 Jens U. Marquardt1,2*
Jens U. Marquardt1,2* Frank Edlich3,4*
Frank Edlich3,4*- 1Department of Medicine I, University Medical Center Schleswig-Holstein, Lübeck, Germany
- 2Department of Medicine, Lichtenberg Research Group, University Mainz, Mainz, Germany
- 3Heisenberg Research Group “Regulation von Bcl-2-Proteinen Durch Konformationelle Flexibilität,” Institute for Biochemistry and Molecular Biology, University of Freiburg, Freiburg, Germany
- 4CIBSS Centre for Integrative Biological Signalling Studies, University of Freiburg, Freiburg, Germany
Hepatocellular carcinoma (HCC) ranks among the most rapidly evolving cancers in the Western world. The majority of HCCs develop on the basis of a chronic inflammatory liver damage that predisposes liver cancer development and leads to deregulation of multiple cellular signaling pathways. The resulting dysbalance between uncontrolled proliferation and impaired predisposition to cell death with consecutive failure to clear inflammatory damage is a key driver of malignant transformation. Therefore, resistance to death signaling accompanied by metabolic changes as well as failed immunological clearance of damaged pre-neoplastic hepatocytes are considered hallmarks of hepatocarcinogenesis. Hereby, the underlying liver disease, the type of liver damage and individual predisposition to apoptosis determines the natural course of the disease as well as the therapeutic response. Here, we will review common and individual aspects of cell death pathways in hepatocarcinogenesis with a particular emphasis on regulatory networks and key molecular alterations. We will further delineate the potential of targeting cell death-related signaling as a viable therapeutic strategy to improve the outcome of HCC patients.
Introduction
The common hallmark of the vast majority of Hepatocellular carcinomas (HCC) is a chronic inflammatory liver damage induced by a diverse spectrum of etiological risk factors (1). Depending on the type of liver injury and persistence of the underlying inflammatory stimulus, HCCs are particularly characterized by a significant phenotypic and molecular heterogeneity. Therefore, HCCs are oncogenic paradigms for inflammation-induced cancers (2). Herein, the underlying causes of the chronic liver disease range from chronic hepatitis B (HBV) and C viruses (HCV) infections over excessive alcohol abuse to metabolic liver diseases. Importantly, the obesity-associated alterations of the hepatic microenvironment resembling non-alcoholic fatty liver disease and, more importantly, steatohepatitis (NAFLD/NASH) are now among the most prominent etiological risk factors for HCC in several Western countries (3). The particular type of inflammatory liver damage induced by NASH is also responsible for a high number of HCCs without underlying cirrhosis (4, 5). Given the rising incidence of the metabolic syndrome worldwide, it is not surprising that HCC currently ranks among the most rapidly evolving and deadliest cancers in the Western world. Further, the impaired liver function and observed molecular heterogeneity renders effective treatments of HCCs particularly challenging (6, 7).
In the context of HCC development and progression, special importance can be assigned to the type of liver damage and associated changes to the hepatic micromilieu that create a pro-oncogenic field effect and precede malignant transformation of hepatocytes (8, 9).
Various types of liver injury and associated chronic cell death responses have been identified to trigger inflammatory liver diseases, fibrosis development and, ultimately, hepatocarcinogenesis (10, 11). Accordingly, major cell death processes as well as signaling pathways are associated with liver cancer development and mainly involve apoptosis and necrosis. However, other forms of cell death, such as autophagy, necroptosis, pyroptosis, ferroptosis, or combinations of these death programs, have also been linked to HCC development and progression (11). Damaged hepatocytes induce activation and cross-talk of other non-parenchymal, immune and stromal cells with subsequent release of cytokine that fuel inflammation-induced damage and prone cancer development (12). Abnormalities in glucose and lipid metabolisms as well as microbiota composition further aggravate the oncogenic process. While the mentioned cell death mechanisms are relevant for hepatocarcinogenesis, regardless of the underlying etiological risk factors, oxidative stress and consecutive impairment of mitochondrial function seem to particularly induce hepatocyte death during metabolic liver damage and lead to signaling through B-cell lymphoma-2 (BCL-2) family proteins and activation of caspases and c-Jun N-terminal kinase during NASH-induced HCC (13). Besides prominent roles of cell death pathways in HCC development, cell death regulation and associated changes are also important for diagnosis and therapy. Several surrogate methods to assess and quantify liver injury, predominant mode of cell death and activation of inflammatory processes have been successfully evaluated in the context of acute and chronic liver diseases (14, 15). However, reliable and non-invasive cell death markers are not available in clinical routine. Cell death and inflammatory markers have also been assessed as prognostic markers or to facilitate monitoring of therapy response in the context of liver cancer (16, 17). In addition, inhibitors of apoptosis, particularly inhibitors of BCL-2 family members or caspases, have recently been introduced to target several chronic inflammatory diseases including NASH. These inhibitors might not only prevent malignant transformation and, thus, be effective as preventive compounds, but also be viable therapeutic strategies for HCC. Together, inflammatory cell death is particular relevant for mechanistic and clinical applications in liver cancer.
The here presented review aims to summarize key cellular and molecular mechanisms involved in liver cell death during hepatocarcinogenesis with a main focus on apoptosis. We will also delineate the importance of predisposition to apoptosis as a key factor for malignant transformation and specify factors that affect differential predisposition to apoptotic stimuli during liver cancer development and therapy. Finally, the impact for personalized medicine and precision oncology will be discussed.
Mechanisms of Cell Death in Hepatocarcinogenesis
Cell death is intrinsically associated with chronic inflammation in various organs including the liver (10). Herein, infectious and metabolic changes induced by the underlying etiological agent prone hepatocytes for further damage. Liver fibrogenesis and carcinogenesis are significantly accelerated by oxidative stress, cell death and inflammation. Thus, HCC is the final and most deadly consequence of all major chronic liver diseases (2). Consistently, continuous inflammatory cell death is one of the hallmarks of hepatocarcinogenesis. Almost all HCC patients show signs of cell death in sera and tissue and their emergence is indicative of adverse biological traits (18).
The apoptosis program governs the cell-autonomous removal of superfluous, infected, or damaged cells (19, 20) and thus constitutes the most prominent defense mechanism against hepatocarcinogenesis. During chronic damage, apoptosis is regulated on the outer mitochondrial membrane (OMM) by BCL-2 proteins. The pro-apoptotic BCL-2 proteins, BCL-2-associated X protein (BAX) and BCL-2 antagonist killer 1 (BAK) permeabilize the OMM and release intermembrane space proteins, such as cytochrome c, into the cytoplasm in order to activate the caspase cascade (21). Therefore, BAX and BAK can commit the cell to apoptosis. The cell is protected from BAX and BAK activity by functionally redundant pro-survival BCL-2 proteins. Although, BAX/BAK activation is usually followed by irreversible cellular commitment to apoptosis, cell survival is possible after limited OMM permeabilization (22). Even cells with the capacity to undergo death receptor-dependent apoptosis without mitochondrial apoptosis signaling enhance their apoptotic response by BAX/BAK activation (23). Therefore, therapeutic success of anti-tumor strategies, including targeted strategies, immune therapies as well as chemotoxic stress, rely on efficient BAX/BAK engagement in targeted cells. Several molecular alterations could be associated with induction or imbalance of pro- and anti-apoptotic BCL-2 proteins in liver cancer. They play an essential role in maintaining genomic integrity of hepatocytes. Disruption of the apoptotic program is frequently observed already during chronic liver diseases (12). Activation of BCL-xL is further observed at high frequencies in human HCC, whereas concomitant downregulation of BAX is a common feature of HCC with p53 alterations and observed at progressed stages of the disease (24). Moreover, inhibition of caspases e.g., by XIAP is also common in human HCC and associated with TGFβ signaling and subsequent acquisition of pro-metastatic properties. In addition to the inhibition of pro-apoptotic proteins or caspases, activation of pro-survival genes as well as pathways contributes to liver cancer development and progression (25).
A prominent molecular alteration detected in a sizable number of HCC patients is NF-kB pathway that is also particularly important in metabolic liver diseases and NASH-induced HCC (26–28). The pathway controls diverse functions in a cell type and context-dependent manner and activity is observed during chronic inflammation, fibrogenesis as well as development and progression of HCC (29, 30). In hepatocytes, NF-κB mainly mediates survival during chronic damage in response to e.g., oxidative stress while suppression contributes to malignant transformation. However, NF-κB activation in non-parenchymal and immune cells can aggravate inflammation and fibrogenesis (31). Tumor necrosis factor-α and interleukin-6 are among the major inflammatory cytokines that induce this pathway. NF-κB downstream signaling resembling c-Jun N-terminal kinase (JNK), and signal transducer and activator of transcription 3 play a major role in inflammation-associated HCC (32). NF-κB activation can also be critically linked to several anti-apoptotic molecules including (cIAP1, cIAP2), XIAP, the BCL-2 family members A1 and BCL- xL, cFLIP, TRAF1, TRAF2, and GADD45β (33). Besides JNK, NF-κB also activates other pro-survival and pro-proliferative pathways, resembling p38 MAPK (mitogen-activated protein kinase) kinase (34, 35). In this context, upstream regulators, such as the NF-κB essential modulator (NEMO), the IKK kinase complex as well as death-domain kinase receptor-interacting protein kinase 1 (RIPK1) are of particular importance. The central regulators of cell death resembling TAK1 and RIPK1 are, consequently, other common findings mechanistically linked to malignant transformation in the liver. TAK1 (MAP3-kinase TGF-β-activated kinase 1) is critically involved in the modulation of innate and adaptive immune responses. Activation of TAK1 in parenchymal cells significantly inhibits apoptosis and demonstrated anti-tumorigenic effects mediated by NF-κB activation via TNF (36). Conversely, deficiency of TAK1 impaired NF-κB activity and induced hepatocyte apoptosis, inflammation as well as HCC development in a NEMO-dependent manner (36). Consistently, alteration of the immune cell composition and impairment of immune-mediated clearance of damaged hepatocytes is an important driver of liver cancer. It has recently been shown that dysregulation of lipid metabolism in NAFLD induces selective ablation of intrahepatic CD4+ cells, which impairs mitochondrial function and generates high levels of oxidative damage, thus, corroborating lipid dysregulation with impaired anti-tumor immune-surveillance (37). Accordingly, impaired senescence surveillance by myeloid cells also induced failure in immune-mediated clearance of damaged hepatocytes and accelerated hepatocarcinogenesis. ER stress induced by metabolic liver damage following a high fat diet further enhanced resulting liver damage, increased immune infiltration, and lipogenesis and, ultimately, led to HCC development (28).
Another form of cell death recently linked to HCC development is necroptosis. Again, the mentioned TAK1 model with liver-specific ablation was employed to clarify the relative contribution of necroptosis during hepatocarcinogenesis. While response to apoptosis in the model promoted inflammation and tumorigenesis, necroptotic response had opposing effects and conferred anti-inflammatory and tumor-suppressive functions. These results indicate the diverse molecular functions of key cell death pathways in mediating apoptotic, necroptotic or other forms of cell death. Detailed dissection of the relative contribution and mechanistic hallmarks are urgently needed (12).
An improved biological understanding of the exact mechanisms driving hepatocyte cell death and, ultimately, cancer growth are not only of particular scientific interest, but also directly imply translational applications. Besides identification of patients at risk for cancer development, biomarkers of cell death might also be instrumental to delineate the biological trait, i.e., prognosis, of a tumor but also for prediction and monitoring of treatment response. Nevertheless, excessive cell death was successfully identified predict the development as well as progression of liver cancer. Furthermore, expression of key markers in cell death and surrogate characteristics were associated with clinical outcome. As such, the new checkpoint molecules RIPK1 and TRAF2 were recently confirmed as independent prognostic markers in liver cancer (38). Furthermore, the serum cell death parameter M65, which detects cleaved and uncleaved CK-18 fragments, was also demonstrated to possess clinical utility as a non-invasive marker for tumor initiation as well as prognosis, corroborating the potential as a new diagnostic tool for HCC (16). Finally, it is well-established that transcriptome profiles conferring to cell death resistance are significantly enriched in HCCs with low differentiation, high invasion and a particularly poor outcome (39). In summary, imbalance of a broad range of molecules with critical function of cell death, including dysregulation of cytokines and inflammatory as well as survival pathways during chronic liver disease, possess high relevance for clinical application and harbor potential as translational biomarkers of malignant transformation as well as progression.
Analysis of BCL-2 Proteins to Predict Tumor Cell Apoptosis
While liver tumors possess molecular characteristics that set them apart from other types of tumors, general mechanisms of apoptosis regulation apply as they have been shown in many different cell types. The discovery of opposing BCL-2 protein activities led to the rheostat model to describe regulatory interactions in mitochondrial apoptosis signaling (40, 41). The model postulates that pro-survival BCL-2 proteins act anti-apoptotic by binding to BAX and BAK. Therefore, mitochondrial apoptosis would largely dependent on different protein expression and degradation rates. In fact, platelets contain a molecular timer that commits them to apoptosis when BAK levels exceed the levels of the predominant pro-survival BCL-2 (42). The rheostat model sparked a body of work suggesting prediction of therapeutic success based on measuring BAX levels (Figure 1). The refined version of this approach investigated the BAX/BCL-2 ratio. However, subsequent research expanded our knowledge on protein localizations and interactions, revealing the absence of the prerequisite of the rheostat model: stable protein complexes. Prediction of apoptotic outcome based on protein levels encountered another major problem with the discovery of new members of the BCL-2 family. Their functional redundancies forsake all educated guesses, whether pro-survival BCL-2 proteins outnumber pro-apoptotic BCL-2 proteins. Therefore, apoptosis predictions based on protein levels, although occasionally attempted, are unrewarding.
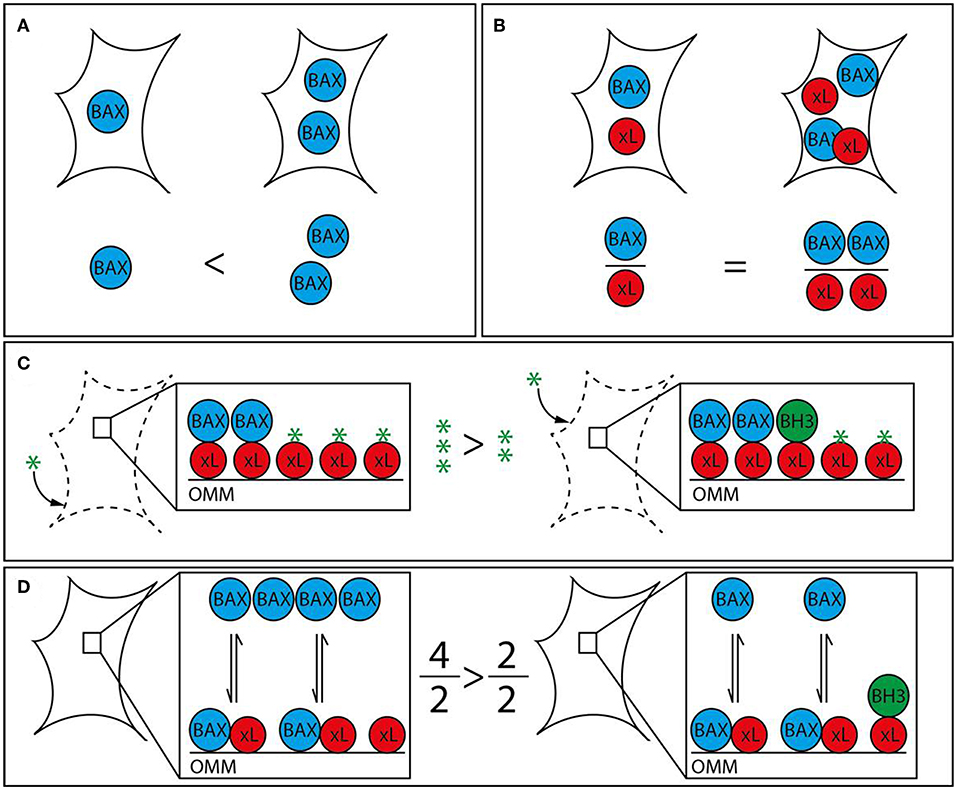
Figure 1. Strategies to analyze apoptotic predisposition based on BCL-2 proteins. (A) BAX level. The right cell contains more BAX (blue) than the left and is therefore considered to have a higher apoptosis predisposition. Other relevant factors are not measured. (B) BAX vs. BCL-xL level. The ratio between BAX and a single pro-survival BCL-2 protein (BCL-xL, red) is similar in both cells. Therefore, both cells would be judged to have the same tendency to initiate apoptosis. The redundancy of the BCL-2 family would require this analysis to be expanded to all BCL-2 proteins in order to be insightful. (C) BH3 profiling. Permeabilized and cultured cells are incubated with BH3 peptides (green star) in order to titrate the amount of free BH3 binding sites on the outer mitochondrial membrane (OMM). BH3-only proteins (green) associated with the OMM following prior cell stress reduce the amount of free BH3 binding sites and thus increase the sensitivity toward BH3 mimetics. The increased capacity of the left cell would translate into a reduced sensitivity toward BH3 mimetics. (D) Relative BAX localization. Determination of the cytosolic and mitochondrial BAX pools in intact cells describes the position of the BAX localization equilibrium and thus the cellular predisposition to apoptosis. While single contributing factors cannot be dissected, all contributing factors, e.g., BCL-2 protein interactions with BH3 motifs and other segments, interacting proteins outside the BCL-2 family, are included. BH3-only proteins reduce the shuttling rate and thus the cytosolic BAX pool. The larger cytosolic pool of the left cell shows reduced predisposition to apoptosis. The analysis can be supplemented with measuring the functionally redundant and similarly regulated BAK, which is usually shifted toward the mitochondria but shows a similar range of localizations in human samples.
A group of proteins that has influenced revised paradigms for apoptosis signaling and predictions of apoptotic outcome contains BH3-only proteins (Figure 2). BH3-only proteins are defined by harboring a single BH3 motif, while the remaining protein structures diverge as much as the type of stress signaled to the OMM, including DNA damage, ER stress, death receptor signaling and other types of stress (43). BH3-only proteins are thought to either inhibit pro-survival BCL-2 proteins and/or directly activate BAX and BAK (44). Inhibition of pro-survival BCL-2 proteins by BH3-only proteins is structurally well-characterized and has led to the development of low molecular weight inhibitors. These targeted anti-cancer small molecule inhibitors called BH3 mimetics bind to and inhibit pro-survival BCL-2 proteins in a manner similar to BH3-only proteins.
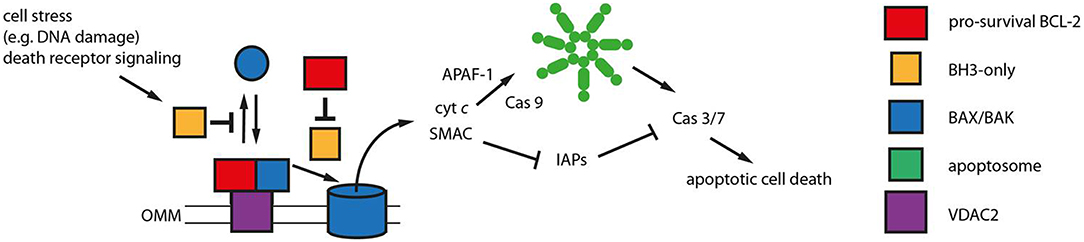
Figure 2. Mitochondrial apoptosis signaling. Mitochondrial apoptosis is regulated by members of the BCL-2 protein family on the outer mitochondrial membrane (OMM). The pro-apoptotic BCL-2 proteins BAX and BAK (blue) constantly translocate to the OMM undergoing a conformational change. The porin voltage-dependent anion channel 2 (VDAC 2, purple) acts as mitochondrial BAX/BAK receptor and as platform for the retrotranslocation of BAX and BAK back into the cytosol dependent on the activities of pro-survival BCL-2 proteins (red). The equilibrium between BAX/BAK translocation and retrotranslocation determines the cellular predisposition to apoptosis. Intrinsic stress as well as death receptor signaling is mediated by BH3-only proteins (yellow) that inhibit BAX/BAK retrotranslocation shifting BAX and BAK toward the mitochondria. The BH3-only proteins tBID, BIM and PUMA are also thought to directly activate BAX and BAK initiating OMM permeabilization and the release of cytochrome c (cyt c) and SMAC into the cytosol. This function can be inhibited by pro-survival BCL-2 proteins. Cytosolic cyt c initiates in turn the formation of the apoptosome (green), an APAF-1 complex activating Caspase 9 (Cas 9). Subsequently, Caspases 3 and 7 are activated that can be inhibited by IAPs in the absence of SMAC in the cytosol. Caspase 3/7 activation leads to the efficient dismantling of the cell into apoptotic bodies that are later phagocytosed.
The concept that BH3-only proteins loaded on the OMM could determine the cellular response to apoptosis has led to the strategy to profile BH3-only proteins. “Mitochondrial priming” in this context is the resulting stress capacity of cells dependent on the presence of pro-survival BCL-2 proteins, OMM-accumulated BH3-only proteins and BAX/BAK (Figure 1). Actually, BH3 profiling is again based on the rheostat model. It expands the model by emphasizing the potential role of BH3-only proteins, but does not take into account the transient nature of BCL-2 protein interactions and interactions among BCL-2 proteins other than through the BH3 motif. Extensive work shows the feasibility of “BH3 profiling” in different cellular settings (45–49). The analysis involves the short culturing of cells, limited cell lysis, incubation with peptides corresponding to BH3 domains and the analysis of OMM permeabilization through a membrane potential-sensitive dye. Cell culturing is prone to changes the apoptotic predisposition of a given tumor clone despite relative genetic stability. In addition, recent research has provided evidence of several secondary binding sites in BCL-2 protein interactions that BH3 profiling cannot account for (50–53). Therefore, BH3 profiling can particularly identify the contribution of pro-survival BCL-2 activities to the survival and therefore support selection of the potentially most effective BH3 mimetic. On the other hand, the appropriate BH3 mimetics could be tested directly, as procedure and readout would be similar.
Apoptosis Predisposition by the Position of the BAX/BAK Localization Equilibrium
Prior stress and stress response influence the apoptotic predisposition but are also reflected in the cellular localization of the pro-apoptotic BCL-2 proteins. Despite their functional redundancy, BAK is found largely on the OMM in many cell types, while BAX resides primarily in the cytoplasm (54, 55). This apparent difference is important, since the sizes of the mitochondrial protein pools prior apoptotic stress determines apoptotic response (56). The mitochondrial BAX pool as much as the corresponding BAK pool is variable because both proteins are inhibited by a dynamic shuttling equilibrium between cytosol and mitochondria (57). Pro-survival BCL-2 proteins constantly retrotranslocate BAX and BAK from the mitochondria and cell stress mediated by BH3-only proteins shifts both pro-apoptotic BCL-2 proteins back onto the mitochondria. The importance of mitochondrial BAX for apoptosis induction implies that (i) the total cell protein population is not critical for apoptosis induction and (ii) accurately measuring mitochondrial BAX (or BAK) fractions or shuttling rates could predict apoptotic outcome in response to stress (Figure 1). Experimental observations have shown that the ratio between cytosolic and mitochondrial BAX/BAK is the best available representation of the average localization dynamics of BAX/BAK molecules (58). The paradigm that relevant protein pool and total protein level are not necessarily connected is true for BAX, BAK, pro-survival proteins, like BCL-2 and BCL-xL, and BH3-only proteins, like BID (53, 59, 60). Relative BAX/BAK localization reflects the combined contributions of all players, known and unknown, to the cellular predisposition to apoptosis. Similar differences in the cellular BAX localization could also be present in HCC and could be associated with distinct molecular and clinical characteristics of the tumors.
Targeting of Cell Death as a Therapeutic Strategy for HCC
Hepatocyte damage and consecutive activation of cell death signaling plays a pivotal role for liver cancer initiation, but is also of particular importance for modulating treatment effects during established therapies. Herein, cell death can be induced by chemotherapeutic as well as targeted approaches (61). Sorafenib and lenvatinib are the only approved first line therapies for advanced stages in liver cancer (62). Both compounds are multi-tyrosine kinase inhibitors with anti-angiogenic properties. It is well-known, that sorafenib is a strong inducer of apoptosis and exposure to hepatoma cells leads to BAX/BAK activation, at least in part through the BH3-only protein PUMA (63). Furthermore, high numbers of objective response rates observed in HCC patients further indicate that considerable cell death follows lenvatinib treatment (64). However, several recent reports suggest that in addition to induction of cell death the treatment effect is also significantly induced by immunomodulation through targeted therapies (65). In consequence, several combination therapies with PD1/PD-L1 therapies are currently under clinical evaluation (62).
Direct targeting of cell death pathways and modulation of the apoptotic response might be a viable preventive strategy in chronic liver diseases but also exert direct anti-tumorigenic properties in HCC (Table 1). Given central role of p53 as a regulator of cell death, restoration of its function was attempted to induce anti-tumor activity in several studies. While adenoviral delivery of recombinant p53 did not reveal promising results, modulation of p53 activity by e.g., ubiquitination through inhibition of COP1, was recently explored (66). Blockade of COP1 by systemic delivery of RNAi decreased in vivo cancer growth and significantly induced apoptosis in several HCC cell lines. Furthermore, several compounds were identified to restore p53 functions. Prominently, p53 reactivation and induction of massive apoptosis (PRIMA-1) and PRIMA-1Met are currently evaluated in several clinical trials (67). In the liver, application of the compounds is currently restricted to preclinical data and shows promising anti-tumor effects when mutant p53 is silenced by siRNA. Other therapeutic strategies aimed to directly target proteins involved in apoptosis to enhance the apoptotic response of cancer therapies. Interestingly, XIAP antisense therapy in combination with sorafenib showed synergistic anti-tumor effects in a recent phase II clinical trial (68). Results showed a moderate increase in progression-free survival (4.0 months vs. 2.6 months), overall survival (6.5 months vs. 5.4 months), and objective response rates (16.1% vs. 9.7%) compared with Sorafenib monotherapy. Notably, drug-related adverse events were moderate.

Table 1. Selected targets of cell death in liver diseases and cancer.
Pro-survival BCL-2 proteins are also under intensive preclinical and clinical evaluation as cancer therapy targets. The use of the BH3 mimetic venetoclax or ABT-199 in chronic lymphocytic leukemia (CLL) has shown the potential of this strategy, as response rates of about 80% can be achieved with single-agent venetoclax even in a relapsed/refractory setting (69). Current efforts explore the combination of venetoclax with rituximab, obinutuzumab or ibrutinib in order to suppress acquired resistance observed during monotherapy (70, 71). Venetoclax in combination with hypomethylating agents (HMAs) has also received special attention for the treatment of acute myeloid leukemia (AML) (72, 73). The combination has been shown to target leukemia stem cells (74). In the liver, recent evidence suggest that BH3-only protein BID significantly contributes to the development of liver cancer (75). Loss of BID was shown to delay hepatocarcinogenesis by reducing cell death, liver inflammation, and compensatory proliferation (76). Thus, modulation of the BCL-2 protein interplay might be a promising therapeutic strategie for liver cancer.
In addition to the therapeutic targeting of critical apoptosis regulators, pan-caspase inhibitors, e.g., Emricasan/IDN-6556, or selective caspase-1,8,9 inhibitors, e.g., GS9450, have been explored in preclinical models as well as clinical trials, mainly in the context of chronic liver diseases (77). While the majority of these trials showed improved liver enzymes as a surrogate for hepatocyte protection, effect on degree of hepatitis and fibrogenesis is still unclear and is currently under evaluation in large phase III trials for the treatment NASH with and without liver cirrhosis (NCT02960204, NCT02686762, NCT03205345). Importantly, caspase inhibition might induce necroptosis or other complications and, thus, require further investigations addressing the safety of long-term administration (12).
Although no clinical trials have yet been initiated to test the clinical efficacy of necroptosis inhibition in liver disease, preclinical studies and early phase clinical trials in inflammatory (auto-immune) disease indicate that inhibition of RIPK1 kinase activity might also be a promising therapeutic strategy and prevent apoptosis in chronic liver diseases (78). However, while the importance of several key proteins including RIPK1, TAK1, and NEMO has been shown, the therapeutic potential for HCC remains to be demonstrated. Based on the regulatory functions of RIPK1, inhibition might even cause paradox reactions depending on the context of inhibition and affected cell type (30). Finally, given the redundancy in the different pathways, combination of different anti-tumor therapies with one or several modulators of cell death pathways might be of particular therapeutic potential.
Summary and Conclusions
Hepatocyte death is a key driver of chronic inflammatory liver diseases and hepatocarcinogenesis. Several lines of evidence suggest that apoptosis and other types of cell death are critically linked to initiation and progression of liver cancer. They participate in shaping the biological trait of the tumor, thus, ultimately determining patient prognosis. Herein, existence and degree of cell death infers several mechanistic and translational implications. While detection of the apoptotic predisposition might be a powerful diagnostic tool, direct targeting of mitochondrial apoptosis might complement the limited therapeutic strategies for HCC. In light of recent advances in immune-oncological approaches, targeting of cell death might also exert synergistic immuno-modulatory properties that could be explored in combination treatment strategies. However, our understanding of the detailed mechanisms and triggers of activation underlying the diverse mechanisms of cell death remains limited. Thus, definition of the actual state of cell death signaling effect in the distinct parenchymal and non-parenchymal cell types within the liver is urgently needed. Furthermore, the relative importance for distinct disease stages, i.e., chronic liver disease, tumor initiation as well as progression should be conclusively dissected to advance the field and before application of specific modulators of cell death in human is warranted. Furthermore, intensive translational research is needed to characterize the molecular hallmarks that operate on the intersection between cell death and inflammation. In this context, individual predisposition to apoptosis of cancer cells or cells within the hepatic microenvironment might be of particular relevance and might require distinct therapeutic strategy, thus, precision oncological approaches. Nevertheless, targeting of apoptosis is a promising avenue of HCC treatment that might yield to novel treatment strategies for this deadly inflammatory-driven cancer.
Author Contributions
JM and FE conceptualized, wrote, and edited the review.
Funding
JM was supported by grants from the German Research Foundation (MA 4443/2-2; SFB1292), the Volkswagen Foundation (Lichtenberg program) and by a grant from the Wilhelm-Sander Foundation (2017.007.1). FE was supported by the DFG Heisenberg program, the Collaborative Research Cluster (CRC) 746, the Else Kröner-Fresenius-Stiftung, the Wilhelm-Sander Foundation, and Germany's Excellence Strategy (CIBSS – EXC-2189 – Project ID 390939984).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Yang JD, Hainaut P, Gores GJ, Amadou A, Plymoth A, Roberts LR. A global view of hepatocellular carcinoma: trends, risk, prevention and management. Nat Rev Gastroenterol Hepatol. (2019) 16:589–60. doi: 10.1038/s41575-019-0186-y
2. Marquardt JU, Andersen JB, Thorgeirsson SS. Functional and genetic deconstruction of the cellular origin in liver cancer. Nature reviews. Cancer. (2015) 15:653–67. doi: 10.1038/nrc4017
3. Xiao J, Wang F, Wong NK, He J, Zhang R, Sun R, et al. Global liver disease burdens and research trends: Analysis from a Chinese perspective. J Hepatol. (2019) 71:212–21. doi: 10.1016/j.jhep.2019.03.004
4. Marquardt JU, Galle PR, Teufel A. Molecular diagnosis and therapy of hepatocellular carcinoma (HCC): an emerging field for advanced technologies. J Hepatol. (2012) 56:267–75. doi: 10.1016/j.jhep.2011.07.007
5. Liu Z, Jiang Y, Yuan H, Fang Q, Cai N, Suo C, et al. The trends in incidence of primary liver cancer caused by specific etiologies: results from the Global Burden of Disease Study 2016 and implications for liver cancer prevention. J Hepatol. (2019) 70:674–83. doi: 10.1016/j.jhep.2018.12.001
6. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. Cancer J Clin. (2018) 68:394–424. doi: 10.3322/caac.21492
7. El-Serag HB. Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology. (2012) 142:1264–73 e1. doi: 10.1053/j.gastro.2011.12.061
8. Hernandez-Gea V, Toffanin S, Friedman SL, Llovet JM. Role of the microenvironment in the pathogenesis and treatment of hepatocellular carcinoma. Gastroenterology. (2013) 144:512–27. doi: 10.1053/j.gastro.2013.01.002
9. Todoric J, Karin M. The fire within: cell-autonomous mechanisms in inflammation-driven cancer. Cancer Cell. (2019) 35:714–20. doi: 10.1016/j.ccell.2019.04.001
10. Kondylis V, Pasparakis M. RIP Kinases in Liver Cell Death, Inflammation and Cancer. Trends Mol Med. (2019) 25:47–63. doi: 10.1016/j.molmed.2018.10.007
11. Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. (2018) 25:486–541. doi: 10.1038/s41418-017-0012-4
12. Schwabe RF, Luedde T. Apoptosis and necroptosis in the liver: a matter of life and death. Nat Rev. (2018) 15:738–52. doi: 10.1038/s41575-018-0065-y
13. Garcia-Ruiz C, Fernandez-Checa JC. Mitochondrial oxidative stress and antioxidants balance in fatty liver disease. Hepatol Commun. (2018) 2:1425–39. doi: 10.1002/hep4.1271
14. Ku NO, Strnad P, Bantel H, Omary MB. Keratins: biomarkers and modulators of apoptotic and necrotic cell death in the liver. Hepatology. (2016) 64:966–76. doi: 10.1002/hep.28493
15. Mazzolini G, Sowa JP, Canbay A. Cell death mechanisms in human chronic liver diseases: a far cry from clinical applicability. Clin Sci. (2016) 130:2121–38. doi: 10.1042/CS20160035
16. Waidmann O, Koberle V, Bettinger D, Trojan J, Zeuzem S, Schultheiss M, et al. Diagnostic and prognostic significance of cell death and macrophage activation markers in patients with hepatocellular carcinoma. J Hepatol. (2013) 59:769–79. doi: 10.1016/j.jhep.2013.06.008
17. Bock B, Hasdemir D, Wandrer F, Rodt T, Manns MP, Schulze-Osthoff K, et al. Serum cell death biomarker mirrors liver cancer regression after transarterial chemoembolisation. Aliment Pharmacol Ther. (2016) 44:747–54. doi: 10.1111/apt.13758
18. Castven D, Fischer M, Becker D, Heinrich S, Andersen JB, Strand D, et al. Adverse genomic alterations and stemness features are induced by field cancerization in the microenvironment of hepatocellular carcinomas. Oncotarget. (2017) 8:48688–700. doi: 10.18632/oncotarget.16231
19. Claveria C, Giovinazzo G, Sierra R, Torres M. Myc-driven endogenous cell competition in the early mammalian embryo. Nature. (2013) 500:39–44. doi: 10.1038/nature12389
20. Sulston JE, Horvitz HR. Post-embryonic cell lineages of the nematode, Caenorhabditis elegans. Dev Biol. (1977) 56:110–56. doi: 10.1016/0012-1606(77)90158-0
21. Eskes R, Antonsson B, Osen-Sand A, Montessuit S, Richter C, Sadoul R, et al. Bax-induced cytochrome C release from mitochondria is independent of the permeability transition pore but highly dependent on Mg2+ ions. J Cell Biol. (1998) 143:217–24. doi: 10.1083/jcb.143.1.217
22. Ichim G, Lopez J, Ahmed SU, Muthalagu N, Giampazolias E, Delgado ME, et al. Limited mitochondrial permeabilization causes DNA damage and genomic instability in the absence of cell death. Mol Cell. (2015) 57:860–72. doi: 10.1016/j.molcel.2015.01.018
23. Hotchkiss RS, Strasser A, McDunn JE, Swanson PE. Cell death. N Eng J Med. (2009) 361:1570–83. doi: 10.1056/NEJMra0901217
24. Farazi PA, DePinho RA. Hepatocellular carcinoma pathogenesis: from genes to environment. Nature reviews. Cancer. (2006) 6:674–87. doi: 10.1038/nrc1934
25. Moreno-Caceres J, Fabregat I. Apoptosis in liver carcinogenesis and chemotherapy. Hepatol Oncol. (2015) 2:381–397. doi: 10.2217/hep.15.27
26. Locatelli I, Sutti S, Vacchiano M, Bozzola C, Albano E. NF-kappaB1 deficiency stimulates the progression of non-alcoholic steatohepatitis (NASH) in mice by promoting NKT-cell-mediated responses. Clin Sci. (2013) 124:279–87. doi: 10.1042/CS20120289
27. Grohmann M, Wiede F, Dodd GT, Gurzov EN, Ooi GJ, Butt T, et al. Obesity drives STAT-1-dependent NASH and STAT-3-dependent HCC. Cell. (2018) 175:1289–306 e20. doi: 10.1016/j.cell.2018.09.053
28. Nakagawa H, Umemura A, Taniguchi K, Font-Burgada J, Dhar D, Ogata H, et al. ER stress cooperates with hypernutrition to trigger TNF-dependent spontaneous HCC development. Cancer Cell. (2014) 26:331–43. doi: 10.1016/j.ccr.2014.07.001
29. Elsharkawy AM, Mann DA. Nuclear factor-kappaB and the hepatic inflammation-fibrosis-cancer axis. Hepatology. (2007) 46:590–7. doi: 10.1002/hep.21802
30. Czauderna C, Castven D, Mahn FL, Marquardt JU. Context-dependent role of NF-kappaB signaling in primary liver cancer-from tumor development to therapeutic implications. Cancers. (2019) 11:E1053. doi: 10.3390/cancers11081053
31. Luedde T, Schwabe RF. NF-kappaB in the liver–linking injury, fibrosis and hepatocellular carcinoma. Nat Rev. (2011) 8:108–18. doi: 10.1038/nrgastro.2010.213
32. Yang YM, Kim SY, Seki E. Inflammation and liver cancer: molecular mechanisms and therapeutic targets. Semin Liver Dis. (2019) 39:26–42. doi: 10.1055/s-0038-1676806
33. Pahl HL. Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene. (1999) 18:6853–66. doi: 10.1038/sj.onc.1203239
34. Heinrichsdorff J, Luedde T, Perdiguero E, Nebreda AR, Pasparakis M. p38 alpha MAPK inhibits JNK activation and collaborates with IkappaB kinase 2 to prevent endotoxin-induced liver failure. EMBO Rep. (2008) 9:1048–54. doi: 10.1038/embor.2008.149
35. Tang G, Minemoto Y, Dibling B, Purcell NH, Li Z, Karin M, et al. Inhibition of JNK activation through NF-kappaB target genes. Nature. (2001) 414:313–7. doi: 10.1038/35104568
36. Bettermann K, Vucur M, Haybaeck J, Koppe C, Janssen J, Heymann F, et al. TAK1 suppresses a NEMO-dependent but NF-kappaB-independent pathway to liver cancer. Cancer Cell. (2010) 17:481–96. doi: 10.1016/j.ccr.2010.03.021
37. Ma C, Kesarwala AH, Eggert T, Medina-Echeverz J, Kleiner DE, Jin P, et al. NAFLD causes selective CD4(+) T lymphocyte loss and promotes hepatocarcinogenesis. Nature. (2016) 531:253–7. doi: 10.1038/nature16969
38. Schneider AT, Gautheron J, Feoktistova M, Roderburg C, Loosen SH, Roy S, et al. RIPK1 suppresses a TRAF2-dependent pathway to liver cancer. Cancer Cell. (2017) 31:94–109. doi: 10.1016/j.ccell.2016.11.009
39. Lee JS, Chu IS, Heo J, Calvisi DF, Sun ZT, Roskams T, et al. Classification and prediction of survival in hepatocellular carcinoma by gene expression profiling. Hepatology. (2004) 40:667–76. doi: 10.1002/hep.20375
40. Oltvai ZN, Milliman CL, Korsmeyer SJ. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell. (1993) 74:609–19. doi: 10.1016/0092-8674(93)90509-O
41. Sedlak TW, Oltvai ZN, Yang E, Wang K, Boise LH, Thompson CB, et al. Multiple Bcl-2 family members demonstrate selective dimerizations with Bax. Proc Natl Acad Sciences USA. (1995) 92:7834–8. doi: 10.1073/pnas.92.17.7834
42. Mason KD, Carpinelli MR, Fletcher JI, Collinge JE, Hilton AA, Ellis S, et al. Programmed anuclear cell death delimits platelet life span. Cell. (2007) 128:1173–86. doi: 10.1016/j.cell.2007.01.037
43. Hinds MG, Smits C, Fredericks-Short R, Risk JM, Bailey M, Huang DC, et al. Bim, Bad and Bmf: intrinsically unstructured BH3-only proteins that undergo a localized conformational change upon binding to prosurvival Bcl-2 targets. Cell Death Differ. (2007) 14:128–36. doi: 10.1038/sj.cdd.4401934
44. Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. (2008) 9:47–59. doi: 10.1038/nrm2308
45. Brunelle JK, Ryan J, Yecies D, Opferman JT, Letai A. MCL-1-dependent leukemia cells are more sensitive to chemotherapy than BCL-2-dependent counterparts. J Cell Biol. (2009) 187:429–42. doi: 10.1083/jcb.200904049
46. Certo M, Del Gaizo Moore V, Nishino M, Wei G, Korsmeyer S, Armstrong SA, V. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell. (2006) 9:351–65. doi: 10.1016/j.ccr.2006.03.027
47. Ni Chonghaile T, Sarosiek KA, Vo TT, Ryan JA, Tammareddi A, Moore Vdel G, V. Pretreatment mitochondrial priming correlates with clinical response to cytotoxic chemotherapy. Science. (2011) 334:1129–33. doi: 10.1126/science.1206727
48. Ryan JA, Brunelle JK, Letai A. Heightened mitochondrial priming is the basis for apoptotic hypersensitivity of CD4+ CD8+ thymocytes. Proc Natl Acad Sci USA. (2010) 107:12895–900. doi: 10.1073/pnas.0914878107
49. Vo TT, Ryan J, Carrasco R, Neuberg D, Rossi DJ, Stone RM, et al. Relative mitochondrial priming of myeloblasts and normal HSCs determines chemotherapeutic success in AML. Cell. (2012) 151:344–55. doi: 10.1016/j.cell.2012.08.038
50. Andreu-Fernandez V, Sancho M, Genoves A, Lucendo E, Todt F, Lauterwasser J, et al. Bax transmembrane domain interacts with prosurvival Bcl-2 proteins in biological membranes. Proc Natl Acad Sci USA. (2017) 114:310–15. doi: 10.1073/pnas.1612322114
51. Barclay LA, Wales TE, Garner TP, Wachter F, Lee S, Guerra RM, et al. Inhibition of pro-apoptotic BAX by a noncanonical interaction mechanism. Mol Cell. (2015) 57:873–86. doi: 10.1016/j.molcel.2015.01.014
52. Gavathiotis E, Suzuki M, Davis ML, Pitter K, Bird GH, Katz SG, et al. BAX activation is initiated at a novel interaction site. Nature. (2008) 455:1076–81. doi: 10.1038/nature07396
53. Liu Q, Osterlund EJ, Chi X, Pogmore J, Leber B, Andrews DW. Bim escapes displacement by BH3-mimetic anti-cancer drugs by double-bolt locking both Bcl-XL and Bcl-2. Elife. (2019) 8:e37689. doi: 10.7554/eLife.37689
54. Griffiths GJ, Dubrez L, Morgan CP, Jones NA, Whitehouse J, Corfe BM, et al. Cell damage-induced conformational changes of the pro-apoptotic protein Bak in vivo precede the onset of apoptosis. J Cell Biol. (1999) 144:903–14. doi: 10.1083/jcb.144.5.903
55. Hsu YT, Youle RJ. Bax in murine thymus is a soluble monomeric protein that displays differential detergent-induced conformations. J Biol Chem. (1998) 273:10777–83. doi: 10.1074/jbc.273.17.10777
56. Todt F, Cakir Z, Reichenbach F, Youle RJ, Edlich F. The C-terminal helix of Bcl-x(L) mediates Bax retrotranslocation from the mitochondria. Cell Death Differ. (2013) 20:333–42. doi: 10.1038/cdd.2012.131
57. Edlich F, Banerjee S, Suzuki M, Cleland MM, Arnoult D, Wang C, Neutzner A, Tjandra N, Youle RJ, Bcl-x(L) retrotranslocates Bax from the mitochondria into the cytosol. Cell. (2011) 145:104–16. doi: 10.1016/j.cell.2011.02.034
58. Reichenbach F, Wiedenmann C, Schalk E, Becker D, Funk K, Scholz-Kreisel P, et al. Mitochondrial BAX determines the predisposition to apoptosis in human AML. Clin Cancer Res. (2017) 23:4805–16. doi: 10.1158/1078-0432.CCR-16-1941
59. Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. (1998) 94:491–501. doi: 10.1016/S0092-8674(00)81590-1
60. Todt F, Cakir Z, Reichenbach F, Emschermann F, Lauterwasser J, Kaiser A, et al. Differential retrotranslocation of mitochondrial Bax and Bak. EMBO J. (2015) 34:67–80. doi: 10.15252/embj.201488806
61. Pinyol R, Nault JC, Quetglas IM, Zucman-Rossi J, Llovet JM. Molecular profiling of liver tumors: classification and clinical translation for decision making. Semin Liver Dis. (2014) 34:363–75. doi: 10.1055/s-0034-1394137
62. Marquardt JU, Saborowski A, Czauderna C, Vogel A. The changing landscape of systemic treatment of advanced hepatocellular carcinoma: new targeted agents and immunotherapies. Target Oncol. (2019) 14:115–23. doi: 10.1007/s11523-019-00624-w
63. Fernando J, Sancho P, Fernandez-Rodriguez CM, Lledo JL, Caja L, Campbell JS, et al. Sorafenib sensitizes hepatocellular carcinoma cells to physiological apoptotic stimuli. J Cell Physiol. (2012) 227:1319–25. doi: 10.1002/jcp.22843
64. Kudo M, Finn RS, Qin S, Han KH, Ikeda K, Piscaglia F, et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial. Lancet. (2018) 391:1163–73. doi: 10.1016/S0140-6736(18)30207-1
65. Kimura T, Kato Y, Ozawa Y, Kodama K, Ito J, Ichikawa K, et al. Immunomodulatory activity of lenvatinib contributes to antitumor activity in the Hepa1-6 hepatocellular carcinoma model. Cancer Sci. (2018) 109:3993–4002. doi: 10.1111/cas.13806
66. Lee YH, Andersen JB, Song HT, Judge AD, Seo D, Ishikawa T, et al. Definition of ubiquitination modulator COP1 as a novel therapeutic target in human hepatocellular carcinoma. Cancer Res. (2010) 70:8264–9. doi: 10.1158/0008-5472.CAN-10-0749
67. Perdrix A, Najem A, Saussez S, Awada A, Journe F, Ghanem G, et al. PRIMA-1 and PRIMA-1(Met) (APR-246): from mutant/wild type p53 reactivation to unexpected mechanisms underlying their potent anti-tumor effect in combinatorial therapies. Cancers. (2017) 9:E172. doi: 10.3390/cancers9120172
68. Lee FA, Zee BC, Cheung FY, Kwong P, Chiang CL, Leung KC, et al. Randomized phase II study of the X-linked Inhibitor of Apoptosis (XIAP) antisense AEG35156 in combination with sorafenib in patients with advanced Hepatocellular Carcinoma (HCC). Am J Clin Oncol. (2016) 39:609–13. doi: 10.1097/COC.0000000000000099
69. Stilgenbauer S, Eichhorst B, Schetelig J, Hillmen P, Seymour JF, Coutre S, et al. Venetoclax for patients with chronic lymphocytic leukemia with 17p deletion: results from the full population of a phase II pivotal trial. J Clin Oncol. (2018) 36:1973–80. doi: 10.1200/JCO.2017.76.6840
70. Kater AP, Seymour JF, Hillmen P, Eichhorst B, Langerak AW, Owen C, et al. Fixed duration of venetoclax-rituximab in relapsed/refractory chronic lymphocyticleukemia eradicates minimal residual disease and prolongs survival: post-treatment follow-up of the MURANO phase III study. J Clin Oncol. (2019) 37:269–77. doi: 10.1200/JCO.18.01580
71. Seymour JF, Kipps TJ, Eichhorst B, Hillmen P, D'Rozario J, Assouline S, et al. Venetoclax-rituximab in relapsed or refractory chronic lymphocytic leukemia. N Eng J Med. (2018) 378:1107–20. doi: 10.1056/NEJMoa1713976
72. DiNardo CD, Pratz K, Pullarkat V, Jonas BA, Arellano M, Becker PS, et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood. (2019) 133:7–17. doi: 10.1182/blood-2018-08-868752
73. Pollyea DA, Jordan CT. Why are hypomethylating agents or low-dose cytarabine and venetoclax so effective? Curr Opin Hematol. (2019) 26:71–6. doi: 10.1097/MOH.0000000000000485
74. Pollyea DA, Stevens BM, Jones CL, Winters A, Pei S, Minhajuddin M, et al. Venetoclax with azacitidine disrupts energy metabolism and targets leukemia stem cells in patients with acute myeloid leukemia. Nat Med. (2018) 24:1859–66. doi: 10.1038/s41591-018-0233-1
75. Orlik J, Schungel S, Buitrago-Molina LE, Marhenke S, Geffers R, Endig J, et al. The BH3-only protein BID impairs the p38-mediated stress response and promotes hepatocarcinogenesis during chronic liver injury in mice. Hepatology. (2015) 62:816–28. doi: 10.1002/hep.27888
76. Wree A, Johnson CD, Font-Burgada J, Eguchi A, Povero D, Karin M, et al. Hepatocyte-specific Bid depletion reduces tumor development by suppressing inflammation-related compensatory proliferation. Cell Death Differ. (2015) 22:1985–94. doi: 10.1038/cdd.2015.46
77. Linton SD, Aja T, Armstrong RA, Bai X, Chen LS, Chen N, et al. First-in-class pan caspase inhibitor developed for the treatment of liver disease. J Med Chem. (2005) 48:6779–82. doi: 10.1021/jm050307e
Keywords: hepatocellular carcinoma, cell death, inflammation, BCL-2 family, BH3, primed to death, BH3 profiling, mitochondrial apoptosis
Citation: Marquardt JU and Edlich F (2019) Predisposition to Apoptosis in Hepatocellular Carcinoma: From Mechanistic Insights to Therapeutic Strategies. Front. Oncol. 9:1421. doi: 10.3389/fonc.2019.01421
Received: 16 September 2019; Accepted: 29 November 2019;
Published: 13 December 2019.
Edited by:
Luis Enrique Gomez-Quiroz, Universidad Autónoma Metropolitana, MexicoReviewed by:
Isabel Fabregat, Biomedical Research Institute of Bellvitge, SpainBarbara Marengo, University of Genoa, Italy
Copyright © 2019 Marquardt and Edlich. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jens U. Marquardt, bWFycXVhcmpAdW5pLW1haW56LmRl; Frank Edlich, ZnJhbmsuZWRsaWNoQGJpb2NoZW1pZS51bmktZnJlaWJ1cmcuZGU=