 Lisa Sevenich
Lisa Sevenich- Georg-Speyer-Haus, Institute for Tumor Biology and Experimental Therapy, Frankfurt, Germany
The development of immunotherapies has revolutionized intervention strategies for a variety of primary cancers. Despite this promising progress, treatment options for primary brain cancer and brain metastasis remain limited and still largely depend on surgical resection, radio- and/or chemotherapy. The paucity in the successful development of immunotherapies for brain cancers can in part be attributed to the traditional view of the brain as an immunologically privileged site. The presence of the blood-brain barrier and the absence of lymphatic drainage were believed to restrict the entry of blood-borne immune and inflammatory cells into the central nervous system (CNS), leading to an exclusion of the brain from systemic immune surveillance. However, recent insight from pre-clinical and clinical studies on the immune landscape of brain cancers challenged this dogma. Recruitment of blood-borne immune cells into the CNS provides unprecedented opportunities for the development of tumor microenvironment (TME)-targeted or immunotherapies against primary and metastatic cancers. Moreover, it is increasingly recognized that in addition to genotoxic effects, ionizing radiation represents a critical modulator of tumor-associated inflammation and synergizes with immunotherapies in adjuvant settings. This review summarizes current knowledge on the cellular and molecular identity of tumor-associated immune cells in primary and metastatic brain cancers and discusses underlying mechanisms by which ionizing radiation modulates the immune response. Detailed mechanistic insight into the effects of radiation on the unique immune landscape of brain cancers is essential for the development of multimodality intervention strategies in which immune-modulatory effects of radiotherapy are exploited to sensitize brain cancers to immunotherapies by converting immunologically “cold” into “hot” environments.
Introduction
Primary and metastatic brain tumors represent a challenging clinical issue. Glioblastoma (GBM) with an incidence of 2–3 per 100,000 population, is the most common primary brain tumor making up 54% of all gliomas and 16% of all primary brain tumors (1). Brain metastases (BrM), that most frequently arise from melanoma, breast- or lung cancers, are the most common intracranial tumor in adults and exceed the number of primary brain tumors by ~5-fold (2, 3). With the advent of improved control of systemic disease and increased life expectancy of cancer patients, the number of patients with brain metastases is rising (4). The development of cerebral tumors is associated with deteriorated quality of life due to headaches, epileptic seizures, and gradual cognitive impairment (5). Surgical resection, chemo- and radiotherapy (RT) remain the standard of care treatment for patients with brain tumors. Despite recent advances in the development of novel therapies against extracranial tumors, only very little progress has been made in the treatment of cerebral cancers. The majority of clinical trials with immunotherapies for GBM or BrM showed only moderate responses and did not significantly improve progression free survival (PFS) and overall survival (OS) (6). The lack of progress in the development of novel therapies for brain tumors can at least in part be attributed to the unique physiology of the central nervous system (CNS) and in consequence the highly complex brain tumor microenvironment (7). Brain tumors establish an immune suppressive tumor microenvironment that is characterized by high myeloid cell content together with relatively low tumor infiltrating lymphocyte (TIL) numbers and signs of T cell exhaustion (7). While immunotherapy alone fails to provide significant survival benefits for brain cancer patients, there is accumulating evidence, that adjuvant radiotherapy increases tumor immunogenicity and sensitizes brain tumors toward immunotherapy (8, 9).
The primary goal of radiotherapy is the induction of DNA damage in rapidly dividing tumor cells to induce different forms of cell death such as apoptosis or mitotic catastrophe (10, 11). In contrast to malignantly transformed tumor cells with impaired DNA repair mechanism, non-transformed stromal cells experience less damage given their post-mitotic state and intact DNA repair machinery (10). Although a link between irradiation and the immune system was proposed already 100 years ago (12), anti-tumor effects of radiotherapy were attributed to genotoxic effects on tumor cells, while effects on bystander cells were largely neglected for decades. Radiation dose and fractionation was therefore chosen to induce maximal damage in tumor cells and to spare bystander cells. However, traditional dose regimens might blunt important immune reactions directed against tumors. The discovery of immunogenic cell death (ICD) and abscopal effects provide formal proofs for immunological effects of radiation (13, 14). Abscopal effects describe the phenomenon that radiotherapy exerts anti-tumor effects in lesions outside the radiation field by triggering systemic anti-tumor effects (15). Therefore, exploiting the immune modulatory functions of radiotherapy represents an attractive tool to convert immunologically “cold” environments into “hot” environments to increase response rates of immunotherapy. This review will discuss preclinical and clinical evidence that support the applicability of radiotherapy as a sensitizer of immunologically inert tumors, such as GBM and BrM toward immunotherapy with a focus on immune checkpoint blockade (ICB). The field of radio-immunology is just at the beginning to understand the complex cellular and molecular effects of ionizing radiation (IR) on tumor cells and tumor-associated stromal cells that lead to more pronounced and long-lasting immune responses. In addition to clinical observations, it is important to employ preclinical models for systematic evaluation of different treatment regimens in terms of scheduling and dosage to maximize the synergy of radio-immunotherapy. Insight into cellular and molecular effects of radio-immunotherapy is critical to provide a strong scientific rationale for the development of multimodality intervention strategies.
Detailed understanding of the immune landscape of the central nervous system (CNS) at steady state and under pathological conditions is critical to appreciate immunological effects of radiotherapy in brain tumors. This review will therefore first summarize current knowledge on immune surveillance in the CNS and discuss how the development of primary and secondary brain tumors modulates the cellular composition of the TME and alters effector functions of tumor-associated immune cells. Based on this knowledge, different immunological aspects of brain tumors will be discussed to provide insight into the molecular basis of immunotherapy and radiotherapy in combination settings with a particular focus on differences in immune modulation depending on dose and fractionation of IR.
Immune Surveillance in the Central Nervous System
The CNS has traditionally been regarded as an immune privileged site that is excluded from systemic immune surveillance (16). Several observations constituted the concept of the immune privileged status of the CNS. First it was noted that the CNS fails to elicit an immune response against immunogenic material that was implanted into the brain parenchyma when avoiding the ventricles and meninges (17, 18). Moreover, the presence of the blood-brain-barrier (BBB) or blood-cerebrospinal fluid barrier (BCB) as well as the absence of lymphatic vessels as a route to the lymph node for antigen presenting cells (APC) further underpinned the concept of the CNS immune privilege (16). However, more detailed insights into the anatomical structures of the brain that represent an interface between the CNS and the periphery led to recent revisiting of the immune privilege of the CNS (16, 19). The use of single cell sequencing and single cell cytometric approaches helped to elucidate the complexity of immune populations in the steady state CNS (20). In this regard it is important to discriminate between brain regions that are excluded from systemic immune surveillance such as the parenchyma and areas at the border between the CNS and the periphery, including the meninges and the choroid plexus. The presence of the BBB and BCB restrict the entry of immune cell-types as well as the exchange of macromolecules into the brain parenchyma under physiological conditions (16, 21). Host defense is therefore performed by microglia, the brain-resident macrophages that constitute the largest population of immune cells in the CNS (22, 23) (Figures 1A,B). Parenchymal microglia are long living myeloid cells with self-renewal capacity that arise exclusively from the yolk sac and populate the brain during embryogenesis before the establishment of the BBB (24–27). As the innate immune cell of the brain, microglia exert key functions in immune surveillance, resolution of infection, wound repair, phagocytosis and debris removal (28). Moreover, microglia are involved in maintaining tissue homeostasis by mediating synaptic pruning, myelo- and neurogenesis as well as neuronal apoptosis (29, 30). However, compared to other cells of the macrophage lineage, microglia show lower antigen-presenting capacity (20, 31). While the brain parenchyma is tightly shielded from the systemic immune system, there are routes that peripheral leukocytes can utilize to enter the cerebral spinal fluid (CSF), choroid plexus, meninges, and the perivascular space (32). Border-associated myeloid cells (BAMs) including meningeal macrophages (mMF), choroid plexus macrophages (cMF), and perivascular macrophages (pvMF) populate those specialized locations in the CNS (33) (Figures 1A,B). While microglia exclusively originate from yolk sac-derived progenitors, BAM progenitors are of mixed ontological origin deriving from the yolk sac during primitive hematopoiesis and the fetal liver or bone marrow during definitive hematopoiesis (33). Single cell sequencing and mass cytometry (CYTOF) approaches indicate that BAMs show distinct gene expression signatures compared to microglia (20). Importantly, certain subsets of BAMs showed high CD38 and MHCII expression indicating a role in antigen presentation (20). In addition to the tissue-resident macrophages, Ly6Chi and Ly6Clow monocytes as well as dendritic cells constitute the myeloid compartment of the CNS (20, 28). Monocytes and DC are primarily localized in the meninges and choroid plexus (34–36). It was recently demonstrated that myeloid cells, i.e., neutrophils migrate through vascular channels in the skull-dura interface, indicating a direct local interaction between the brain and the skull bone marrow through the meninges as a route for immediate response to brain damage (37).
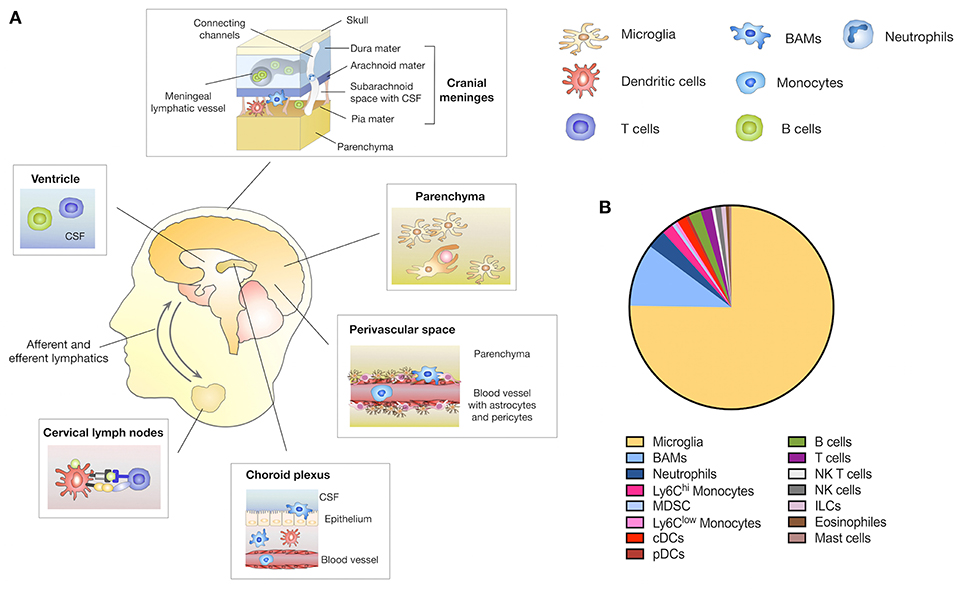
Figure 1. Immune compartments in the CNS. (A) The brain parenchyma is tightly shielded from the systemic immune system by the BBB and BCB that restricts the entry of peripheral immune and inflammatory cells. Yolk sac-derived microglia represent in the innate immune cell type within the brain parenchyma and exert key functions in host defense, immune surveillance and tissue homeostasis. In addition to the brain parenchyma, the CNS comprises structural interfaces that connect the CNS with the periphery, such as the choroid plexus, the meninges and the perivascular spaces. Border-associated macrophages (BAMs) are located in each of these niches. Circulating immune cells such as dendritic cells (DC), B- and T-cells are found in the CSF predominately in the meninges, ventricles and choroid plexus. A meningeal lymphatic system allows DC trafficking from the CNS to CNS draining lymph nodes i.e., cervical lymph nodes. Direct channels that connect the skull bone marrow with the dura enable neutrophils to migrate to the CNS as immediate responders to CNS damage. (B) Relative amount of distinct CNS immune cell populations located in the parenchyma and border areas of the CNS. Depicted amounts are based on data published in Mrdjen et al. (20).
Moreover, a range of lymphoid cells including B- and T cells as well as innate lymphoid cells (ILC), natural killer (NK), and natural killer T (NKT) cells constitute the lymphoid compartment in the CNS (20). Lymphocytes are absent from the brain parenchyma but can be found within the CSF of the meninges, choroid plexus and the ventricles. Importantly, the recent discovery that the CNS is directly connected to secondary cervical lymph nodes via a standard lymphatic drainage system fundamentally changed the concept of peripheral immune responses within the CNS (38, 39). There are three known routes by which intracranial antigens can traffic to CNS draining lymph nodes (40). The first is via ventricular and subarachnoid CSF that is able to cross the cribriform plate and enter the lymphatics draining into the deep cervical lymph nodes (41). Secondly, CSF is able to enter meningeal lymphatics located in the dura that also drain to the deep cervical lymph nodes (39). The third route results from parenchymal interstitial fluid trafficking through the basement membrane of the wall of capillaries and arteries of the brain (42). The first two routes are accessible to immune cells such as T cells, monocytes and DC as well as soluble antigens, while the third route is limited to soluble antigens (40).
The immune landscape in the CNS under steady state has recently been shown to be more complex than previously noted. Based on recent observations it was proposed to refer to the brain as an immunologically distinct rather than privileged site. Despite the description of the cellular constituents of the immune landscape in the CNS, it will be critical to evaluate to which extent the presence of lymphoid and myeloid cells in border-associated areas affects the immune privileged state of the brain parenchyma. In this regard, it is important to identify pathological stimuli that trigger infiltration of immune cells from border-associated areas as a route for immediate response or lead to recruitment of immune cells from the periphery and induction of a systemic response. There is accumulating evidence that different pathological conditions, including neurodegenerative disorders as well as cerebral cancers, induce fundamental changes in the cellular composition of the immune infiltrate and activation state of key players in neuro-inflammation (43). Importantly, recent studies revealed that in particular cells that are recruited from the periphery are implicated in the generation of an immune suppressive and cancer permissive environment, while brain-resident cells rather maintain host defense functions (31). This review will focus on tumor-associated inflammation in primary and metastatic brain cancers and highlight similarities and unique characteristics of immune responses that are provoked in the CNS during tumor progression. Understanding the complex immune landscape of brain cancers is critical to develop strategies to overcome the generation of an immune-suppressive environment and perturb traits of tumor cells to escape immune surveillance.
Brain Tumors Establish an Immune-Suppressive Environment
The development of primary and metastatic brain tumors disrupts the BBB leading to pronounced influx of blood-borne myeloid and lymphoid cells that are usually absent from the brain parenchyma. Tumor-associated macrophages (TAM) represent the most abundant stromal cell type in GBM and BrM often constituting up to 30% of the tumor mass (44, 45). Microglia and BMDMs share many phenotypic and functional similarities. The discrimination of both cell types in the context of brain cancers was previously challenging due to their overlapping marker expression and similar morphology in brain tumors. Lineage tracing approaches (46) and the recent discovery of specific markers (31, 47, 48) significantly contributed to our understanding of cell type specific functions of microglia and BMDMs during disease progression. It was long believed that tumor-associated microglia (TAM-MG) and tumor-associated bone marrow-derived macrophages (TAM-BMDM) exert similar functions in brain tumors. However, gene expression analysis of GBM-associated microglia and macrophages revealed that TAM-MG maintain gene signatures that are associated with house-keeping functions such as synaptic pruning and host defense and induce pro-inflammatory responses. In contrast, recruited TAM-BMDM showed gene signatures that are associated with wound healing, antigen presentation and immune suppression (31, 49) (Figure 2). Hence, functional differences of TAMs based on their ontological origin affect their contribution to disease progression and the ratio of TAM-BMDM to TAM-MG is expected to determine prognosis and therapeutic response especially of intervention strategies that aim to reinstate an effective anti-tumor immune response. In addition to evidence from the mouse models, single cell RNAseq analysis confirmed functional differences between TAM-BMDM and TAM-MG based on their ontological origin in human GBM (50). Interestingly, Müller et al. found that TAM-BMDM signatures correlate with significantly shorter survival in low-grade glioma (LGG) with similar trends in GBM, while there is no correlation between survival and TAM-MG signatures (50). Similar to GBM, it was also reported, that TAM-BMDM infiltrate BrM, although to a lesser extent (31). However, it remains unclear if BrM induce similar gene signatures in TAM-MG and TAM-BMDM as described in GBM. In addition to TAM-BMDM, tumor-infiltrating dendritic cells (DC) represent the most important antigen presenting cell type in brain tumors (51). The ability of DC to collect antigens in peripheral organs and to migrate to draining lymph nodes to activate and prime T cells is fundamental for cytotoxic T cell responses directed against specific antigens (52, 53). Given the immune privileged status of the brain, it remained unclear whether tumor-specific antigens in the CNS are surveyed by the immune system involving trafficking of DCs from CNS tumors to draining lymph nodes as well as trafficking of primed T cells from cervical lymph nodes into CNS tumors. To address the question on T cell trafficking, Prins et al., performed cell-tracking experiments to follow tumor antigen specific T cells after adoptive transfer of in vitro activated Pmel T cells (54). Imaging of T cell trafficking in this experimental system that is based on systemic vaccination revealed an early accumulation of T cells in all lymphoid organs including the cervical lymph nodes that drain the CNS and a subsequent accumulation in the bone marrow and brain tumors (54). Moreover, Garzon-Muvdi et al. employed an OVA-expressing GBM model with adaptive transfer of OT-1 T cells to identify the site of antigen presentation of tumor-antigens (55). The authors found proliferating OT1 T cells in cervical lymph nodes indicating that antigen presentation and T cell priming against tumor antigens might take place in the CNS draining lymph nodes (Figure 2). T cell expansion in the lymph nodes and anti-tumor effects were most pronounced in response to DC activation by the Toll-like Receptor (TLR)-3 agonist poly(I:C) in combination with PD-1 mediated immunotherapy (55), indicating that a strong proliferative stimulus is needed for effective T cell expansion. Moreover, it remains to be elucidated whether endogenous tumor antigens can elicit T cell priming or if this only occurs in response to highly immunogenic epitopes such as OVA. While the studies by Prins et al. and Garzon-Muvdi et al. provide evidence that under experimental conditions the proposed route of DC migration, T cell priming and local expansion might take place, it is important to acknowledge that the systems are based on strong experimental stimuli that are not expected in naturally grown CNS tumors. Hence, the formal proof of the route in which DCs migrate to cervical lymph nodes to prime T cells that subsequently traffic to CNS tumors to exert cytotoxic functions is still missing to date. While the question on the natural route for DCs and T cells remains to be addressed in CNS tumors, there is strong evidence that activated, cytotoxic T cells that infiltrate CNS tumors encounter a highly immune-suppressive milieu (56–59). Immune-suppression is particularly well-documented in GBM that are almost completely devoid of T cells (60). Quantitative deficits in the T cell compartment i.e., lymphopenia have been described for GBM patients since the late 1970 (61). It was recently demonstrated that glioblastoma as well as other intracranial tumors induce lymphopenia through sequestration of T cells in the bone marrow leading to a decline in T cell numbers at the tumor site and in lymphoid organs (62). Moreover, T cell apoptosis is induced through interactions of tumor cells via CD70-CD27 signaling (63, 64) or through astrocytes-derived FasL (65) at the tumor site. In addition to quantitative effects on T cells, qualitative deficits of T cells are a common phenomenon in patients with intracranial tumors (66). T cell dysfunction in brain tumors can be induced by a variety of mechanisms (67). High levels of immune-suppressive cytokines such as IL6, IL10, and TGFβ dampen T cell proliferation and effector functions (68). Tumor infiltrating lymphocytes (TILs) show high levels of PD-1, CTLA-4, LAG3, TIM3, TIGIT, and CD39 indicating T cell exhaustion (69–73). Regulatory T cells (Treg) comprise up to 30% of the TILs in GBM that further suppress T cell responses (57, 74). Tumor-associated macrophages and microglia have also been shown to support immune suppression and inhibit the expansion of CD4+ and CD8+ T cell expansion while inducing Treg production (31, 49, 75, 76).
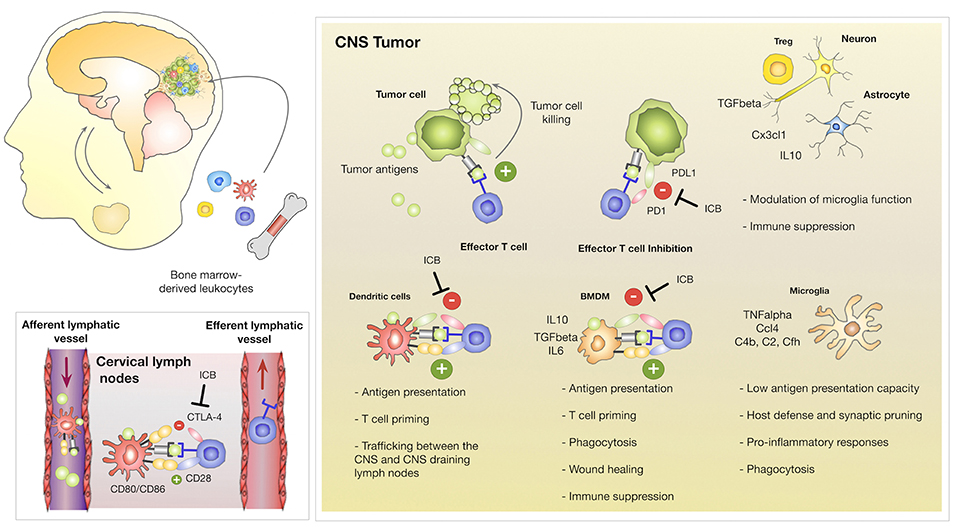
Figure 2. Innate and adoptive immune responses in CNS tumors. Brain tumors induce the recruitment of myeloid and lymphoid cells. Brain-resident and recruited cell types exert different functions within tumor-associated inflammation. Brain resident microglia maintain functions associated to their role as the innate immune cell of the CNS including host defense and synaptic pruning while bone marrow derived macrophages are associated with antigen-presentation, immune suppression and wound healing/tumor promoting functions. TAM-BMDMs express high levels of checkpoint molecules including PD-L1 to inactivate T cells. Dendritic cells traffic between CNS tumors and the cervical lymph nodes to prime T cells against tumor neo-antigens. T cells receive activating signals through interactions of the T cell receptor with antigens presented on MHC molecules and co-stimulation through interactions with CD28 and CD80/CD86. DC express the checkpoint molecule CTLA-4 that binds to CD28 on T cells to prevent activation of auto-reactive T cells. It remains unclear to which extent DCs activate or inhibit cytotoxic T cell functions in CNS tumors. Cytotoxic T cell responses are further inhibited by aberrant expression of checkpoint molecules on tumor cells as well as the secretion of immune-suppressive cytokines by Tregs, astrocytes and neurons. Checkpoint inhibitors (Immune Checkpoint Blockade; ICB) that block CTLA-4, PD-1, or PD-L1 unleash T cell effector functions to induce cytotoxic activity against tumor cells.
The immune landscape of BrM and its consequences on systemic and CNS immunity are less well-characterized compared to primary brain cancers. The question, if immune responses in BrM are predominately driven by the tissue environment or if the cellular identity of the tumor of origin (e.g., melanoma, lung, renal or breast cancer) shapes the mode of inflammation, is currently a field of active research. In contrast to GBM, BrM show moderate or even pronounced T cell infiltration. Several studies reported that the extent and pattern of T cell infiltration depends on the primary tumor entity that metastasizes to the brain (77, 78). T cell density is highest in melanoma with a diffuse pattern throughout the metastatic lesion, while renal-, lung- and breast cancer lead to a moderate T cell influx and T cells are dominantly localized within the stromal compartments of the tumor (77). Data from different studies indicate that T cell exhaustion also appears in brain metastasis. For example, expression of PD1 has been found in ~63% of TILs in melanoma brain metastasis (79, 80). A study by Harter et al. demonstrated that high TIL level, PD1+/CD8+ and PDL1 staining were associated with smaller lesions, however there was no significant association with survival (77). In contrast, a study by Berghoff et al., reported a significant correlation of the density of CD3+, CD8+, and CD45RO+ TILs with favorable median survival (78). As observed in GBM, immune suppressive cell types such as Tregs have also been shown to infiltrate experimental models of metastatic melanoma, breast and colon cancer within the brain and were found in patient brain metastases (57, 81–84).
Taken together, thorough investigation of the tumor microenvironment in GBM and BrM indicate that brain cancers contain the cellular and molecular constituents for therapeutic intervention by checkpoint inhibition. However, clinical data revealed that checkpoint inhibitors as mono-therapy often fail to significantly improve survival rates (85, 86). A possible explanation for the inability of checkpoint inhibitors to reinstate an anti-tumor response might be that the majority of infiltrating T cells are bystander cells that are not directed against specific tumor antigens (87). However, it is also possible that T cells with anti-tumor activity are present within brain tumors, but local immune suppression efficiently blunts their cytotoxic activity even in the presence of checkpoint inhibitors (Figure 2). If this is the case, therapeutic strategies that block immune suppression are required to sensitize brain tumors toward immunotherapy. In this context, immunological effects of radiotherapy recently attracted attention and a series of clinical trials have been initiated to test the efficacy of radiotherapy in combination with immunotherapy. The next paragraph will summarize the current status of standard of care and discuss insights from clinical trials with a focus on trials with ICB in GBM and BrM.
Clinical Management of GBM and BrM
Standard of Care
The current standard of care for newly diagnosed GBM is maximal surgical resection with concurrent radiotherapy and temozolomide (TMZ) chemotherapy followed by 6 months of adjuvant TMZ treatment (88). However, despite multimodality therapeutic intervention, GBM has an almost 100% relapse rate with a median time to recurrence of 7 months (89). The clinical situation for patients with recurrent GBM is extremely dire. Surgery is only considered for ~25% of the patients and re-irradiation is only possible as a palliative option in rare cases (90). Moreover, response rates to chemotherapy including TMZ rarely exceed 10% and no effects on OS have been reported (91–93).
Previous radiation regimen for brain metastasis patients involved whole brain radiotherapy (WBRT) with fractionated doses of 30 Gy in 10 fractions or 20 Gy in 5 fractions. Different clinical trials report a cerebral response following WBRT in 60% of patients and tumor volume reduction after WBRT has been associated with better neurocognitive function and prolonged survival (94). Median survival following WBRT alone in patients with multiple brain metastasis ranges from 3 to 6 months, with 10–15% of patients still alive at 1 year. However, numerous detrimental effects of WBRT in terms of acute and delayed neurotoxicity such as leuko-encephalopathy and loss of memory function as well as radiation necrosis have been described (95, 96). Given the lack of survival benefit and both short- and long-term toxicities associated with WBRT, recent guidelines from the European Association of Neuro-Oncology (EANO) recommend a deferment and replacement of WBRT by stereotactic radiosurgery (SRS) for the treatment of patients with a limited number of brain metastases and/or favorable prognostic factors (97). SRS is a single high dose radiation treatment with high accuracy in placing the irradiation field on tumor lesions and improved protection of surrounding tissue. The treatment efficacy of SRS is similar to surgical resection of brain metastases, with local control rates ranging from 80–85% (98). Clinical data also show, that the use of SRS after surgical resection significantly lowers local recurrence compared to surgery alone and that it is associated with a decreased risk of cognitive decline compared to WBRT (99, 100). Controversy remains over potential differences of SRS plus WBRT compared to SRS or WBRT alone. A recent meta-analysis that compared the outcome of patients with one or more brain metastases revealed no differences on survival for patients with multiple metastases, while a WBRT plus SRS improved survival in patients with single metastasis. Moreover, WBRT plus SRS resulted in significantly better local tumor control than WBRT alone (101).
Given the dismal prognosis for GBM and BrM patients, in particular patients with recurrent GBM or patients with multiple BrM it is evident that improved intervention strategies are urgently needed to provide better care for brain cancer patients. The introduction of immunotherapy into the clinics for select cancer types led to new hope for an improved management of primary and metastatic brain cancers.
Immunotherapy
Immune checkpoints are an important component of immune responses to keep cytotoxic activity of T cells under control to prevent autoimmunity. Cancers exploit this safety mechanism by up-regulation of checkpoint components on their cell surface to block T cell activity or by co-opting cells of the tumor microenvironment to establish an immune suppressive environment by dampening T cell responses. Checkpoint inhibitors unleash T cells from their inactive or exhausted state to induce anti-tumor responses (102) (Figure 2). To date, the most prominent examples have been antibodies that block the inhibitory immune checkpoint proteins cytotoxic T lymphocyte antigen 4 (CTLA-4), and PD-1 that are expressed predominantly on T cells, or PD-L1 that is expressed on different immune cells as well as aberrantly on tumor cells (103, 104). Consequently, successful immunotherapy by checkpoint inhibition relies on the natural ability of T cells to recognize and destroy malignant cells. While GBM and BrM are both characterized by highly immunosuppressive environments, GBM is further characterized by T cell exclusion and low mutational burden resulting in minimal neo-antigen generation (105). In contrast, BrM show moderate to high T cell content depending on the primary tumor entity and the majority of tumors that metastasize to the brain show high mutational load. However, it has to be taken into account that mutations that are found in brain metastasis are often not present in matched primary tumors (106). Data on brain metastasis patients are limited since those patients are often excluded from clinical trials. However, immunotherapies have demonstrated survival benefits for patients with tumors that frequently metastasize to the brain such as melanoma and NSCLC (107, 108). The use of checkpoint inhibitors in those patient cohorts allows for retrospective studies to evaluate the efficacy of checkpoint inhibitors against brain metastasis (109), which indicated efficacy of ICB in BrM. Given the potential beneficial effect of ICB in brain metastasis, a limited number of prospective trials have now been initiated to test the efficacy of immunotherapies in the treatment of brain metastases. First clinical trials to evaluate the efficacy of ipilimumab in patients with melanoma brain metastasis reported intracranial responses in 18% of patients with asymptomatic BrM without corticosteroid treatment while only 5% of symptomatic BrM patients on corticosteroid treatment showed intracranial responses (110). This finding further underpins the need that patients are not treated with corticosteroids at the time of ipilimumab treatment. Following clinical trials such as the ABC trial (NCT02374242) (111) and CheckMate-204 (112) aimed to test the efficacy of combining nivolumab and ipilimumab. Both trials report significant intracranial response rates of 46% in the combined treatment group compared to 20% in the nivolumab group in the ABC trial and 57% in the CheckMate-204 trial. Novel combinations are currently explored in clinical trials to further increase the intracranial response and to reduce adverse effects. For example, the activity and safety of the VEGF neutralizing antibody bevacizumab in combination with pembrolizumab or atezolizumab is tested in clinical trial for patients with untreated BrM (NCT02681549; melanoma and NSCLC and NCT03175432 BEAT-MBM; melanoma). Besides the effects of bevacizumab on angiogenesis, there is accumulating evidence, that VEGF blockade leads to reprogramming of the tumor microenvironment from an immunosuppressive to an immune permissive milieu, thus representing a promising combination together with immune checkpoint inhibitors (113). Results from these trials are still pending.
Although multiple factors indicate that GBM harbors intrinsic resistance against checkpoint inhibition as mono-therapy, pre-clinical testing showed promising results (114). The CheckMate143 trial (NCT0207717) was the first large-scale randomized clinical trial of PD pathway inhibition in GBM. However, treatment with the PD1 blocking antibody nivolumab failed to extent OS in patients with recurrent GBM, leading to a termination of this trial arm (86). In particular in the context of GBM it is important to take into account that in addition to the immunosuppressive tumor microenvironment, GBM patients often receive TMZ that is known to cause lymphopenia and permanently affect numbers of memory T cells (115). Moreover, corticosteroids such as dexamethasone are commonly used in the treatment of GBM patients to control cerebral edema. However, as already mentioned for BrM patients, corticosteroids are known to adversely affect the efficacy of immunotherapies (116).
In sum, the extent of immune suppression that is established in GBM and BrM together with effects from standard of care treatment that further dampens immune responses might ultimately prevent effective immunotherapies. Therefore, it is important to develop improved intervention strategies that overcome current obstacles to successful immunotherapy in cerebral tumors. In addition to the recently initiated clinical trials that aim to test the efficacy of the combination of different checkpoint inhibitors, there is increasing interest in the potential synergy of radiotherapy and immunotherapy.
Radio-Immunotherapy
Data from retrospective trials suggest that combinations of immunotherapy and radiotherapy significantly increase response rates and show effects on overall survival. For example, Knisely et al. reported that melanoma patients that received ipilimumab plus WBRT achieved longer median survival compared to WBRT alone [21.3 vs. 4.9 months] and a greater 2-year survival rate [47.2 vs. 19.7%] (117). Similarly, ipilimumab plus SRS was shown to increase OS from 5.3 to 18.3 months, while in this study no survival benefits for the combination of ipilimumab plus WBRT was reported (118). Sharverdian et al. reported that within the patient cohort that was enrolled in the KEYNOTE-001 trial (NCT01295827), NSCLC patients that received radiotherapy before pembrolizumab showed better PFS and OS compared to patients who did not receive radiotherapy (119). Ahmed et al. recently reported data from melanoma BrM patients that received nivolumab plus SRS demonstrating high rates of local BrM control of 91% and 85% at the 6 and 12 months follow-up (120). A central question that remains to be addressed in combination trials is the timing of each component for optimal outcome. Possible regimens comprise concurrent, sequential or neo-adjuvant application of the treatment modules (121). So far, the results suggest that the optimal schedule is tumor type and immunotherapy dependent. However, to date, the majority of trials report data that provide evidence for a benefit of concurrent schedules (122) and lowest response rate if radiotherapy is given after the immunotherapy. For example, a study of patients with melanoma brain metastasis showed that concurrent immunotherapy with anti-PD-L1 and anti-CTLA-4 showed improved response rates if immunotherapy was given within a time frame of 4 weeks after radiation compared to treatments that were more than 4 weeks apart (122). Dovedi et al. demonstrated that acquired resistance to fractionated radiation could be overcome by PD-L1 blockade using syngeneic mouse models of melanoma, colorectal and triple-negative breast cancer. However, the effect was only apparent if treatments were applied either concomitantly with or at the end of radiation. The effect was lost if PD-L1 blockade was given 1 week after radiotherapy (123). In contrast, a retrospective analysis of data from 758 patients suggested an improvement of OS with concurrent ICB and RT and hypo-fractionated RT particularly when immune checkpoint inhibition is started at least 1 month before RT, implying a benefit to commence ICB prior to RT (124). To date, pre-clinical and clinical evidence that optimal scheduling of radio-immuno-therapy critically affects the therapeutic response largely stems from studies on extracranial tumors and must be carefully considered for individual cancer types and different checkpoint inhibitors. Defining the optimal schedule for primary and metastatic brain tumors will require carefully designed prospective clinical trials in combination with systematic preclinical testing or mathematical modeling approaches as recently proposed by Serre et al. (125). While there are several ongoing clinical trials that aim to compare the efficacy of immune checkpoint inhibitors in combination with either WBRT or SRS, there are only few trials that are specifically designed to evaluate how different schedules affect safety and efficacy of combined treatment. One phase II clinical trial at the University of Michigan Cancer Center (NCT02097732) that considers the timing of immunotherapy is evaluating the efficacy of an “induction” of ICB prior to SRS [2 doses of ipilimumab prior to SRS, 2 doses of ipilimumab after SRS] vs. “no induction” [SRS first, followed by 4 doses of ipilimumab (3 mg/kg)]. In the future, more clinical trials that address the question on optimal scheduling will be required for conclusive results.
In sum, data from clinical and pre-clinical studies indicate that radiotherapy can act as a sensitizer for immunotherapy. Conventional fractionation takes advantage of the higher radio-sensitivity of tumor cells compared to normal cells with respect to DNA repair and cell cycle regulation. However, whether conventional fractionation represents the optimal strategy to maximize synergy with immunotherapy remains unclear to date. Moreover, it will be critical to evaluate whether radiation dose and fractionation that is optimal to induce an immune response in the CNS, is tolerated by the sensitive brain tissue. In order to optimize radiation dose and fractionation as well as scheduling for therapeutic application, it is essential to gain detailed insight into the molecular basis of genotoxic and immune modulatory effects of radiotherapy. The following paragraph will summarize current knowledge on effects of radiotherapy that result from direct damage on tumor cells (i.e., different forms of cell death) with subsequent effects on the inflammatory response against tumors. Moreover, local and systemic immune responses can also be modulated by radiation-induced changes in different cell populations of the tumor microenvironment (Figure 3). Direct and indirect effects on tumor cells and tumor-associated immune cells together determine the extent by which radiotherapy increases immunogenicity of tumors and the synergy between radio- and immunotherapy.
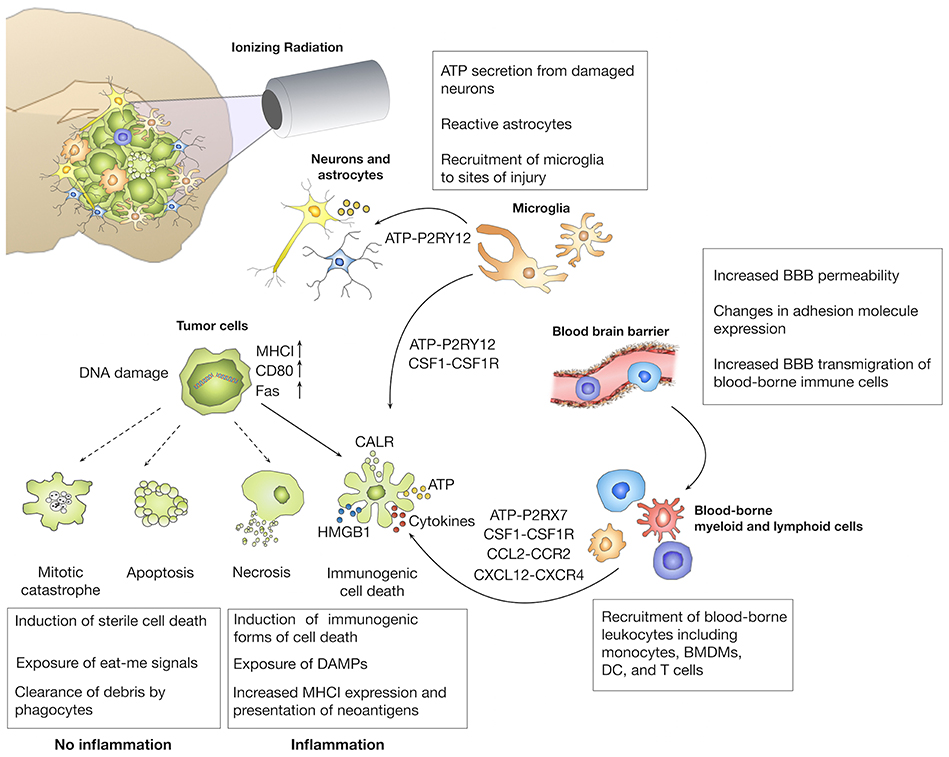
Figure 3. Immune modulatory effects of radiotherapy. The primary aim of radiotherapy is the induction of DNA damage in malignant cells. Depending on the extent of DNA damage, different forms of cell death are induced. The induction of immunogenic cell death (ICD) has been linked to the exposure of danger-associated molecular patterns (DAMP) that induce the recruitment and affect effector functions of brain-resident and recruited immune cells. Radiotherapy can also increase the immunogenicity of tumors by inducing presentation of neo-antigens by upregulation of MHC I molecules. In addition to effects on tumor cells, ionizing radiation also affects tumor-associated stromal cells including cells of the blood vessels and immune cells that further contribute to the establishment of radiation-induced immune responses.
Molecular Basis of Immune Modulatory Effects of IR
Radiation-Induced Immune Responses Depend on the Type of Cell Death by Which Tumor Cells Are Killed
The central dogma of traditional radiobiology states that effects of radiation on tumor cells are primarily due to the generation of double strand breaks that lead to the induction of different forms of cell death including apoptosis, necrosis, autophagy, or mitotic catastrophe (Figure 3). Apart from the notion that necrosis elicits inflammation due to the release of cellular content, radiotherapy has long been regarded as an immunologically inert process. While immunological effects of radiotherapy were neglected for decades, several discoveries established a link between the immune system and the ability of radiotherapy to achieve tumor control. Stone et al. demonstrated already end of the 70s, that the radiation dose that is required to control tumor growth was twice as high in immune-compromised mice compared to immune-competent mice (126). Moreover, the occurrence of abscopal effects in which tumor control is achieved in lesions outside the radiation field provides further proof for the contribution of the immune system in tumor control (127). The discovery of immunogenic cell death (ICD) as a molecularly defined processes that leads to priming and activation of immune cells recently led to a paradigm shift. ICD is characterized by the cell surface translocation of calreticulin (CRT), the extracellular release of HMGB1 (High motility group box 1) and of adenosine triphosphate (ATP) (128). Radiotherapy has been shown to induce all three arms of ICD and is therefore regarded as a potent inducer of ICD (129). Different doses or fractionations are believed to induce different forms of cell death (130), and thus modulate downstream cellular responses. Radiation regimens that induce immunologically silent forms of cell death, i.e., apoptotic cell death are therefore not expected to synergize with ICB, while doses and fractionation that trigger inflammatory responses could be used as immune-modulators to induce additive effects of radiotherapy and immunotherapy (131).
Radiation Increases Recognition of Tumor Antigens by the Immune System
Within the process of immune evasion, tumor cells acquire traits that mask the tumor from immune surveillance and destruction. The immune escape stage is characterized by up-regulation of inhibitory ligands and cytokines, reduced MHCI expression, and increased number of suppressive cell types such as Tregs (132). Radiotherapy has been reported to unmask the tumor and thus make it visible again for the innate and adaptive immune system (133). Radiation can up-regulate MHCI expression on the tumor cell surface to enable better antigen presentation of tumor-specific peptides for recognition by cytotoxic T cells (134). Mutational load of tumors is known to correlate with therapeutic response rates to immunotherapy (135–138). Identification of mutations that drive anti-tumor responses would therefore be of great benefit to identify patients with a high chance of responding to immunotherapy. Moreover, radiation-induced DNA damage can cause an increase in mutational load (139). While an increase in mutational burden might enhance tumor aggressiveness, it might also generate neoantigens that can be recognized and targeted by the immune system (140). Indeed it was demonstrated that IR induces novel peptide synthesis in tumor cells and enhances antigen presentation by MHC class I molecules (134, 141). On the other hand, there is evidence that brain tumors show higher systemic tolerance than tumors at extracranial sites. For example, Jackson et al. employed the B16 melanoma model to compare cytotoxic responses against tumors in the CNS and in the periphery. The study showed that CNS melanomas were more tolerogenic than tumors in extracranial sites due to antigen-specific CD8 T cell depletion leading to impaired systemic antitumor immunity (142). The authors concluded, that the observed T cell dysfunction was mainly caused by elevated levels of microglia-derived TGF-β (142). Interestingly, it was demonstrated that the effect of systemic tolerance was reversible by radiotherapy and vaccination.
The cGas-STING Axis in Anti-tumor Immunity
Radiation-induced DNA damage that causes leakage of DNA into the cytosol is known to be sensed by the stimulator of interferon gens (STING) leading to the activation of innate and adaptive immune responses (143). The STING pathway has originally been described as a host defense mechanism to protect organisms against infection with DNA pathogens. When cytosolic DNA is detected, the product of cyclic GMP-AMP synthase (cGAS), cyclic GMP-AMP (cGAMP) activates STING. STING induces the transcription of type I interferon genes via a cascade that involves the STING downstream factors Tank binding kinase (TBK), interferon regulatory factor 3 (IRF3) and nuclear factor kappa light chain enhancer of B cells (NFkB) (144). A growing body of literature suggests that the STING pathway plays a central role in anti-tumor immunity and its expression is lost in several cancer types including colorectal cancer and melanoma (145, 146). While several studies linked radiation-induced STING activity to type-I-interferon mediated anti-tumor immunity, there is also evidence that STING activation could drive immune-suppression and radio-resistance via CCR2 mediated recruitment of MDSCs (147). Given the complex cellular and molecular interactions of STING mediated immunological effects, clearly more systemic studies are needed to gain comprehensive mechanistic insight.
Dose and Fractionation Are Critical Parameters for Effective Induction of Immunogenicity
Determining the optimal scheduling for radio-immunotherapy is a major challenge for the field and requires carefully designed prospective clinical studies together with comprehensive studies in animal models to test effects of different treatment regimens (148, 149). As discussed above, data from retrospective and prospective clinical trials suggests that treatment schedules in which radiotherapy was given as concurrent, sequential, or neoadjuvant therapy lead to different therapeutic efficacy. Several preclinical studies compared single high-dose with fractionated radiation for their ability to induce immune responses. For example, in a B16-OVA model, both single dose (15 Gy) and fractionated radiation (5x3 Gy) increased the generation of antigen-specific T cells. However, the single 15 Gy dose generated more tumor-infiltrating T cells than conventional fractionation (150). Later it was demonstrated, that the immune response triggered by ablative radiation doses was abrogated by conventional fractionation (151). Moreover, Camphausen et al. demonstrated in a model of Lewis lung carcinoma that 5 × 10 Gy induced more robust abscopal effects than 12 × 2 Gy (152). However, hypo-fractionation might not be favorable when combined with immunotherapy. Dewan et al. demonstrated in a breast cancer model that an abscopal effect was only induced in response to fractionated radiation, not single dose radiation when combined with CTLA-4 inhibition (153). A potential explanation for dose dependent effects of radiation was recently provided by the balance between activation of cGAS-STING signaling vs. Trex1 activation (154). Extremely high single doses (20-30Gy) were shown to blunt immunogenicity by the induction of the DNA exonuclease Trex1. Trex1-mediated degradation of cytosolic DNA consequently abrogates cGAS-STING activation and downstream IFN type1 production. In this study, CTLA-4 blockade did not synergize with high dose irradiation to induce abscopal effects. However, knockdown of Trex1 reinstated synergistic effects of anti-CTLA-4 in combination with high dose radiation (20Gy) (155).
Radiation Modulates the Cellular Composition of the Tumor Microenvironment and Affects Effector Functions of Immune Cells
Another important factor that determines synergy of radio-immunotherapy is the cellular composition of the tumor microenvironment (156). As discussed in the paragraph above, in particular the tumor microenvironment of brain tumors represents a highly complex milieu with brain resident and recruited immune cells (7). Brain tumors are known to establish immune-suppressive environments that are characterized by high myeloid cell content and low percentage of CD8+ effector T cells. Several studies demonstrated that radiotherapy induces increased influx of immune cells into brain tumors. This effect can in part be attributed to effects on the vasculature (157, 158). IR also has profound effects on the secretion of cytokines that serve as chemo-attractants for different immune cells including DC and macrophages (159). In addition, IR has been shown to affect key effector functions such as phagocytosis, antigen presentation, and cytotoxicity and alters activation states of immune cells (160–162). Moreover, radio-sensitivity of T cells has to be taken into account when testing optimal dosage and fractionation. Since immunotherapies rely on functional T cells, their ablation or inactivation is expected to abrogate critical anti-tumor immune responses. Tumor-infiltrating T cells are exposed to radiation and it has been shown that conventional 2 Gy doses given once daily can inactivate T cells (163). This effect is also evidenced by the occurrence of lymphopenia as a common adverse effect associated with whole brain radiotherapy (164) that could significantly dampen anti-tumor immune responses.
Taken together, radiation dose and fractionation have profound effects on the induction of genotoxic and immunogenic effects. Systematic interrogation of the dose dependency of immune responses directed against different cancer types is needed to determine optimal regimens to increase the immunogenicity of tumors and boost the immune system for effective anti-tumor responses that synergize with immunotherapy.
Combination of IR and ICB—Future Perspectives
To date, clinical and pre-clinical data suggest that combining radiotherapy with immunotherapy show higher efficacy compared to mono-therapies. These results represent promising first steps in the quest for improved treatment options for brain cancer patients. However, it is also evident that many hurdles exist that prevent higher response rates and more sustainable anti-tumor reactions. While individual patients show prominent cerebral responses, significant effects on overall survival are rarely reported. This indicates that the pressure of the CNS to establish an immunosuppressive environment is dominant over the attempt to unleash the immune system by immune checkpoint blockade. Based on our current mechanistic understanding of the cellular and molecular drivers of immune-suppression in the steady-state CNS and in the context of cerebral cancers, different approaches appear as viable strategies to overcome the highly immune-suppressive environment in the CNS. Based on the results that TAM-BMDM rather than TAM-MG are implicated in tumor-promotion and immune-suppression, selective depletion or blockade of TAM-BMDM recruitment could lead to more effective T cell activation and execution of anti-tumor effector functions. In addition, systems that would allow more efficient recruitment of T cells into CNS tumors could significantly boost cytotoxic T cell responses directed against tumor neo-antigens. A recent study by Samaha et al. reported the engineering of T cells with an Activated Leukocyte Cell Adhesion Molecule (ALCAM) homing system (HS) (165). In this approach, CD6 (the ligand for ALCAM) was re-engineered to trigger initial anchorage to ALCAM followed by adhesion to ICAM1 expressed on cancer endothelium. Cytotoxic HS T cells infiltrated brain tumors after intravenous injection and showed potent anti-tumor activity. Other strategies that aim to convert immune-suppressive milieus into inflamed environments might utilize neutralizing antibodies against suppressive cytokines such as TGFβ, IL10, or IL6. Moreover, activation of adenosine signaling has been associated with immune-suppression and acquisition of resistance against immunotherapy in different cancer types including melanoma (166–169). Pharmacological inhibition of enzymes that process ATP into adenosine, i.e., CD39/Entpd1 and CD73/Nt5e or targeting of adenosine receptors are currently evaluated for their potential to block the conversion of a purine-driven, pro-inflammatory environment into an adenosine-driven, immune-suppressive milieu (170–173). Another promising strategy could employ Trex1 inhibitors to prevent the degradation of cytosolic DNA to more efficiently induce cGAS-STING-IRF signaling to trigger innate immune responses (155). Overcoming the immune-suppressive environment appears to be one of the limiting factors for successful immunotherapy against brain cancers. However, it is also important to keep in mind, that immune suppression is an important safety mechanism that protects the brain from excessive inflammation. Inflammatory responses are often associated with swelling that would harm the delicate structures of the CNS and ultimately lead to brain damage. The increased risk of auto-immunity has been reported in several clinical trials with combination of different ICB. The most recent data on the CheckMate143 trial report that 9 of 10 patients treated with a combination of nivolumab and ipilimumab experienced grade 3 or 4 adverse events with 4 of 10 patients discontinuing therapy due to side effects (83). Therapeutic strategies that aim to convert immune suppressive milieus into inflamed environments should therefore be carefully considered and the potential risk of inducing autoimmunity should be evaluated. Detailed mechanistic insight into pathways that are implicated in cancer-associated immune-suppression and inherent or acquired resistance against brain tumors will hopefully lead to the development of novel multimodality intervention strategies that meet the safety and efficacy criteria for the induction of more efficient and long-lasting anti-tumor immune responses in GBM and BrM patients.
Concluding Remarks
A close link between radiotherapy and the immune system has been proposed already 100 years ago by Russ and Murphy (12). Murphy's observation that “large doses of x-rays, by destroying the immune conditions, will favor the growth of tumors while small doses, by producing immune conditions will help to overcome the tumor” closely resembles the current view on effects of RT on cancer-associated inflammation (174). Yet, immunogenic effects of radiotherapy have been neglected for decades. The introduction of immunotherapy into the clinic and more detailed molecular insights into the underlying mechanism of immunogenic effects of radiation have recently attracted attention to radiotherapy as a potent modulator of cancer-associated inflammation. While immunotherapy is highly effective for patients with inflamed or hot tumor environments, large patient cohort remain unresponsive to immunotherapy due to intrinsic or acquired resistance. Although we are just at the beginning to understand the cellular and molecular basis that distinguish responders from non-responders, accumulating clinical and pre-clinical data indicate that immunogenic effects of radiotherapy convert cold into hot environments and thus sensitize unresponsive tumors toward immunotherapy. Our current knowledge on radio-immunotherapy against brain cancers is largely based on retrospective clinical trials or pre-clinical studies on animal models. The initiation of clinical trials and pre-clinical studies that aim to systematically evaluate the effects of different fractionation and treatment regimens is needed to provide deeper insight into optimal schedules to induce synergy between immunotherapy and radiotherapy. While there are certainly indications that favor specific treatment regimens, it is still too early to draw conclusions on effects on PFS or OS. To further improve the response rate, it will be critical to identify molecular pathways that determine the mode of immune responses and to develop strategies that efficiently induce anti-tumor immune responses without the risk of inducing auto-immunity.
Author Contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
LS is grateful to support by the Max-Eder Junior Group Leader Program (German Cancer Aid, Deutsche Krebshilfe).
References
1. Ostrom QT, Gittleman H, Farah P, Ondracek A, Chen Y, Wolinsky Y, et al. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2006-2010. Neuro Oncol. (2013) 15(Suppl 2):ii1–56. doi: 10.1093/neuonc/not151
2. Cagney DN, Martin AM, Catalano PJ, Redig AJ, Lin NU, Lee EQ, et al. Incidence and prognosis of patients with brain metastases at diagnosis of systemic malignancy: a population-based study. Neuro Oncol. (2017) 19:1511–21. doi: 10.1093/neuonc/nox077
3. Stelzer KJ. Epidemiology and prognosis of brain metastases. Surg Neurol Int. (2013) 4 (Suppl 4):S192–202. doi: 10.4103/2152-7806.111296
4. Tabouret E, Chinot O, Metellus P, Tallet A, Viens P, Goncalves A. Recent trends in epidemiology of brain metastases: an overview. Anticancer Res. (2012) 32:4655–4662.
5. Patchell RA. The management of brain metastases. Cancer Treat Rev. (2003) 29:533–40. doi: 10.1016/S0305-7372(03)00105-1
6. Maxwell R, Jackson CM, Lim M. Clinical trials investigating immune checkpoint blockade in glioblastoma. Curr Treat Options Oncol. (2017) 18:51. doi: 10.1007/s11864-017-0492-y
7. Quail DF, Joyce JA. The microenvironmental landscape of brain tumors. Cancer Cell. (2017) 31:326–41. doi: 10.1016/j.ccell.2017.02.009
8. Sahebjam S, Sharabi A, Lim M, Kesarwani P, Chinnaiyan P. Immunotherapy and radiation in glioblastoma. J Neurooncol. (2017) 134:531–9. doi: 10.1007/s11060-017-2413-0
9. Margolin K. Ipilimumab in a Phase II trial of melanoma patients with brain metastases. Oncoimmunology. (2012) 1:1197–9. doi: 10.4161/onci.20687
10. Baskar R, Dai J, Wenlong N, Yeo R, Yeoh KW. Biological response of cancer cells to radiation treatment. Front Mol Biosci. (2014) 1:24. doi: 10.3389/fmolb.2014.00024
11. Eriksson D, Stigbrand T. Radiation-induced cell death mechanisms. Tumour Biol. (2010) 31:363–72. doi: 10.1007/s13277-010-0042-8
12. Schaue D. A Century of radiation therapy and adaptive immunity. Front Immunol. (2017) 8:431. doi: 10.3389/fimmu.2017.00431
13. Demaria S, Ng B, Devitt ML, Babb JS, Kawashima N, Liebes L, et al. Ionizing radiation inhibition of distant untreated tumors (abscopal effect) is immune mediated. Int J Radiat Oncol Biol Phys. (2004) 58:862–70. doi: 10.1016/j.ijrobp.2003.09.012
14. Tesniere A, Panaretakis T, Kepp O, Apetoh L, Ghiringhelli F, Zitvogel L, et al. Molecular characteristics of immunogenic cancer cell death. Cell Death Differ. (2008) 15:3–12. doi: 10.1038/sj.cdd.4402269
15. Hu ZI, McArthur HL, Ho AY. The abscopal effect of radiation therapy: what is it and how can we use it in breast cancer? Curr Breast Cancer Rep. (2017) 9:45–51. doi: 10.1007/s12609-017-0234-y
16. Galea I, Bechmann I, Perry VH. What is immune privilege (not)? Trends Immunol. (2007) 28:12–8. doi: 10.1016/j.it.2006.11.004
18. Mason DW, Charlton HM, Jones AJ, Lavy CB, Puklavec M, Simmonds SJ. The fate of allogeneic and xenogeneic neuronal tissue transplanted into the third ventricle of rodents. Neuroscience. (1986) 19:685–694
19. Engelhardt B, Vajkoczy P, Weller RO. The movers and shapers in immune privilege of the CNS. Nat Immunol. (2017) 18:123–31. doi: 10.1038/ni.3666
20. Mrdjen D, Pavlovic A, Hartmann FJ, Schreiner B, Utz SG, Leung BP, et al. High-dimensional single-cell mapping of central nervous system immune cells reveals distinct myeloid subsets in health, aging, and disease. Immunity. (2018) 48:380–395 e6. doi: 10.1016/j.immuni.2018.01.011
21. Forrester JV, McMenamin PG, Dando SJ. CNS infection and immune privilege. Nat Rev Neurosci. (2018) 19:655–71. doi: 10.1038/s41583-018-0070-8
22. Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. (2005) 308:1314–8. doi: 10.1126/science.1110647
24. Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. (2010) 330:841–5. doi: 10.1126/science.1194637
25. Kierdorf K, Erny D, Goldmann T, Sander V, Schulz C, Perdiguero EG, et al. Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nat Neurosci. (2013) 16:273–280. doi: 10.1038/nn.3318
26. Gomez Perdiguero E, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature. (2015) 518:547–51. doi: 10.1038/nature13989
27. Schulz C, Gomez Perdiguero E, Chorro L, Szabo-Rogers H, Cagnard N, Kierdorf K, et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science. (2012) 336:86–90. doi: 10.1126/science.1219179
28. Herz J, Filiano AJ, Smith A, Yogev N, Kipnis J. Myeloid cells in the central nervous system. Immunity. (2017) 46:943–56. doi: 10.1016/j.immuni.2017.06.007
29. Colonna M, Butovsky O. Microglia function in the central nervous system during health and neurodegeneration. Annu Rev Immunol. (2017) 35:441–68. doi: 10.1146/annurev-immunol-051116-052358
30. Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. (2012) 74:691–705. doi: 10.1016/j.neuron.2012.03.026
31. Bowman RL, Klemm F, Akkari L, Pyonteck SM, Sevenich L, Quail DF, et al. Macrophage ontogeny underlies differences in tumor-specific education in brain malignancies. Cell Rep. (2016) 17:2445–59. doi: 10.1016/j.celrep.2016.10.052
32. Ransohoff RM, Kivisakk P, Kidd G. Three or more routes for leukocyte migration into the central nervous system. Nat Rev Immunol. (2003) 3:569–81. doi: 10.1038/nri1130
33. Goldmann T, Wieghofer P, Jordao MJ, Prutek F, Hagemeyer N, Frenzel K, et al. Origin, fate and dynamics of macrophages at central nervous system interfaces. Nat Immunol. (2016) 17:797–805. doi: 10.1038/ni.3423
34. Mildner A, Djukic M, Garbe D, Wellmer A, Kuziel WA, Mack M, et al. Ly-6G+CCR2- myeloid cells rather than Ly-6ChighCCR2+ monocytes are required for the control of bacterial infection in the central nervous system. J Immunol. (2008) 181:2713–2722. doi: 10.4049/jimmunol.181.4.2713
35. Anandasabapathy N, Victora GD, Meredith M, Feder R, Dong B, Kluger C, et al. Flt3L controls the development of radiosensitive dendritic cells in the meninges and choroid plexus of the steady-state mouse brain. J Exp Med. (2011) 208:1695–705. doi: 10.1084/jem.20102657
36. Chinnery HR, Ruitenberg MJ, McMenamin PG. Novel characterization of monocyte-derived cell populations in the meninges and choroid plexus and their rates of replenishment in bone marrow chimeric mice. J Neuropathol Exp Neurol. (2010) 69:896–909. doi: 10.1097/NEN.0b013e3181edbc1a
37. Herisson F, Frodermann V, Courties G, Rohde D, Sun Y, Vandoorne K, et al. Direct vascular channels connect skull bone marrow and the brain surface enabling myeloid cell migration. Nat Neurosci. (2018) 21:1209–17. doi: 10.1038/s41593-018-0213-2
38. Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD, et al. Structural and functional features of central nervous system lymphatic vessels. Nature. (2015) 523:337–41. doi: 10.1038/nature14432
39. Aspelund A, Antila S, Proulx ST, Karlsen TV, Karaman S, Detmar M, et al. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J Exp Med. (2015) 212:991–9. doi: 10.1084/jem.20142290
40. Laman JD, Weller RO. Drainage of cells and soluble antigen from the CNS to regional lymph nodes. J Neuroimmune Pharmacol. (2013) 8:840–56. doi: 10.1007/s11481-013-9470-8
41. Kida S, Pantazis A, Weller RO. CSF drains directly from the subarachnoid space into nasal lymphatics in the rat. Anatomy, histology and immunological significance Neuropathol Appl Neurobiol. (1993) 19:480–8.
42. Engelhardt B, Carare RO, Bechmann I, Flugel A, Laman JD, Weller RO. Vascular, glial, and lymphatic immune gateways of the central nervous system. Acta Neuropathol. (2016) 132:317–38. doi: 10.1007/s00401-016-1606-5
43. Sevenich L. Brain-resident microglia and blood-borne macrophages orchestrate central nervous system inflammation in neurodegenerative disorders and brain cancer. Front Immunol. (2018) 9:697. doi: 10.3389/fimmu.2018.00697
44. Graeber MB, Moran LB. Mechanisms of cell death in neurodegenerative diseases: fashion, fiction, and facts. Brain Pathol. (2002) 12:385–90. doi: 10.1111/j.1750-3639.2002.tb00452.x
45. Sevenich L, Bowman RL, Mason SD, Quail DF, Rapaport F, Elie BT, et al. Analysis of tumour- and stroma-supplied proteolytic networks reveals a brain-metastasis-promoting role for cathepsin S. Nat Cell Biol. (2014) 16:876–88. doi: 10.1038/ncb3011
46. Goldmann T, Wieghofer P, Muller PF, Wolf Y, Varol D, Yona S, et al. A new type of microglia gene targeting shows TAK1 to be pivotal in CNS autoimmune inflammation. Nat Neurosci. (2013) 16:1618–26. doi: 10.1038/nn.3531
47. Buttgereit A, Lelios I, Yu X, Vrohlings M, Krakoski NR, Gautier EL, et al. Sall1 is a transcriptional regulator defining microglia identity and function. Nat Immunol. (2016) 17:1397–406. doi: 10.1038/ni.3585
48. Bennett ML, Bennett FC, Liddelow SA, Ajami B, Zamanian JL, Fernhoff NB, et al. New tools for studying microglia in the mouse and human CNS. Proc Natl Acad Sci USA. (2016) 113:E1738–E1746. doi: 10.1073/pnas.1525528113
49. Chen Z, Feng X, Herting CJ, Garcia VA, Nie K, Pong WW, et al. Cellular and molecular identity of tumor-associated macrophages in glioblastoma. Cancer Res. (2017) 77:2266–78. doi: 10.1158/0008-5472.CAN-16-2310
50. Müller S, Kohanbash G, Liu SJ, Alvarado B, Carrera D, Bhaduri A, et al. Single-cell profiling of human gliomas reveals macrophage ontogeny as a basis for regional differences in macrophage activation in the tumor microenvironment. Genome Biol. (2017) 18:234. doi: 10.1186/s13059-017-1362-4
51. Ricard C, Tchoghandjian A, Luche H, Grenot P, Figarella-Branger D, Rougon G, et al. Phenotypic dynamics of microglial and monocyte-derived cells in glioblastoma-bearing mice. Sci Rep. (2016) 6:26381. doi: 10.1038/srep26381
52. Banchereau J, Palucka AK. Dendritic cells as therapeutic vaccines against cancer. Nat Rev Immunol. (2005) 5:296–306. doi: 10.1038/nri1592
53. Tomura M, Hata A, Matsuoka S, Shand FH, Nakanishi Y, Ikebuchi R, et al. Tracking and quantification of dendritic cell migration and antigen trafficking between the skin and lymph nodes. Sci Rep. (2014) 4:6030. doi: 10.1038/srep06030
54. Prins RM, Shu CJ, Radu CG, Vo DD, Khan-Farooqi H, Soto H, et al. Anti-tumor activity and trafficking of self, tumor-specific T cells against tumors located in the brain. Cancer Immunol Immunother. (2008) 57:1279–89. doi: 10.1007/s00262-008-0461-1
55. Garzon-Muvdi T, Theodros D, Luksik AS, Maxwell R, Kim E, Jackson CM, et al. Dendritic cell activation enhances anti-PD-1 mediated immunotherapy against glioblastoma. Oncotarget. (2018) 9:20681–97. doi: 10.18632/oncotarget.25061
56. Nduom EK, Weller M, Heimberger AB. Immunosuppressive mechanisms in glioblastoma. Neuro Oncol. (2015) 17 (Suppl 7) vii9–vii14. doi: 10.1093/neuonc/nov151
57. Jacobs JF, Idema AJ, Bol KF, Nierkens S, Grauer OM, Wesseling P, et al. Regulatory T cells and the PD-L1/PD-1 pathway mediate immune suppression in malignant human brain tumors. Neuro Oncol. (2009) 11:394–402. doi: 10.1215/15228517-2008-104
58. Jacobs JF, Idema AJ, Bol KF, Grotenhuis JA, de Vries IJ, Wesseling P, et al. Prognostic significance and mechanism of Treg infiltration in human brain tumors. J Neuroimmunol. (2010) 225:195–9. doi: 10.1016/j.jneuroim.2010.05.020
59. Grauer OM, Nierkens S, Bennink E, Toonen LW, Boon L, Wesseling P, et al. CD4+FoxP3+ regulatory T cells gradually accumulate in gliomas during tumor growth and efficiently suppress antiglioma immune responses in vivo. Int J Cancer. (2007) 121:95–105. doi: 10.1002/ijc.22607
60. Han S, Ma E, Wang X, Yu C, Dong T, Zhan W, et al. Rescuing defective tumor-infiltrating T-cell proliferation in glioblastoma patients. Oncol Lett. (2016) 12:2924–9. doi: 10.3892/ol.2016.4944
61. Brooks WH, Roszman TL, Mahaley MS, Woosley RE. Immunobiology of primary intracranial tumours. II Analysis of lymphocyte subpopulations in patients with primary brain tumours. Clin Exp Immunol. (1977) 29:61–6.
62. Chongsathidkiet P, Jackson C, Koyama S, Loebel F, Cui X, Farber SH, et al. Sequestration of T cells in bone marrow in the setting of glioblastoma and other intracranial tumors. Nat Med. (2018) 24:1459–68. doi: 10.1038/s41591-018-0135-2
63. Chahlavi A, Rayman P, Richmond AL, Biswas K, Zhang R, Vogelbaum M, et al. Glioblastomas induce T-lymphocyte death by two distinct pathways involving gangliosides and CD70. Cancer Res. (2005) 65:5428–38. doi: 10.1158/0008-5472.CAN-04-4395
64. Wischhusen J, Jung G, Radovanovic I, Beier C, Steinbach JP, Rimner A, et al. Identification of CD70-mediated apoptosis of immune effector cells as a novel immune escape pathway of human glioblastoma. Cancer Res. (2002) 62:2592–9.
65. Bechmann I, Steiner B, Gimsa U, Mor G, Wolf S, Beyer M, et al. Astrocyte-induced T cell elimination is CD95 ligand dependent. J Neuroimmunol. (2002) 132:60–5. doi: 10.1016/S0165-5728(02)00311-9
66. Brooks WH, Roszman TL, Rogers AS. Impairment of rosette-forming T lymphocytes in patients with primary intracranial tumors. Cancer. (1976) 37:1869–73.
67. Woroniecka KI, Rhodin KE, Chongsathidkiet P, Keith KA, Fecci PE. T-cell dysfunction in glioblastoma: applying a new framework. Clin Cancer Res. (2018) 24:3792–802. doi: 10.1158/1078-0432.CCR-18-0047
68. Hao C, Parney IF, Roa WH, Turner J, Petruk KC, Ramsay DA. Cytokine and cytokine receptor mRNA expression in human glioblastomas: evidence of Th1, Th2 and Th3 cytokine dysregulation. Acta Neuropathol. (2002) 103:171–8. doi: 10.1007/s004010100448
69. Mirzaei R, Sarkar S, Yong VW. T cell exhaustion in glioblastoma: intricacies of immune checkpoints. Trends Immunol. (2017) 38:104–15. doi: 10.1016/j.it.2016.11.005
70. Woroniecka K, Chongsathidkiet P, Rhodin K, Kemeny H, Dechant C, Farber SH, et al. T-Cell exhaustion signatures vary with tumor type and are severe in glioblastoma. Clin Cancer Res. (2018) 24:4175–86. doi: 10.1158/1078-0432.CCR-17-1846
71. Liu Z, Han H, He X, Li S, Wu C, Yu C, et al. Expression of the galectin-9-Tim-3 pathway in glioma tissues is associated with the clinical manifestations of glioma. Oncol Lett. (2016) 11:1829–34. doi: 10.3892/ol.2016.4142
72. Liu Z, Meng Q, Bartek JJr, Poiret T, Persson O, Rane L, et al. Tumor-infiltrating lymphocytes (TILs) from patients with glioma. Oncoimmunology. (2017) 6:e1252894. doi: 10.1080/2162402X.2016.1252894
73. Mohme M, Schliffke S, Maire CL, Runger A, Glau L, Mende KC, et al. Immunophenotyping of newly diagnosed and recurrent glioblastoma defines distinct immune exhaustion profiles in peripheral and tumor-infiltrating lymphocytes. Clin Cancer Res. (2018) 24:4187–200. doi: 10.1158/1078-0432.CCR-17-2617
74. Fecci PE, Mitchell DA, Whitesides JF, Xie W, Friedman AH, Archer GE, et al. Increased regulatory T-cell fraction amidst a diminished CD4 compartment explains cellular immune defects in patients with malignant glioma. Cancer Res. (2006) 66:3294–302. doi: 10.1158/0008-5472.CAN-05-3773
75. Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med. (2013) 19:1264–72. doi: 10.1038/nm.3337
76. Hussain SF, Yang D, Suki D, Aldape K, Grimm E, Heimberger AB. The role of human glioma-infiltrating microglia/macrophages in mediating antitumor immune responses. Neuro Oncol. (2006) 8:261–79. doi: 10.1215/15228517-2006-008
77. Harter PN, Bernatz S, Scholz A, Zeiner PS, Zinke J, Kiyose M, et al. Distribution and prognostic relevance of tumor-infiltrating lymphocytes (TILs) and PD-1/PD-L1 immune checkpoints in human brain metastases. Oncotarget. (2015) 6:40836–49. doi: 10.18632/oncotarget.5696
78. Berghoff AS, Fuchs E, Ricken G, Mlecnik B, Bindea G, Spanberger T, et al. Density of tumor-infiltrating lymphocytes correlates with extent of brain edema and overall survival time in patients with brain metastases. Oncoimmunology. (2016) 5:e1057388. doi: 10.1080/2162402X.2015.1057388
79. Berghoff AS, Ricken G, Widhalm G, Rajky O, Dieckmann K, Birner P, et al. Tumour-infiltrating lymphocytes and expression of programmed death ligand 1 (PD-L1) in melanoma brain metastases. Histopathology. (2015) 66:289–99. doi: 10.1111/his.12537
80. Berghoff AS, Ricken G, Wilhelm D, Rajky O, Widhalm G, Dieckmann K, et al. Tumor infiltrating lymphocytes and PD-L1 expression in brain metastases of small cell lung cancer (SCLC). J Neurooncol. (2016) 130:19–29. doi: 10.1007/s11060-016-2216-8
81. Du Four S, Maenhout SK, De Pierre K, Renmans D, Niclou SP, Thielemans K, et al. Axitinib increases the infiltration of immune cells and reduces the suppressive capacity of monocytic MDSCs in an intracranial mouse melanoma model. Oncoimmunology. (2015) 4:e998107. doi: 10.1080/2162402X.2014.998107
82. Kong LY, Abou-Ghazal MK, Wei J, Chakraborty A, Sun W, Qiao W, et al. A novel inhibitor of signal transducers and activators of transcription 3 activation is efficacious against established central nervous system melanoma and inhibits regulatory T cells. Clin Cancer Res. (2008) 14:5759–68. doi: 10.1158/1078-0432.CCR-08-0377
83. Sugihara AQ, Rolle CE, Lesniak MS. Regulatory T cells actively infiltrate metastatic brain tumors. Int J Oncol. (2009) 34:1533–40. doi: 10.3892/ijo_00000282
84. Farber SH, Tsvankin V, Narloch JL, Kim GJ, Salama AK, Vlahovic G, et al. Embracing rejection: Immunologic trends in brain metastasis. Oncoimmunology. (2016) 5:e1172153. doi: 10.1080/2162402X.2016.1172153
85. Reardon DA, Omuro A, Brandes AA, Rieger J, Wick A, Sepulveda J, et al. OS10.3 Randomized Phase 3 study evaluating the efficacy and safety of nivolumab vs bevacizumab in patients with recurrent glioblastoma: CheckMate 143. Neuro-oncology. (2017) 19:iii21. doi: 10.1093/neuonc/nox036.071
86. Filley AC, Henriquez M, Dey M. Recurrent glioma clinical trial, CheckMate-143: the game is not over yet. Oncotarget. (2017) 8:91779–94. doi: 10.18632/oncotarget.21586
87. Simoni Y, Becht E, Fehlings M, Loh CY, Koo SL, Teng KWW, et al. Bystander CD8(+) T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature. (2018) 557:575–9. doi: 10.1038/s41586-018-0130-2
88. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. (2005) 352:987–996. doi: 10.1056/NEJMoa043330
89. Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. (2009) 10:459–466. doi: 10.1016/S1470-2045(09)70025-7
90. Weller M, Cloughesy T, Perry JR, Wick W. Standards of care for treatment of recurrent glioblastoma–are we there yet? Neuro Oncol. (2013) 15:4–27. doi: 10.1093/neuonc/nos273
91. Perry JR, Belanger K, Mason WP, Fulton D, Kavan P, Easaw J, et al. Phase II trial of continuous dose-intense temozolomide in recurrent malignant glioma: RESCUE study. J Clin Oncol. (2010) 28:2051–7. doi: 10.1200/JCO.2009.26.5520
92. Lamborn KR, Yung WK, Chang SM, Wen PY, Cloughesy TF, DeAngelis LM, et al. Progression-free survival: an important end point in evaluating therapy for recurrent high-grade gliomas. Neuro Oncol. (2008) 10:162–70. doi: 10.1215/15228517-2007-062
93. Chamberlain MC. Bevacizumab for the treatment of recurrent glioblastoma. Clin Med Insights Oncol. (2011) 5:117–29. doi: 10.4137/CMO.S7232
94. McTyre E, Scott J, Chinnaiyan P. Whole brain radiotherapy for brain metastasis. Surg Neurol Int. (2013) 4(Suppl 4):S236–44. doi: 10.4103/2152-7806.111301
95. Ahluwalia MS, Vogelbaum MV, Chao ST, Mehta MM. Brain metastasis and treatment. F1000Prime Rep. (2014) 6:114. doi: 10.12703/P6-114
96. Dietrich J, Monje M, Wefel J, Meyers C. Clinical patterns and biological correlates of cognitive dysfunction associated with cancer therapy. Oncologist. (2008) 13:1285–95. doi: 10.1634/theoncologist.2008-0130
97. Soffietti R, Abacioglu U, Baumert B, Combs SE, Kinhult S, Kros JM, et al. Diagnosis and treatment of brain metastases from solid tumors: guidelines from the European Association of Neuro-Oncology (EANO). Neuro Oncol. (2017) 19:162–74. doi: 10.1093/neuonc/now241
98. Ramakrishna N, Margolin KA. Multidisciplinary approach to brain metastasis from melanoma; local therapies for central nervous system metastases. Am Soc Clin Oncol Educ Book. (2013) 399–403. doi: 10.1200/EdBook_AM.2013.33.399
99. Brown PD, Ballman KV, Cerhan JH, Anderson SK, Carrero XW, Whitton AC, et al. Postoperative stereotactic radiosurgery compared with whole brain radiotherapy for resected metastatic brain disease (NCCTG N107C/CEC.3): a multicentre, randomised, controlled, phase 3 trial. Lancet Oncol. (2017) 18:1049–1060. doi: 10.1016/S1470-2045(17)30441-2
100. Mahajan A, Ahmed S, McAleer MF, Weinberg JS, Li J, Brown P, et al. Post-operative stereotactic radiosurgery versus observation for completely resected brain metastases: a single-centre, randomised, controlled, phase 3 trial. Lancet Oncol. (2017) 18:1040–8. doi: 10.1016/S1470-2045(17)30414-X
101. Khan M, Lin J, Liao G, Li R, Wang B, Xie G, et al. Comparison of WBRT alone, SRS alone, and their combination in the treatment of one or more brain metastases: Review and meta-analysis. Tumour Biol. (2017) 39:1010428317702903. doi: 10.1177/1010428317702903
102. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. (2012) 12:252–64. doi: 10.1038/nrc3239
103. Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. (1996) 271:1734–6.
104. Ishida Y, Agata Y, Shibahara K, Honjo T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. (1992) 11:3887–95.
105. Hodges TR, Ott M, Xiu J, Gatalica Z, Swensen J, Zhou S, et al. Mutational burden, immune checkpoint expression, and mismatch repair in glioma: implications for immune checkpoint immunotherapy. Neuro Oncol. (2017) 19:1047–57. doi: 10.1093/neuonc/nox026
106. Brastianos PK, Carter SL, Santagata S, Cahill DP, Taylor-Weiner A, Jones RT, et al. Genomic characterization of brain metastases reveals branched evolution and potential therapeutic targets. Cancer Discov. (2015) 5:1164–77. doi: 10.1158/2159-8290.CD-15-0369
107. Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. (2010) 363:711–23. doi: 10.1056/NEJMoa1003466
108. Wolchok JD, Neyns B, Linette G, Negrier S, Lutzky J, Thomas L, et al. Ipilimumab monotherapy in patients with pretreated advanced melanoma: a randomised, double-blind, multicentre, phase 2, dose-ranging study. Lancet Oncol. (2010) 11:155–64. doi: 10.1016/S1470-2045(09)70334-1
109. Parakh S, Park JJ, Mendis S, Rai R, Xu W, Lo S, et al. Efficacy of anti-PD-1 therapy in patients with melanoma brain metastases. Br J Cancer. (2017) 116:1558–63. doi: 10.1038/bjc.2017.142
110. Margolin K, Ernstoff MS, Hamid O, Lawrence D, McDermott D, Puzanov I, et al. Ipilimumab in patients with melanoma and brain metastases: an open-label, phase 2 trial. Lancet Oncol. (2012) 13:459–65. doi: 10.1016/S1470-2045(12)70090-6
111. Long GV, Atkinson V, Lo S, Sandhu S, Guminski AD, Brown MP, et al. Combination nivolumab and ipilimumab or nivolumab alone in melanoma brain metastases: a multicentre randomised phase 2 study. Lancet Oncol. (2018) 19:672–81. doi: 10.1016/S1470-2045(18)30139-6
112. Tawbi HA, Forsyth PA, Algazi A, Hamid O, Hodi FS, Moschos SJ, et al. Combined nivolumab and ipilimumab in melanoma metastatic to the brain. N Engl J Med. (2018) 379:722–30. doi: 10.1056/NEJMoa1805453
113. Hegde PS, Wallin JJ, Mancao C. Predictive markers of anti-VEGF and emerging role of angiogenesis inhibitors as immunotherapeutics. Semin Cancer Biol. (2018)52(Pt 2):117–24. doi: 10.1016/j.semcancer.2017.12.002
114. Reardon DA, Gokhale PC, Klein SR, Ligon KL, Rodig SJ, Ramkissoon SH, et al. Glioblastoma eradication following immune checkpoint blockade in an orthotopic, immunocompetent model. Cancer Immunol Res. (2016) 4:124–35. doi: 10.1158/2326-6066.CIR-15-0151
115. Mathios D, Kim JE, Mangraviti A, Phallen J, Park CK, Jackson CM, et al. Anti-PD-1 antitumor immunity is enhanced by local and abrogated by systemic chemotherapy in GBM. Sci Transl Med. (2016) 8:370ra180. doi: 10.1126/scitranslmed.aag2942
116. Horvat TZ, Adel NG, Dang TO, Momtaz P, Postow MA, Callahan MK, et al. Immune-related adverse events, need for systemic immunosuppression, and effects on survival and time to treatment failure in patients with melanoma treated with ipilimumab at memorial sloan kettering cancer center. J Clin Oncol. (2015) 33:3193–8. doi: 10.1200/JCO.2015.60.8448
117. Knisely JP, Yu JB, Flanigan J, Sznol M, Kluger HM, Chiang VL. Radiosurgery for melanoma brain metastases in the ipilimumab era and the possibility of longer survival. J Neurosurg. (2012) 117:227–33. doi: 10.3171/2012.5.JNS111929
118. Silk AW, Bassetti MF, West BT, Tsien CI, Lao CD. Ipilimumab and radiation therapy for melanoma brain metastases. Cancer Med. (2013) 2:899–906. doi: 10.1002/cam4.140
119. Shaverdian N, Lisberg AE, Bornazyan K, Veruttipong D, Goldman JW, Formenti SC, et al. Previous radiotherapy and the clinical activity and toxicity of pembrolizumab in the treatment of non-small-cell lung cancer: a secondary analysis of the KEYNOTE-001 phase 1 trial. Lancet Oncol. (2017) 18:895–903. doi: 10.1016/S1470-2045(17)30380-7
120. Ahmed KA, Stallworth DG, Kim Y, Johnstone PA, Harrison LB, Caudell JJ, et al. Clinical outcomes of melanoma brain metastases treated with stereotactic radiation and anti-PD-1 therapy. Ann Oncol. (2016) 27:434–41. doi: 10.1093/annonc/mdv622
121. Wang Y, Deng W, Li N, Neri S, Sharma A, Jiang W, et al. Combining immunotherapy and radiotherapy for cancer treatment: current challenges and future directions. Front Pharmacol. (2018) 9:185. doi: 10.3389/fphar.2018.00185
122. Qian JM, Yu JB, Kluger HM, Chiang VL. Timing and type of immune checkpoint therapy affect the early radiographic response of melanoma brain metastases to stereotactic radiosurgery. Cancer. (2016) 122:3051–8. doi: 10.1002/cncr.30138
123. Dovedi SJ, Adlard AL, Lipowska-Bhalla G, McKenna C, Jones S, Cheadle EJ, et al. Acquired resistance to fractionated radiotherapy can be overcome by concurrent PD-L1 blockade. Cancer Res. (2014) 74:5458–68. doi: 10.1158/0008-5472.CAN-14-1258
124. Samstein R, Rimmer A, Barker CA, Yamada Y. Combined immune checkpoint blockade and radiation therapy: timing and dose fractionation associated with greatest survival duration among over 750 treated patients. Int J Radiat Oncol Biol Phys. (2017) 99:129–30. doi: 10.1016/j.ijrobp.2017.06.303
125. Serre R, Benzekry S, Padovani L, Meille C, Andre N, Ciccolini J, et al. Mathematical modeling of cancer immunotherapy and its synergy with radiotherapy. Cancer Res. (2016) 76:4931–40. doi: 10.1158/0008-5472.CAN-15-3567
126. Stone HB, Peters LJ, Milas L. Effect of host immune capability on radiocurability and subsequent transplantability of a murine fibrosarcoma. J Natl Cancer Inst. (1979) 63:1229–35.
127. Rodriguez-Ruiz ME, Vanpouille-Box C, Melero I, Formenti SC, Demaria S. Immunological mechanisms responsible for radiation-induced abscopal effect. Trends Immunol. (2018) 39:644–55. doi: 10.1016/j.it.2018.06.001
128. Kepp O, Senovilla L, Vitale I, Vacchelli E, Adjemian S, Agostinis P, et al. Consensus guidelines for the detection of immunogenic cell death. Oncoimmunology. (2014) 3:e955691. doi: 10.4161/21624011.2014.955691
129. Golden EB, Apetoh L. Radiotherapy and immunogenic cell death. Semin Radiat Oncol. (2015) 25:11–7. doi: 10.1016/j.semradonc.2014.07.005
130. Hellevik T, Martinez-Zubiaurre I. Radiotherapy and the tumor stroma: the importance of dose and fractionation. Front Oncol. (2014) 4:1. doi: 10.3389/fonc.2014.00001
131. Brown JS, Sundar R, Lopez J. Combining DNA damaging therapeutics with immunotherapy: more haste, less speed. Br J Cancer. (2018) 118:312–24. doi: 10.1038/bjc.2017.376
132. Beatty GL, Gladney WL. Immune escape mechanisms as a guide for cancer immunotherapy. Clin Cancer Res. (2015) 21:687–92. doi: 10.1158/1078-0432.CCR-14-1860
133. Jiang W, Chan CK, Weissman IL, Kim BYS, Hahn SM. Immune priming of the tumor microenvironment by radiation. Trends Cancer. (2016) 2:638–45. doi: 10.1016/j.trecan.2016.09.007
134. Reits EA, Hodge JW, Herberts CA, Groothuis TA, Chakraborty M, Wansley EK, et al. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. J Exp Med. (2006) 203:1259–71. doi: 10.1084/jem.20052494
135. Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. (2014) 371:2189–99. doi: 10.1056/NEJMoa1406498
136. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. (2015) 348:124–8. doi: 10.1126/science.aaa1348
137. McGranahan N, Furness AJ, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. (2016) 351:1463–9. doi: 10.1126/science.aaf1490
138. Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, et al. Genomic and transcriptomic features of response to Anti-PD-1 therapy in metastatic melanoma. Cell. (2016) 165:35–44. doi: 10.1016/j.cell.2016.02.065
139. Behjati S, Gundem G, Wedge DC, Roberts ND, Tarpey PS, Cooke SL, et al. Mutational signatures of ionizing radiation in second malignancies. Nat Commun. (2016) 7:12605. doi: 10.1038/ncomms12605
140. Lauss M, Donia M, Harbst K, Andersen R, Mitra S, Rosengren F, et al. Mutational and putative neoantigen load predict clinical benefit of adoptive T cell therapy in melanoma. Nat Commun. (2017) 8:1738. doi: 10.1038/s41467-017-01460-0
141. Sharabi AB, Tran PT, Lim M, Drake CG, Deweese TL. Stereotactic radiation therapy combined with immunotherapy: augmenting the role of radiation in local and systemic treatment. Oncology. (2015) 29:331–40.
142. Jackson CM, Kochel CM, Nirschl CJ, Durham NM, Ruzevick J, Alme A, et al. Systemic tolerance mediated by melanoma brain tumors is reversible by radiotherapy and vaccination. Clin Cancer Res. (2016) 22:1161–72. doi: 10.1158/1078-0432.CCR-15-1516
143. Barber GN. STING: infection, inflammation and cancer. Nat Rev Immunol. (2015) 15:760–70. doi: 10.1038/nri3921
144. Chen Q, Sun L, Chen ZJ. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat Immunol. (2016) 17:1142–9. doi: 10.1038/ni.3558
145. Xia T, Konno H, Ahn J, Barber GN. Deregulation of STING signaling in colorectal carcinoma constrains DNA damage responses and correlates with tumorigenesis. Cell Rep. (2016) 14:282–97. doi: 10.1016/j.celrep.2015.12.029
146. Xia T, Konno H, Barber GN. Recurrent loss of STING signaling in melanoma correlates with susceptibility to viral oncolysis. Cancer Res. (2016) 76:6747–59. doi: 10.1158/0008-5472.CAN-16-1404
147. Liang H, Deng L, Hou Y, Meng X, Huang X, Rao E, et al. Host STING-dependent MDSC mobilization drives extrinsic radiation resistance. Nat Commun. (2017) 8:1736. doi: 10.1038/s41467-017-01566-5
148. Gandhi SJ, Minn AJ, Vonderheide RH, Wherry EJ, Hahn SM, Maity A. Awakening the immune system with radiation: optimal dose and fractionation. Cancer Lett. (2015) 368:185–90. doi: 10.1016/j.canlet.2015.03.024
149. Haikerwal SJ, Hagekyriakou J, MacManus M, Martin OA, Haynes NM. Building immunity to cancer with radiation therapy. Cancer Lett. (2015) 368:198–208. doi: 10.1016/j.canlet.2015.01.009
150. Lugade AA, Moran JP, Gerber SA, Rose RC, Frelinger JG, Lord EM. Local radiation therapy of B16 melanoma tumors increases the generation of tumor antigen-specific effector cells that traffic to the tumor. J Immunol. (2005) 174:7516–23. doi: 10.4049/jimmunol.174.12.7516
151. Lee Y, Auh SL, Wang Y, Burnette B, Wang Y, Meng Y, et al. Therapeutic effects of ablative radiation on local tumor require CD8+ T cells: changing strategies for cancer treatment. Blood. (2009) 114:589–95. doi: 10.1182/blood-2009-02-206870
152. Camphausen K, Moses MA, Menard C, Sproull M, Beecken WD, Folkman J, et al. Radiation abscopal antitumor effect is mediated through p53. Cancer Res. (2003) 63:1990–1993. doi: 10.1016/S0360-3016(02)03449-1
153. Dewan MZ, Galloway AE, Kawashima N, Dewyngaert JK, Babb JS, Formenti SC, et al. Fractionated but not single-dose radiotherapy induces an immune-mediated abscopal effect when combined with anti-CTLA-4 antibody. Clin Cancer Res. (2009) 15:5379–88. doi: 10.1158/1078-0432.CCR-09-0265
154. Vanpouille-Box C, Formenti SC, Demaria S. TREX1 dictates the immune fate of irradiated cancer cells. Oncoimmunology. (2017) 6:e1339857. doi: 10.1080/2162402X.2017.1339857
155. Vanpouille-Box C, Alard A, Aryankalayil MJ, Sarfraz Y, Diamond JM, Schneider RJ, et al. DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat Commun. (2017) 8:15618. doi: 10.1038/ncomms15618
156. Barker HE, Paget JT, Khan AA, Harrington KJ. The tumour microenvironment after radiotherapy: mechanisms of resistance and recurrence. Nat Rev Cancer. (2015) 15:409–25. doi: 10.1038/nrc3958
157. Ahn GO, Tseng D, Liao CH, Dorie MJ, Czechowicz A, Brown JM. Inhibition of Mac-1 (CD11b/CD18) enhances tumor response to radiation by reducing myeloid cell recruitment. Proc Natl Acad Sci USA. (2010) 107:8363–8. doi: 10.1073/pnas.0911378107
158. Russell JS, Brown JM. The irradiated tumor microenvironment: role of tumor-associated macrophages in vascular recovery. Front Physiol. (2013) 4:157. doi: 10.3389/fphys.2013.00157
159. Schaue D, Kachikwu EL, McBride WH. Cytokines in radiobiological responses: a review. Radiat Res. (2012) 178:505–23. doi: 10.1667/RR3031.1
160. Coates PJ, Rundle JK, Lorimore SA, Wright EG. Indirect macrophage responses to ionizing radiation: implications for genotype-dependent bystander signaling. Cancer Res. (2008) 68:450–6. doi: 10.1158/0008-5472.CAN-07-3050
161. Klug F, Prakash H, Huber PE, Seibel T, Bender N, Halama N, et al. Low-dose irradiation programs macrophage differentiation to an iNOS(+)/M1 phenotype that orchestrates effective T cell immunotherapy. Cancer Cell. (2013) 24:589–602. doi: 10.1016/j.ccr.2013.09.014
162. Teresa Pinto A, Laranjeiro Pinto M, Patricia Cardoso A, Monteiro C, Teixeira Pinto M, Filipe Maia A, et al. Ionizing radiation modulates human macrophages towards a pro-inflammatory phenotype preserving their pro-invasive and pro-angiogenic capacities. Sci Rep. (2016) 6:18765. doi: 10.1038/srep18765
163. Deschavanne PJ, Fertil B. A review of human cell radiosensitivity in vitro. Int J Radiat Oncol Biol Phys. (1996) 34:251–266
164. Cao KI, Lebas N, Gerber S, Levy C, Le Scodan R, Bourgier C, et al. Phase II randomized study of whole-brain radiation therapy with or without concurrent temozolomide for brain metastases from breast cancer. Ann Oncol. (2015) 26:89–94. doi: 10.1093/annonc/mdu488
165. Samaha H, Pignata A, Fousek K, Ren J, Lam FW, Stossi F, et al. A homing system targets therapeutic T cells to brain cancer. Nature. (2018) 561:331–7. doi: 10.1038/s41586-018-0499-y
166. Ohta A. A metabolic immune checkpoint: adenosine in tumor microenvironment. Front Immunol. (2016) 7:109. doi: 10.3389/fimmu.2016.00109
167. Leone RD, Emens LA. Targeting adenosine for cancer immunotherapy. J Immunother Cancer. (2018) 6:57. doi: 10.1186/s40425-018-0360-8
168. Reinhardt J, Landsberg J, Schmid-Burgk JL, Ramis BB, Bald T, Glodde N, et al. MAPK signaling and inflammation link melanoma phenotype switching to induction of CD73 during Immunotherapy. Cancer Res. (2017) 77:4697–709. doi: 10.1158/0008-5472.CAN-17-0395
169. Monteiro I, Vigano S, Faouzi M, Treilleux I, Michielin O, Menetrier-Caux C, et al. CD73 expression and clinical significance in human metastatic melanoma. Oncotarget. (2018) 9:26659–69. doi: 10.18632/oncotarget.25426
170. Young A, Ngiow SF, Barkauskas DS, Sult E, Hay C, Blake SJ, et al. Co-inhibition of CD73 and A2AR adenosine signaling improves anti-tumor immune responses. Cancer Cell. (2016) 30:391–403. doi: 10.1016/j.ccell.2016.06.025
171. Mediavilla-Varela M, Castro J, Chiappori A, Noyes D, Hernandez DC, Allard B, et al. A novel antagonist of the immune checkpoint protein adenosine A2a receptor restores tumor-infiltrating lymphocyte activity in the context of the tumor microenvironment. Neoplasia. (2017) 19:530–6. doi: 10.1016/j.neo.2017.02.004
172. Allard B, Longhi MS, Robson SC, Stagg J. The ectonucleotidases CD39 and CD73: Novel checkpoint inhibitor targets. Immunol Rev. (2017) 276:121–44. doi: 10.1111/imr.12528
173. Allard D, Turcotte M, Stagg J. Targeting A2 adenosine receptors in cancer. Immunol Cell Biol. (2017) 95:333–9. doi: 10.1038/icb.2017.8
Keywords: GBM, brain metastasis, tumor immunology, immune therapy, immune checkpoint blockade, ionizing radiation, radiotherapy
Citation: Sevenich L (2019) Turning “Cold” Into “Hot” Tumors—Opportunities and Challenges for Radio-Immunotherapy Against Primary and Metastatic Brain Cancers. Front. Oncol. 9:163. doi: 10.3389/fonc.2019.00163
Received: 29 October 2018; Accepted: 25 February 2019;
Published: 19 March 2019.
Edited by:
Udo S. Gaipl, University Hospital Erlangen, GermanyReviewed by:
Amanda W. Lund, Oregon Health & Science University, United StatesBozena Kaminska, Nencki Institute of Experimental Biology (PAS), Poland
Copyright © 2019 Sevenich. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lisa Sevenich, c2V2ZW5pY2hAZ3NoLnVuaS1mcmFua2Z1cnQuZGU=