 Lizhi Li1,2†
Lizhi Li1,2† Jian Ma
Jian Ma- 1Hunan Cancer Hospital and the Affiliated Cancer Hospital of Xiangya School of Medicine, Central South University, Changsha, China
- 2Cancer Research Institute, School of Basic Medical Science, Xiangya School of Medicine, Central South University, Changsha, China
- 3Hunan Key Laboratory of Nonresolving Inflammation and Cancer, National Health Commission Key Laboratory of Carcinogenesis, Key Laboratory of Carcinogenesis and Cancer Invasion of Ministry of Education, Changsha, China
In recent years, oncolytic virotherapy has emerged as a promising anticancer therapy. Oncolytic viruses destroy cancer cells, without damaging normal tissues, through virus self-replication and antitumor immunity responses, showing great potential for cancer treatment. However, the clinical guidelines for administering oncolytic virotherapy remain unclear. Delivery routes for oncolytic virotherapy to patients vary in existing studies, depending on the tumor sites and the objective of studies. Moreover, the biosafety of oncolytic virotherapy, including mainly uncontrolled adverse events and long-term complications, remains a serious concern that needs to be accurately measured. This review provides a comprehensive and detailed overview of the delivery and biosafety of oncolytic virotherapy.
Oncolytic virotherapy has been recognized as a promising new treatment for cancer in recent years. Oncolytic viruses are genetically modified or naturally occurring viruses that selectively replicate in cancer cells and kill them without damaging normal cells (1). The idea of using viruses to treat cancer patients was originated in the 1950s (2). Many cancer patients were treated with oncolytic virus preparations administered by almost every feasible route, and some of them had tumor regression over different time scales (3). In a study from Osaka University, tumor regressions were reported in 37 of 90 terminal cancer patients who received non-attenuated mumps virus treatment (4). Since then, researchers have increasingly focused on oncolytic viruses for cancer treatment. The mechanism of oncolytic virotherapy includes two main aspects: (1) after infection, oncolytic viruses inhibit protein synthesis of cancer cells and destroy infected cancer cells by virus self-replication, and (2) oncolytic viruses recruit and activate tumor-infiltrating immune cells by promoting the release of a large amount of tumor antigens and cytokines, thus inducing strong antitumor immunity responses (5–7). As a new cancer therapy strategy, oncolytic virotherapy has immeasurable application potential, bringing new hope to cancer patients. This review summarizes the delivery of oncolytic viruses to patients based on a scan of existing preclinical and clinical studies, including those on intratumoral, intravenous, intraperitoneal, limb perfusion, aerosol delivery, etc. (8). Currently, the commonly used oncolytic viruses include herpes simplex virus type 1 (HSV-1), oncolytic adenovirus, oncolytic pox virus, Newcastle disease virus, and reovirus. A large number of natural and genetically modified oncolytic viruses have been developed and have reached the clinical research stages (9). However, biosafety issues remain a matter of serious concern. The primary problem in oncolytic virotherapy is the risk of uncontrolled replication in vivo and possible transmission to patients' contacts, such as other patients and health care workers (10). In recent years, clinical trials to address these concerns have been conducted. In this review, the route of delivery and the biosafety of oncolytic virus are discussed. All oncolytic viruses included in this review are summarized in Supplementary Table 1.
Oncolytic Virotherapy Emerged as a New Weapon Against Cancer
To date, three oncolytic virus drugs have been approved for cancer therapy. Rigvir (Riga virus) is an unmodified Echo virus that became the first approved oncolytic virus in the world for the treatment of melanoma in 2004 (11); Oncorine is an attenuated adenovirus that became the first clinically approved oncolytic virus in China in 2005 and the first approved recombinant oncolytic virus in the world for the treatment of head and neck tumors combined with chemotherapy (12). T-VEC, a recombinant human HSV-1, was approved by the U.S. Food and Drug Administration (FDA) in 2015 for the treatment of unresectable metastatic melanoma and was subsequently approved in the European Union for the treatment of locally advanced or metastatic cutaneous melanoma (13). The efficacy of oncolytic viruses on other types of tumors, such as lung cancer, liver cancer, pancreatic cancer, ovarian cancer, breast cancer, prostate cancer, bladder cancer, glioma, etc., is currently being addressed in clinical research and remains largely unknown (9). Recent clinical studies have shown the benefit of oncolytic virotherapy on some refractory malignant tumors, such as glioblastoma and triple-negative breast cancer (14–16).
Oncolytic viruses can also be used for tumor imaging with molecular imaging techniques. An oncolytic virus carrying a reporter gene can selectively replicate and express the reporter gene in the tumor cells such that the tumor cells emit fluorescence and absorb exogenous radionuclides. The tumors can be accurately imaged by bioluminescent detection systems such as CT (17). Human sodium iodide synergistic transporter protein (hNIS) was combined with human somatostatin receptor 2 (hSSR2) to engineer oncolytic viruses. After systemic administration of this virus, radioisotopes (99Tc and 131I) were administered, resulting in accumulation of the isotopes in the tumor mass, thereby enabling the tumor to be observed and located in a mouse model using a SPECT/CT imaging system (18). A combination of an exogenous lysine-rich protein (LRP) gene with the HSV genome can be used to image tumors by MRI because this construct changes the magnetic field associated with the metabolism rate of the tumors (19). The accurate imaging of tumors by oncolytic viruses has shown broad application prospects for early diagnosis and localization and visualization of tumors (18, 19).
Oncolytic viruses are thought to mediate antitumor activity through two different mechanisms: selective replication within tumor cells, which results in a direct lytic effect on the tumor cells, and induction of a systemic antitumor immunity response. After an oncolytic virus infects normal cells, it activates intracellular Toll-like receptors (TLRs) through pathogen-associated molecular patterns (PAMPs, including elements of viral capsids, DNAs, RNAs and protein products), thus activating the JAK–STAT or NF-κB pathway, inducing type I interferon (IFN) transcription and release (6). The interferon-induced double-stranded RNA-dependent protein kinase (PKR) can be activated by type I interferon and TLR and is essential for regulating cell proliferation and innate cellular antiviral responses. The activation of PKR inhibits cellular protein synthesis, which subsequently blocks cell proliferation and inhibits viral propagation (20). In cancer cells, interferon signaling and PKR activity are inhibited; thus, virus clearance is blocked, enabling virus replication (6, 20) (Figure 1). Following virus replication, most oncolytic viruses induce cell death, triggering not only the release of tumor-associated antigens that can promote an adaptive immune response but also viral PAMPs, cellular danger-associated molecular pattern signals (DAMPs; for example, heat shock proteins, HMGB1 protein, calreticulin, ATP and uric acid), and cytokines (for example, type I interferon, TNFα and IL-12). These released molecules recruit antigen-presenting cells (APCs) and promote their maturation, subsequently activating antigen-specific CD4+ and CD8+ T cell responses, enabling CD8+ T cells to expand into cytotoxic effector cells and mediating antitumor immunity (6, 15). The local release of interferons, chemokines and DAMP and PAMP factors activates tumor-infiltrating immune cells, which contribute to the reversal of the immune suppressive state of the tumor microenvironment and promote effective antitumor responses (5, 7, 15).
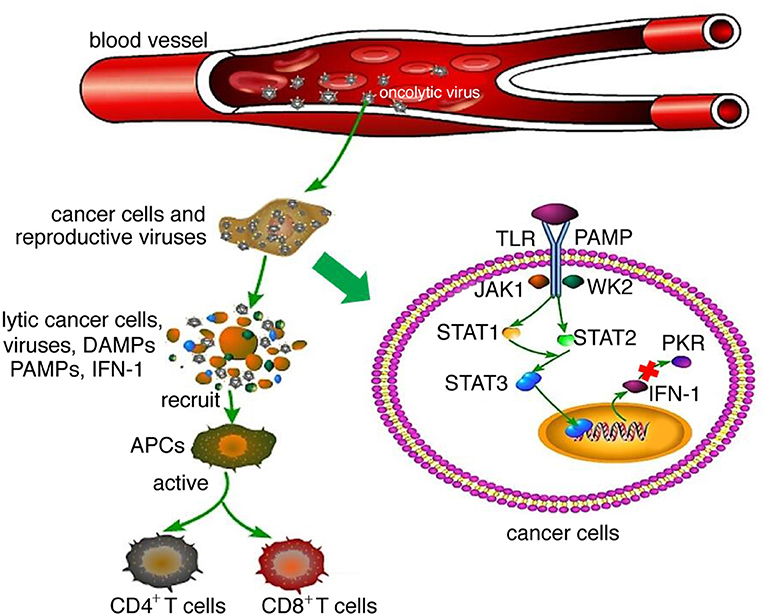
Figure 1. Mechanism of oncolytic virotherapy. When oncolytic viruses attack a normal cell, viruses activate JAK-STAT or NF-κB pathways through interaction between TLRs and PAMPs, which induce type I IFN transcription and release. Then, type I IFN activates PKR, which is essential for regulating abnormal cell proliferation and innate cellular antiviral responses. However, when oncolytic viruses attack cancer cells, interferon signaling and PKR activity are inhibited; thus, virus clearance is blocked, enabling virus replication. Following virus replication, most oncolytic viruses can induce cell death, at which time they release not only tumor-associated antigens that can promote an adaptive immune response but also viral PAMPs and additional cellular DAMPs and cytokines. These released molecules recruit antigen-presenting cells (APCs) and promote their maturation, subsequently activating antigen-specific CD4+ and CD8+ T cell responses, enabling CD8+ T cells to expand into cytotoxic effector cells and mediate antitumor immunity.
Since the antitumor activity of an oncolytic virus is not enough to effectively eliminate tumors, various strategies have been designed to improve their efficacy. The main strategies have been genetic modification, combined treatment and increasing the extent of virus replication and transmission.
Genetical Modification of Oncolytic Viruses
Genetic manipulation of the viral genome to create non-pathogenic viruses has become the main technique of oncolytic virus development with the goals of weakening virus pathogenicity, enhancing target selectivity, reducing adverse reactions, and/or inserting exogenous therapeutic genes into the virus genome, thereby increasing their expression in tumors and enhancing the treatment effect of the oncolytic virus (5). Deletion of ICP6 in the HSV-1 genome can weaken the pathogenicity of the virus; deletion of the gamma 34.5 gene can reduce the neurotoxicity of HSV-1 and enhance its selective replication in tumor cells. Introduction of microRNA targeting sites in the HSV-1 genome can inhibit viral gene expression and translation into normal cells that express specific microRNAs and improve the tumor cell selectivity of the virus (21). The oncolytic virus expressing PGE2-inactivating enzyme HPGD after genetic modification can reduce the level of myeloid-derived suppressor cells (MDSCs), thereby breaking down the immunosuppressive microenvironment of tumors and enhancing the sensitivity of oncolytic virotherapy (22). A genetically modified oncolytic adenovirus rich in CpG sites can overstimulate TLR9, thereby activating innate and adaptive immune responses and enhancing antitumor activity (23). Introducing oncogenesis-related gene-specific siRNA into an adenovirus genome can suppress the expression of oncogenes and inhibit tumor growth. Construction of adenoviruses expressing specific cytokines (such as GM-SF, IL-2, IL-12, etc.) can induce an antitumor immune response and enhance the oncolytic effects of the virus (24).
Combination Therapies With Oncolytic Viruses
An attractive feature of an oncolytic virus is that it can be combined with other immunotherapy approaches, among which immunological checkpoint inhibitor-combined therapy has become a mainstream strategy. The elevated expression of PD-L1 in the tumor microenvironment inhibits the infiltration of immune cells, which results in an immunosuppressed tumor microenvironment, which restricts the antitumor effect of oncolytic viruses. The combined approach of an oncolytic virus and a PD-1 or PD-L1 blockade can enhance antitumor immunity and the oncolytic effect (25–28). Additionally, the combination of oncolytic virotherapy and CAR-T immunotherapy produces a synergistic effect in cancer patients. Oncolytic viruses can induce tumor cell lysis and the release of tumor-associated antigens, thus stimulating the immune response to tumors and overcoming the obstacles associated with CAR-T applied to solid tumors. At the same time, the CAR-T antitumor effect on metastatic tumor sites overcomes the limitations of the oncolytic virus. The combination of these two approaches enhances the antitumor activity and has great application prospects (29, 30). Oncolytic viruses combined with chemotherapy have shown a promising effect. It is possible to combine oncolytic viruses with different immune characteristics to overcome antiviral immune responses and exert synergistic effects. Similarly, it has been demonstrated that bacteria can synergize with oncolytic viruses (31). For example, Le Boeuf et al. demonstrated that VSV (Vesicular Stomatitis Virus) combined with VACV (Vacienia Virus) improved antitumor response in immunodeficient and immunocompetent mouse tumor models (32). Cronin et al. showed that intravenous application of nonpathogenic E. coli expressing the vaccinia type 1 IFN antagonist B18R augmented subsequent therapy with oncolytic VSV by overcoming innate immunity against oncolytic viruses in an athymic nude mouse model (33).
Improvement in the Replication and Transmission of Oncolytic Viruses
Multiple strategies are used to overcome the obstacle of immune clearance, which challenges oncolytic virus therapy. Using stem cells as oncolytic virus carriers can reduce the immunogenicity of the viruses, while modifying the surface of oncolytic viruses with polymers and liposomes can increase the transmission of the viruses and enhance their antitumor effect (34, 35). The extracellular matrix (ECM), as a physical barrier, interferes with the transmission of oncolytic virus in a solid tumor mass, therefore, reconfiguration of the ECM can enhance the transmission of a virus into tumors. For instance, relaxin can inhibit the expression of collagen and the formation of ECM, and a decolorant can change the structure of ECM-residing collagen to remodel the ECM (24). Oncolytic adenoviruses expressing relaxin, which selectively degrades aberrant ECM, generated a potent antitumor effect through the effective induction of apoptosis (36). Most viruses can be engineered to encode exogenous genes. Expression of the transcription inhibitor PRd1-bf1/blimp1 induced by vascular endothelial growth factor (VEGF) can decrease the expression of type I interferon, thus weakening the antiviral immunity of vascular endothelial cells and promoting virus replication and transmission in tumors (37). Expression of interferon antagonists by gene modification can reduce the expression level of interferon, which weakens antiviral immunity, thereby promoting virus replication and enhancing the antitumor activity and transmission of oncolytic viruses (38).
Delivery Routes of Oncolytic Viruses
Suboptimal delivery is a potential cause of treatment failure. Multiple routes of delivery have been investigated for oncolytic virotherapy (Figure 2), the selection of which is critical for therapy efficiency. Researchers choose different routes of delivery according to their research objectives and the available and necessary experimental materials.
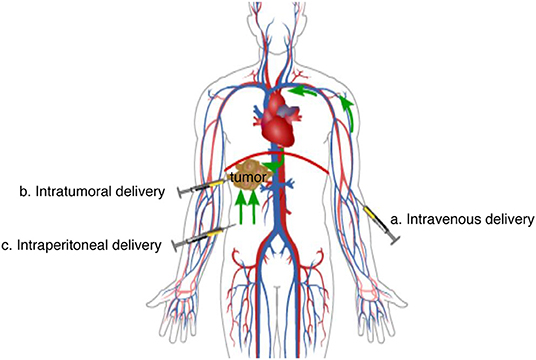
Figure 2. Main delivery routes of oncolytic virotherapy. (a) Intravenous delivery: When oncolytic viruses are injected into the peripheral vein, they reach tumor lesions in non-specific organs and systems through the circulation system. (b) Intratumoral delivery: When oncolytic viruses are injected into tumors, they have a direct therapeutic effect on the lesion. (c) Intraperitoneal delivery: When oncolytic viruses are injected into the peritoneal cavity, they will be absorbed into the veins of the peritoneum and then reach tumor lesions of some organs or systems through the circulation system, or they will diffuse directly to tumor lesions in the peritoneal cavity.
Direct Intratumoral Delivery
Intratumoral delivery is the most common route of oncolytic virus delivery. The concentration of oncolytic virus in the target site can be accurately controlled, and at the same time, the side effects caused by the virus being mistargeted to other organs can be prevented. Intratumoral delivery is much more suitable for surface tumors such as melanoma than it is for deep tumors such as glioblastoma due to operational difficulties in delivery.
Maintaining the concentration of oncolytic virus in the target site is a crucial advantage of intratumoral delivery, and researchers tend to observe more definite therapeutic effects with this method (39–41). Intratumoral delivery enables researchers to control the precise concentration of the oncolytic virus at the tumor site and to compare the results of in vitro experiments with those of in vivo studies (26, 42, 43). However, the risks and expenses associated with the complex procedures involved in intratumoral delivery make repeat dosing in vivo difficult.
At the level of cellular and animal models, Streby et al. demonstrated that HSV1716 (an oncolytic herpesvirus) was safely applied by direct intratumoral delivery (44). Dimethyl fumarate and various other fumaric acid and maleate (FMAE) compounds can enhance the ability of an oncolytic virus to infect melanoma cancer cells by direct intratumoral delivery (39). Warner et al. demonstrated that early expression of hNIS in colon cancer cells made viral replication reliably imageable via positron emission tomography (PET) of I-124 uptake (45). Direct intratumoral injection of oncolytic adenovirus VCN-01 for the treatment of retinoblastoma has also been shown to be effective (46). Choi et al. proved that CF33 (a novel chimeric orthopoxvirus encoding luciferase, enabling real-time view of cell infection), was effective in vitro with potent cytotoxicity and efficient intracellular replication observed in triple-negative breast cancer (TNBC, an aggressive subtype of breast cancer with high recurrence rate and poor prognosis) lines with PI3K/AKT pathway mutations (47). Jung et al. constructed two mathematical models to analyze the spread and expression of oncolytic measles viruses administered by direct intratumoral injection in vivo (42).
At the preclinical level, Bartee et al. demonstrated that tumors can secrete a soluble form of programmed cell death protein 1 (PD1) upon intratumoral injection of a novel recombinant myxoma virus (vPD1), thus enhancing the effect of the oncolytic virotherapy (26). Kicielinski et al. reported a multicenter study which demonstrated the approach of delivering intratumoral infusion of reovirus to patients with recurrent malignant glioma to be safe and well-tolerated (48). Intratumorally injected oncolytic adenovirus that had been engineered to encode a bispecific antibody (T-cell-targeted substances), combined with direct virolysis, induced endogenous T-cell activation to attack stromal fibroblasts, providing a multimodal treatment strategy within a single therapeutic agent (43). Blockade of immune checkpoints, immunogenic chemotherapy and IFN-α suppression can promote the local therapeutic effect of oncolytic viruses by direct intratumoral injection (41). Antagonizing the glycolysis and carboxylation of glutamine can enhance the activity of an oncolytic adenovirus directly injected into tumors by promoting its lysis in the cancer cells (49).
At the clinical trial level, Hirooka et al. conducted a phase I clinical trial by intratumoral injection. It was shown that HF10 injection of oncolytic virus was effective for unresectable locally advanced pancreatic cancer (50). On this basis, Nakao et al. conducted a phase I clinical trial of oncolytic virus HF10 with increasing doses for pancreatic cancer (51). Intratumoral injection of reovirus is effective in patients with recurrent malignant glioma, which has been histologically confirmed in a phase I clinical trial (52).
Intravenous Delivery
Many researchers prefer using intravenous injection to intratumoral injection in the clinical trial stage, which may be related to the complexity of the operation as well as the hurdles for distant metastasis of intratumoral delivery. Intravenous delivery of oncolytic virus represents a more simplified administration route for physicians.
For central nervous system tumors, Samson et al. demonstrated that oncolytic reovirus (ORV) vaccination by intravenous administration can block subsequent immune checkpoints in patients with brain tumors (53). The combination of oncolytic virus with CXCR4 antagonism can enhance the antitumor effect of dendritic cells in the context of neuroblastoma by intravenous administration (54). Tang et al. explored the selectivity of the oncolytic virus poxvirus to central nervous system tumors by intravenous administration (55). The therapeutic effect of oncolytic virus HSV G207 is obvious for children with progressive or recurrent malignant supratentorial brain tumors in a phase I clinical trial by means of intravenous delivery (56). These clinical trials confirmed that some oncolytic viruses can reach brain tumor tissues by bypassing the blood-brain barrier.
In addition to central nervous system tumors, intravenous administration has been applied to tumors in other organs and systems of the body. Saito et al. demonstrated that intravenous delivery of oncolytic adenovirus-carrying tumor vaccine into mouse squamous cell carcinoma models can inhibit the growth of multiple lung tumors (57). Oncolytic virus M1 was used to treat invasive bladder cancer by intravenous administration (58). Intravenous administration of oncolytic virus can modify the tumor microenvironment of prostate cancer and thus inhibit the growth of prostate cancer (59). Paclitaxel combined with oncolytic reovirus is effective in the treatment of recurrent ovarian cancer, fallopian tube cancer and peritoneal cancer through intravenous administration in a phase III clinical trial (60). Moreover, Nguyen et al. evaluated the polymer shielding effect of oncolytic adenovirus (Ad6) used to treat human prostate cancer by intravenous administration (61). Huang et al. explored the possibility of editing oncolytic virus vaccines using functional peptides by intravenous administration (62). Intravenous administration of oncolytic virus T-VEC combined with ipilimumab was successfully used to treat stage IIIb-IV melanoma, which could not be treated or resected in the past (63). The effectiveness of intravenous injection of oncolytic measles virus was also observed in the treatment of atypical teratoid rhabdomyoma in a xenotransplantation mouse model (64).
Other Routes of Delivery
In addition to the above two main methods, researchers also apply other routes of delivery. Chen et al. conducted an experiment using intraperitoneal injection of oHSV-1 to mice, suggesting that the combination of a PD-1 blockade and oHSV-1 may be an effective treatment strategy for childhood soft-tissue sarcoma (65). Besides, low dose of CF33 was confirmed to treat pancreatic cancer by intraperitoneal injection in vivo experiments (66). Kuryk et al. mainly used subcutaneous administration to indicate the clinical safety of application for Phase I clinical studies of ONCOS-102 (Ad5/3-D24-GM-CSF) for therapy for advanced cancers (67). Ochiai et al. administered PVS-RIPO into the spinal cord of transgenic mice, suggesting that intrathecal treatment with PVS-RIPO may be useful for the treatment of neoplastic meningitis in patients with glioblastoma multiforme (68).
Due to the large peritoneal area, intraperitoneal injected drugs can be absorbed faster than drugs administered the rough subcutaneous injection but slower than those delivered by intravenous injection. Because it is relatively easy to administer, intraperitoneal injection requires few specialty skills. Intraperitoneal injection is an ideal choice for targeting the organs in the abdominal cavity. Subcutaneous injection is the common method for administrating oncolytic viruses, but it is applied only to small animals in which veins are difficult to find. In addition, the scope of intrathecal injection is limited to central nervous system tumors. In other words, these delivery routes are used less frequently and are mainly limited to animal experiments mostly because of their low efficiency and narrow range of effectiveness.
Table 1 summarizes the different administration methods currently used for different types of tumors. The scientific community has not yet established a clear rubric to determine the advantages and disadvantages of using different delivery routes, which means that the best criteria for choosing the routes of delivery are debated.
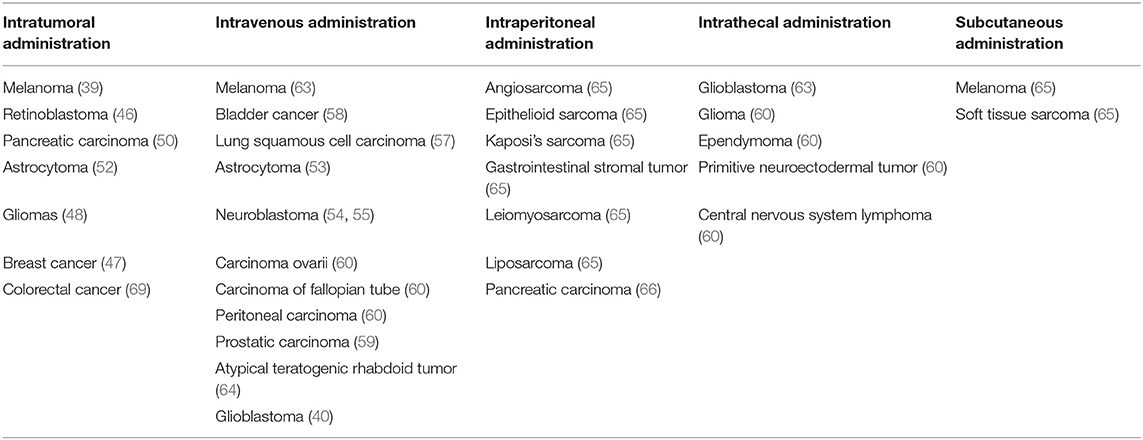
Table 1. Delivery routes of oncolytic viruses in multiple tumors.
As shown in Table 2, there are advantages and disadvantages to the five routes of delivery. Intratumoral delivery can maintain the concentration of the oncolytic virus in the target site, and researchers tend to observe more definitive therapeutic effects (39–41). Furthermore, the application of intratumoral injection enables researchers to control the precise concentration of oncolytic virus in the tumor site and to compare the results of in vitro experiments with those of in vivo studies (26, 42, 43). However, the risks and expense associated with the complex procedures involved in intratumoral delivery make repeat dosing difficult.
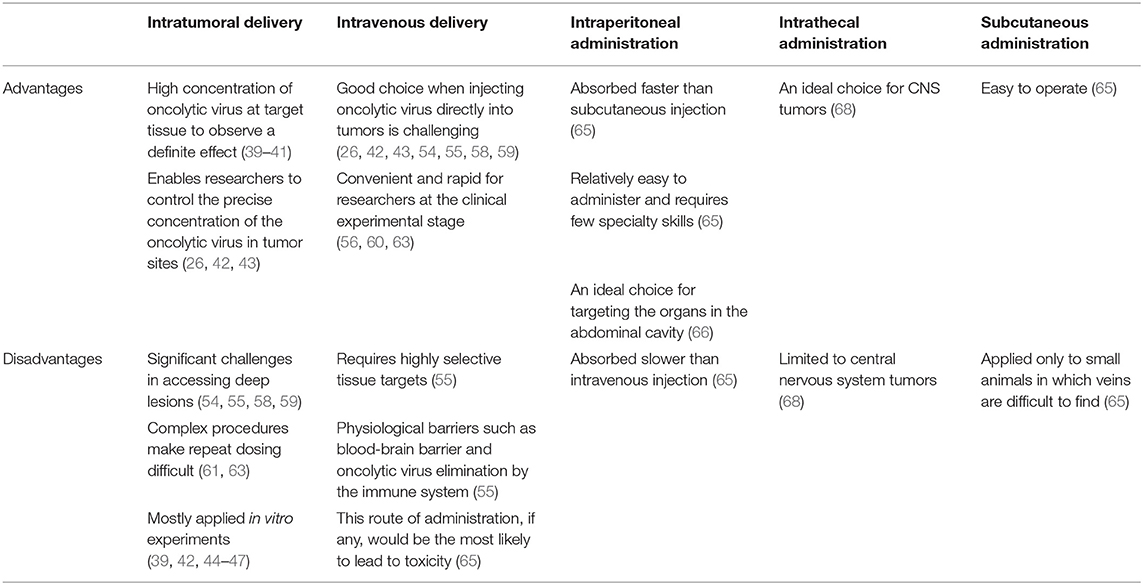
Table 2. Advantages and disadvantages of two different routes for delivering oncolytic viruses.
In some in vivo studies of tumors, researchers have difficulty injecting oncolytic an virus directly into tumors, such as astrocytoma; therefore, intravenous administration is a favorable choice (54, 55, 58, 59). Additionally, intravenous administration of an oncolytic virus has the advantages of convenience and rapidity, which are more suitable at the clinical trial stage (56, 60, 63). However, intravenous administration of oncolytic viruses requires highly selective target tissues (55). That is to say, this route of administration, if any, would be the most likely to lead to toxicity (65). Thus, blindly increasing the application concentration of the virus to compensate for its lack of selectivity will inevitably increase the public concern about the safety of oncolytic virotherapy.
Additionally, those considering use of intravenous administration of an oncolytic virus also need to consider the existence of physiological barriers, such as blood-brain barrier, and the elimination of oncolytic virus by the immune system (55). Is it possible that a wide variety of oncolytic viruses can bypass the blood-brain barrier? In what proportion can the viruses penetrate the blood-brain barrier? How can the immune system of the host be prevented from completely eliminating the injected oncolytic viruses?
The remaining three delivery routes are used less frequently and are mainly limited to animal experiments mostly because of their low efficiency and narrow range of effectiveness. Intraperitoneal injected drugs are absorbed slower than drugs delivered by intravenous injection, although it is an ideal choice for targeting the organs in the abdominal cavity. And subcutaneous injection is applied only to small animals whose veins are difficult to find. Similarly, the scope of intrathecal injection is limited to central nervous system tumors.
In summary, the current choice of the routes for delivery of oncolytic viruses is mainly based on the research purpose and material. No clear criteria or guidelines for choosing between the intratumor and intravenous administration of oncolytic virus have been established, and these approaches need to be further explored by researchers to provide more conclusive evidence for establishing selection criteria.
Biosafety of Oncolytic Virotherapy
Adverse Events Induced by Oncolytic Viruses
Oncolytic virotherapy was first used in a clinical trial of cervical cancer in 1956 (70). Since then, in pace with the success of tumor immunotherapy, scientists have paid more attention to oncolytic virotherapy. There is growing recognition that oncolytic virotherapy has the potential to be a safe treatment for cancer patients.
This review consulted 104 clinical trials of oncolytic virotherapy in the PubMed database. After summarizing the results of these 104 clinical trials, we found that the most common adverse events associated with oncolytic virotherapy were mild flulike symptoms and local reactions at the injection sites. Flulike symptoms caused by oncolytic virotherapy often manifested as fever, chills, myalgia, fatigue, nausea, diarrhea, vomiting, headache, etc. (63, 71–73), primarily a grade I–II flulike syndrome. Few patients experienced grade III-IV flulike syndrome (71, 74, 75). Some flulike symptoms disappeared spontaneously during the treatment process, and patients responded well to non-steroidal anti-inflammatory drugs. In addition, a predose of acetaminophen before initiation of the oncolytic virotherapy could reduce these symptoms (76). Local reactions often manifested as pain, rash, erythema, peripheral edema, etc. (77, 78), which spontaneously disappear a few days later or after symptom treatment, and most of the treatments did not induce dose-limiting toxicity (79). Moreover, some common adverse events, including anemia, leukopenia, lymphopenia, neutropenia, thrombocytopenia, liver dysfunction, and hematological abnormalities, specifically emerged in the trials of reovirus, HSV, and adenovirus (80–83). Some patients experienced liver dysfunction as a result of the liver and spleen tropism of the adenovirus (84–86).
Few oncolytic virotherapies cause severe adverse events that harm patients' health, and those that were induced could be managed, rarely causing severe damage to patients. However, oncolytic HSV caused severe hypotension, tachycardia, pleural effusion, herpes virus infection, central nervous system symptoms (such as brain edema, speech disorder, encephalitis, seizures, etc.) in clinical trials (44, 77, 87–89). In addition, oncolytic adenovirus caused pleural effusion, dehydration, hypokalemia, severe liver dysfunction, and sepsis in clinical trials (90–92). Severe hematological abnormalities (leukopenia, lymphopenia, and neutropenia), hypokalemia and pancreatitis were observed in the trials of oncolytic pox virus (93–96). All of the abovementioned virus treatments posed a health-threatening risk to patients who participated in these clinical trials. Pleural effusion could lead to dyspnea and even asphyxia. Fortunately, most of these severe adverse events were managed after withdrawal of the treatment or symptomatic treatment, rarely threatening patients' lives (93, 94, 97–99). Moreover, some preventive measures adopted before oncolytic virotherapy prevented patients from experiencing severe adverse events, as proved by the successful prevention of hypotension by patients drinking high volumes of water or being infused with saline (93, 94). The reasons for some severe events could be attributed to the primary diseases of the patients or to tumor progression (50, 100, 101).
Few oncolytic virotherapies caused virus infection symptoms in trials. Oncolytic HSV led to herpes in patients, and pustules were observed in trials of pox virus (44, 102–104). However, the herpes caused by HSV-based treatments could be managed by acyclovir or ganciclovir, which indicated that specific infection symptoms could be treated by antiviral drugs (78, 83).
The common adverse events of oncolytic virotherapies are summarized in Table 3.
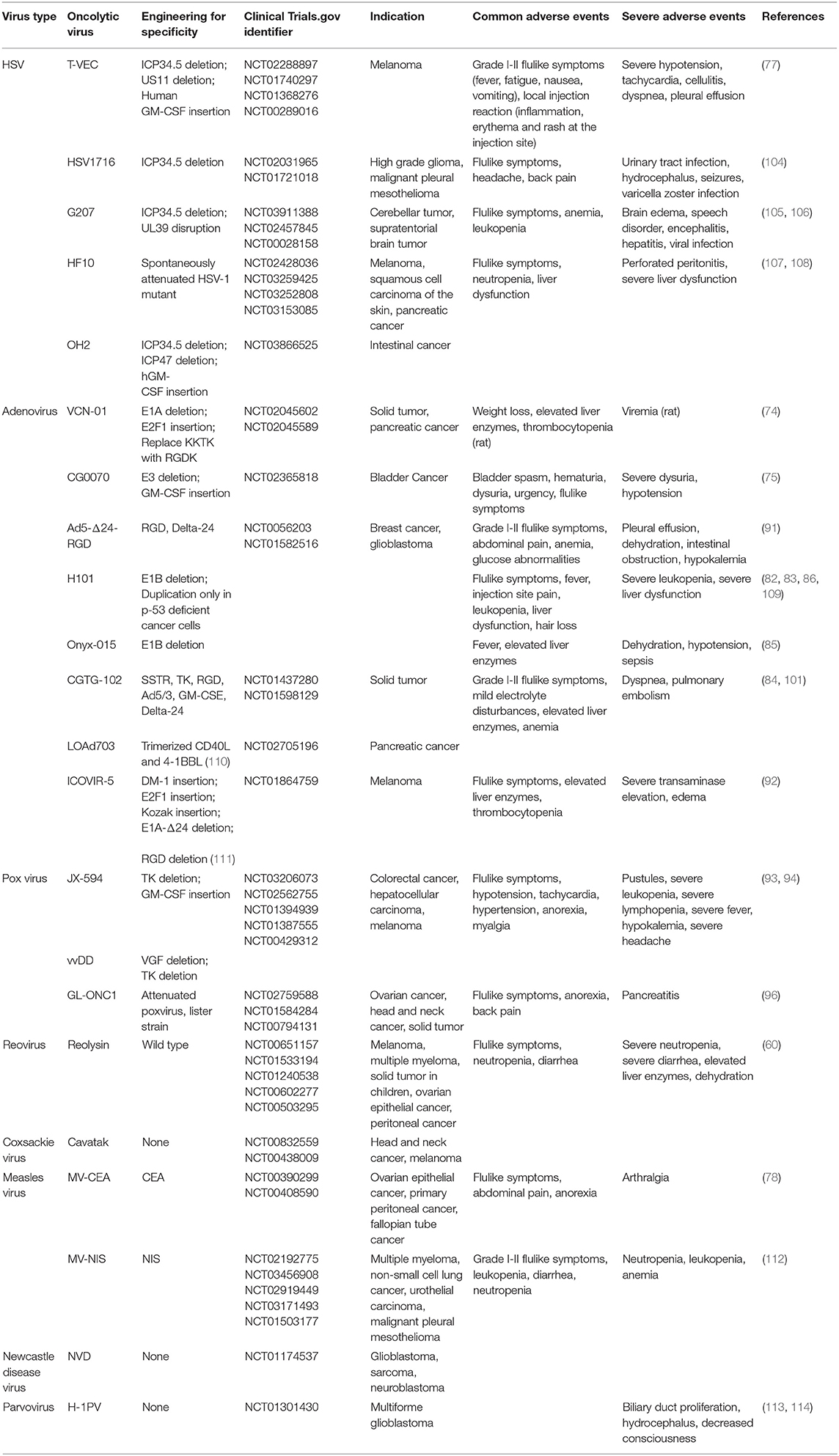
Table 3. Common adverse reactions to oncolytic virotherapies.
Potential Biosafety Issues of Oncolytic Virotherapies
Although current clinical trials of oncolytic virotherapies have led to severe uncontrollable adverse events, further oncolytic virotherapies needs to be promoted with caution. T-VEC, the first oncolytic virus approved by the FDA after a phase III clinical trial, has been used as a novel cancer therapy modality for only 4 years. Moreover, the potential safety issues and long-term adverse events of oncolytic virotherapies remain unclear.
Oncolytic virotherapies can cause latent infections and more severe potential safety problems that may manifest as long-term adverse events in the future. T-VEC is an oncolytic viral drug developed from HSV-1 that can be latent in nerves and thus induce latent infection. Corrigan et al. indicated that the DNA of oncolytic HSV may persistently remain in neuron bodies surrounding the injection site and may induce severe neurological HSV infection from a long-term perspective (88, 97).
Shedding and transmission of oncolytic viruses during therapy have also caused potential safety issues. Currently, T-VECs kill tumor cells through intratumoral delivery and are mainly used for melanoma treatment. However, during T-VEC therapy, the virus may transfer to other body parts of patients or to people in close contact with patients (87, 115). In clinical trials of Ad5-Δ24-RGD, Kimball et al. found frequent virus shedding in patients who received high doses of oncolytic viruses, which could be detected in serum, urine and saliva, and most commonly in saliva, and the shedding proportion seems to have been interrelated with the dose of the oncolytic virotherapy (90). However, intra-arterial hepatic injection did not result in detectable environmental shedding (116). The infectious shedding virus can be transferred throughout patients' own body and to people who are most likely to be exposed to these patients' body fluids, especially patients' family members and health care providers. Viral shedding was detected for HSV, adenovirus, poxvirus, and reovirus treatments, while it was rarely observed in treatments administered with intravenous VSV or poliovirus (117). However, there was no shedding virus detected in body fluids away from the injection site in HSV1617, H-1PV, or REO-10 therapy (44, 80, 118). In addition to infection, viral shedding can cause homologous recombination between an oncolytic virus and a residual-wild type virus. There is a high risk of homologous recombination when two similar viruses infect the same cell, which could produce a pathogenic transgenic virus. This mechanism has not been observed in the administration of oncolytic virotherapies but has been found during vaccine manufacture and usage (119). Still, shedding viruses observed in past studies are quite limited and highly attenuated, which is hard to cause damage to cancer patients, and the dose of oncolytic viruses for cancer patients is too small to cause shedding. In order to minimize the environmental viral shedding, the exposure of healthcare providers should be controlled when administrating oncolytic viruses (119).
Studies on the safety of oncolytic virotherapies for specific populations are currently insufficient. The T-VEC guidelines clearly indicate that people with low immunity or pregnancy should avoid using T-VECs. Wild-type HSV-1 that infects pregnant may cross the placental barrier and influence the fetus (71, 87). Preclinical studies of H-1PV showed that H-1PV induces embryonic and fetal toxicity in rodents and harmful effects on progeny, usually leading to the death of a fetus infected during the second trimester. When pregnant women are infected in the third trimester or a few days before birth, the progeny often develop “osteolytic syndrome” characterized by dwarfism and various down syndrome-like features (120, 121). Cancer patients who received radiotherapy and chemotherapy usually show low immunity to virus infection. Whether it is safe for these patients to be administered oncolytic virotherapy is debated (73). Children with severe combined immunodeficiency (SCID) who received an oncolytic retrovirus were found to have viral genes integrated into the LMO2 proto-oncogene region, which triggered the development of leukemia, reducing the survival rate of these patients with this compromised condition (122).
Methods to Improve the Biosafety of Oncolytic Virotherapy
To improve the biosafety of oncolytic virotherapies, the following three aspects may be considered. One approach involves selecting viruses that are not infectious to normal tissues. The natural host of parvovirus is rat, so parvovirus is non-pathogenic to humans because of anticellulosic selectivity, resulting in low selectivity for non-malignant tumor cells in humans. The overexpression of cytokines and transcription factors in tumors can active metabolic pathway which regulate function of Non-structural protein 1 (NS1, an essential protein for viral DNA replication, gene expression, and virus-induced cytotoxic effects), which can increase the tumor selectivity of parvoviruses (114, 118). In addition, reovirus has no or low pathogenicity in humans, and its pathogenicity in normal cells can be attenuated by repeated subculturing (80, 119). Kaid et al. reported that ZIKVBR can kill central nervous system (CNS) tumor cells specifically and effectively without causing damage to normal cells and other kinds of tumor cells, which means ZIKVBR have possibility to treat CNS embryonal tumors, and ZIKVBR caused very few infective cases of infants and adults in clinical trials (123).
The second approach involves attenuating the pathogenicity of oncolytic viruses to normal cells by genetic modification of the viruses, many of which have been used in studies. To develop oncolytic HSV-1 drugs, such as T-VEC and G207, the ICP-34.5 gene in HSV1716, which is a neurovirulence factor of HSV, was deleted, attenuating the infectivity of HSV in normal neurons (73, 117, 122, 124, 125). Mutation and deletion of the E1 gene can reduce adenovirus selectivity of normal cells, and this genetic modification was made to the Onyx-015 and H10 viruses (90, 126). Moreover, through genetic manipulation, the arg-gly-asp (RGD) sequence was integrated into the HI loop of the adenovirus capsid protein to enhance the infectivity of the oncolytic adenovirus in tumor cells (90, 127). It was reported that the enhanced liver infection by adenovirus may be caused by the binding between coagulation factor (F) X and hypervariable regions (HVR). Thus, inserting mutations in the FX-binding domain of the HVR and replacing them with HVRs of other serotypes of the original adenovirus can significantly reduce the liver tropism of the oncolytic adenovirus (128). Deleting genes such as TK, VGF, hemagglutinin, and B18R in the oncolytic pox virus can notably reduce its virulence in normal cells (81, 95, 103).
The third approach involves the recombination of different kinds of oncolytic viruses for therapy. The recombination of vesicular stomatitis virus (VSV) and Newcastle disease virus (NDV), named recombinant VSV-NDV (rVSV-NDV), greatly reduced cytotoxicity in healthy hepatocytes and neurons and was not pathogenic to the embryonated eggs. In the rVSV-NDV, the backbone of the VSV is retained. However, its glycoprotein is replaced by hemagglutinin-neuraminidase (HN) and the envelope proteins of the NVD. The adverse events caused by the off-target effects in brain and liver, which were observed in the trials of wild-type VSV, were significantly decreased because of the replacement of the glycoprotein (129). Adenoviruses are widely used in recombinant oncolytic virotherapies, such as those based on adenovirus-coxsackie virus and adenovirus-parvovirus (130). Recombinant adenoviruses and parvoviruses retain the infectivity of the adenovirus and the harmlessness of the parvovirus in normal cells, thereby killing the tumor cells and exempting the normal cells (131).
After analysis of the clinical trials of different types of oncolytic virotherapies, we concluded that oncolytic virotherapies are generally safe, and have a low incidence of adverse events, and cause only slight damage, which can be controlled or spontaneously regress (132). Clinical trials of oncolytic viruses such as G207, HSV1716, NV1020, Ad [I/PPT-E1A] and reovirus have proven their biosafety and effectiveness in tumor therapy (44, 73, 80, 106, 125). The HSV-2 and measles viruses were also proven non-toxic to humans in a number of mammalian experiments (124, 133–135). However, oncolytic virotherapies have unpredictability problems, such as long-term adverse events, which still need to be closely observed. As clinical trials expand and more patients participate, more long-term or short-term adverse events will likely be reported and analyzed. In addition, with new discoveries of oncolytic virotherapies, increasing numbers of genetic modifications to oncolytic viruses, and additional recombinant viruses, microRNAs, and viral vectors found, the safety of oncolytic viruses in tumor immunotherapy will be further guaranteed (117, 136, 137).
Conclusion and Perspective
In the past decade, oncolytic virotherapy, as a tumor immunotherapy, has emerged as a promising approach because of its selective killing of tumors (summarized in Supplementary Table 1). Currently, many clinical trials are ongoing, and most studies focus on how to improve the efficiency of oncolytic virotherapy, for which genetic modification, combination therapy and increasing viral replication and spreading are of specific interest. Moreover, the application of oncolytic viruses to tumor imaging is also under investigation.
This review summarizes the pros and cons of various routes of virus delivery and the biosafety of oncolytic virotherapy. Currently, oncolytic viruses are primarily administered through intratumoral and intravenous delivery, with each having advantages and disadvantages. The specific choice of which route of delivery is made without a clear standard or criterion and is mainly selected to reduce adverse events and enhance efficacy. Because of the advancement of virus recombination and genetic modification, as well as the specific mechanisms of oncolytic virotherapy, severe adverse events caused by oncolytic virotherapy have rarely been reported, while milder adverse events can generally be controlled or disappear spontaneously. Therefore, oncolytic virotherapy is currently generally safe, potential safety issues that are not currently presented or detected cannot be eliminated.
For virus delivery, first, more attention should be paid to maximizing the effective viral load in tumor lesions, thereby improving oncolytic virotherapy efficacy, which is also based on improved tumor selectivity. Second, similar to the that of drug combination therapy, different routes of delivery can be combined to enhance oncolytic virotherapy efficacy. In recent years, relevant clinical trials have been carried out (NCT01301430). However, the best application of oncolytic virotherapy is related to personalized medicine; that is, specific viral delivery to specific tumors forms a one-to-one precision therapeutic mode and may be the direction of oncolytic virotherapy development.
With regard to biosafety issues, a great majority of oncolytic virotherapy remains in phase II/III clinical trials. There is still a long road ahead before virotherapy is widely applied in tumor therapy. Although oncolytic virotherapy rarely caused fatal adverse events in recent clinical trials, continued studies should actively seek a balance between enhancing efficacy and reducing adverse events through virus modification or other means, with the goal of minimizing the adverse events as much as possible on the basis of ensuring treatment efficacy, which would also show the absolute advantage of oncolytic virotherapy over traditional tumor therapy such as surgical resection and adjuvant chemoradiation. Future studies should increasingly focus on the specific mechanism of the interaction between oncolytic viruses and the human immune system and the tumor immune microenvironment, thus preventing adverse events from the source of these viruses and latent virus infection and virus shedding and transmission, which would result in overall improved patient safety. In addition, more attention should be paid to the tumor selectivity of specific oncolytic virotherapies. By developing new viral vectors targeting tumor cells, oncolytic viruses can attach to tumor lesions with affinity but not to normal tissues, thereby eliminating adverse events and ensuring safety.
The need to choose the delivery mode and ensure the biosafety of the oncolytic virotherapy provides an important impetus to transform existing research results into clinical translations and plays a decisive role in guiding future clinical applications. It is expected that oncolytic virotherapy will become a powerful weapon for the therapy of malignant tumors. In the future, if oncolytic virotherapy can be developed into an oral preparation instead of an injection, delivered through a novel vector, specified by genetic modification or recombination, and effectively reach tumor lesions or reach an effective concentration through intestinal absorption, such that it exerts resistance against cancer cells and does not harm to normal tissues, then oncolytic virotherapy will be a revolutionary development in the new generation of tumor immunotherapy.
Author Contributions
LL, SL, DH, BT, and JM analyzed the literatures and studies and wrote the manuscript.
Funding
This work was supported by National Natural Science Foundation of China (81874170, 81672889), China 111 Project (111-2-12), National College Students' Innovation and Entrepreneurship Training Program of China (GS201910533237).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2020.00475/full#supplementary-material
References
1. Fukuhara H, Ino Y, Todo T. Oncolytic virus therapy: a new era of cancer treatment at dawn. Cancer Sci. (2016) 107:1373–9. doi: 10.1111/cas.13027
2. Kelly E, Russell SJ. History of oncolytic viruses: genesis to genetic engineering. Mol. Therap. (2007) 15:651–9. doi: 10.1038/sj.mt.6300108
3. Southam CM. Present status of oncolytic virus studies. Trans N Y Acad Sci. (1960) 22:657–73. doi: 10.1111/j.2164-0947.1960.tb00739.x
5. Bai Y, Hui P, Du X, Su X. Updates to the antitumor mechanism of oncolytic virus. Thorac Cancer. (2019) 10:1031–5. doi: 10.1111/1759-7714.13043
6. Kaufman HL, Kohlhapp FJ, Zloza A. Oncolytic viruses: a new class of immunotherapy drugs. Nat Rev Drug Discov. (2015) 14:642–62. doi: 10.1038/nrd4663
7. Gujar S, Bell J, Diallo JS. SnapShot: cancer immunotherapy with oncolytic viruses. Cell. (2019) 176:1240–0 e1241. doi: 10.1016/j.cell.2019.01.051
8. Chan WM, McFadden G. Oncolytic poxviruses. Annu Rev Virol. (2014) 1:119–41. doi: 10.1146/annurev-virology-031413-085442
9. Russell SJ, Peng KW, Bell JC. Oncolytic virotherapy. Nat Biotechnol. (2012) 30:658–70. doi: 10.1038/nbt.2287
10. Robilotti EV, Kumar A, Glickman MS, Kamboj M. Viral oncolytic immunotherapy in the war on cancer: Infection control considerations. Infect Control Hosp Epidemiol. (2019) 40:350–4. doi: 10.1017/ice.2018.358
11. Alberts P, Tilgase A, Rasa A, Bandere K, Venskus D. The advent of oncolytic virotherapy in oncology: The Rigvir® story. Eur Pharmacol J. (2018) 837:117–26. doi: 10.1016/j.ejphar.2018.08.042
12. Russell L, Peng KW. The emerging role of oncolytic virus therapy against cancer. Chin Clin Oncol. (2018) 7:16. doi: 10.21037/cco.2018.04.04
13. Bommareddy PK, Patel A, Hossain S, Kaufman HL. Talimogene Laherparepvec (T-VEC) and other oncolytic viruses for the treatment of melanoma. Am J Clin Dermatol. (2017). 18:1–15. doi: 10.1007/s40257-016-0238-9
14. Delwar ZM, Kuo Y, Wen YH, Rennie PS, Jia W. Oncolytic virotherapy blockade by microglia and macrophages requires STAT1/3. Cancer Res. (2018) 78:718–30. doi: 10.1158/0008-5472.CAN-17-0599
15. Bourgeois-Daigneault MA-O, Roy DA-O, Aitken AS, El Sayes N, Martin NT, Varette OA-OX, et al. Neoadjuvant oncolytic virotherapy before surgery sensitizes triple-negative breast cancer to immune checkpoint therapy. Sci Transl Med. (2018) 10:eaao1641. doi: 10.1126/scitranslmed.aao1641
16. Martikainen M, Essand M. Virus-based immunotherapy of glioblastoma. Cancers. (2019) 11:186. doi: 10.3390/cancers11020186
17. Wu ZJ, Tang FR, Ma ZW, Peng XC, Xiang Y, Zhang Y, et al. Oncolytic viruses for tumor precision imaging and radiotherapy. Hum Gene Ther. (2018) 29:204–22. doi: 10.1089/hum.2017.189
18. Wang J, Arulanandam R, Wassenaar R, Falls T, Petryk J, Paget J, et al. Enhancing expression of functional human sodium iodide symporter and somatostatin receptor in recombinant oncolytic vaccinia virus for in vivo imaging of tumors. J Nucl Med. (2017) 58:221–7. doi: 10.2967/jnumed.116.180463
19. Farrar CT, Buhrman JS, Liu G, Kleijn A, Lamfers ML, McMahon MT, et al. Establishing the Lysine-rich Protein CEST Reporter Gene as a CEST MR imaging detector for oncolytic virotherapy. Radiology. (2015) 275:746–54. doi: 10.1148/radiol.14140251
20. Kohlhapp FJ, Kaufman HL. Molecular pathways: mechanism of action for talimogene laherparepvec, a new oncolytic virus immunotherapy. Clin. Cancer Res. (2016) 22:1048–54. doi: 10.1158/1078-0432.CCR-15-2667
21. Bommareddy PK, Peters C, Saha D, Rabkin SD, Kaufman HL. Oncolytic herpes simplex viruses as a paradigm for the treatment of cancer. Ann Rev Cancer Biol. (2018) 2:155–73. doi: 10.1146/annurev-cancerbio-030617-050254
22. Hou W, Sampath P, Rojas JJ, Thorne SH. Oncolytic virus-mediated targeting of PGE2 in the tumor alters the immune status and sensitizes established and resistant tumors to immunotherapy. Cancer Cell. (2016) 30:108–19. doi: 10.1016/j.ccell.2016.05.012
23. Cerullo V, Diaconu I, Romano V, Hirvinen M, Ugolini M, Escutenaire S, et al. An oncolytic adenovirus enhanced for toll-like receptor 9 stimulation increases antitumor immune responses and tumor clearance. Mol Therapy. (2012) 20:2076–86. doi: 10.1038/mt.2012.137
24. Choi JW, Lee JS, Kim SW, Yun CO. Evolution of oncolytic adenovirus for cancer treatment. Adv Drug Deliv Rev. (2012) 64:720–9. doi: 10.1016/j.addr.2011.12.011
25. Ribas A, Dummer R, Puzanov I, VanderWalde A, Andtbacka RHI, Michielin O, et al. Oncolytic virotherapy promotes intratumoral T cell infiltration and improves anti-PD-1 immunotherapy. Cell. (2017) 170:1109–19.e1110. doi: 10.1016/j.cell.2017.08.027
26. Bartee MY, Dunlap KM, Bartee E. Tumor-localized secretion of soluble PD1 enhances oncolytic virotherapy. Cancer Res. (2017) 77:2952–63. doi: 10.1158/0008-5472.CAN-16-1638
27. Jiang H, Rivera-Molina Y, Gomez-Manzano C, Clise-Dwyer K, Bover L, Vence LM, et al. Oncolytic adenovirus and tumor-targeting immune modulatory therapy improve autologous cancer vaccination. Cancer Res. (2017) 77:3894–907. doi: 10.1158/0008-5472.CAN-17-0468
28. Zamarin D, Ricca JM, Sadekova S, Oseledchyk A, Yu Y, Blumenschein WM, et al. PD-L1 in tumor microenvironment mediates resistance to oncolytic immunotherapy. J Clin Invest. (2018) 128:1413–28. doi: 10.1172/JCI98047
29. Rosewell Shaw A, Suzuki M. Oncolytic viruses partner with T-cell therapy for solid tumor treatment. Front Immunol. (2018) 9:2103. doi: 10.3389/fimmu.2018.02103
30. Ajina A, Maher J. Prospects for combined use of oncolytic viruses and CAR T-cells. J ImmunoTherap Cancer. (2017) 5:90. doi: 10.1186/s40425-017-0294-6
31. Martin NT, Bell JC. Oncolytic virus combination therapy: killing one bird with two stones. Mol Ther. (2018) 26:1414–22. doi: 10.1016/j.ymthe.2018.04.001
32. Le Boeuf F, Diallo J-S, McCart JA, Thorne S, Falls T, Stanford M, et al. Synergistic interaction between oncolytic viruses augments tumor killing. Mol Therapy. (2010) 18:888–95. doi: 10.1038/mt.2010.44
33. Cronin M, Le Boeuf F, Murphy C, Roy DG, Falls T, Bell JC, et al. Bacterial-mediated knockdown of tumor resistance to an oncolytic virus enhances therapy. Mol Therap. (2014) 22:1188–97. doi: 10.1038/mt.2014.23
34. Zendedel E, Atkin SL, Sahebkar A. Use of stem cells as carriers of oncolytic viruses for cancer treatment. J Cell Physiol. (2019) 234:14906–13. doi: 10.1002/jcp.28320
35. Liu XQ, Xin HY, Lyu YN, Ma ZW, Peng XC, Xiang Y, et al. Oncolytic herpes simplex virus tumor targeting and neutralization escape by engineering viral envelope glycoproteins. Drug Deliv. (2018) 25:1950–62. doi: 10.1080/10717544.2018.1534895
36. Jung KH, Choi IK, Lee HS, Yan HH, Son MK, Ahn HM, et al. Oncolytic adenovirus expressing relaxin (YDC002) enhances therapeutic efficacy of gemcitabine against pancreatic cancer. Cancer Lett. (2017) 396:155–66. doi: 10.1016/j.canlet.2017.03.009
37. Arulanandam R, Batenchuk C, Angarita FA, Ottolino-Perry K, Cousineau S, Mottashed A, et al. VEGF-mediated induction of PRD1-BF1/Blimp1 expression sensitizes tumor vasculature to oncolytic virus infection. Cancer Cell. (2015) 28:210–24. doi: 10.1016/j.ccell.2015.06.009
38. Le Boeuf F, Batenchuk C, Vaha-Koskela M, Breton S, Roy D, Lemay C, et al. Model-based rational design of an oncolytic virus with improved therapeutic potential. Nat Commun. (2013) 4:1974. doi: 10.1038/ncomms2974
39. Selman M, Ou P, Rousso C, Bergeron A, Krishnan R, Pikor L, et al. Dimethyl fumarate potentiates oncolytic virotherapy through NF-kappaB inhibition. Sci Transl Med. (2018) 10:eaao1613. doi: 10.1126/scitranslmed.aao1613
40. Xiao X, Liang J, Huang C, Li K, Xing F, Zhu W, et al. DNA-PK inhibition synergizes with oncolytic virus M1 by inhibiting antiviral response and potentiating DNA damage. Nat Commun. (2018) 9:4342. doi: 10.1038/s41467-018-06771-4
41. Fend L, Yamazaki T, Remy C, Fahrner C, Gantzer M, Nourtier V, et al. Immune checkpoint blockade, immunogenic chemotherapy or IFN-alpha blockade boost the local and abscopal effects of oncolytic virotherapy. Cancer Res. (2017) 77:4146–57. doi: 10.1158/0008-5472.CAN-16-2165
42. Jung MY, Offord CP, Ennis MK, Kemler I, Neuhauser C, Dingli D. In vivo estimation of oncolytic virus populations within tumors. Cancer Res. (2018) 78:5992–6000. doi: 10.1158/0008-5472.CAN-18-0447
43. Freedman JD, Duffy MR, Lei-Rossmann J, Muntzer A, Scott EM, Hagel J, et al. An oncolytic virus expressing a T-cell engager simultaneously targets cancer and immunosuppressive stromal cells. Cancer Res. (2018) 78:6852–65. doi: 10.1158/0008-5472.CAN-18-1750
44. Streby KA, Geller JI, Currier MA, Warren PS, Racadio JM, Towbin AJ, et al. Intratumoral injection of HSV1716, an oncolytic herpes virus, is safe and shows evidence of immune response and viral replication in young cancer patients. Clin Cancer Res. (2017) 23:3566–74. doi: 10.1158/1078-0432.CCR-16-2900
45. Warner SG, Kim S-I, Chaurasiya S, O'Leary MP, Lu J, Sivanandam V, et al. A novel chimeric poxvirus encoding hNIS is tumor-tropic, imageable, and synergistic with radioiodine to sustain colon cancer regression. Mol Therap Oncol. (2019) 13:82–92. doi: 10.1016/j.omto.2019.04.001
46. Pascual-Pasto G, Bazan-Peregrino M, Olaciregui NG, Restrepo-Perdomo CA, Mato-Berciano A, Ottaviani D, et al. Therapeutic targeting of the RB1 pathway in retinoblastoma with the oncolytic adenovirus VCN-01. Sci Transl Med. (2019) 11:eaat9321. doi: 10.1126/scitranslmed.aat9321
47. Choi AH, O'Leary MP, Lu J, Kim S-I, Fong Y, Chen NG. Endogenous Akt activity promotes virus entry and predicts efficacy of novel chimeric orthopoxvirus in triple-negative breast cancer. Mol Therap Oncol. (2018) 9:22–9. doi: 10.1016/j.omto.2018.04.001
48. Kicielinski KP, Chiocca EA, Yu JS, Gill GM, Coffey M, Markert JM. Phase 1 clinical trial of intratumoral reovirus infusion for the treatment of recurrent malignant gliomas in adults. Mol Therap. (2014) 22:1056–62. doi: 10.1038/mt.2014.21
49. Dyer A, Schoeps B, Frost S, Jakeman P, Scott EM, Freedman J, et al. Antagonism of glycolysis and reductive carboxylation of glutamine potentiates activity of oncolytic adenoviruses in cancer cells. Cancer Res. (2019) 79:331–45. doi: 10.1158/0008-5472.CAN-18-1326
50. Hirooka Y, Kasuya H, Ishikawa T, Kawashima H, Ohno E, Villalobos IB, et al. A Phase I clinical trial of EUS-guided intratumoral injection of the oncolytic virus, HF10 for unresectable locally advanced pancreatic cancer. BMC Cancer. (2018) 18:596. doi: 10.1186/s12885-018-4453-z
51. Nakao A, Kasuya H, Sahin TT, Nomura N, Kanzaki A, Misawa M, et al. A phase I dose-escalation clinical trial of intraoperative direct intratumoral injection of HF10 oncolytic virus in non-resectable patients with advanced pancreatic cancer. Cancer Gene Ther. (2011) 18:167–75. doi: 10.1038/cgt.2010.65
52. Forsyth P, Roldan G, George D, Wallace C, Palmer CA, Morris D, et al. A phase I trial of intratumoral administration of reovirus in patients with histologically confirmed recurrent malignant gliomas. Mol Ther. (2008) 16:627–32. doi: 10.1038/sj.mt.6300403
53. Samson A, Scott KJ, Taggart D, West EJ, Wilson E, Nuovo GJ, et al. Intravenous delivery of oncolytic reovirus to brain tumor patients immunologically primes for subsequent checkpoint blockade. Sci Transl Med. (2018) 10:eaam7577. doi: 10.1126/scitranslmed.aam7577
54. Komorowski M, Tisonczyk J, Kolakowska A, Drozdz R, Kozbor D. Modulation of the tumor microenvironment by CXCR4 antagonist-armed viral oncotherapy enhances the antitumor efficacy of dendritic cell vaccines against neuroblastoma in syngeneic mice. Viruses. (2018) 10:455. doi: 10.3390/v10090455
55. Tang B, Guo ZS, Bartlett DL, Liu J, McFadden G, Shisler JL, et al. A cautionary note on the selectivity of oncolytic poxviruses. Oncol Virotherap. (2019) 8:3–8. doi: 10.2147/OV.S189832
56. Waters AM, Johnston JM, Reddy AT, Fiveash J, Madan-Swain A, Kachurak K, et al. Rationale and design of a phase I clinical trial to evaluate HSV G207 alone or with a single radiation dose in children with progressive or recurrent malignant supratentorial brain tumors. Hum Gene Therap Clin Dev. (2017) 28:7–16. doi: 10.1089/hum.2017.002
57. Saito A, Morishita N, Mitsuoka C, Kitajima S, Hamada K, Lee K-M, et al. Intravenous injection of irradiated tumor cell vaccine carrying oncolytic adenovirus suppressed the growth of multiple lung tumors in a mouse squamous cell carcinoma model. J Gene Med. (2011) 13:353–61. doi: 10.1002/jgm.1578
58. Hu C, Liu Y, Lin Y, Liang J-K, Zhong W-W, Li K, et al. Intravenous injections of the oncolytic virus M1 as a novel therapy for muscle-invasive bladder cancer. Cell Death Disease. (2018) 9:274. doi: 10.1038/s41419-018-0325-3
59. Atherton MJ, Stephenson KB, Tzelepis F, Bakhshinyan D, Nikota JK, Son HH, et al. Transforming the prostatic tumor microenvironment with oncolytic virotherapy. OncoImmunology. (2018) 7:e1445459. doi: 10.1080/2162402X.2018.1445459
60. Cohn DE, Sill MW, Walker JL, O'Malley D, Nagel CI, Rutledge TL, et al. Randomized phase IIB evaluation of weekly paclitaxel versus weekly paclitaxel with oncolytic reovirus (Reolysin(R)) in recurrent ovarian, tubal, or peritoneal cancer: an NRG Oncology/Gynecologic Oncology Group study. Gynecol Oncol. (2017) 146:477–83. doi: 10.1016/j.ygyno.2017.07.135
61. Nguyen TV, Heller GJ, Barry ME, Crosby CM, Turner MA, Barry MA. Evaluation of polymer shielding for adenovirus serotype 6 (Ad6) for systemic virotherapy against human prostate cancers. Mol Ther Oncol. (2016) 3:15021. doi: 10.1038/mto.2015.21
62. Huang L-L, Li X, Liu K, Zou B, Xie H-Y. Engineering oncolytic vaccinia virus with functional peptides through mild and universal strategy. Anal Bioanal Chem. (2018) 411:925–33. doi: 10.1007/s00216-018-1519-3
63. Puzanov I, Milhem MM, Minor D, Hamid O, Li A, Chen L, et al. Talimogene laherparepvec in combination with ipilimumab in previously untreated, unresectable stage IIIB-IV melanoma. J Clin Oncol. (2016) 34:2619–26. doi: 10.1200/JCO.2016.67.1529
64. Studebaker AW, Hutzen B, Pierson CR, Shaffer TA, Raffel C, Jackson EM. Oncolytic measles virus efficacy in murine xenograft models of atypical teratoid rhabdoid tumors. Neuro Oncol. (2015) 17:1568–77. doi: 10.1093/neuonc/nov058
65. Chen CY, Wang PY, Hutzen B, Sprague L, Swain HM, Love JK, et al. Cooperation of oncolytic herpes virotherapy and PD-1 blockade in murine rhabdomyosarcoma models. Sci Rep. (2017) 7:2396. doi: 10.1038/s41598-017-02503-8
66. O'Leary MP, Choi AH, Kim S-I, Chaurasiya S, Lu J, Park AK, et al. Novel oncolytic chimeric orthopoxvirus causes regression of pancreatic cancer xenografts and exhibits abscopal effect at a single low dose. J Transl Med. (2018) 16:110. doi: 10.1186/s12967-018-1483-x
67. Kuryk L, Vassilev L, Ranki T, Hemminki A, Karioja-Kallio A, Levalampi O, et al. Toxicological and bio-distribution profile of a GM-CSF-expressing, double-targeted, chimeric oncolytic adenovirus ONCOS-102 - Support for clinical studies on advanced cancer treatment. PLoS ONE. (2017) 12:e0182715. doi: 10.1371/journal.pone.0182715
68. Ochiai H, Campbell SA, Archer GE, Chewning TA, Dragunsky E, Ivanov A, et al. Targeted therapy for glioblastoma multiforme neoplastic meningitis with intrathecal delivery of an oncolytic recombinant poliovirus. Clin Cancer Res. (2006) 12:1349–54. doi: 10.1158/1078-0432.CCR-05-1595
69. O'Leary MP, Warner SG, Kim S-I, Chaurasiya S, Lu J, Choi AH, et al. A novel oncolytic chimeric orthopoxvirus encoding luciferase enables real-time view of colorectal cancer cell infection. Mol Therap Oncol. (2018) 9:13–21. doi: 10.1016/j.omto.2018.03.001
70. Igase M, Shousu K, Fujiki N, Sakurai M, Bonkobara M, Hwang CC, et al. Anti-tumour activity of oncolytic reovirus against canine histiocytic sarcoma cells. Vet Comp Oncol. (2019) 17:184–93. doi: 10.1111/vco.12468
71. Gangi A, Zager JS. The safety of talimogene laherparepvec for the treatment of advanced melanoma. Expert Opin Drug Saf. (2017) 16:265–9. doi: 10.1080/14740338.2017.1274729
72. Chesney J, Awasthi S, Curti B, Hutchins L, Linette G, Triozzi P, et al. Phase IIIb safety results from an expanded-access protocol of talimogene laherparepvec for patients with unresected, stage IIIB-IVM1c melanoma. Melanoma Res. (2018) 28:44–51. doi: 10.1097/CMR.0000000000000399
73. Varghese S, Newsome JT, Rabkin SD, McGeagh K, Mahoney D, Nielsen P, et al. Preclinical safety evaluation of G207, a replication-competent herpes simplex virus type 1, inoculated intraprostatically in mice and nonhuman primates. Hum Gene Ther. (2001) 12:999–1010. doi: 10.1089/104303401750195944
74. Rodriguez-Garcia A, Gimenez-Alejandre M, Rojas JJ, Moreno R, Bazan-Peregrino M, Cascallo M, et al. Safety and efficacy of VCN-01, an oncolytic adenovirus combining fiber HSG-binding domain replacement with RGD and hyaluronidase expression. Clin Cancer Res. (2015) 21:1406–18. doi: 10.1158/1078-0432.CCR-14-2213
75. Packiam VT, Lamm DL, Barocas DA, Trainer A, Fand B, Davis RL, et al. An open label, single-arm, phase II multicenter study of the safety and efficacy of CG0070 oncolytic vector regimen in patients with BCG-unresponsive non-muscle-invasive bladder cancer: Interim results. Urol Oncol. (2018) 36:440–7. doi: 10.1016/j.urolonc.2017.07.005
76. Lawler SE, Speranza M-C, Cho C-F, Chiocca EA. Oncolytic viruses in cancer treatment: a reviewoncolytic viruses in cancer treatmentoncolytic viruses in cancer treatment. JAMA Oncol. (2017) 3:841–9. doi: 10.1001/jamaoncol.2016.2064
77. Hu JCC, Coffin RS, Davis CJ, Graham NJ, Groves N, Guest PJ, et al. A Phase I Study of OncoVEXGM-CSF, a second-generation oncolytic herpes simplex virus expressing granulocyte macrophage colony-stimulating factor. Clin Cancer Res. (2006) 12:6737–47. doi: 10.1158/1078-0432.CCR-06-0759
78. Galanis E, Hartmann LC, Cliby WA, Long HJ, Peethambaram PP, Barrette BA, et al. Phase I trial of intraperitoneal administration of an oncolytic measles virus strain engineered to express carcinoembryonic antigen for recurrent ovarian cancer. Cancer Res. (2010) 70:875–82. doi: 10.1158/0008-5472.CAN-09-2762
79. Breitbach CJ, Burke J, Jonker D, Stephenson J, Haas AR, Chow LQ, et al. Intravenous delivery of a multi-mechanistic cancer-targeted oncolytic poxvirus in humans. Nature. (2011) 477:99–102. doi: 10.1038/nature10358
80. Comins C, Spicer J, Protheroe A, Roulstone V, Twigger K, White CM, et al. REO-10: a phase I study of intravenous reovirus and docetaxel in patients with advanced cancer. Clin Cancer Res. (2010) 16:5564–72. doi: 10.1158/1078-0432.CCR-10-1233
81. Waters AM, Friedman GK, Ring EK, Beierle EA. Oncolytic virotherapy for pediatric malignancies: future prospects. Oncolytic Virother. (2016) 5:73–80. doi: 10.2147/OV.S96932
82. Xu RH, Yuan ZY, Guan ZZ, Cao Y, Wang HQ, Hu XH, et al. [Phase II clinical study of intratumoral H101, an E1B deleted adenovirus, in combination with chemotherapy in patients with cancer]. Ai Zheng. (2003) 22:1307–10.
83. Xia ZJ, Chang JH, Zhang L, Jiang WQ, Guan ZZ, Liu JW, et al. [Phase III randomized clinical trial of intratumoral injection of E1B gene-deleted adenovirus (H101) combined with cisplatin-based chemotherapy in treating squamous cell cancer of head and neck or esophagus]. Ai Zheng. (2004) 23:1666–70.
84. Pesonen S, Diaconu I, Kangasniemi L, Ranki T, Kanerva A, Pesonen SK, et al. Oncolytic immunotherapy of advanced solid tumors with a CD40L-expressing replicating adenovirus: assessment of safety and immunologic responses in patients. Cancer Res. (2012) 72:1621–31. doi: 10.1158/0008-5472.CAN-11-3001
85. Nemunaitis J, Cunningham C, Buchanan A, Blackburn A, Edelman G, Maples P, et al. Intravenous infusion of a replication-selective adenovirus (ONYX-015) in cancer patients: safety, feasibility and biological activity. Gene Therap. (2001) 8:746–59. doi: 10.1038/sj.gt.3301424
86. Lu W, Zheng S, Li XF, Huang JJ, Zheng X, Li Z. Intra-tumor injection of H101, a recombinant adenovirus, in combination with chemotherapy in patients with advanced cancers: a pilot phase II clinical trial. World J Gastroenterol. (2004) 10:3634–8. doi: 10.3748/wjg.v10.i24.3634
87. Harrington KJ, Michielin O, Malvehy J, Pezzani Gruter I, Grove L, Frauchiger AL, et al. A practical guide to the handling and administration of talimogene laherparepvec in Europe. Onco Targets Ther. (2017) 10:3867–80. doi: 10.2147/OTT.S133699
88. Corrigan PA, Beaulieu C, Patel RB, Lowe DK. Talimogene laherparepvec: an oncolytic virus therapy for melanoma. Ann Pharmacother. (2017) 51:675–81. doi: 10.1177/1060028017702654
89. Todo T, Feigenbaum F, Rabkin SD, Lakeman F, Newsome JT, Johnson PA, et al. Viral shedding and biodistribution of G207, a multimutated, conditionally replicating herpes simplex virus type 1, after intracerebral inoculation in aotus. Mol Ther. (2000) 2:588–95. doi: 10.1006/mthe.2000.0200
90. Kimball KJ, Preuss MA, Barnes MN, Wang M, Siegal GP, Wan W, et al. A phase I study of a tropism-modified conditionally replicative adenovirus for recurrent malignant gynecologic diseases. Clin Cancer Res. (2010) 16:5277–87. doi: 10.1158/1078-0432.CCR-10-0791
91. Kim KH, Ryan MJ, Estep JE, Miniard BM, Rudge TL, Peggins JO, et al. A new generation of serotype chimeric infectivity-enhanced conditionally replicative adenovirals: the safety profile of ad5/3-Delta24 in advance of a phase I clinical trial in ovarian cancer patients. Hum Gene Ther. (2011) 22:821–8. doi: 10.1089/hum.2010.180
92. Margarita G, Rafael M, Marta G-M, Manel C, Ochoa dOM, Carmen C, et al. A Phase 1 Trial of oncolytic adenovirus ICOVIR-5 administered intravenously to cutaneous and uveal melanoma patients. Hum Gene Therap. (2019) 30:352–64. doi: 10.1089/hum.2018.107
93. Cripe TP, Ngo MC, Geller JI, Louis CU, Currier MA, Racadio JM, et al. Phase 1 study of intratumoral Pexa-Vec (JX-594), an oncolytic and immunotherapeutic vaccinia virus, in pediatric cancer patients. Mol Ther. (2015) 23:602–8. doi: 10.1038/mt.2014.243
94. Park SH, Breitbach CJ, Lee J, Park JO, Lim HY, Kang WK, et al. Phase 1b Trial of Biweekly Intravenous Pexa-Vec (JX-594), an oncolytic and immunotherapeutic vaccinia virus in colorectal cancer. Mol Ther. (2015) 23:1532–40. doi: 10.1038/mt.2015.109
95. Zeh HJ, Downs-Canner S, McCart JA, Guo ZS, Rao UN, Ramalingam L, et al. First-in-man study of western reserve strain oncolytic vaccinia virus: safety, systemic spread, and antitumor activity. Mol Ther. (2015) 23:202–14. doi: 10.1038/mt.2014.194
96. Downs-Canner S, Guo ZS, Ravindranathan R, Breitbach CJ, O'Malley ME, Jones HL, et al. Phase 1 study of intravenous oncolytic poxvirus (vvDD) in patients with advanced solid cancers. Mol. Ther. (2016) 24:1492–501. doi: 10.1038/mt.2016.101
97. Buijs PR, Verhagen JH, van Eijck CH, van den Hoogen BG. Oncolytic viruses: from bench to bedside with a focus on safety. Hum Vaccin Immunother. (2015) 11:1573–84. doi: 10.1080/21645515.2015.1037058
98. Lam HY, Yeap SK, Pirozyan MR, Omar AR, Yusoff K, Suraini AA, et al. Safety and clinical usage of newcastle disease virus in cancer therapy. J Biomed Biotechnol. (2011) 2011:718710. doi: 10.1155/2011/718710
99. Cho E, Ryu EJ, Jiang F, Jeon UB, Cho M, Kim CH, et al. Preclinical safety evaluation of hepatic arterial infusion of oncolytic poxvirus. Drug Des Devel Ther. (2018) 12:2467–74. doi: 10.2147/DDDT.S171269
100. Kohno S, Luo C, Goshima F, Nishiyama Y, Sata T, Ono Y. Herpes simplex virus type 1 mutant HF10 oncolytic viral therapy for bladder cancer. Urology. (2005) 66:1116–21. doi: 10.1016/j.urology.2005.05.041
101. Kanerva A, Nokisalmi P, Diaconu I, Koski A, Cerullo V, Liikanen I, et al. Antiviral and antitumor T-cell immunity in patients treated with GM-CSF-coding oncolytic adenovirus. Clin. Cancer Res. (2013) 19:2734–44. doi: 10.1158/1078-0432.CCR-12-2546
102. Voigt EA, Kennedy RB, Poland GA. Defending against smallpox: a focus on vaccines. Expert Rev Vaccines. (2016) 15:1197–211. doi: 10.1080/14760584.2016.1175305
103. Futami M, Sato K, Miyazaki K, Suzuki K, Nakamura T, Tojo A. Efficacy and safety of doubly-regulated vaccinia virus in a mouse xenograft model of multiple myeloma. Mol Ther Oncol. (2017). 6:57–68. doi: 10.1016/j.omto.2017.07.001
104. Harrow S, Papanastassiou V, Harland J, Mabbs R, Petty R, Fraser M, et al. HSV1716 injection into the brain adjacent to tumour following surgical resection of high-grade glioma: safety data and long-term survival. Gene Therapy. (2004) 11:1648–58. doi: 10.1038/sj.gt.3302289
105. Markert JM, Razdan SN, Kuo HC, Cantor A, Knoll A, Karrasch M, et al. A phase 1 trial of oncolytic HSV-1, G207, given in combination with radiation for recurrent GBM demonstrates safety and radiographic responses. Mol Ther. (2014) 22:1048–55. doi: 10.1038/mt.2014.22
106. Aghi MK, Chiocca EA. Phase ib trial of oncolytic herpes virus G207 shows safety of multiple injections and documents viral replication. Mol Ther. (2009) 17:8–9. doi: 10.1038/mt.2008.275
107. Fujimoto Y, Mizuno T, Sugiura S, Goshima F, Kohno S, Nakashima T, et al. Intratumoral injection of herpes simplex virus HF10 in recurrent head and neck squamous cell carcinoma. Acta Otolaryngol. (2006) 126:1115–7. doi: 10.1080/00016480600702100
108. Kasuya H, Kodera Y, Nakao A, Yamamura K, Gewen T, Zhiwen W, et al. Phase I dose-escalation clinical trial of HF10 oncolytic herpes virus in 17 Japanese patients with advanced cancer. Hepatogastroenterology. (2014) 61:599–605. doi: 10.5754/hge14104
109. Wei D, Xu J, Liu XY, Chen ZN, Bian H. Fighting cancer with viruses: oncolytic virus therapy in China. Hum Gene Ther. (2018) 29:151–9. doi: 10.1089/hum.2017.212
110. Eriksson E, Milenova I, Wenthe J, Ståhle M, Leja-Jarblad J, Ullenhag G, et al. Shaping the tumor stroma and sparking immune activation by CD40 and 4-1BB signaling induced by an armed oncolytic virus. Clin. Cancer Res. (2017) 23:5846–57. doi: 10.1158/1078-0432.CCR-17-0285
111. Garcia M, Moreno R, Gil M, Cascallo M, de Olza MO, Cuadra C, et al. A phase I trial of oncolytic adenovirus ICOVIR-5 administered intravenously to melanoma patients. Hum Gene Ther Clin Dev. (2018) 30:352–64. doi: 10.1089/humc.2018.107
112. Dispenzieri A, Tong C, LaPlant B, Lacy MQ, Laumann K, Dingli D, et al. Phase I trial of systemic administration of Edmonston strain of measles virus genetically engineered to express the sodium iodide symporter in patients with recurrent or refractory multiple myeloma. Leukemia. (2017) 31:2791–98. doi: 10.1038/leu.2017.120
113. Geletneky K, Hajda J, Angelova AL, Leuchs B, Capper D, Bartsch AJ, et al. Oncolytic H-1 parvovirus shows safety and signs of immunogenic activity in a first Phase I/IIa glioblastoma trial. Mol Ther. (2017) 25:2620–34. doi: 10.1016/j.ymthe.2017.08.016
114. Geletneky K, Huesing J, Rommelaere J, Schlehofer JR, Leuchs B, Dahm M, et al. Phase I/IIa study of intratumoral/intracerebral or intravenous/intracerebral administration of Parvovirus H-1 (ParvOryx) in patients with progressive primary or recurrent glioblastoma multiforme: ParvOryx01 protocol. BMC Cancer. (2012) 12:99. doi: 10.1186/1471-2407-12-99
115. Baldo A, Galanis E, Tangy F, Herman P. Biosafety considerations for attenuated measles virus vectors used in virotherapy and vaccination. Hum Vaccin Immunother. (2016) 12:1102–16. doi: 10.1080/21645515.2015.1122146
116. Geevarghese SK, Geller DA, de Haan HA, Horer M, Knoll AE, Mescheder A, et al. Phase I/II study of oncolytic herpes simplex virus NV1020 in patients with extensively pretreated refractory colorectal cancer metastatic to the liver. Hum Gene Ther. (2010) 21:1119–28. doi: 10.1089/hum.2010.020
117. Maroun J, Munoz-Alia M, Ammayappan A, Schulze A, Peng KW Russell Designing S, and building oncolytic viruses. Future Virol. (2017) 12:193–213. doi: 10.2217/fvl-2016-0129
118. Angelova AL, Geletneky K, Nuesch JP, Rommelaere J. Tumor selectivity of oncolytic parvoviruses: from in vitro and animal models to cancer patients. Front Bioeng Biotechnol. (2015) 3:55. doi: 10.3389/fbioe.2015.00055
119. Yamaguchi T, Uchida E. Oncolytic virus: regulatory aspects from quality control to clinical studies. Curr Cancer Drug Targets. (2018) 18:202–8. doi: 10.2174/1568009617666170222142650
120. Hajda J, Lehmann M, Krebs O, Kieser M, Geletneky K, Jager D, et al. A non-controlled, single arm, open label, phase II study of intravenous and intratumoral administration of ParvOryx in patients with metastatic, inoperable pancreatic cancer: ParvOryx02 protocol. BMC Cancer. (2017) 17:576. doi: 10.1186/s12885-017-3604-y
121. Geletneky K, Leoni AL, Pohlmeyer-Esch G, Loebhard S, Leuchs B, Hoefer C, et al. Bioavailability, biodistribution, and CNS toxicity of clinical-grade parvovirus H1 after intravenous and intracerebral injection in rats. Comp Med. (2015) 65:36–45.
122. Lundstrom K. New frontiers in oncolytic viruses: optimizing and selecting for virus strains with improved efficacy. Biologics. (2018) 12:43–60. doi: 10.2147/BTT.S140114
123. Kaid C, Goulart E, Caires-Júnior LC, Araujo BHS, Soares-Schanoski A, Bueno HMS, et al. Zika Virus selectively kills aggressive human embryonal CNS tumor cells. Cancer Res. (2018) 78:3363–74. doi: 10.1158/0008-5472.CAN-17-3201
124. Wang Y, Zhou X, Wu Z, Hu H, Jin J, Hu Y, et al. Preclinical safety evaluation of oncolytic herpes simplex virus type 2. Hum Gene Ther. (2019) 30:651–60. doi: 10.1089/hum.2018.170
125. Kelly KJ, Wong J, Fong Y. Herpes simplex virus NV1020 as a novel and promising therapy for hepatic malignancy. Expert Opin Investig Drugs. (2008) 17:1105–13. doi: 10.1517/13543784.17.7.1105
126. Wei N, Fan JK, Gu JF, He LF, Tang WH, Cao X, et al. A double-regulated oncolytic adenovirus with improved safety for adenocarcinoma therapy. Biochem Biophys Res Commun. (2009) 388:234–9. doi: 10.1016/j.bbrc.2009.07.142
127. Jung SJ, Kasala D, Choi JW, Lee SH, Hwang JK, Kim SW, et al. Safety profiles and antitumor efficacy of oncolytic adenovirus coated with bioreducible polymer in the treatment of a CAR negative tumor model. Biomacromolecules. (2015) 16:87–96. doi: 10.1021/bm501116x
128. Yamamoto Y, Nagasato M, Yoshida T, Aoki K. Recent advances in genetic modification of adenovirus vectors for cancer treatment. Cancer Sci. (2017) 108:831–7. doi: 10.1111/cas.13228
129. Abdullahi S, Jakel M, Behrend SJ, Steiger K, Topping G, Krabbe T, et al. A novel chimeric oncolytic virus vector for improved safety and efficacy as a platform for the treatment of hepatocellular carcinoma. J Virol. (2018) 92:e01386–18. doi: 10.1128/JVI.01386-18
130. Coughlan L, Vallath S, Gros A, Gimenez-Alejandre M, Van Rooijen N, Thomas GJ, et al. Combined fiber modifications both to target alpha(v)beta(6) and detarget the coxsackievirus-adenovirus receptor improve virus toxicity profiles in vivo but fail to improve antitumoral efficacy relative to adenovirus serotype 5. Hum Gene Ther. (2012) 23:960–79. doi: 10.1089/hum.2011.218
131. Marchini A, Bonifati S, Scott EM, Angelova AL, Rommelaere J. Oncolytic parvoviruses: from basic virology to clinical applications. Virol J. (2015) 12:6. doi: 10.1186/s12985-014-0223-y
132. Schenk E, Essand M, Kraaij R, Adamson R, Maitland NJ, Bangma CH. Preclinical safety assessment of Ad[I/PPT-E1A], a novel oncolytic adenovirus for prostate cancer. Hum Gene Ther Clin Dev. (2014) 25:7–15. doi: 10.1089/humc.2013.181
133. Jing Y, Zaias J, Duncan R, Russell SJ, Merchan JR. In vivo safety, biodistribution and antitumor effects of uPAR retargeted oncolytic measles virus in syngeneic cancer models. Gene Ther. (2014) 21:289–97. doi: 10.1038/gt.2013.84
134. Lal S, Peng KW, Steele MB, Jenks N, Ma H, Kohanbash G, et al. Safety Study: intraventricular Injection of a Modified Oncolytic Measles Virus into Measles-Immune, hCD46-Transgenic, IFNalphaRko Mice. Hum Gene Ther Clin Dev. (2016) 27:145–51. doi: 10.1089/humc.2016.062
135. Ayala-Breton C, Suksanpaisan L, Mader EK, Russell SJ, Peng KW. Amalgamating oncolytic viruses to enhance their safety, consolidate their killing mechanisms, and accelerate their spread. Mol Ther. (2013) 21:1930–7. doi: 10.1038/mt.2013.164
136. Zhang L, Steele MB, Jenks N, Grell J, Suksanpaisan L, Naik S, et al. Safety studies in tumor and non-tumor-bearing mice in support of clinical trials using oncolytic VSV-IFNbeta-NIS. Hum Gene Ther Clin Dev. (2016) 27:111–22. doi: 10.1089/humc.2016.061
Keywords: oncolytic virotherapy, tumor, immunotherapy, delivery route, biosafety
Citation: Li L, Liu S, Han D, Tang B and Ma J (2020) Delivery and Biosafety of Oncolytic Virotherapy. Front. Oncol. 10:475. doi: 10.3389/fonc.2020.00475
Received: 10 January 2020; Accepted: 16 March 2020;
Published: 16 April 2020.
Edited by:
Edward Wenge Wang, City of Hope National Medical Center, United StatesReviewed by:
Er Yue, City of Hope National Medical Center, United StatesZhifang Zhang, Beckman Research Institute, United States
Xiaobing Tian, Brown University, United States
Copyright © 2020 Li, Liu, Han, Tang and Ma. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jian Ma, bWFqaWFuQGNzdS5lZHUuY24=
†These authors have contributed equally to this work