 Qiongqiong Wu
Qiongqiong Wu Jing Huang
Jing Huang Renrong Wu
Renrong Wu- Department of Psychiatry, National Clinical Research Center for Mental Disorders, The Second Xiangya Hospital of Central South University, Changsha, China
Treatments for negative symptoms and cognitive dysfunction in schizophrenia remain issues that psychiatrists around the world are trying to solve. Their mechanisms may be associated with N-methyl-D-aspartate receptors (NMDARs). The NMDAR hypofunction hypothesis for schizophrenia was brought to the fore mainly based on the clinical effects of NMDAR antagonists and anti-NMDAR encephalitis pathology. Drugs targeted at augmenting NMDAR function in the brain seem to be promising in improving negative symptoms and cognitive dysfunction in patients with schizophrenia. In this review, we list NMDAR-targeted drugs and report on related clinical studies. We then summarize their effects on negative symptoms and cognitive dysfunction and analyze the unsatisfactory outcomes of these clinical studies according to the improved glutamate hypothesis that has been revealed in animal models. We aimed to provide perspectives for scientists who sought therapeutic strategies for negative symptoms and cognitive dysfunction in schizophrenia based on the NMDAR hypofunction hypothesis.
Introduction
Schizophrenia is a psychiatric disorder afflicting approximately 1% of the population worldwide (Perala et al., 2007). It has multiple overlapping symptom clusters including positive symptoms (i.e., delusions, hallucinations), negative symptoms (i.e., social withdrawal, anhedonia), and cognitive impairments (i.e., deficits in attention, working memory, and executive function) (van Os and Kapur, 2009). Among them, negative symptoms and cognitive dysfunction have recently attracted psychiatrists’ attention. The majority of positive symptoms have been relieved by mainstream antipsychotics (the first and second generation of antipsychotics), while most negative symptoms and cognitive dysfunction remain unaffected and may even worsen with the development of schizophrenia (Lieberman et al., 2005; Lieberman, 2007; Leucht et al., 2009).
Mechanisms of Negative Symptoms and Cognitive Dysfunction in Schizophrenia
The general mechanisms underlying negative symptoms are associated with deficient cortical dopamine transmission in mesocortical pathways and deficient serotonergic and noradrenergic transmission (Galderisi et al., 2018). There are five negative symptom domains: (1) blunted affect (diminished emotional expression); (2) alogia (diminished quantity of words spoken and spontaneous elaboration); (3) asociality (diminished social interactions and initiative); (4) avolition (diminished initiation and persistence of goal-directed activity); (5) anhedonia (diminished ability to experience pleasure) (Galderisi et al., 2018). Different domains are associated with different mechanisms in neurobiology. For example, the avolition/apathy domain involves two mechanisms, the motivational value system and the motivational salience circuit. The dysfunctional dopamine transmission in the reward circuit, especially in areas of the basal ganglia such as the ventral striatum, may cause apathy (Bortolon et al., 2018). Other domains of negative symptoms such as blunted affect and alogia remained unclear with very few revealed mechanisms (Galderisi et al., 2018).
Cognitive dysfunction, a core feature of schizophrenia, is characterized to emerge before the first episode of psychosis (FEP) and is regarded as one of the strongest predictors of functional recovery in patients with schizophrenia (Kahn and Sommer, 2015). There are eight domains in cognition: (1) attention/vigilance, (2) cognitive control/executive function, (3) reasoning/problem solving, (4) social cognition, (5) speed of processing, (6) verbal learning, (7) visual learning, and (8) working memory. Cognition is generally regulated by the hippocampus, basal ganglia, the dorsolateral prefrontal cortex, the thalamus, and regions of the motor cortex (Robison et al., 2020). Cognitive dysfunction in schizophrenia is associated with the proactive control mechanism resulting from impaired function of the dorsolateral prefrontal cortex prefrontal cortex (DLPFC) (Guo et al., 2019). The weakened interaction between DLPFC and other brain regions, as well as the neurotransmitter system disorder, such as dopamine and glutamate, contribute to cognitive dysfunction in schizophrenia (Barch and Ceaser, 2012).
The mechanisms of both negative symptoms and cognitive dysfunction are associated with NMDARs (Kahn and Sommer, 2015). First, the animal models associated with NMDARs (NMDAR antagonists induced model, gene engineered model) exhibited negative symptoms (Lee and Zhou, 2019) and impairments in cognition (recognition memory, attention, and working memory) (Thomas et al., 2017). The NMDAR hypofunction was linked to negative symptoms via mismatch negativity (MMN), a negative event-related potential response to a deviant stimulus in the repetitive auditory stimuli (Thiebes et al., 2017). MMN might be a biomarker for negative symptoms in schizophrenia due to glutamatergic deficits (Thiebes et al., 2017). Negative symptoms seemed to be controlled by the D3 receptors in reward and motivation circuits, which can interact with NMDARs directly (Sokoloff and Le Foll, 2017). When it comes to cognition, NMDARs in hippocampus CA1 play a predominant role in the generation of long-term plasticity (long-term potentiation and depression), and NMDAR-dependent long-term plasticity is critical in neurochemical foundations of learning and memory (Lau and Zukin, 2007; Volianskis et al., 2015). Aberrant glutamatergic inputs in dopamine circuits (mesolimbic circuit and mesocortical circuit) may induce cognitive dysfunction in schizophrenia (Robison et al., 2020). Thus, NMDAR hypofunction is closely associated with cognitive dysfunction in schizophrenia (Henneberger et al., 2010).
NMDAR Hypofunction and Schizophrenia
There are two types of glutamate receptors: ionotropic (iGluRs) and metabotropic (mGluRs). The iGluRs are composed of α-amino-3-hydroxy-5-methylisoxazole-4-propionate receptor (AMPARs), kainate receptors (KARs), and NMDARs (also named as GluNs) (Uno and Coyle, 2019). NMDAR is highly permeable to Ca2+, thus playing an important role in the excitatory synapse transmission and long-term neural structural plasticity (Cull-Candy et al., 2001; Paoletti and Neyton, 2007). NMDAR is a heterotetrameric complex composed of two GluN1 subunits, which are obligatory, and with either two GluN2 subunits or a combination of GluN2/3 subunits (Paoletti and Neyton, 2007; Hansen et al., 2018). GluN2 subunits have four variants (GluN2A-D) and GluN3 subunits have two variants (GluN3A-B). There is a glycine modulatory site (GMS) on the GluN1 subunit, which binds co-agonists glycine and D-serine (Paoletti and Neyton, 2007). The GluN2 subunit has a glutamate binding site and other allosteric modulatory sites (Paoletti and Neyton, 2007). GluN1 and GluN2 subunits both have redox modulatory sites composed of the disulfide bond of cysteine residues (Lipton et al., 2002). The activation of the NMDAR requires: (1) post-synaptic depolarization induced by the activation of the AMPA receptor, which relieves the Mg2+ blockade of the channel; (2) glutamate binding to the GluN2 subunit and glycine or D-serine binding to the GMS on the GluN1 subunit (Paoletti et al., 2013).
The NMDAR hypofunction hypothesis emerged when the noncompetitive antagonists of the NMDAR (phencyclidine/PCP, ketamine) were found to induce nearly all three symptom clusters of schizophrenia in healthy people (Luisada, 1978; Krystal et al., 1994). Anti-NMDAR encephalitis with autoantibodies against NMDAR also supported this hypothesis because patients with anti-NMDAR encephalitis presented schizophrenia-like symptoms (Zandi et al., 2011; Kayser and Dalmau, 2016). The abundant evidence for the NMDAR hypofunction hypothesis has been extensively reviewed (Coyle, 2012; Uno and Coyle, 2019), including genetic findings (Neale et al., 2014; Yu et al., 2018), clinical findings, neuroimaging (1H-MRS, PET/SPECT), neurophysiological findings (mismatch negativity/P300/gamma band oscillations) (Wacongne, 2016; Greenwood et al., 2018; Javitt et al., 2018), and postmortem neurochemical findings.
NMDAR-Targeted Drugs for Negative Symptoms and Cognitive Dysfunction
Drugs based on the NMDAR hypofunction hypothesis and their effects on patients with schizophrenia are summarized in Table 1.
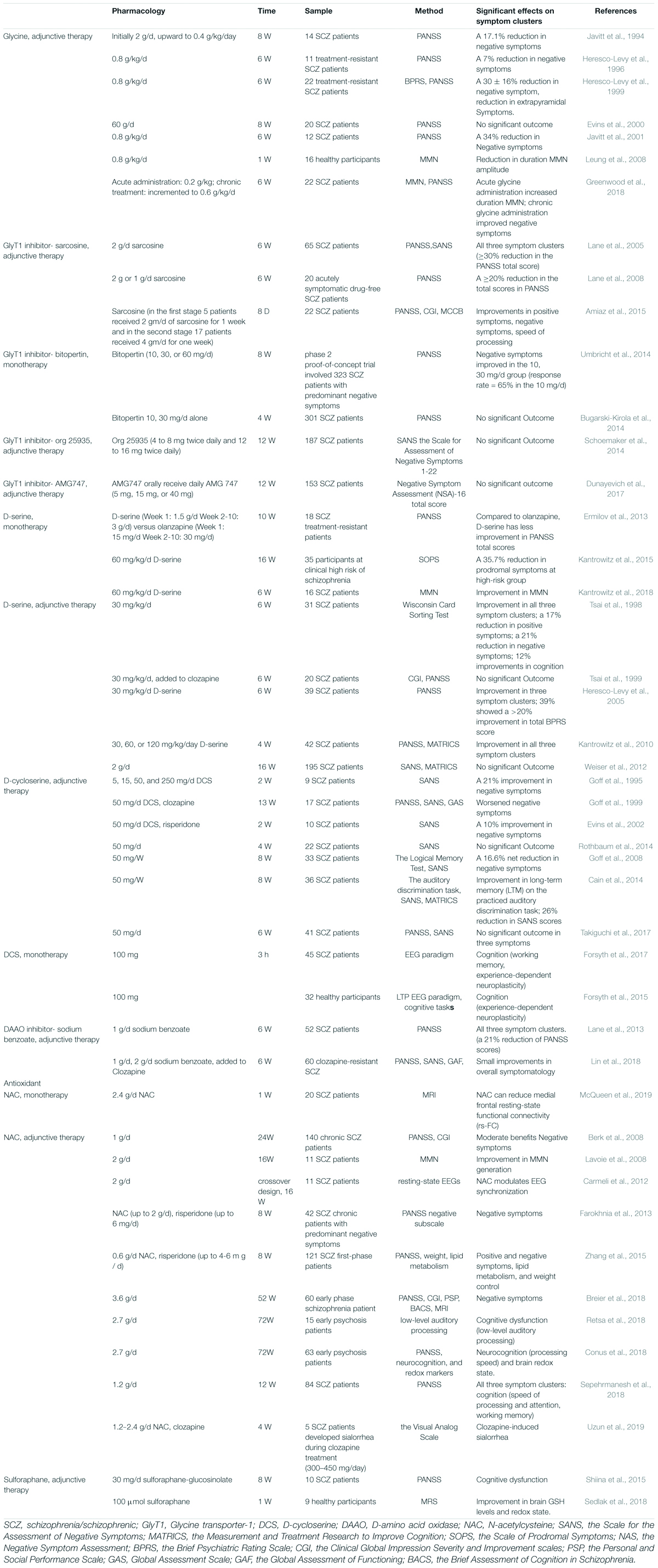
Table 1. The summary of clinical researches on drugs augmenting NMDAR function in schizophrenia.
Targeting the Glycine Modulatory Site (GMS)
Glycine, d-serine, D-cycloserine (DCS), and kynurenic acid (KYNA) can bind to the GMS on the GluN1 subunit. Glycine and d-serine function as co-agonists, and DCS is a partial agonist while KYNA is a competitive antagonist. The GMS is not saturated in brains and thus the NMDAR function can be augmented via stimulating the GMS (Balu and Coyle, 2015). Drugs targeting GMS on GluN1 have been discovered, as is shown in Figure 1. On the contrary, glutamate as an agonist for GluN2 has a tendency to induce excitotoxicity and epilepsy (Uno and Coyle, 2019), thus few drugs targeting GluN2 have been discovered (Uno and Coyle, 2019).
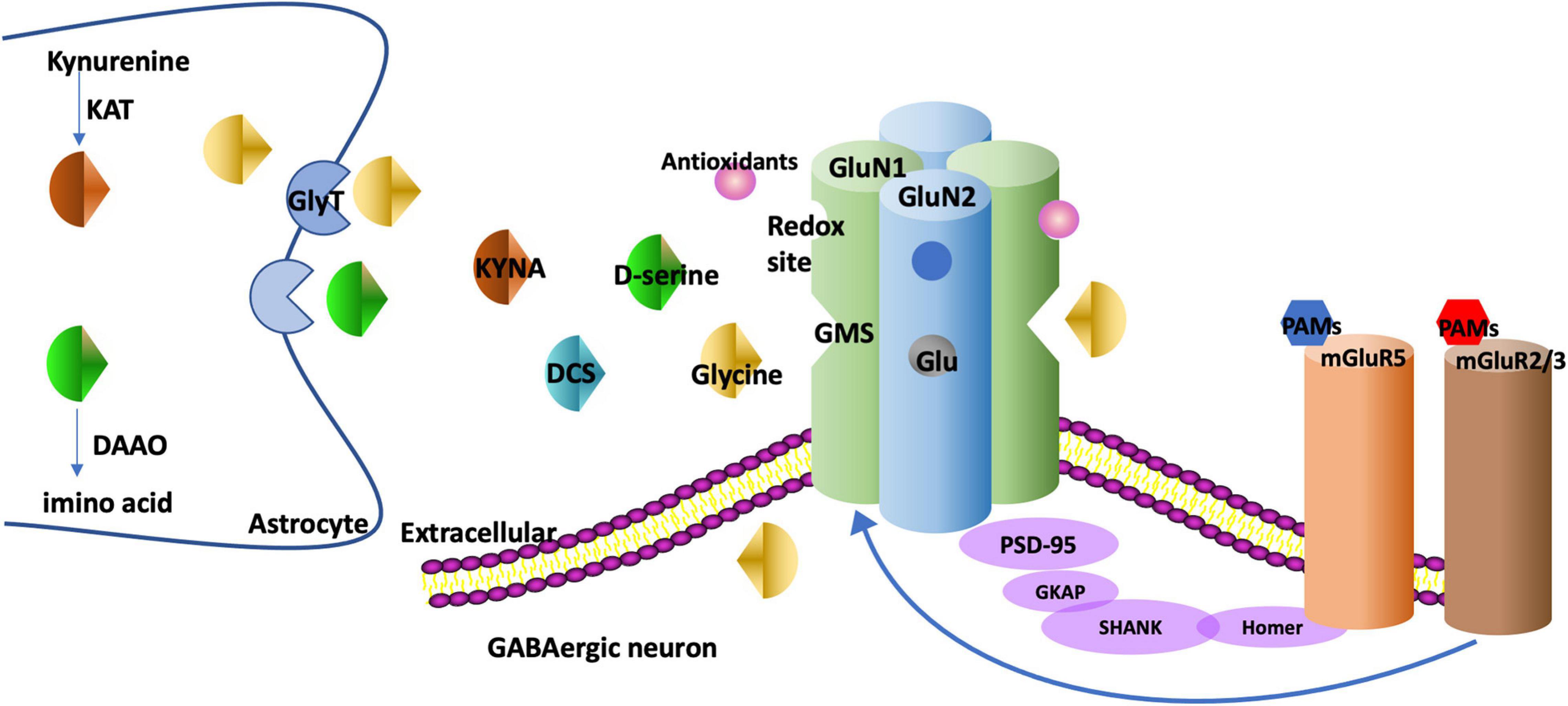
Figure 1. The targets of drugs augmenting NMDAR function. NMDAR is generally composed of two GluN1 and two GluN2 subunits. There is a GMS in GluN1. Glycine and d-serine bind to the GMS as co-agonists. DCS binds to the GMS as a partial agonist and KYNA binds to the GMS as a competitive antagonist. GlyT-1, the glycine transporter type, is expressed at glutamatergic synapses to modulate glycine uptake. DAAO is an intracellular enzyme degrading d-serine into amino acids. KAT regulates the production of KYNA in astrocytes. The metabotropic glutamate receptors such as mGluR5s and mGluR2/3s have multiple interactions with NMDARs. For example, mGluR5s and NMDARs are linked physically via scaffolding proteins Homer, SHANK. The PAMs of these receptors regulate metabotropic glutamate receptors and NMDARs as well. NMDARs have redox sites which can be regulated by antioxidants. GMS, glycine modulatory site; DCS, D-cycloserine; KYNA, Kynurenic acid; GlyT, glycine transporter; DAAO, D-amino acid oxidase; KAT, Kynurenine aminotransferase; PAM, positive allosteric modulator; PANSS, the Positive and Negative Syndrome Scale; SANS, Scale for the Assessment of Negative Symptoms.
Glycine and Glycine Transporter-1 (GlyT-1) Inhibitors
Glycine has been administrated with mainstream antipsychotics since 1994. The neural function has an inverted-U relationship with the synaptic glycine concentration (Greenwood et al., 2018), which is associated with the internalization of NMDARs from the membrane to the cytoplasm (Nong et al., 2003). The efficacy of glycine in improving negative symptoms has been confirmed in schizophrenia (Javitt et al., 1994, 2001; Heresco-Levy et al., 1996, 1999). A significant improvement (around 30% reduction rates) in negative symptoms has been revealed by comparing the PANSS scores after glycine administration (Heresco-Levy et al., 1996; Javitt et al., 2001). Furthermore, glycine is effective for negative symptoms in treatment-resistant schizophrenic patients (Heresco-Levy et al., 1996, 1999). In addition, acute glycine administration improved MMN both in healthy controls and patients with schizophrenia (Leung et al., 2008; Greenwood et al., 2018). However, some studies reported no significant improvements in negative symptoms or cognitive dysfunction (Evins et al., 2000; Buchanan et al., 2007). Glycine has some disadvantages. It requires a high dose (0.8 g/kg/day) to have an effect, and it may induce long-term depression (Greenwood et al., 2018) and digestive dysfunction (Heresco-Levy et al., 1999).
Glycine transporter-1 is a predominant modulator for the intra/extra-cellular glycine concentration via regulating glycine reuptake (Hashimoto, 2010). GlyT-1, a subtype of GlyT, co-localized with NMDAR and is primarily expressed in the synapse of glutamatergic neurons (Hashimoto, 2011). When GlyT-1 is inhibited, the extra-cellular glycine concentration rises, and subsequently the NMDAR function is augmented (Hashimoto, 2010). Thus, GlyT-1 inhibitors (such as sarcosine, bitopertin, org 25935, AMG747) are promising for improving negative symptoms and cognitive dysfunction in schizophrenia. They are classified into competitive antagonists (sarcosine) and noncompetitive antagonists (bitopertin, org 25935, AMG747) (Hashimoto, 2010).
Bitopertin/RG1678, as the first potent GlyT-1 inhibitor, has reported marked effects on negative symptoms in preclinical studies (Pinard et al., 2018). However, the third phase of the clinical trial of bitopertin (Bugarski-Kirola et al., 2014; Umbricht et al., 2014; Pinard et al., 2018) failed, and another two noncompetitive GlyT-1 inhibitors org 25935 (Schoemaker et al., 2014), AMG747 (Dunayevich et al., 2017) failed to have positive results in their phase II trials as well. It was suspected that the modest effect of GlyT1 inhibitors may be associated with the preferential action of glycine on extra-synaptic NMDAR function rather than synaptic NMDAR function (Papouin et al., 2012). However, there are still some GlyT-1 inhibitors such as PF-03463275 (D’Souza et al., 2018) and BI-425809 (Moschetti et al., 2018) in phase II trials and some have exhibited the potential to improve memorization and cognitive dysfunction in the preclinical phase (Harada et al., 2012; D’Souza et al., 2018).
Sarcosine (N-methylglycine), an endogenous competitive GlyT-1 antagonist, is generated as an intermediate in glycine synthesis and degeneration (Lane et al., 2008). After its pharmacokinetic evaluations and safety was confirmed in humans (Amiaz et al., 2015), sarcosine exhibited improvements in positive symptoms, negative symptoms, and general psychopathology (Amiaz et al., 2015). The cognitive dysfunction (especially speed of processing), was significantly improved after sarcosine administration in patients with schizophrenia (Amiaz et al., 2015). In addition, sarcosine was found to be beneficial in both the chronic (Lane et al., 2010; Amiaz et al., 2015) and acute phase of schizophrenia (Lane et al., 2005, 2008).
D-Serine, DCS, D-Amino Acid Oxidase (DAAO) Inhibitors
D-serine is localized especially in GABAergic neurons and astrocytes (Wolosker et al., 2008). It is generated by the serine racemases (SRs) in neurons and degenerated by DAAOs or SRs in astrocytes. Compared with glycine, D-serine presented closer associations with schizophrenia and induced a more powerful activation of synaptic NMDARs because D-serine can serve as a monotherapy in schizophrenia (MacKay et al., 2019). The first study that added D-serine with the non-clozapine mainstream antipsychotics revealed that D-serine improved all three symptom clusters in chronic treatment-refractory schizophrenia (Tsai et al., 1998). However, whether the mainstream antipsychotic is clozapine or not can affect the efficacy of adjunctive D-serine. When follow-up studies administered D-serine with clozapine, the results were negative (Tsai et al., 1999). In the studies that followed, some found that adjunctive D-serine was effective in all three symptom clusters (Heresco-Levy et al., 2005; Kantrowitz et al., 2010) while some found it to be ineffective (Lane et al., 2005; Weiser et al., 2012). These negative results may be due to the narrow therapeutic window of D-serine. A high dose (60 mg/kg/day) of D-serine is required to achieve adequate brain concentration and potentiate NMDAR activation (MacKay et al., 2019), because its oral bioavailability is low (Iwata et al., 2015). However, a high dose of D-serine can lead to nephrotoxicity (Iwata et al., 2015). These features of D-serine limit its application in patients with schizophrenia. Owing to its possible effects on positive symptoms, the effects of D-serine monotherapy were explored in patients with schizophrenia (Tsai et al., 1998). The studies that followed compared the efficacy of D-serine monotherapy and high-dose olanzapine monotherapy, because olanzapine is recognized as a recommended medication for schizophrenia (Ermilov et al., 2013). However, the D-serine has less reduction in PANSS total scores compared to high-dose olanzapine (Ermilov et al., 2013). Of note, D-serine can improve prodromal symptoms in participants who are at a clinically high risk of schizophrenia (Kantrowitz et al., 2015). In addition, a recent study reported an improvement in MMN in patients with schizophrenia when treated with D-serine for just 1 week (Kantrowitz et al., 2018).
D-amino acid oxidase is a catalyzing flavoprotein in the oxidative deamination of neutral D-amino acids, and the inhibition of DAAOs can augment NMDAR function as a treatment for schizophrenia. Multiple DAAO inhibitors were discovered with wide structural variety (Szilagyi et al., 2018). The first-generation of DAAO inhibitors is carboxylic acids. Sodium benzoate, a food preservative with good safety, is a DAAO inhibitor of the first generation with moderate activation (Lin et al., 2018). When sodium benzoate was administrated with risperidone or haloperidol, a wide variety of symptom domains, including neurocognition, were improved in patients (Lane et al., 2013). When added to clozapine, sodium benzoate improved three symptom clusters, and the 2 g/day dose had better results than the 1 g/day dose in clozapine-resistant schizophrenia (Lin et al., 2018). However, the D-serine level in the brain was not altered when the patients were administered sodium benzoate (Lin et al., 2018). It challenged the expected working mechanism of benzoate as a DAAO inhibitor in brain. In addition to DAAO inhibition, sodium benzoate can serve as an antioxidant, protecting neurons from oxidative stress (Lin et al., 2018) and it may affect the D-serine/L-serine cycle as well (Matsuura et al., 2015). Another clinical trial to explore the efficacy of sodium benzoate in early psychosis is ongoing (Ryan et al., 2017). In the discovery of other generations of DAAO inhibitors, bioisosteric moiety replaced the carboxylic group in second-generation inhibitors and cyclic carboxylic acid bioisosters connected to bulky nonpolar moieties with a flexible linker in third-generation inhibitors (Szilagyi et al., 2018). Other novel DAAO inhibitors, such as PGM030756 (Howley et al., 2017), are still in pre-clinical studies and need further confirmation of their efficacy in humans for species difference in D-serine metabolism (Szilagyi et al., 2018).
D-Cycloserine (DCS)
D-cycloserine, a partial agonist for GMS, increases the NMDAR channel open time and possibility at low doses (Dravid et al., 2010). It is noted that DCS has an inverted-U dose response curve similar to glycine, and chronic DCS administration may facilitate NMDAR internalization and become ineffective (Goff et al., 1995; Goff, 2016). A high dose of DCS functions as an antagonist of GMS competing with other full agonists like D-serine and glycine (Uno and Coyle, 2019). The benefits of adjunctive DCS in negative symptoms was reported early in 1995, and 50 mg/day was reported as the best dose with significant outcomes (Goff et al., 1995, 1996). However, the studies that followed found that when DCS was added with clozapine, the negative symptoms worsened in a 13-week follow-up (Goff et al., 1999). It is probably because clozapine differs from other antipsychotics in releasing D-serine and glutamate to enhance NMDARs, and DCS may attenuate the positive effects of clozapine via competing for GMS (Goff et al., 1999). When DCS was added with non-clozapine antipsychotics like risperidone, negative symptoms were improved (Evins et al., 2002). The intermittent DCS administration (per week) proved to be effective as well (Goff, 2016). It was revealed that taking DCS 50 mg per week improved cognitive tasks such as memory consolidation (Goff et al., 2008) and auditory discrimination task (Cain et al., 2014). Cognitive behavioral therapy for positive symptoms was improved if DCS was taken 1 h prior to an intervention (Gottlieb et al., 2011). Recent studies revealed the working memory and experience-dependent plasticity were enhanced in healthy participants (Forsyth et al., 2015) or patients with schizophrenia (Forsyth et al., 2017). However, some negative results have also been reported in the administration of DCS as adjunctive therapy, and a high dose (100 mg) of DSC aggravated symptoms of early-onset schizophrenia (van Berckel et al., 1999; Buchanan et al., 2007; Takiguchi et al., 2017), because a high dose can function as antagonists of GMS. Thus, the therapeutic window of DCS seems to be crucial in the application of DCS (Cascella et al., 1994). It was revealed that the effects of DCS were associated with the onset age of schizophrenia and the white matter integrity (Takiguchi et al., 2017). In conclusion, DCS features in improving negative symptoms (10-26%) and part of cognitive dysfunction (working memory, experience-dependent neuroplasticity) in patients with schizophrenia (Goff, 2017).
Kynurenine Aminotransferase (KAT) Inhibitors Targeting KYNA
Kynurenic acid is an endogenous competitive antagonist for GMS, and its production is catalyzed by KATs in astrocytes (Plitman et al., 2017). The association between the increased brain KYNA levels and subsequent cognitive dysfunction in schizophrenia was revealed by both human and animal studies (Bortz et al., 2017; Plitman et al., 2017). The underlying mechanisms may be the dysregulation of kynurenine metabolism which leads to dorsolateral prefrontal cortex volume loss and attention impairment in patients with schizophrenia (Kindler et al., 2020). Thus, the inhibition of KATs is supposed to enhance NMDAR function as a therapeutic target for schizophrenia (Nematollahi et al., 2016). But this strategy may involve another controversial mechanism independent from NMDARs, which is associated with nicotinic acetylcholine receptors (nAChRs): KYNA is also a non-competitive antagonist of the α7nAChR and the decreased KYNA can reverse α7nAChRs dysfunction in schizophrenia (Albuquerque and Schwarcz, 2013; Plitman et al., 2017). However, most evidence supported kynurenate acting via NMDARs (Stone, 2020). The potent KAT-I inhibitors include phenylhydrazone hexanoic acid derivatives and KAT-II inhibitors include a pyrazole series of compounds (Jayawickrama et al., 2015). Although KAT inhibitors (such as BFF816 Bortz et al., 2017) were reported to have good efficacy in cognitive dysfunction in animal models, this strategy has not been confirmed in humans yet.
Positive Allosteric Modulators (PAMs) for Metabotropic Glutamate Receptors (mGluRs)
The direct therapeutic targets for NMDARs were challenged for the widespread distribution of NMDARs in the brain and the potential toxicity induced by the hyperglutamatergic condition. Thus, PAMs for mGluRs were discovered because they reversed NMDAR hypofunction but did not induce glutamatergic overexcitation (Field et al., 2011). The mGluRs have eight subtypes and they can be classified into three groups: Group I includes mGluR1 and mGluR5; Group II includes mGluR2 and mGluR3; Group III includes mGluR4, mGluR6-8 (Lundstrom et al., 2016). Nearly all of the subtypes of mGluRs have been recognized as promising therapeutic targets for schizophrenia. Among them, the mGlu5Rs and Group II mGluRs have been extensively researched, which we will discuss in the following section (Maksymetz et al., 2017; Stansley and Conn, 2018). PAMs refer to the agents binding to allosteric binding sites to potentiate the targeted receptor function (Ellaithy et al., 2015). They have apparent advantages over traditional agonists because they activate the receptors indirectly and they are less likely to cause NMDAR internalization which interferes with the effects of glycine and DCS (Field et al., 2011).
PAMs of mGlu5 Receptors
The mGluR5 has functional associations with NMDARs in GABAergic interneurons as they have some overlapping intracellular signaling pathways (Matosin and Newell, 2013). mGluR5s and NMDARs are linked physically via scaffolding proteins Homer, SHANK, guanylate-kinase-associated (GKAP), and post-synaptic density 95 (PSD95) as is shown in Figure 1 (Matosin and Newell, 2013). The mGluR5 PAMs can subsequently activate mGluR5s and NMDARs. VU0409551 (Conde-Ceide et al., 2015), a well-developed PAM, was reported to augment NMDAR function and improve cognitive dysfunction in the animal models of schizophrenia (Balu et al., 2016). mGluR5 PAMs may function via selectively potentiating mGluR5 coupling to Gαq-mediated signaling independent of NMDAR (Rook et al., 2015; Ghoshal et al., 2017). However, the safety and efficacy of mGluR5 PAMs have not yet been confirmed in clinical studies.
PAMs of Group II mGlu Receptors
Group II mGlu receptors have been shown to modulate glutamatergic activity in brain synapses (Fell et al., 2012). The activation of mGluR2/3s in the postsynaptic membrane promoted NMDAR currents via Src kinase (Trepanier et al., 2013), protein kinase C (Tyszkiewicz et al., 2004), or SNARE protein (Cheng et al., 2013). Furthermore, Group II agonists decreased excessive glutamate release in animal models of schizophrenia (Marek, 2010). Furthermore, mGluR2s was reported to have interaction with 5-HT2A receptors, which are the targets of second generation antipsychotics as well (Ellaithy et al., 2015). A mixed mGlu2/3 receptor agonist LY2140023 failed in Phase II trials when it was administered with olanzapine (Kinon et al., 2011), even though it had shown good efficacy in positive and negative symptoms of schizophrenia in a previous study (Patil et al., 2007). Two mGlu2 PAMs have reached clinical trials: AZD8529 from AstraZeneca and JNJ-40411813/ADX71149 from Janssen Pharmaceuticals, Inc. and Addex Therapeutics (Field et al., 2011). The Phase IIa study of ADX71149 demonstrated the efficacy of adjunctive ADX71149 in improving the residual negative symptoms in patients with schizophrenia1. But AZD8529 failed to have a significant difference to the placebo in a phase IIa proof of concept (POC) study2, 3.
Antioxidants
Oxidative stress is defined as an imbalance between pro-oxidants and antioxidants. It was speculated that redox state and NMDAR activity interacted with each other in schizophrenia (Hardingham and Do, 2016) and these two individual factors both had developmental effects on parvalbumin-expressing GABAergic interneurons in schizophrenia (Korotkova et al., 2010; Hasam-Henderson et al., 2018). The underlying mechanisms are that the redox state regulates the disulfide bond formation of cysteine residues in GluN1 and GluN2A (also called the redox modulatory site) and subsequently regulates NMDAR activity (Lipton et al., 2002). Glutathione (GSH), protective from reactive oxidative stress, can bind to the redox modulatory site of NMDARs. GSH deficits and oxidative stress may induce the NMDAR hypofunction state in schizophrenia (Steullet et al., 2006). In addition, the conventional antipsychotics such as clozapine and risperidone, can induce oxidative stress and cause neural injury. Thus, the adjunctive therapy of antioxidant has the potential to reverse oxidative stress and NMDAR hypofunction in patients with schizophrenia (Hardingham and Do, 2016). Although there are various types of antioxidants that are beneficial to negative symptoms and cognitive dysfunction in schizophrenia (Magalhaes et al., 2016), few antioxidants have been reported to have an effect associated with NMDARs, except for N-acetylcysteine (NAC) and Sulforaphane.
N-Acetylcysteine (NAC)
N-acetylcysteine (NAC) is a precursor of L-cysteine, contributing to glutathione synthesis (Chen et al., 2016). It passes the blood-brain barrier easily and is involved in diverse pathways in the pathology of schizophrenia, such as brain GSH regulation, glutamatergic transmission, inflammatory pathways, and mitochondrial function (Minarini et al., 2017). In NMDAR hypofunction models, NAC protected mice from mitochondrial and synaptic injury (Phensy et al., 2017a). A two-hit model (the exposure to perinatal infection, peripubertal unpredictable stress) of schizophrenia confirmed the benefits of NAC as well (Monte et al., 2020). The synaptic deficits of schizophrenia can be reversed by NAC as revealed in cortical interneurons derived from human induced stem cells (Kathuria et al., 2019).
In clinical studies, NAC as adjunctive therapy was found to ameliorate severity of all three symptom clusters in patients with schizophrenia (Berk et al., 2008; Farokhnia et al., 2013; Zhang et al., 2015; Breier et al., 2018; Conus et al., 2018; Retsa et al., 2018; Sepehrmanesh et al., 2018; McQueen et al., 2019; Uzun et al., 2019). Four of these studies recruited first-phase or early phase patients with schizophrenia (Zhang et al., 2015; Breier et al., 2018; Conus et al., 2018; Retsa et al., 2018) and the others studied the effects of NAC in chronic schizophrenia (Sepehrmanesh et al., 2018). Another study on the administration of NAC for 26 weeks will be recruiting 162 young people in first-episode psychosis (Cotton et al., 2019). NAC has been added to different antipsychotics such as risperidone (Farokhnia et al., 2013) and clozapine (Uzun et al., 2019) to explore its efficacy in schizophrenia, and all the combinations reported improvements in negative symptoms and cognitive dysfunction. Of note, NAC was discovered to ameliorate clozapine-induced sialorrhea in a case series (Uzun et al., 2019). A significant improvement in lipid metabolism and weight control was noted as it was difficult for patients to maintain weight and spare them from metabolic disorders during the administration of the second-generation antipsychotics (Zhang et al., 2015). Sepehrmanesh Z found that NAC had positive effects on positive symptoms as well (Sepehrmanesh et al., 2018). A meta-analysis in 2018 included three randomized controlled trials (RCTs) on NAC in schizophrenia and confirmed the efficacy of the adjunctive NAC (Zheng et al., 2018). In addition, NAC can alter MMN and EEG synchronization (Carmeli et al., 2012), which linked NMDAR augmentation (Lavoie et al., 2008). Recently, the efficacy of NAC monotherapy on functional signatures was explored, and it reduced medial frontal resting-state functional connectivity (McQueen et al., 2019).
Sulforaphane
Sulforaphane is a natural compound found in the seeds and sprouts of cruciferous plants. It is protective of inflammation and oxidative stress (Fahey and Talalay, 1999) via binding to kelch-like ECH-associated protein 1 (KEAP1) and activating the transcription factor NF-E2-related factor 2 (Nrf2) (Keum, 2011; Cardozo et al., 2013). Sulforaphane has advantages over NAC, because NAC reduces oxidative stress mainly via cysteine, but cysteine cannot be fully utilized to generate GSH while sulforaphane is functioning via KEAP1/Nrf2 which affects GSH more efficiently (Hardingham and Do, 2016). Furthermore, the action of sulforaphane was linked to NMDAR as sulforaphane prevented the alteration of NMDAR activity in rats (Feng et al., 2017). However, there have only been two published studies on the administration of sulforaphane in human studies associated with schizophrenia (Shiina et al., 2015; Sedlak et al., 2018). Sulforaphane-rich broccoli sprout extract (30 mg/d) improved cognitive dysfunction in patients with schizophrenia (Shiina et al., 2015). The 7-day administration of sulforaphane increased brain GSH levels both in the animal model and human sample (Sedlak et al., 2018). In a PCP-induced animal model for schizophrenia, taking sulforaphane-rich broccoli sprout extracts during juvenile and adolescence rescued the cognitive dysfunction in adulthood (Shirai et al., 2015).
Discussion
The NMDAR hypofunction hypothesis produces novel targets for therapeutic interventions for patients with schizophrenia, especially for negative symptoms and cognitive dysfunction. Therapeutic targets to augment NMDAR function include compounds targeting the GMS on GluN1, PAMs for mGlu receptors, and antioxidants. Compounds targeting the GMS on GluN1 include glycine & GlyT-1 inhibitors, D-serine & D-amino acid oxidase (DAAO) inhibitors, DCS, and KAT inhibitors.
Most of the drugs discussed above can improve some of the negative symptoms in patients with schizophrenia. We listed the net reduction rates of PANSS-negative scores/SANS scores if they were reported in Table 1. Glycine seemed to have had the strongest effects on negative symptoms, reducing about 30% of the Positive and Negative Syndrome Scale (PANSS)-negative score. In addition, other drugs such as D-serine and D-cycloserine were reported to have >20% reduction rates on PANSS -negative scores or the Scale for the Assessment of Negative Symptoms (SANS) scores, where 20% is recognized as the cutoff threshold for meaningful improvement (Heresco-Levy et al., 2005). The meta-analyses reported that the overall effect size for NMDAR-targeted adjunctive drugs was small to moderate for negative symptoms. A meta-analysis including studies on DCS and D-serine in 2013 reported a moderate effect-size improvement while a meta-analysis in Singh and Singh (2011) and a meta-analysis in Tsai and Lin (2010) both reported a small effect-size.
Compared with their effects on negative symptoms, NMDAR-targeted drugs had less improvements for cognitive dysfunction in patients with schizophrenia. The improved cognition domains after the administration of NMDAR-targeted drugs are limited to one or two. For example, DCS was recognized as a robust agent to increase neuroplasticity and to improve working memory as revealed in DCS monotherapy studies (Forsyth et al., 2015, 2017). NAC can improve more cognition domains than DCS, which includes speed of processing, attention, and working memory (Conus et al., 2018; Sepehrmanesh et al., 2018). There are four meta-analyses exploring the effects of NMDAR-targeted drugs on cognition. Only one meta-analysis in 2010 reported positive results, while the others reported non-significant results. The meta-analysis in 2010 reported that NMDAR-targeted drugs exhibited a small effect-size improvement in cognition (Tsai and Lin, 2010). A meta-analysis including D-serine, DCS, benzoate, and NAC in 2019 demonstrated that these drugs had a small but non-significant effect on overall cognitive function, but had significant positive effects in the 30-39 year old subgroup (Chang et al., 2019). These drugs presented a significant improvement in working memory among all eight domains (Chang et al., 2019). However, another two meta-analyses both reported their non-significant effects on overall cognitive function in patients with schizophrenia (Choi et al., 2013; Iwata et al., 2015).
Most of the NMDAR-targeted drug administrations were reported as safe and well-tolerated throughout the studies without significant clinical or laboratory side-effects. Sodium benzoate as a food additive and antioxidant sulforaphane extracted from food are safe for humans. However, high-dose glycine had slight side-effects on gastrointestinal functions (Heresco-Levy et al., 1999).
Altogether, the overall effects of these NMDAR-targeted drugs had little to moderate negative symptoms and cognitive dysfunction, see Table 2. The development of NMDAR-targeted drugs has resulted in many unexpected outcomes. For example, some potential new drugs such as GlyT-1 inhibitors bitopertin/RG1678 (Bugarski-Kirola et al., 2014; Umbricht et al., 2014; Pinard et al., 2018) and GlyT-1 inhibitors (org 25935 Schoemaker et al., 2014, AMG747 Dunayevich et al., 2017) failed in their clinical trials. However, these drugs (such as sodium benzoate, D-serine) have potential in clozapine-resistant/treatment-resistance schizophrenia, and dual activation of GMS and antioxidants is viewed as a novel treatment for treatment-resistance schizophrenia (Lin et al., 2020).
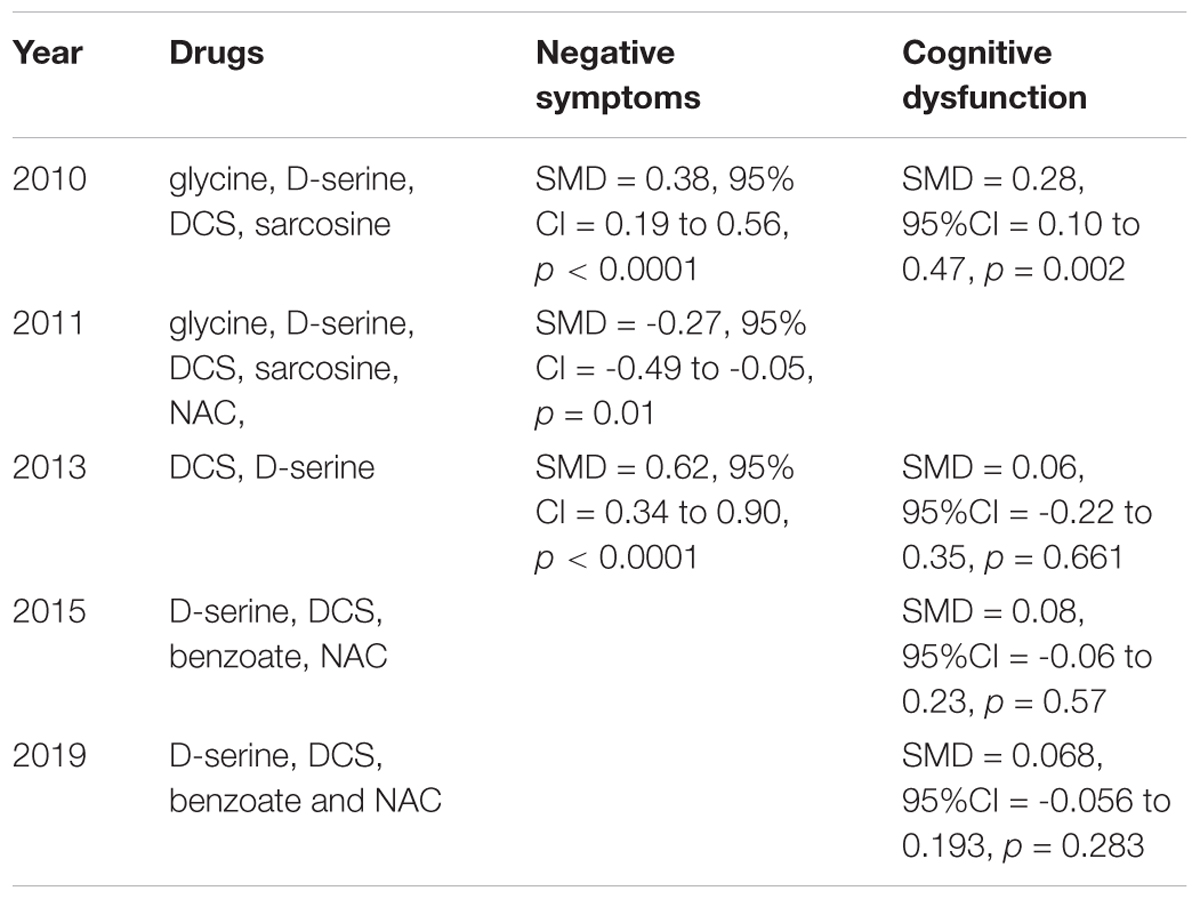
Table 2. Summary of published meta-analyses on NMDAR-targeted drugs.
The Possible Reasons for Unsatisfactory Clinical Results
These unsatisfactory clinical results challenged the NMDAR hypofunction hypothesis in schizophrenia pathology. There are some conflicting results in this hypothesis. (1) The strongest evidence for this hypothesis is that PCP and ketamine induce all symptoms in schizophrenia. However, the latest studies have revealed that PCP and ketamine do not function exclusively as NMDAR antagonists in the brain (Seillier and Giuffrida, 2009; Zanos and Gould, 2018; Fujigaki et al., 2019; Wang et al., 2020). (2) Early-life NMDAR ablation eliciting schizophrenia-like symptoms were revealed in an animal model (Belforte et al., 2010) but has not been confirmed in humans. The neurodevelopmental differences among species may be overcame via models from human induced pluripotent stem cells—a research direction that is worth exploring (Brennand et al., 2011; Noh et al., 2017). (3) Many studies found that NMDAR antagonists can also relieve the symptoms in patients with schizophrenia, which is in contrast to the NMDAR hypofunction hypothesis (Matsuda et al., 2013).
We should however, analyze the pharmacological mechanisms of these drugs in detail before challenging the NMDAR hypofunction hypothesis. In a genetically engineered NMDAR hypofunction model for schizophrenia, NMDAR hypofunction had spatial and temporal boundaries (Nakazawa et al., 2017): (1) The origin of NMDAR hypofunction was reported in cortical and hippocampal GABAergic neurons, particularly in parvalbumin-positive fast-spiking interneurons (Nakazawa and Sapkota, 2020). (2) NMDAR hypofunction happened in the postnatal period, which is before the maturation of parvalbumin-expressing interneurons, and which can cause schizophrenia-like behaviors (Belforte et al., 2010; Lewis et al., 2012; Hardingham and Do, 2016). A new hypothesis named Dual NMDAR hypofunction has been brought up by Nakazawa et al. (2017). It suggests that NMDAR hypofunction in cortical and hippocampal GABAergic neurons in early postnatal development serve as a vital starting point, causing cortical circuitry maturation defects, an oxidative stress increase, dopamine dysregulation, etc. The first NMDAR hypofunction elicited the emergence of the three symptom clusters in schizophrenia. A second NMDAR hypofunction in pyramidal neurons then results from the Excitation/Inhibition imbalance and glutamate spillover, which may worsen the negative symptoms and cognitive dysfunction in the progression of schizophrenia (Nakazawa et al., 2017).
First, these NMDAR-targeted drugs may have the imprecise targets for brain regions and cell types. NMDARs are distributed universally in the brain. They are localized mainly in post-synaptic regions with a small percentage being localized in extra-synaptic and pre-synaptic regions (Hardingham and Bading, 2010; Parsons and Raymond, 2014). The impaired NMDARs in schizophrenia pathology were supposed to be synaptic GluN2A-containing NMDARs in cortical and hippocampal GABAergic interneurons (Paoletti et al., 2013). However, these NMDAR-targeted drugs were all systematically administered and few have preference for the cortex and hippocampus. Some drugs such as glycine and GlyT-1 inhibitors failed to augment synaptic NMDARs but target extra-synaptic NMDARs (Papouin et al., 2012). On the contrary, D-serine serves as a synaptic NMDAR co-agonist (Papouin et al., 2012), and endogenous D-serine is co-localized with NMDARs and mainly in GABAergic neurons (Hashimoto et al., 1993). Thus, drugs associated with D-serine elicited stronger efficacy in clinical studies than those associated with glycine. The mGluRs and KATIIs are mainly expressed in GABAergic interneurons in the cortex, which are consistent with the origin of NMDAR hypofunction in schizophrenia (Alexander, 2007; Plitman et al., 2017). Accordingly, the mGluR PAMs and KATII inhibitors elicited good outcomes in pre-clinical studies and have potential to have good results in patients with schizophrenia (Matosin and Newell, 2013; Ellaithy et al., 2015). DAAOs are expressed mainly in cerebellum with little expression in the frontal cortex (Jagannath et al., 2017), so, DAAO inhibitors may not precisely target schizophrenia. GlyT-1s are expressed mainly in glial cells and in glutamatergic neurons rather than GABAergic interneurons (Borowsky et al., 1993), which may account for the failure of many GlyT-1 inhibitors. GABAergic interneurons, especially parvalbumin-positive neurons, have susceptibility to oxidative stress, thus the antioxidants had robust results in reversing the three symptom clusters in patients with schizophrenia (Steullet et al., 2010; Phensy et al., 2017a).
Second, the current treatment may be too late for intervention. NMDAR hypofunction in early-life may be associated with genetic and environmental risk factors. Genetic factors leading to NMDAR hypofunction include rare mutations in NMDAR-encoding genes (Yu et al., 2018) and gene sets associated with NMDAR-mediated synaptic currents (Wang et al., 2018). Environmental risk factors associated with NMDAR hypofunction include stress events or virus infections that occur in early-life or in the prenatal period, which can induce oxidative stress and neuroinflammation in GABAergic interneurons (Nakazawa and Sapkota, 2020). In animal models, NMDAR hypofunction reversed in early-life as NMDAR-targeted drugs can reverse the schizophrenia-like behavioral abnormalities (Phensy et al., 2017a,b). However, in studies, most NMDAR-targeted drugs were administered after the onset of schizophrenia, except for D-serine which showed good efficacy in prodromal symptoms in a clinical high-risk group (Kantrowitz et al., 2015). The effects of early-life administration of NMDAR-targeted drugs in genetic or clinical high-risk groups require further exploration.
Conclusion
The general effects of NMDAR-targeted drugs were small to moderate in negative symptoms, and small or nonsignificant in cognitive dysfunction in schizophrenia. When comparing the NMDAR-targeted drugs with mainstream antipsychotics, both the NMDAR-targeted drugs and mainstream antipsychotics cannot improve all three symptom clusters of schizophrenia. Can we conclude that the starting point in schizophrenia pathology is neither dopamine disorder nor NMDAR hypofunction? Not yet. However, these two hypotheses interacted with each other. In animal models, GluN1 ablation in GABAergic interneurons at the prenatal period can elicit a hypodopaminergic state in the cortex and a hyperdopaminergic state in the mesolimbic pathway, which is consistent with the dopamine disorder in schizophrenia (Field et al., 2011; Nakazawa et al., 2017). The main dopamine receptor involved in schizophrenia pathology, D2 dopamine receptors (D2Rs), can regulate the NMDAR function and then reverse cognitive dysfunction. In the D2R-expressed pyramidal neuron, the deletion of GSK3β, which took part in the downstream signaling for D2Rs, can upregulate the expression and function of NMDARs (Li et al., 2009, 2020).
Further exploration of the NMDAR hypothesis and therapeutic strategies based on it may bring forward a breakthrough in improving negative symptoms and cognitive dysfunction of schizophrenia. It may provide patients suffering from schizophrenia functional recoveries and normal lives.
Author Contributions
QW, JH, and RW wrote and revised the review. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Key R&D Program Projects, National Science Foundation of China (Grant No. 2016YFC1306900) and the National Natural Science Foundation of China (Grant No. 82072096).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank all leaders, teachers, and working staff. Without their help, it would have been much harder for us to have completed this manuscript.
Abbreviations
MMN, mismatch negativity; GMS, glycine modulatory site; DCS, D-cycloserine; KYNA, kynurenic acid; GlyT-1, glycine transporter-1; PAMs, Positive allosteric modulators; mGluRs, metabotropic glutamate receptors.
Footnotes
- ^ http://clinicaltrials.gov/show/NCT01323205
- ^ http://clinicaltrials.gov/show/NCT00921804
- ^ http://clinicaltrials.gov/show/NCT00986531
References
Albuquerque, E. X., and Schwarcz, R. (2013). Kynurenic acid as an antagonist of alpha7 nicotinic acetylcholine receptors in the brain: facts and challenges. Biochem. Pharmacol. 85, 1027–1032. doi: 10.1016/j.bcp.2012.12.014
Alexander, S. P. H. (2007). “Metabotropic glutamate receptors,” in xPharm: The Comprehensive Pharmacology Reference, eds S. J. Enna and D. B. Bylund (New York: Elsevier), 1–4.
Amiaz, R., Kent, I., Rubinstein, K., Sela, B. A., Javitt, D., and Weiser, M. (2015). Safety, tolerability and pharmacokinetics of open label sarcosine added on to anti-psychotic treatment in schizophrenia – preliminary study. Isr. J. Psychiatry Relat. Sci. 52, 12–15.
Balu, D. T., and Coyle, J. T. (2015). The NMDA receptor ‘glycine modulatory site’ in schizophrenia: D-serine, glycine, and beyond. Curr. Opin. Pharmacol. 20, 109–115. doi: 10.1016/j.coph.2014.12.004
Balu, D. T., Li, Y., Takagi, S., Presti, K. T., Ramikie, T. S., Rook, J. M., et al. (2016). An mGlu5-positive allosteric modulator rescues the neuroplasticity deficits in a genetic model of NMDA receptor hypofunction in schizophrenia. Neuropsychopharmacology 41, 2052–2061. doi: 10.1038/npp.2016.2
Barch, D. M., and Ceaser, A. (2012). Cognition in schizophrenia: core psychological and neural mechanisms. Trends Cogn. Sci. 16, 27–34. doi: 10.1016/j.tics.2011.11.015
Belforte, J. E., Zsiros, V., Sklar, E. R., Jiang, Z., Yu, G., Li, Y., et al. (2010). Postnatal NMDA receptor ablation in corticolimbic interneurons confers schizophrenia-like phenotypes. Nat. Neurosci. 13, 76–83. doi: 10.1038/nn.2447
Berk, M., Copolov, D., Dean, O., Lu, K., Jeavons, S., Schapkaitz, I., et al. (2008). N-acetyl cysteine as a glutathione precursor for schizophrenia–a double-blind, randomized, placebo-controlled trial. Biol. Psychiatry 64, 361–368. doi: 10.1016/j.biopsych.2008.03.004
Borowsky, B., Mezey, E., and Hoffman, B. J. (1993). Two glycine transporter variants with distinct localization in the CNS and peripheral tissues are encoded by a common gene. Neuron 10, 851–863. doi: 10.1016/0896-6273(93)90201-2
Bortolon, C., Macgregor, A., Capdevielle, D., and Raffard, S. (2018). Apathy in schizophrenia: a review of neuropsychological and neuroanatomical studies. Neuropsychologia 118(Pt B), 22–33. doi: 10.1016/j.neuropsychologia.2017.09.033
Bortz, D. M., Wu, H. Q., Schwarcz, R., and Bruno, J. P. (2017). Oral administration of a specific kynurenic acid synthesis (KAT II) inhibitor attenuates evoked glutamate release in rat prefrontal cortex. Neuropharmacology 121, 69–78. doi: 10.1016/j.neuropharm.2017.04.023
Breier, A., Liffick, E., Hummer, T. A., Vohs, J. L., Yang, Z., Mehdiyoun, N. F., et al. (2018). Effects of 12-month, double-blind N-acetyl cysteine on symptoms, cognition and brain morphology in early phase schizophrenia spectrum disorders. Schizophr. Res. 199, 395–402. doi: 10.1016/j.schres.2018.03.012
Brennand, K. J., Simone, A., Jou, J., Gelboin-Burkhart, C., Tran, N., Sangar, S., et al. (2011). Modelling schizophrenia using human induced pluripotent stem cells. Nature 473, 221–225.
Buchanan, R. W., Javitt, D. C., Marder, S. R., Schooler, N. R., Gold, J. M., McMahon, R. P., et al. (2007). The cognitive and negative symptoms in schizophrenia trial (CONSIST): the efficacy of glutamatergic agents for negative symptoms and cognitive impairments. Am. J. Psychiatry 164, 1593–1602. doi: 10.1176/appi.ajp.2007.06081358
Bugarski-Kirola, D., Wang, A., Abi-Saab, D., and Blattler, T. (2014). A phase II/III trial of bitopertin monotherapy compared with placebo in patients with an acute exacerbation of schizophrenia – results from the CandleLyte study. Eur. Neuropsychopharmacol. 24, 1024–1036. doi: 10.1016/j.euroneuro.2014.03.007
Cain, C. K., McCue, M., Bello, I., Creedon, T., Tang, D. I., Laska, E., et al. (2014). d-Cycloserine augmentation of cognitive remediation in schizophrenia. Schizophr. Res. 153, 177–183. doi: 10.1016/j.schres.2014.01.016
Cardozo, L. F., Pedruzzi, L. M., Stenvinkel, P., Stockler-Pinto, M. B., Daleprane, J. B., and Leite, M. Jr., et al. (2013). Nutritional strategies to modulate inflammation and oxidative stress pathways via activation of the master antioxidant switch Nrf2. Biochimie 95, 1525–1533. doi: 10.1016/j.biochi.2013.04.012
Carmeli, C., Knyazeva, M. G., Cuenod, M., and Do, K. Q. (2012). Glutathione precursor N-acetyl-cysteine modulates EEG synchronization in schizophrenia patients: a double-blind, randomized, placebo-controlled trial. PLoS One 7:e29341. doi: 10.1371/journal.pone.0029341
Cascella, N. G., Macciardi, F., Cavallini, C., and Smeraldi, E. (1994). d-cycloserine adjuvant therapy to conventional neuroleptic treatment in schizophrenia: an open-label study. J. Neural Transm. Gen. Sect. 95, 105–111. doi: 10.1007/bf01276429
Chang, C. H., Lane, H. Y., Tseng, P. T., Chen, S. J., Liu, C. Y., and Lin, C. H. (2019). Effect of N-methyl-D-aspartate-receptor-enhancing agents on cognition in patients with schizophrenia: a systematic review and meta-analysis of double-blind randomised controlled trials. J. Psychopharmacol. 33, 436–448. doi: 10.1177/0269881118822157
Chen, A. T., Chibnall, J. T., and Nasrallah, H. A. (2016). Placebo-controlled augmentation trials of the antioxidant NAC in schizophrenia: a review. Ann. Clin. Psychiatry 28, 190–196.
Cheng, J., Liu, W., Duffney, L. J., and Yan, Z. (2013). SNARE proteins are essential in the potentiation of NMDA receptors by group II metabotropic glutamate receptors. J. Physiol. 591, 3935–3947. doi: 10.1113/jphysiol.2013.255075
Choi, K. H., Wykes, T., and Kurtz, M. M. (2013). Adjunctive pharmacotherapy for cognitive deficits in schizophrenia: meta-analytical investigation of efficacy. Br. J. Psychiatry 203, 172–178. doi: 10.1192/bjp.bp.111.107359
Conde-Ceide, S., Martinez-Viturro, C. M., Alcazar, J., Garcia-Barrantes, P. M., Lavreysen, H., Mackie, C., et al. (2015). Discovery of VU0409551/JNJ-46778212: an mGlu5 positive allosteric modulator clinical candidate targeting schizophrenia. ACS Med. Chem. Lett. 6, 716–720. doi: 10.1021/acsmedchemlett.5b00181
Conus, P., Seidman, L. J., Fournier, M., Xin, L., Cleusix, M., Baumann, P. S., et al. (2018). N-acetylcysteine in a double-blind randomized placebo-controlled trial: toward biomarker-guided treatment in early psychosis. Schizophr. Bull. 44, 317–327. doi: 10.1093/schbul/sbx093
Cotton, S. M., Berk, M., Watson, A., Wood, S., Allott, K., Bartholomeusz, C. F., et al. (2019). ENACT: a protocol for a randomised placebo-controlled trial investigating the efficacy and mechanisms of action of adjunctive N-acetylcysteine for first-episode psychosis. Trials 20:658.
Coyle, J. T. (2012). NMDA receptor and schizophrenia: a brief history. Schizophr. Bull. 38, 920–926. doi: 10.1093/schbul/sbs076
Cull-Candy, S., Brickley, S., and Farrant, M. (2001). NMDA receptor subunits: diversity, development and disease. Curr. Opin. Neurobiol. 11, 327–335. doi: 10.1016/s0959-4388(00)00215-4
Dravid, S. M., Burger, P. B., Prakash, A., Geballe, M. T., Yadav, R., Le, P., et al. (2010). Structural determinants of D-cycloserine efficacy at the NR1/NR2C NMDA receptors. J. Neurosci. 30, 2741–2754. doi: 10.1523/jneurosci.5390-09.2010
D’Souza, D. C., Carson, R. E., Driesen, N., Johannesen, J., Ranganathan, M., and Krystal, J. H. (2018). Dose-related target occupancy and effects on circuitry, behavior, and neuroplasticity of the glycine transporter-1 inhibitor PF-03463275 in healthy and schizophrenia subjects. Biol. Psychiatry 84, 413–421. doi: 10.1016/j.biopsych.2017.12.019
Dunayevich, E., Buchanan, R. W., Chen, C. Y., Yang, J., Nilsen, J., Dietrich, J. M., et al. (2017). Efficacy and safety of the glycine transporter type-1 inhibitor AMG 747 for the treatment of negative symptoms associated with schizophrenia. Schizophr. Res. 182, 90–97. doi: 10.1016/j.schres.2016.10.027
Ellaithy, A., Younkin, J., González-Maeso, J., and Logothetis, D. E. (2015). Positive allosteric modulators of metabotropic glutamate 2 receptors in schizophrenia treatment. Trends Neurosci. 38, 506–516. doi: 10.1016/j.tins.2015.06.002
Ermilov, M., Gelfin, E., Levin, R., Lichtenberg, P., Hashimoto, K., Javitt, D. C., et al. (2013). A pilot double-blind comparison of d-serine and high-dose olanzapine in treatment-resistant patients with schizophrenia. Schizophr. Res. 150, 604–605. doi: 10.1016/j.schres.2013.09.018
Evins, A. E., Amico, E., Posever, T. A., Toker, R., and Goff, D. C. (2002). D-cycloserine added to risperidone in patients with primary negative symptoms of schizophrenia. Schizophr. Res. 56, 19–23. doi: 10.1016/s0920-9964(01)00220-1
Evins, A. E., Fitzgerald, S. M., Wine, L., Rosselli, R., and Goff, D. C. (2000). Placebo-controlled trial of glycine added to clozapine in schizophrenia. Am. J. Psychiatry 157, 826–828. doi: 10.1176/appi.ajp.157.5.826
Fahey, J. W., and Talalay, P. (1999). Antioxidant functions of sulforaphane: a potent inducer of Phase II detoxication enzymes. Food Chem. Toxicol. 37, 973–979. doi: 10.1016/s0278-6915(99)00082-4
Farokhnia, M., Azarkolah, A., Adinehfar, F., Khodaie-Ardakani, M. R., Hosseini, S. M., Yekehtaz, H., et al. (2013). N-acetylcysteine as an adjunct to risperidone for treatment of negative symptoms in patients with chronic schizophrenia: a randomized, double-blind, placebo-controlled study. Clin. Neuropharmacol. 36, 185–192. doi: 10.1097/wnf.0000000000000001
Fell, M. J., McKinzie, D. L., Monn, J. A., and Svensson, K. A. (2012). Group II metabotropic glutamate receptor agonists and positive allosteric modulators as novel treatments for schizophrenia. Neuropharmacology 62, 1473–1483. doi: 10.1016/j.neuropharm.2011.06.007
Feng, S., Xu, Z., Wang, F., Yang, T., Liu, W., Deng, Y., et al. (2017). Sulforaphane prevents methylmercury-induced oxidative damage and excitotoxicity through activation of the Nrf2-ARE pathway. Mol. Neurobiol. 54, 375–391. doi: 10.1007/s12035-015-9643-y
Field, J. R., Walker, A. G., and Conn, P. J. (2011). Targeting glutamate synapses in schizophrenia. Trends Mol. Med. 17, 689–698. doi: 10.1016/j.molmed.2011.08.004
Forsyth, J. K., Bachman, P., Mathalon, D. H., Roach, B. J., and Asarnow, R. F. (2015). Augmenting NMDA receptor signaling boosts experience-dependent neuroplasticity in the adult human brain. Proc. Natl. Acad. Sci. U.S.A. 112, 15331–15336. doi: 10.1073/pnas.1509262112
Forsyth, J. K., Bachman, P., Mathalon, D. H., Roach, B. J., Ye, E., and Asarnow, R. F. (2017). Effects of augmenting N-methyl-D-aspartate receptor signaling on working memory and experience-dependent plasticity in schizophrenia: an exploratory study using acute d-cycloserine. Schizophr. Bull. 43, 1123–1133. doi: 10.1093/schbul/sbw193
Fujigaki, H., Mouri, A., Yamamoto, Y., Nabeshima, T., and Saito, K. (2019). Linking phencyclidine intoxication to the tryptophan-kynurenine pathway: therapeutic implications for schizophrenia. Neurochem. Int. 125, 1–6. doi: 10.1016/j.neuint.2019.02.001
Galderisi, S., Mucci, A., Buchanan, R. W., and Arango, C. (2018). Negative symptoms of schizophrenia: new developments and unanswered research questions. Lancet Psychiatry 5, 664–677. doi: 10.1016/s2215-0366(18)30050-6
Ghoshal, A., Moran, S. P., Dickerson, J. W., Joffe, M. E., Grueter, B. A., Xiang, Z., et al. (2017). Role of mGlu5 receptors and inhibitory neurotransmission in M1 dependent muscarinic LTD in the prefrontal cortex: implications in schizophrenia. ACS Chem. Neurosci. 8, 2254–2265. doi: 10.1021/acschemneuro.7b00167
Goff, D. (2016). The therapeutic role of d-cycloserine in schizophrenia. Adv. Pharmacol. 76, 39–66. doi: 10.1016/bs.apha.2016.02.001
Goff, D. C. (2017). D-cycloserine in schizophrenia: new strategies for improving clinical outcomes by enhancing plasticity. Curr. Neuropharmacol. 15, 21–34. doi: 10.2174/1570159x14666160225154812
Goff, D. C., Cather, C., Gottlieb, J. D., Evins, A. E., Walsh, J., Raeke, L., et al. (2008). Once-weekly D-cycloserine effects on negative symptoms and cognition in schizophrenia: an exploratory study. Schizophr. Res. 106, 320–327. doi: 10.1016/j.schres.2008.08.012
Goff, D. C., Henderson, D. C., Evins, A. E., and Amico, E. (1999). A placebo-controlled crossover trial of D-cycloserine added to clozapine in patients with schizophrenia. Biol. Psychiatry 45, 512–514. doi: 10.1016/s0006-3223(98)00367-9
Goff, D. C., Tsai, G., Manoach, D. S., and Coyle, J. T. (1995). Dose-finding trial of D-cycloserine added to neuroleptics for negative symptoms in schizophrenia. Am. J. Psychiatry 152, 1213–1215. doi: 10.1176/ajp.152.8.1213
Goff, D. C., Tsai, G., Manoach, D. S., Flood, J., Darby, D. G., and Coyle, J. T. (1996). D-cycloserine added to clozapine for patients with schizophrenia. Am. J. Psychiatry 153, 1628–1630. doi: 10.1176/ajp.153.12.1628
Gottlieb, J. D., Cather, C., Shanahan, M., Creedon, T., Macklin, E. A., and Goff, D. C. (2011). D-cycloserine facilitation of cognitive behavioral therapy for delusions in schizophrenia. Schizophr. Res. 131, 69–74. doi: 10.1016/j.schres.2011.05.029
Greenwood, L. M., Leung, S., Michie, P. T., Green, A., Nathan, P. J., Fitzgerald, P., et al. (2018). The effects of glycine on auditory mismatch negativity in schizophrenia. Schizophr. Res. 191, 61–69. doi: 10.1016/j.schres.2017.05.031
Guo, J. Y., Ragland, J. D., and Carter, C. S. (2019). Memory and cognition in schizophrenia. Mol. Psychiatry 24, 633–642.
Hansen, K. B., Yi, F., Perszyk, R. E., Furukawa, H., Wollmuth, L. P., Gibb, A. J., et al. (2018). Structure, function, and allosteric modulation of NMDA receptors. J. Gen. Physiol. 150, 1081–1105.
Harada, K., Nakato, K., Yarimizu, J., Yamazaki, M., Morita, M., Takahashi, S., et al. (2012). A novel glycine transporter-1 (GlyT1) inhibitor, ASP2535 (4-[3-isopropyl-5-(6-phenyl-3-pyridyl)-4H-1,2,4-triazol-4-yl]-2,1,3-benzoxadiazol e), improves cognition in animal models of cognitive impairment in schizophrenia and Alzheimer’s disease. Eur. J. Pharmacol. 685, 59–69. doi: 10.1016/j.ejphar.2012.04.013
Hardingham, G. E., and Bading, H. (2010). Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat. Rev. Neurosci. 11, 682–696. doi: 10.1038/nrn2911
Hardingham, G. E., and Do, K. Q. (2016). Linking early-life NMDAR hypofunction and oxidative stress in schizophrenia pathogenesis. Nat. Rev. Neurosci. 17, 125–134. doi: 10.1038/nrn.2015.19
Hasam-Henderson, L. A., Gotti, G. C., Mishto, M., Klisch, C., Gerevich, Z., Geiger, J. R. P., et al. (2018). NMDA-receptor inhibition and oxidative stress during hippocampal maturation differentially alter parvalbumin expression and gamma-band activity. Sci. Rep. 8:9545.
Hashimoto, A., Nishikawa, T., Oka, T., and Takahashi, K. (1993). Endogenous D-serine in rat brain: N-methyl-D-aspartate receptor-related distribution and aging. J. Neurochem. 60, 783–786. doi: 10.1111/j.1471-4159.1993.tb03219.x
Hashimoto, K. (2010). Glycine transport inhibitors for the treatment of schizophrenia. Open Med. Chem. J. 4, 10–19. doi: 10.2174/1874104501004010010
Hashimoto, K. (2011). Glycine transporter-1: a new potential therapeutic target for schizophrenia. Curr. Pharm. Des. 17, 112–120. doi: 10.2174/138161211795049598
Henneberger, C., Papouin, T., Oliet, S. H., and Rusakov, D. A. (2010). Long-term potentiation depends on release of D-serine from astrocytes. Nature 463, 232–236. doi: 10.1038/nature08673
Heresco-Levy, U., Javitt, D. C., Ebstein, R., Vass, A., Lichtenberg, P., Bar, G., et al. (2005). D-serine efficacy as add-on pharmacotherapy to risperidone and olanzapine for treatment-refractory schizophrenia. Biol. Psychiatry 57, 577–585. doi: 10.1016/j.biopsych.2004.12.037
Heresco-Levy, U., Javitt, D. C., Ermilov, M., Mordel, C., Horowitz, A., and Kelly, D. (1996). Double-blind, placebo-controlled, crossover trial of glycine adjuvant therapy for treatment-resistant schizophrenia. Br. J. Psychiatry 169, 610–617. doi: 10.1192/bjp.169.5.610
Heresco-Levy, U., Javitt, D. C., Ermilov, M., Mordel, C., Silipo, G., and Lichtenstein, M. (1999). Efficacy of high-dose glycine in the treatment of enduring negative symptoms of schizophrenia. Arch. Gen. Psychiatry 56, 29–36. doi: 10.1001/archpsyc.56.1.29
Howley, E., Bestwick, M., Fradley, R., Harrison, H., Leveridge, M., Okada, K., et al. (2017). Assessment of the target engagement and D-serine biomarker profiles of the D-amino acid oxidase inhibitors sodium benzoate and PGM030756. Neurochem. Res. 42, 3279–3288. doi: 10.1007/s11064-017-2367-9
Iwata, Y., Nakajima, S., Suzuki, T., Keefe, R. S., Plitman, E., Chung, J. K., et al. (2015). Effects of glutamate positive modulators on cognitive deficits in schizophrenia: a systematic review and meta-analysis of double-blind randomized controlled trials. Mol. Psychiatry 20, 1151–1160. doi: 10.1038/mp.2015.68
Jagannath, V., Marinova, Z., Monoranu, C.-M., Walitza, S., and Grünblatt, E. (2017). Expression of D-amino acid oxidase (DAO/DAAO) and D-amino acid oxidase activator (DAOA/G72) during development and aging in the human post-mortem brain. Front. Neuroanat. 11:31. doi: 10.3389/fnana.2017.00031
Javitt, D. C., Lee, M., Kantrowitz, J. T., and Martinez, A. (2018). Mismatch negativity as a biomarker of theta band oscillatory dysfunction in schizophrenia. Schizophr. Res. 191, 51–60. doi: 10.1016/j.schres.2017.06.023
Javitt, D. C., Silipo, G., Cienfuegos, A., Shelley, A. M., Bark, N., Park, M., et al. (2001). Adjunctive high-dose glycine in the treatment of schizophrenia. Int. J. Neuropsychopharmacol. 4, 385–391.
Javitt, D. C., Zylberman, I., Zukin, S. R., Heresco-Levy, U., and Lindenmayer, J. P. (1994). Amelioration of negative symptoms in schizophrenia by glycine. Am. J. Psychiatry 151, 1234–1236. doi: 10.1176/ajp.151.8.1234
Jayawickrama, G. S., Sadig, R. R., Sun, G., Nematollahi, A., Nadvi, N. A., Hanrahan, J. R., et al. (2015). Kynurenine aminotransferases and the prospects of inhibitors for the treatment of schizophrenia. Curr. Med. Chem. 22, 2902–2918. doi: 10.2174/0929867322666150608094054
Kahn, R. S., and Sommer, I. E. (2015). The neurobiology and treatment of first-episode schizophrenia. Mol. Psychiatry 20, 84–97. doi: 10.1038/mp.2014.66
Kantrowitz, J. T., Epstein, M. L., Lee, M., Lehrfeld, N., Nolan, K. A., Shope, C., et al. (2018). Improvement in mismatch negativity generation during d-serine treatment in schizophrenia: correlation with symptoms. Schizophr. Res. 191, 70–79. doi: 10.1016/j.schres.2017.02.027
Kantrowitz, J. T., Malhotra, A. K., Cornblatt, B., Silipo, G., Balla, A., Suckow, R. F., et al. (2010). High dose D-serine in the treatment of schizophrenia. Schizophr. Res. 121, 125–130. doi: 10.1016/j.schres.2010.05.012
Kantrowitz, J. T., Woods, S. W., Petkova, E., Cornblatt, B., Corcoran, C. M., Chen, H., et al. (2015). D-serine for the treatment of negative symptoms in individuals at clinical high risk of schizophrenia: a pilot, double-blind, placebo-controlled, randomised parallel group mechanistic proof-of-concept trial. Lancet Psychiatry 2, 403–412. doi: 10.1016/s2215-0366(15)00098-x
Kathuria, A., Lopez-Lengowski, K., Watmuff, B., McPhie, D., Cohen, B. M., and Karmacharya, R. (2019). Synaptic deficits in iPSC-derived cortical interneurons in schizophrenia are mediated by NLGN2 and rescued by N-acetylcysteine. Transl. Psychiatry 9:321.
Kayser, M. S., and Dalmau, J. (2016). Anti-NMDA receptor encephalitis, autoimmunity, and psychosis. Schizophr. Res. 176, 36–40. doi: 10.1016/j.schres.2014.10.007
Keum, Y. S. (2011). Regulation of the Keap1/Nrf2 system by chemopreventive sulforaphane: implications of posttranslational modifications. Ann. N. Y. Acad. Sci. 1229, 184–189. doi: 10.1111/j.1749-6632.2011.06092.x
Kindler, J., Lim, C. K., Weickert, C. S., Boerrigter, D., Galletly, C., Liu, D., et al. (2020). Dysregulation of kynurenine metabolism is related to proinflammatory cytokines, attention, and prefrontal cortex volume in schizophrenia. Mol. Psychiatry 25, 2860–2872. doi: 10.1038/s41380-019-0401-9
Kinon, B. J., Zhang, L., Millen, B. A., Osuntokun, O. O., Williams, J. E., Kollack-Walker, S., et al. (2011). A multicenter, inpatient, phase 2, double-blind, placebo-controlled dose-ranging study of LY2140023 monohydrate in patients with DSM-IV schizophrenia. J. Clin. Psychopharmacol. 31, 349–355. doi: 10.1097/jcp.0b013e318218dcd5
Korotkova, T., Fuchs, E. C., Ponomarenko, A., von Engelhardt, J., and Monyer, H. (2010). NMDA receptor ablation on parvalbumin-positive interneurons impairs hippocampal synchrony, spatial representations, and working memory. Neuron 68, 557–569. doi: 10.1016/j.neuron.2010.09.017
Krystal, J. H., Karper, L. P., Seibyl, J. P., Freeman, G. K., Delaney, R., Bremner, J. D., et al. (1994). Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch. Gen. Psychiatry 51, 199–214. doi: 10.1001/archpsyc.1994.03950030035004
Lane, H. Y., Chang, Y. C., Liu, Y. C., Chiu, C. C., and Tsai, G. E. (2005). Sarcosine or D-serine add-on treatment for acute exacerbation of schizophrenia: a randomized, double-blind, placebo-controlled study. Arch. Gen. Psychiatry 62, 1196–1204. doi: 10.1001/archpsyc.62.11.1196
Lane, H. Y., Lin, C. H., Green, M. F., Hellemann, G., Huang, C. C., Chen, P. W., et al. (2013). Add-on treatment of benzoate for schizophrenia: a randomized, double-blind, placebo-controlled trial of D-amino acid oxidase inhibitor. JAMA Psychiatry 70, 1267–1275. doi: 10.1001/jamapsychiatry.2013.2159
Lane, H. Y., Lin, C. H., Huang, Y. J., Liao, C. H., Chang, Y. C., and Tsai, G. E. (2010). A randomized, double-blind, placebo-controlled comparison study of sarcosine (N-methylglycine) and D-serine add-on treatment for schizophrenia. Int. J. Neuropsychopharmacol. 13, 451–460. doi: 10.1017/s1461145709990939
Lane, H. Y., Liu, Y. C., Huang, C. L., Chang, Y. C., Liau, C. H., Perng, C. H., et al. (2008). Sarcosine (N-methylglycine) treatment for acute schizophrenia: a randomized, double-blind study. Biol. Psychiatry 63, 9–12. doi: 10.1016/j.biopsych.2007.04.038
Lau, C. G., and Zukin, R. S. (2007). NMDA receptor trafficking in synaptic plasticity and neuropsychiatric disorders. Nat. Rev. Neurosci. 8, 413–426. doi: 10.1038/nrn2153
Lavoie, S., Murray, M. M., Deppen, P., Knyazeva, M. G., Berk, M., Boulat, O., et al. (2008). Glutathione precursor, N-acetyl-cysteine, improves mismatch negativity in schizophrenia patients. Neuropsychopharmacology 33, 2187–2199. doi: 10.1038/sj.npp.1301624
Lee, G., and Zhou, Y. (2019). NMDAR hypofunction animal models of schizophrenia. Front. Mol. Neurosci. 12:185. doi: 10.3389/fnmol.2019.00185
Leucht, S., Corves, C., Arbter, D., Engel, R. R., Li, C., and Davis, J. M. (2009). Second-generation versus first-generation antipsychotic drugs for schizophrenia: a meta-analysis. Lancet 373, 31–41. doi: 10.1016/s0140-6736(08)61764-x
Leung, S., Croft, R. J., O’Neill, B. V., and Nathan, P. J. (2008). Acute high-dose glycine attenuates mismatch negativity (MMN) in healthy human controls. Psychopharmacology 196, 451–460. doi: 10.1007/s00213-007-0976-8
Lewis, D. A., Curley, A. A., Glausier, J. R., and Volk, D. W. (2012). Cortical parvalbumin interneurons and cognitive dysfunction in schizophrenia. Trends Neurosci. 35, 57–67. doi: 10.1016/j.tins.2011.10.004
Li, Y. C., Panikker, P., Xing, B., Yang, S. S., Alexandropoulos, C., McEachern, E. P., et al. (2020). Deletion of glycogen synthase kinase-3β in D(2) receptor-positive neurons ameliorates cognitive impairment via NMDA receptor-dependent synaptic plasticity. Biol. Psychiatry 87, 745–755. doi: 10.1016/j.biopsych.2019.10.025
Li, Y. C., Xi, D., Roman, J., Huang, Y. Q., and Gao, W. J. (2009). Activation of glycogen synthase kinase-3 beta is required for hyperdopamine and D2 receptor-mediated inhibition of synaptic NMDA receptor function in the rat prefrontal cortex. J. Neurosci. 29, 15551–15563. doi: 10.1523/jneurosci.3336-09.2009
Lieberman, J. A. (2007). Effectiveness of antipsychotic drugs in patients with chronic schizophrenia: efficacy, safety and cost outcomes of CATIE and other trials. J. Clin. Psychiatry 68:e04. doi: 10.4088/jcp.0207e04
Lieberman, J. A., Stroup, T. S., McEvoy, J. P., Swartz, M. S., Rosenheck, R. A., Perkins, D. O., et al. (2005). Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N. Engl. J. Med. 353, 1209–1223.
Lin, C. H., Chen, Y. M., and Lane, H. Y. (2020). Novel treatment for the hardest-to-treat schizophrenia: dual activation of NMDA receptor and antioxidant. Curr. Drug Targets 21, 610–615. doi: 10.2174/1389450120666191011163539
Lin, C. H., Lin, C. H., Chang, Y. C., Huang, Y. J., Chen, P. W., Yang, H. T., et al. (2018). Sodium benzoate, a D-amino acid oxidase inhibitor, added to clozapine for the treatment of schizophrenia: a randomized, double-blind, placebo-controlled trial. Biol. Psychiatry 84, 422–432. doi: 10.1016/j.biopsych.2017.12.006
Lipton, S. A., Choi, Y. B., Takahashi, H., Zhang, D., Li, W., Godzik, A., et al. (2002). Cysteine regulation of protein function–as exemplified by NMDA-receptor modulation. Trends Neurosci. 25, 474–480. doi: 10.1016/s0166-2236(02)02245-2
Luisada, P. V. (1978). The phencyclidine psychosis: phenomenology and treatment. NIDA Res. Monogr. 21, 241–253.
Lundstrom, L., Bissantz, C., Beck, J., Dellenbach, M., Woltering, T. J., Wichmann, J., et al. (2016). Pharmacological and molecular characterization of the positive allosteric modulators of metabotropic glutamate receptor 2. Neuropharmacology 111, 253–265. doi: 10.1016/j.neuropharm.2016.08.032
MacKay, M. B., Kravtsenyuk, M., Thomas, R., Mitchell, N. D., Dursun, S. M., and Baker, G. B. (2019). D-serine: potential therapeutic agent and/or biomarker in schizophrenia and depression? Front. Psychiatry 10:25. doi: 10.3389/fpsyt.2019.00025
Magalhaes, P. V., Dean, O., Andreazza, A. C., Berk, M., and Kapczinski, F. (2016). Antioxidant treatments for schizophrenia. Cochrane Database Syst. Rev. 2:Cd008919.
Maksymetz, J., Moran, S. P., and Conn, P. J. (2017). Targeting metabotropic glutamate receptors for novel treatments of schizophrenia. Mol. Brain 10:15.
Marek, G. J. (2010). Metabotropic glutamate2/3 (mGlu2/3) receptors, schizophrenia and cognition. Eur. J. Pharmacol. 639, 81–90. doi: 10.1016/j.ejphar.2010.02.058
Matosin, N., and Newell, K. A. (2013). Metabotropic glutamate receptor 5 in the pathology and treatment of schizophrenia. Neurosci. Biobehav. Rev. 37, 256–268. doi: 10.1016/j.neubiorev.2012.12.005
Matsuda, Y., Kishi, T., and Iwata, N. (2013). Efficacy and safety of NMDA receptor antagonists augmentation therapy for schizophrenia: an updated meta-analysis of randomized placebo-controlled trials. J. Psychiatr. Res. 47, 2018–2020. doi: 10.1016/j.jpsychires.2013.09.002
Matsuura, A., Fujita, Y., Iyo, M., and Hashimoto, K. (2015). Effects of sodium benzoate on pre-pulse inhibition deficits and hyperlocomotion in mice after administration of phencyclidine. Acta Neuropsychiatr. 27, 159–167. doi: 10.1017/neu.2015.1
McQueen, G., Lay, A., Lally, J., Gabay, A. S., Collier, T., Lythgoe, D. J., et al. (2019). Effect of single dose N-acetylcysteine administration on resting state functional connectivity in schizophrenia. Psychopharmacology 237, 443–451. doi: 10.1007/s00213-019-05382-1
Minarini, A., Ferrari, S., Galletti, M., Giambalvo, N., Perrone, D., Rioli, G., et al. (2017). N-acetylcysteine in the treatment of psychiatric disorders: current status and future prospects. Expert Opin. Drug Metab. Toxicol. 13, 279–292. doi: 10.1080/17425255.2017.1251580
Monte, A. S., da Silva, F. E. R., Lima, C. N. C., Vasconcelos, G. S., Gomes, N. S., Miyajima, F., et al. (2020). Sex influences in the preventive effects of N-acetylcysteine in a two-hit animal model of schizophrenia. J. Psychopharmacol. 34, 125–136. doi: 10.1177/0269881119875979
Moschetti, V., Desch, M., Goetz, S., Liesenfeld, K. H., Rosenbrock, H., Kammerer, K. P., et al. (2018). Safety, tolerability and pharmacokinetics of oral BI 425809, a glycine transporter 1 inhibitor, in healthy male volunteers: a partially randomised, single-blind, placebo-controlled, first-in-human study. Eur. J. Drug Metab. Pharmacokinet. 43, 239–249. doi: 10.1007/s13318-017-0440-z
Nakazawa, K., and Sapkota, K. (2020). The origin of NMDA receptor hypofunction in schizophrenia. Pharmacol. Ther. 205:107426. doi: 10.1016/j.pharmthera.2019.107426
Nakazawa, K., Jeevakumar, V., and Nakao, K. (2017). Spatial and temporal boundaries of NMDA receptor hypofunction leading to schizophrenia. NPJ Schizophr. 3:7.
Neale, B. M., Corvin, A., Walters, J. T. R., Farh, K. H., Holmans, P. A., Lee, P., et al. (2014). Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427. doi: 10.1038/nature13595
Nematollahi, A., Sun, G., Jayawickrama, G. S., and Church, W. B. (2016). Kynurenine Aminotransferase isozyme inhibitors: a review. Int. J. Mol. Sci. 17:946. doi: 10.3390/ijms17060946
Noh, H., Shao, Z., Coyle, J. T., and Chung, S. (2017). Modeling schizophrenia pathogenesis using patient-derived induced pluripotent stem cells (iPSCs). Biochim. Biophys. Acta Mol. Basis Dis. 1863, 2382–2387. doi: 10.1016/j.bbadis.2017.06.019
Nong, Y., Huang, Y. Q., Ju, W., Kalia, L. V., Ahmadian, G., Wang, Y. T., et al. (2003). Glycine binding primes NMDA receptor internalization. Nature 422, 302–307. doi: 10.1038/nature01497
Paoletti, P., and Neyton, J. (2007). NMDA receptor subunits: function and pharmacology. Curr. Opin. Pharmacol. 7, 39–47. doi: 10.1016/j.coph.2006.08.011
Paoletti, P., Bellone, C., and Zhou, Q. (2013). NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci. 14, 383–400. doi: 10.1038/nrn3504
Papouin, T., Ladépêche, L., Ruel, J., Sacchi, S., Labasque, M., Hanini, M., et al. (2012). Synaptic and extrasynaptic NMDA receptors are gated by different endogenous coagonists. Cell 150, 633–646. doi: 10.1016/j.cell.2012.06.029
Parsons, M. P., and Raymond, L. A. (2014). Extrasynaptic NMDA receptor involvement in central nervous system disorders. Neuron 82, 279–293. doi: 10.1016/j.neuron.2014.03.030
Patil, S. T., Zhang, L., Martenyi, F., Lowe, S. L., Jackson, K. A., Andreev, B. V., et al. (2007). Activation of mGlu2/3 receptors as a new approach to treat schizophrenia: a randomized phase 2 clinical trial. Nat. Med. 13, 1102–1107. doi: 10.1038/nm1632
Perala, J., Suvisaari, J., Saarni, S. I., Kuoppasalmi, K., Isometsa, E., Pirkola, S., et al. (2007). Lifetime prevalence of psychotic and bipolar I disorders in a general population. Arch. Gen. Psychiatry 64, 19–28. doi: 10.1001/archpsyc.64.1.19
Phensy, A., Driskill, C., Lindquist, K., Guo, L., Jeevakumar, V., Fowler, B., et al. (2017a). Antioxidant treatment in male mice prevents mitochondrial and synaptic changes in an NMDA receptor dysfunction model of schizophrenia. eNeuro 4: ENEURO.0081-17.2017.
Phensy, A., Duzdabanian, H. E., Brewer, S., Panjabi, A., Driskill, C., Berz, A., et al. (2017b). Antioxidant treatment with N-acetyl cysteine prevents the development of cognitive and social behavioral deficits that result from perinatal ketamine treatment. Front. Behav. Neurosci. 11:106. doi: 10.3389/fnbeh.2017.00106
Pinard, E., Borroni, E., Koerner, A., Umbricht, D., and Alberati, D. (2018). Glycine transporter type I (GlyT1) inhibitor, bitopertin: a journey from lab to patient. Chimia 72, 477–484. doi: 10.2533/chimia.2018.477
Plitman, E., Iwata, Y., Caravaggio, F., Nakajima, S., Chung, J. K., Gerretsen, P., et al. (2017). Kynurenic acid in schizophrenia: a systematic review and meta-analysis. Schizophr. Bull. 43, 764–777. doi: 10.1093/schbul/sbw221
Retsa, C., Knebel, J. F., Geiser, E., Ferrari, C., Jenni, R., Fournier, M., et al. (2018). Treatment in early psychosis with N-acetyl-cysteine for 6months improves low-level auditory processing: Pilot study. Schizophr. Res. 191, 80–86. doi: 10.1016/j.schres.2017.07.008
Robison, A. J., Thakkar, K. N., and Diwadkar, V. A. (2020). Cognition and reward circuits in schizophrenia: synergistic, not separate. Biol. Psychiatry 87, 204–214. doi: 10.1016/j.biopsych.2019.09.021
Rook, J. M., Xiang, Z., Lv, X., Ghoshal, A., Dickerson, J. W., Bridges, T. M., et al. (2015). Biased mGlu5-positive allosteric modulators provide in vivo efficacy without potentiating mGlu5 modulation of NMDAR currents. Neuron 86, 1029–1040. doi: 10.1016/j.neuron.2015.03.063
Rothbaum, B. O., Price, M., Jovanovic, T., Norrholm, S. D., Gerardi, M., Dunlop, B., (2014). A randomized, double-blind evaluation of D-cycloserine or alprazolam combined with virtual reality exposure therapy for posttraumatic stress disorder in Iraq and Afghanistan War veterans. Am. J. Psychiatry. 171, 640-648. doi: 10.1176/appi.ajp.2014.13121625
Ryan, A., Baker, A., Dark, F., Foley, S., Gordon, A., Hatherill, S., et al. (2017). The efficacy of sodium benzoate as an adjunctive treatment in early psychosis – CADENCE-BZ: study protocol for a randomized controlled trial. Trials 18:165.
Schoemaker, J. H., Jansen, W. T., Schipper, J., and Szegedi, A. (2014). The selective glycine uptake inhibitor org 25935 as an adjunctive treatment to atypical antipsychotics in predominant persistent negative symptoms of schizophrenia: results from the GIANT trial. J. Clin. Psychopharmacol. 34, 190–198. doi: 10.1097/jcp.0000000000000073
Sedlak, T. W., Nucifora, L. G., Koga, M., Shaffer, L. S., Higgs, C., Tanaka, T., et al. (2018). Sulforaphane augments glutathione and influences brain metabolites in human subjects: a clinical pilot study. Mol. Neuropsychiatry 3, 214–222. doi: 10.1159/000487639
Seillier, A., and Giuffrida, A. (2009). Evaluation of NMDA receptor models of schizophrenia: divergences in the behavioral effects of sub-chronic PCP and MK-801. Behav. Brain Res. 204, 410–415. doi: 10.1016/j.bbr.2009.02.007
Sepehrmanesh, Z., Heidary, M., Akasheh, N., Akbari, H., and Heidary, M. (2018). Therapeutic effect of adjunctive N-acetyl cysteine (NAC) on symptoms of chronic schizophrenia: a double-blind, randomized clinical trial. Prog. Neuropsychopharmacol. Biol. Psychiatry 82, 289–296. doi: 10.1016/j.pnpbp.2017.11.001
Shiina, A., Kanahara, N., Sasaki, T., Oda, Y., Hashimoto, T., Hasegawa, T., et al. (2015). An open study of sulforaphane-rich broccoli sprout extract in patients with schizophrenia. Clin. Psychopharmacol. Neurosci. 13, 62–67. doi: 10.9758/cpn.2015.13.1.62
Shirai, Y., Fujita, Y., Hashimoto, R., Ohi, K., Yamamori, H., Yasuda, Y., et al. (2015). Dietary intake of sulforaphane-rich broccoli sprout extracts during juvenile and adolescence can prevent phencyclidine-induced cognitive deficits at adulthood. PLoS One 10:e0127244. doi: 10.1371/journal.pone.0127244
Singh, S. P., and Singh, V. (2011). Meta-analysis of the efficacy of adjunctive NMDA receptor modulators in chronic schizophrenia. CNS Drugs 25, 859–885. doi: 10.2165/11586650-000000000-00000
Sokoloff, P., and Le Foll, B. (2017). The dopamine D3 receptor, a quarter century later. Eur. J. Neurosci. 45, 2–19. doi: 10.1111/ejn.13390
Stansley, B. J., and Conn, P. J. (2018). The therapeutic potential of metabotropic glutamate receptor modulation for schizophrenia. Curr. Opin. Pharmacol. 38, 31–36. doi: 10.1016/j.coph.2018.02.003
Steullet, P., Cabungcal, J. H., Kulak, A., Kraftsik, R., Chen, Y., Dalton, T. P., et al. (2010). Redox dysregulation affects the ventral but not dorsal hippocampus: impairment of parvalbumin neurons, gamma oscillations, and related behaviors. J. Neurosci. 30, 2547–2558. doi: 10.1523/jneurosci.3857-09.2010
Steullet, P., Neijt, H. C., Cuenod, M., and Do, K. Q. (2006). Synaptic plasticity impairment and hypofunction of NMDA receptors induced by glutathione deficit: relevance to schizophrenia. Neuroscience 137, 807–819. doi: 10.1016/j.neuroscience.2005.10.014
Stone, T. W. (2020). Does kynurenic acid act on nicotinic receptors? An assessment of the evidence. J. Neurochem. 152, 627–649. doi: 10.1111/jnc.14907
Szilagyi, B., Ferenczy, G. G., and Keseru, G. M. (2018). Drug discovery strategies and the preclinical development of D-amino-acid oxidase inhibitors as antipsychotic therapies. Expert Opin. Drug Discov. 13, 973–982. doi: 10.1080/17460441.2018.1524459
Takiguchi, K., Uezato, A., Itasaka, M., Atsuta, H., Narushima, K., Yamamoto, N., et al. (2017). Association of schizophrenia onset age and white matter integrity with treatment effect of D-cycloserine: a randomized placebo-controlled double-blind crossover study. BMC Psychiatry 17:249. doi: 10.1186/s12888-017-1410-3
Thiebes, S., Leicht, G., Curic, S., Steinmann, S., Polomac, N., Andreou, C., et al. (2017). Glutamatergic deficit and schizophrenia-like negative symptoms: new evidence from ketamine-induced mismatch negativity alterations in healthy male humans. J. Psychiatry Neurosci. 42, 273–283. doi: 10.1503/jpn.160187
Thomas, E. H. X., Bozaoglu, K., Rossell, S. L., and Gurvich, C. (2017). The influence of the glutamatergic system on cognition in schizophrenia: a systematic review. Neurosci. Biobehav. Rev. 77, 369–387. doi: 10.1016/j.neubiorev.2017.04.005
Trepanier, C., Lei, G., Xie, Y. F., and MacDonald, J. F. (2013). Group II metabotropic glutamate receptors modify N-methyl-D-aspartate receptors via Src kinase. Sci. Rep. 3:926.
Tsai, G. E., and Lin, P. Y. (2010). Strategies to enhance N-methyl-D-aspartate receptor-mediated neurotransmission in schizophrenia, a critical review and meta-analysis. Curr. Pharm. Des. 16, 522–537. doi: 10.2174/138161210790361452
Tsai, G. E., Yang, P., Chung, L. C., Tsai, I. C., Tsai, C. W., and Coyle, J. T. (1999). D-serine added to clozapine for the treatment of schizophrenia. Am. J. Psychiatry 156, 1822–1825.
Tsai, G., Yang, P., Chung, L. C., Lange, N., and Coyle, J. T. (1998). D-serine added to antipsychotics for the treatment of schizophrenia. Biol. Psychiatry 44, 1081–1089. doi: 10.1016/s0006-3223(98)00279-0
Tyszkiewicz, J. P., Gu, Z., Wang, X., Cai, X., and Yan, Z. (2004). Group II metabotropic glutamate receptors enhance NMDA receptor currents via a protein kinase C-dependent mechanism in pyramidal neurones of rat prefrontal cortex. J. Physiol. 554(Pt 3), 765–777. doi: 10.1113/jphysiol.2003.056812
Umbricht, D., Alberati, D., Martin-Facklam, M., Borroni, E., Youssef, E. A., Ostland, M., et al. (2014). Effect of bitopertin, a glycine reuptake inhibitor, on negative symptoms of schizophrenia: a randomized, double-blind, proof-of-concept study. JAMA Psychiatry 71, 637–646. doi: 10.1001/jamapsychiatry.2014.163
Uno, Y., and Coyle, J. T. (2019). Glutamate hypothesis in schizophrenia. Psychiatry Clin. Neurosci. 73, 204–215.
Uzun, O., Bolu, A., and Celik, C. (2019). Effect of N-acetylcysteine on clozapine-induced sialorrhea in schizophrenic patients: a case series. Int. Clin. Psychopharmacol. 35, 229–231.
van Berckel, B. N., Evenblij, C. N., van Loon, B. J., Maas, M. F., van der Geld, M. A., Wynne, H. J., et al. (1999). D-cycloserine increases positive symptoms in chronic schizophrenic patients when administered in addition to antipsychotics: a double-blind, parallel, placebo-controlled study. Neuropsychopharmacology 21, 203–210. doi: 10.1016/s0893-133x(99)00014-7
Volianskis, A., France, G., Jensen, M. S., Bortolotto, Z. A., Jane, D. E., and Collingridge, G. L. (2015). Long-term potentiation and the role of N-methyl-D-aspartate receptors. Brain Res. 1621, 5–16. doi: 10.1016/j.brainres.2015.01.016
Wacongne, C. (2016). A predictive coding account of MMN reduction in schizophrenia. Biol. Psychol. 116, 68–74. doi: 10.1016/j.biopsycho.2015.10.011
Wang, C., Inselman, A., Liu, S., and Liu, F. (2020). Potential mechanisms for phencyclidine/ketamine-induced brain structural alterations and behavioral consequences. Neurotoxicology 76, 213–219. doi: 10.1016/j.neuro.2019.12.005
Wang, Q., Wu, H. M., Yue, W., Yan, H., Zhang, Y., Tan, L., et al. (2018). Effect of damaging rare mutations in synapse-related gene sets on response to short-term antipsychotic medication in Chinese patients with schizophrenia: a randomized clinical trial. JAMA Psychiatry 75, 1261–1269. doi: 10.1001/jamapsychiatry.2018.3039
Weiser, M., Heresco-Levy, U., Davidson, M., Javitt, D. C., Werbeloff, N., Gershon, A. A., et al. (2012). A multicenter, add-on randomized controlled trial of low-dose d-serine for negative and cognitive symptoms of schizophrenia. J. Clin. Psychiatry 73, e728–e734.
Wolosker, H., Dumin, E., Balan, L., and Foltyn, V. N. (2008). D-amino acids in the brain: D-serine in neurotransmission and neurodegeneration. FEBS J. 275, 3514–3526. doi: 10.1111/j.1742-4658.2008.06515.x
Yu, Y., Lin, Y., Takasaki, Y., Wang, C., Kimura, H., Xing, J., et al. (2018). Rare loss of function mutations in N-methyl-D-aspartate glutamate receptors and their contributions to schizophrenia susceptibility. Transl. Psychiatry 8:12.
Zandi, M. S., Irani, S. R., Lang, B., Waters, P., Jones, P. B., McKenna, P., et al. (2011). Disease-relevant autoantibodies in first episode schizophrenia. J. Neurol. 258, 686–688. doi: 10.1007/s00415-010-5788-9
Zanos, P., and Gould, T. D. (2018). Mechanisms of ketamine action as an antidepressant. Mol. Psychiatry 23, 801–811. doi: 10.1038/mp.2017.255
Zhang, J.-H., Chen, B., and Lu, J.-R. (2015). Treatment effect of risperidone alone and combined with N-acetly-cysteine for first-episode schizophenic patients. J. Clin. Psychiatry 1005-3220, 394–396.
Keywords: NMDA receptor, glutamate, schizophrenia, cognitive dysfunction, negative symptoms
Citation: Wu Q, Huang J and Wu R (2021) Drugs Based on NMDAR Hypofunction Hypothesis in Schizophrenia. Front. Neurosci. 15:641047. doi: 10.3389/fnins.2021.641047
Received: 13 December 2020; Accepted: 12 March 2021;
Published: 12 April 2021.
Edited by:
Helene Tricoire-Leignel, Université d’Angers, FranceReviewed by:
Neil M. Fournier, Trent University, CanadaJean-Marie Billard, INSERM U1075 Université de Caen Normandie - Pôle des Formations et de Recherche en Santé, France
Copyright © 2021 Wu, Huang and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Renrong Wu, d3VyZW5yb25nQGNzdS5lZHUuY24=