 Yukti Vyas
Yukti Vyas Juliette E. Cheyne
Juliette E. Cheyne Kevin Lee
Kevin Lee Yewon Jung
Yewon Jung Pang Ying Cheung1
Pang Ying Cheung1 Johanna M. Montgomery
Johanna M. Montgomery- 1Department of Physiology, Faculty of Medical and Health Sciences, Centre for Brain Research, University of Auckland, Auckland, New Zealand
- 2Department of Pharmacology, Faculty of Medical and Health Sciences, University of Auckland, Auckland, New Zealand
The SHANK family of proteins play critical structural and functional roles in the postsynaptic density (PSD) at excitatory glutamatergic synapses. Through their multidomain structure they form a structural platform across the PSD for protein–protein interactions, as well as recruiting protein complexes to strengthen excitatory synaptic transmission. Mutations in SHANKs reflect their importance to synapse development and plasticity. This is evident in autism spectrum disorder (ASD), a neurodevelopmental disorder resulting in behavioural changes including repetitive behaviours, lack of sociability, sensory issues, learning, and language impairments. Human genetic studies have revealed ASD mutations commonly occur in SHANKs. Rodent models expressing these mutations display ASD behavioural impairments, and a subset of these deficits are rescued by reintroduction of Shank in adult animals, suggesting that lack of SHANK during key developmental periods can lead to permanent changes in the brain’s wiring. Here we explore the differences in synaptic function and plasticity from development onward in rodent Shank ASD models. To date the most explored brain regions, relate to the behavioural changes observed, e.g., the striatum, hippocampus, sensory, and prefrontal cortex. In addition, less-studied regions including the hypothalamus, cerebellum, and peripheral nervous system are also affected. Synaptic phenotypes include weakened but also strengthened synaptic function, with NMDA receptors commonly affected, as well as changes in the balance of excitation and inhibition especially in cortical brain circuits. The effects of shankopathies in activity-dependent brain wiring is an important target for therapeutic intervention. We therefore highlight areas of research consensus and identify remaining questions and challenges.
Introduction
Autism spectrum disorders (ASDs) are characterised by deficits in social communication and interactions, as well as repetitive and restrictive behaviours (American Psychiatric Association, 2013). The prevalence of ASD is 1 in 54 (Maenner et al., 2020). When genetic mutations implicated in ASD are functionally grouped, many converge on specific biological pathways involved in neuronal connectivity and synaptic plasticity, thus leading to the synaptic hypothesis of ASD (Bourgeron, 2009). The SRC homology 3 (SH3) and multiple Ankyrin repeat domain (SHANK) family of proteins are known as organisers of excitatory glutamatergic synapses (Boeckers et al., 1999a,b; Naisbitt et al., 1999). There are three known SHANK isoforms: SHANK1 (ProSAP3; chromosome 19q13.33), SHANK2 (ProSAP1; c.11q13.3), and SHANK3 (ProSAP2; c.22q13.3). However, multiple intragenic promoters and alternate splicing lead to an extensive array of SHANK protein isoforms (Lim et al., 1999; Sheng and Kim, 2000; Jiang and Ehlers, 2013; Wang et al., 2014), that show specific protein interactions (Boeckers et al., 2005; Shi et al., 2017). Structurally, SHANK proteins have 63–87% amino acid sequence homology and a characteristic domain sequence including the SHANK/ProSAP N-terminal domain, multiple Ankyrin (ANK) repeats, SH3 domain, PDZ domain, a proline-rich region, and a sterile alpha motile (SAM) domain (Sheng and Kim, 2000; Mameza et al., 2013). Through these protein binding domains, SHANKS interact with receptors, ion channels, cytoskeletal proteins, and scaffolding proteins, thereby contributing to the integrity and molecular composition of excitatory glutamatergic synapses (Jiang and Ehlers, 2013). For example, the PDZ domain in SHANKs can bind to the adaptor protein guanylate kinase-associated protein (GKAP; Naisbitt et al., 1999), which in turn, through the interaction with the postsynaptic density molecule 95 (PSD95; Naisbitt et al., 1999), associate with glutamatergic receptors, such as NMDARs (Kornau et al., 1995) and AMPARs (Uemura et al., 2004; for review see Boeckers et al., 2002).
SHANK1 is exclusively expressed in the brain, whereas SHANK2 and SHANK3 are predominantly found in the brain but are also expressed in the kidneys, liver, testis, pancreas, spleen, and heart (Lim et al., 1999; Yao et al., 1999; Zitzer et al., 1999). Within the brain, SHANK1 is highly expressed in the hippocampus, cortex, and amygdala, at moderate levels in the substantia nigra and thalamus, and at very low levels in the striatum, cerebellum and subthalamic nucleus (Zitzer et al., 1999). SHANK2 and SHANK3 are abundantly expressed in the cortex, hippocampus, and olfactory bulb. In the striatum, SHANK3 is most abundant, but there is also SHANK2 expression (Peça et al., 2011; Schmeisser et al., 2012; Monteiro and Feng, 2017). Within the cerebellum, SHANK2 is expressed in Purkinje cells whereas SHANK3 is found only in the granule cell layer (Boeckers et al., 1999a,b; Kleijer et al., 2014).
SHANK2 is among the first proteins to be expressed at the postsynaptic density (PSD), suggesting a key role in synapse development, followed by the recruitment of SHANK3, and then SHANK1 (Boeckers et al., 1999a,b; Grabrucker et al., 2011) which are both implicated in synapse maturation (Sala et al., 2001; Roussignol et al., 2005; Arons et al., 2016). Once localised within the PSD, SHANKs act as major structural and functional scaffolding proteins regulating other PSD proteins, glutamatergic receptors, and proteins in the presynapse through the neuroligin–neurexin trans-synaptic complex (Arons et al., 2012). Predictably ASD-associated mutations in SHANK genes have detrimental effects on synaptic function and overall neurological development (Figure 1). Many isoform-specific SHANK mutations have been identified in ASD patients (Durand et al., 2007; Berkel et al., 2010; Leblond et al., 2012; Sato et al., 2012), and other neuropsychiatric disorders (for a review see Wan et al., 2021). Here we review the influence of Shankopathies in ASD, in particular how Shank mutations impact synapse structure and function in different regions of the rodent brain.
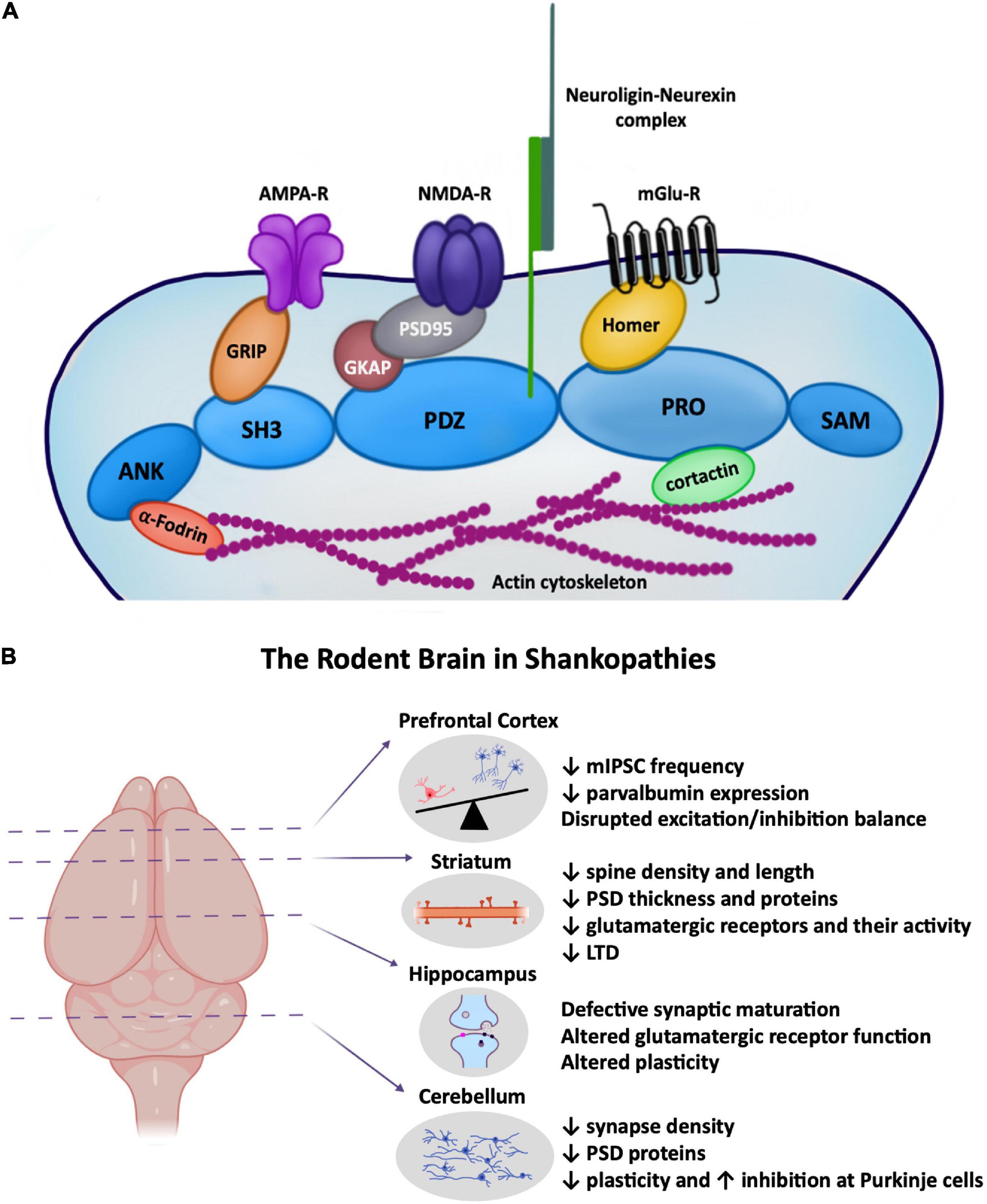
Figure 1. (A) Cartoon depiction highlighting the major role of SHANKs in the postsynaptic structure of glutamatergic synapses, linking proteins across the depth of the PSD from the actin cytoskeleton to surface AMPA and NMDA receptors. (B) ASD-associated synaptic changes in Shank rodent models known to occur in the prefrontal cortex, striatum, hippocampus, and cerebellum. (Left) Brain regions highlighted in this minireview where Shankopathies have been most studied. (Right) Cartoon depictions of some of the synaptic changes occurring in each region. Created with Biorender.
Hippocampus
The hippocampus plays a critical role in processing multisensory information, memory, cognitive function, and modulating emotion, stress, and social behaviour (Bannerman et al., 2014; Hitti and Siegelbaum, 2014; Zemla and Basu, 2017). Therefore, disruption to the development of hippocampal circuits might underlie social memory impairments and cognitive decline observed in individuals with ASD (Solomon et al., 2016; Cooper et al., 2017; Habib et al., 2019).
SHANK2 is highly expressed in the developing hippocampus, especially during spinogenesis (Boeckers et al., 1999a,b, 2004). Consistently, ASD-associated Shank2-specific mouse models show impairments in spatial and working memory, which is dependent on the hippocampus [but also includes other brain regions such as the prefrontal cortex (PFC)] (Schmeisser et al., 2012; Won et al., 2012; Pappas et al., 2017). However, both anatomical and electrophysiological studies have described differential effects of ASD-associated Shank2 mutations on spine number and morphology, NMDA/AMPA ratio, and synaptic plasticity (Table 1; Schmeisser et al., 2012; Won et al., 2012; Pappas et al., 2017). For example, at Schaffer collateral-CA1 synapses, Shank2-Δe6–7–/– mice fail to maintain long-term potentiation (LTP) or express long-term depression (LTD) (Won et al., 2012; Lim et al., 2017), whereas Shank2-Δe7–/– mice display increased LTP with no alterations in LTD (Schmeisser et al., 2012; Wegener et al., 2018). Likewise, mice with mutations in other SHANK homologues showed varying changes in cognition, synaptic structure, function, and plasticity in the hippocampus (Table 1; Hung et al., 2008; Kouser et al., 2013; Wang et al., 2014; Lee E.-J. et al., 2015; Jaramillo et al., 2016). Given the heterogeneity in isoform-specific protein–protein interactions (Boeckers et al., 2005; Shi et al., 2017), distinct spatio-temporal SHANK expression (Boeckers et al., 2004; Grabrucker et al., 2011; Wang et al., 2014), and cell type specific functional outcomes at different synapses (Lim et al., 2017; Kim et al., 2018), the development of differential hippocampal phenotypes in Shank mutant animals are not unexpected.
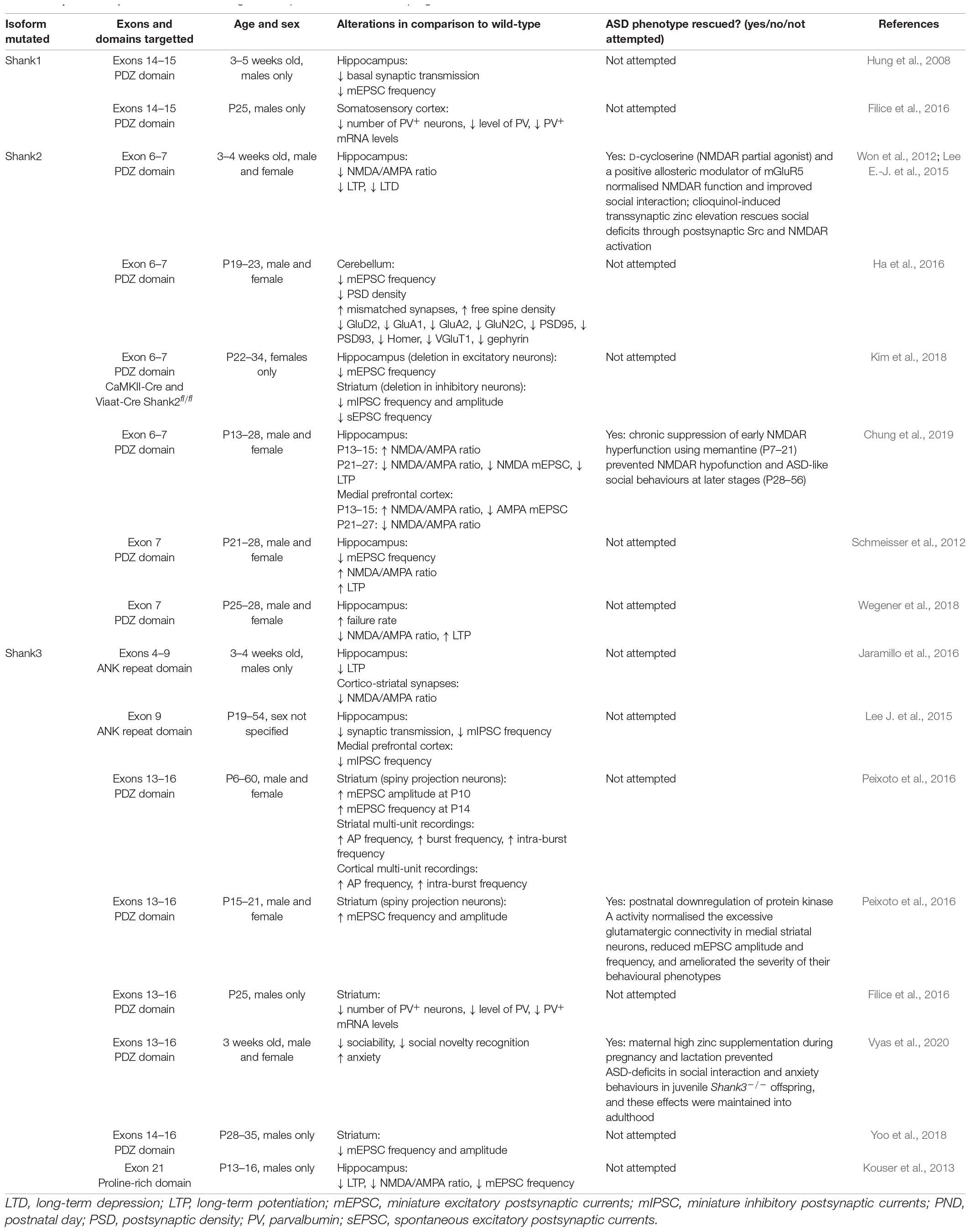
Table 1. Summary of studies examining shankopathies in the developing brain.
To date, hippocampal synaptic physiology has typically been examined in ASD-associated Shank mutant animals post-weaning [> postnatal day 21 (P21)] (Hung et al., 2008; Lee E.-J. et al., 2015; Lim et al., 2017), with fewer studies examining developing hippocampal synapses in pre-weaned animals (Kouser et al., 2013; Jaramillo et al., 2016; Wegener et al., 2018; Chung et al., 2019). Shank3-Δe13–16–/– mice show enhanced glutamatergic network activity in CA3 pyramidal neurons from birth to the second postnatal week (Chiesa et al., 2019). Shank2-Δe6–7–/– mice exhibit a developmental switch in NMDAR function at Schaffer collateral-CA1 synapses, from hyperfunction at P14 to hypofunction beyond P21, that was not observed in other Shank2 mutant mice such as Shank2-Δe7–/– or Shank2-Δe24–/– mice (Chung et al., 2019). Interestingly, Schaffer collateral-CA1 synapses in Shank2-Δe7–/– mice showed reduced AMPAR-mediated synaptic transmission but an enhanced NMDA/AMPA ratio throughout development (Wegener et al., 2018). This suggests that synapse maturation is defective as immature silent synapses, which lack AMPARs but not NMDARs, increase their synaptic strength by incorporating AMPARs (Isaac et al., 1995; Liao et al., 1995; Montgomery et al., 2001). Such disparate developmental alterations in the hippocampus signify that future studies deciphering Shank-related synaptopathy need to be conducted in an age-dependent manner.
Multiple treatment strategies have been tested to reverse hippocampal deficits, particularly targetting NMDAR function: D-cycloserine, a partial NMDAR agonist, normalised spatial learning in Shank2-Δe6–7–/– mice (Won et al., 2012). Moreover, early correction of NMDAR hyperfunction observed during development in Shank2-Δe6–7–/– mice with the NMDAR antagonist memantine (P7–21) prevented autistic-like social behaviours post-weaning (Chung et al., 2019). Zinc has also been a major focus for reversal of ASD-related synaptic and behavioural deficits (Skalny et al., 2021): acute zinc elevation via clioquinol also effectively restored NMDAR function in the hippocampus and prevented spatial memory deficits in Shank2-Δe6–7–/– mice (Lee E.-J. et al., 2015). Together with the work by Vyas et al. (2020) showing prenatal maternal dietary zinc supplementation can prevent the development of some ASD deficits, these data suggest that amending hippocampal deficits during early brain development may be key to the successful development of effective therapeutic interventions.
Striatum
The striatum is part of the basal ganglia and in humans consists of the caudate nucleus, putamen, and globus pallidus (Rundles and Papez, 1937). In rodents the dorsolateral striatum (posterior lateral putamen in humans) receives strong projections from sensorimotor cortical regions and has been implicated in mediating the characteristic stereotypic repetitive behaviours in ASD patients (Langen et al., 2009; Lewis and Kim, 2009). SHANK3 is the predominant isoform expressed in the striatum, and mice lacking Shank3 demonstrate ASD-associated repetitive grooming behaviour (Peça et al., 2011; Wang et al., 2014; Wang et al., 2016; Jaramillo et al., 2016). However, targetted Shank3 deficiency in the striatum failed to exhibit a repetitive grooming phenotype, although this could be a developmental effect as this phenotype worsens with ageing (Vyas et al., 2020), whereas perseverative exploratory behaviour was observed in mice with Shank3 disruption in striatal inhibitory neurons (Bey et al., 2018). Examination of striatal synapse structure and function has revealed altered striatal synapse morphologies including decreased spine density and length, and decreased PSD thickness in Shank3–/– mice. Shank3 deletion-associated reductions in GluA2, GluN2A, GluN2B, SAPAP, Homer1, and PSD93, and increased mGluR5 expression have also been noted in the striatum (Figure 1 and Table 1; Peça et al., 2011; Mei et al., 2016; Wang et al., 2016). Specifically, complete Shank3 deletion (Shank3Δe4–22–/–) causes a significant disruption of the mGluR5-Homer scaffold that decreases mGluR5-mediated signalling but enhances mGluR5 function, highlighting mGluR5 manipulation as a key therapeutic target (Wang et al., 2016).
Similar to what was observed in the hippocampus, different synaptic phenotypes are evident in Shank mutant mice. Specifically, during early postnatal development (P13–14), cortical hyperactivity results in increased striatal mEPSC frequency in Shank3–/– mice (Peixoto et al., 2016). Beyond this developmental time-point, however, the corticostriatal pathway demonstrates reduced field population spike amplitude, NMDAR/AMPAR ratio, NMDAR-mediated EPSCs, mEPSC amplitude and frequency, and impaired LTD (Jaramillo et al., 2016; Wang et al., 2016; Zhou et al., 2016). In comparison to global Shank3 deletion, GABA neuron-specific Shank3 deletion showed similar decreases in striatal mEPSC amplitude and frequency suggesting that the suppressed excitatory input onto dorsolateral striatal neurons is induced by the deletion of Shank3 in striatal GABAergic neurons (Yoo et al., 2018). In addition, targetted disruption of Shank3 in striatal D1 and D2 neurons resulted in hyper-excitability (Bey et al., 2018), and global Shank3 loss demonstrated early hyper-excitability which can disrupt the development of corticostriatal circuits (Peixoto et al., 2016). Furthermore, comparisons of the D1 receptor expressing striatonigral neurons (direct projection pathway) and D2 receptor expressing striatopallidal neurons (indirect projection pathway) demonstrated a preferential hypoactivity of the indirect pathway in Shank3–/– mice (Wang et al., 2017). These structural and functional alterations in striatal synapses, and the imbalance between the direct and indirect pathway of the basal ganglia, are thought to underlie the repetitive grooming behaviour in mouse models of ASD.
Considering the significant deficits caused by the lack of SHANK3 in the striatum (Figure 1), several studies have focused on rescuing neurological and behavioural deficits in Shank3–/– mice both in adulthood and during development. Adult Shank3 restoration was found to increase striatal synaptic Homer1b/c, SAPAP3, NR2A, NR2B, and GluA2 levels in Shank3–/– mice to a level comparable to wild-type mice, as well as significantly increase spine density and mEPSC frequency (Mei et al., 2016). Additionally, Shank3-restored mice displayed reduced repetitive grooming and improved social interaction in comparison to Shank3–/– mice (Mei et al., 2016). Therefore, adult Shank3 restoration improves striatal neurotransmission and rescues ASD-associated behavioural deficits in Shank3–/– mice, showing that some of the developmental neural deficits caused by lack of Shank3 can be reversed. However, complete rescue of ASD behaviours was not achieved, with anxiety and motor coordination deficits remaining after Shank3 restoration. Recent work has examined whether therapeutic strategies targetted during brain development can also influence ASD-associated behaviours and striatal synaptic physiology: supplementation of dietary zinc, which is known to ameliorate ASD-associated behaviours post-weaning (Fourie et al., 2018), is also effective during brain development in Shank3–/– pups when given to female mice during pregnancy and lactation. Maternal dietary zinc supplementation regulated NMDAR-mediated glutamatergic function, rescued pre-synaptic deficits, and prevented LTP in the cortico-striatal pathway in Shank3–/– offspring mice. ASD-associated repetitive grooming behaviours were also rescued in these mice (Vyas et al., 2020). Furthermore, postnatal downregulation of protein kinase A (PKA) activity normalised the excessive glutamatergic connectivity in medial striatal neurons in Shank3–/– mice and ameliorated the severity of their behavioural phenotypes (Peixoto et al., 2016). These studies exhibit that Shank3-deletion associated striatal deficits have the potential to be prevented when strategies are applied during brain development.
Sensory Cortex
For each of our senses there are dedicated parts of the cortex that process each type of information (visual, auditory, somatosensory, olfactory, and gustatory). In ASD, sensory disturbances present as a complex set of symptoms ranging from hypersensitivity to hyposensitivity. As the sensory cortex is readily accessible for in vivo recording, several studies have examined sensory processing in Shank models of ASD. Multiunit recordings in adult Shank3 heterozygote rats revealed that auditory responses in the primary auditory cortex and secondary auditory areas were reduced (Engineer et al., 2018). Similarly, auditory responses to speech sounds were weaker in the primary auditory cortex (Engineer et al., 2018). Interestingly, these responses could be enhanced through reward-based speech sound discrimination training, suggesting that these deficits can be corrected with speech therapy. In contrast, adult Shank3B–/– mice showed normal auditory processing and enhanced pitch discrimination (Rendall et al., 2019), and postnatal Shank+/– mice showed normal startle responses to loud sounds (Orefice et al., 2016), suggesting that auditory processing is largely normal in Shank mutant mice both during development and in adulthood. When tactile stimulation was included, however, startle responses were enhanced in postnatal Shank mutant mice (Orefice et al., 2016). In this tactile prepulse inhibition test, where a tactile stimulus in the form of an airpuff directed at the skin precedes an auditory stimulus, postnatal ASD mutant mice showed increased responses. Furthermore, the ASD mice showed enhanced startle responses to the airpuff alone, and an inability to discriminate in a textured version of novel object recognition. This suggests hypersensitivity to touch plays an important role in ASD sensory behaviours from early development.
Somatosensory hypersensitivity has also been observed in the barrel cortex in response to whisker stimulation in adult Shank3B–/– mice (Chen et al., 2020). Using in vivo calcium imaging in excitatory and inhibitory layer 2/3 neurons this was found to be due to reduced responses in GABAergic neurons, which in turn increased responses in cortical pyramidal neurons. Interestingly, lack of Shank3B specifically in GABAergic neurons in the somatosensory cortex leads to increased pyramidal responses and hypersensitivity to whisker stimulation. This study reveals that SHANK3 in GABAergic interneurons functions to strengthen the excitatory input to these neurons, increasing inhibitory network participation and balancing the network. Without SHANK3 in interneurons, they respond less to tactile stimuli and excitation dominates, leading to hypersensitivity (Chen et al., 2020), demonstrating the importance of correctly functioning SHANK3-containing synapses for balancing excitation/inhibition for normal sensory responses and behaviour. Furthermore, Shank3B–/– mice show reductions in the number, intensity, and size of parvalbumin-positive terminals on pyramidal neurons in cortex (Gogolla et al., 2014).
During early development, the somatosensory cortex of Shank3B–/– mice is hyperactive (Peixoto et al., 2016). This heightened cortical activity feeds forward into the striatum, resulting in increased corticostriatal connectivity in Shank3B–/– mice. This shows that cortical hyperactivity can have feedforward effects that contribute to ASD behavioural phenotypes that aren’t typically considered to be sensory, such as increased grooming.
Prefrontal Cortex
The PFC interconnects subcortical areas including the nucleus accumbens, limbic cortex, ventral tegmental area (VTA), amygdala, and hypothalamus, underpinning cognitive processes, regulation of emotions, sociability, and motivation (Miller and Cohen, 2001). The medial prefrontal cortex (mPFC) is specifically involved in higher cognitive function and social behaviour, and is implicated in many neurological disorders such as ASD and schizophrenia (Alexander and Brown, 2011).
SHANK1, 2, and 3 are highly expressed in the PFC (Monteiro and Feng, 2017). Shank1 and Shank3 deficient mice exhibit reduced expression of parvalbumin in the mPFC early in development (Filice et al., 2016), and a reduced number of parvalbumin interneurons is also observed in postmortem tissue from ASD patients (Hashemi et al., 2017). The frequency of spontaneous inhibitory synaptic activity is decreased in the mPFC in young Shank3Δe9–/– mice (P19–25), resulting in enhanced excitatory activity (Lee J. et al., 2015). The disruption in the balance between neuronal excitation and inhibition in the mPFC has been elucidated as one of the major cellular mechanisms contributing to social deficits in ASD model animals (Figure 1; Xu et al., 2019), and indeed social interaction deficits are prevalent in Shank ASD mice (Table 1). Moreover, increasing the excitability of parvalbumin interneurons in the mPFC has been shown to rescue social deficits (Selimbeyoglu et al., 2017; Cao et al., 2018).
Several mechanistic studies have now demonstrated downstream elements involved in the improvement of social deficits and/or synaptic dysfunction in the mPFC caused by Shank deficiency in young mice. This includes restoration of F-actin by inhibiting cofilin, resulting in reversal of NMDAR hypofunction and sociability deficits (Duffney et al., 2015). Other targets include histone deacetylase 2 (HDAC2) transcription, and the Shank3 gene itself, with HDAC inhibitors or re-expression of Shank3 alleviating social deficits (Qin et al., 2018; Lee et al., 2021). Restoration of Shank3 in the anterior cingulate cortex, which is the association cortices like PFC and its role is emphasised in sociability, also improved social deficits (Guo et al., 2019). Targetting pathways into the mPFC from the hippocampus with oxytocin treatment, which is required for the formation of social recognition memory, also attenuates the changes in synaptic plasticity as well as social behaviour deficits during development (Harony-Nicolas et al., 2017). Using designer receptors exclusively activated by designer drugs (DREADDs), chemogenic activation of pyramidal neurons in the mPFC can rescue NMDAR hypofunction and social deficits in a Shank3 haploinsufficiency model (Qin et al., 2019). These findings together identify future therapeutic strategies for ASD specifically in relation to PFC dysfunction and social deficits.
Other Brain Regions
While the majority of Shank-related ASD research has focussed on the hippocampus, cortex, and striatum, it is clear that other brain regions are also affected, reflecting the heterogeneous nature of ASD. The VTA modulates cortical and striatal networks and as such contributes to social deficits in ASD. Downregulation of SHANK3 specifically in the VTA during early development slows excitatory synapse maturation, reducing the excitability of dopaminergic neurons and altering excitation/inhibition balance (Bariselli et al., 2016). Potentiation of mGluR1 activity during the developmental period rescues dopaminergic neuron function and prevents social deficits into adulthood, and this effect can be mimicked by direct optogenetic activation of these neurons, highlighting the importance of VTA dopaminergic neuron activity in social behavioural deficits in ASD.
Other lesser studied brain regions affected by ASD-associated mutations in Shanks include the cerebellum and hypothalamus. The cerebellum is a site of pathology in ASD aetiology, with behavioural deficits in motor learning, motor coordination, and social interaction dependent on cerebellar changes. Lack of Shank2 reduces excitatory synapse density, levels of postsynaptic proteins, and reduced plasticity at Purkinje cell synapses (Figure 1; Ha et al., 2016; Peter et al., 2016). Excitation/inhibitory balance is also altered – with an increase in inhibitory input onto Purkinje cells observed. Restricting Shank2 deletion to the cerebellum results in enhanced repetitive and anxiety behaviours (Ha et al., 2016), linking ASD-associated mutations in Shank2 in the cerebellum with specific ASD behaviours. In human patients, brain imaging studies have revealed cerebellar vermis hypoplasia with Phelan–McDermid Syndrome, a neurodevelopmental disorder with ASD-like features that is associated with the loss of one copy of SHANK3 (22q13.3 deletion). However, this was not observed in all 22q13.3 patients, suggesting SHANK3 disruption is not causative for abnormal development of posterior fossa structures (Aldinger et al., 2013).
The hypothalamus is another brain region implicated in the development of ASD, especially in social behaviour and pup rearing deficits. Indeed lack of Shank2 results in lack of attachment and decreases social bonding between mother and pups as evidenced by offspring neglect (Grabrucker et al., 2021). Low levels of neuronal activation were observed in the medial preoptic region of the hypothalamus and the associated regions of these mice. Increasing activity in this region using DREADDs re-established social bonding in ASD mice, providing strong anatomical and physiological information on the biological origin of the development of this ASD deficit. Altered neurotransmitter systems are evident early during postnatal development in the hypothalamus, with neonatal mice lacking Shank3 showing decreases in GABAergic and glutamatergic markers, but increases in cholinergic, dopaminergic, and serotonergic markers that may underpin adult ASD phenotypes (Bukatova et al., 2021).
Beyond synaptic effects, electrophysiological studies in multiple brain regions have shown ASD-related Shank mutations also influence hyperpolarization-activated cyclic nucleotide-gated channel proteins (HCN proteins) that form Ih channels (Yi et al., 2016; Zhu et al., 2018). In the ventrobasal (VB) complex of the thalamus, VB thalamocortical relay neurons highly express HCN channels which enable switching between tonic and burst firing modes. These neurons also highly express SHANK3, and ASD-related SHANK3 mutations reduce Ih current and alter action potential firing (Zhu et al., 2018). SHANK3 mutations expressed in embryonic-stem cell derived human neurons also have been shown to specifically impair Ih channels (Yi et al., 2016). These SHANK-related inductions of Ih channelopathy influence essential neuronal properties such as resting membrane potential and action potential firing and show an important function of SHANKs in regulating electrical properties outside of the synapse that may underpin ASD-related cellular mechanisms.
The expression of SHANKs in the peripheral nervous system (PNS; Raab et al., 2010) raises the idea that some ASD phenotypes will include peripheral synaptic pathology, however, this is an understudied area. Shank2/3 expression has been described at the neuromuscular junction as well as at synapses of the superior cervical and myenteric ganglion (Raab et al., 2010). Growing evidence suggests that ASD-genetic mutations also alter peripheral mechanisms that contribute to ASD-associated sensory abnormalities including tactile hypersensitivity (Orefice et al., 2019; Schaffler et al., 2019). Peripheral GABAA receptor agonist treatment can reduce tactile over-reactivity, as well as improve anxiety behaviours, body condition, and some social impairments (Orefice et al., 2019). Interestingly, memory and motor impairments, and repetitive behaviours were not altered, strengthening the evidence that the PNS plays a strong role in a subset of ASD-associated behaviours.
Future Challenges
Multiple brain regions are clearly significantly affected in ASD from the synaptic to the network level (Figure 1). The resulting effects on neural activity potentially result in miswiring of the developing brain, altering both output and communication between these regions to collectively elicit the complex social behavioural deficits in ASD (Fourie et al., 2018; Han et al., 2020). Significant challenges exist for the development of therapeutics in ASD, including (1) the differential effects of ASD on neuronal structure and function across brain regions, even within specific genetic variants, (2) the differential effects of varying ASD-associated mutations, (3) fewer cellular studies examining ASD effects during early brain development, and (4) the different effects observed in neuronal function across developmental stages when these have been examined. Added to this is the often-unrecognised limitation that many studies perform functional experiments during the postnatal development but compare these to structural changes occurring in adult tissue. Together these temporal, functional, and location-specific differences will likely necessitate multiple therapeutic options that are specifically targetted to counterbalance differences in Shank mutation, affected brain region, and developmental time points.
Author Contributions
YV, JC, KL, YJ, and JM developed the writing plan and drafted the manuscript. YV and PC developed the figure. All authors approved the final manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Aldinger, K. A., Kogan, J., Kimonis, V., Fernandez, B., Horn, D., Klopocki, E., et al. (2013). Cerebellar and posterior fossa malformations in patients with autism-associated chromosome 22q13 terminal deletion. Am. J. Med. Genet. A 161A, 131–136. doi: 10.1002/ajmg.a.35700
Alexander, W. H., and Brown, J. W. (2011). Medial prefrontal cortex as an action-outcome predictor. Nat. Neurosci. 14, 1338–1344. doi: 10.1038/nn.2921
American Psychiatric Association (2013). American Psychiatric Association: Diagnostic and Statistical Manual of Mental Disorders, 5th Edn. Washington, DC: American Psychiatric Association.
Arons, M. H., Lee, K., Thynne, C. J., Kim, S. A., Schob, C., Kindler, S., et al. (2016). shank3 is part of a zinc-sensitive signaling system that regulates excitatory synaptic strength. J. Neurosci. 36, 9124–9134. doi: 10.1523/JNEUROSCI.0116-16.2016
Arons, M. H., Thynne, C. J., Grabrucker, A. M., Li, D., Schoen, M., Cheyne, J. E., et al. (2012). Autism-associated mutations in ProSAP2/Shank3 impair synaptic transmission and Neurexinf_neuroligin-mediated transsynaptic signaling. J. Neurosci. 32, 14966–14978. doi: 10.1523/JNEUROSCI.2215-12.2012
Bannerman, D. M., Sprengel, R., Sanderson, D. J., McHugh, S. B., Rawlins, J. N. P., Monyer, H., et al. (2014). Hippocampal synaptic plasticity, spatial memory and anxiety. Nat. Rev. Neurosci. 15, 181–192. doi: 10.1038/nrn3677
Bariselli, S., Tzanoulinou, S., Glangetas, C., Prévost-Solié, C., Pucci, L., Viguié, J., et al. (2016). SHANK3 controls maturation of social reward circuits in the VTA. Nat. Neurosci. 19, 926–934. doi: 10.1038/nn.4319
Berkel, S., Marshall, C. R., Weiss, B., Howe, J., Roeth, R., Moog, U., et al. (2010). Mutations in the SHANK2 synaptic scaffolding gene in autism spectrum disorder and mental retardation. Nat. Genet. 42, 489–491. doi: 10.1038/ng.589
Bey, A. L., Wang, X., Yan, H., Kim, N., Passman, R. L., Yang, Y., et al. (2018). Brain region-specific disruption of Shank3 in mice reveals a dissociation for cortical and striatal circuits in autism-related behaviors. Transl. Psychiatry 8:94. doi: 10.1038/s41398-018-0142-6
Boeckers, T. M., Bockmann, J., Kreutz, M. R., and Gundelfinger, E. D. (2002). ProSAP/Shank proteins - a family of higher order organizing molecules of the postsynaptic density with an emerging role in human neurological disease. J. Neurochem. 81, 903–910. doi: 10.1046/j.1471-4159.2002.00931.x
Boeckers, T. M., Kreutz, M. R., Winter, C., Zuschratter, W., Smalla, K.-H., Sanmarti-Vila, L., et al. (1999a). Proline-rich synapse-associated protein-1/cortactin binding protein 1 (ProSAP1/CortBP1) is a PDZ-domain protein highly enriched in the postsynaptic density. J. Neurosci. 19, 6506–6518. doi: 10.1523/jneurosci.19-15-06506.1999
Boeckers, T. M., Winter, C., Smalla, K.-H., Kreutz, M. R., Bockmann, J., Seidenbecher, C., et al. (1999b). Proline-rich synapse-associated proteins ProSAP1 and ProSAP2 interact with synaptic proteins of the SAPAP/GKAP family. Biochem. Biophys. Res. Commun. 264, 247–252. doi: 10.1006/bbrc.1999.1489
Boeckers, T. M., Liedtke, T., Spilker, C., Dresbach, T., Bockmann, J., Kreutz, M. R., et al. (2005). C-terminal synaptic targeting elements for postsynaptic density proteins ProSAP1/Shank2 and ProSAP2/Shank3. J. Neurochem. 92, 519–524. doi: 10.1111/j.1471-4159.2004.02910.x
Boeckers, T. M., Segger-Junius, M., Iglauer, P., Bockmann, J., Gundelfinger, E. D., Kreutz, M. R., et al. (2004). Differential expression and dendritic transcript localization of Shank family members: identification of a dendritic targeting element in the 3′ untranslated region of Shank1 mRNA. Mol. Cell. Neurosci. 26, 182–190. doi: 10.1016/j.mcn.2004.01.009
Bourgeron, T. (2009). A synaptic trek to autism. Curr. Opin. Neurobiol. 19, 231–234. doi: 10.1016/j.conb.2009.06.003
Bukatova, S., Renczes, E., Reichova, A., Filo, J., Sadlonova, A., Mravec, B., et al. (2021). Shank3 deficiency is associated with altered profile of neurotransmission markers in pups and adult mice. Neurochem. Res. 46, 3342–3355. doi: 10.1007/s11064-021-03435-6
Cao, W., Lin, S., Xia, Q.-Q., Du, Y.-L., Yang, Q., Zhang, M.-Y., et al. (2018). Gamma oscillation dysfunction in mPFC leads to social deficits in neuroligin 3 R451C knockin mice. Neuron 97, 1253–1260.
Chen, Q., Deister, C. A., Gao, X., Guo, B., Lynn-Jones, T., Chen, N., et al. (2020). Dysfunction of cortical GABAergic neurons leads to sensory hyper-reactivity in a Shank3 mouse model of ASD. Na. Neurosci. 23, 520–532. doi: 10.1038/s41593-020-0598-6
Chiesa, M., Nardou, R., Lozovaya, N., Eftekhari, S., Tyzio, R., Guimond, D., et al. (2019). Enhanced glutamatergic currents at birth in Shank3 KO Mice. Neural Plast. 2019:2382639. doi: 10.1155/2019/2382639
Chung, C., Ha, S., Kang, H., Lee, J., Um, S. M., Yan, H., et al. (2019). Early correction of N-Methyl-D-aspartate receptor function improves autistic-like social behaviors in adult Shank2–/– Mice. Biol. Psychiatry 85, 534–543. doi: 10.1016/j.biopsych.2018.09.025
Cooper, R. A., Richter, F. R., Bays, P. M., Plaisted-Grant, K. C., Baron-Cohen, S., and Simons, J. S. (2017). Reduced hippocampal functional connectivity during episodic memory retrieval in autism. Cereb. Cortex 27, 888–902. doi: 10.1093/cercor/bhw417
Duffney, L. J., Zhong, P., Wei, J., Matas, E., Cheng, J., Qin, L., et al. (2015). Autism-like deficits in shank3-deficient mice are rescued by targeting actin regulators. Cell Rep. 11, 1400–1413. doi: 10.1016/j.celrep.2015.04.064
Durand, C. M., Betancur, C., Boeckers, T. M., Bockmann, J., Chaste, P., Fauchereau, F., et al. (2007). Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat. Genet. 39, 25–27. doi: 10.1038/ng1933
Engineer, C. T., Rahebi, K. C., Borland, M. S., Buell, E. P., Im, K. W., Wilson, L. G., et al. (2018). Shank3-deficient rats exhibit degraded cortical responses to sound. Autism Res. 11, 59–68. doi: 10.1002/aur.1883
Filice, F., Vörckel, K. J., Sungur, A. Ö, Wöhr, M., and Schwaller, B. (2016). Reduction in parvalbumin expression not loss of the parvalbumin expressing GABA interneuron subpopulation. Mol. Brain 9:10. doi: 10.1186/s13041-016-0192-8
Fourie, C., Vyas, Y., Lee, K., Jung, Y., Garner, C. C., and Montgomery, J. M. (2018). Dietary zinc supplementation prevents autism related behaviors and striatal synaptic dysfunction in Shank3 Exon 13–16 mutant mice. Front. Cell. Neurosci. 12:374. doi: 10.3389/fncel.2018.00374
Gogolla, N., Takesian, A. E., Feng, G., Fagiolini, M., and Hensch, T. K. (2014). Sensory integration in mouse insular cortex reflects GABA circuit maturation. Neuron 83, 894–905. doi: 10.1016/j.neuron.2014.06.033
Grabrucker, A. M., Knight, M. J., Proepper, C., Bockmann, J., Joubert, M., Rowan, M., et al. (2011). Concerted action of zinc and ProSAP/Shank in synaptogenesis and synapse maturation. EMBO J. 30, 569–581. doi: 10.1038/emboj.2010.336
Grabrucker, S., Pagano, J., Schweizer, J., Urrutia-Ruiz, C., Schön, M., Thome, K., et al. (2021). Activation of the medial preoptic area (MPOA) ameliorates loss of maternal behavior in a Shank2 mouse model for autism. EMBO J. 40:e104267. doi: 10.15252/embj.2019104267
Guo, B., Chen, J., Chen, Q., Ren, K., Feng, D., Mao, H., et al. (2019). Anterior cingulate cortex dysfunction underlies social deficits in Shank3 mutant mice. Nat. Neurosci. 22, 1223–1234. doi: 10.1038/s41593-019-0445-9
Ha, S., Lee, D., Cho, Y. S., Chung, C., Yoo, Y. E., Kim, J., et al. (2016). Cerebellar shank2 regulates excitatory synapse density, motor coordination, and specific repetitive and anxiety-like behaviors. J. Neurosci. 36, 12129–12143. doi: 10.1523/JNEUROSCI.1849-16.2016
Habib, A., Harris, L., Pollick, F., and Melville, C. (2019). A meta-analysis of working memory in individuals with autism spectrum disorders. PLoS One 14:e0216198. doi: 10.1371/journal.pone.0216198
Han, K. A., Yoon, T. H., Shin, J., Um, J. W., and Ko, J. (2020). Differentially altered social dominance- and cooperative-like behaviors in Shank2- and Shank3-mutant mice. Mol. Autism 11:87. doi: 10.1186/s13229-020-00392-9
Harony-Nicolas, H., Kay, M., du Hoffmann, J., Klein, M. E., Bozdagi-Gunal, O., Riad, M., et al. (2017). Oxytocin improves behavioural and electrophysiological deficits in a novel Shank3 deficient rat. eLife 31:e18904. doi: 10.7554/eLife.18904
Hashemi, E., Ariza, J., Rogers, H., Noctor, S. C., and Martínez-Cerdeño, V. (2017). The number of parvalbumin expressing interneurons is decreased in the prefrontal cortex in autism. Cereb. Cortex 27, 1931–1943.
Hitti, F. L., and Siegelbaum, S. A. (2014). The hippocampal CA2 region is essential for social memory. Nature 508, 88–92. doi: 10.1038/nature13028
Hung, A. Y., Futai, K., Sala, C., Valtschanoff, J. G., Ryu, J., Woodworth, M. A., et al. (2008). Smaller dendritic spines, weaker synaptic transmission, but enhanced spatial learning in mice lacking Shank1. J. Neurosci. 28, 1697–1708. doi: 10.1523/jneurosci.3032-07.2008
Isaac, J. T. R., Nicoll, R. A., and Malenka, R. C. (1995). Evidence for silent synapses: implications for the expression of LTP. Neuron 15, 427–434. doi: 10.1016/0896-6273(95)90046-2
Jaramillo, T. C., Speed, H. E., Xuan, Z., Reimers, J. M., Liu, S., and Powell, C. M. (2016). Altered striatal synaptic function and abnormal behaviour in shank3 exon4-9 deletion mouse model of autism. Autism Res. 9, 350–375. doi: 10.1002/aur.1529
Jiang, Y.-H., and Ehlers, M. D. (2013). Modeling autism by SHANK gene mutations in mice. Neuron 78, 8–27. doi: 10.1016/j.neuron.2013.03.016
Kim, R., Kim, J., Chung, C., Ha, S., Lee, S., Lee, E., et al. (2018). Cell type-specific Shank2 deletion in mice leads to differential synaptic and behavioral phenotypes. J. Neurosci. 38, 2684–2617. doi: 10.1523/jneurosci.2684-17.2018
Kleijer, K. T., Schmeisser, M. J., Krueger, D. D., Boeckers, T. M., Scheiffele, P., Bourgeron, T., et al. (2014). Neurobiology of autism gene products: towards pathogenesis and drug targets. Psychopharmacology 231, 1037–1062. doi: 10.1007/s00213-013-3403-3
Kornau, H. C., Schenker, L. T., Kennedy M. B., and Seeburg, P. H. (1995). Domain interaction between NMDA receptor subunits and the postsynaptic density protein PSD-95. Science. 269, 1737–1740. doi: 10.1126/science.7569905
Kouser, M., Speed, H. E., Dewey, C. M., Reimers, J. M., Widman, A. J., Gupta, N., et al. (2013). Loss of predominant Shank3 isoforms results in hippocampus-dependent impairments in behavior and synaptic transmission. J. Neurosci. 33, 18448–18468. doi: 10.1523/jneurosci.3017-13.2013
Langen, M., Schnack, H. G., Nederveen, H., Bos, D., Lahuis, B. E., de Jonge, M. V., et al. (2009). Changes in the developmental trajectories of striatum in autism. Biol. Psychiatry 66, 327–333. doi: 10.1016/j.biopsych.2009.03.017
Leblond, C. S., Heinrich, J., Delorme, R., Proepper, C., Betancur, C., Huguet, G., et al. (2012). Genetic and functional analyses of SHANK2 mutations suggest a multiple hit model of autism spectrum disorders. PLoS Genet. 8:e1002521. doi: 10.1371/journal.pgen.1002521
Lee, D. K., Li, S. W., Bounni, F., Friedman, G., Jamali, M., Strahs, L., et al. (2021). Reduced sociability and social agency encoding in adult Shank3-mutant mice are restored through gene re-expression in real time. Nat. Neurosci. 24, 1243–1255. doi: 10.1038/s41593-021-00888-4
Lee, E.-J., Lee, H., Huang, T.-N., Chung, C., Shin, W., Kim, K., et al. (2015). Trans-synaptic zinc mobilization improves social interaction in two mouse models of autism through NMDAR activation. Nat. Commun. 6:7168. doi: 10.1038/ncomms8168
Lee, J., Chung, C., Ha, S., Lee, D., Kim, D.-Y., Kim, H., et al. (2015). Shank3-mutant mice lacking exon 9 show altered excitation/inhibition balance, enhanced rearing, and spatial memory deficit. Front. Cell. Neurosci. 9:94. doi: 10.3389/fncel.2015.00094
Lewis, M., and Kim, S.-J. (2009). The pathophysiology of restricted repetitive behavior. J. Neurodev. Disord. 1, 114–132.
Liao, D., Hessler, N. A., and Malinow, R. (1995). Activation of postsynaptically silent synapses during pairing-induced LTP in CA1 region of hippocampal slice. Nature 375, 400–404. doi: 10.1038/375400a0
Lim, C.-S., Kim, H., Yu, N.-K., Kang, S. J., Kim, T., Ko, H.-G., et al. (2017). Enhancing inhibitory synaptic function reverses spatial memory deficits in Shank2 mutant mice. Neuropharmacology 112, 104–112. doi: 10.1016/j.neuropharm.2016.08.016
Lim, S., Naisbitt, S., Yoon, J., Hwang, J.-I., Suh, P.-G., Sheng, M., et al. (1999). Characterization of the shank family of synaptic proteins multiple genes, alternative splicing, and differential expression in brain and development. J. Biol. Chem. 274, 29510–29518. doi: 10.1074/jbc.274.41.29510
Maenner, M. J. Shaw, K. A., Baio, J., Washington, A., Patrick, M., DiRienzo, M., et al. (2020). Prevalence of autism spectrum disorder among children aged 8 years — autism and developmental disabilities monitoring network, 11 Sites, United States, 2016. MMWR Surveill Summ 69, 1–12. doi: 10.15585/mmwr.ss6904a1
Mameza, M. G., Dvoretskova, E., Bamann, M., Hönck, H.-H., Güler, T., Boeckers, T. M., et al. (2013). SHANK3 gene mutations associated with autism facilitate ligand binding to the Shank3 ankyrin repeat region. J. Biol. Chem. 288, 26697–26708. doi: 10.1074/jbc.M112.424747
Mei, Y., Monteiro, P., Zhou, Y., Kim, J.-A., Gao, X., Fu, Z., et al. (2016). Adult restoration of Shank3 expression rescues selective autistic-like phenotypes. Nature 530, 481–484. doi: 10.1038/nature16971
Miller, E. K., and Cohen, J. D. (2001). An integrative theory of prefrontal cortex function. Annu. Rev. Neurosci. 24, 167–202. doi: 10.1146/annurev.neuro.24.1.167
Monteiro, P., and Feng, G. (2017). SHANK proteins: roles at the synapse and in autism spectrum disorder. Nat. Rev. 18, 147–157. doi: 10.1038/nrn.2016.183
Montgomery, J. M., Pavlidis, P., and Madison, D. V. (2001). Pair recordings reveal all-silent synaptic connections and the postsynaptic expression of long-term potentiation. Neuron 29, 691–701. doi: 10.1016/S0896-6273(01)00244-6
Naisbitt, S., Kim, E., Tu, J. C., Xiao, B., Sala, C., Valtschanoff, J., et al. (1999). Shank, a novel family of postsynaptic density proteins that binds to the NMDA receptor/PSD-95/GKAP complex and cortactin. Neuron 23, 569–582. doi: 10.1016/s0896-6273(00)80809-0
Orefice, L. L., Mosko, J. R., Morency, D. T., Wells, M. F., Tasnim, A., Mozeika, S. M., et al. (2019). Targeting peripheral somatosensory neurons to improve tactile-related phenotypes in ASD models. Cell 178, 867.e24–886.e24. doi: 10.1016/j.cell.2019.07.024
Orefice, L. L., Zimmerman, A. L., Chirila, A. M., Sleboda, S. J., Head, J. P., and Ginty, D. D. (2016). Peripheral mechanosensory neuron dysfunction underlies tactile and behavioral deficits in mouse models of ASDs. Cell 166, 299–313. doi: 10.1016/j.cell.2016.05.033
Pappas, A. L., Bey, A. L., Wang, X., Rossi, M., Kim, Y. H., Yan, H., et al. (2017). Deficiency of Shank2 causes mania-like behavior that responds to mood stabilizers. JCI Insight 2:e92052. doi: 10.1172/jci.insight.92052
Peça, J., Feliciano, C., Ting, J. T., Wang, W., Wells, M. F., Venkatraman, T. N., et al. (2011). Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature 472, 437–442. doi: 10.1038/nature09965
Peixoto, R. T., Wang, W., Croney, D. M., Kozorovitskiy, Y., and Sabatini, B. L. (2016). Early hyperactivity and precocious maturation of corticostriatal circuits in Shank3B(-/-) mice. Nat. Neurosci. 19, 716–724. doi: 10.1038/nn.4260
Peter, S., Ten Brinke, M. M., Stedehouder, J., Reinelt, C. M., Wu, B., Zhou, H., et al. (2016). Dysfunctional cerebellar purkinje cells contribute to autism-like behaviour in Shank2-deficient mice. Nat. Commun. 7:12627. doi: 10.1038/ncomms12627
Qin, L., Ma, K., Wang, Z.-J., Hu, Z., Matas, E., Wei, J., et al. (2018). Social deficits in Shank3-deficient mouse models of autism are rescued by histone deacetylase (HDAC) inhibition. Nat. Neurosci. 21, 564–575.
Qin, L., Ma, K., and Yan, Z. (2019). Chemogenetic activation of prefrontal cortex in Shank3-deficient mice ameliorates social deficits, NMDAR hypofunction, and Sgk2 downregulation. iScience 17, 24–35. doi: 10.1016/j.isci.2019.06.014
Raab, M., Boeckers, T. M., and Neuhuber, W. L. (2010). Proline-rich synapse-associated protein-1 and 2 (ProSAP1/Shank2 and ProSAP2/Shank3)-scaffolding proteins are also present in postsynaptic specializations of the peripheral nervous system. Neuroscience 171, 421–433. doi: 10.1016/j.neuroscience.2010.08.041
Rendall, A. R., Perrino, P. A., Buscarello, A. N., and Fitch, R. H. (2019). Shank3B mutant mice display pitch discrimination enhancements and learning deficits. Int. J. Dev. Neurosci. 72, 13–21. doi: 10.1016/j.ijdevneu.2018.10.003
Roussignol, G., Ango, F., Romorini, S., Tu, J. C., Sala, C., Worley, P. F., et al. (2005). Shank expression is sufficient to induce functional dendritic spine synapses in aspiny neurons. J. Neurosci. 25, 3560–3570. doi: 10.1523/JNEUROSCI.4354-04.2005
Rundles, R. W., and Papez, J. W. (1937). Connections between the striatum and the substantia nigra in a human brain. Arch. Neurol. Psychiatry 38, 550–563. doi: 10.1001/archneurpsyc.1937.02260210116008
Sala, C., Piëch, V., Wilson, N. R., Passafaro, M., Liu, G., and Sheng, M. (2001). Regulation of dendritic spine morphology and synaptic function by Shank and Homer. Neuron 31, 115–130. doi: 10.1016/s0896-6273(01)00339-7
Sato, D., Lionel, A. C., Leblond, C. S., Prasad, A., Pinto, D., Walker, S., et al. (2012). SHANK1 deletions in males with autism spectrum disorder. Am. J. Hum. Genet. 90, 879–887. doi: 10.1016/j.ajhg.2012.03.017
Schaffler, M. D., Middleton, L. J., and Abdus-Saboor, I. (2019). Mechanisms of tactile sensory phenotypes in autism: current understanding and future directions for research. Curr. Psychiatry Rep. 21:134. doi: 10.1007/s11920-019-1122-0
Schmeisser, M. J., Ey, E., Wegener, S., Bockmann, J., Stempel, A. V., Kuebler, A., et al. (2012). Autistic-like behaviours and hyperactivity in mice lacking ProSAP1/Shank2. Nature 486, 256–260. doi: 10.1038/nature11015
Selimbeyoglu, A., Kim, C. K., Inoue, M., Lee, S. Y., Hong, A. S. O., Kauvar, I., et al. (2017). Modulation of prefrontal cortex excitation/inhibition balance rescues social behavior in CNTNAP2-deficient mice. Sci. Trans. Med. 9:eaah6733. doi: 10.1126/scitranslmed.aah6733
Sheng, M., and Kim, E. (2000). The Shank family of scaffold proteins. J. Cell Sci. 113, 1851–1856. doi: 10.1242/jcs.113.11.1851
Shi, R., Redman, P., Ghose, D., Hwang, H., Liu, Y., Ren, X., et al. (2017). Shank proteins differentially regulate synaptic transmission. eNeuro 4:ENEURO.0163-15.2017. doi: 10.1523/eneuro.0163-15.2017
Skalny, A. V., Aschner, M., and Tinkov, A. A. (2021). Zinc. Adv. Food Nutr. Res. 96, 251–310. doi: 10.1016/bs.afnr.2021.01.003
Solomon, M., McCauley, J. B., Iosif, A.-M., Carter, C. S., and Ragland, J. D. (2016). Cognitive control and episodic memory in adolescents with autism spectrum disorders. Neuropsychologia 89, 31–41. doi: 10.1016/j.neuropsychologia.2016.05.013
Uemura, T., Mori, H., and Mishina, M. (2004). Direct interaction of GluRdelta2 with Shank scaffold proteins in cerebellar Purkinje cells. Mol. Cell. Neurosci. 26, 330–341. doi: 10.1016/j.mcn.2004.02.007
Vyas, Y., Lee, K., Jung, Y., and Montgomery, J. M. (2020). Influence of maternal zinc supplementation on the development of autism-associated behavioural and synaptic deficits in offspring Shank3-knockout mice. Mol. Brain 13:110. doi: 10.1186/s13041-020-00650-0
Wan, L., Liu, D., Xiao, W.-B., Zhang, B.-X., Yan, X.-X., Luo, Z.-H., et al. (2021). Association of SHANK family with neuropsychiatric disorders: an update on genetic and animal model discoveries. Cell Mol. Neurobiol. doi: 10.1007/s10571-021-01054-x [Epub ahead of print].
Wang, W., Li, C., Chen, Q., Goes, M.-S., van der Hawrot, J., Yao, A. Y., et al. (2017). Striatopallidal dysfunction underlies repetitive behavior in Shank3-deficient model of autism. J. Clin. Invest. 127, 1978–1990. doi: 10.1172/JCI87997
Wang, X., Bey, A. L., Katz, B. M., Badea, A., Kim, N., David, L. K., et al. (2016). Altered mGluR5-Homer scaffolds and corticostriatal connectivity in a Shank3 complete knockout model of autism. Nat. Commun. 7:11459. doi: 10.1038/ncomms11459
Wang, X., Xu, Q., Bey, A. L., Lee, Y., and Jiang, Y.-H. (2014). Transcriptional and functional complexity of Shank3 provides a molecular framework to understand the phenotypic heterogeneity of SHANK3 causing autism and Shank3 mutant mice. Mol. Autism 5:30. doi: 10.1186/2040-2392-5-30
Wegener, S., Buschler, A., Stempel, A. V., Kang, S. J., Lim, C.-S., Kaang, B.-K., et al. (2018). Defective synapse maturation and enhanced synaptic plasticity in shank2 Δex7 Mice. eNeuro 5:ENEURO.0398-17.2018. doi: 10.1523/eneuro.0398-17.2018
Won, H., Lee, H.-R., Gee, H. Y., Mah, W., Kim, J.-I., Lee, J., et al. (2012). Autistic-like social behaviour in Shank2-mutant mice improved by restoring NMDA receptor function. Nature 486, 261–265. doi: 10.1038/nature11208
Xu, P., Chen, A., Li, Y., Xing, X., and Lu, H. (2019). Medial prefrontal cortex in neurological disorder. Physiol. Genomics 51, 432–442.
Yao, I., Hata, Y., Hirao, K., Deguchi, M., Ide, N., Takeuchi, M., et al. (1999). Synamon, a novel neuronal protein interacting with synapse-associated protein 90/postsynaptic density-95-associated protein. J. Biol. Chem. 274, 27463–27466. doi: 10.1074/jbc.274.39.27463
Yi, F., Danko, T., Botelho, S. C., Patzke, C., Pak, C., Wernig, M., et al. (2016). Autism-associated SHANK3 haploinsufficiency causes Ih channelopathy in human neurons. Science 352:aaf2669. doi: 10.1126/science.aaf2669
Yoo, T., Cho, H., Lee, J., Park, H., Yoo, Y.-E., Yang, E., et al. (2018). GABA neuronal deletion of Shank3 Exons 14-16 in mice suppresses striatal excitatory synaptic input and induces social and locomotor abnormalities. Front. Cell Neurosci. 12:341. doi: 10.3389/fncel.2018.00341
Zemla, R., and Basu, J. (2017). Hippocampal function in rodents. Curr. Opin. Neurobiol. 43, 187–197. doi: 10.1016/j.conb.2017.04.005
Zhou, Y., Kaiser, T., Monteiro, P., Zhang, X., Van der Goes, M. S., Wang, D., et al. (2016). Mice with Shank3 mutations associated with ASD and schizophrenia display both shared and distinct defects. Neuron 89, 147–162. doi: 10.1016/j.neuron.2015.11.023
Zhu, M., Idikuda, V. K., Wang, J., Wei, F., Kumar, V., Shah, N., et al. (2018). Shank3-deficient thalamocortical neurons show HCN channelopathy and alterations in intrinsic electrical properties. J Physiol. 596, 1259–1276. doi: 10.1113/JP275147
Keywords: synapse, autism (ASD), shank, development, glutamate
Citation: Vyas Y, Cheyne JE, Lee K, Jung Y, Cheung PY and Montgomery JM (2021) Shankopathies in the Developing Brain in Autism Spectrum Disorders. Front. Neurosci. 15:775431. doi: 10.3389/fnins.2021.775431
Received: 14 September 2021; Accepted: 22 November 2021;
Published: 22 December 2021.
Edited by:
Tommas J. Ellender, University of Oxford, United KingdomReviewed by:
Rui Peixoto, University of Pittsburgh, United StatesJosé Vicente Negrete Díaz, University of Guanajuato, Mexico
Copyright © 2021 Vyas, Cheyne, Lee, Jung, Cheung and Montgomery. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Johanna M. Montgomery, am0ubW9udGdvbWVyeUBhdWNrbGFuZC5hYy5ueg==
†These authors have contributed equally to this work
†Present address: Yukti Vyas, INSERM, Neurocentre Magendie, U1215, Bordeaux, France