 Rakesh Kumar1 Nobuo Horikoshi1 Mayank Singh1 Arun Gupta1 Hari S. Misra1,2 Kevin Albuquerque1 Clayton R. Hunt1
Rakesh Kumar1 Nobuo Horikoshi1 Mayank Singh1 Arun Gupta1 Hari S. Misra1,2 Kevin Albuquerque1 Clayton R. Hunt1 Tej K. Pandita1*
Tej K. Pandita1*- 1Department of Radiation Oncology, University of Texas Southwestern Medical Center, Dallas, TX, USA
- 2Homi Bhabha National Institute, Molecular Genetics Section, Molecular Biology Division, Bhabha Atomic Research Centre, Mumbai, India
In order to survive, cells have evolved highly effective repair mechanisms to deal with the potentially lethal DNA damage produced by exposure to endogenous as well as exogenous agents. Ionizing radiation exposure induces highly lethal DNA damage, especially DNA double-strand breaks (DSBs), that is sensed by the cellular machinery and then subsequently repaired by either of two different DSB repair mechanisms: (1) non-homologous end joining, which re-ligates the broken ends of the DNA and (2) homologous recombination, that employs an undamaged identical DNA sequence as a template, to maintain the fidelity of DNA repair. Repair of DSBs must occur within the natural context of the cellular DNA which, along with specific proteins, is organized to form chromatin, the overall structure of which can impede DNA damage site access by repair proteins. The chromatin complex is a dynamic structure and is known to change as required for ongoing cellular processes such as gene transcription or DNA replication. Similarly, during the process of DNA damage sensing and repair, chromatin needs to undergo several changes in order to facilitate accessibility of the repair machinery. Cells utilize several factors to modify the chromatin in order to locally open up the structure to reveal the underlying DNA sequence but post-translational modification of the histone components is one of the primary mechanisms. In this review, we will summarize chromatin modifications by the respective chromatin modifying factors that occur during the DNA damage response.
Introduction
Cells derived from individuals with ataxia-telangiectasia are radiation sensitive and have a higher rate of conversion of DNA double-strand breaks (DSBs) into chromosome breaks post irradiation (Pandita and Hittelman, 1992a,b; Pandita and Richardson, 2009). The increased frequency of chromosome aberrations observed in ataxia-telangiectasia mutated (ATM) defective cells has been attributed to an altered chromatin status which is observed at both the global chromatin level as well as specifically at telomeric chromatin (Pandita and Hittelman, 1995; Smilenov et al., 1999; Pandita, 2001, 2002, 2003). It is now evident that DNA damage renders chromatin more sensitive to micrococal nuclease digestion (Telford and Stewart, 1989) and that induction of DSB results in decondensation of chromatin at the local as well as global level (Kruhlak et al., 2006; Dellaire et al., 2009), an essential requirement for activation of the DNA damage response (DDR) and subsequent DSB repair. The basic repeating unit of chromatin, the nucleosome, consists of a histone octamer complex containing four types of histones, namely H2A, H2B, H3, and H4 (Luger et al., 1997), around which is wound 146 bp of DNA. Nucleosome formation compacts naked double helical DNA by approximately six fold its linear dimension. DSB formation has been reported to lead to a localized disruption of nucleosomes, a process that depends on NBS1 and ATM (Berkovich et al., 2007). Histones consist structurally of a globular core domain with flexible tail regions at both the N-terminal and C-terminal regions. The globular core domain interacts with other histones while the termini contain highly conserved, charged lysine and arginine amino acid residues involved in DNA–histone interaction. In addition to histones, non-histone proteins are also involved in developing special chromatin structures. The known histone modifications induced following ionizing radiation exposure are phosphorylation, acetylation, methylation, and ubiquitination (Pandita and Richardson, 2009; Deem et al., 2012). Among these major chromatin modifications, post-damage-induced histone phosphorylation has been the most thoroughly studied. Recent studies have provided evidence that histone acetylation of lysine is also critical for the DDR. Post-translational modifications (PTMs) of histone proteins are critical for cellular recognition of DNA damage and subsequent recruitment of repair protein complexes to the damage sites. Moreover, evidence is emerging suggesting that pre-existing chromatin modifications also play an important role in the DDR (Sharma et al., 2010).
Histone Modifications in DNA DSB Repair
Histone modifications provide support for critical repair proteins that act on the DNA damage site by helping in the recruitment of specific protein DNA repair factors and also play a role in sensing the initial DNA damage. Histone modifications have been detected on lysine as acetylation, methylation, or ubiquitination modification, on arginine as methylation and on serine/threonine as phosphorylation. Except for ubiquitination, all these modifications alter histone/DNA electrostatic interactions and ultimately change chromatin dynamics and function by altering access of the cellular factors involved in DDR.
Histone Phosphorylation
Phosphorylation of all histones has been reported to affect transcription, DNA repair, apoptosis, and chromosome condensation during mitosis (Hanks et al., 1983; Van Hooser et al., 1998). H2A phosphorylation has been extensively studied in response to DNA damaging agents (Paull et al., 2000) and phosphorylation specifically of the H2A variant H2AX occurs immediately (within a few minutes) after exposure to ionizing radiation. Phosphorylation of H2AX (Rogakou et al., 1998), the product being referred to as γ-H2AX, has been shown to be an essential event in the response to DNA strand breaks and is thought to modify the higher order chromatin structure at the damage site. In yeast, H2A is phosphorylated at Ser129 by the Tel1 and Mec1 kinases in response to various DNA damaging agents (Kouzarides, 2000, 2007; Paull et al., 2000; Foster and Downs, 2005) while in humans ATM and ATR (human ortholog for yeast Tel1 and Mec1, respectively) phosphorylate the corresponding Ser139 of H2A. This phosphorylation event is critical for the recruitment of signaling/repair proteins to DNA damage sites since ionizing radiation induced formation of γ-H2AX foci is rapid, precedes repair factor assembly into repairosome foci and is required for subsequent foci formation by numerous factors including 53BP1, NBS1, BRCA1, and MDC1 (reviewed in Fernandez-Capetillo et al., 2004b). In addition, γ-H2AX has been shown to physically interact with NBS1, 53BP1, and MDC1 and deficiency of H2AX results in enhanced genomic instability, further supporting the role of γ-H2AX in DDR (reviewed in Fernandez-Capetillo et al., 2004b). Ionizing radiation (IR)-induced phosphorylation of H2AX occurs in all phases of the cell cycle, which is consistent with the fact that IR-induced ATM activation occurs in all phases of the cell cycle (Pandita et al., 2000). Phosphorylation of H2AX facilitates damage site recruitment of the DDR component MDC1 which binds to γ-H2AX via its BRCT domain (Yuan et al., 2010). γ-H2AX-recruited MDC1 is phosphorylated by ATM as well as by casein kinase 2 (Stucki et al., 2005; Lou et al., 2006). Binding of MDC1 to γ-H2AX is further modulated by males absent on the first (MOF)-dependent H4K16 acetylation (Li et al., 2010). Consistent with this, H2AX phosphorylation has been shown to facilitate the recruitment of SW1/SNF and RSC remodeling complexes and several repair proteins including RAD16, CSB/RAD26, RAD5, and RAD54 that belong to the SWI2/SNF2 family of helicases involved in DNA repair (Widlak et al., 2006). In addition to H2AX, other histones also undergo phosphorylation as part of the DDR, e.g., phosphorylation of H2B at Ser14 and N-terminal phosphorylation of H4 (Fernandez-Capetillo et al., 2004a). Phosphorylation of H2B and H4 has been reported to be abundant in close proximity to endonuclease-induced double-strand breaks (Cheung et al., 2005; Utley et al., 2005) and is mediated by sterile 20 kinase (Mst1; Ahn et al., 2005) and casein kinase 2 (Cheung et al., 2005; Utley et al., 2005). The major types of phosphorylated histones in chromatin and the associated kinases are summarized in Table 1.

TABLE 1. Phosphorylation.
Histone Methylation
Histone methylation on lysine and arginine (Grant, 2001; Zhang and Reinberg, 2001; Fischle et al., 2005; Kouzarides, 2007) was discovered 40 years ago (Murray, 1964). Specific methylation sites linked to the DDR are summarized in Table 2. Multiple methylations of H3 and H4 are reported (mono-, di-, and trimethyl groups per residue) at K4, K9, K27, K36, K79 and R2, R8, R17, R26 for H3 and K20 and R3 for H4. Histone methyl transferases use S-adenosyl-methionine as a methyl donor for the catalytic process. Lysine methylation is mainly commenced by the SET domain containing proteins [Dm Su(var)3-9, Enhancer of zeste (E(z)), and trithorax (trx)] while arginine methylation is carried out by co-activator arginine methyltransferase (CARM1) and the protein arginine methyl transferase (PRMT1). Additionally, as histone acetylation can be detected by bromodomain containing proteins, methylated sites are detected by proteins containing a chromodomain motif. Although dimethylation of histone H4 lysine 20 (H4K20me2), mediated by the histone methyltransferase MMSET (also known as NSD2 or WHSC1) in mammals (Pei et al., 2011), does not seem to increase globally after DNA damage, it is critical for 53BP1 recruitment to double-strand breaks (Pei et al., 2011) but does increase in the vicinity of DSBs. Not surprisingly then, MMSET depletion significantly decreases H4K20 methylation at DSBs and the subsequent accumulation of 53BP1. Recruitment of MMSET to DSBs requires the γ-H2AX–MDC1 pathway; specifically, the interaction between the MDC1 BRCT domain and phosphorylated Ser102 of MMSET (Pei et al., 2011). Based on such observations, it is possible that a pathway involving γ-H2AX–MDC1–MMSET regulates the induction of H4K20 methylation on histones around DSBs, which, in turn, facilitates 53BP1 recruitment.
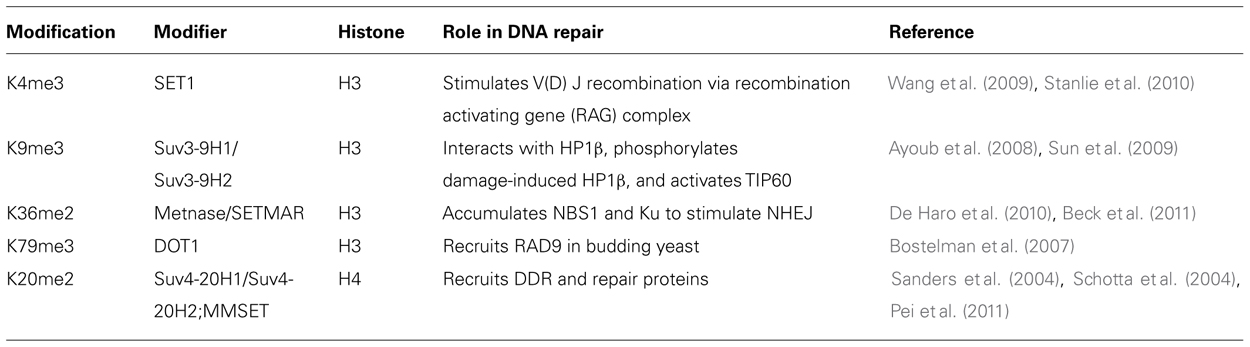
TABLE 2. Methylation.
Histone Acetylation
Histone acetylation neutralizes positively charged lysine residues, altering the chromatin fiber intra- and inter-nucleosomal interactions to facilitate decondensation and enhance access to nucleosomal DNA (Kouzarides, 2000; Anderson et al., 2001). Table 3 summarizes the histone acetylation enzymes with known connections to the DDR and their specific histone substrates. Acetylation is a dynamic histone marker regulated by the balance between histone acetyltransferases (HATs), which transfer an acetyl moiety from acetyl-coenzyme A to the ε-amino group of lysine, and histone deacetylases (HDACs) which remove the acetyl-group from histones (Shahbazian and Grunstein, 2007). Histone modification occurs in both the nucleus and cytoplasm by Type-A and Type-B HATs, respectively. Type-A HATs are responsible for chromatin dynamics in the nucleus. There are multiple Type A HAT families including the GNAT superfamily, MYST family, p300/CBP, nuclear receptor co-activators, TAFII250 and TFIIIC. Individual histones can contain modifications that differentially regulate chromatin functions. While acetylation of H3 and H4 histones increases interactions with components of the transcriptional machinery containing bromodomains, methylation of lysine 9 on H3 (H3K9) allows heterochromatin protein 1 (HP1) binding via the chromodomain to the chromatin, thereby inhibiting binding of the transcription machinery to DNA. The acetylation at the N-terminuses of H3 and H4 and at H3K56 plays an important role in genomic stability, DNA replication, and in the binding of chromatin assembly factor (CF1)–PCNA complex (Chen and Tyler, 2008). Acetylation has been detected at four lysines (K5, K8, K12, and K16) in the N-terminal tail of the H4 histone. Acetylation at K16 in H4 was observed on the Drosophila male X chromosome (Turner et al., 1992) and correlated with gene dosage compensation. The modification has also been implicated in the control of chromatin structure responsible for interaction of other proteins (Shogren-Knaak et al., 2006). The acetyl-transferase responsible for histone H4 acetylation at K16 is MOF (Gupta et al., 2005, 2008; Smith et al., 2005; Sharma et al., 2010). Acetylation at H4 K16 (H4K16ac) has been implicated in the proper compaction of chromatin 30-nm fibers (Shogren-Knaak et al., 2006). More importantly, lack of MOF also influences ATM activation (Gupta et al., 2005) and results in delayed appearance of IR-induced γ-H2AX foci (Sharma et al., 2010). Consistent with the influence of histone H4 K16 acetylation on ATM activation, HDAC inhibitor treatment results in global ATM activation even in the absence of DNA damage (Bakkenist and Kastan, 2003).
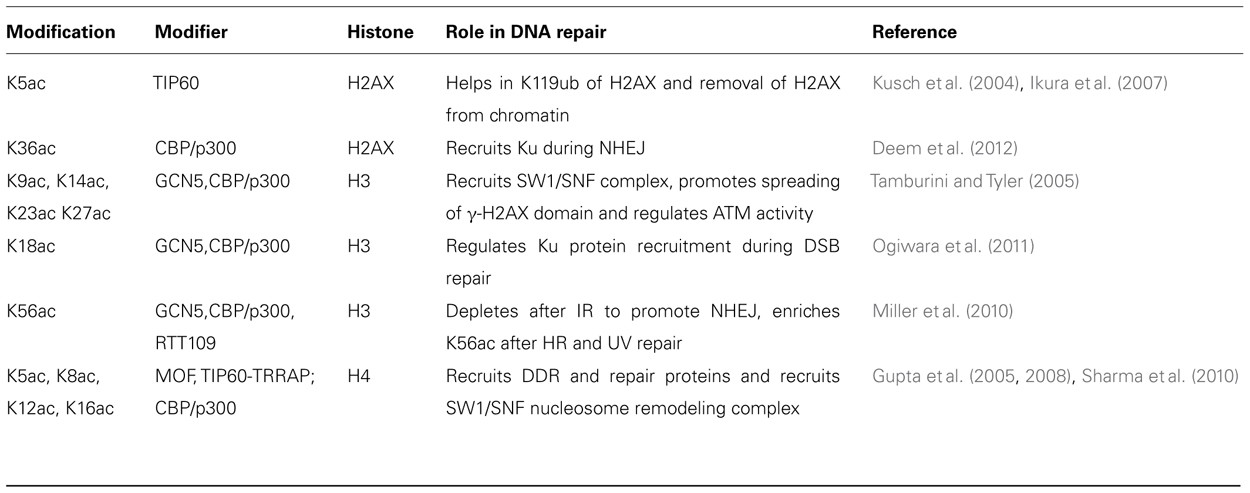
TABLE 3. Histone acetylation.
Histone Ubiquitination
Ubiquitination is a cellular process that conjugates a 76 amino acid protein, ubiquitin, to the lysine ε-amino group of specific proteins as a result of which it acts as a signaling molecule to regulate protein function and stability. Ubiquitination of H2A at K119 is associated with transcriptional repression via polycomb repressive complex (de Napoles et al., 2004; Fang et al., 2004) and the E3 ubiquitin-protein ligase RNF2 or RING2 responsible for this ubiquitination is stimulated by RING finger-domain containing proteins like BMI-1 and RINGIA (Cao et al., 2005). Interestingly, BMI-1, RINGIA, and RINGIB are involved in DSB-associated H2A ubiquitination (Cao and Yan, 2012). Ionizing radiation induces ubiquitination of nuclear H2A and H2AX histones. Monoubiquitinated H2A is enriched in the satellite regions of genome where as the ubiquitination of H2B is mostly in transcriptionally active genes. Histone H2A and H2AX can also be polyubiquitinated by ubiquitin ligase complex. During DNA repair at the break site, RNF8 and RNF168 catalyze formation of lysine 63 linked polyubiquitination chains on histones H2A and H2AX (Doil et al., 2009; Stewart et al., 2009; Campbell et al., 2012). RNF8 is rapidly recruited to the sites of DNA damage in an MDC1-dependent manner through its functional FHA domain and RNF8 is required to recruit other repair factors (Huen et al., 2007; Kolas et al., 2007; Mailand et al., 2007; Shi et al., 2008; Marteijn et al., 2009; Mok and Henderson, 2012). RNF8-catalyzed ubiquitin modification does not lead to protein degradation because the polyubiquitin synthesized in the RNF8/UBC13-mediated pathways is a lysine-63 linkage, rather than the lysine-48 canonical signal for protein degradation. Recent studies have revealed that K63Ub synthesis is regulated by the deubiquitinating enzyme, OTUB1 (OTU domain-containing ubiquitin aldehyde-binding protein 1; Wiener et al., 2012). It is thought that RNF8 executed ubiquitination has a role in maintaining genomic integrity, however, the role of post-damage monoubiquitylation in chromatin reassembly needs to be elucidated (Deem et al., 2012). It has been reported that RNF8 is inactive toward nucleosomal histone H2A. In contrast RNF168 catalyzes the monoubiquitination of the histones (H2A/H2AX) specifically on K13-15 (Mattiroli et al., 2012). Interestingly, E3 ligases like BRCA1 promote BRCA2 recruitment that appears in turn to promote the recruitment of RAD51 involved in homologous recombination (HR; Qing et al., 2011). The major histone sites of ubiquitination, the enzymes required and the role of the modification in the DDR are summarized in Table 4.
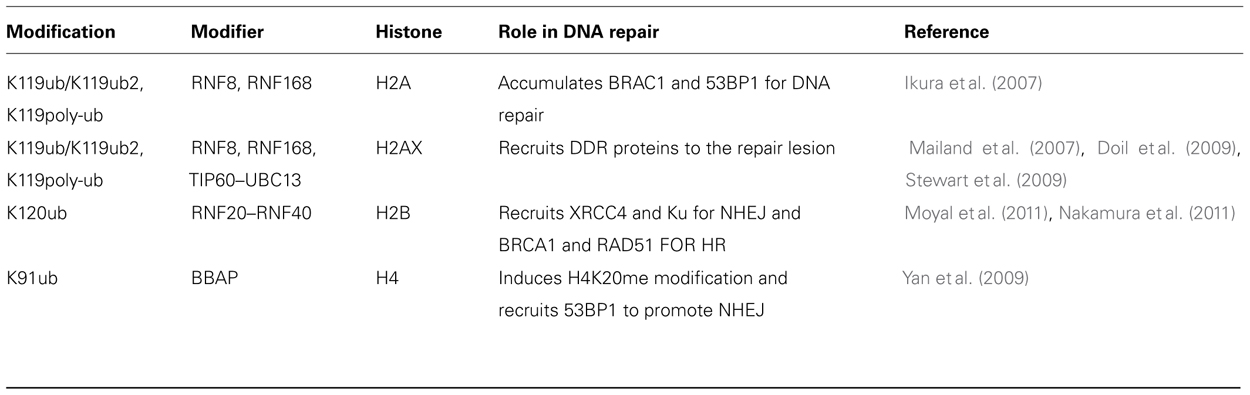
TABLE 4. Ubiquitination.
Chromatin Modifications During Non-Homologous End Joining Repair
A majority of the genes responsible for DSB repair in human cells have been identified and have been assigned to one of several different repair pathways. DNA DSBs induced by IR are mostly repaired by either non-homologous end joining (NHEJ) or HR (Cartwright et al., 1998; Jeggo, 1998; Haber, 2000; Rothkamm et al., 2003; Scott and Pandita, 2006; Sonoda et al., 2006; Pandita and Richardson, 2009; Chapman et al., 2012). For DNA DSB repair by NHEJ, a very little or no DNA sequence homology at the damaged ends is required. NHEJ in mammalian cells is thought to be the major pathway for DNA damage repair after ionizing radiation exposure. There are two types of NHEJ repair: (1) Canonical end joining pathway (C-NHEJ) and the (2) Alternative NHEJ (A-NHEJ) pathway, the later being a minor DSB repair pathway that works in specific situations such as the absence of Ku. C-NHEJ is essential for both somatic cell survival after IR treatment and V(D)J recombination, which generates the antibody and T cell receptor diversity required for lymphocyte maturation (Boboila et al., 2012). Binding of the ring shaped Ku70/Ku80 heterodimer (Ku) to both ends of a DSB initiates C-NHEJ at damaged DNA sites followed by loading of DNA-dependent protein kinase catalytic subunit (DNA-PKcs) onto the DNA-Ku complexes. Prior to loading of the repair proteins, histone modifications occur which facilitate the recruitment of repair proteins at the damage site. DNA-bound Ku proteins serve as a template for loading DNA-PKcs which help translocate into the duplex by one helical turn, leaving DNA-PKcs near the DNA terminus to assist in tethering the broken ends together and prevent exonucleolytic degradation of the DNA ends (Gapud and Sleckman, 2011). This also helps DNA strands search for micro homologies. Activated DNA-PKcs phosphorylates different components of the NHEJ machinery to facilitate end-processing; DNA-PKcs itself undergoes autophosphorylation, which helps release DNA-PKcs from the break site so that subsequent steps of C-NHEJ can occur. DNA-PK holoenzyme (Ku/DNA-PKcs) recognizes, protects and bridges the DNA ends. DNA-PK conformational autophosphorylation is necessary for activation of end-processing enzymes, such as the Artemis nuclease. This complex serves as a docking platform for DNA polymerases and ligation factors like DNA ligase IV and X-ray cross complementing factor 4 (XRCC4), which help in DNA joining. Cell deficient for the canonical NHEJ proteins undergo DSB repair by an alternative form of NHEJ (Alt NHEJ), which operates at telomeres in telomerase deficient cells or in the absence of Ku or DNA-PKcs. This pathway involves proteins different from those involved in the C-NHEJ pathway including poly(ADP-ribose) polymerase 1 (PARP-1), XRCC1, DNA ligase III (LIG3), polynucleotide kinase, or Flap endonuclease 1. Though this pathway is relatively poorly characterized, certain features of A-NHEJ are well characterized such as a slower kinetics of DSB repair than in C-NHEJ and competitive repression of A-NHEJ by Ku under normal conditions. Whether C-NHEJ and Alt-NHEJ require any pathway specific chromatin modifications is not yet known.
The first visible chromatin modification that occurs immediately after cellular exposure to IR is phosphorylation of histone H2AX on Ser139, the product being termed γ-H2AX, which functions to recruit repair proteins. Besides γ-H2AX, other modifications involved in the NHEJ are acetylation, methylation, phosphorylation, and ubiquitination of histone H2A, H2B, H3, and H4. These histone modifications may play a role in either marking the site of a DNA break to facilitate the recruitment of DNA repair proteins or relaxing the chromatin so the repair proteins can access the damaged DNA site. For example, Metnase, a methylase which dimethylates histone H3 residue K36 (H3K36me2) in the DNA DSB region, and the levels of H3K36me2 have been found to correspond positively to DSB repair efficiency (reviewed in Williamson et al., 2012). Another modification, H3K4me3, carried out by Set1p methyltransferase has been found at DSBs and the absence of this modification has been linked to poor DNA DSB repair (Faucher and Wellinger, 2010).
During NHEJ repair, histones H3 and H4 are reportedly acetylated on the N-terminal lysines (reviewed in Williamson et al., 2012). NuA4–TIP60 acetylates histone H4 at DSBs and this acetylation has been linked with improved DSB repair (Bird et al., 2002; Murr et al., 2006). Similarly, the base levels of H4K16ac prior to DNA DSB induction influences repair as cells with decreased levels of H4K16ac have reduced efficiency of DNA DSB repair by NHEJ (Sharma et al., 2010). Other histone modifications by INO80, SWR1, and RSC complexes are necessary for recruitment of Ku70/80 at DNA DSB sites (Shim et al., 2007; van Attikum et al., 2007). It is interesting to note that at DSBs, RSC complex recruits Mre11, which is followed by ATPase remodeling to facilitate access to the site by proteins required for NHEJ-mediated repair (Shim et al., 2007). Furthermore, acetylation of H3 and H4 by CBP and p300 cooperate with the SW1/SNF complex to facilitate recruitment of Ku70/80 (Ogiwara et al., 2011). The function of histone acetylation therefore seems to be during the early stages of NHEJ to facilitate chromatin relaxation and subsequently allow the repair proteins to access the DNA DSB.
Chromatin Modifications During Homologous Recombination Repair
During the DNA DSB repair by HR, nucleotide sequences are exchanged between two similar or identical molecules of DNA, allowing the cells to accurately repair DNA. HR type repair is predominant during meiosis and in the S- and G2-phases of the cell cycle, whereas NHEJ repair is predominant in G1-phase cells. HR repair is a complex process, which requires significant alterations in chromatin structure in order to allow repair proteins access to damaged DNA. During this process, the first step is the processing of the exposed DNA ends to produce 3′-overhanging DNA ends for RAD51 binding. Resection is followed by RAD51 filament formation on the resulting single-stranded DNA, homologous sequence search, heteroduplex formation, repair synthesis, and resolution of the heteroduplex, all steps that require major changes in the chromatin structure. While HR makes a significant contribution to cell survival after IR exposure only in the S- and G2-phases, replication-associated one-ended DSBs are also efficiently and primarily repaired by HR.
The initiating step for HR is end-resection to create a 3′-overhang, which is subsequently coated by replication protein A (RPA). After formation of a DSB end, these ends are resected in vertebrates by the MRE11/RAD50/NBS1(MRN) complex or the MRX complex in yeast. Exo1 along with DNA2 nuclease then digest the 5′-end in eukaryotes. In yeast, the MRX complex, in cooperation with Sae2, is important for initiating DNA resection. The MRN complex serve as a multipurpose DNA clamp that acts to directly bridge severed DNA ends. DNA resection is followed by a critical step, which is recognition of homologous DNA sequences followed by generation of a joint molecule between single-stranded DNA and its duplex repair template. DNA strand exchange leads to switching of base-paired partners between these DNAs. Homology recognition and strand exchange are mediated by recombinase proteins such as RecA in prokaryotes and RAD51 in eukaryotes. RecA and RAD51 assembled into protein filaments on single-stranded DNA catalyze DNA rearrangement aided by accessory/mediator proteins. Subsequent steps in HR can include the engagement of the second DNA end, DNA branch migration, and eventual resolution of repaired DNA strands.
Upon joint molecule formation, a critical common step in the HR pathways is the hand off of the 3′-end of the incoming DNA molecule to a DNA polymerase, such that recombination will result in two restored duplex DNAs. At this stage of the reaction the partner DNA molecules are physically joined via branched DNA structures, which upon ligation can be converted into so-called Holliday junctions. There appear to be many structure-specific endonucleases that can resolve these structures resulting in the completion of DNA repair by HR. Enzymes such as the Escherichia coli RuvABC complex and the eukaryotic Gen1 protein incise Holliday junctions producing directly ligatable crossover and non-crossover products. Alternatively, a DNA helicase, BLM, in combination with a type I topoisomerase, can resolve Holliday junctions. Branched DNA intermediates in HR can also be acted upon by evolutionary-conserved structure-specific endonucleases, including Mus81/Eme1 and Slx1/Slx4. During the HR process, a number of acetylation events occur on histones H3 and H4 with the proteins implicated in the modification being GCN5, NuA4, and HAT1 (Tamburini and Tyler, 2005; Murr et al., 2006). GCN5 also plays some role in pathway choice for DSB repair as DNA-PKcs phosphorylates GCN5 to inactivate its HAT domain. In addition, GCN5 also interacts with BRCA1 through a mechanism that is dependent upon its HAT activity, suggesting a role in HR repair of DNA DSBs (Oishi et al., 2006).
During HR repair, MDC1 recruits RNF8 (E3) to ubiquitinate H2AX (Kolas et al., 2007; Mohammad and Yaffe, 2009). Subsequently, recruited BRCA1 further maintains H2AX ubiquitination in order to recruit downstream DDR and DSB repair components. Furthermore, at the completion of HR repair chromatin structure must be restored to the pre-damage state, a process that starts with dephosphorylation of γ-H2AX by protein phosphatase 2A (PP2A) or PP4C in mammals (Hanks et al., 1983). Removal of acetylation marks occurs by HDACs, which subsequently results in the condensation of chromatin back to its native configuration.
Besides the chromatin modifying factors, a non-histone chromatin protein known as HP1, initially identified in Drosophila and named for its predominant localization to pericentric heterochromatin (Li and Smerdon, 2002), plays a role in genomic stability (Sharma et al., 2003). There are three mammalian HP1 isoforms termed HP1α, HP1β (M31) (Cbx1), and HP1γ (M32) (Singh et al., 1991; Eissenberg and Elgin, 2000; Jones et al., 2000). HP1β is a dosage-dependent modifier of pericentric heterochromatin-induced silencing (Festenstein et al., 1999). HP1β plays a critical role in maintaining genomic stability in mammalian cells (Sharma et al., 2003; Aucott et al., 2008) with both negative (Ayoub et al., 2008; Goodarzi et al., 2008) as well as positive (Luijsterburg et al., 2009) effects on DNA damage repair. First, damage-dependent phosphorylation of HP1β decreases its chromodomain-dependent affinity for H3K9me3 leading to transient displacement of HP1β from DNA damage sites (Ayoub et al., 2008). In addition, HP1β depletion alleviates the ATM requirement for efficient DSB repair in heterochromatic regions (Goodarzi et al., 2008), suggesting HP1β suppresses repair in heterochromatic DNA regions. However, HP1β was also found to accumulate at DNA damage sites, indicating a more active involvement in DNA repair (Luijsterburg et al., 2009). In human cells, HP1β overexpression increased IR-induced chromosomal damage (Sharma et al., 2003). HP1β mobilization during DNA repair is regulated by ATM-dependent KAP-1 Ser473 phosphorylation (Ziv et al., 2006; Bolderson et al., 2012).
Conclusion and Future Directions
While the mechanisms by which cells activate the DDR and repair DNA damage are becoming clear, much less is known about how the cell restores the pre-damage original chromatin structure once damage is repaired. In addition, there is growing evidence that besides post DNA damage alteration of histone modifications, pre-existing histone PTMs play a critical role in the DDR and subsequent repair. Histone modification patterns change during the induction of DNA DSB as well as during the repair process. These changing modifications allow the chromatin to open at the sites of DNA damage but some modifications can hold the chromatin in a condensed state. Pre-existing chromatin modifications at the histone level can directly affect the initial DDR and subsequent pathway choice for NHEJ and HR. Modifications like H4K16ac or H4K20me2,regulated by MOF and MMSET, respectively, are epigenetic markers that could act as a histone code since the absence of appropriate markers results both in the loss of a DDR and subsequent repair (Gupta et al., 2005, 2008; Sharma et al., 2010; Hajdu et al., 2011; Bhadra et al., 2012). It will be important to establish the relative impact of epigenetic changes in the context of DNA sequence and their role in DNA DSB repair. Modulating histone acetylation with HDAC inhibitors can alter the IR-induced response resulting in radioprotection as well as radiosensitization (Camphausen and Tofilon, 2007). Small molecule inhibitors of histone modifying enzymes, therefore, represent a promising means for modulating radiotherapy response. As with other areas of basic research, understanding the significance of chromatin modifications as histone code will help to tailor radiotherapy based on patient and tumor epigenetic phenotype.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Jessica Tyler for her insightful thoughts and usage of her work. The work in Tej K. Pandita’s laboratory is supported by National Institutes of Health National Cancer Institute Grants R01CA123232, R01CA129537, R01CA154320, and U19A1091175.
References
Ahn, S. H., Cheung, W. L., Hsu, J. Y., Diaz, R. L., Smith, M. M., and Allis, C. D. (2005). Sterile 20 kinase phosphorylates histone H2B at serine 10 during hydrogen peroxide-induced apoptosis in S. cerevisiae. Cell 120, 25–36.
Anderson, L., Henderson, C., and Adachi, Y. (2001). Phosphorylation and rapid relocalization of 53BP1 to nuclear foci upon DNA damage. Mol. Cell. Biol. 21, 1719–1729.
Aucott, R., Bullwinkel, J., Yu, Y., Shi, W., Billur, M., Brown, J. P., et al. (2008). HP1-beta is required for development of the cerebral neocortex and neuromuscular junctions. J. Cell Biol. 183, 597–606.
Ayoub, N., Jeyasekharan, A. D., Bernal, J. A., and Venkitaraman, A. R. (2008). HP1-beta mobilization promotes chromatin changes that initiate the DNA damage response. Nature 453, 682–686.
Bakkenist, C. J., and Kastan, M. B. (2003). DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 421, 499–506.
Beck, B. D., Lee, S. S., Williamson, E., Hromas, R. A., and Lee, S. H. (2011). Biochemical characterization of metnase’s endonuclease activity and its role in NHEJ repair. Biochemistry 50, 4360–4370.
Berkovich, E., Monnat, R. J. Jr., and Kastan, M. B. (2007). Roles of ATM and NBS1 in chromatin structure modulation and DNA double-strand break repair. Nat. Cell Biol. 9, 683–690.
Bhadra, M. P., Horikoshi, N., Pushpavallipvalli, S. N., Sarkar, A., Bag, I., Krishnan, A., et al. (2012). The role of MOF in the ionizing radiation response is conserved in Drosophila melanogaster. Chromosoma 121, 79–90.
Bird, A. W., Yu, D. Y., Pray-Grant, M. G., Qiu, Q., Harmon, K. E., Megee, P. C., et al. (2002). Acetylation of histone H4 by Esa1 is required for DNA double-strand break repair. Nature 419, 411–415.
Boboila, C., Alt, F. W., and Schwer, B. (2012). Classical and alternative end-joining pathways for repair of lymphocyte-specific and general DNA double-strand breaks. Adv. Immunol. 116, 1–49.
Bolderson, E., Savage, K. I., Mahen, R., Pisupati, V., Graham, M. E., Richard, D. J., et al. (2012). Kruppel-associated Box (KRAB)-associated co-repressor (KAP-1) Ser-473 phosphorylation regulates heterochromatin protein 1beta (HP1-beta) mobilization and DNA repair in heterochromatin. J. Biol. Chem. 287, 28122–28131.
Bostelman, L. J., Keller, A. M., Albrecht, A. M., Arat, A., and Thompson, J. S. (2007). Methylation of histone H3 lysine-79 by Dot1p plays multiple roles in the response to UV damage in Saccharomyces cerevisiae. DNA Repair (Amst.) 6, 383–395.
Campbell, S. J., Edwards, R. A., Leung, C. C., Neculai, D., Hodge, C. D., Dhe-Paganon, S., et al. (2012). Molecular insights into the function of RING finger (RNF)-containing proteins hRNF8 and hRNF168 in Ubc13/Mms2-dependent ubiquitylation. J. Biol. Chem. 287, 23900–23910.
Camphausen, K., and Tofilon, P. J. (2007). Inhibition of histone deacetylation: a strategy for tumor radiosensitization. J. Clin. Oncol. 25, 4051–4056.
Cao, J., and Yan, Q. (2012). Histone ubiquitination and deubiquitination in transcription, DNA damage response, and cancer. Front. Oncol. 2:26. doi: 10.3389/fonc.2012.00026
Cao, R., Tsukada, Y., and Zhang, Y. (2005). Role of Bmi-1 and Ring1A in H2A ubiquitylation and Hox gene silencing. Mol. Cell 20, 845–854.
Cartwright, R., Dunn, A. M., Simpson, P. J., Tambini, C. E., and Thacker, J. (1998). Isolation of novel human and mouse genes of the recA/RAD51 recombination-repair gene family. Nucleic Acids Res. 26, 1653–1659.
Chapman, J. R., Taylor, M. R., and Boulton, S. J. (2012). Playing the end game: DNA double-strand break repair pathway choice. Mol. Cell 47, 497–510.
Chen, C. C., and Tyler, J. (2008). Chromatin reassembly signals the end of DNA repair. Cell Cycle 7, 3792–3797.
Cheung, W. L., Turner, F. B., Krishnamoorthy, T., Wolner, B., Ahn, S. H., Foley, M., et al. (2005). Phosphorylation of histone H4 serine 1 during DNA damage requires casein kinase II in S. cerevisiae. Curr. Biol. 15, 656–660.
Cook, P. J., Ju, B. G., Telese, F., Wang, X., Glass, C. K., and Rosenfeld, M. G. (2009). Tyrosine dephosphorylation of H2AX modulates apoptosis and survival decisions. Nature 458, 591–596.
Deem, A. K., Li, X., and Tyler, J. K. (2012). Epigenetic regulation of genomic integrity. Chromosoma 121, 131–151.
De Haro, L. P., Wray, J., Williamson, E. A., Durant, S. T., Corwin, L., Gentry, A. C., et al. (2010). Metnase promotes restart and repair of stalled and collapsed replication forks. Nucleic Acids Res. 38, 5681–5691.
Dellaire, G., Kepkay, R., and Bazett-Jones, D. P. (2009). High resolution imaging of changes in the structure and spatial organization of chromatin, gamma-H2A.X and the MRN complex within etoposide-induced DNA repair foci. Cell Cycle 8, 3750–3769.
de Napoles, M., Mermoud, J. E., Wakao, R., Tang, Y. A., Endoh, M., Appanah, R., et al. (2004). Polycomb group proteins Ring1A/B link ubiquitylation of histone H2A to heritable gene silencing and X inactivation. Dev. Cell 7, 663–676.
Doil, C., Mailand, N., Bekker-Jensen, S., Menard, P., Larsen, D. H., Pepperkok, R., et al. (2009). RNF168 binds and amplifies ubiquitin conjugates on damaged chromosomes to allow accumulation of repair proteins. Cell 136, 435–446.
Eissenberg, J. C., and Elgin, S. C. (2000). The HP1 protein family: getting a grip on chromatin. Curr. Opin. Genet. Dev. 10, 204–210.
Fang, J., Chen, T., Chadwick, B., Li, E., and Zhang, Y. (2004). Ring1b-mediated H2A ubiquitination associates with inactive X chromosomes and is involved in initiation of X inactivation. J. Biol. Chem. 279, 52812–52815.
Faucher, D., and Wellinger, R. J. (2010). Methylated H3K4, a transcription-associated histone modification, is involved in the DNA damage response pathway. PLoS Genet. 6:e1001082. doi:10.1371/journal.pgen.1001082
Fernandez-Capetillo, O., Allis, C. D., and Nussenzweig, A. (2004a). Phosphorylation of histone H2B at DNA double-strand breaks. J. Exp. Med. 199, 1671–1677.
Fernandez-Capetillo, O., Lee, A., Nussenzweig, M., and Nussenzweig, A. (2004b). H2AX: the histone guardian of the genome. DNA Repair (Amst.) 3, 959–967.
Festenstein, R., Sharghi-Namini, S., Fox, M., Roderick, K., Tolaini, M., Norton, T., et al. (1999). Heterochromatin protein 1 modifies mammalian PEV in a dose- and chromosomal-context-dependent manner. Nat. Genet. 23, 457–461.
Fischle, W., Tseng, B. S., Dormann, H. L., Ueberheide, B. M., Garcia, B. A., Shabanowitz, J., et al. (2005). Regulation of HP1-chromatin binding by histone H3 methylation and phosphorylation. Nature 438, 1116–1122.
Foster, E. R., and Downs, J. A. (2005). Histone H2A phosphorylation in DNA double-strand break repair. FEBS J. 272, 3231–3240.
Gapud, E. J., and Sleckman, B. P. (2011). Unique and redundant functions of ATM and DNA-PKcs during V(D)J recombination. Cell Cycle 10, 1928–1935.
Goodarzi, A. A., Noon, A. T., Deckbar, D., Ziv, Y., Shiloh, Y., Lobrich, M., et al. (2008). ATM signaling facilitates repair of DNA double-strand breaks associated with heterochromatin. Mol. Cell 31, 167–177.
Gupta, A., Guerin-Peyrou, T. G., Sharma, G. G., Park, C., Agarwal, M., Ganju, R. K., et al. (2008). The mammalian ortholog of Drosophila MOF that acetylates histone H4 lysine 16 is essential for embryogenesis and oncogenesis. Mol. Cell. Biol. 28, 397–409.
Gupta, A., Sharma, G. G., Young, C. S., Agarwal, M., Smith, E. R., Paull, T. T., et al. (2005). Involvement of human MOF in ATM function. Mol. Cell. Biol. 25, 5292–5305.
Haber, J. E. (2000). Partners and pathways repairing a double-strand break. Trends Genet. 16, 259–264.
Hajdu, I., Ciccia, A., Lewis, S. M., and Elledge, S. J. (2011). Wolf–Hirschhorn syndrome candidate 1 is involved in the cellular response to DNA damage. Proc. Natl. Acad. Sci. U.S.A. 108, 13130–13134.
Hanks, S. K., Rodriguez, L. V., and Rao, P. N. (1983). Relationship between histone phosphorylation and premature chromosome condensation. Exp. Cell Res. 148, 293–302.
Huen, M. S., Grant, R., Manke, I., Minn, K., Yu, X., Yaffe, M. B., et al. (2007). RNF8 transduces the DNA-damage signal via histone ubiquitylation and checkpoint protein assembly. Cell 131, 901–914.
Ikura, T., Tashiro, S., Kakino, A., Shima, H., Jacob, N., Amunugama, R., et al. (2007). DNA damage-dependent acetylation and ubiquitination of H2AX enhances chromatin dynamics. Mol. Cell. Biol. 27, 7028–7040.
Jones, D. O., Cowell, I. G., and Singh, P. B. (2000). Mammalian chromodomain proteins: their role in genome organisation and expression. BioEssays 22, 124–137.
Kolas, N. K., Chapman, J. R., Nakada, S., Ylanko, J., Chahwan, R., Sweeney, F. D., et al. (2007). Orchestration of the DNA-damage response by the RNF8 ubiquitin ligase. Science 318, 1637–1640.
Kouzarides, T. (2000). Acetylation: a regulatory modification to rival phosphorylation? EMBO J. 19, 1176–1179.
Kruhlak, M. J., Celeste, A., Dellaire, G., Fernandez-Capetillo, O., Muller, W. G., McNally, J. G., et al. (2006). Changes in chromatin structure and mobility in living cells at sites of DNA double-strand breaks. J. Cell Biol. 172, 823–834.
Kusch, T., Florens, L., Macdonald, W. H., Swanson, S. K., Glaser, R. L., Yates, J. R. III, et al. (2004). Acetylation by Tip60 is required for selective histone variant exchange at DNA lesions. Science 306, 2084–2087.
Li, S., and Smerdon, M. J. (2002). Nucleosome structure and repair of N-methylpurines in the GAL1-10 genes of Saccharomyces cerevisiae. J. Biol. Chem. 277, 44651–44659.
Li, X., Corsa, C. A., Pan, P. W., Wu, L., Ferguson, D., Yu, X., et al. (2010). MOF and H4 K16 acetylation play important roles in DNA damage repair by modulating recruitment of DNA damage repair protein Mdc1. Mol. Cell. Biol. 30, 5335–5347.
Lou, Z., Minter-Dykhouse, K., Franco, S., Gostissa, M., Rivera, M. A., Celeste, A., et al. (2006). MDC1 maintains genomic stability by participating in the amplification of ATM-dependent DNA damage signals. Mol. Cell 21, 187–200.
Luger, K., Mader, A. W., Richmond, R. K., Sargent, D. F., and Richmond, T. J. (1997). Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 389, 251–260.
Luijsterburg, M. S., Dinant, C., Lans, H., Stap, J., Wiernasz, E., Lagerwerf, S., et al. (2009). Heterochromatin protein 1 is recruited to various types of DNA damage. J. Cell Biol. 185, 577–586.
Mailand, N., Bekker-Jensen, S., Faustrup, H., Melander, F., Bartek, J., Lukas, C., et al. (2007). RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell 131, 887–900.
Marteijn, J. A., Bekker-Jensen, S., Mailand, N., Lans, H., Schwertman, P., Gourdin, A. M., et al. (2009). Nucleotide excision repair-induced H2A ubiquitination is dependent on MDC1 and RNF8 and reveals a universal DNA damage response. J. Cell Biol. 186, 835–847.
Mattiroli, F., Vissers, J. H., van Dijk, W. J., Ikpa, P., Citterio, E., Vermeulen, W., et al. (2012). RNF168 ubiquitinates K13-15 on H2A/H2AX to drive DNA damage signaling. Cell 150, 1182–1195.
Miller, K. M., Tjeertes, J. V., Coates, J., Legube, G., Polo, S. E., Britton, S., et al. (2010). Human HDAC1 and HDAC2 function in the DNA-damage response to promote DNA nonhomologous end-joining. Nat. Struct. Mol. Biol. 17, 1144–1151.
Mohammad, D. H., and Yaffe, M. B. (2009). 14-3-3 proteins, FHA domains and BRCT domains in the DNA damage response. DNA Repair (Amst.) 8, 1009–1017.
Mok, M. T., and Henderson, B. R. (2012). Three-dimensional imaging reveals the spatial separation of gammaH2AX–MDC1–53BP1 and RNF8–RNF168–BRCA1-A complexes at ionizing radiation-induced foci. Radiother. Oncol. 103, 415–420.
Moyal, L., Lerenthal, Y., Gana-Weisz, M., Mass, G., So, S., Wang, S. Y., et al. (2011). Requirement of ATM-dependent monoubiquitylation of histone H2B for timely repair of DNA double-strand breaks. Mol. Cell 41, 529–542.
Murr, R., Loizou, J. I., Yang, Y. G., Cuenin, C., Li, H., Wang, Z. Q., et al. (2006). Histone acetylation by Trrap–Tip60 modulates loading of repair proteins and repair of DNA double-strand breaks. Nat. Cell Biol. 8, 91–99.
Nakamura, K., Kato, A., Kobayashi, J., Yanagihara, H., Sakamoto, S., Oliveira, D. V., et al. (2011). Regulation of homologous recombination by RNF20-dependent H2B ubiquitination. Mol. Cell 41, 515–528.
Ogiwara, H., Ui, A., Otsuka, A., Satoh, H., Yokomi, I., Nakajima, S., et al. (2011). Histone acetylation by CBP and p300 at double-strand break sites facilitates SWI/SNF chromatin remodeling and the recruitment of non-homologous end joining factors. Oncogene 30, 2135–2146.
Oishi, H., Kitagawa, H., Wada, O., Takezawa, S., Tora, L., Kouzu-Fujita, M., et al. (2006). An hGCN5/TRRAP histone acetyltransferase complex co-activates BRCA1 transactivation function through histone modification. J. Biol. Chem. 281, 20–26.
Pandita, T. K. (2001). The role of ATM in telomere structure and function. Radiat. Res. 156, 642–647.
Pandita, T. K. (2003). A multifaceted role for ATM in genome maintenance. Expert Rev. Mol. Med. 5, 1–21.
Pandita, T. K., and Hittelman, W. N. (1992a). The contribution of DNA and chromosome repair deficiencies to the radiosensitivity of ataxia-telangiectasia. Radiat. Res. 131, 214–223.
Pandita, T. K., and Hittelman, W. N. (1992b). Initial chromosome damage but not DNA damage is greater in ataxia telangiectasia cells. Radiat. Res 130, 94–103.
Pandita, T. K., and Hittelman, W. N. (1995). Evidence of a chromatin basis for increased mutagen sensitivity associated with multiple primary malignancies of the head and neck. Int. J. Cancer 61, 738–743.
Pandita, T. K., Lieberman, H. B., Lim, D. S., Dhar, S., Zheng, W., Taya, Y., et al. (2000). Ionizing radiation activates the ATM kinase throughout the cell cycle. Oncogene 19, 1386–1391.
Pandita, T. K., and Richardson, C. (2009). Chromatin remodeling finds its place in the DNA double-strand break response. Nucleic Acids Res. 37, 1363–1377.
Paull, T. T., Rogakou, E. P., Yamazaki, V., Kirchgessner, C. U., Gellert, M., and Bonner, W. M. (2000). A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr. Biol. 10, 886–895.
Pei, H., Zhang, L., Luo, K., Qin, Y., Chesi, M., Fei, F., et al. (2011). MMSET regulates histone H4K20 methylation and 53BP1 accumulation at DNA damage sites. Nature 470, 124–128.
Qing, Y., Yamazoe, M., Hirota, K., Dejsuphong, D., Sakai, W., Yamamoto, K. N., et al. (2011). The epistatic relationship between BRCA2 and the other RAD51 mediators in homologous recombination. PLoS Genet. 7:e1002148. doi: 10.1371/journal.pgen.1002148
Rogakou, E. P., Pilch, D. R., Orr, A. H., Ivanova, V. S., and Bonner, W. M. (1998). DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 273, 5858–5868.
Rothkamm, K., Kruger, I., Thompson, L. H., and Lobrich, M. (2003). Pathways of DNA double-strand break repair during the mammalian cell cycle. Mol. Cell. Biol. 23, 5706–5715.
Sanders, S. L., Portoso, M., Mata, J., Bahler, J., Allshire, R. C., and Kouzarides, T. (2004). Methylation of histone H4 lysine 20 controls recruitment of Crb2 to sites of DNA damage. Cell 119, 603–614.
Schotta, G., Lachner, M., Sarma, K., Ebert, A., Sengupta, R., Reuter, G., et al. (2004). A silencing pathway to induce H3-K9 and H4-K20 trimethylation at constitutive heterochromatin. Genes Dev. 18, 1251–1262.
Scott, S. P., and Pandita, T. K. (2006). The cellular control of DNA double-strand breaks. J. Cell. Biochem. 99, 1463–1475.
Shahbazian, M. D., and Grunstein, M. (2007). Functions of site-specific histone acetylation and deacetylation. Annu. Rev. Biochem. 76, 75–100.
Sharma, G. G., Hwang, K. K., Pandita, R. K., Gupta, A., Dhar, S., Parenteau, J., et al. (2003). Human heterochromatin protein 1 isoforms HP1(Hsalpha) and HP1(Hsbeta) interfere with hTERT–telomere interactions and correlate with changes in cell growth and response to ionizing radiation. Mol. Cell. Biol. 23, 8363–8376.
Sharma, G. G., So, S., Gupta, A., Kumar, R., Cayrou, C., Avvakumov, N., et al. (2010). MOF and histone H4 acetylation at lysine 16 are critical for DNA damage response and double-strand break repair. Mol. Cell. Biol. 30, 3582–3595.
Shi, W., Ma, Z., Willers, H., Akhtar, K., Scott, S. P., Zhang, J., et al. (2008). Disassembly of MDC1 foci is controlled by ubiquitin-proteasome-dependent degradation. J. Biol. Chem. 283, 31608–31616.
Shim, E. Y., Hong, S. J., Oum, J. H., Yanez, Y., Zhang, Y., and Lee, S. E. (2007). RSC mobilizes nucleosomes to improve accessibility of repair machinery to the damaged chromatin. Mol. Cell. Biol. 27, 1602–1613.
Shogren-Knaak, M., Ishii, H., Sun, J. M., Pazin, M. J., Davie, J. R., and Peterson, C. L. (2006). Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science 311, 844–847.
Singh, P. B., Miller, J. R., Pearce, J., Kothary, R., Burton, R. D., Paro, R., et al. (1991). A sequence motif found in a Drosophila heterochromatin protein is conserved in animals and plants. Nucleic Acids Res. 19, 789–794.
Smilenov, L. B., Dhar, S., and Pandita, T. K. (1999). Altered telomere nuclear matrix interactions and nucleosomal periodicity in ataxia telangiectasia cells before and after ionizing radiation treatment. Mol. Cell. Biol. 19, 6963–6971.
Smith, E. R., Cayrou, C., Huang, R., Lane, W. S., Cote, J., and Lucchesi, J. C. (2005). A human protein complex homologous to the Drosophila MSL complex is responsible for the majority of histone H4 acetylation at lysine 16. Mol. Cell. Biol. 25, 9175–9188.
Sonoda, E., Hochegger, H., Saberi, A., Taniguchi, Y., and Takeda, S. (2006). Differential usage of non-homologous end-joining and homologous recombination in double strand break repair. DNA Repair (Amst.) 5, 1021–1029.
Stanlie, A., Aida, M., Muramatsu, M., Honjo, T., and Begum, N. A. (2010). Histone3 lysine4 trimethylation regulated by the facilitates chromatin transcription complex is critical for DNA cleavage in class switch recombination. Proc. Natl. Acad. Sci. U.S.A. 107, 22190–22195.
Stewart, G. S., Panier, S., Townsend, K., Al-Hakim, A. K., Kolas, N. K., Miller, E. S., et al. (2009). The RIDDLE syndrome protein mediates a ubiquitin-dependent signaling cascade at sites of DNA damage. Cell 136, 420–434.
Stucki, M., Clapperton, J. A., Mohammad, D., Yaffe, M. B., Smerdon, S. J., and Jackson, S. P. (2005). MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell 123, 1213–1226.
Sun, Y., Jiang, X., Xu, Y., Ayrapetov, M. K., Moreau, L. A., Whetstine, J. R., et al. (2009). Histone H3 methylation links DNA damage detection to activation of the tumour suppressor Tip60. Nat. Cell Biol. 11, 1376–1382.
Tamburini, B. A., and Tyler, J. K. (2005). Localized histone acetylation and deacetylation triggered by the homologous recombination pathway of double-strand DNA repair. Mol. Cell. Biol. 25, 4903–4913.
Telford, D. J., and Stewart, B. W. (1989). Micrococcal nuclease: its specificity and use for chromatin analysis. Int. J. Biochem. 21, 127–137.
Turner, B. M., Birley, A. J., and Lavender, J. (1992). Histone H4 isoforms acetylated at specific lysine residues define individual chromosomes and chromatin domains in Drosophila polytene nuclei. Cell 69, 375–384.
Utley, R. T., Lacoste, N., Jobin-Robitaille, O., Allard, S., and Cote, J. (2005). Regulation of NuA4 histone acetyltransferase activity in transcription and DNA repair by phosphorylation of histone H4. Mol. Cell. Biol. 25, 8179–8190.
van Attikum, H., Fritsch, O., and Gasser, S. M. (2007). Distinct roles for SWR1 and INO80 chromatin remodeling complexes at chromosomal double-strand breaks. EMBO J. 26, 4113–4125.
Van Hooser, A., Goodrich, D. W., Allis, C. D., Brinkley, B. R., and Mancini, M. A. (1998). Histone H3 phosphorylation is required for the initiation, but not maintenance, of mammalian chromosome condensation. J. Cell Sci. 111(Pt 23), 3497–3506.
Wang, G. G., Song, J., Wang, Z., Dormann, H. L., Casadio, F., Li, H., et al. (2009). Haematopoietic malignancies caused by dysregulation of a chromatin-binding PHD finger. Nature 459, 847–851.
Widlak, P., Pietrowska, M., and Lanuszewska, J. (2006). The role of chromatin proteins in DNA damage recognition and repair. Histochem. Cell Biol. 125, 119–126.
Wiener, R., Zhang, X., Wang, T., and Wolberger, C. (2012). The mechanism of OTUB1-mediated inhibition of ubiquitination. Nature 483, 618–622.
Williamson, E. A., Wray, J. W., Bansal, P., and Hromas, R. (2012). Overview for the histone codes for DNA repair. Prog. Mol. Biol. Transl. Sci. 110, 207–227.
Xiao, A., Li, H., Shechter, D., Ahn, S. H., Fabrizio, L. A., Erdjument-Bromage, H., et al. (2009). WSTF regulates the H2A.X DNA damage response via a novel tyrosine kinase activity. Nature 457, 57–62.
Yan, Q., Dutt, S., Xu, R., Graves, K., Juszczynski, P., Manis, J. P., et al. (2009). BBAP monoubiquitylates histone H4 at lysine 91 and selectively modulates the DNA damage response. Mol. Cell. 36, 110–120.
Yuan, J., Adamski, R., and Chen, J. (2010). Focus on histone variant H2AX: to be or not to be. FEBS Lett. 584, 3717–3724.
Zhang, Y., and Reinberg, D. (2001). Transcription regulation by histone methylation: interplay between different covalent modifications of the core histone tails. Genes Dev. 15, 2343–2360.
Keywords: histone modifications, DNA repair
Citation: Kumar R, Horikoshi N, Singh M, Gupta A, Misra HS, Albuquerque K, Hunt CR and Pandita TK (2013) Chromatin modifications and the DNA damage response to ionizing radiation. Front. Oncol. 2:214. doi: 10.3389/fonc.2012.00214
Received: 21 October 2012; Paper pending published: 05 December 2012;
Accepted: 29 December 2012; Published online: 22 January 2013
Edited by:
Mira Jung, Georgetown University, USACopyright: © 2013 Kumar, Horikoshi, Singh, Gupta,Misra, Albuquerque, Hunt and Pandita. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Tej K. Pandita, Department of Radiation Oncology, University of Texas Southwestern Medical Center, 5323 Harry Hines Boulevard, Dallas, TX 75390, USA. e-mail:dGVqLnBhbmRpdGFAdXRzb3V0aHdlc3Rlcm4uZWR1