



- 1 Department of Pediatrics, Memorial Sloan-Kettering Cancer Center, New York, NY, USA
- 2 Medical College of Georgia, Augusta, GA, USA
- 3 Eureka Therapeutics, Inc., Emeryville, CA, USA
Targeted therapies require cellular protein expression that meets specific requirements that will maximize effectiveness, minimize off-target toxicities, and provide an opportunity for a therapeutic effect. The receptor tyrosine kinase-like orphan receptors (ROR) are possible targets for therapy that may meet such requirements. RORs are transmembrane proteins that are part of the receptor tyrosine kinase (RTK) family. The RORs have been shown to play a role in tumor-like behavior, such as cell migration and cell invasiveness and are normally not expressed in normal adult tissue. As part of the large effort in target discovery, ROR proteins have recently been found to be expressed in human cancers. Their unique expression profiles may provide a novel class of therapeutic targets for small molecules against the kinase or for antibody-based therapies against these receptors. Being restricted on tumor cells and not on most normal tissues, RORs are excellent targets for the treatment of minimal residual disease, the final hurdle in the curative approach to many cancers, including solid tumors such as neuroblastoma. In this review, we summarize the biology of RORs as they relate to human cancer, and highlight the therapeutic approaches directed toward them.
Introduction
The receptor tyrosine kinase-like orphan receptors (ROR) are transmembrane proteins that are part of the receptor tyrosine kinase (RTK) family. They are related to the Trk–RTK, muscle-specific kinase (MuSK), and neurotrophic tyrosine kinase (NTRTK) receptor families (Masiakowski and Carroll, 1992; Forrester et al., 1999). The ROR1 and ROR2 genes, first identified in a neuroblastoma cell line, are located on chromosomes 1 and 9, respectively, both encoding a 104-kDa proteins (Reddy et al., 1997). Homozygous mutations in ROR2 have been shown to be responsible for the Robinow syndrome, a skeletal dysplasia syndrome characterized by generalized limb shortening, segmental defects of the spine, and dysmorphic facial appearance, while heterozygous mutations have been found in patients with dominant brachydactyly B1, characterized by terminal deficiency of fingers and toes (Afzal et al., 2000; Afzal and Jeffery, 2003). The RORs are made up of an extracellular domain consisting of an immunoglobulin-like motif, a cysteine-rich frizzled domain, a kringle domain, and an intracellular tyrosine kinase domain (Figure 1). They were named orphan receptors because their endogenous ligands had not yet been discovered. However, recent studies have shown that the frizzle-domain of ROR2 associates with the Wnt5a protein and is involved with the non-canonical Wnt pathways (McDonald and Silver, 2009), but the definitive ligand for ROR1 is yet to be determined. The RORs have been shown to play a role in establishing cellular polarity (Green et al., 2008) and in tumor-like behavior, such as cell migration and cell invasiveness (Morioka et al., 2009). These receptors are normally expressed at high levels during development, playing a key role in skeletal and neural organogenesis, but then become repressed in adult tissues (Al-Shawi et al., 2001). Interestingly, ROR1 is upregulated in B-cell chronic lymphocytic leukemia (B-CLL; Baskar et al., 2008), B-cell acute lymphocytic leukemia (B-ALL; Shabani et al., 2008), and mantle cell leukemia (MCL; Hudecek et al., 2010), while ROR2 is overexpressed in osteosarcoma (OS; Morioka et al., 2009), and renal cell carcinoma (RCC; Wright et al., 2009). This unique expression profile may provide a novel family of therapeutic targets for small molecules against the kinase or for antibody-based therapies against these receptors (Hudecek et al., 2010). In this review, we summarize the biology of RORs as they relate to human cancer, and highlight the therapeutic approaches directed toward them.
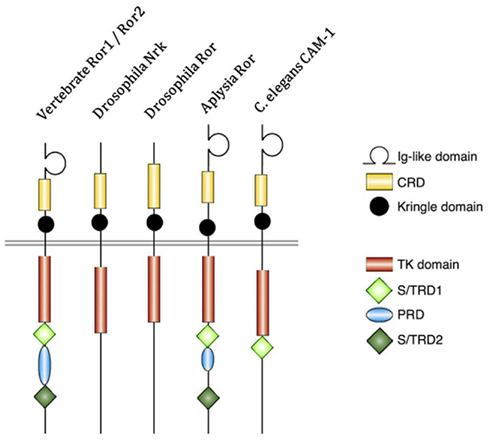
Figure 1. Structure of receptor tyrosine kinase-like orphan receptor (ROR) in different species. Type 1 receptor tyrosine kinase evolutionarily conserved, co-receptor with Frizzled-2/4, with immunoglobulin (Ig) domain, cysteine-rich domain (CRD), and Kringle domain. The intracellular portion contains tyrosine kinase (TK) domain, proline-rich domain (PRD) flanked by Ser/Thr rich domains (S/TRD1 and 2; Green et al., 2008; Minami et al., 2010).
Receptor Properties of ROR Proteins
Receptor tyrosine kinase-like orphan receptors are membrane proteins composed of three extracellular domains, one transmembrane domain, and one intracellular domain. Comparative genomic studies have found that the three extracellular domains and the one intracellular domain are conserved from drosophila and Caenorhabditis elegans to mice to humans (Figure 1; Katoh, 2005). Two different splice variants have been identified for ROR1, one lacking all the extracellular domains known as truncated-ROR1 (t-ROR1) and one lacking both the intracellular domain and transmembrane domains (Reddy et al., 1996). So far most studies have focused on the full length form of ROR1. Some reports have claimed that the intracellular tyrosine kinase domain lacks biological activity (Gentile et al., 2011), while others suggest its critical role in signal transduction to downstream proteins (Mikels et al., 2009). The kinase activity of ROR2, on the other hand, has been well-established (Kani et al., 2004; Yamamoto et al., 2007; Liu et al., 2008). We will discuss ROR2 kinase activity in more details later.
Intriguingly, vertebrate ROR proteins seem to have acquired additional cytosolic domains important for downstream signaling. In addition to the tyrosine kinase domain, vertebrate RORs contain a serine/threonine-rich domain (S/TRD1), a proline-rich domain (PRD), and an additional serine/threonine-rich domain (S/TRD2; Minami et al., 2010). Current studies have mainly focused on the extracellular cysteine-rich domain (CRD) in Frizzle since this domain has been shown to bind Wnt ligands for other cell surface receptors (Rehn et al., 1998). Studies in drosophila and mice have identified Wnt5a to be a ligand for ROR2 by comparing expression levels and loss-of-function phenotypes between ROR2 and Wnt5a homologs (Oishi et al., 2003; Green et al., 2007). Co-expression and co-immunoprecipitation studies have shown that Wnt5b could also bind to ROR2 in osteosarcoma cells (Morioka et al., 2009). Yet, the definitive ligand for ROR1 is still uncertain. The properties of the immunoglobulin domain and kringle extracellular domains have not been well characterized; thus, the understanding of the key biologic function of ROR proteins remains incomplete.
Mikels et al. (2009) has shown that ROR2 and its ligand, Wnt5a might be involved in the non-canonical Wnt pathway. In vivo studies in mice have shown that when mROR2 or Wnt5a expression is knocked down, Wnt/β-catenin signaling is enhanced, consistent with ROR2’s function as an inhibitor of canonical Wnt pathways. Furthermore, the CRD, immunoglobulin-like extracellular domains, and intracellular tyrosine kinase domain all seem necessary for inhibition to occur, since truncated forms of ROR2 lose their inhibitory function. The canonical pathway could also be inhibited through the Wnt/calcium pathway via CamKII (Ishitani et al., 2003). When Wnt5a binds to ROR2, CamKII is activated, and through the mitogen-activated protein kinase (MAPK) pathway, negatively regulates the canonical Wnt/β-catenin signaling. These inhibitory pathways against canonical signaling may function as fail-safes to prevent aberrant β-catenin-induced gene expression from cancer-promoting activity. This tumor-suppressing effect has been proposed for high risk neuroblastoma since Wnt5a is down-regulated in neuroblastoma cell lines (Blanc et al., 2005).
Expression Pattern during Normal Development
Receptor tyrosine kinase-like orphan receptors play a significant role in embryonic development. Studies of ROR orthologs found in C. elegans, Drosophila melanogaster, Xenopus, and mice have mapped the expression profiles of RORs during development demonstrating a striking conservation during evolution to human (Wilson et al., 1993; Yoda et al., 2003). When the C. elegans ROR ortholog, CAM-1, was mutated, normal migration of canal-associated neurons was disrupted, suggesting a critical role of RORs in neuronal development. In mice, mRor1 and mRor2 are highly expressed during the early stages of development, represented in most of the major systems in tissues derived from all three germ layers, but most prominently seen in neural crest cells and their derivatives, such as special sense organs (Al-Shawi et al., 2001). While mRor2 is broadly expressed in the developing nervous system, mRor1 is restricted primarily to the head mesenchyme during fetal development (Oishi et al., 1999; Al-Shawi et al., 2001). In earlier report, knockout phenotype could not be studied since mRor1−/− mice do not survive beyond 24 h of life, dying from respiratory dysfunction, highly suggestive of the role of ROR1 in normal lung development (Al-Shawi et al., 2001; Nomi et al., 2001). A recent study showed that ROR1−/− mutants could survive, but with growth retardation. These mutant mice had multiple skeletal defects and urogenital abnormalities, resulting in reduced life expectancy and female infertility (Lyashenko et al., 2010). Ror2-deficient mice are viable, but have well-defined skeletal and heart abnormalities, modulated to some extent by mRor1 (Nomi et al., 2001). Although RORs are critical during normal development, they become undetectable in normal adult tissues, including humans. ROR1 is not expressed in mature B-cells, T-cells, monocytes, and natural killer cells; but transient expression was found at intermediate stage of normal B-cell development and the earliest B-cell precursors in the bone marrow. ROR1 was expressed in undifferentiated ES cells and expression was down-regulated upon differentiation in conditioned medium (Hudecek et al., 2010).
Aberrant Expression of RORs in Cancer
Although expression of RORs is embryonal, limited to the normal stages of development, their aberrant expression in certain cancers is of interest. As an oncofetal antigen, high levels of ROR1 expression have been found as mRNA and as protein in B-CLL (Baskar et al., 2008; Daneshmanesh et al., 2008; Fukuda et al., 2008), while totally absent in healthy donor peripheral blood mononuclear cells (PBMC) and all other normal tissues except for tonsillar B-cells (Daneshmanesh et al., 2008). ROR1 is preferentially expressed over ROR2 in B-CLL cells. Furthermore, it is constitutively expressed on B-CLL cells despite B-cell activation via CD40L and IL-4. The pattern of expression did not differ between IgVH mutated and unmutated B-CLL cases (Daneshmanesh et al., 2008). In addition to B-CLL, ROR1 was also upregulated at the mRNA level in B-ALL (Shabani et al., 2008). ROR1 was only overexpressed in mature leukemic B-cells but not in immature forms (Shabani et al., 2008). IL-6 could regulate ROR1 expression through Stat3 in CLL cells (Li et al., 2010). ROR1 was extensively modified by N-linked glycosylation (Kaucka et al., 2011), producing variants with electrophoretic migration of approximately 100, 115, and 130 kDa. Inhibition of glycosylation interfered with cell surface localization of the 130-kDa variant of ROR1 and prevented Ror1-induced formation of filopodia. The 130-kDa form was mono-ubiquitinated. Due to various glycosylation patterns, ROR1 from individual CLL patients showed striking differences in the electrophoretic mobility.
Among solid tumors, ROR1 had increased expression in gastric carcinoma and non-small cell carcinoma cell lines (Gentile et al., 2011). Here ROR1 functions as a pseudokinase crucial for Met-driven tumorigenesis. On the other hand, ROR2 was upregulated in osteosarcoma and RCC cell lines, as well as primary tumors, while not on normal tissues except for thyroid and stomach (Morioka et al., 2009; Wright et al., 2009). In sum, this differential expression of RORs among human cancers and the low to absent expression in normal tissues support their potential as therapeutic targets both in leukemia and in select solid tumors. In neuroblastoma, ROR1 and ROR2 were expressed in all tumor stages. Among patients, high levels of ROR1 or ROR2 gene expression correlated significantly with poorer survival (Asgharzadeh et al., 2006).
Oncogenic Properties of RORs
Although in neuroblastoma, Wnt5a/ROR signaling was hypothesized to have tumor-suppressing effects, several studies using other human cancers have shown the complete opposite, in which Wnt/ROR signaling induces tumor proliferation and tumor invasiveness. The ROR2/Wnt5a pathway has been shown to involve signaling through Wnt-c-Jun N-terminal kinase (Jnk) via activation of the actin binding protein, filamin A, which binds to the PRD intracellular domain in vertebrates (Oishi et al., 2003; Nomachi et al., 2008). Through activation of this pathway, mice fibroblasts could be induced to undergo cellular migration and invasion, features pathognomonic of malignancy. siRNA studies of osteosarcoma cells have shown a strong correlation between ROR2 knockdown and decreased cell migration in transwell assays (Enomoto et al., 2009). Furthermore, introduction of ROR2 into human fibroblast and kidney cells greatly enhanced the invasive activity of these cells. When knocked down by siRNA, the viability and survival of these cells decreased (Morioka et al., 2009). This effect of gene knockdown was also seen in RCC cells, where the suppression of ROR2 expression not only decreased the migration of RCC cells, but also inhibited anchorage-independent growth in soft agar and growth in an orthotopic xenograft mouse model (Wright et al., 2009). Furthermore, ROR2 knockdown by siRNA inhibited invasion of melanoma cells in a murine in vivo tumor model (O’Connell et al., 2010). All these studies implies ROR2 role in tumorigenesis and progression.
ROR1 was reported to be upregulated in B-CLL. Studies conducted by Choudhury et al. (2010) showed that silencing of ROR1 with siRNA results in apoptosis of primary B-CLL cells from patients, but not PBMC from healthy donors. It was found that ROR1 is constitutively phosphorylated in the B-CLL cells of al patients tested. Using a siRNA screening method, ROR1 was identified as a key factor for survival of t(1:19) acute lymphoblastic leukemia (ALL) primary cells and cell lines in vitro (Bicocca et al., 2011). Interestingly, ROR1 was elevated only in the t(1:19) ALL and not other ALL subtypes. It remains to be investigated whether ROR1 is phosphorylated in t(1:19) ALL cells. In a transgenic mice model, studies suggested that ROR1 can accelerate the development of CLL. In these studies, transgenic mice expressing human ROR1 under control of a B-cell specific promoter developed hROR1+/CD5+/B220 low B-cell leukemia resembling human CLL at 15–18 months of age. These human ROR1 transgenic mice were further crossed with TCL1 transgenic mice, which at 7 months of age develop CD5 + B220 low leukemia B-cells. The F1 mice with both transgenes (hROR1 × TCL1) developed hROR1+/CD5+/B220 low leukemia B-cell CLL at a significantly younger median age than did littermate-control mice having either transgene alone (Cui et al., 2011).
To further explore the invasive character of these cells upon ROR activation, invadopodia, and extracellular matrix (ECM) remodeling proteins, such as matrix metalloproteinases (MMPs), were studied to see if these components were co-expressed with ROR. In order for cells to migrate, the ECM has to be remodeled to allow for the cell to migrate through the extracellular milieu. Invadopodia are actin-rich protrusions that degrade and extend into the ECM, remodeling the ECM for tumor migration and invasion (Bowden et al., 1999), while MMPs are enzymes that degrade collagen and other components of the ECM. Through immunostaining and ECM degrading assays, ROR2 was seen to be concentrated in regions where invadopodia were located, as determined by the location of cortactin, an essential component of invadopodia that is expressed in areas where the ECM has been degraded (Enomoto et al., 2009). Furthermore, a comparison between several MMPs and ROR2 expression in osteosarcoma cells was studied and found that when ROR2 was silenced via transfection of siROR2, MMP13, a collagenase, was also decreased. The same effect was seen when siWnt5a was introduced into OS cells, suggesting that MMP13 is activated through Wnt5a/ROR2 signaling (Enomoto et al., 2009; Minami et al., 2010). Staining of invadopodia for MMP13 also indicated its presence in this ECM remodeling complex. Closer analysis of the signaling pathway indicated that the tyrosine kinase domain of ROR2 was essential for MMP13 activation and subsequent ECM remodeling and that Src-family protein tyrosine kinases (SFKs), a series of kinases shown to play an essential role in invadopodia production (Weaver, 2006), were shown to be activated through Wnt5a/ROR2 signaling. This then suggested that Wnt5a/ROR2 signaling activated a SFK, leading to subsequent activation of MMP13, overall leading to ECM remodeling to allow for tumor migration and invasion. A similar finding was found in RCC cells, in which ROR2 regulated MMP2, shown to be overexpressed in many metastatic cancers (Kurban et al., 2006) and play a important role in the renal tubular cell epithelial to mesenchymal transformation during wound healing, leading to ECM remodeling and subsequent invasive growth (Wright et al., 2009).
Therapeutic Approach Against ROR
Small Molecule Inhibitors
Given their restricted tissue distribution and implication in cancer, RORs are potential targets for cancer therapy (see Table 1). The TK domain of RORs is homologous to the discoidin-like domain receptor (Ddr), muscle-specific kinase (MuSK), and most of all, to the tropomyosin receptor kinase (Trk) family of receptors (Masiakowski and Carroll, 1992; Green et al., 2008). These receptors recognize the Src homology domain 2 (SH2) on phosphotyrosine substrates that constitute phosphorylation sites that lead to downstream transduction of the signal (Sadowski et al., 1986). Furthermore, SH3 substrates recognize PRDs, which are found in vertebrate ROR proteins. Since these domains are very specific for interaction with the phosphorylated portions of the TK domain, blocking their activity could be an effective way to truncate the signal transduction responsible for oncogenesis (Vidal et al., 2001). However, recent work (Gentile et al., 2011) on the tyrosine kinase domain of ROR1 showed that ROR1 is a pseudokinase. Overexpression of ROR1 failed to lead to kinase autophosphorylation, nor was it able to phosphorylate exogenous substrates. Moreover, the intracellular kinase domain of ROR1 contains amino acid substitutions in positions known to be critical for the enzymatic function of tyrosine kinase. On the contrary, constitutively activated MET receptor tyrosine kinase leads to ROR1 phosphorylation in gastric carcinoma cells (HS746T) and non-small cell lung carcinoma cells (NCI-H1993). Knockdown of ROR1 by siRNA in these two cell lines induces apoptosis in vitro and inhibits tumor growth in mouse tumor models. Noticeably, ROR1 knockdown has no effect on cell survival in cancer cells where ROR1 is overexpressed but not phosphorylated.
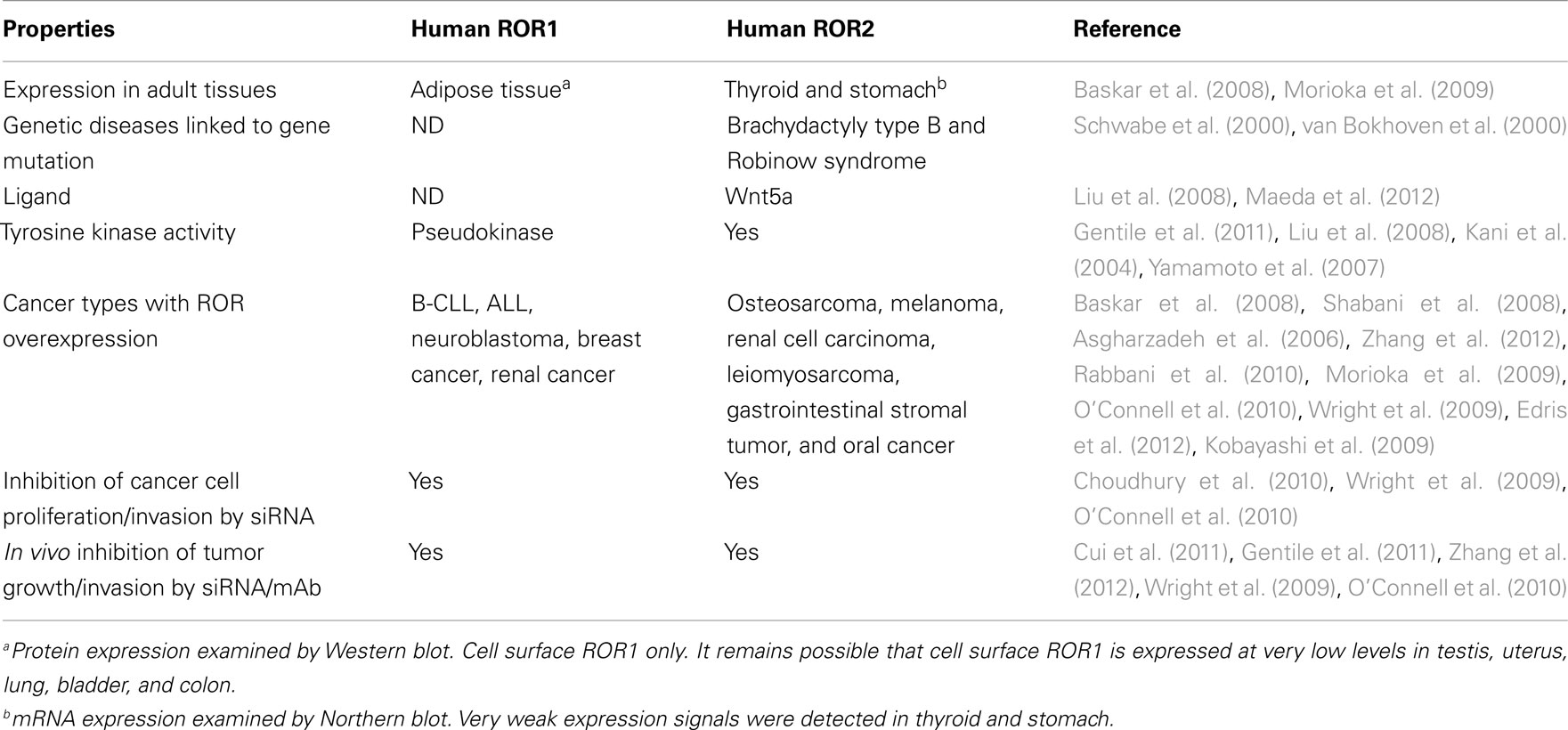
Table 1. Summary of evidence for human ROR1/2 as cancer therapeutic targets.
It was reported that in t(1:19) ALL cells, combination of ROR1 knockdown and a kinase inhibitor, dasatinib, resulted in additive impairment of cell viability compared with dasatinib alone, implying that therapies blocking ROR1 function might sensitize cancer cells to small molecule kinase inhibitors. Dasatinib was known to inhibit the kinase activities of Bruton’s tyrosine kinase (BTK) and tyrosine-protein kinase Lyn, which in turn leads to downregulating activity of AKT. In support of the hypothesis that ROR1 also serve to regulate AKT activity, ROR1 knockdown results in AKT inhibition in t(1:19) ALL cells (Bicocca et al., 2011).
The kinase activity of ROR2 is better understood. Using HEK293 transient expression system as a model system, Kani et al. has shown that ROR2 is phosphorylated by casein kinase Iε, a crucial regulator of the canonical Wnt signaling. Casein kinase Iε phosphorylates ROR2 on serine/threonine residues, which in turn resulted in autophosphorylation of ROR2 on tyrosine residues. Moreover, phosphorylated ROR2 might further phosphorylate G protein-coupled receptor kinase 2 (Kani et al., 2004). It was later shown in NIH3T3 and HeLa-S3 cells that simulation with Wnt5a results in ROR2 phosphorylation on serine/threonine residues. And this phosphorylation is mediated by GSK-3α (and GSK-3β; Yamamoto et al., 2007). Interestingly, Liu et al. (2008) found in human U2OS osteosarcoma cells that Wnt5a was able to induce homodimerization of ROR2 and autophosphorylation of its tyrosine residues without phosphorylation of serine/threonine residues. However, despite the strong evidence of ROR2 involvement in tumorigenesis and progress, there are no published reports on small molecule inhibitors against ROR2.
Antibody-Based Targeted Therapy
As surface receptors, RORs are potential targets for monoclonal antibodies (MoAb). Among non-hematopoietic tissues, only adipose tissue express cell surface ROR1 (Baskar et al., 2008). The restricted pattern of expression among normal tissues makes ROR1 an attractive target for MoAbs. High affinity MoAb have been derived from a chimeric rabbit/human Fab library and applied to the CLL and MCL lymphoma preclinical models (Yang et al., 2011). These antibodies have rabbit variable domains and human constant domains. Affinities of the Fab by BIACORE ranged from 0.56 to 8.8 nM. As full IgG1, the virtual affinities were 0.04–0.7 nM. Antibodies with the highest affinity and slowest rate of internalization could mediate antibody-dependent cell-mediated cytotoxicity (ADCC), but not complement mediated cytotoxicity (CDC). Using lymphoma cell line as target, ADCC was observed only for the high affinity antibody at 5 μg/ml, substantially weaker than anti-CD20 antibody rituximab. Antibody internalization appeared slower with the high affinity antibody. None of the antibodies induced apoptosis in lymphoma cells. The advantage of these antibodies compared to other therapeutic B-cell specific MoAbs is the absence of ROR1 in mature B-cells (Hudecek et al., 2010). However, given the relatively low cell surface density, ROR1 was proposed as a preferred target for armed rather than naked MoAb (Yang et al., 2011). Since ROR1 mediates partial internalization of bound antibody by endocytosis, it also provides a means to deliver cytotoxic agents into tumor cells (Baskar et al., 2008).
Other efforts, however, identified ROR1 MoAbs that are cytotoxic to hROR1-expressing leukemia cells (Cui et al., 2011; Daneshmanesh et al., 2012). In one study, over 70 unique mAbs against human ROR1 extracellular domain were generated through immunizing mice with various formats of full length hROR1–ECD peptides. Most mAbs recognize epitopes within the ROR1 Ig-like domain; others recognize epitopes within the kringle domain. One mAb binds to a unique epitope between the Ig and CRD domain, and is directly cytotoxic to hROR1-expressing leukemia cells in vitro. This mAb can also significantly decrease the basal levels of phosphorylated AKT in primary CLL cells and hROR1-expressing leukemic cells. Moreover, treatment of this mAb significantly decreased the number of primary CLL cells transferred into the peritoneal cavity of NSG mice, and inhibited CLL cell engraftment in the hROR1 × TCL1 double transgenic adoptive transfer model mentioned previously in the review. CDC activity against primary CLL cells and hROR1-expressing cancer cell lines was also observed for this mAb. In the other study, Mellstedt’ lab has generated five mouse by immunizing 15- to 23-amino-acid-long peptides derived from Ig domain, CRD domain, and kringle domain of human ROR1. All the five mAbs induce apoptosis of primary B-CLL cells but not of normal B-cells. Most effective were two mAbs against CRD and kringle domains, respectively. They showed significant superiority to rituximab. Two of the mAbs induced CDC similar to that of rituximab, and one mAb against the kringle domain showed ADCC activity against ROR1-expressing cancer cells. The discrepancy among the three studies might be explained by the different binding epitope of unique hROR1-specific MoAbs. Table 2 summarizes reported studies of ROR1 MoAbs.
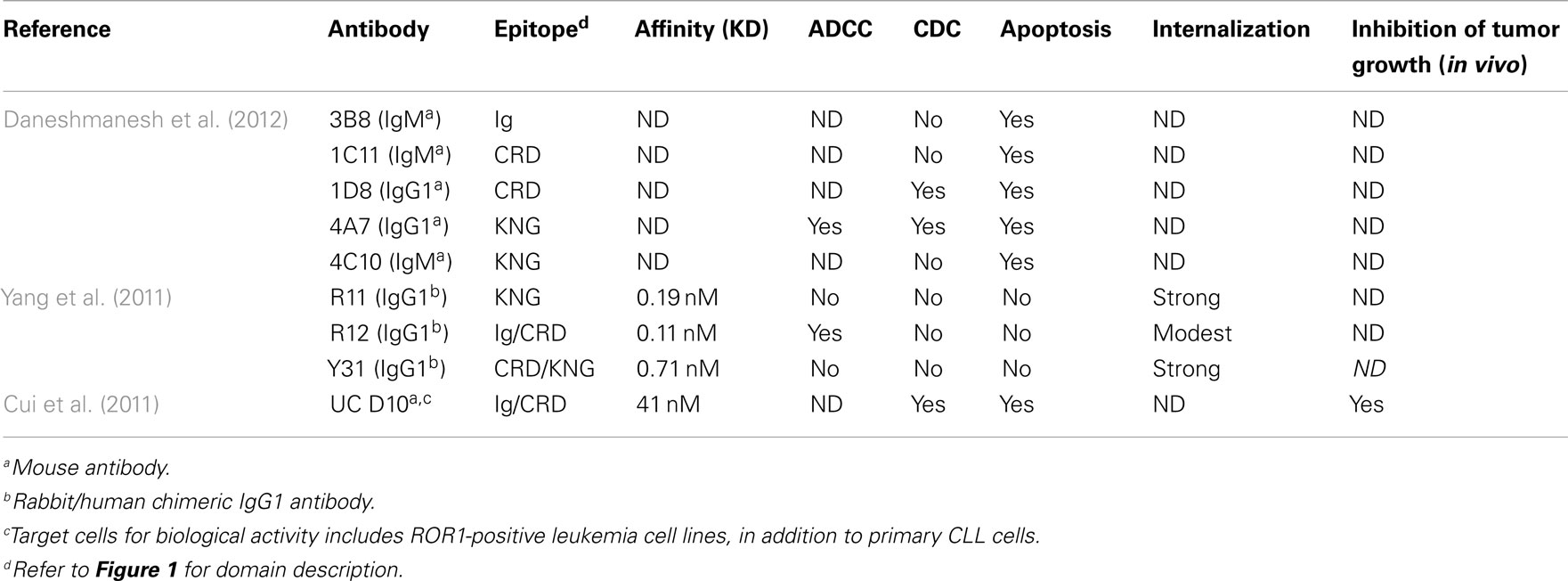
Table 2. Biological activity of ROR1-specific MoAbs against human primary chronic lymphocytic leukemia (CLL).
Another approach to overcome the low receptor density of ROR1 was to exploit T-cells. By genetic modification, T-cells can be made to express a chimeric antigen receptor (CAR) specific for ROR1, retargeting T-cell-mediated cytotoxicity against tumor cells (Hudecek et al., 2010). Since these CD8+ modified T-cells are efficient killers, probably requiring few target antigens, and no longer restricted to HLA or HLA type, this approach can potentially tackle three significant issues simultaneously, i.e., low target density, low HLA expression, and HLA type restriction of natural T cell epitopes. ROR1–CAR transduced T-cells efficiently lysed primary B-CLL, primary MCL, and ROR1-positive tumor cell lines including the rare subset of CLL cells that efflux chemotherapy, but not normal resting or activated B-cells. These T-cells also produced effector cytokines including TNF-α, IFN-γ, and IL-2, and were capable of proliferating in response to ROR1-expressing tumor cells (Hudecek et al., 2010). Whether the T-cells were derived from normal volunteers or from CLL patients, CAR-modification made them equally effective against ROR1-positive targets. Another obvious alternative may be the use of bispecific antibodies such as Bispecific T-cell engaging antibodies (BiTE; Bargou et al., 2008; Choi et al., 2011). However, for monovalent constructs, high affinity ROR-specific antibodies may be needed.
Future Directions
As part of the large effort in target discovery, ROR proteins have recently been characterized as unique markers for several cancers. Being restricted on tumor cells and not on most normal tissues, RORs are excellent targets for the treatment of minimal residual disease, the final hurdle in the curative approach to many cancers, including solid tumors such as neuroblastoma. Currently, ROR1 is used as a diagnostic tool for CLL (Uhrmacher et al., 2011), and is likely a critical receptor in the evolution of other non-Hodgkin lymphomas, such as MCL and marginal zone lymphoma (Hudecek et al., 2010; Barna et al., 2011). Most attention has been focused on ROR1 for targeted measures (Gentile et al., 2011), however ROR2 remains a viable target candidate. A deeper understanding of the signaling pathway of both ROR1 and ROR2 will go a long way in designing rational therapy. Clarity on the duality of Wnt5a signaling as a tumor-suppressor and an oncogenic protein is needed, with full characterization of the cellular environment and conditions that promote either fate. Several reports point to the receptor profile on the cell surface as a marker to determine which Wnt signal dictates the cell, showing that Wnt5a could induce clathrin-mediated internalization when bound to a Frizzle2-containing receptor, suppressing the ability of Wnt3a, also a putative ligand for ROR2 that leads to canonical β-catenin accumulation, to bind to ROR2. This establishes a dose-dependent environment for whichever Wnt ligand can first bind to ROR2 and dictates the signaling pathway (Liu et al., 2008; Sato et al., 2010). There is further suggestion that the presence of the Frizzle4-containing receptor could also bind Wnt5a and induce the canonical β-catenin-dependent pathway, suggesting that Wnt signaling is dictated by receptor availability as opposed to the Wnt ligands themselves (Mikels and Nusse, 2006). This interesting dichotomy could provide new insight into the mechanism behind tumor metastasis and invasion via Wnt/ROR signaling. Another issue that needs to be further resolved is to determine the number of ROR molecules present on the cell surface in order for future antibody therapies to be effective. For efficient antibody-dependent cell-mediated cytotoxicity (ADCC) and complement dependent cytotoxicity (CDC), there needs to be enough molecules of the antigen present on the cell surface to actively engage these cellular processes. In B-CLL cells and neuroblastoma, the antigen density was on the lower end around 103–104 molecules per cell, thereby limiting the capabilities of ADCC and CDC (Baskar et al., 2008; Yang et al., 2011). On the other hand, the antigen density needed for NK or T-cell-mediated cytotoxicities could be much less stringent (Tassev et al., 2012). Strategies to increase RORs expression on tumor cells using cytokines may be a possible solution. Alternatively, highly potent toxin immunoconjugates, radioimmunoconjugates, bispecific antibodies, or CAR-retargeted T-cells or NK cells could overcome these antigen density limitations.
Conflict of Interest Statement
Dr. Cheng Liu is founder and CEO of Eureka Therapeutics, Inc., which owns intellectual property and interests related to ROR1/ROR2 antibodies. Eureka Therapeutics, Inc., could profit financially from successful development of ROR1/ROR2 targeted therapies. Dr. Nai-Kong Cheung has a contractual research relationship with Eureka Therapeutics, Inc.; he owns no rights and has no commercial interest in ROR1 or ROR2 antibodies.
Acknowledgments
We would like to thank Joseph Olechnowicz for editing this manuscript and Chelsea Chang for Figure 1 drawing.
References
Afzal, A. R., and Jeffery, S. (2003). One gene, two phenotypes: ROR2 mutations in autosomal recessive Robinow syndrome and autosomal dominant brachydactyly type B. Hum. Mutat. 22, 1–11.
Afzal, A. R., Rajab, A., Fenske, C. D., Oldridge, M., Elanko, N., Ternes-Pereira, E., Tuysuz, B., Murday, V. A., Patton, M. A., Wilkie, A. O., and Jeffery, S. (2000). Recessive Robinow syndrome, allelic to dominant brachydactyly type B, is caused by mutation of ROR2. Nat. Genet. 25, 419–422.
Al-Shawi, R., Ashton, S. V., Underwood, C., and Simons, J. P. (2001). Expression of the Ror1 and Ror2 receptor tyrosine kinase genes during mouse development. Dev. Genes Evol. 211, 161–171.
Asgharzadeh, S., Pique-Regi, R., Sposto, R., Wang, H., Yang, Y., Shimada, H., Matthay, K., Buckley, J., Ortega, A., and Seeger, R. C. (2006). Prognostic significance of gene expression profiles of metastatic neuroblastomas lacking MYCN gene amplification. J. Natl. Cancer Inst. 98, 1193–1203.
Bargou, R., Leo, E., Zugmaier, G., Klinger, M., Goebeler, M., Knop, S., Noppeney, R., Viardot, A., Hess, G., Schuler, M., Einsele, H., Brandl, C., Wolf, A., Kirchinger, P., Klappers, P., Schmidt, M., Riethmuller, G., Reinhardt, C., Baeuerle, P. A., and Kufer, P. (2008). Tumor regression in cancer patients by very low doses of a T cell-engaging antibody. Science 321, 974–977.
Barna, G., Mihalik, R., Timar, B., Tombol, J., Csende, Z., Sebestyen, A., Bodor, C., Csernus, B., Reiniger, L., Petak, I., and Matolcsy, A. (2011). ROR1 expression is not a unique marker of CLL. Hematol. Oncol. 29, 17–21.
Baskar, S., Kwong, K. Y., Hofer, T., Levy, J. M., Kennedy, M. G., Lee, E., Staudt, L. M., Wilson, W. H., Wiestner, A., and Rader, C. (2008). Unique cell surface expression of receptor tyrosine kinase ROR1 in human B-cell chronic lymphocytic leukemia. Clin. Cancer Res. 14, 396–404.
Bicocca, V. T., Chang, B. H., Muschen, M., Druker, B. J., and Tyner, J. W. (2011). “Compensatory signaling from ROR1 and the pre-B cell receptor promote survival of t(1;19) acute lymphoblastic leukemia blood,” in ASH Annual Meeting Abstracts, San Diego, Vol. 118, :2466.
Blanc, E., Roux, G. L., Benard, J., and Raguenez, G. (2005). Low expression of Wnt-5a gene is associated with high-risk neuroblastoma. Oncogene 24, 1277–1283.
Bowden, E. T., Barth, M., Thomas, D., Glazer, R. I., and Mueller, S. C. (1999). An invasion-related complex of cortactin, paxillin and PKCmu associates with invadopodia at sites of extracellular matrix degradation. Oncogene 18, 4440–4449.
Choi, B. D., Cai, M., Bigner, D. D., Mehta, A. I., Kuan, C. T., and Sampson, J. H. (2011). Bispecific antibodies engage T cells for antitumor immunotherapy. Expert Opin. Biol. Ther. 11, 843–853.
Choudhury, A., Derkow, K., Daneshmanesh, A. H., Mikaelsson, E., Kiaii, S., Kokhaei, P., Osterborg, A., and Mellstedt, H. (2010). Silencing of ROR1 and FMOD with siRNA results in apoptosis of CLL cells. Br. J. Haematol. 151, 327–335.
Cui, B., Widhopf, G. F. II., Martinez, J. Y. D., Avery, E., Zhang, S., Chen, L., Wu, R., Wu, C. C. N., Prussak, C., and Kipps, T. J. (2011). “Targeting of chronic lymphocytic leukemia B cells with a novel monoclonal antibody to ror1 blood,” in ASH Annual Meeting Abstracts, San Diego, Vol. 118, :984.
Daneshmanesh, A. H., Hojjat-Farsangi, M., Khan, A. S., Jeddi-Tehrani, M., Akhondi, M. M., Bayat, A. A., Ghods, R., Mahmoudi, A. R., Hadavi, R., Osterborg, A., Shokri, F., Rabbani, H., and Mellstedt, H. (2012). Monoclonal antibodies against ROR1 induce apoptosis of chronic lymphocytic leukemia (CLL) cells. Leukemia. doi:10.1038/leu.2011.362. [Epub ahead of print].
Daneshmanesh, A. H., Mikaelsson, E., Jeddi-Tehrani, M., Bayat, A. A., Ghods, R., Ostadkarampour, M., Akhondi, M., Lagercrantz, S., Larsson, C., Osterborg, A., Shokri, F., Mellstedt, H., and Rabbani, H. (2008). Ror1, a cell surface receptor tyrosine kinase is expressed in chronic lymphocytic leukemia and may serve as a putative target for therapy. Int. J. Cancer 123, 1190–1195.
Edris, B., Espinosa, I., Mühlenberg, T., Mikels, A., Lee, C. H., Steigen, S. E., Zhu, S., Montgomery, K. D., Lazar, A. J., Lev, D., Fletcher, J. A., Beck, A. H., West, R. B., Nusse, R., and van de Rijn, M. (2012). ROR2 is a novel prognostic biomarker and a potential therapeutic target in leiomyosarcoma and gastrointestinal stromal tumour. J. Pathol. doi:10.1002/path.3986. [Epub ahead of print].
Enomoto, M., Hayakawa, S., Itsukushima, S., Ren, D. Y., Matsuo, M., Tamada, K., Oneyama, C., Okada, M., Takumi, T., Nishita, M., and Minami, Y. (2009). Autonomous regulation of osteosarcoma cell invasiveness by Wnt5a/Ror2 signaling. Oncogene 28, 3197–3208.
Forrester, W. C., Dell, M., Perens, E., and Garriga, G. (1999). A C. elegans Ror receptor tyrosine kinase regulates cell motility and asymmetric cell division. Nature 400, 881–885.
Fukuda, T., Chen, L., Endo, T., Tang, L., Lu, D., Castro, J. E., Widhopf, G. F. II, Rassenti, L. Z., Cantwell, M. J., Prussak, C. E., Carson, D. A., and Kipps, T. J. (2008). Antisera induced by infusions of autologous Ad-CD154-leukemia B cells identify ROR1 as an oncofetal antigen and receptor for Wnt5a. Proc. Natl. Acad. Sci. U.S.A. 105, 3047–3052.
Gentile, A., Lazzari, L., Benvenuti, S., Trusolino, L., and Comoglio, P. M. (2011). Ror1 is a pseudokinase that is crucial for Met-driven tumorigenesis. Cancer Res. 71, 3132–3141.
Green, J. L., Inoue, T., and Sternberg, P. W. (2007). The C. elegans ROR receptor tyrosine kinase, CAM-1, non-autonomously inhibits the Wnt pathway. Development 134, 4053–4062.
Green, J. L., Kuntz, S. G., and Sternberg, P. W. (2008). Ror receptor tyrosine kinases: orphans no more. Trends Cell Biol. 18, 536–544.
Hudecek, M., Schmitt, T. M., Baskar, S., Lupo-Stanghellini, M. T., Nishida, T., Yamamoto, T. N., Bleakley, M., Turtle, C. J., Chang, W. C., Greisman, H. A., Wood, B., Maloney, D. G., Jensen, M. C., Rader, C., and Riddell, S. R. (2010). The B-cell tumor-associated antigen ROR1 can be targeted with T cells modified to express a ROR1-specific chimeric antigen receptor. Blood 116, 4532–4541.
Ishitani, T., Kishida, S., Hyodo-Miura, J., Ueno, N., Yasuda, J., Waterman, M., Shibuya, H., Moon, R. T., Ninomiya-Tsuji, J., and Matsumoto, K. (2003). The TAK1-NLK mitogen-activated protein kinase cascade functions in the Wnt-5a/Ca(2+) pathway to antagonize Wnt/beta-catenin signaling. Mol. Cell. Biol. 23, 131–139.
Kani, S., Oishi, I., Yamamoto, H., Yoda, A., Suzuki, H., Nomachi, A., Iozumi, K., Nishita, M., Kikuchi, A., Takumi, T., and Minami, Y. (2004). The receptor tyrosine kinase Ror2 associates with and is activated by casein kinase Iepsilon. J. Biol. Chem. 279, 50102–50109.
Kaucka, M., Krejci, P., Plevova, K., Pavlova, S., Prochazkova, J., Janovska, P., Valnohova, J., Kozubik, A., Pospisilova, S., and Bryja, V. (2011). Post-translational modifications regulate signalling by Ror1. Acta Physiol. (Oxf). 203, 351–362.
Kobayashi, M., Shibuya, Y., Takeuchi, J., Murata, M., Suzuki, H., Yokoo, S., Umeda, M., Minami, Y., and Komori, T. (2009). Ror2 expression in squamous cell carcinoma and epithelial dysplasia of the oral cavity. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod. 107, 398–406.
Kurban, G., Hudon, V., Duplan, E., Ohh, M., and Pause, A. (2006). Characterization of a von Hippel Lindau pathway involved in extracellular matrix remodeling, cell invasion, and angiogenesis. Cancer Res. 66, 1313–1319.
Li, P., Harris, D., Liu, Z., Liu, J., Keating, M., and Estrov, Z. (2010). Stat3 activates the receptor tyrosine kinase like orphan receptor-1 gene in chronic lymphocytic leukemia cells. PLoS ONE 5, e11859. doi:10.1371/journal.pone.0011859
Liu, Y., Rubin, B., Bodine, P. V., and Billiard, J. (2008). Wnt5a induces homodimerization and activation of Ror2 receptor tyrosine kinase. J. Cell. Biochem. 105, 497–502.
Lyashenko, N., Weissenböck, M., Sharir, A., Erben, R. G., Minami, Y., and Hartmann, C. (2010). Mice lacking the orphan receptor ror1 have distinct skeletal abnormalities and are growth retarded. Dev. Dyn. 239, 2266–2277.
Maeda, K., Kobayashi, Y., Udagawa, N., Uehara, S., Ishihara, A., Mizoguchi, T., Kikuchi, Y., Takada, I., Kato, S., Kani, S., Nishita, M., Marumo, K., Martin, T. J., Minami, Y., and Takahashi, N. (2012). Wnt5a-Ror2 signaling between osteoblast-lineage cells and osteoclast precursors enhances osteoclastogenesis. Nat. Med. 18, 405–12.
Masiakowski, P., and Carroll, R. D. (1992). A novel family of cell surface receptors with tyrosine kinase-like domain. J. Biol. Chem. 267, 26181–26190.
McDonald, S. L., and Silver, A. (2009). The opposing roles of Wnt-5a in cancer. Br. J. Cancer 101, 209–214.
Mikels, A., Minami, Y., and Nusse, R. (2009). Ror2 receptor requires tyrosine kinase activity to mediate Wnt5A signaling. J. Biol. Chem. 284, 30167–30176.
Mikels, A. J., and Nusse, R. (2006). Purified Wnt5a protein activates or inhibits beta-catenin-TCF signaling depending on receptor context. PLoS Biol. 4, e115. doi:10.1371/journal.pbio.0040115
Minami, Y., Oishi, I., Endo, M., and Nishita, M. (2010). Ror-family receptor tyrosine kinases in noncanonical Wnt signaling: their implications in developmental morphogenesis and human diseases. Dev. Dyn. 239, 1–15.
Morioka, K., Tanikawa, C., Ochi, K., Daigo, Y., Katagiri, T., Kawano, H., Kawaguchi, H., Myoui, A., Yoshikawa, H., Naka, N., Araki, N., Kudawara, I., Ieguchi, M., Nakamura, K., Nakamura, Y., and Matsuda, K. (2009). Orphan receptor tyrosine kinase ROR2 as a potential therapeutic target for osteosarcoma. Cancer Sci. 100, 1227–1233.
Nomachi, A., Nishita, M., Inaba, D., Enomoto, M., Hamasaki, M., and Minami, Y. (2008). Receptor tyrosine kinase Ror2 mediates Wnt5a-induced polarized cell migration by activating c-Jun N-terminal kinase via actin-binding protein filamin A. J. Biol. Chem. 283, 27973–27981.
Nomi, M., Oishi, I., Kani, S., Suzuki, H., Matsuda, T., Yoda, A., Kitamura, M., Itoh, K., Takeuchi, S., Takeda, K., Akira, S., Ikeya, M., Takada, S., and Minami, Y. (2001). Loss of mRor1 enhances the heart and skeletal abnormalities in mRor2-deficient mice: redundant and pleiotropic functions of mRor1 and mRor2 receptor tyrosine kinases. Mol. Cell. Biol. 21, 8329–8335.
O’Connell, M. P., Fiori, J. L., Xu, M., Carter, A. D., Frank, B. P., Camilli, T. C., French, A. D., Dissanayake, S. K., Indig, F. E., Bernier, M., Taub, D. D., Hewitt, S. M., and Weeraratna, A. T.. (2010). The orphan tyrosine kinase receptor, ROR2, mediates Wnt5A signaling in metastatic melanoma. Oncogene 29, 34–44.
Oishi, I., Suzuki, H., Onishi, N., Takada, R., Kani, S., Ohkawara, B., Koshida, I., Suzuki, K., Yamada, G., Schwabe, G. C., Mundlos, S., Shibuya, H., Takada, S., and Minami, Y. (2003). The receptor tyrosine kinase Ror2 is involved in non-canonical Wnt5a/JNK signalling pathway. Genes Cells 8, 645–654.
Oishi, I., Takeuchi, S., Hashimoto, R., Nagabukuro, A., Ueda, T., Liu, Z. J., Hatta, T., Akira, S., Matsuda, Y., Yamamura, H., Otani, H., and Minami, Y. (1999). Spatio-temporally regulated expression of receptor tyrosine kinases, mRor1, mRor2, during mouse development: implications in development and function of the nervous system. Genes Cells 4, 41–56.
Rabbani, H., Ostadkarampour, M., Danesh Manesh, A. H., Basiri, A., Jeddi-Tehrani, M., and Forouzesh, F. (2010). Expression of ROR1 in patients with renal cancer–a potential diagnostic marker. Iran. Biomed. J. 14, 77–82.
Reddy, U. R., Phatak, S., Allen, C., Nycum, L. M., Sulman, E. P., White, P. S., and Biegel, J. A. (1997). Localization of the human Ror1 gene (NTRKR1) to chromosome 1p31-p32 by fluorescence in situ hybridization and somatic cell hybrid analysis. Genomics 41, 283–285.
Reddy, U. R., Phatak, S., and Pleasure, D. (1996). Human neural tissues express a truncated Ror1 receptor tyrosine kinase, lacking both extracellular and transmembrane domains. Oncogene 13, 1555–1559.
Rehn, M., Pihlajaniemi, T., Hofmann, K., and Bucher, P. (1998). The frizzled motif: in how many different protein families does it occur? Trends Biochem. Sci. 23, 415–417.
Sadowski, I., Stone, J. C., and Pawson, T. (1986). A noncatalytic domain conserved among cytoplasmic protein-tyrosine kinases modifies the kinase function and transforming activity of Fujinami sarcoma virus P130gag-fps. Mol. Cell. Biol. 6, 4396–4408.
Sato, A., Yamamoto, H., Sakane, H., Koyama, H., and Kikuchi, A. (2010). Wnt5a regulates distinct signalling pathways by binding to Frizzled2. EMBO J. 29, 41–54.
Schwabe, G. C., Tinschert, S., Buschow, C., Meinecke, P., Wolff, G., Gillessen-Kaesbach, G., Oldridge, M., Wilkie, A. O., Kömec, R., and Mundlos, S. (2000). Distinct mutations in the receptor tyrosine kinase gene ROR2 cause brachydactyly type B. Am. J. Hum. Genet. 67, 822–31.
Shabani, M., Asgarian-Omran, H., Vossough, P., Sharifian, R. A., Faranoush, M., Ghragozlou, S., Khoshnoodi, J., Roohi, A., Jeddi-Tehrani, M., Mellstedt, H., Rabbani, H., and Shokri, F. (2008). Expression profile of orphan receptor tyrosine kinase (ROR1) and Wilms’ tumor gene 1 (WT1) in different subsets of B-cell acute lymphoblastic leukemia. Leuk. Lymphoma 49, 1360–1367.
Tassev, D. V., Cheng, M., and Cheung, N. K. (2012). Retargeting NK92 cells using an HLA-A2-restricted EBNA3C-specific chimeric antigen receptor. Cancer Gene Ther. 19, 84–100.
Uhrmacher, S., Schmidt, C., Erdfelder, F., Poll-Wolbeck, S. J., Gehrke, I., Hallek, M., and Kreuzer, K. A. (2011). Use of the receptor tyrosine kinase-like orphan receptor 1 (ROR1) as a diagnostic tool in chronic lymphocytic leukemia (CLL). Leuk. Res. 35, 1360–1366.
van Bokhoven, H., Celli, J., Kayserili, H., van Beusekom, E., Balci, S., Brussel, W., Skovby, F., Kerr, B., Percin, E. F., Akarsu, N., and Brunner, H. G. (2000). Mutation of the gene encoding the ROR2 tyrosine kinase causes autosomal recessive Robinow syndrome. Nat. Genet. 25, 423–426.
Vidal, M., Gigoux, V., and Garbay, C. (2001). SH2 and SH3 domains as targets for anti-proliferative agents. Crit. Rev. Oncol. Hematol. 40, 175–186.
Weaver, A. M. (2006). Invadopodia: specialized cell structures for cancer invasion. Clin. Exp. Metastasis 23, 97–105.
Wilson, C., Goberdhan, D. C., and Steller, H. (1993). Dror, a potential neurotrophic receptor gene, encodes a Drosophila homolog of the vertebrate Ror family of Trk-related receptor tyrosine kinases. Proc. Natl. Acad. Sci. U.S.A. 90, 7109–7113.
Wright, T. M., Brannon, A. R., Gordan, J. D., Mikels, A. J., Mitchell, C., Chen, S., Espinosa, I., Van De Rijn, M., Pruthi, R., Wallen, E., Edwards, L., Nusse, R., and Rathmell, W. K. (2009). Ror2, a developmentally regulated kinase, promotes tumor growth potential in renal cell carcinoma. Oncogene 28, 2513–2523.
Yamamoto, H., Yoo, S. K., Nishita, M., Kikuchi, A., and Minami, Y. (2007). Wnt5a modulates glycogen synthase kinase 3 to induce phosphorylation of receptor tyrosine kinase Ror2. Genes Cells 12, 1215–1223.
Yang, J., Baskar, S., Kwong, K. Y., Kennedy, M. G., Wiestner, A., and Rader, C. (2011). Therapeutic potential and challenges of targeting receptor tyrosine kinase ROR1 with monoclonal antibodies in B-cell malignancies. PLoS ONE 6, e21018. doi:10.1371/journal.pone.0021018
Yoda, A., Oishi, I., and Minami, Y. (2003). Expression and function of the Ror-family receptor tyrosine kinases during development: lessons from genetic analyses of nematodes, mice, and humans. J. Recept. Signal Transduct. Res. 23, 1–15.
Keywords: solid tumor, immunotherapy, receptor tyrosine kinase orphan receptors (ROR1/ROR2), Wnt/beta-catenin
Citation: Rebagay G, Yan S, Liu C and Cheung N-K (2012) ROR1 and ROR2 in human malignancies: potentials for targeted therapy. Front. Oncol. 2:34. doi: 10.3389/fonc.2012.00034
Received: 13 December 2011; Accepted: 22 March 2012;
Published online: 18 April 2012.
Edited by:
Silvia Giordano, University of Torino, ItalyReviewed by:
Anna Rita Migliaccio, Mount Sinai School of Medicine, USALuca Tamagnone, University of Torino, Italy
Livio Trusolino, Institute for Cancer Research and Treatment, Italy
Copyright: © 2012 Rebagay, Yan, Liu and Cheung. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Nai-Kong Cheung, Department of Pediatrics, Memorial Sloan-Kettering Cancer Center, 1275 York Avenue, New York, NY 10065, USA. e-mail:Y2hldW5nbkBtc2tjYy5vcmc=