 Patrick W. Sheehan
Patrick W. Sheehan Erik S. Musiek
Erik S. Musiek- 1Department of Neurology, Washington University School of Medicine in St. Louis, St. Louis, MO, United States
- 2Knight Alzheimer’s Disease Research Center, Washington University School of Medicine in St. Louis, St. Louis, MO, United States
Circadian dysfunction has been described in patients with symptomatic Alzheimer’s disease (AD), as well as in presymptomatic phases of the disease. Modeling this circadian dysfunction in mouse models would provide an optimal platform for understanding mechanisms and developing therapies. While numerous studies have examined behavioral circadian function, and in some cases clock gene oscillation, in mouse models of AD, the results are variable and inconsistent across models, ages, and conditions. Ultimately, circadian changes observed in APP/PS1 models are inconsistent across studies and do not always replicate circadian phenotypes observed in human AD. Other models, including the 3xTG mouse, tau transgenic lines, and the accelerated aging SAMP8 line, show circadian phenotypes more consistent with human AD, although the literature is either inconsistent or minimal. We summarize these data and provide some recommendations to improve and standardize future studies of circadian function in AD mouse models.
Introduction
Numerous human studies have demonstrated that changes in circadian function are common in Alzheimer’s disease (AD) patients and contribute to disease morbidity. Circadian changes observed in AD patients include circadian fragmentation and decreased amplitude of circadian rhythms, which generally manifest as increased wakefulness at night and increased napping during the day (Satlin et al., 1995; Ancoli-Israel et al., 1997). These changes are observed in patients with preclinical AD pathology, meaning that they harbor amyloid plaque and/or tau pathology but do not yet have cognitive symptoms (Musiek et al., 2018). They are also evident in the mild cognitive impairment stage and worsen with disease severity. In symptomatic AD patients, a phase delay has been described, which means that the peak of activity occurs later in the day (Satlin et al., 1995; Ancoli-Israel et al., 1997). This phase delay has been hypothesized as a possible cause of “sundowning” in AD, or increased confusion and agitation in the afternoon and evening (Volicer et al., 2001). As the disease progresses, some patients develop severe fragmentation of circadian rhythms, leading to poor behavioral differentiation between day and night (Ancoli-Israel et al., 1997; Hatfield et al., 2004). The underlying mechanisms governing this circadian dysfunction are not well understood, creating a need for accurate mouse models which replicate some of these phenotypes.
Circadian biology can be a daunting field for AD researchers. Thus, a brief introduction to common circadian parameters may be helpful (Banks and Nolan, 2011). Circadian period is the length of time of a full circadian cycle. This can only be measured under “constant conditions” (usually meaning constant darkness for mice, indicated as “DD”). Most mice have a period just under 24 h in constant darkness; shortening or lengthening of period can be indicative of changes in the circadian clock itself. Amplitude indicates the difference between the peak and nadir of activity, averaged over several days. In general, mice should have minimal activity during the light phase (as they are nocturnal and sleeping), but are very active during the dark phase. Increased activity during the light phase and decreased activity in the dark would indicate blunted circadian amplitude, as is seen in AD patients. However, generally hyperactive animals can be overactive during the dark phase and have a high amplitude, while hypoactive animals may be inactive during both phases and show artificially blunted amplitude. If these differences in baseline activity levels are altered in transgenic mouse models, it may lead to difficulty interpreting circadian behavioral analyses. Phase indicates the time of day of peak activity, averaged over several days. A later peak (phase delay) is seen in AD patients. Fragmentation indicates a breakdown of circadian timing to drive consolidated periods of rest and activity. Fragmentation usually leads to a blunted amplitude, or can be measured by a non-parametric test called intradaily variability (IV) (Huang et al., 2002). Of note, circadian rhythms in behavior are largely driven by the activity of the suprachiasmatic nucleus (SCN) of the hypothalamus, the “master clock” of the body. Degeneration or dysfunction of the SCN, as has been described in human AD (Swaab et al., 1985), can lead to weaker circadian synchronization, increased fragmentation, and decreased amplitude (Nakamura et al., 2011). At a molecular level, circadian rhythms are driven by oscillations of circadian clock genes in the SCN and other tissues. These circadian clock genes include Bmal1 and Clock, which function as transcription factors to drive transcription of their own repressors: Per1, Per2, Per3, Cry1, Cry2, and Rev-erbα and β. Levels of these clock gene transcripts oscillate with a 24-h period in most tissue and are entrained to daily light cycles (Buhr and Takahashi, 2013). Thus, measurement of clock gene oscillations in SCN and other tissues can be a molecular marker of clock function.
Over the past 20 years, a considerable literature has arisen in which circadian parameters have been examined in various AD models. Many of these studies have described modest changes in AD model mice, though these changes vary considerably between studies. The sheer number of distinct AD mouse models, as well as the age- and sex-dependency of pathology in these models, has complicated things further. Thus, it remains difficult to identify AD models which consistently and faithfully recapitulate the findings observed in human AD. Below, we have attempted to divide AD mouse models into their most common categories, and to review the existing literature on circadian rhythms in these models. These data are compiled in Table 1. While not an exhaustive or systematic review, the included studies represent many of the vast majority of published studies, as well as the most commonly cited and most thorough studies of circadian function in AD mouse models (other species were excluded), and thus provide what we believe is a representative sample of the literature.
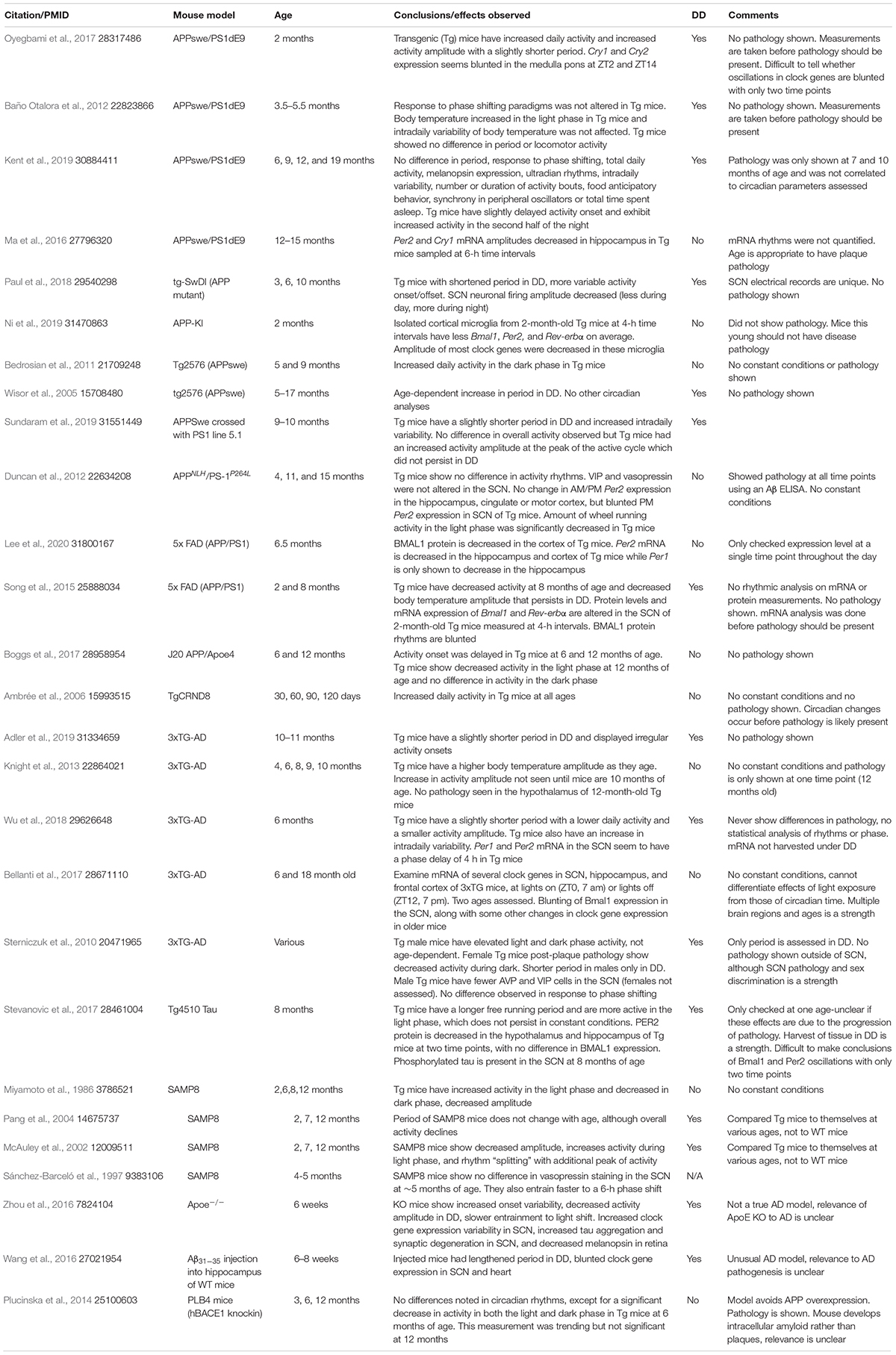
Table 1. Summary of studies of circadian function in mouse models of AD.
APP and APP/PS1 Mice
There are a multitude of different transgenic mouse lines which express human amyloid precursor protein (APP) and in some cases human presenilin 1 (PS1), both harboring a variety of AD-associated mutations which promote amyloid-beta (Aβ) generation and aggregation. These mice accumulate amyloid plaque pathology at variable ages and in varying brain regions depending on the specific transgene and sex of the animal. APP/PS1 mice also vary in the biochemical character of the plaques that are formed, as well as the presence or absence of intracellular amyloid. Presumably, APP and APP/PS1 mice model the earliest stage of AD, that being amyloid plaque deposition, as most of these models develop minimal tau pathology or overt neurodegeneration. Humans with preclinical AD or MCI have been shown to have decreased circadian amplitude, increased fragmentation, and, in MCI, a phase delay (Naismith et al., 2014; Ortiz-Tudela et al., 2014). However, results of circadian studies in APP and APP/PS1 mice do not clearly demonstrate consistent AD-like phenotypes. Several studies have shown increased activity in these mouse lines during the dark phase (the active phase of mice), as well as increases in circadian amplitude (Ambrée et al., 2006; Bedrosian et al., 2011; Baño Otalora et al., 2012; Oyegbami et al., 2017). Aside from this, the wide variety of subtle and oft-conflicting circadian phenotypes in APP and APP/PS1 mouse studies can be appreciated in Table 1. Thus, a clear circadian phenotype which models early human AD has not emerged from the literature. Recently, Kent et al. conducted a very thorough study of APPswe/PS1dE9 mice, a commonly used APP/PS1 model which developed plaques around 6 months old (mo). This study, which examined mice at multiple ages and included a variety of light manipulations and Per2-luciferase rhythm recordings in SCN and various peripheral tissues (such as liver, lung, and spleen), revealed only a minimal delay in activity onset after lights-off in transgenic mice (Kent et al., 2019). Moreover, several studies demonstrate circadian changes which precede amyloid plaque pathology, suggesting possible strain or transgene effects, though an effect of soluble Aβ cannot be excluded (Bedrosian et al., 2011; Ortiz-Tudela et al., 2014; Oyegbami et al., 2017; Ni et al., 2019). While not a true circadian study, Roh et al. (2012) demonstrated blunting of amplitude in diurnal rhythms in sleep and brain lactate levels in APPswe/PS1dE9 mice, which was reversible with immunization of an anti-Aβ antibody, suggesting that amyloid pathology may indeed contribute to some aspects of rhythm dysfunction. Paul et al. (2018) examined the TgSwDI APP mutant mouse and observed shortened period and increased variability in activity onset/offset that were associated with blunted amplitude of neuronal firing in the SCN, providing an electrophysiological basis for rhythm disturbance. A human BACE1 knockin mouse, which drives intracellular amyloid accumulation without APP overexpression, showed decreased dark-phase activity but no other major circadian alterations, though assessment was limited (Plucinska et al., 2014). An issue with the wide array of APP and APP/PS1 is that these transgenes are driven by specific promoters (including the Thy1 and prion protein promoters) which may not express in all relevant cell types and regions (such as the SCN and circadian output pathways). This variability of transgene expression may account for some of the inconsistency in circadian phenotypes across APP/PS1 models. While APP and APP/PS1 models have been the most studied in terms of circadian function, the wide array of unique APP and APP/PS1 lines, as well as the possible amyloid-independent effects of these transgenes on activity level, has led to a murky literature which undermines the utility of these models for examining circadian changes.
Tauopathy Models
Tau transgenic mice overexpress the human MAPT gene with disease-causing mutations (often P301S or P301L) and are often used to model tauopathy associated with AD. Unlike APP mice, tau transgenics generally develop striking neurodegeneration and premature death. The Tg4510 mouse, an aggressive model of tauopathy which expresses MAPTP301L (Ramsden et al., 2005), develops a lengthened circadian period and decreased circadian amplitude, as well as seemingly blunted circadian clock gene expression in the hypothalamus and hippocampus when measured at two times of day (Stevanovic et al., 2017). These changes were observed at 8 mo, when severe tau pathology is present. SCN tau pathology was also noted in this study. A study of MAPTP301S PS19 mice, which focused exclusively on sleep, showed a decrease in sleep and an increase in wakefulness, suggesting a possible underlying circadian deficit, very late in the disease progression (Holth et al., 2017). While promising, further studies of circadian function in tauopathy models are needed to determine how robust and consistent these changes are across tau models and studies.
3xTG Mice
The 3xTG mouse was introduced in 2003 as a murine model of both of the hallmark neuropathologies of human AD: amyloid plaques and tau tangles. 3xTG mice express three transgenes (APP Swedish, MAPT P301L, and PS1 M146V) and develop amyloid plaque pathology starting around 4 months, with aggregated tau pathology beginning around 12 months (Oddo et al., 2003). Several studies have examined circadian function in 3xTG mice, some of which describe phenotypes which more closely resemble human AD. However, results are fraught with inconsistency across different studies. Wu et al. (2018) show a striking decrease in amplitude, less daily activity, and fragmentation (increased IV) in 6 mo 3xTG mice, reminiscent of human AD circadian dysfunction. Knight et al. (2013) report no changes in activity profile in 6 mo 3xTG mice and go on to show an increased amplitude in daily activity and temperature rhythm by 10 months. Moreover, Sterniczuk et al. (2010) show increased amplitude and daytime activity in four month old male mice while female mice show a decreased amplitude by 11 months. Finally, Adler et al. (2019) showed a shortened period and decreased amplitude in 3xTG mice which was ameliorated by inhibiting casein kinase 1δ and 1ε, enzymes that are important for the degradation of PER proteins and maintenance of core clock function. Thus, while the 3xTG mouse shows some promise as a more accurate model of AD-like circadian changes, the inconsistency across studies makes interpretation difficult. It is also notable that these circadian changes were all observed at ages prior to the accumulation of considerable tau pathology. Of note, a different triple-transgenic mouse expressing APP, hTau, and PS1, specifically in the forebrain (PLB1 mice), did not show obvious circadian deficits, suggesting that SCN-dependent expression of pathology may be important (Platt et al., 2011).
Other AD Models
The Senescence Accelerated Mouse, line P8 (SAMP8 mouse) is a unique, non-transgenic mouse line that has been selectively bred to promote accelerated aging (Butterfield and Poon, 2005; Ramsden et al., 2005; Holth et al., 2017; Stevanovic et al., 2017). SAMP8 mice are usually compared to another SAM mouse line which is aging-resistant (SAMR1) as a control. SAMP8 mice spontaneously develop mild amyloid-beta accumulation, mild tauopathy, synapse loss, and cognitive impairment early in life, and have been used as a model of AD (Morley et al., 2012). SAMP8 mice show striking circadian changes, including increased activity during light phase, decreased circadian amplitude, and fragmentation (Miyamoto et al., 1986; McAuley et al., 2002; Pang et al., 2004), although this has not always been reported (Sánchez-Barceló et al., 1997). Like APP mice, they show a general hyperactivity phenotype, which complicates interpretation. However, the striking circadian changes observed in several studies suggest that SAMP8 mice have potential as a model of age and AD related circadian dysfunction.
Apolipoprotein E (ApoE) genotype is the major genetic risk factor for sporadic AD, with the E4 allele imparting increased risk. Apoe−/− mice have been reported to have more variable activity rhythms in DD, impaired entrainment, and alterations of SCN clock gene expression rhythms (Zhou et al., 2016). However, APP mice expressing human ApoE4 do not have more severe circadian changes than APP with wild type mouse ApoE (Graybeal et al., 2015; Boggs et al., 2017). This is an unexpected finding, as ApoE4 increases amyloid pathology in mice (Castellano et al., 2011) and might be expected to exacerbate circadian dysfunction. More detailed studies which incorporate pathology assessments are needed. Injection of Aβ peptide into the brain of wild type mice has also been used to model AD, and has been reported to lengthen period and blunt clock gene rhythms in the SCN (Navigatore-Fonzo et al., 2017), and to alter expression patterns of Apoe and other mRNAs in the hippocampus (Wang et al., 2016), though this model is not widely used.
Impact of AD Pathology on Clock Gene Expression
Differences in circadian outputs at the level of sleep-wake cycles can generally be correlated to changes in the molecular clock. However, differences in clock gene expression in studies of AD models are generally difficult to interpret. For example, some studies have suggested decreased expression of clock genes in AD models before the onset of pathology. Oyegbami et al. (2017) show a decrease in Cry1 and Cry2 in the medulla pons of 2-month-old APPswe/PS1dE9 mice, while Ni et al. (2019) show a decrease in Bmal1, Per2, and Rev-erbα in isolated cortical microglia from 2-month-old APP-KI mice (both well before plaque deposition occurs). Song et al. (2015) showed blunting of rhythmic BMAL1 protein levels prior to disease pathology in the SCN of 2 mo 5xFAD mice (an APP/PS1 mutant model). These may indicate an effect of soluble Aβ, or effects of the transgene/mutation introduced, highlighting the importance of these assays at multiple ages over the course of disease pathology. Wu et al. (2018) showed a slight phase delay and decreased expression of Per1/2 in the SCN of 3xTG mice, while Ma et al. (2016) showed altered rhythms in hippocampal Bace2 and Apoe mRNA. Other studies in AD models have examined clock gene expression at one or two time points throughout the circadian cycle, usually early morning and early evening, in some case showing a loss of variation in AD mice (Duncan et al., 2012; Bellanti et al., 2017; Stevanovic et al., 2017; Lee et al., 2020). While these can be used to make general conclusions, it is difficult to interpret if clock genes are changing amplitude, or if there is just a difference in phase of expression of that particular gene. Detailed time courses of circadian gene expression in different brain regions of AD mouse models at pathology-bearing ages are needed to thoroughly address this issue.
Recommendations for Circadian Studies in AD Mouse Models
Considering the complexity of this literature, we offer here some recommendations for future studies of circadian rhythms in AD mouse models:
Constant conditions: Mouse activity must be recorded under constant conditions (usually constant darkness or dim light) in order to make firm conclusions about the circadian system. Many studies of behavioral rhythms in AD model mice are conducted in 12:12 h light:dark, which introduces factors related to light intensity, as well as the issue of “masking,” or behavioral suppression by light which occurs independently of the circadian system. Furthermore, previous studies have shown some rhythm abnormalities in AD models in 12:12 h light:dark cycles that resolve in constant conditions (Stevanovic et al., 2017; Sundaram et al., 2019). Moreover, tissue harvests for clock gene expression studies should also be conducted in DD, as light can impact clock gene expression (Moriya et al., 2000).
Correlation with pathology: Many studies of circadian function in AD models do not describe or quantify the degree of pathology in their mice. It is important to demonstrate the degree of pathology (amyloid plaque burden, tau pathology, etc.) in the mice at the same age and in the same sex that are used for behavioral studies. While previous papers can be used as general guidelines, mice of a similar genotype can have very different pathology when raised in different colonies and facilities. Moreover, circadian deficits arising in AD mice well before pathology may suggest behavioral effects of transgene overexpression, rather than true pathology-driven changes. Indeed, behavioral changes in 3xTG can be dependent on genetic background (Pardossi-Piquard et al., 2016). Finally, it is potentially important to assess the degree of pathology in the SCN in these mouse models, to see if there is a direct effect on the central pacemaker. SCN pathology has only been analyzed in a few models, as 3xTG mice do not have plaques or tangles at 12 months, while Tg4510 mice do have SCN tau pathology at 8 months (Knight et al., 2013; Stevanovic et al., 2017). Even in the absence of clear plaque pathology, it is possible that soluble Aβ species (such as oligomers) may impact SCN function, though levels and dynamics of soluble Aβ species in the SCN have not been examined. Finally, human post-mortem AD SCN generally shows neuronal loss and tau pathology, rather than plaques, which may not be recapitulated in amyloid-based models (Stopa et al., 1999).
Multiple endpoints: Most studies focus on rhythms in wheel running as the primary endpoint. While some have included temperature or some limited clock gene investigation, little is known about other circadian endpoints. Peripheral clocks, for instance, could be disrupted even in the face of normal-appearing wheel running behavior. Some studies do examine Per2-luciferase rhythms in SCN explants (Kent et al., 2019), clock gene expression in SCN (Song et al., 2015; Stevanovic et al., 2017), or SCN firing rate (Paul et al., 2018). Integration of multiple endpoints in future studies may reveal important phenotypes.
Sex and age considerations: Sex has a strong effect on both circadian rhythms, as well as on pathology in AD models. In general, female APP and APP/PS1 mouse lines develop amyloid plaques faster than males, while this relationship is reversed for the accumulation of tau pathology in MAPT mice (Carroll et al., 2010; Sun et al., 2020). Furthermore, there is potentially conflicting effects of sex on circadian biology that could further complicate these studies (Bailey and Silver, 2014). Thus, sex must always be considered. Age also has strong effects on circadian function, but is less clearly tied to amyloid/tau pathology in mouse models, as specific transgene/mutations can drive pathology at vastly different ages. In some cases, mice develop plaques when they are still young, while in others age and amyloid pathology come together and may interact (Wisor et al., 2005). Thus, the effects of age itself, and the rate of pathology accumulation, must be considered.
Conclusion
In summary, while many AD mouse models exhibit alterations in circadian behavioral rhythms and/or gene expression, these changes are generally not consistent across studies or models, or have questionable relevance to human AD. As such, it is still unclear if specific protein pathologies (such as amyloid or tau aggregation) directly drive circadian changes, or if these observed changes are due to other factors (such as transgene overexpression, genetic background, sex, or age). As new AD models are developed, circadian studies must consider basic study design principles, degree of pathology, age, and sex, in order to provide interpretable and consistent results.
Author Contributions
PS and EM assessed the literature and wrote the manuscript. PS created Table 1. PS and EM edited the manuscript. Both authors contributed to the article and approved the submitted version.
Funding
Funding was provided by the NIA grants 5T32AG058518 (PS) and 1RF1AG061776 (EM).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
A β, amyloid-beta peptide; AD, Alzheimer’s disease; ApoE, apolipoprotein E; APP, amyloid precursor protein; DD, constant darkness; LD, 12 h light:dark cycle; IV, intradaily variability; PS1, presenilin 1; SAMP8, senescence accelerated mouse P8; SCN, suprachiasmatic nucleus; 3xTG, APP/PS1/tau triple transgenic mouse.
References
Adler, P., Mayne, J., Walker, K., Ning, Z., and Figeys, D. (2019). Therapeutic targeting of casein kinase 1delta/epsilon in an Alzheimer’s disease mouse model. J. Proteome Res. 18, 3383–3393. doi: 10.1021/acs.jproteome.9b00312
Ambrée, O., Touma, C., Gortz, N., Keyvani, K., Paulus, W., Palme, R., et al. (2006). Activity changes and marked stereotypic behavior precede Abeta pathology in TgCRND8 Alzheimer mice. Neurobiol. Aging 27, 955–964. doi: 10.1016/j.neurobiolaging.2005.05.009
Ancoli-Israel, S., Klauber, M. R., Jones, D. W., Kripke, D. F., Martin, J., Mason, W., et al. (1997). Variations in circadian rhythms of activity, sleep, and light exposure related to dementia in nursing-home patients. Sleep 20, 18–23.
Bailey, M., and Silver, R. (2014). Sex differences in circadian timing systems: implications for disease. Front. Neuroendocrinol. 35, 111–139. doi: 10.1016/j.yfrne.2013.11.003
Banks, G. T., and Nolan, P. M. (2011). Assessment of circadian and light-entrainable parameters in mice using wheel-running activity. Curr. Protoc. Mouse Biol. 1, 369–381. doi: 10.1002/9780470942390.mo110123
Baño Otalora, B., Popovic, N., Gambini, J., Popovic, M., Vina, J., Bonet-Costa, V., et al. (2012). Circadian system functionality, hippocampal oxidative stress, and spatial memory in the APPswe/PS1dE9 transgenic model of Alzheimer disease: effects of melatonin or ramelteon. Chronobiol. Int. 29, 822–834. doi: 10.3109/07420528.2012.699119
Bedrosian, T. A., Herring, K. L., Weil, Z. M., and Nelson, R. J. (2011). Altered temporal patterns of anxiety in aged and amyloid precursor protein (APP) transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 108, 11686–11691. doi: 10.1073/pnas.1103098108
Bellanti, F., Iannelli, G., Blonda, M., Tamborra, R., Villani, R., Romano, A., et al. (2017). Alterations of clock gene RNA expression in brain regions of a triple transgenic model of Alzheimer’s disease. J. Alzheimers Dis. 59, 615–631. doi: 10.3233/jad-160942
Boggs, K. N., Kakalec, P. A., Smith, M. L., Howell, S. N., and Flinn, J. M. (2017). Circadian wheel running behavior is altered in an APP/E4 mouse model of late onset Alzheimer’s disease. Physiol. Behav. 182, 137–142. doi: 10.1016/j.physbeh.2017.09.021
Buhr, E. D., and Takahashi, J. S. (2013). Molecular components of the Mammalian circadian clock. Handb. Exp. Pharmacol. 217, 3–27. doi: 10.1007/978-3-642-25950-0_1
Butterfield, D. A., and Poon, H. F. (2005). The senescence-accelerated prone mouse (SAMP8): a model of age-related cognitive decline with relevance to alterations of the gene expression and protein abnormalities in Alzheimer’s disease. Exp. Gerontol. 40, 774–783. doi: 10.1016/j.exger.2005.05.007
Carroll, J. C., Rosario, E. R., Kreimer, S., Villamagna, A., Gentzschein, E., Stanczyk, F. Z., et al. (2010). Sex differences in beta-amyloid accumulation in 3xTg-AD mice: role of neonatal sex steroid hormone exposure. Brain Res. 1366, 233–245. doi: 10.1016/j.brainres.2010.10.009
Castellano, J. M., Kim, J., Stewart, F. R., Jiang, H., DeMattos, R. B., Patterson, B. W., et al. (2011). Human apoE isoforms differentially regulate brain amyloid-beta peptide clearance. Sci. Transl. Med. 3:89ra57. doi: 10.1126/scitranslmed.3002156
Duncan, M. J., Smith, J. T., Franklin, K. M., Beckett, T. L., Murphy, M. P., St Clair, D. K., et al. (2012). Effects of aging and genotype on circadian rhythms, sleep, and clock gene expression in APPxPS1 knock-in mice, a model for Alzheimer’s disease. Exp. Neurol. 236, 249–258. doi: 10.1016/j.expneurol.2012.05.011
Graybeal, J. J., Bozzelli, P. L., Graybeal, L. L., Groeber, C. M., McKnight, P. E., Cox, D. N., et al. (2015). Human ApoE epsilon4 alters circadian rhythm activity, IL-1beta, and GFAP in CRND8 mice. J. Alzheimers Dis. 43, 823–834. doi: 10.3233/jad-132009
Hatfield, C. F., Herbert, J., van Someren, E. J., Hodges, J. R., and Hastings, M. H. (2004). Disrupted daily activity/rest cycles in relation to daily cortisol rhythms of home-dwelling patients with early Alzheimer’s dementia. Brain 127(Pt 5), 1061–1074. doi: 10.1093/brain/awh129
Holth, J. K., Mahan, T. E., Robinson, G. O., Rocha, A., and Holtzman, D. M. (2017). Altered sleep and EEG power in the P301S Tau transgenic mouse model. Ann. Clin. Transl. Neurol. 4, 180–190. doi: 10.1002/acn3.390
Huang, Y. L., Liu, R. Y., Wang, Q. S., Van Someren, E. J., Xu, H., and Zhou, J. N. (2002). Age-associated difference in circadian sleep-wake and rest-activity rhythms. Physiol. Behav. 76, 597–603. doi: 10.1016/s0031-9384(02)00733-3
Kent, B. A., Michalik, M., Marchant, E. G., Yau, K. W., Feldman, H. H., Mistlberger, R. E., et al. (2019). Delayed daily activity and reduced NREM slow-wave power in the APPswe/PS1dE9 mouse model of Alzheimer’s disease. Neurobiol. Aging 78, 74–86. doi: 10.1016/j.neurobiolaging.2019.01.010
Knight, E. M., Brown, T. M., Gumusgoz, S., Smith, J. C., Waters, E. J., Allan, S. M., et al. (2013). Age-related changes in core body temperature and activity in triple-transgenic Alzheimer’s disease (3xTgAD) mice. Dis. Model Mech. 6, 160–170. doi: 10.1242/dmm.010173
Lee, J., Kim, D. E., Griffin, P., Sheehan, P. W., Kim, D. H., Musiek, E. S., et al. (2020). Inhibition of REV-ERBs stimulates microglial amyloid-beta clearance and reduces amyloid plaque deposition in the 5XFAD mouse model of Alzheimer’s disease. Aging Cell 19:e13078.
Ma, Z., Jiang, W., and Zhang, E. E. (2016). Orexin signaling regulates both the hippocampal clock and the circadian oscillation of Alzheimer’s disease-risk genes. Sci. Rep. 6:36035.
McAuley, J. D., Miller, J. P., Beck, E., Nagy, Z. M., and Pang, K. C. (2002). Age-related disruptions in circadian timing: evidence for “split” activity rhythms in the SAMP8. Neurobiol. Aging. 23, 625–632. doi: 10.1016/s0197-4580(01)00344-x
Miyamoto, M., Kiyota, Y., Yamazaki, N., Nagaoka, A., Matsuo, T., Nagawa, Y., et al. (1986). Age-related changes in learning and memory in the senescence-accelerated mouse (SAM). Physiol. Behav. 38, 399–406. doi: 10.1016/0031-9384(86)90112-5
Moriya, T., Horikawa, K., Akiyama, M., and Shibata, S. (2000). Correlative association between N-methyl-D-aspartate receptor-mediated expression of period genes in the suprachiasmatic nucleus and phase shifts in behavior with photic entrainment of clock in hamsters. Mol. Pharmacol. 58, 1554–1562. doi: 10.1124/mol.58.6.1554
Morley, J. E., Farr, S. A., Kumar, V. B., and Armbrecht, H. J. (2012). The SAMP8 mouse: a model to develop therapeutic interventions for Alzheimer’s disease. Curr. Pharm. Des. 18, 1123–1130. doi: 10.2174/138161212799315795
Musiek, E. S., Bhimasani, M., Zangrilli, M. A., Morris, J. C., Holtzman, D. M., and Ju, Y. E. (2018). Circadian rest-activity pattern changes in aging and preclinical Alzheimer disease. JAMA Neurol. 75, 582–590.
Naismith, S. L., Hickie, I. B., Terpening, Z., Rajaratnam, S. M., Hodges, J. R., Bolitho, S., et al. (2014). Circadian misalignment and sleep disruption in mild cognitive impairment. J. Alzheimers Dis. 38, 857–866. doi: 10.3233/jad-131217
Nakamura, T. J., Nakamura, W., Yamazaki, S., Kudo, T., Cutler, T., Colwell, C. S., et al. (2011). Age-related decline in circadian output. J. Neurosci. 31, 10201–10205. doi: 10.1523/jneurosci.0451-11.2011
Navigatore-Fonzo, L., Castro, A., Pignataro, V., Garraza, M., Casais, M., and Anzulovich, A. C. (2017). Daily rhythms of cognition-related factors are modified in an experimental model of Alzheimer’s disease. Brain Res. 1660, 27–35. doi: 10.1016/j.brainres.2017.01.033
Ni, J., Wu, Z., Meng, J., Saito, T., Saido, T. C., Qing, H., et al. (2019). An impaired intrinsic microglial clock system induces neuroinflammatory alterations in the early stage of amyloid precursor protein knock-in mouse brain. J. Neuroinflamm. 16:173.
Oddo, S., Caccamo, A., Shepherd, J. D., Murphy, M. P., Golde, T. E., Kayed, R., et al. (2003). Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron 39, 409–421.
Ortiz-Tudela, E., Martinez-Nicolas, A., Diaz-Mardomingo, C., Garcia-Herranz, S., Pereda-Perez, I., Valencia, A., et al. (2014). The characterization of biological rhythms in mild cognitive impairment. Biomed. Res. Int. 2014:524971.
Oyegbami, O., Collins, H. M., Pardon, M. C., Ebling, F. J., Heery, D. M., and Moran, P. M. (2017). Abnormal clock gene expression and locomotor activity rhythms in two month-old female APPSwe/PS1dE9 mice. Curr. Alzheimer Res. 14, 850–860.
Pang, K. C., Miller, J. P., and McAuley, J. D. (2004). Circadian rhythms in SAMP8: a longitudinal study of the effects of age and experience. Neurobiol. Aging 25, 111–123. doi: 10.1016/s0197-4580(03)00029-0
Pardossi-Piquard, R., Lauritzen, I., Bauer, C., Sacco, G., Robert, P., and Checler, F. (2016). Influence of genetic background on apathy-like behavior in triple transgenic AD mice. Curr. Alzheimer Res. 13, 942–949. doi: 10.2174/1567205013666160404120106
Paul, J. R., Munir, H. A., van Groen, T., and Gamble, K. L. (2018). Behavioral and SCN neurophysiological disruption in the Tg-SwDI mouse model of Alzheimer’s disease. Neurobiol. Dis. 114, 194–200. doi: 10.1016/j.nbd.2018.03.007
Platt, B., Drever, B., Koss, D., Stoppelkamp, S., Jyoti, A., Plano, A., et al. (2011). Abnormal cognition, sleep, EEG and brain metabolism in a novel knock-in Alzheimer mouse, PLB1. PLoS One 6:e27068. doi: 10.1371/journal.pone.0027068
Plucinska, K., Crouch, B., Koss, D., Robinson, L., Siebrecht, M., Riedel, G., et al. (2014). Knock-in of human BACE1 cleaves murine APP and reiterates Alzheimer-like phenotypes. J. Neurosci. 34, 10710–10728. doi: 10.1523/jneurosci.0433-14.2014
Ramsden, M., Kotilinek, L., Forster, C., Paulson, J., McGowan, E., SantaCruz, K., et al. (2005). Age-dependent neurofibrillary tangle formation, neuron loss, and memory impairment in a mouse model of human tauopathy (P301L). J. Neurosci. 25, 10637–10647. doi: 10.1523/jneurosci.3279-05.2005
Roh, J. H., Huang, Y., Bero, A. W., Kasten, T., Stewart, F. R., Bateman, R. J., et al. (2012). Disruption of the sleep-wake cycle and diurnal fluctuation of beta-amyloid in mice with Alzheimer’s disease pathology. Sci. Transl. Med. 4:150ra22.
Sánchez-Barceló, E. J., Megias, M., Verduga, R., and Crespo, D. (1997). Differences between the circadian system of two strains of senescence-accelerated mice (SAM). Physiol. Behav. 62, 1225–1229. doi: 10.1016/s0031-9384(97)00208-4
Satlin, A., Volicer, L., Stopa, E. G., and Harper, D. (1995). Circadian locomotor activity and core-body temperature rhythms in Alzheimer’s disease. Neurobiol. Aging 16, 765–771. doi: 10.1016/0197-4580(95)00059-n
Song, H., Moon, M., Choe, H. K., Han, D. H., Jang, C., Kim, A., et al. (2015). Abeta-induced degradation of BMAL1 and CBP leads to circadian rhythm disruption in Alzheimer’s disease. Mol. Neurodegener. 10:13.
Sterniczuk, R., Dyck, R. H., Laferla, F. M., and Antle, M. C. (2010). Characterization of the 3xTg-AD mouse model of Alzheimer’s disease: part 1. Circadian changes. Brain Res. 1348, 139–148. doi: 10.1016/j.brainres.2010.05.013
Stevanovic, K., Yunus, A., Joly-Amado, A., Gordon, M., Morgan, D., Gulick, D., et al. (2017). Disruption of normal circadian clock function in a mouse model of tauopathy. Exp. Neurol. 294, 58–67. doi: 10.1016/j.expneurol.2017.04.015
Stopa, E. G., Volicer, L., Kuo-Leblanc, V., Harper, D., Lathi, D., Tate, B., et al. (1999). Pathologic evaluation of the human suprachiasmatic nucleus in severe dementia. J. Neuropathol. Exp. Neurol. 58, 29–39. doi: 10.1097/00005072-199901000-00004
Sun, Y., Guo, Y., Feng, X., Jia, M., Ai, N., Dong, Y., et al. (2020). The behavioural and neuropathologic sexual dimorphism and absence of MIP-3alpha in tau P301S mouse model of Alzheimer’s disease. J. Neuroinflamm. 17:72.
Sundaram, S., Nagaraj, S., Mahoney, H., Portugues, A., Li, W., Millsaps, K., et al. (2019). Inhibition of casein kinase 1delta/epsilonimproves cognitive-affective behavior and reduces amyloid load in the APP-PS1 mouse model of Alzheimer’s disease. Sci. Rep. 9:13743.
Swaab, D. F., Fliers, E., and Partiman, T. S. (1985). The suprachiasmatic nucleus of the human brain in relation to sex, age and senile dementia. Brain Res. 342, 37–44. doi: 10.1016/0006-8993(85)91350-2
Volicer, L., Harper, D. G., Manning, B. C., Goldstein, R., and Satlin, A. (2001). Sundowning and circadian rhythms in Alzheimer’s disease. Am. J. Psychiatry 158, 704–711. doi: 10.1176/appi.ajp.158.5.704
Wang, X., Wang, L., Yu, Q., Xu, Y., Zhang, L., Zhao, X., et al. (2016). Alterations in the expression of Per1 and Per2 induced by Abeta31-35 in the suprachiasmatic nucleus, hippocampus, and heart of C57BL/6 mouse. Brain Res. 1642, 51–58. doi: 10.1016/j.brainres.2016.03.026
Wisor, J. P., Edgar, D. M., Yesavage, J., Ryan, H. S., McCormick, C. M., Lapustea, N., et al. (2005). Sleep and circadian abnormalities in a transgenic mouse model of Alzheimer’s disease: a role for cholinergic transmission. Neuroscience 131, 375–385. doi: 10.1016/j.neuroscience.2004.11.018
Wu, M., Zhou, F., Cao, X., Yang, J., Bai, Y., Yan, X., et al. (2018). Abnormal circadian locomotor rhythms and Per gene expression in six-month-old triple transgenic mice model of Alzheimer’s disease. Neurosci. Lett. 676, 13–18. doi: 10.1016/j.neulet.2018.04.008
Keywords: circadian, Alzheimer’s disease, clock, tau, amyloid
Citation: Sheehan PW and Musiek ES (2020) Evaluating Circadian Dysfunction in Mouse Models of Alzheimer’s Disease: Where Do We Stand? Front. Neurosci. 14:703. doi: 10.3389/fnins.2020.00703
Received: 07 April 2020; Accepted: 10 June 2020;
Published: 07 July 2020.
Edited by:
Sigrid Veasey, University of Pennsylvania, United StatesReviewed by:
Karen L. Gamble, The University of Alabama at Birmingham, United StatesLisa Carlson Lyons, Florida State University, United States
Copyright © 2020 Sheehan and Musiek. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Erik S. Musiek, bXVzaWVrZUB3dXN0bC5lZHU=