2012.04.009
Kirkeby, A., Nolbrant, S., Tiklova, K., Heuer, A., Kee, N., Cardoso, T., et al. (2017). Predictive markers guide differentiation to improve graft outcome in clinical translation of hESC-Based Therapy for Parkinson’s Disease. Cell Stem Cell 20, 135–148. doi: 10.1016/j.stem.2016.09.004
Klein, C. J., Botuyan, M. V., Wu, Y., Ward, C. J., Nicholson, G. A., Hammans, S., et al. (2011). Mutations in DNMT1 cause hereditary sensory neuropathy with dementia and hearing loss. Nat. Genet. 43, 595–600. doi: 10.1038/ng.830
Kozlenkov, A., Roussos, P., Timashpolsky, A., Barbu, M., Rudchenko, S., Bibikova, M., et al. (2014). Differences in DNA methylation between human neuronal and glial cells are concentrated in enhancers and non-CpG sites. Nucleic Acids Res. 42, 109–127. doi: 10.1093/nar/gkt838
Kozlenkov, A., Wang, M., Roussos, P., Rudchenko, S., Barbu, M., Bibikova, M., et al. (2015). Substantial DNA methylation differences between two major neuronal subtypes in human brain. Nucleic Acids Res. 44, 2593–2612. doi: 10.1093/nar/gkv1304
Kriks, S., Shim, J.-W., Piao, J., Ganat, Y. M., Wakeman, D. R., Xie, Z., et al. (2011). Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson’s disease. Nature 480, 547–551. doi: 10.1038/nature10648
Ladewig, J., Mertens, J., Kesavan, J., Doerr, J., Poppe, D., Glaue, F., et al. (2012). Small molecules enable highly efficient neuronal conversion of human fibroblasts. Nat. Methods 9, 575–578. doi: 10.1038/nmeth.1972
Landgrave-Gomez, J., Mercado-Gomez, O., and Guevara-Guzman, R. (2015). Epigenetic mechanisms in neurological and neurodegenerative diseases. Front. Cell. Neurosci. 9:58. doi: 10.3389/fncel.2015.00058
Lang, C., Campbell, K. R., Ryan, B. J., Carling, P., Attar, M., Vowles, J., et al. (2018). Single-cell sequencing of iPSC-dopamine neurons reconstructs disease progression and identifies HDAC4 as a regulator of parkinson cell phenotypes. Cell Stem Cell 24, 93–106.e6. doi: 10.1016/j.stem.2018.10.023
Liu, X. S., Wu, H., Krzisch, M., Wu, X., Graef, J., Muffat, J., et al. (2018). Rescue of fragile X syndrome neurons by DNA methylation editing of the FMR1 gene. Cell 172, 979–991.e6. doi: 10.1016/j.cell.2018.01.012
Löhle, M., and Reichmann, H. (2010). Clinical neuroprotection in Parkinson’s disease - Still waiting for the breakthrough. J. Neurol. Sci. 289, 104–114. doi: 10.1016/j.jns.2009.08.025
Lopez de Maturana, R., Lang, V., Zubiarrain, A., Sousa, A., Vazquez, N., Gorostidi, A., et al. (2016). Mutations in LRRK2 impair NF-kappaB pathway in iPSC-derived neurons. J. Neuroinflammation 13:295. doi: 10.1186/s12974-016-0761-x
Lotharius, J., Falsig, J., van Beek, J., Payne, S., Dringen, R., Brundin, P., et al. (2005). Progressive degeneration of human mesencephalic neuron-derived cells triggered by dopamine-dependent oxidative stress is dependent on the mixed-lineage kinase pathway. J. Neurosci. 25, 6329–6342. doi: 10.1523/JNEUROSCI.1746-05.2005
Lowdon, R. F., Jang, H. S., and Wang, T. (2016). Evolution of epigenetic regulation in vertebrate genomes. Trends Genet. 32, 269–283. doi: 10.1016/j.tig.2016.03.001
Lowenstine, L. J., McManamon, R., and Terio, K. A. (2016). Comparative pathology of aging great apes: bonobos, chimpanzees, gorillas, and orangutans. Vet. Pathol. 53, 250–276. doi: 10.1177/0300985815612154
Marras, C., and Lang, A. (2013). Parkinson’s disease subtypes: lost in translation? J. Neurol. Neurosurg. Psychiatry 84, 409–415. doi: 10.1136/jnnp-2012-303455
Mastroeni, D., McKee, A., Grover, A., Rogers, J., and Coleman, P. D. (2009). Epigenetic differences in cortical neurons from a pair of monozygotic twins discordant for Alzheimer’s disease. PLoS One 4:e6617. doi: 10.1371/journal.pone.0006617
Matsumoto, L., Takuma, H., Tamaoka, A., Kurisaki, H., Date, H., Tsuji, S., et al. (2010). CpG demethylation enhances alpha-synuclein expression and affects the pathogenesis of Parkinson’s disease. PLoS One 5:e15522. doi: 10.1371/journal.pone.0015522
Mazzulli, J. R., Zunke, F., Tsunemi, T., Toker, N. J., Jeon, S., Burbulla, L. F., et al. (2016). Activation of beta-glucocerebrosidase reduces pathological alpha-synuclein and restores lysosomal function in Parkinson’s patient midbrain neurons. J. Neurosci. 36, 7693–7706. doi: 10.1523/JNEUROSCI.0628-16.2016
Mendizabal, I., Keller, T. E., Zeng, J., and Yi, S. V. (2014). Integrative and comparative biology epigenetics and evolution. Integr. Comp. Biol. 54, 31–42. doi: 10.1093/icb/icu040
Mertens, J., Paquola, A. C. M., Ku, M., Hatch, E., Bohnke, L., Ladjevardi, S., et al. (2015). Directly reprogrammed human neurons retain aging-associated transcriptomic signatures and reveal age-related nucleocytoplasmic defects. Cell Stem Cell 17, 705–718. doi: 10.1016/j.stem.2015.09.001
Miller, J. D., Ganat, Y. M., Kishinevsky, S., Bowman, R. L., Liu, B., Tu, E. Y., et al. (2013). Human iPSC-based modeling of late-onset disease via progerin-induced aging. Cell Stem Cell 13, 691–705. doi: 10.1016/j.stem.2013.11.006
Montaño, C. M., Irizarry, R. A., Kaufmann, W. E., Talbot, K., Gur, R. E., Feinberg, A. P., et al. (2013). Measuring cell-type specific differential methylation in human brain tissue. Genome Biol. 14:R94. doi: 10.1186/gb-2013-14-8-r94
Nakagawa, M., Koyanagi, M., Tanabe, K., Takahashi, K., Ichisaka, T., Aoi, T., et al. (2008). Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat. Biotechnol. 26, 101–106. doi: 10.1038/nbt1374
Nguyen, H. N., Byers, B., Cord, B., Shcheglovitov, A., Byrne, J., Gujar, P., et al. (2011). LRRK2 mutant iPSC-derived DA neurons demonstrate increased susceptibility to oxidative stress. Cell Stem Cell 8, 267–280. doi: 10.1016/j.stem.2011.01.013
Oh, S. E., Park, H. J., He, L., Skibiel, C., Junn, E., and Mouradian, M. M. (2018). The Parkinson’s disease gene product DJ-1 modulates miR-221 to promote neuronal survival against oxidative stress. Redox Biol. 19, 62–73. doi: 10.1016/j.redox.2018.07.021
Olanow, C. W., Kieburtz, K., and Schapira, A. H. V. (2008). Why have we failed to achieve neuroprotection in Parkinson’s disease? Ann. Neurol. 64, S101–S110. doi: 10.1002/ana.21461
Oliveira, L. M. A., Falomir-Lockhart, L. J., Botelho, M. G., Lin, K.-H., Wales, P., Koch, J. C., et al. (2015). Elevated alpha-synuclein caused by SNCA gene triplication impairs neuronal differentiation and maturation in Parkinson’s patient-derived induced pluripotent stem cells. Cell Death Dis. 6:e1994. doi: 10.1038/cddis.2015.318
Paiva, I., Pinho, R., Pavlou, M. A., Hennion, M., Wales, P., Schutz, A.-L., et al. (2017). Sodium butyrate rescues dopaminergic cells from alpha-synuclein-induced transcriptional deregulation and DNA damage. Hum. Mol. Genet. 26, 2231–2246. doi: 10.1093/hmg/ddx114
Pavlou, M. A. S., Colombo, N., Fuertes-Alvarez, S., Nicklas, S., Cano, L. G., Marin, M. C., et al. (2017). Expression of the Parkinson’s Disease-associated gene alpha-synuclein is regulated by the neuronal cell fate determinant TRIM32. Mol. Neurobiol. 54, 4257–4270. doi: 10.1007/s12035-016-9989-9
Petrif, F., Giles, R. H., Dauwerse, H. G., Saris, J. J., Hennekam, R. C. M., Masuno, M., et al. (1995). Rubinstein-Taybi syndrome caused by mutations in the transcriptional co-activator CBP. Nature 376, 348–351. doi: 10.1038/376348a0
Pierce, S. E., Tyson, T., Booms, A., Prahl, J., and Coetzee, G. A. (2018). Parkinson’s disease genetic risk in a midbrain neuronal cell line. Neurobiol. Dis. 114, 53–64. doi: 10.1016/j.nbd.2018.02.007
Portela, A., and Esteller, M. (2010). Epigenetic modifications and human disease. Nat. Biotechnol. 28, 1057–1068. doi: 10.1038/nbt.1685
Prasad, A., Manivannan, J., Loong, D. T. B., Chua, S. M., Gharibani, P. M., and All, A. H. (2016). A review of induced pluripotent stem cell, direct conversion by trans-differentiation, direct reprogramming and oligodendrocyte differentiation. Regen. Med. 11, 181–191. doi: 10.2217/rme.16.5
Qiu, L., Liao, M.-C., Chen, A. K., Wei, S., Xie, S., Reuveny, S., et al. (2017). Immature midbrain dopaminergic neurons derived from floor-plate method improve cell transplantation therapy efficacy for Parkinson’s Disease. Stem Cells Transl. Med. 6, 1803–1814. doi: 10.1002/sctm.16-0470
Qureshi, I. A., and Mehler, M. F. (2018). Epigenetic mechanisms underlying nervous system diseases. Handb. Clin. Neurol. 147, 43–58. doi: 10.1016/B978-0-444-63233-3.00005-1
Rando, T. A., and Chang, H. Y. (2012). Aging, rejuvenation, and epigenetic reprogramming: resetting the aging clock. Cell 148, 46–57. doi: 10.1016/j.cell.2012.01.003
Ren, Y., Jiang, H., Hu, Z., Fan, K., Wang, J., Janoschka, S., et al. (2015). Parkin mutations reduce the complexity of neuronal processes in iPSC-derived human neurons. Stem Cells 33, 68–78. doi: 10.1002/stem.1854
Reyes, J. F., Olsson, T. T., Lamberts, J. T., Devine, M. J., Kunath, T., and Brundin, P. (2015). A cell culture model for monitoring alpha-synuclein cell-to-cell transfer. Neurobiol. Dis. 77, 266–275. doi: 10.1016/j.nbd.2014.07.003
Richards, E. J. (2008). Population epigenetics. Curr. Opin. Genet. Dev. 18, 221–226. doi: 10.1016/j.gde.2008.01.014
Sandor, C., Robertson, P., Lang, C., Heger, A., Booth, H., Vowles, J., et al. (2017). Transcriptomic profiling of purified patient-derived dopamine neurons identifies convergent perturbations and therapeutics for Parkinson’s disease. Hum. Mol. Genet. 26, 552–566. doi: 10.1093/hmg/ddw412
Sato, F., Tsuchiya, S., Meltzer, S. J., and Shimizu, K. (2011). MicroRNAs and epigenetics. FEBS J. 278, 1598–1609. doi: 10.1111/j.1742-4658.2011.08089.x
Schondorf, D. C., Aureli, M., McAllister, F. E., Hindley, C. J., Mayer, F., Schmid, B., et al. (2014). iPSC-derived neurons from GBA1-associated Parkinson’s disease patients show autophagic defects and impaired calcium homeostasis. Nat. Commun. 5:4028. doi: 10.1038/ncomms5028
Schwab, A. J., and Ebert, A. D. (2015). Neurite aggregation and calcium dysfunction in iPSC-derived sensory neurons with Parkinson’s disease-related LRRK2 G2019S Mutation. Stem Cell Rep. 5, 1039–1052. doi: 10.1016/j.stemcr.2015.11.004
Schwab, A. J., Sison, S. L., Meade, M. R., Broniowska, K. A., Corbett, J. A., and Ebert, A. D. (2017). Decreased sirtuin deacetylase activity in LRRK2 G2019S iPSC-Derived Dopaminergic Neurons. Stem Cell Rep. 9, 1839–1852. doi: 10.1016/j.stemcr.2017.10.010
Schwamborn, J. C., Berezikov, E., and Knoblich, J. A. (2009). The TRIM-NHL protein TRIM32 activates microRNAs and prevents self-renewal in mouse neural progenitors. Cell 136, 913–925. doi: 10.1016/j.cell.2008.12.024
Sheng, C., Jungverdorben, J., Wiethoff, H., Lin, Q., Flitsch, L., Eckert, D., et al. (2018). A stably self-renewing adult blood-derived induced neural stem cell exhibiting patternability and epigenetic rejuvenation. Nat. Commun. 9:4047. doi: 10.1038/s41467-018-06398-5
Siddiqui, A., Bhaumik, D., Chinta, S. J., Rane, A., Rajagopalan, S., Lieu, C. A., et al. (2015). Mitochondrial quality control via the PGC1alpha-TFEB signaling pathway is compromised by parkin Q311X mutation but independently restored by rapamycin. J. Neurosci. 35, 12833–12844. doi: 10.1523/JNEUROSCI.0109-15.2015
Siegmund, K. D., Connor, C. M., Campan, M., Long, T. I., Weisenberger, D. J., Biniszkiewicz, D., et al. (2007). DNA methylation in the human cerebral cortex is dynamically regulated throughout the life span and involves differentiated neurons. PLoS One 2:e895. doi: 10.1371/journal.pone.0000895
Singh, M., Kakkar, A., Sharma, R., Kharbanda, O. P., Monga, N., Kumar, M., et al. (2017). Synergistic Effect of BDNF and FGF2 in efficient generation of functional dopaminergic neurons from human mesenchymal stem cells. Sci. Rep. 7:10378. doi: 10.1038/s41598-017-11028-z
Singleton, A. B., Farrer, M., Johnson, J., Singleton, A. B., Hague, S., Kachergus, J., et al. (2003). Alpha-Synuclein locus triplication causes Parkinson’s disease. Science 302:841. doi: 10.1126/science.1090278
Spillantini, M. G., Schmidt, M. L., Lee, V. M., Trojanowski, J. Q., Jakes, R., and Goedert, M. (1997). Alpha-synuclein in Lewy bodies. Nature 388, 839–840. doi: 10.1038/42166
Straniero, L., Rimoldi, V., Samarani, M., Goldwurm, S., Di Fonzo, A., Kruger, R., et al. (2017). The GBAP1 pseudogene acts as a ceRNA for the glucocerebrosidase gene GBA by sponging miR-22-3p. Sci. Rep. 7:12702. doi: 10.1038/s41598-017-12973-5
Subramaniam, S. R., and Federoff, H. J. (2017). Targeting microglial activation states as a Therapeutic Avenue in Parkinson’s disease. Front. Aging Neurosci. 9:176. doi: 10.3389/fnagi.2017.00176
Tagliafierro, L., Glenn, O. C., Zamora, M. E., Beach, T. G., Woltjer, R. L., and Lutz, M. W. (2018). Genetic analysis of SNCA 3’UTR and its corresponding miRNAs in relation to Parkinson’s compared to Dementia with Lewy Bodies. Alzheimers Dement. 13, 1237–1250. doi: 10.1016/j.jalz.2017.03.001.Genetic
Tanner, C. M., and Goldman, S. M. (1996). Epidemiology of Parkinson’s disease. Neurol. Clin. 14, 317–335. doi: 10.1016/S0733-8619(05)70259-0
Teschendorff, A. E., West, J., and Beck, S. (2013). Age-associated epigenetic drift: implications, and a case of epigenetic thrift? Hum. Mol. Genet. 22, R7–R15. doi: 10.1093/hmg/ddt375
Thomson, J. A., Itskovitz-Eldor, J., Shapiro, S. S., Waknitz, M. A., Swiergiel, J. J., Marshall, V. S., et al. (1998). Embryonic stem cell lines derived from human blastocysts. Science 282, 1145–1147. doi: 10.1126/science.282.5391.1145
Tolosa, E., Botta-Orfila, T., Morató, X., Calatayud, C., Ferrer-Lorente, R., Martí, M. J., et al. (2018). MicroRNA alterations in iPSC-derived dopaminergic neurons from Parkinson disease patients. Neurobiol. Aging 69, 283–291. doi: 10.1016/j.neurobiolaging.2018.05.032
Tsai, E.-M., Wang, Y.-C., Lee, T. T.-Y., Tsai, C.-F., Chen, H.-S., Lai, F.-J., et al. (2015). Dynamic Trk and G protein signalings regulate dopaminergic neurodifferentiation in human trophoblast stem cells. PLoS One 10:e0143852. doi: 10.1371/journal.pone.0143852
Wen, K.-X., Milic, J., El-Khodor, B., Dhana, K., Nano, J., Pulido, T., et al. (2016). The Role of DNA methylation and histone modifications in neurodegenerative diseases: a systematic review. PLoS One 11:e0167201. doi: 10.1371/journal.pone.0167201
Woodard, C. M., Campos, B. A., Kuo, S.-H., Nirenberg, M. J., Nestor, M. W., Zimmer, M., et al. (2014). iPSC-derived dopamine neurons reveal differences between monozygotic twins discordant for Parkinson’s disease. Cell Rep. 9, 1173–1182. doi: 10.1016/j.celrep.2014.10.023
Wulansari, N., Kim, E.-H., Sulistio, Y. A., Rhee, Y.-H., Song, J.-J., and Lee, S.-H. (2017). Vitamin C-induced epigenetic modifications in donor NSCs Establish midbrain marker expressions critical for cell-based therapy in parkinson’s disease. Stem Cell Rep. 9, 1192–1206. doi: 10.1016/j.stemcr.2017.08.017
Wullner, U., Kaut, O., deBoni, L., Piston, D., and Schmitt, I. (2016). DNA methylation in Parkinson’s disease. J. Neurochem. 139(Suppl. 1), 108–120. doi: 10.1111/jnc.13646
Xia, N., Zhang, P., Fang, F., Wang, Z., Rothstein, M., Angulo, B., et al. (2016). Transcriptional comparison of human induced and primary midbrain dopaminergic neurons. Sci. Rep. 6:20270. doi: 10.1038/srep20270
Xu, Z., Chu, X., Jiang, H., Schilling, H., Chen, S., and Feng, J. (2017). Induced dopaminergic neurons: a new promise for Parkinson’s disease. Redox Biol. 11, 606–612. doi: 10.1016/j.redox.2017.01.009
Yang, S.-Y., Beavan, M., Chau, K.-Y., Taanman, J.-W., and Schapira, A. H. V. (2017). A human neural crest stem cell-derived dopaminergic neuronal model recapitulates biochemical abnormalities in GBA1 mutation carriers. Stem Cell Rep. 8, 728–742. doi: 10.1016/j.stemcr.2017.01.011
Zeng, J., Konopka, G., Hunt, B. G., Preuss, T. M., Geschwind, D., and Yi, S. V. (2012). Divergent whole-genome methylation maps of human and chimpanzee brains reveal epigenetic basis of human regulatory evolution. Am. J. Hum. Genet. 91, 455–465. doi: 10.1016/j.ajhg.2012.07.024
Keywords: stem cells, iPSC, neurons, human, epigenetics, Parkinson’s disease
Citation: de Boni L and Wüllner U (2019) Epigenetic Analysis in Human Neurons: Considerations for Disease Modeling in PD. Front. Neurosci. 13:276. doi: 10.3389/fnins.2019.00276
Received: 05 July 2018; Accepted: 08 March 2019;
Published: 05 April 2019.
Edited by:
Aideen M. Sullivan, University College Cork, IrelandReviewed by:
Maeve Ann Caldwell, Trinity College Dublin, IrelandMario Ezquerra, August Pi i Sunyer Biomedical Research Institute (IDIBAPS), Spain
Copyright © 2019 de Boni and Wüllner. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ullrich Wüllner, VWxscmljaC53dWVsbG5lckB1a2Jvbm4uZGU=