 Qingda Meng1
Qingda Meng1 Martin Rao
Martin Rao Markus Maeurer
Markus Maeurer- 1Division of Therapeutic Immunology (TIM), Department of Laboratory Medicine (LABMED), Karolinska Institutet, Stockholm, Sweden
- 2Centre for Allogeneic Stem Cell Transplantation (CAST), Karolinska University Hospital, Stockholm, Sweden
KRAS is a driver mutation for malignant transformation. It is found in 30% of all cancers and in 90% of pancreatic cancers. The identification of small molecules selectively inhibiting KRAS mutants has been challenging, yet mutant KRAS has recently been shown to be targeted by tumor-infiltrating lymphocyte (TIL)-derived T cells that confer tumor regression upon adoptive transfer. Furthermore, a human IgG1 monoclonal antibody interfering with mutant KRAS function inside the cell has been described to inhibit growth of KRAS-mutant xenografts in tumor-bearing mice. B cells have been described to infiltrate pancreatic cancer and may be associated with tertiary lymphoid structures associated with good prognosis, or, in contrast, promote tumor growth. However, their function, nor their antigen-specificity has been clearly defined. We discuss here the presence of tumor-infiltrating B cells (TIB) in patients with pancreatic cancer that produce KRAS-mutant specific IgG, underlining that intratumoral T and B cells may exclusively target mutant KRAS. KRAS-specific IgG may, therefore, serve as a readout of the activation of both arms of the anti-tumor adaptive immune armament although some B-cell populations may promote tumor progression.
Background
The Kirsten Rat Sarcoma 2 Viral Oncogene Homolog (KRAS) gene and its product have had their renaissance several times. The most recent highlight came from the unsuspected identification of the association of mutant KRAS (in endothelial cells) driving arteriovenous malformation (AVM) in the CNS (1). The first KRAS renaissance, after the original description of mutant KRAS forms as a protooncogene (2), is manifested in the observation that mutant KRAS is found in approximately 30% of all human cancers. Lung (3) and gastrointestinal cancers commonly harbor KRAS mutations, with 90–95% of pancreatic cancer lesions (particularly pancreatic ductal adenocarcinoma (PDAC)) carrying a point mutation at the G12 locus (4), the most common being KRASG12D and KRASG12V. With the advent of novel therapies targeting the epidermal growth factor receptor (EGFR), the focus once again shifted to KRAS as a predictor of response to cancer therapy. Patients with KRAS-wild type colorectal cancer (CRC) will most likely, with a few reported exceptions, become refractory to targeted therapy (5). Nakadate and colleague reported that CRC cells harboring a KRAS-G13 mutation display resistance to cetuximab (anti-EGFR IgG1)-mediated antibody-dependent cellular cytotoxicity (ADCC) which involves Fas-FasL interaction (6). The heterogeneity and evolution of intra-tumoral KRAS mutations remains to be addressed in greater depth. Detailed analysis of tumor heterogeneity, measured as the tumor mutational burden (TMB), showed that up to 69% of all somatic mutations are not detectable across different tumor regions (7). Not surprisingly, tumor cell subpopulations with diverse KRAS mutations exist either within the same tumor lesion, or at different anatomical sites [for a review see Ref (8)]. KRAS mutations undermine EGFR-targeted therapies (5, 9) and variant KRAS-mutant tumor cells contribute to the outgrowth of EGFR-resistant tumor cell populations. Yet there is a different aspect. KRAS has been considered a viable tumor vaccine target, since mutant KRAS is exclusively present in cancer cells except for rare, dispersed cells within healthy colon tissue, although the direct effect of mutant KRAS in the oncogenesis of human and murine pancreatic cancer was recently addressed using CRISPR/Cas9-genome editing (10). In this study, Muzumdar et al. showed that KRAS deficiency in pancreatic cancer cells, achieved by inactivation of the gene by means of CRISPR/Cas9 excision, reduced cell growth kinetics in some PDAC cells lines and increased their dependence on phosphoinositide-3-kinase (PI3K) and downstream the mitogen-activated protein kinase (MAPK) activity for proliferation. As such, KRAS absence was also shown to sensitize PDAC cells to pharmacological PI3K/mammalian target of rapamycin (mTOR) inhibition.
To date, anti-KRAS vaccine studies have yet to deliver clinically promising results. However, a pivotal report from the Steven Rosenberg group has offered strong evidence for KRAS-directed immunotherapy: a patient with CRC exhibited clinically relevant remission upon infusion of tumor-infiltrating lymphocytes (TIL) targeting a mutant KRAS epitope (harboring the G12D mutation) restricted by HLA-C*w0802 (11). Furthermore, transgenic expression of the nominal T-cell receptor (TCR) targeting the KRAS mutant in recipient immune effector cells resulted in observable tumor regression: anti-KRASG12D T-cell responses cleared all mutation-positive metastatic lesions expect for one which lost the HLA-C*08:02 gene. Thus, mutant KRAS-specific T cells residing within tumor lesions can elicit clinically beneficial effects if the matching mutant KRAS epitope is naturally processed and presented (11). The patient's own immune cells have, therefore, been shown to target mutant KRAS, which was previously considered a “non-druggable” target. In addition to T cells, B cells can also target transformed cells expressing tumor-associated antigens (TAA) i.e. NY-ESO-1. This TAA was identified using serological analysis of recombinant cDNA expression libraries (SEREX), where antibodies are used to screen a protein expression library for specific reactivity (12–14). In vitro-cultivated, tumor-infiltrating B cells (TIBs) from non-small cell lung cancer (NSCLC)—which commonly harbors KRAS and EGFR mutations (3)–are able to present antigen to and activate CD4 TIL, while some TIB subsets can have an equally immunosuppressive effect on CD4+ T cells (15). Strong B-cell responses are often associated with strong T-cell responses, as in the context of anti-NY-ESO-1 reactivity (16–18) and as clinically observed in gastric cancer (19, 20). As such, the role of B cells in mediating anti-tumor immune responses due to their ability to produce cytokines as well as differentiate into antibody-secreting plasma cells in addition to professional antigen-presenting capacity has more recently received increased attention, even as a viable candidate for immunotherapy (21–24).
Unlike NY-ESO-1, efforts have been disappointing in identifying serum antibodies against KRAS in patients with a KRAS-positive malignancy. This may be possibly due to lower amounts of mutant KRAS protein expressed and subsequent lower antigen turnover in the tumor as compared to NY-ESO-1 expression, which is quite high based on immunohistological studies (25). Nevertheless, a proof-of-concept study in mice harboring various KRAS-mutant tumor xenografts showed that an anti-mutant KRAS antibody (RT11) in able to penetrate tumor cells and inhibit KRAS-mutated tumor-cell proliferation and disease progression (26).
The PDAC Tumor Microenvironment (TME)
Cancer cells proliferate and sustain growth in a complex network of non-transformed cells that may inhibit or promote cancer progression. In PDAC, this phenomenon engages a multilayer interaction between the tumor cells, stromal compartment as well as immune cells which infiltrate the tumor (27, 28). Antigen-specific CD4 and CD8 lymphocyte infiltration into pancreatic tumor lesions are generally viewed to be a positive prognostic factor for survival (29). In contrast, the accumulation of CD4+ CD25hi regulatory T cells (Tregs) contribute to immunosuppression in the TME and blunt productive anti-tumor responses, as demonstrated by depletion studies in preclinical models of PDAC (30–32). Transforming growth factor beta (TGF-β), a crucial biological mediator associated with Tregs, promotes fibrosis in the TME, which in turn inhibits the cytolytic function of cytotoxic CD8 T cells in addition to promoting tumor growth (33). There is also evidence that PD-L1-expressing TCR gamma delta (γδ) T cells can abrogate the tumor-directed activity of CD4 and CD8 T cells, which can be reversed by immune checkpoint blockade using anti-PD-L1 antibodies (34). Among non-immune cells are fibroblasts and different types of epithelial cells that contribute to the tumor architecture, maintain close reciprocation with immune and tumor cells and are associated with prognosis of patients with cancer (35).
An important characteristic of chronic immune activation in chronic infections as well as cancer is the development of tertiary lymphoid structures or organs (TLS or TLOs, respectively) containing B and T cells. This has been identified also in patients with pancreatic cancer; intra-tumoral rather than peri-tumoral TLOs with cellular infiltrates encompassing B and T cell populations in conjunction with lower immunosuppressive Tregs and increased expression of IFN-γ, IL-12, TBX21 (Th1-specific transcription factor) and IL-23 were associated with improved disease-free survival (p = 0.016) (36). However, CD1dhi CD5+ B cells which produce IL-35, in fact, contribute to the pathogenesis of pancreatic cancer and tumor progression, as shown in a mouse model of PDAC (37). Although PDAC is thought to exhibit an immunosuppressive microenvironment, the presence of cytotoxic immune cells proximal to transformed cells has been reported to be associated with increased patient survival despite desmoplastic histology in the tumor texture (38). T cells directed against pancreatic cancer cells by recognizing the patient's “private mutations” (private neoantigens) (39, 40) or against cancer-associated antigens, e.g., mesothelin (41, 42) and survivin (43), have been reported by various laboratories and demonstrate the possibility to successfully isolate and expand tumor-reactive T cells from pancreatic cancer specimens (39, 43, 44). Collectively, these findings suggest that although present in the TME, tumor-reactive T cells are unable to contain tumor progression due to unfavorable conditions therein.
Antibodies Against KRAS in Pancreatic Cancer-Associated B Cells
Anti-tumor cellular immune responses may be directed against: (i) non-mutant but overexpressed targets i.e. mesothelin, NY-ESO-1, survivin, (ii) private mutated proteins (neoantigens) as well as (iii) mutant targets that are shared among different individuals (shared neoantigen), such as KRAS (39). T cells infiltrating into PDAC and can recognize TAAs tend to be positively associated with patient survival—this observation is likely applicable to T cells targeting private mutations also (39, 45). However, the question of whether anti-PDAC antibodies are present—either circulating (serum-bound) or within the TME—remains to be fully addressed. Of note, bona-fide mutant KRAS-specific T cells have been primarily identified in situ, i.e., in the tumor lesion, and not in the peripheral circulation. This notion can now be complemented with anti-KRAS antibodies in situ.
TIL encompass not only T cells, but also B cells: their contribution to anti-tumor immune responses, however, was described to be negative. More recent studies show that some TIB subsets appear to be involved in promoting pancreatic tumorigenesis and cancer development in association with co-expression of CD1d and CD5 on their surface, IL-35 production as well as loss of intracellular expression of hypoxia-inducing factor 1 alpha (HIF-1α) (37, 46). Furthermore, TIBs expressing Bruton tyrosine kinase (BTK) can impede productive T-cell responses in the TME, although this immunosuppressive activity is pharmacologically reversible with ibrutinib (47). Thus, one might ask if there is a positive side to B cells in PDAC. We, therefore, tested the antigen specificity of PDAC-infiltrating B cells (TIB) by first isolating them from tumor tissue, immortalizing them using Epstein-Barr virus (EBV) infection and assessing their IgG production against wild type and mutant KRAS targets. Tumor tissue samples were obtained from eight patients with PDAC for generating TIL cultures comprising B and T cells separately, and the study was approved by Regional Ethics Review Board (Regionala etikprövningsnämnden) at Karolinska Institutet, Sweden (EPN: 2013/576-31 & 2013/977-31/1). In addition, the patients also provided written informed consent. TIB supernatant samples in serum-free medium (no human serum or FBS) were incubated in three 12-plex peptide microarray chips [format described in (48), which was previously reported from our group for human serum IgG reactivity to the H1N1 influenza proteome] embedded with 12-mer peptides from the human KRAS protein (UniProt ID: P01116) covering the most frequently occurring point mutations associated with cancer: G12D, G12V, G12R, G12S, G12C, G13D, Q61K, Q61H, Q61R, and A146T. The corresponding wild type/non-mutated sequences were also included. PDAC-infiltrating T cells had been expanded in vitro with IL-2, IL-15, and IL-21 and were later tested for KRAS wild type and mutant recognition defined by IFN-γ production. Genetic analysis, using a commercial kit, showed that 7/8 pancreatic cancer specimens (87.5%) carried a mutation at the G12 position. 4/7 patients (57.1%) had the KRASG12D mutation, two patients carried the KRASG12R mutation (25%), one patient tested positive for the KRASG12V mutation and tumor material from another patient harbored the KRASQ61H mutation. Wild type KRAS peptides were not recognized by any of the TIB-IgG samples. Only a single patient had TIB-IgG that recognized the endogenous KRAS mutation (PanTT41, KRASG12D; Figure 1A). TIB-IgG from the same patient also recognized two other KRAS mutations, namely KRASQ61K and KRASG12C. Although TIB-IgG from patient PanTT43 did not recognize the autologous KRAS mutation (G12R), reactivity to five other KRAS mutations was observed. TIB-IgG from patient PanTT55, positive for the KRASG12V mutation, also recognized four non-endogenous KRAS mutations (G12R, G12S, G13D, and Q61R). Only a single mutated KRAS target was recognized by TIB-IgG from 4/8 patients, while none of the KRAS targets tested were recognized by TIB-IgG from patient PanTT68. In general, mutations occurring at position G12, which cover approximately 98% of human PDAC tumors, were most frequently recognized by TIB-IgG from 5/8 patients tested. Representative photographs of immuno-histological staining from two patients with PDAC showed that T- and B-cell infiltration into the center of the pancreatic tumor tissue cluster together, indicating these cells are in close contact with the tumor parenchymal cells (Figure 1B). Only 1/3 T-cell cultures (expanded from TIL) recognized mutant KRASG12C (PanTT24) but not the patient's endogenous KRASG12D mutation after repeated exposure to the autologous tumor cell line (Meng et al. manuscript accepted for publication in the British Journal of Cancer, July 2018).
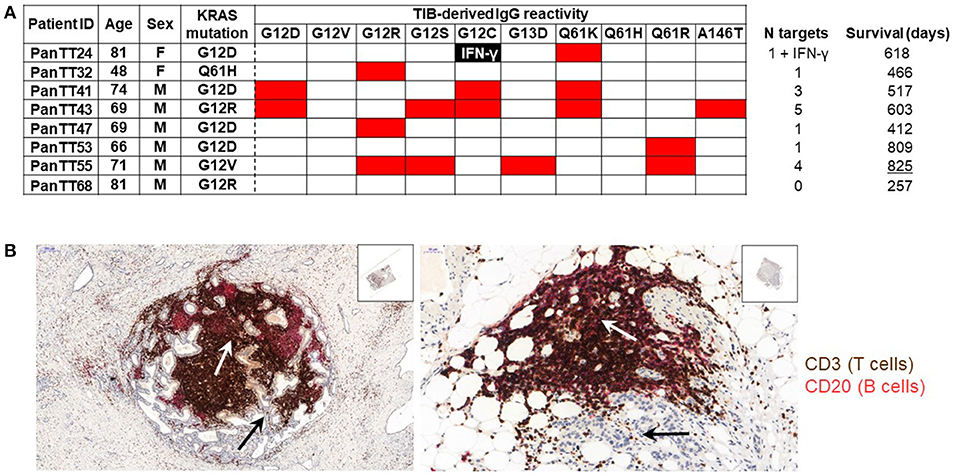
Figure 1. Recognition of mutated KRAS targets by patients' TIB-derived IgG. (A) TIB supernatants from eight patients with pancreatic adenocarcinoma (PDAC) were tested for IgG reactivity to KRAS mutated peptides (and the corresponding wildtype sequences) on a peptide microarray chip. Shown is a heat map, wherein red indicates a positive response while the blank boxes indicate the absence of a response. TIB-IgG reactivity to mutated KRAS targets appeared to be generally diverse. None of the TIB supernatants showed reactivity to any of the wildtype KRAS sequences, while only TIB-IgG from patient PanTT41 recognized the endogenous KRAS mutation. (B) Immunohistochemistry photos from representative pancreatic cancer tissue sections, focusing on the center of the tumor mass. The white arrows point to the tumor parenchyma (thus, the tumor cells within the PDAC lesion), while the black arrows point to clustering of T (brown) and B (pink/red) cells at the same location within the tumor parenchyma, indicating that lymphocytes are in close physical contact with the cancer cells. Details of the immunohistochemical staining protocol are included in the “Materials and Methods” section of the Supplementary Material. The Table indicates the patients ID (right column), gender and age, as well as the summary of the number of mutant KRAS targets recognized / TIB IgG sample and the survival time (after primary diagnosis). IFNγ production directed against a mutant KRAS epitope was observed in a single patient. Isolation and in vitro expansion of pancreatic cancer TIL in the presence of IL-2, IL-15, and IL-21 was performed as previously described (43, 56). TIB cultures were generated by culturing each tumor tissue fragment in 24-well plates containing 1 ml TIB medium (70% Cellgro GMP-grade serum-free medium (CellGenix), 20% B95-8 supernatant containing EBV virus (filtered with 0.22um filter), 10% FBS, penicillin (100 IU/mL), streptomycin (100 mg/mL; Life Technologies) and amphotericin B (2.5 mg/L; Sigma-Aldrich). Once a stable TIB cell line was established, the cultures were replenished with and maintained in TIB medium without the B95-8 supernatant.
Perspective For KRAS-Directed Ig in Tumor-Infiltrating B Cells
These data show that, akin to tumor-infiltrating T cells, B-cell responses exclusively targeting mutant and not wildtype KRAS are present in the TME yet not detectable in serum. Whether the recognition of non-endogenous (except in 1/8 specimen) KRAS mutations represents an ongoing or a past evolution of cancer cells with mutant KRAS (8) has to be evaluated. Like T-cell responses (11), immunological fine-tuning of antibody reactivity to cancer-associated mutated proteins in the TME may qualify TIBs as a viable source for the molecular blueprint of potential anti-tumor antibodies targeting neoantigens. TIBs bearing KRAS-specific BCRs could represent the cross-activation of the humoral immune response in patients with KRAS-mutant cancer the same way as the NY-ESO-1 paradigm in melanoma (12, 13, 16, 17, 25, 49, 50). The potentially tumor-promoting role of PDAC could be outsmarted, particularly if KRAS-directed B cells promote tumor progression upon binding of their BCR to the nominal target antigens and subsequent production of tumorigenic cytokines. Importantly, mutant KRAS-directed IgG responses may also indicate co-amplification of the T-cell response reflecting antigen processing and presentation by specific B cells to enhance immune surveillance even when antigen concentration may be limiting (51). The single-chain variable fragment (scFv) of specific anti-mutant KRAS antibodies may also serve as a scaffold for intracellular immunotoxins (26), which can be delivered via genes encoding antibody sequences packaged in liposomes or attached to nanoparticles (52). In a phase I/II clinical trial involving patients with locally advanced pancreatic cancer, RNA interference-based molecular therapy targeting KRASG12D as an adjunct to first-line chemotherapy was shown to be well-tolerated, with all patients displaying stable disease and two individuals even exhibiting a partial response (53). Furthermore, increased sensitivity to EGFR inhibition (54) and “druggability” of the focal adhesion kinase (FAK)—a crucial determinant of PDAC growth and aggressiveness (55)—is also achievable by targeting mutant KRAS. More cycles of KRAS renaissance are to be expected as biologically relevant tools for the targeted specific immunological therapy of patients with KRAS mutant cancer.
Author Contributions
QM performed experiments. DV was responsible for statistical analysis. MR wrote the initial draft, compiled and interpreted data, and assisted with the revision. MM initiated the work, conceptualized the information in the draft and wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are indebted to Prof. Matthias Löhr for KRAS determination, to Drs. Marco del Chiaro and Elena Rangelova for tissue procurement, Dr. Ernest Dodoo, Karolinska Hospital, Prof Elke Jäger, KHNW, Frankfurt, Germany for discussions; we are indebted to Dr. Carlos Fernando-Moro, Dept of Pathology, Karolinska Hospital for performing immunohistological analysis of tissue from patients with pancreatic cancer.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2018.00384/full#supplementary-material
References
1. Nikolaev SI, Vetiska S, Bonilla X, Boudreau E, Jauhiainen S, Rezai Jahromi B, et al. Somatic activating KRAS mutations in arteriovenous malformations of the brain. N Engl J Med. (2018) 378:250–61. doi: 10.1056/NEJMoa1709449
2. Der CJ, Krontiris TG, Cooper GM, Transforming genes of human bladder and lung carcinoma cell lines are homologous to the ras genes of Harvey and Kirsten sarcoma viruses. Proc Natl Acad Sci USA. (1982) 79:3637–40. doi: 10.1073/pnas.79.11.3637
3. Román M, Baraibar I, López I, Nadal E, Rolfo C, Vicent S, Gil-Bazo I. KRAS oncogene in non-small cell lung cancer: clinical perspectives on the treatment of an old target. Mol Cancer (2018) 17:33. doi: 10.1186/s12943-018-0789-x
4. Almoguera C, Shibata D, Forrester K, Martin J, Arnheim N, Perucho M. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell (1988) 53:549–54. doi: 10.1016/0092-8674(88)90571-5
5. Siravegna G, Mussolin B, Buscarino M, Corti G, Cassingena A, Crisafulli G, et al. Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. Nat Med. (2015) 21:795–801. doi: 10.1038/nm.3870
6. Yusuke N, Yasuo K, Yuka K, Senji S, Taro T, Tomohide T, et al. KRAS mutation confers resistance to antibody-dependent cellular cytotoxicity of cetuximab against human colorectal cancer cells. Int J Cancer (2014) 134:2146–55. doi: 10.1002/ijc.28550
7. Gerlinger M, Rowan AJ, Horswell S, Math M, Larkin J, Endesfelder D, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Eng J Med. (2012) 366:883–92. doi: 10.1056/NEJMoa1113205
8. Parsons BL, Myers MB. KRAS mutant tumor subpopulations can subvert durable responses to personalized cancer treatments. Per Med. (2013) 10:191–9. doi: 10.2217/pme.13.1
9. Pao W, Wang TY, Riely GJ, Miller VA, Pan Q, Ladanyi M, et al. KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med. (2005) 2:e17. doi: 10.1371/journal.pmed.0020017
10. Muzumdar MD, Chen PY, Dorans KJ, Chung KM, Bhutkar A, Hong E, et al. Survival of pancreatic cancer cells lacking KRAS function. Nat Commun. (2017) 8:1090. doi: 10.1038/s41467-017-00942-5
11. Tran E, Robbins PF, Lu YC, Prickett TD, Gartner JJ, Jia L, et al. Rosenberg SA, T-cell transfer therapy targeting mutant KRAS in cancer. N Eng J Med. (2016) 375:2255–62. doi: 10.1056/NEJMoa1609279
12. Chen YT, Scanlan MJ, Sahin U, Tureci O, Gure AO, Tsang S, et al. A testicular antigen aberrantly expressed in human cancers detected by autologous antibody screening. Proc Natl Acad Sci USA. (1997) 94:1914–8. doi: 10.1073/pnas.94.5.1914
13. Jager E, Chen YT, Drijfhout JW, Karbach J, Ringhoffer M, Jager D, et al. Simultaneous humoral and cellular immune response against cancer-testis antigen NY-ESO-1: definition of human histocompatibility leukocyte antigen (HLA)-A2-binding peptide epitopes. J Exp Med. (1998) 187:265–70. doi: 10.1084/jem.187.2.265
14. Neumann F, Wagner C, Kubuschok B, Stevanovic S, Rammensee HG, Pfreundschuh M. Identification of an antigenic peptide derived from the cancer-testis antigen NY-ESO-1 binding to a broad range of HLA-DR subtypes. Cancer Immunol Immunother. (2004) 53:589–99. doi: 10.1007/s00262-003-0492-6
15. Bruno TC, Ebner PJ, Moore BL, Squalls OG, Waugh KA, Eruslanov EB, et al. Antigen-presenting intratumoral B cells affect CD4(+) TIL phenotypes in non-small cell lung cancer patients. Cancer Immunol Res. (2017) 5:898–907. doi: 10.1158/2326-6066.CIR-17-0075
16. Jager E, Nagata Y, Gnjatic S, Wada H, Stockert E, Karbach J, et al. Monitoring CD8 T cell responses to NY-ESO-1: correlation of humoral and cellular immune responses. Proc Natl Acad Sci USA. (2000) 97:4760-5. doi: 10.1073/pnas.97.9.4760
17. Zeng G, Wang X, Robbins PF, Rosenberg SA, and Wang RF, CD4(+) T cell recognition of MHC class II-restricted epitopes from NY-ESO-1 presented by a prevalent HLA DP4 allele: association with NY-ESO-1 antibody production. Proc Natl Acad Sci USA. (2001) 98:3964-9. doi: 10.1073/pnas.061507398
18. Chen JL, Dawoodji A, Tarlton A, Gnjatic S, Tajar A, Karydis I, et al. NY-ESO-1 specific antibody and cellular responses in melanoma patients primed with NY-ESO-1 protein in ISCOMATRIX and boosted with recombinant NY-ESO-1 fowlpox virus. Int J Cancer (2015) 136:E590–601. doi: 10.1002/ijc.29118
19. Garnelo M, Tan A, Her Z, Yeong J, Lim CJ, Chen J, et al. Interaction between tumour-infiltrating B cells and T cells controls the progression of hepatocellular carcinoma. Gut (2017) 66:342–51. doi: 10.1136/gutjnl-2015-310814
20. Sakimura C, Tanaka H, Okuno T, Hiramatsu S, Muguruma K, Hirakawa K, et al. B cells in tertiary lymphoid structures are associated with favorable prognosis in gastric cancer. J Surg Res. (2017) 215:74–82. doi: 10.1016/j.jss.2017.03.033
21. Sabbatini P, Tsuji T, Ferran L, Ritter E, Sedrak C, Tuballes K, et al. Phase I trial of overlapping long peptides from a tumor self-antigen and poly-ICLC shows rapid induction of integrated immune response in ovarian cancer patients. Clin cancer Res. (2012) 18:6497–508. doi: 10.1158/1078-0432.CCR-12-2189
22. Teillaud JL, Dieu-Nosjean MC. Tertiary lymphoid structures: an anti-tumor school for adaptive immune cells and an antibody factory to fight cancer? Front. Immunol. (2017) 8:830. doi: 10.3389/fimmu.2017.00830
23. Germain C, Gnjatic S, Tamzalit F, Knockaert S, Remark R, Goc J, et al. Presence of B cells in tertiary lymphoid structures is associated with a protective immunity in patients with lung cancer. Am J Respir Crit Care Med. (2014) 189:832–44. doi: 10.1164/rccm.201309-1611OC
24. Fremd C, Stefanovic S, Beckhove P, Pritsch M, Lim H, Wallwiener M, et al. Mucin 1-specific B cell immune responses and their impact on overall survival in breast cancer patients. Oncoimmunology (2016) 5:e1057387. doi: 10.1080/2162402X.2015.1057387
25. Gnjatic S, Nishikawa H, Jungbluth AA, Gure AO, Ritter G, Jager E, et al. NY-ESO-1: review of an immunogenic tumor antigen. Adv Cancer Res. (2006) 95:1–30. doi: 10.1016/S0065-230X(06)95001-5
26. Shin SM, Choi DK, Jung K, Bae J, Kim JS, Park SW, et al. Antibody targeting intracellular oncogenic Ras mutants exerts anti-tumour effects after systemic administration. Nat Commun. (2017) 8:15090. doi: 10.1038/ncomms15090
27. Wachsmann MB, Pop LM, Vitetta ES. Pancreatic ductal adenocarcinoma: a review of immunologic aspects. J Invest Med. (2012) 60:643–63. doi: 10.2310/JIM.0b013e31824a4d79
28. Vonderheide RH, Bayne LJ. Inflammatory networks and immune surveillance of pancreatic carcinoma. Curr Opin Immunol. (2013) 25:200–5. doi: 10.1016/j.coi.2013.01.006
29. Wang WQ, Liu L, Xu HX, Wu CT, Xiang JF, Xu J, et al. Infiltrating immune cells and gene mutations in pancreatic ductal adenocarcinoma. Br J Surg. (2016) 103:1189–99. doi: 10.1002/bjs.10187
30. Nummer D, Suri-Payer E, Schmitz-Winnenthal H, Bonertz A, Galindo L, Antolovich D, et al. Role of tumor endothelium in CD4+ CD25+ regulatory T cell infiltration of human pancreatic carcinoma. J Natl Cancer Inst. (2007) 99:1188–99. doi: 10.1093/jnci/djm064
31. Leao IC, Ganesan P, Armstrong TD, Jaffee EM. Effective depletion of regulatory T cells allows the recruitment of mesothelin-specific CD8 T cells to the antitumor immune response against a mesothelin-expressing mouse pancreatic adenocarcinoma. Clin Trans Sci. (2008) 1:228–39. doi: 10.1111/j.1752-8062.2008.00070.x
32. Shevchenko I, Karakhanova S, Soltek S, Link J, Bayry J, Werner J, et al. Low-dose gemcitabine depletes regulatory T cells and improves survival in the orthotopic Panc02 model of pancreatic cancer. Int J Cancer (2013) 133:98–107. doi: 10.1002/ijc.27990
33. Principe DR, DeCant B, Mascariñas E, Wayne EA, Diaz AM, Diaz N, et al. TGFβ signaling in the pancreatic tumor microenvironment promotes fibrosis and immune evasion to facilitate tumorigenesis. Cancer Res. (2016) 76:2525–39. doi: 10.1158/0008-5472.CAN-15-1293
34. Daley D, Zambirinis CP, Seifert L, Akkad N, Mohan N, Werba G, et al. Gammadelta T cells support pancreatic oncogenesis by restraining alphabeta T cell activation. Cell (2016) 166:1485–99 e15. doi: 10.1016/j.cell.2016.07.046
35. Protti MP, De Monte L. Immune infiltrates as predictive markers of survival in pancreatic cancer patients. Front. Physiol. (2013) 4:210. doi: 10.3389/fphys.2013.00210
36. Hiraoka N, Ino Y, Yamazaki-Itoh R, Kanai Y, Kosuge T, Shimada K. Intratumoral tertiary lymphoid organ is a favourable prognosticator in patients with pancreatic cancer. Br J Cancer (2015) 112:1782–90. doi: 10.1038/bjc.2015.145
37. Pylayeva-Gupta Y, Das S, Handler JS, Hajdu CH, Coffre M, Koralov SB, et al. IL35-Producing B cells promote the development of pancreatic neoplasia. Cancer Disc. (2016) 6:247–55. doi: 10.1158/2159-8290.CD-15-0843
38. Carstens JL, Correa de Sampaio P, Yang D, Barua S, Wang H, Rao A, et al. Spatial computation of intratumoral T cells correlates with survival of patients with pancreatic cancer. Nat Commun. (2017) 8:15095. doi: 10.1038/ncomms15095
39. Tran E, Robbins PF, Rosenberg SA. “Final common pathway” of human cancer immunotherapy: targeting random somatic mutations. Nat Immunol. (2017) 18:255–62. doi: 10.1038/ni.3682
40. Balachandran VP, Luksza M, Zhao JN, Makarov V, Moral JA, Remark R, et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature (2017) 551:512–6. doi: 10.1038/nature24462
41. Meng Q, Valentini D, Rao M, Liu Z, Xie S, Morgell A, et al. Prediction of improved survival in patients with pancreatic cancer via IL-21 enhanced detection of mesothelin epitope-reactive T-cell responses. Oncotarget (2018) 9:22451–9. doi: 10.18632/oncotarget.25121
42. Thomas AM, Santarsiero LM, Lutz ER, Armstrong TD, Chen YC, Huang LQ, et al. Mesothelin-specific CD8(+) T cell responses provide evidence of in vivo cross-priming by antigen-presenting cells in vaccinated pancreatic cancer patients. J Exp Med. (2004) 200:297–306. doi: 10.1084/jem.20031435
43. Meng Q, Liu Z, Rangelova E, Poiret T, Ambati A, Rane L, et al. Expansion of tumor-reactive T cells from patients with pancreatic cancer. J Immunother. (2016) 39:81–9. doi: 10.1097/CJI.0000000000000111
44. Poschke I, Faryna M, Bergmann F, Flossdorf M, Lauenstein C, Hermes J, et al. Identification of a tumor-reactive T-cell repertoire in the immune infiltrate of patients with resectable pancreatic ductal adenocarcinoma. Oncoimmunology (2016) 5:e1240859. doi: 10.1080/2162402X.2016.1240859
45. Lohneis P, Sinn M, Bischoff S, Juhling A, Pelzer U, Wislocka L, et al. Cytotoxic tumour-infiltrating T lymphocytes influence outcome in resected pancreatic ductal adenocarcinoma. Eur J Cancer (2017) 83:290–301. doi: 10.1016/j.ejca.2017.06.016
46. Lee KE, Spata M, Bayne LJ, Buza EL, Durham AC, Allman D, et al. Hif1a Deletion reveals pro-neoplastic function of B cells in pancreatic neoplasia. Cancer Disc. (2016) 6:256–69. doi: 10.1158/2159-8290.CD-15-0822
47. Gunderson AJ, Kaneda MM, Tsujikawa T, Nguyen AV, Affara NI, Ruffell B, et al. Bruton tyrosine kinase-dependent immune cell cross-talk drives pancreas Cancer. Cancer Disc. (2016) 6:270–85. doi: 10.1158/2159-8290.CD-15-0827
48. Ambati A, Valentini D, Montomoli E, Lapini G, Biuso F, Wenschuh H, et al. H1N1 viral proteome peptide microarray predicts individuals at risk for H1N1 infection and segregates infection versus Pandemrix((R)) vaccination. Immunology (2015) 145:357–66. doi: 10.1111/imm.12448
49. Jager E, Stockert E, Zidianakis Z, Chen YT, Karbach J, Jager D, et al. Humoral immune responses of cancer patients against “Cancer-Testis” antigen NY-ESO-1: correlation with clinical events. Int J Cancer (1999) 84:506–10. doi: 10.1002/(SICI)1097-0215(19991022)84:5<506::AID-IJC10>3.0.CO;2-6
50. Jager E, Gnjatic S, Nagata Y, Stockert E, Jager D, Karbach J, et al. Induction of primary NY-ESO-1 immunity: CD8+ T lymphocyte and antibody responses in peptide-vaccinated patients with NY-ESO-1+ cancers. Proc Natl Acad Sci USA. (2000) 97:12198–203. doi: 10.1073/pnas.220413497
51. Rodríguez-Pinto D. B cells as antigen presenting cells. Cell Immunol. (2005) 238:67–75. doi: 10.1016/j.cellimm.2006.02.005
52. Trenevska I, Li D, Banham AH. Therapeutic antibodies against intracellular tumor antigens. Front Immunol. (2017) 8:1001. doi: 10.3389/fimmu.2017.01001
53. Golan T, Khvalevsky EZ, Hubert A, Gabai RM, Hen N, Segal A, et al. RNAi therapy targeting KRAS in combination with chemotherapy for locally advanced pancreatic cancer patients. Oncotarget (2015) 6:24560–70. doi: 10.18632/oncotarget.4183
54. Knickelbein K, Zhang L. Mutant KRAS as a critical determinant of the therapeutic response of colorectal cancer. Genes Dis. (2015) 2:4–12. doi: 10.1016/j.gendis.2014.10.002
55. Jiang H, Hegde S, Knolhoff BL, Zhu Y, Herndon JM, Meyer MA, et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat Med. (2016) 22:851–60. doi: 10.1038/nm.4123
Keywords: pancreatic cancer, KRAS mutations, antibody reactivity, tumor-infiltrating lymphocytes, immunotherapy, B cells
Citation: Meng Q, Valentini D, Rao M and Maeurer M (2018) KRAS RENAISSANCE(S) in Tumor Infiltrating B Cells in Pancreatic Cancer. Front. Oncol. 8:384. doi: 10.3389/fonc.2018.00384
Received: 23 June 2018; Accepted: 28 August 2018;
Published: 19 September 2018.
Edited by:
Fabrizio Mattei, Istituto Superiore di Sanità (ISS), ItalyReviewed by:
Kawaljit Kaur, University of California, Los Angeles, United StatesYong Qin, University of Texas MD Anderson Cancer Center, United States
Copyright © 2018 Meng, Valentini, Rao and Maeurer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Markus Maeurer, bWFya3VzLm1hZXVyZXJAZ21haWwuY29t
†Present Address: Markus Maeurer, Krankenhaus Nordwest, Frankfurt, Germany and Department of Immunotherapy, Champalimaud Foundation, Lisbon, Portugal