 Giuseppe Schepisi1*
Giuseppe Schepisi1* Vincenza Conteduca1
Vincenza Conteduca1 Chiara Casadei1
Chiara Casadei1 Giorgia Gurioli2
Giorgia Gurioli2 Lorena Rossi1Valentina Gallà3
Lorena Rossi1Valentina Gallà3 Maria Concetta Cursano4Nicole Brighi1
Maria Concetta Cursano4Nicole Brighi1 Cristian Lolli1Cecilia Menna1
Cristian Lolli1Cecilia Menna1 Alberto Farolfi1Salvatore Luca Burgio1
Alberto Farolfi1Salvatore Luca Burgio1 Amelia Altavilla1
Amelia Altavilla1 Giovanni Martinelli1
Giovanni Martinelli1 Ugo De Giorgi1
Ugo De Giorgi1- 1Department of Medical Oncology, Istituto Scientifico Romagnolo per lo Studio e la Cura dei Tumori, Istituto di Ricovero e Cura a Carattere Scientifico, Meldola, Italy
- 2Biosciences Laboratory, Istituto Scientifico Romagnolo per lo Studio e la Cura dei Tumori, Istituto di Ricovero e Cura a Carattere Scientifico, Meldola, Italy
- 3Unit of Biostatistics and Clinical Trials, Istituto Scientifico Romagnolo per lo Studio e la Cura dei Tumori, Istituto di Ricovero e Cura a Carattere Scientifico, Meldola, Italy
- 4Department of Medical Oncology, Campus Bio-Medico University of Rome, Rome, Italy
Currently, renal cell carcinoma is characterized by encouraging benefits from immunotherapy that have led to significant results in treatment outcome. The approval of nivolumab primarily as second-line monotherapy and, more recently, the approval of new combination therapies as first-line treatment have confirmed the importance of immunotherapy in this type of tumor. In this context, the chimeric antigen receptor (CAR)-T represents a further step forward in the field of immunotherapy. Initially tested on hematological malignancies, this new therapeutic approach is also becoming a topic of great interest for solid tumors. Although the treatment has several advantages over previous T-cell receptor-dependent immunotherapy, it is facing some obstacles in solid tumors such as a hostile tumor microenvironment and on-tumor/off-tumor toxicities. Several strategies are under investigation to overcome these problems, but the approval of CAR-T cell therapy is still some way off. In renal cancer, the significant advantages obtained from immune checkpoint inhibitors represent a good starting point, but the potential nephrological toxicity of CAR-T cell therapy represents an important risk. In this review, we provide the rationale and preliminary results of CAR-T cell therapy in renal cell malignancies.
Introduction
Renal cell carcinoma (RCC) represents the 9th and 14th most common tumor worldwide in males and females, respectively. Incidence rates are higher in Europe and the United States than in Africa or South-Eastern Asia and are increasing in other countries especially in Latin America (1). Epidemiological studies have demonstrated that smoking, chronic kidney diseases, hypertension, and obesity are risk factors for RCC development (2). For decades, the only effective treatment against RCC was surgery because of its well-known chemoresistance. The subsequent approval of cytokines [interferon (IFN) and interleukin (IL)-2 (3)] and tyrosine kinase inhibitors (TKIs) led to an advantage in survival in patients with metastatic disease (4–7) [>26 months using vascular endothelial growth factor (VEGF) inhibitors (8)].
The approval of TKIs minimized the use of cytokines due to the differences in terms of survival and toxicity. Notwithstanding, the concept that IL-2 induces an immune response against tumor-mediated immune suppression (9) has, over the years, focused our attention on the possibility that RCC may be sensitive to immunological treatments. In recent years, the new Immunotherapeutic Era has led to the approval of several drugs for the treatment of urological tumors (including RCC) (3). Nivolumab (10), nivolumab + ipilimumab (11), pembrolizumab in combination with axitinib (12), and avelumab plus axitinib (13) have demonstrated the most important advantages in RCC in terms of survival and response with respect to TKIs (12–14). However, these advantages were obtained most frequently in two patient subgroups, defined as intermediate- and poor-risk cohorts according to the International Metastatic Renal Cell Carcinoma Database Consortium (IMDC). Research into the molecular reasons for this gap among patient cohorts is currently ongoing to drive clinical decisions.
More recently, the development of chimeric antigen receptors (CARs) has led to a new modality of immunotherapy. The astounding results in terms of responses demonstrated by this strategy against hematological neoplasms have turned the attention toward solid tumors, including RCC.
In this review, we will focus our attention on CAR-T cell therapy in patients with RCC.
CARs: Structure and Function
Chimeric antigen receptor-T cells are T cells that are genetically engineered to express antigen-specific, non-major histocompatibility complex (MHC)-restricted receptors on their membranes. They are classified into four generations based on molecular complexity, all composed of different domains: (1) a single-chain antibody fragment (scFV) located in the extracellular part of the cell, representing the antigen-binding region; (2) a hinge domain linked to a (3) transmembrane region; and (4) an intracellular domain composed of the signal transduction part of the T-cell receptor (TCR), called the cluster of differentiation (CD)3ζ, linked with one (second and fourth CAR generations) or two (only third CAR generation) costimulatory domains. Moreover, the fourth CAR-T generation, also known as TRUCKs (T cells redirected for universal cytokine killing), are CAR-T cells combined with immune stimulatory molecules, such as cytokines [interleukin (IL)-12, IL-15, IL-18, IL-7R), multiantigen-targeting combinations [human epidermal growth factor receptor 2 (HER2), interleukin-13 receptor subunit alpha-2 (IL-13Rα2), and ephrin-A2 (EphA2)], knock-in genes, such as C-X-C chemokine receptor type 4 (CXCR4) and TCR α constant (TRAC), knock-out genes, e.g., programmed death-1 (PD1) and diacylglycerol kinase (DGK), or controlled and inducible systems, such as synthetic Notch receptor (Syn/Notch). Given that TRUCKs present both costimulatory element and proinflammatory factor, this characteristic increases T-cell efficacy with respect to previous CAR generations (Figure 1A). Those molecules have several advantages over the previous modalities of adoptive cell therapy, i.e., TCR and tumor-infiltrating lymphocytes (TIL) (15, 16). First and foremost, their immune activity is not MHC restricted because it is dependent on a surface–antigen interaction. This difference is crucial in treating tumors with low MHC expression, which, being TCR or TIL resistant, may be sensitive to CAR-T cell therapy (9, 17). Second, TCRs commonly have low antigen affinity, which can lead to off-target toxicities (18). Third, CAR-T cells not only have the antigen-binding activity of T cells (like monoclonal antibodies) but also their lytic property (19).
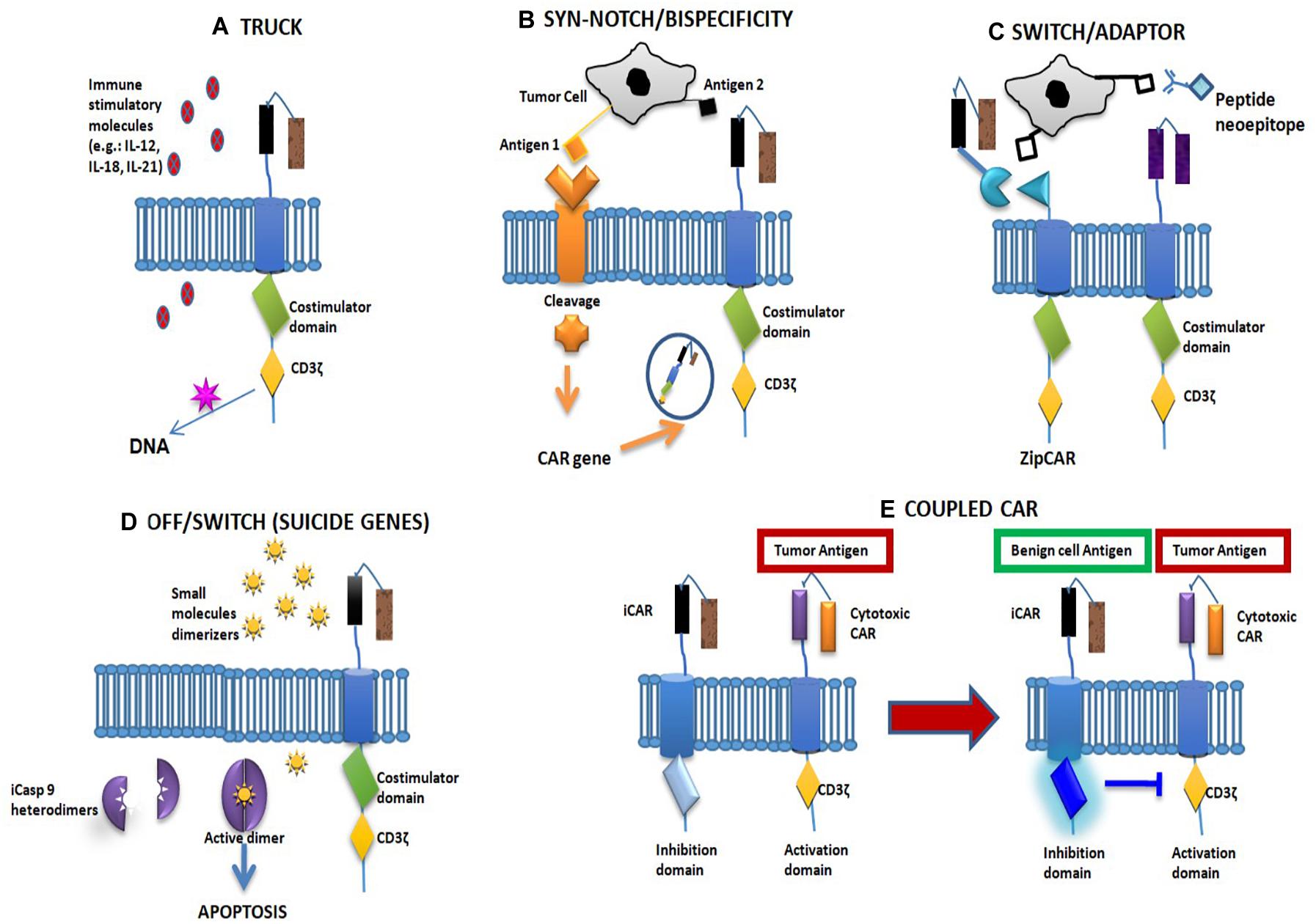
Figure 1. Fourth-generation chimeric antigen receptor (CAR) and main solutions to CAR toxicity. (A) The fourth-generation CAR structure. (B,C) CAR-related solutions. (D) Non-CAR-related solutions. (E) Coupled CAR. (A) The fourth CAR-T generation, also known as TRUCKs (T cells redirected for universal cytokine killing), is a CAR-T cell combined with immune stimulatory molecules such as cytokines [interleukin (IL)-12, IL-15, IL-18, IL-7R], multiantigen-targeting combinations [human epidermal growth factor receptor 2 (HER2), interleukin-13 receptor subunit alpha-2 (IL13Rα2), ephrin-A2 (EphA2)], knock-in genes (CXCR4, TRAC) and knock-out genes (PD1, DGK), or controlled and inducible systems (Syn/Notch). (B) “Synthetic Notch receptor”: the single-chain antibody fragment (scFV), connected with a Notch receptor fragment, is cleaved after antigen binding, allowing the intracellular domain to drive the expression of a second tumor antigen. This option improves tumor specificity of the CAR-T cells. Its double specificity controls off-tumor toxicities by depleting transferred cells or increasing specificity against tumor targets. (C) In “switchable CARs,” the use of adaptor molecules represents a tool to modulate CAR activation and longevity. The CAR molecule only binds a neoepitope, promoting T-cell activity after antibody–antigen binding. (D) The insertion of a truncated form of caspase-9 (iCasp9) with a binding domain specific for a “dimerizer” molecule could lead to a dimerization-related T-cell destruction in around 30 min. (E) Combination of a stimulatory CAR and an inhibitory CAR: the latter contains an intracellular domain with an immune checkpoint molecule (CTLA4 or PD1). In the event of contact with a normal tissue antigen, the inhibitory CAR irreversibly blocks T-cell activity.
The first results of CAR-T cell therapy were obtained in hematological malignancies, where around 90% of complete responses were obtained in CD19-positive B-acute lymphoblastic leukemia. Conversely, only 26% of the patients with chronic lymphoblastic leukemia benefited from CD19 CAR-T therapy (20). This discrepancy seems to be related to the development of T-cell exhaustion induced by coinhibitory pathways. This exhaustion is, in turn, the cause of poor T-cell expansion and short-term persistence of T cells (21). In fact, by analyzing CAR-T cells from non-responders, an upregulation was found in pathways involved in exhaustion and apoptosis (22). In CAR-T cells, expression levels of T-cell coinhibitory receptors, e.g., PD-1, T-cell immunoglobulin and mucin domain-3 (Tim-3), and lymphocyte activation gene-3 (LAG-3) were found to be upregulated, probably to inhibit T-cell activity (23). For these reasons, some studies were conducted combining CAR-T cell therapy and immune checkpoint inhibitors (ICIs) in hematological malignancies, with interesting results (24).
Challenges Facing the Use of CAR-T Cells in Treating Solid Tumors
Chimeric antigen receptor-T cell therapies were initially developed for the treatment of hematologic neoplasms but have not shown the same efficacy in solid tumors. In the latter, better results have been obtained from the use of TIL-dependent immunotherapeutic agents. This discrepancy is due to several conditions: (a) immune-mediated tumor antigen selection, which can improve the proliferation of tumor cells whose membrane does not express that specific target (25); (b) poor intra- and peritumoral trafficking (26); (c) limited CAR-T persistence in the host (27, 28). Little is also known about potential surrogate markers of T-cell persistence. One study on RCC patients demonstrated a correlation between CAR-T persistence and IFN-γ and IL-6, whereas the same correlation was not shown in relation to CAR-T toxicities (29); (d) CAR-T destruction mediated by the hostile tumor microenvironment (30); and (e) absence of a cancer-specific antigen in several tumor types that is suitable for inclusion in the CAR structure (31). This relative aspecificity may increase the risk of immune-related toxicity, as recently reported in literature (31–34).
CAR-T-Related Toxicities
Several CAR-T-related toxicities have been reported. In particular, this therapy may induce severe toxicities, potentially affecting kidney function, including cytokine release syndrome (CRS), acute kidney injury (AKI), and tumor lysis syndrome (TLS). These toxicities are of primary importance for all cancer patients, especially for RCC patients, who have often undergone nephrectomy.
Several trials on hematological malignancies reported that CRS occurred in 40–50% of the patients. Its characteristic signs were that of hyperpyrexia for about a week followed by organ dysfunction (with cardiac failure in around 25% of the cases); high levels of C-reactive protein (>20 mg/dl) and ferritin; and hypotension, hypoxia, and neurologic symptoms, such as obtundation and seizures (35). This problem is related to higher levels of IL-6, which determines vasodilation, hypotension, hypoperfusion and, consequently, AKI. The fundamental role of IL-6 in the development of this syndrome has also been confirmed by the fact that the use of the anti-IL-6 receptor antibody tocilizumab restored organ function in several cases (36) in whom an alteration in electrolyte levels was frequently observed. However, it is still not clear whether this was directly due to CAR-T cell therapy or CLS.
Tumor lysis syndrome, another syndrome potentially related to the use of CAR-T cell therapy, is characterized by an elevation of lactate dehydrogenase and uric acid levels around 3 weeks after CAR-T infusion (37).
Chimeric antigen receptor-T cell therapy can also cause other non-renal adverse events, such as neurological and “on-target/off-tumor” toxicity. The former, as previously stated, is characterized by seizures, confusion, myoclonus, delirium, and expressive aphasia. These symptoms have been reported in patients specifically treated with a CD19-specific CAR-T, and it is still not known whether they could be caused by other antigen-specific treatments. Neurological toxicity is probably related to CRS but may also be related to a central nervous system-directed toxicity.
“On-target/off-tumor” toxicity is linked to antigen engagement in non-cancer tissues. First demonstrated in hematological malignancies, it has also been reported in solid neoplasms, such as gastrointestinal and lung cancer. More recently, the use of a carboxyanhydrase-IX (CAIX)-specific CAR-T cell for RCC caused the development of cholestasis because of CAIX expression in biliary duct epithelium (38, 39). This toxicity seems to be correlated with dosage, as shown in cases of HER-2-specific CAR-T infusion: high doses have led to patient death (33), but lower doses have proven safe (40).
Anaphylaxis and graft-versus-host disease are toxicities described in CAR-T cell trials. The former is often due to the use of murine domains in developing CAR molecules (36, 41). For the latter, two types of strategies are being pursued to counteract the potential alloreactivity linked to the infusion of non-host CAR-T cells, CAR-transduced viral-specific cells and endogenous TCR silencing (42–44).
Potential Solutions to Reduce CAR Toxicity
As mentioned above, CAR-related toxicity represents an important limit to the development of this strategy in solid tumors, and several studies are ongoing to evaluate potential solutions to the problem (45). These can be divided into three groups: (1) CAR related, (2) non-CAR related, and (3) coupled CARs.
The first group comprises studies exploring the possibility of modifying the CAR structure to avoid toxicity. For example, it has been demonstrated that designing a CAR with a reduced antigen affinity in its scFV can spare normal tissues from immune-mediated consequences (46–48). The same effect has been achieved by modifying the extramembrane spacer length between the scFV and the cellular membrane (18, 49). Another option consists in the development of a “synthetic Notch receptor”: the scFV, connected to a Notch receptor fragment, is cleaved after antigen binding, allowing the intracellular domain to drive the expression of a second tumor antigen. This option improves the tumor specificity of CAR-T cells (49) (Figure 1B). Other options included in this group are the development of “split CARs” in which the CAR domains are only linked in the presence of a small molecule with dimerizing activity and the development of “switchable CARs” in which the CAR molecule only binds a neoepitope that can activate T-cell activity after antibody–antigen binding (50, 51) (Figure 1C).
The second group is composed of solutions not involving the CAR structure but which act at different levels. For example, some studies have inserted inducible suicide gene cassettes to induce apoptosis in the T cell in cases of immune-related toxicity. Two strategies have been tested: (a) the insertion of a truncated form of caspase IX (iCasp9) with a binding domain specific for a “dimerizer” molecule, which can lead to a dimerization-related T-cell destruction in around 30 min (52) (Figure 1D) and (b) the inclusion of the gene for herpes simplex virus tyrosine kinase. In this case, the administration of ganciclovir can induce T-cell apoptosis (53). Using a similar procedure, an “elimination gene” can also be inserted, leading to antibody-mediated T-cell destruction (in the event of toxicity) (54).
The third group is composed of studies evaluating the possibility of using two CAR molecules with different functions: (a) a combination of a CAR with an intracellular TCR portion, while the stimulatory portions are located on the second CAR. The T-cell immune activity can only start when both CARs bind their antigen. Results, to date, have been somewhat contradictory, and in one study this, procedure was also shown to destroy healthy cells expressing only one antigen (52, 55); (b) a combination of a stimulatory CAR and an inhibitory CAR: the latter contains an intracellular domain for an immune checkpoint molecule (CTLA4 or PD1). In the event of contact with a normal tissue antigen, the inhibitory CAR blocks T-cell activity. This block is reversible, and so T cells can be reactivated by another tumor antigen (56) (Figure 1E).
Inflammation, CAR-T Treatment, and Its Rationale in RCC
The role of immunotherapy has always been considered in RCC, and this approach has obtained significant results from the IL-2 era to the latest ICIs. The rationale for its efficacy lies in the involvement of the immune system in RCC, and several studies have investigated this correlation. Trials investigating the role of ICIs in RCC patients demonstrated a >1% PD-L1 expression ranging from 24% in the CheckMate-214 study (57) to 63.2% in the JAVELIN Renal 101 study (13). In both trials, the overall response rate (ORR) and survival (OS) were sharply in favor of immunotherapy, especially in the presence of PD-L1 expression. The JAVELIN Renal 101 study demonstrated a 55.2% ORR from the combination of avelumab plus axitinib compared to 25.5% from sunitinib (the data reported were only from PD-L1-positive cases). Conversely, the combination of nivolumab + ipilimumab in the CheckMate-214 study showed a 42% ORR compared to 26% in the sunitinib arm. In the same study, the immunotherapy combination obtained a median OS gain of 20 months compared to the sunitinib arm (47 versus 26.6 months, respectively) (58).
Both studies highlighted a difference between the IMDC prognostic groups, showing that the intermediate- and high-risk groups tended to benefit more from immunotherapy, contrary to what happens in good prognosis cases, which appear to do better with oral TKIs (58). Recently, McDermott et al. evaluated the impact of immunotherapy according to the IMDC risk criteria, selecting a heatmap of genes previously established as angiogenesis-related and immune biology-related genes. They identified subgroups with different biological features, the differences based on the expression (high/low) of angiogenesis (Angio), immune system (T effector), and myeloid inflammation-associated genes. The subgroup expressing a high T-effector gene signature responded best to immunotherapy, whereas the cases with a high Angio signature benefited more from TKIs. Patients with a high myeloid gene signature treated with immunotherapy showed a poorer survival than those with a low expression of the gene (59).
In parallel to this type of study, the correlation between the immune system and RCC was also assessed from a prognostic/predictive point of view. In the cytokine era, absolute neutrophil count was considered as a prognostic factor in a prognostic model for RCC validated by the Groupe Francais d’Immunotherapie (60). Subsequently, several studies validated the role of other inflammation parameters such as neutrophil/lymphocyte ratio (NLR) (61), systemic inflammation index (SII) (62), and CRP (63) as independent prognostic factors in RCC. With the discovery of inhibitory checkpoints, greater attention was paid to more specific prognostic and predictive markers. The PD1/PD-L1 axis is also the most widely studied parameter in RCC. PD-L1 expression is known to be significantly related to poor response to antiangiogenic treatments and has shown an independent association with shorter survival in stage IV RCC pretreated with VEGF–TKIs (64, 65). Overall, PD-1, PD-L1, and PD-L2 expression is associated with poor outcome in TKI-pretreated RCC patients (66). As mentioned above, the most recent studies on ICI combinations as first-line treatment reported better patient outcomes than those on sunitinib in intermediate and poor prognostic groups, suggesting a correlation between prognosis and “immuno-susceptibility” in RCC patients (14).
Interest has recently been aroused in the role of inflammasome complexes in solid tumors. These multimolecular complexes are known for their ability to control the activation of caspase-1, a proteolytic enzyme involved in the maturation of proinflammatory cytokines (IL-1β and IL-18), and in inducing inflammatory-like apoptosis (pyroptosis) against pathogens and endogenous danger signals. In brief, inflammasome may play a role in neoplastic development via the regulation of tumor inflammation (67). Some studies on RCC have shown that enhancing inflammasome activation blocks tumor proliferation, promoting pyroptosis. Wang et al. demonstrated that the nuclear receptor liver X receptor alpha (LXRα) is upregulated and associated with a poor prognosis in RCC patients (68). In fact, LXRα downregulates the NLRP3 inflammasome, leading to metastatization. Tan et al. showed that tumor proliferation and epithelial mesenchymal transition (EMT) in RCC patients are inhibited by BRD4 inhibition (69). The authors demonstrated that this molecule, an epigenetic reader, exerts an antitumor effect by activating pyroptosis. Chai et al. reported that absence in melanoma-2 (AIM-2), a tumor suppressor, influences inflammasome activity in RCC (70).
Based on what has been reported so far, especially for the role that the modulation of the immune system has always shown in the treatment of renal neoplasms, it is clear that RCC represents one of the most interesting test beds for the development of CAR-T technology in solid tumors.
CAR-T Cell Therapy in RCC Patients: Pros and Cons
At this point, a list of pros and cons can be compiled on the development and subsequent use of this new therapeutic approach in patients with RCC. It is undoubtedly an innovative and interesting therapy because of its high response rates obtained in hematological diseases and would also appear to be a promising strategy in RCC patients. It is a non-MHC-restricted approach and so has several advantages over TCR, as previously mentioned. Furthermore, unlike MHC-restricted immunotherapy, CAR-T cell therapy is susceptible to the modulation of T-cell function to improve efficacy and reduce toxicity. This last aspect is both an advantage (the ability to self-modulate antitumor activity is certainly an improvement compared to the past) and a disadvantage because, as previously mentioned, this type of treatment can cause particularly severe toxicities that were not induced by previous therapeutic approaches. In particular, the risk of renal toxicity, such as AKI, must be accurately evaluated in RCC patients who frequently undergo nephrectomy. We must also not forget the high cost of the drug, as well as the time required (a few weeks) for its preparation. It may not always be possible to wait so long before starting a treatment. In addition, CART-T cell therapy requires apheresis and adequate lymphocyte count and function, which may exclude some patients.
For these reasons, if an efficacy of CAR-T cell therapy similar to that observed in hematological malignancies is proven in RCC, the pros and cons of its use will need to be carefully evaluated in each individual patient.
CAR-T and Radiotherapy in RCC Patients
The association of immunotherapy with radiation therapy has been under investigation for some time, some studies hypothesizing its potential usefulness for the treatment of different cancers, including RCC (71–74).
The recent development of CAR-T cell therapy in solid tumors has led to the hypothesis of its combination with radiation therapy. In fact, the latter would appear to play a role in stimulating cancer antigenicity, promoting CAR-T cell chemotaxis and making the tumor microenvironment more sensitive to immune activation (75). In particular, it has been shown that γ-irradiation can enhance CAR-T efficacy by increasing tumor antigen expression on cancer cell surface and by stimulating IFN-γ secretion. Secreted by cancer cells, IFN-γ is known to promote immune infiltration into the tumor microenvironment, and radiation therapy influences tumor vasculature, facilitating the diffusion of lymphocytes within the tumor (75). However, there are still very few data on the combination of radiotherapy and CAR-T cell therapy in RCC. Given the solid rationale for this combination, further research is warranted.
CAR-T Cell Therapy: Ongoing Trials in RCC
In recent years, several articles have been published on the role of CAR-T cell therapy in solid tumors. Some studies are ongoing in the area of RCC (Table 1): a dose escalation and dose expansion trial is being carried out to assess the efficacy of autologous CAR-T cells CCT 301-38 or CCT 301-59 in recurrent/refractory stage IV RCC. The authors are simultaneously evaluating the effectiveness of two CARs directed against two different molecular targets. CCT301-59 is a CAR-targeting tyrosine kinase-like orphan receptor 2 (ROR2), an atypical receptor of the tyrosine kinase family involved in several human diseases. In RCC patients, ROR2 expression is correlated with other genes associated with mytosis and migration, including PCNA, CDK1, TWIST, and MMP-2 (76). CCT301-38 is another CAR directed against AXL, a cell surface tyrosine kinase receptor, which is part of the TAM kinase family. AXL, the high-affinity ligand growth arrest-specific protein 6 (GAS6), is involved in multiple tumor processes, including proliferation, angiogenesis, invasion, metastatization, immune regulation, stem cell maintenance, EMT, and drug resistance. Aberrant Gas6/AXL expression has been described in several tumor types, including RCC (77).
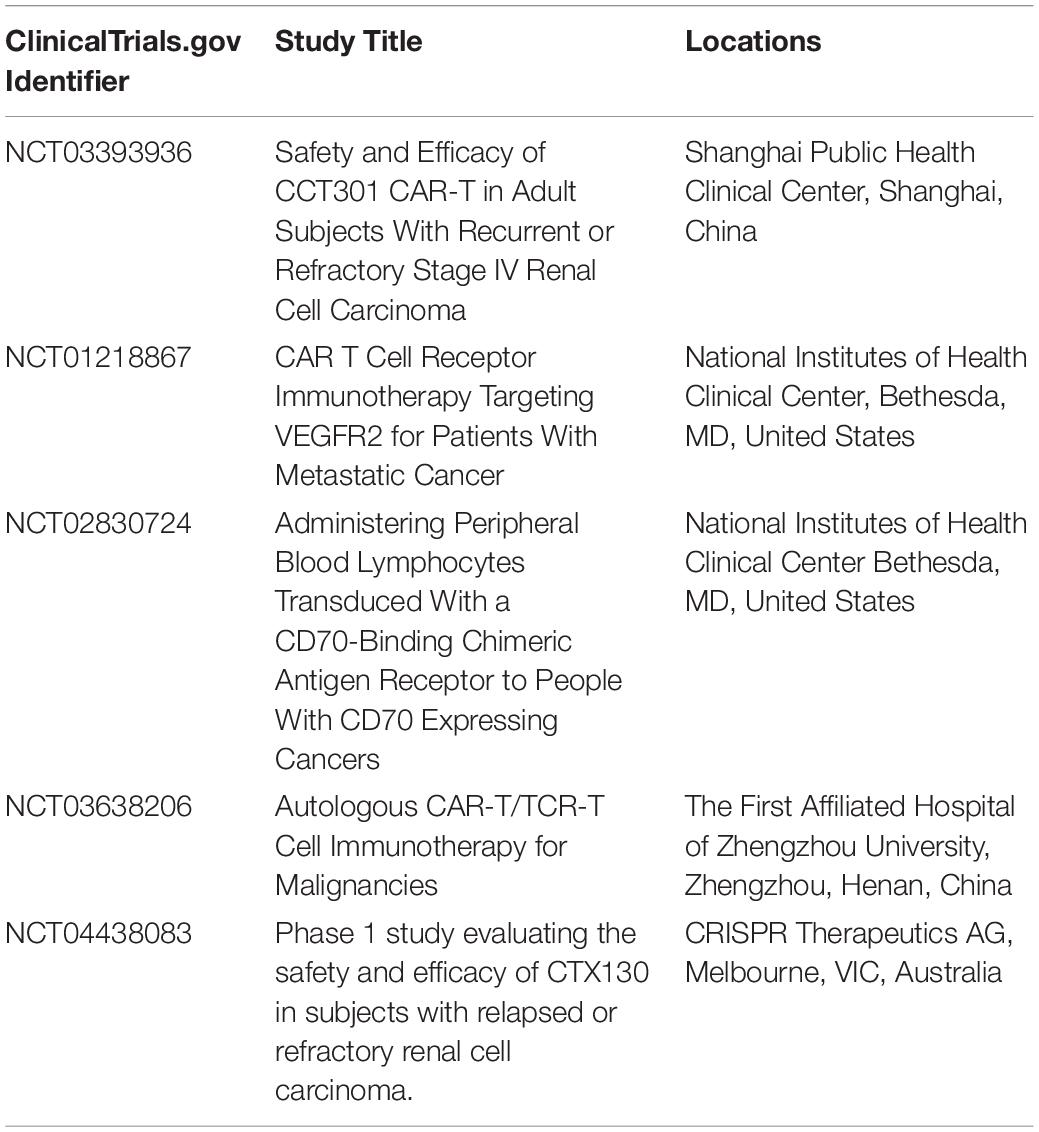
Table 1. Clinical trials of chimeric antigen receptor (CAR)-T cell therapy in renal cell carcinoma (RCC).
In the ongoing trial, patients with a ROR2-positive biopsy will receive CCT301-59, while those with an AXL tyrosine kinase receptor-positive but ROR2-negative biopsy will receive CCT301-38. A blood sample will be taken from patients to isolate peripheral blood mononuclear cells (PBMCs) for the production of CCT301-48 or CCT301-59. During the procedure, patients will undergo a cyclophosphamide plus fludarabine conditioning regimen to deplete lymphocytes, after which one intravenous cycle of CCT301-48 or CCT301-59 will be administered. A 3 + 3 dose escalation model will investigate the safety and efficacy of these molecules. Three different CAR T dosages will be evaluated: 1 × 105/kg, 1 × 106/kg, and 1 × 107/kg CAR+ T cells. This study is currently active but not recruiting patients. The primary completion date of the study is scheduled for the first months of 2021 (NCT03393936).
Another study is evaluating the safety and effectiveness of the anti-VEGFR2 gene-modified CD8 cells in mRCC patients (NCT01218867). Patients undergo lymphocyte-depleting chemotherapy (cyclophosphamide and fludarabine) followed by CAR gene-transduced CD8+ PBMC in combination with aldesleukin. Participants are divided into two cohorts according to histology: cohort 1 includes metastatic melanoma and RCC patients and cohort 2, patients with other metastatic tumor types. Preliminary results have failed to show any objective responses.
The same lymphodepleting preparative regimen is also being used in another study currently ongoing to test peripheral blood lymphocytes transduced with anti-hCD70 CAR in combination with aldesleukin. CD70, normally expressed in B, T, and NK cells, is a transmembrane receptor with a costimulatory role in immune cell activation. CD70 is upregulated in several tumors where it stimulates immune escape by promoting cytotoxic effects on B and T lymphocytes. It is highly expressed in different RCC histologies (clear cell, sarcomatoid, and some papillary tumors), and this condition is correlated with decreased survival, thus representing a potential target for therapies against RCC.
Two separate cohorts will be included, one with patients with CD70-expressing RCC and another with patients with CD70-expressing non-RCC solid tumors (NCT02830724).
More recently, a phase I study has begun enrollment of patients with advanced, relapsed, or refractory clear-cell RCC to test a new allogeneic therapy (CTX130). CTX130 consists of an allogeneic CRISPR/Cas9 gene-edited CAR-T cell therapy targeting CD70, which is currently under development for the treatment of both solid tumors and hematologic malignancies. The theoretical advantages of this type of CAR (allogeneic) are: (1) immediate availability; (2) increased potency (because it is derived from healthy donors); (3) greater consistency (many doses from healthy donors); (4) no need for patient apheresis; and (5) flexibility to titrate dose or re-dose. The final data on the primary outcome measure is expected in February 2027 (NCT04438083).
Overall, despite the interesting ongoing research, conclusive results have yet to be obtained.
Conclusion
Unlike other genitourinary tumor histotypes, RCC is a neoplasm for which numerous drugs have been approved. After decades in which no effective therapies were available because of its known chemoresistance, the advent of biological therapies and then immunotherapy a little over a decade ago has brought about a marked improvement in terms of survival. In the wake of these data, the unexpectedly promising results obtained from CAR-T therapy in hematological tumors has prompted research into the possibility of also using this treatment for RCC.
To date, however, there are still few results from few studies. Furthermore, the peculiarities of solid tumors pose different challenges with respect to hematological malignancies. The presence of stroma and various inhibitory factors within the tissue, the problems related to T-cell trafficking, and the non-high antigenic selectivity represent just a few of the obstacles to the successful outcome of the treatment. In addition, the risk of side effects, including the aforementioned “on-target/off-tumor” toxicity justifies the delay in the use of CAR-T cell therapy in solid tumors, including RCC. Further research is thus needed to resolve these problems before being able to claim an efficacy comparable to that achieved in hematological tumors.
Author Contributions
GS and UD conceived and designed the article. GS created the table and figures and drafted the manuscript. CC was responsible for compiling the “References” section. VC, GG, LR, VG, MC, NB, CL, CM, AF, SB, AA, and GM analyzed the data. All authors read and approved the final version of the manuscript for submission.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Gráinne Tierney for the editing assistance.
References
1. Rossi SH, Klatte T, Usher-Smith J, Stewart GD. Epidemiology and screening for renal cancer. World J Urol. (2018) 36:1341–53. doi: 10.1007/s00345-018-2286-7
2. Capitanio U, Bensalah K, Bex A, Boorjian SA, Bray F, Coleman J, et al. Epidemiology of renal cell carcinoma [Figure presented]. Eur Urol. (2019) 75:74–84. doi: 10.1016/j.eururo.2018.08.036
3. Fisher RI, Rosenberg SA, Fyfe G. Long-term survival update for high-dose recombinant interleukin-2 in patients with renal cell carcinoma. Cancer J Sci Am. (2000) 6(Suppl. 1):S55–7.
4. Motzer RJ, Mazumdar M, Bacik J, Russo P, Berg WJ, Metz EM. Effect of cytokine therapy on survival for patients with advanced renal cell carcinoma. J Clin Oncol. (2000) 18:1928–35. doi: 10.1200/JCO.2000.18.9.1928
5. Escudier B, Pluzanska A, Koralewski P, Ravaud A, Bracarda S, Szczylik C, et al. Bevacizumab plus interferon alfa-2a for treatment of metastatic renal cell carcinoma: a randomised, double-blind phase III trial. Lancet. (2007) 370:2103–11. doi: 10.1016/S0140-6736(07)61904-7
6. Motzer RJ, Hutson TE, Tomczak P, Michaelson D, Bukowski RM, Rixe O, et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N Engl J Med. (2007) 356:115–24. doi: 10.1056/NEJMoa065044
7. Sternberg CN, Davis ID, Mardiak J, Szczylik C, Lee E, Wagstaff J, et al. Pazopanib in locally advanced or metastatic renal cell carcinoma: results of a randomized phase III trial. J Clin Oncol. (2010) 28:1061–8. doi: 10.1200/JCO.2009.23.9764
8. Rodriguez-Vida A, Hutson TE, Bellmunt J, Strijbos MH. New treatment options for metastatic renal cell carcinoma. ESMO Open. (2017) 2:e000185. doi: 10.1136/esmoopen-2017-000185
9. Pantuck M, Palaskas N, Drakaki A. Next generation T-cell therapy for genitourinary malignancies, part A: introduction and current state of the art. Cancer Treat Res Commun. (2018) 17:8–12. doi: 10.1016/j.ctarc.2018.08.001
10. Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S, et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med. (2015) 373:1803–13. doi: 10.1056/NEJMoa1510665
11. Motzer RJ, Tannir NM, McDermott DF, Arén Frontera O, Melichar B, Choueiri TK, et al. Nivolumab plus Ipilimumab versus Sunitinib in advanced renal-cell carcinoma. N Engl J Med. (2018) 378:1277–90. doi: 10.1056/NEJMoa1712126
12. Rini BI, Plimack ER, Stus V, Gafanov R, Hawkins R, Nosov D, et al. Pembrolizumab plus axitinib versus sunitinib for advanced renal-cell carcinoma. N Engl J Med. (2019) 380:1116–27. doi: 10.1056/NEJMoa1816714
13. Motzer RJ, Penkov K, Haanen J, Rini B, Albiges L, Campbell MT, et al. Avelumab plus axitinib versus sunitinib for advanced renal-cell carcinoma. N Engl J Med. (2019) 380:1103–15. doi: 10.1056/NEJMoa1816047
14. Motzer RJ, Rini BI, McDermott DF, Arén Frontera O, Hammers HJ, Carducci MA, et al. Nivolumab plus ipilimumab versus sunitinib in first-line treatment for advanced renal cell carcinoma: extended follow-up of efficacy and safety results from a randomised, controlled, phase 3 trial. Lancet Oncol. (2019) 20:1370–85. doi: 10.1016/S1470-2045(19)30413-9
15. Harris DT, Hager MV, Smith SN, Cai Q, Stone JD, Kruger P, et al. Comparison of T cell activities mediated by human TCRs and CARs that use the same recognition domains. J Immunol. (2018) 200:1088–100. doi: 10.4049/jimmunol.1700236
16. Rohaan MW, Wilgenhof S, Haanen JBAG. Adoptive cellular therapies: the current landscape. Virchows Arch. (2019) 474:449–61. doi: 10.1007/s00428-018-2484-0
17. June C, Rosenberg SA, Sadelain M, Weber JS. T-cell therapy at the threshold. Nat Biotechnol. (2012) 30:611–4. doi: 10.1038/nbt.2305
18. Figueroa JA, Reidy A, Mirandola L, Trotter K, Suvorava N, Figueroa A, et al. Chimeric antigen receptor engineering: a right step in the evolution of adoptive cellular immunotherapy. Int Rev Immunol. (2015) 34:154–87. doi: 10.3109/08830185.2015.1018419
19. Katari UL, Keirnan JM, Worth AC, Hodges SE, Leen AM, Fisher WE, et al. Engineered T cells for pancreatic cancer treatment. HPB. (2011) 13:643–50. doi: 10.1111/j.1477-2574.2011.00344.x
20. Wang H, Kaur G, Sankin AI, Chen F, Guan F, Zang X. Immune checkpoint blockade and CAR-T cell therapy in hematologic malignancies. J Hematol Oncol. (2019) 12:59. doi: 10.1186/s13045-019-0746-1
21. Rafiq S, Yeku OO, Jackson HJ, Purdon TJ, van Leeuwen DG, Drakes DJ, et al. Targeted delivery of a PD-1-blocking scFV by CAR-T cells enhances anti-tumor efficacy in vivo. Nat Biotechnol. (2018) 36:847–58. doi: 10.1038/nbt.4195
22. Fraietta JA, Lacey SF, Orlando EJ, Pruteanu-Malinici I, Gohil M, Lundh S, et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat Med. (2018) 24:563–71. doi: 10.1038/s41591-018-0010-1
23. Zolov SN, Rietberg SP, Bonifant CL. Programmed cell death protein 1 activation preferentially inhibits CD28.CAR–T cells. Cytotherapy. (2018) 20:1259–66. doi: 10.1016/j.jcyt.2018.07.005
24. Li AM, Hucks GE, Dinofia AM, Seif AE, Teachey DT, Baniewicz D, et al. Checkpoint inhibitors augment CD19-directed chimeric antigen receptor (CAR) T cell therapy in relapsed B-Cell acute lymphoblastic leukemia. Blood. (2018) 132:556–556. doi: 10.1182/blood-2018-99-112572
25. O’Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med. (2017) 9:eaaa0984. doi: 10.1126/scitranslmed.aaa0984
26. Pule MA, Savoldo B, Myers GD, Rossig C, Russell HV, Dotti G, et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med. (2008) 14:1264–70. doi: 10.1038/nm.1882
27. Lamers CHJ, Gratama JW, Pouw NMC, Langeveld SCL, Van Krimpen BA, Kraan J, et al. Parallel detection of transduced T lymphocytes after immunogene therapy of renal cell cancer by flow cytometry and real-time polymerase chain reaction: implications for loss of transgene expression. Hum Gene Ther. (2005) 16:1452–62. doi: 10.1089/hum.2005.16.1452
28. Thistlethwaite FC, Gilham DE, Guest RD, Rothwell DG, Pillai M, Burt DJ, et al. The clinical efficacy of first-generation carcinoembryonic antigen (CEACAM5)-specific CAR T cells is limited by poor persistence and transient pre-conditioning-dependent respiratory toxicity. Cancer Immunol Immunother. (2017) 66:1425–36. doi: 10.1007/s00262-017-2034-7
29. Klaver Y, van Steenbergen SCL, Sleijfer S, Debets R, Lamers CHJ. Plasma IFN-γ and IL-6 levels correlate with peripheral T-cell numbers but not toxicity in RCC patients treated with CAR T-cells. Clin Immunol. (2016) 169:107–13. doi: 10.1016/j.clim.2016.06.014
30. Scarfò I, Maus MV. Current approaches to increase CAR T cell potency in solid tumors: targeting the tumor microenvironment. J Immunother Cancer. (2017) 5:28. doi: 10.1186/s40425-017-0230-9
31. Knochelmann HM, Smith AS, Dwyer CJ, Wyatt MM, Mehrotra S, Paulos CM. CAR T cells in solid tumors: blueprints for building effective therapies. Front Immunol. (2018) 9:1740. doi: 10.3389/fimmu.2018.01740
32. Martinez M, Moon EK. CAR T cells for solid tumors: new strategies for finding, infiltrating, and surviving in the tumor microenvironment. Front Immunol. (2019) 10:128. doi: 10.3389/fimmu.2019.00128
33. Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of t cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. (2010) 18:843–51. doi: 10.1038/mt.2010.24
34. Richman SA, Nunez-Cruz S, Moghimi B, Li LZ, Gershenson ZT, Mourelatos Z, et al. High-Affinity GD2-specific CAR T cells induce fatal encephalitis in a preclinical neuroblastoma model. Cancer Immunol Res. (2018) 6:36–46. doi: 10.1158/2326-6066.CIR-17-0211
35. Fitzgerald JC, Weiss SL, Maude SL, Barrett DM, Lacey SF, Melenhorst JJ, et al. Cytokine release syndrome after chimeric antigen receptor T cell therapy for acute lymphoblastic leukemia. Crit Care Med. (2017) 45:e124–5. doi: 10.1097/CCM.0000000000002053
36. Bonifant CL, Jackson HJ, Brentjens RJ, Curran KJ. Toxicity and management in CAR T-cell therapy. Mol Ther Oncolytics. (2016) 3:16011. doi: 10.1038/mto.2016.11
37. Jhaveri KD, Rosner MH. Chimeric antigen receptor T cell therapy and the kidney: what the nephrologist needs to know. Clin J Am Soc Nephrol. (2018) 13:796–8. doi: 10.2215/CJN.12871117
38. Lamers CHJ, Sleijfer S, Vulto AG, Kruit WHJ, Kliffen M, Debets R, et al. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J Clin Oncol. (2006) 24:e20–2. doi: 10.1200/JCO.2006.05.9964
39. Lamers CHJ, Sleijfer S, Van Steenbergen S, Van Elzakker P, Van Krimpen B, Groot C, et al. Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: clinical evaluation and management of on-target toxicity. Mol Ther. (2013) 21:904–12. doi: 10.1038/mt.2013.17
40. Ahmed N, Brawley VS, Hegde M, Robertson C, Ghazi A, Gerken C, et al. Human epidermal growth factor receptor 2 (HER2)–specific chimeric antigen receptor–modified T cells for the immunotherapy of HER2-positive sarcoma. J Clin Oncol. (2015) 33:1688–96. doi: 10.1200/JCO.2014.58.0225
41. Curran KJ, Pegram HJ, Brentjens RJ. Chimeric antigen receptors for T cell immunotherapy: current understanding and future directions. J Gene Med. (2012) 14:405–15. doi: 10.1002/jgm.2604
42. Cruz CRY, Micklethwaite KP, Savoldo B, Ramos CA, Lam S, Ku S, et al. Infusion of donor-derived CD19-redirected virus-specific T cells for B-cell malignancies relapsed After allogeneic stem cell transplant: a phase 1 study. Blood. (2013) 122:2956–73. doi: 10.1182/blood-2013-06-506741
43. Wang X, Wong CW, Urak R, Mardiros A, Budde LE, Chang WC, et al. CMVpp65 vaccine enhances the antitumor efficacy of adoptively transferred CD19-redirected CMV-specific T cells. Clin Cancer Res. (2015) 21:2993–3002. doi: 10.1158/1078-0432.CCR-14-2920
44. Philip LPB, Schiffer-Mannioui C, Le Clerre D, Chion-Sotinel I, Derniame S, Potrel P, et al. Multiplex genome-edited T-cell manufacturing platform for “off-the-shelf” adoptive T-cell immunotherapies. Cancer Res. (2015) 75:3853–64. doi: 10.1158/0008-5472.CAN-14-3321
45. Pantuck M, Palaskas N, Drakaki A. Next generation T-cell therapy for genitourinary malignancies, part B: overcoming obstacles and future strategies for success. Cancer Treat Res Commun. (2018) 17:1–7. doi: 10.1016/j.ctarc.2018.08.002
46. Priceman SJ, Gerdts EA, Tilakawardane D, Kennewick KT, Murad JP, Park AK, et al. Co-stimulatory signaling determines tumor antigen sensitivity and persistence of CAR T cells targeting PSCA+ metastatic prostate cancer. Oncoimmunology. (2018) 7:e1380764. doi: 10.1080/2162402X.2017.1380764
47. Liu X, Jiang S, Fang C, Yang S, Olalere D, Pequignot EC, et al. Affinity-tuned ErbB2 or EGFR chimeric antigen receptor T cells exhibit an increased therapeutic index against tumors in mice. Cancer Res. (2015) 75:3596–607. doi: 10.1158/0008-5472.CAN-15-0159
48. Park S, Shevlin E, Vedvyas Y, Zaman M, Park S, Hsu YMS, et al. Micromolar affinity CAR T cells to ICAM-1 achieves rapid tumor elimination while avoiding systemic toxicity. Sci Rep. (2017) 7:14366. doi: 10.1038/s41598-017-14749-3
49. Esensten JH, Bluestone JA, Lim WA. Engineering therapeutic T cells: from synthetic biology to clinical trials. Annu Rev Pathol Mech Dis. (2017) 12:305–30. doi: 10.1146/annurev-pathol-052016-100304
50. Wu CY, Roybal KT, Puchner EM, Onuffer J, Lim WA. Remote control of therapeutic T cells through a small molecule-gated chimeric receptor. Science. (2015) 350:aab4077. doi: 10.1126/science.aab4077
51. Arcangeli S, Magnani CF, Tettamanti S, Biagi E. Switchable chimeric antigen receptor T cells: a novel universal chimeric antigen receptor platform for a safe control of T-cell activation. Transl Cancer Res. (2016) 5:S174–7. doi: 10.21037/tcr.2016.07.23
52. Zhang E, Xu H. A new insight in chimeric antigen receptor-engineered T cells for cancer immunotherapy. J Hematol Oncol. (2017) 10:1–11. doi: 10.1186/s13045-016-0379-6
53. Makita S, Yoshimura K, Tobinai K. Clinical development of anti-CD19 chimeric antigen receptor T-cell therapy for B-cell non-Hodgkin lymphoma. Cancer Sci. (2017) 108:1109–18. doi: 10.1111/cas.13239
54. Philip B, Kokalaki E, Mekkaoui L, Thomas S, Straathof K, Flutter B, et al. A highly compact epitope-based marker/suicide gene for easier and safer T-cell therapy. Blood. (2014) 124:1277–87. doi: 10.1182/blood-2014-01-545020
55. Kloss CC, Condomines M, Cartellieri M, Bachmann M, Sadelain M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotechnol. (2013) 31:71–5. doi: 10.1038/nbt.2459
56. Fedorov VD, Themeli M, Sadelain M. PD-1- and CTLA-4-based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Sci Transl Med. (2013) 5:215ra172. doi: 10.1126/scitranslmed.3006597
57. Tannir NM, McDermott DF, Escudier B, Hammers HJ, Aren OR, Plimack ER, et al. Overall survival and independent review of response in CheckMate 214 with 42-month follow-up: first-line nivolumab + ipilimumab (N+I) versus sunitinib (S) in patients (pts) with advanced renal cell carcinoma (aRCC). J Clin Oncol. (2020) 38:609–609. doi: 10.1200/jco.2020.38.6_suppl.609
58. Argentiero A, Solimando AG, Krebs M, Leone P, Susca N, Brunetti O, et al. Anti-angiogenesis and Immunotherapy: novel Paradigms to Envision Tailored Approaches in Renal Cell-Carcinoma. J Clin Med. (2020) 9:1594. doi: 10.3390/jcm9051594
59. McDermott DF, Huseni MA, Atkins MB, Motzer RJ, Rini BI, Escudier B, et al. Clinical activity and molecular correlates of response to atezolizumab alone or in combination with bevacizumab versus sunitinib in renal cell carcinoma. Nat Med. (2018) 24:749–57. doi: 10.1038/s41591-018-0053-3
60. Négrier S, Escudier B, Gomez F, Douillard JY, Ravaud A, Chevreau C, et al. Prognostic factors of survival and rapid progression in 782 patients with metastatic renal carcinomas treated by cytokines: a report from the Groupe Français d’Immunothérapie. Ann Oncol. (2002) 13:1460–8. doi: 10.1093/annonc/mdf257
61. Park YH, Ku JH, Kwak C, Kim HH. Post-treatment neutrophil-to-lymphocyte ratio in predicting prognosis in patients with metastatic clear cell renal cell carcinoma receiving sunitinib as first line therapy. Springerplus. (2014) 3:1–6. doi: 10.1186/2193-1801-3-243
62. Lolli C, Basso U, Derosa L, Scarpi E, Sava T, Santoni M, et al. Systemic immune-inflammation index predicts the clinical outcome in patients with metastatic renal cell cancer treated with sunitinib. Oncotarget. (2016) 7:54564–71. doi: 10.18632/oncotarget.10515
63. Yasuda Y, Saito K, Yuasa T, Uehara S, Kawamura N, Yokoyama M, et al. Early response of C-reactive protein as a predictor of survival in patients with metastatic renal cell carcinoma treated with tyrosine kinase inhibitors. Int J Clin Oncol. (2017) 22:1081–6. doi: 10.1007/s10147-017-1166-2
64. Ueda K, Suekane S, Kurose H, Chikui K, Nakiri M, Nishihara K, et al. Prognostic value of PD-1 and PD-L1 expression in patients with metastatic clear cell renal cell carcinoma. Urol Oncol Semin Orig Investig. (2018) 36:.e9–499. doi: 10.1016/j.urolonc.2018.07.003
65. Shin S, Jeon YK, Cho YM, Lee J, Chung DH, Park JY, et al. The association between PD−L1 expression and the clinical outcomes to vascular endothelial growth factor−targeted therapy in patients with metastatic clear cell renal cell carcinoma. Oncologist. (2015) 20:1253–60. doi: 10.1634/theoncologist.2015-0151
66. Hara T, Miyake H, Fujisawa M. Expression pattern of immune checkpoint–associated molecules in radical nephrectomy specimens as a prognosticator in patients with metastatic renal cell carcinoma treated with tyrosine kinase inhibitors. Urol Oncol Semin Orig Investig. (2017) 35:363–9. doi: 10.1016/j.urolonc.2017.01.002
67. Rathinam VAK, Fitzgerald KA. Inflammasome complexes: emerging mechanisms and effector functions. Cell. (2016) 165:792–800. doi: 10.1016/j.cell.2016.03.046
68. Wang KS, Xu TB, Ruan HL, Xiao HB, Liu J, Song ZS, et al. LXRα promotes cell metastasis by regulating the NLRP3 inflammasome in renal cell carcinoma. Cell Death Dis. (2019) 10:159. doi: 10.1038/s41419-019-1345-3
69. Tan YF, Wang M, Chen ZY, Wang L, Liu XH. Inhibition of BRD4 prevents proliferation and epithelial–mesenchymal transition in renal cell carcinoma via NLRP3 inflammasome-induced pyroptosis. Cell Death Dis. (2020) 11:239. doi: 10.1038/s41419-020-2431-2
70. Chai D, Liu N, Li H, Wang G, Song J, Fang L, et al. H1/pAIM2 nanoparticles exert anti-tumour effects that is associated with the inflammasome activation in renal carcinoma. J Cell Mol Med. (2018) 22:5670–81. doi: 10.1111/jcmm.13842
71. Seyedin SN, Schoenhals JE, Lee DA, Cortez MA, Wang X, Niknam S, et al. Strategies for combining immunotherapy with radiation for anticancer therapy. Immunotherapy. (2015) 7:967–80. doi: 10.2217/imt.15.65
72. Ngiow SF, McArthur GA, Smyth MJ. Radiotherapy complements immune checkpoint blockade. Cancer Cell. (2015) 27:437–8. doi: 10.1016/j.ccell.2015.03.015
73. Tang C, Welsh JW, De Groot P, Massarelli E, Chang JY, Hess KR, et al. Ipilimumab with stereotactic ablative radiation therapy: phase i results and immunologic correlates from peripheral T cells. Clin Cancer Res. (2017) 23:1388–96. doi: 10.1158/1078-0432.CCR-16-1432
74. Formenti SC, Rudqvist NP, Golden E, Cooper B, Wennerberg E, Lhuillier C, et al. Radiotherapy induces responses of lung cancer to CTLA-4 blockade. Nat Med. (2018) 24:1845–51. doi: 10.1038/s41591-018-0232-2
75. Santoni M, Heng DYC, Aurilio G, Iozzelli A, Servi L, Fabiani A, et al. Combining radiotherapy with immunocheckpoint inhibitors or CAR-T in renal cell carcinoma. Curr Drug Targets. (2019) 21:416–23. doi: 10.2174/1389450120666191017113051
76. Yang CM, Ji S, Li Y, Fu LY, Jiang T, Meng FD. Ror2, a developmentally regulated kinase, is associated with tumor growth, apoptosis, migration, and invasion in renal cell carcinoma. Oncol Res. (2017) 25:195–205. doi: 10.3727/096504016X14732772150424
Keywords: CAR (chimeric antigen receptor) T cells, RCC, immunotherapy, inflammation, toxicity
Citation: Schepisi G, Conteduca V, Casadei C, Gurioli G, Rossi L, Gallà V, Cursano MC, Brighi N, Lolli C, Menna C, Farolfi A, Burgio SL, Altavilla A, Martinelli G and De Giorgi U (2020) Potential Application of Chimeric Antigen Receptor (CAR)-T Cell Therapy in Renal Cell Tumors. Front. Oncol. 10:565857. doi: 10.3389/fonc.2020.565857
Received: 26 May 2020; Accepted: 24 August 2020;
Published: 23 September 2020.
Edited by:
Walter J. Storkus, University of Pittsburgh, United StatesReviewed by:
Stephen Gottschalk, St. Jude Children’s Research Hospital, United StatesRasha Abu Eid, University of Aberdeen, United Kingdom
Copyright © 2020 Schepisi, Conteduca, Casadei, Gurioli, Rossi, Gallà, Cursano, Brighi, Lolli, Menna, Farolfi, Burgio, Altavilla, Martinelli and De Giorgi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Giuseppe Schepisi, Z2l1c2VwcGUuc2NoZXBpc2lAaXJzdC5lbXIuaXQ=