 Plabon Kumar Das
Plabon Kumar Das Farhadul Islam
Farhadul Islam Robert A. Smith3,4
Robert A. Smith3,4 Alfred K. Lam
Alfred K. Lam- 1Department of Biochemistry and Molecular Biology, University of Rajshahi, Rajshahi, Bangladesh
- 2Institute for Glycomics, Griffith University, Gold Coast, QLD, Australia
- 3Centre for Genomics and Personalised Health, Genomics Research Centre, School of Biomedical Sciences, Institute of Health and Biomedical Innovation, Queensland University of Technology (QUT), Kelvin Grove, QLD, Australia
- 4Cancer Molecular Pathology, School of Medicine, Griffith University, Gold Coast, QLD, Australia
- 5Faculty of Medicine, The University of Queensland, Brisbane, QLD, Australia
Cancer stem cells (CSCs) in esophageal cancer have a key role in tumor initiation, progression and therapy resistance. Novel therapeutic strategies to target CSCs are being tested, however, more in-depth research is necessary. Eradication of CSCs can result in successful therapeutic approaches against esophageal cancer. Recent evidence suggests that targeting signaling pathways, miRNA expression profiles and other properties of CSCs are important strategies for cancer therapy. Wnt/β-catenin, Notch, Hedgehog, Hippo and other pathways play crucial roles in proliferation, differentiation, and self-renewal of stem cells as well as of CSCs. All of these pathways have been implicated in the regulation of esophageal CSCs and are potential therapeutic targets. Interference with these pathways or their components using small molecules could have therapeutic benefits. Similarly, miRNAs are able to regulate gene expression in esophageal CSCs, so targeting self-renewal pathways with miRNA could be utilized to as a potential therapeutic option. Moreover, hypoxia plays critical roles in esophageal cancer metabolism, stem cell proliferation, maintaining aggressiveness and in regulating the metastatic potential of cancer cells, therefore, targeting hypoxia factors could also provide effective therapeutic modalities against esophageal CSCs. To conclude, additional study of CSCs in esophageal carcinoma could open promising therapeutic options in esophageal carcinomas by targeting hyper-activated signaling pathways, manipulating miRNA expression and hypoxia mechanisms in esophageal CSCs.
Introduction
Esophageal cancer (EC) is the seventh most common malignancy around the world and the sixth most leading cause of cancer-related mortalities with an estimated 572,000 new incidences and 509,000 deaths in 2018 (1, 2). There are two histopathological subtypes of esophageal cancer such as esophageal squamous cell carcinoma (OSCC) and esophageal adenocarcinoma (OAC) (3–5). The incidence of OAC has been escalating in the Western world, whereas OSCC is more common in the Asia-Pacific region (1). Currently, patients with either subtype receive similar treatment, which is a neoadjuvant chemo-radiotherapy followed by surgery (5). The clinical outcome of the standard therapeutic regimen is, however, limited, as much as 20% of tumors do not respond to chemo-radiotherapy at all, and more than 50% do not respond sufficiently. Furthermore, even after complete responses to adjuvant therapy, early and distant relapse occurs in most cases (5). Therefore, in-depth research is required to investigate the underlying mechanisms of therapy resistance and the subpopulation of cancer cells causing therapy failure needs to be thoroughly investigated.
Accumulating information from research has revealed that a subpopulation of cancer cells known as cancer stem cells (CSCs) are associated with clinical features such as drug resistance, self-renewal, and tumorigenicity in esophageal cancer (6–10). Several pathways e.g. Wnt/beta-catenin, Hedgehog, Notch, JAK-STAT3 and Hippo pathways are hyper-activated in both OSCC and OAC, especially in esophageal CSCs. These pathways drive proliferation, differentiation, stemness, and resistance to therapy in the tumors in which they are activated (11–16). For example, the Wnt/beta-catenin pathway was found to contribute to CSC renewal, whereas the Hedgehog pathway has been found to play profound roles in regulating proliferation, not only of normal embryonic cells, but also of cancer cells (11, 13). In addition, altered expression of micro-RNAs; tumor microenvironmental factors such as autophagy, and hypoxia; and reactivation of epithelial-mesenchymal transition (EMT) alone or in combination can trigger the pool of CSCs by aberrant activation of signaling pathways, resulting in the development of cancer recurrences and treatment resistance in esophageal cancer (17–19).Therefore, further investigation regarding the function of CSCs or their associated pathways could provide new potential therapeutic options against esophageal cancers.
Novel therapeutics targeting CSCs rather than bulk-cancer cells or later differentiated progenitors could provide many benefits in patients with esophageal cancer. Traditional cytotoxic agents cannot target CSCs properly as a majority of anti-tumor drugs at present are DNA damage inducing agents (20). They induce tumor cell death most effectively during cell division, while CSCs are usually dormant and do not enter the cell cycle. Thus, DNA damaging agents have little capacity to not induce the death of CSCs (20). Moreover, several mechanisms have been identified in CSCs to avoid DNA damage-induced cell death. For example, CSCs enhance ROS scavenging to inhibit oxidative DNA damage, promote DNA repair capability through ATM and CHK1/CHK2phosphorylation, and activate anti-apoptotic signaling pathways, such as PI3K/Akt, WNT/b-catenin, and Notch signaling pathways to escape DNA damaging agent mediated insults (21).
Interestingly, several therapies that specifically target CSCs or their components in the tumor microenvironment are making their way into clinics. Thus, in this review, we undertake a comprehensive overview of the literature regarding the role of CSCs in esophageal cancer. Moreover, the review also discusses potential therapies targeting aberrantly activated signaling pathways, miRNA expression and hypoxia regulated signaling in esophageal CSCs.
The Role of Cancer Stem Cells in Esophageal Cancer
Cancer stem cells (CSCs) harbor unique properties, such as self-renewal, tumor maintenance (proliferation), invasion and migration, immune evasion, and therapy resistance (22, 23). Virchow and Conheim first proposed that CSCs exist as a subpopulation of cancer cells, which possess the traits of embryonic cells, including the ability to proliferate different lineages and renew themselves (24). They further assumed that cancer is derived from dormant stem-like cells of the same tissue (24). An experimental approach using leukemia stem cells provided the first evidence of the existence of a cell population having the capacity to initiate a secondary tumor, confirming the presence of CSCs (25). In general, there are two hypotheses that have been proposed regarding the origin of CSCs (5). Firstly, normal stem cells can be transformed into CSCs because of genetic and epigenetic alterations. Secondly, dedifferentiated cancer cells acquire the capabilities of CSCs by the process called cellular plasticity (22, 23, 25–27). CSCs often display resistance to therapy, the exact mechanisms of which are not clear, however, a number of underlying mechanisms have been identified i.e. enhanced DNA repair efficiency, increased expression of detoxification enzymes (ALDH), increased expression of drug resistance proteins, up-regulation of anti-apoptotic proteins (Bcl-2, Bcl-xL, Mcl-l, Bcl-w), mutations in key signaling molecules, and overexpression of drug efflux pumps (P glycoprotein 1, ABCG2) etc. in CSCs (28, 29).
Esophageal CSCs directly regulate cancer initiation, progression, metastasis, therapy resistance and recurrence both in esophageal adenocarcinomas (OAC) and esophageal squamous cell carcinomas (OSCC) (26, 30, 31). CSCs of esophageal cancer can be identified and isolated by specific cell surface and intracellular markers. For example, cell surface and intracellular markers such as CD44, ALDH, Pygo2, MAML1, Twist1, Musashi1, CD271, and CD90, are used to identify CSCs, whereas, stem cell markers including ALDH1, HIWI, Oct3/4, ABCG2, SOX2, SALL4, BMI-1, NANOG, CD133, and podoplanin were associated with the enrichment of CSCs in OSCC (26, 30, 31). In addition, isolation of side population (SP), a subpopulation of cells with the ability to exclude dyes such as Hoechst 33342, are enriched with stem cells and SP isolation can be used to identify CSCs in OSCC (31). According to several studies, side population has been utilized in the isolation of CSCs from esophageal cancer (32–34). For example, isolation of side population in different esophageal cancer cells such as OSCC (OE21) and OAC (OE19, OE33, PT1590, and LN1590) revealed that the proportions of side population cells are varied among the cell lines and they are resistant to chemotherapy (34). Also, SP cells exhibited stem-like cell phenomena such as epithelial-mesenchymal transition (EMT) (34). The stem-like esophageal cells also become more radio-resistant than parental cells (35). The radio-resistant property of esophageal CSCs is attributed to the overexpression of β-catenin, Oct3/4, and β1-integrin (36). Moreover, esophageal CSCs dictate intrinsic and acquired chemotherapy resistance to 5-fluorouracil (5-FU) and cisplatin in OAC (22). This therapy resistance is associated with changes in the regulation of EMT (22). Additionally, recent studies demonstrated a relationship between the expression of miRNAs, for example, miR-296 (37) and miR-200c (38) and chemoresistance in esophageal CSCs. Furthermore, overexpression of WNT10A, a member of the Wnt gene family, increases self-renewal capabilities of CSCs and induces a larger population of CSCs in OSCC (39). Most importantly, CSCs with increased tumorigenicity were formed when tumors multiply and experience treatment threats such as targeted agents, cytotoxic agents or radiation (19). Therefore, it is plausible that eradication of CSCs or, alternatively, reduction of their malignant and stemness properties can result in more successful therapeutic approaches.
Targeting Signaling Pathways in Esophageal Cancer Stem Cells
The signaling pathways which trigger embryogenesis also play a significant role in oncogenesis (40). The pathways highly associated with the maintenance of esophageal CSCs include Wnt/β-catenin, Notch, Hh, and Hippo pathways (39). These pathways are involved in maintaining tissue homeostasis and normal stem cell renewal and dysregulation of these signaling pathways drives esophageal CSCs formation (39). For example, a Wnt/β-catenin activator WNT10A is highly expressed in OSCC tissue. Consistently cells with the expression of WNT10A showed enrichment for CD44+/CD24−, and these cells showed increased self-renewal, invasive and metastatic potential (40, 41). Notch signaling is another prominent driver of cancer stemness in OAC. Experimental work shows, for example, that inhibiting Notch pathway by γ-secretase inhibitors reduces the size of patient-derived xenograft tumors of OAC in mice (42). Furthermore, aberrant activation of these pathways can result from autophagy, hypoxia, anti-cancer therapy and EMT, alone or in combination with each other, which subsequently leads to an enrichment of CSCs and development of recurrences, metastasis and increasing treatment resistance (39). These phenomena can be manipulated by novel therapeutics targeting specific components involving the stemness of cancer cells to offset their role in treatment resistance.
Targeting the Wnt/β-Catenin Pathway in Esophageal Cancer Stem Cells
The Wnt/β-catenin signaling pathway plays a pivotal role in oncogenesis through different mechanisms (43). In normal physiological conditions, the Wnt/β-catenin pathway controls the expression of downstream genes, which are involved in basic cellular and biological functions including proliferation, differentiation, apoptosis, and cell death (44). Thus, in order to exert normal physiological functions, activation of Wnt/β-catenin signaling should be kept at the normal level. However, aberrant activation of this pathway is associated with many cancers including esophageal cancer. For instance, over-activation of the Wnt/β-catenin pathway can be an underlying factor of progression, metastasis, and invasion in OSCC by inducing a CSC phenotype (40). Therefore, targeting the Wnt/β-catenin pathway has potential for the inhibition of CSC growth. Though Wnt/β-catenin inhibitors are in clinical trials for various solid tumors, inhibitors are yet to reach clinical trials in esophageal cancer (39). Emerging molecules inhibiting Wnt/β-catenin signaling have provided promising preclinical outcome against esophageal cancer (Figure 1, Table 1). For example, Icaritin, an alkaloid extracted from Herba epimedii, was found to reduce the growth of CSCs derived from the OSCC cell line ECA109 by inhibiting Wnt/β-catenin and Hedgehog pathway (45). Icaritin inhibited proliferation, migration, and invasion of CD133+ esophageal CSCs in a dose-dependent manner and enhanced the apoptosis of these stem cells. In addition, Icaritin induced up-regulation of GSK3β and down-regulation of Wnt and β-catenin, Hedgehog, Smo, and Gli proteins in Wnt/β-catenin and Hedgehog pathways, respectively (45).
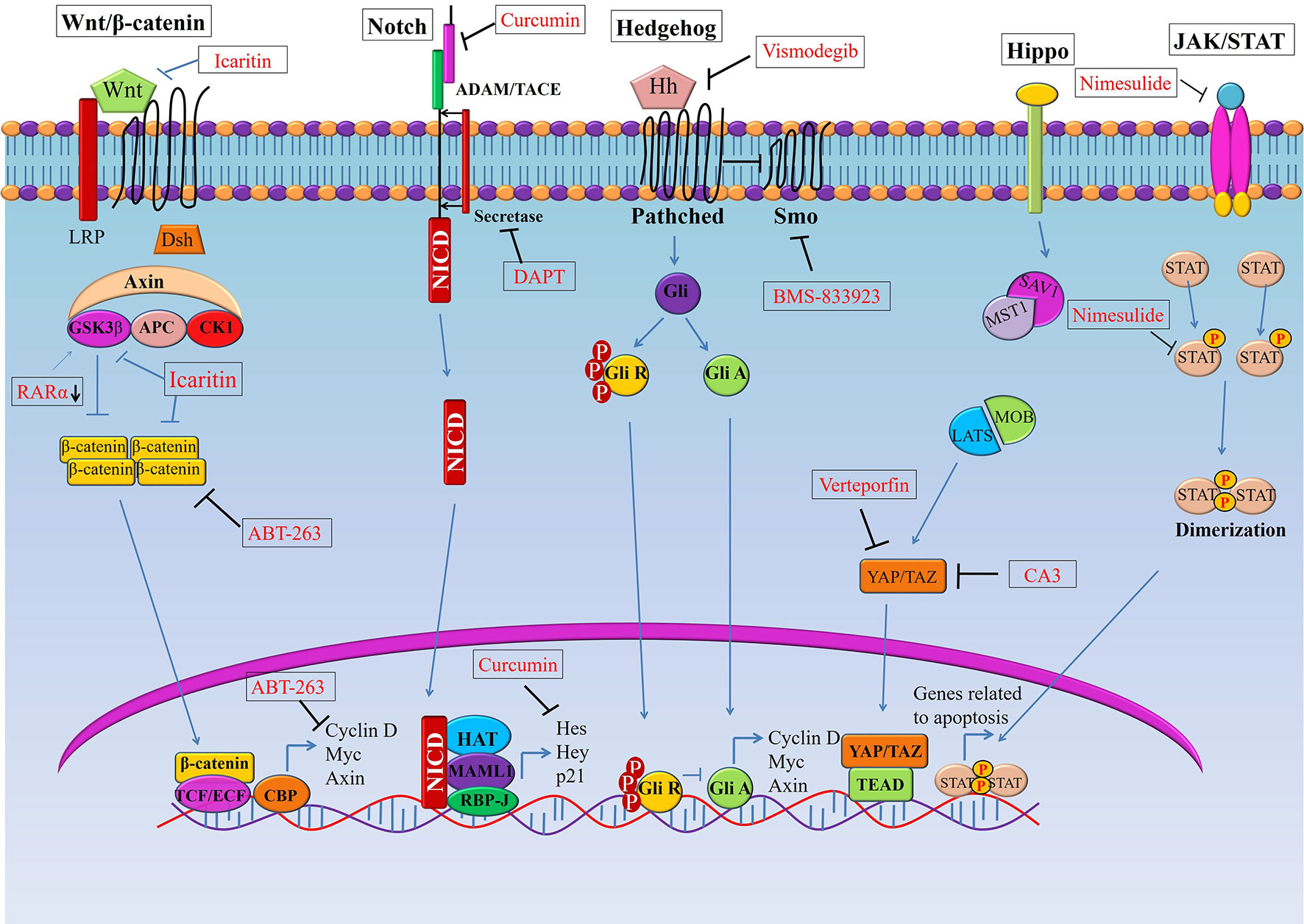
Figure 1 Targeting signaling pathways in esophageal cancer stem cells (CSCs). Schematic representation of the Wnt, Notch, Hedgehog (Hh), and Hippo pathways in esophageal CSCs. Novel therapeutics (synthetic and natural) kill CSCs by targeting these signalling pathways or their components.GSK3β, Glycogen synthase kinase 3 beta; Dsh, Disheveled; APC, Adenomatous polyposis coli; CK1, Casein kinase 1; TCF, T-cell factor/lymphoid enhancer factor; SMO, Smoothened; YAP, (Yes-associated protein); TAZ, Transcriptional coactivator with PDZ-binding motif.
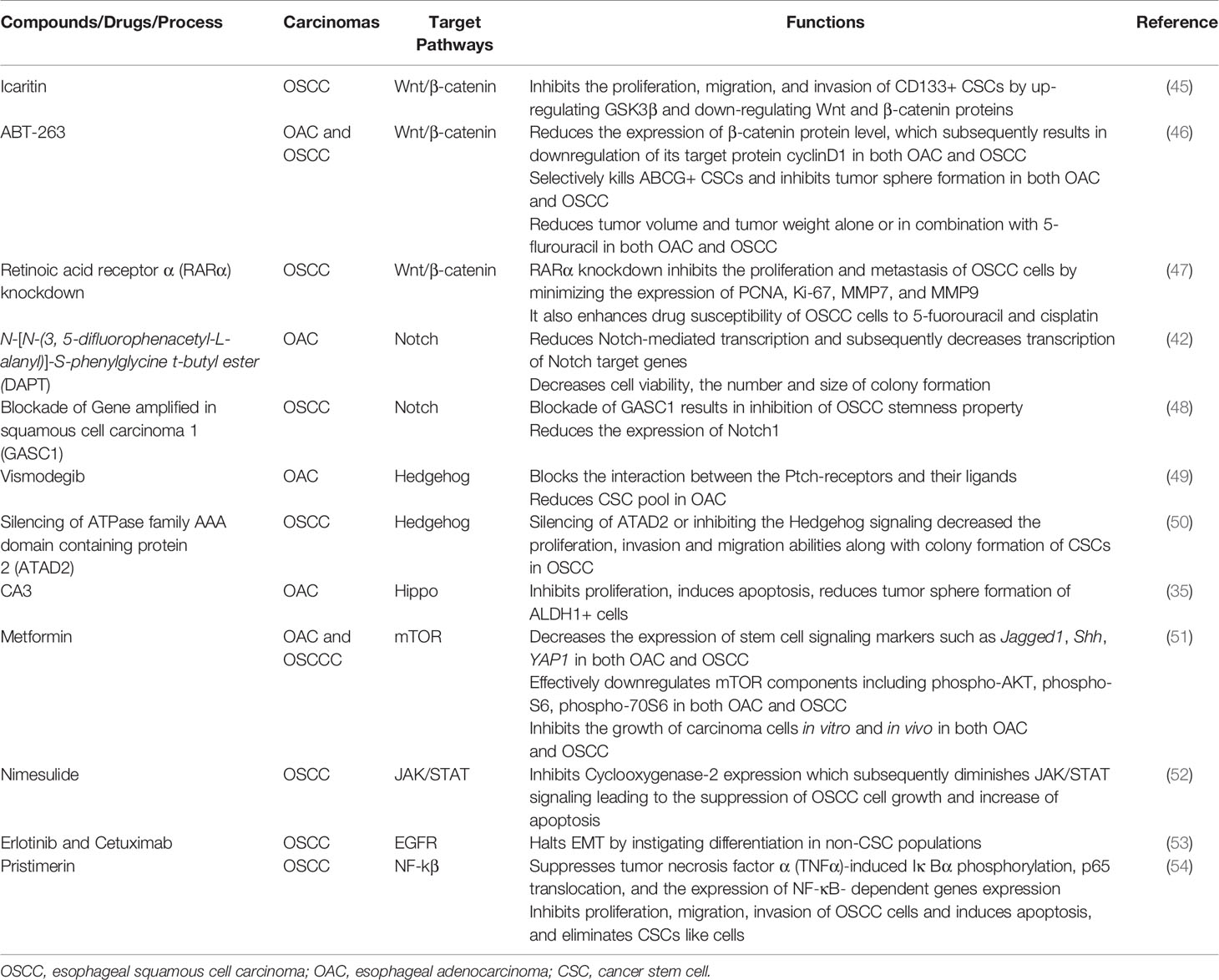
Table 1 Targeting signaling pathways in esophageal cancer stem cells.
ABT-263, a potent Bcl-2 family inhibitor inhibits cell proliferation and induces apoptosis of human esophageal cells, especially CSCs derived from OAC cell lines (FLO-1, SKGT-4, BE3 and OE33) and OSCC cell lines (YES-6 and KATO-TN) (46, 55). ABT263 reduces the expression of many oncogenes, including genes associated with stemness pathways such as Wnt and YAP/SOX9 axes. Treatment of esophageal CSCs with ABT-263 alone and in combination with 5-FUresulted in the reduction of β-catenin and its target cyclinD1, as well asYAP-1 and its target SOX9 in a dose-dependent manner (46). In addition, ABT-263 selectively kills ABCG+ CSCs and inhibits tumor sphere formation of esophageal CSCs (both OSCC and OAC). Also, ABT-263 alone or in combination with 5-fluorouracil reduced tumor volume and tumor weight in a xenograft model. These treatments dramatically reduced the level of YAP1, SOX9 and the proliferation marker Ki-67 in xenotransplanted tumors of both OSCC and OAC cells (46).
Retinoic acids play a crucial role in embryogenesis, differentiation, and tumorigenesis, which are controlled by retinoic acid receptors (RARs) and retinoid X receptors (RXRs) (56). RARα knockdown suppresses the proliferation and metastasis of OSCC cells by minimizing the expression of proliferative markers (PCNA, Ki-67) and matrix metallo-proteinases (MMP7 and MMP9) (47). Not only that, RARα knockdown also enhances drug susceptibility of OSCC cells to 5-fluorouracil and cisplatin (47). On top of that, RARα knockdown results in inhibition of Wnt/β-catenin pathway by decreasing GSK3βphosphorylation at Ser-9 and inducing phosphorylation at Tyr-216, which subsequently results in reduced expression of its downstream targets such as MMP7, MMP9, and P-glycoprotein. Therefore, targeting Wnt/β-catenin or their components to inhibit the pathway should be effective to halt the growth of CSCs in OSCC (47). Moreover, a few Wnt inhibitors such as PRI-724, LGK-974, Vantictumab and OMP-54F28 are in clinical trials as a single agent or in combination with conventional therapy for many solid cancers (57).
Targeting Notch Signaling in Esophageal Cancer Stem Cells
Notch signaling is highly activated in less differentiated tumors and drives CSC phenotypes and carcinogenesis in both OSCC and OAC (39, 42). This signaling helps to maintain a robust population of CSCs, thereby resulting in therapy resistance and cancer recurrence (38, 40). Notch inhibition depletes CSC populations in tumors and sensitizes cancer cells to chemotherapeutic agents, which leads to promising response toward neoadjuvant chemotherapy (NAC) in patients with both OSCC and OAC (Figure 1, Table 1). For example, blocking Notch pathway by DAPT (N-[N-(3, 5-difluorophenacetyl-L-alanyl)]-S-phenylglycine t-butyl ester), a commonly used gamma-secretase inhibitors (GSI), is effective in downsizing tumor growth of OAC. Efficacy of the treatment was shown by a dramatic reduction of the intracellular domain of the notch protein (NICD) in esophageal adenocarcinoma cells (OE33). There was also a reduction in Notch-mediated transcription and a subsequent decrease in the transcription of Notch target genes (42). Treatment of OAC cells with DAPT caused a decrease in cell viability, as well as reducing the number and size of colonies formed by OAC (OE33 and JH-EsoAd1) cells. The inhibition of the Notch pathway caused a significant reduction in transcription of several stem cell marker genes, including ALDH, CD24, LGR5, SOX2 and TWIST1. Furthermore, patient-derived xenograft models clearly demonstrated that inhibition of Notch signaling by gamma-secretase inhibitors is efficacious in downsizing tumor growth (42). Thus, inhibition of Notch signaling by DAPT could impair the stemness of OAC cells i.e. esophageal CSCs, resulting in reduced tumor growth in both in vitro and in vivo.
Gene amplified in squamous cell carcinoma 1 (GASC1), plays a critical role in maintaining self-renewal and differentiation potential of embryonic stem cells (48). GASC1 epigenetically controls the stemness of OSCC by regulation of Notch1. Examination of the expression of GASC1 in OSCC cells and tissues indicated that GASC1 expression is increased in poorly differentiated OSCC (48). Consistent with this observation, patients with OSCCs expressing GASC1 presented a significantly worse survival rate than those without. Most importantly, GASC1 expression in purified CSCs (ALDH+) cells was higher than that in non-CSCs (ALDH−) cells. Several stemness phenotypes of CSCs from OSCC were dramatically decreased after GASC1 blockade, which subsequently resulted in reduced Notch1 expression via demethylation of Notch1 promoters (H3K9me2 and H3K9me3). However, the impaired stemness property of CSCs from OSCC followed by GASC1 inhibition was reversed with exogenous Notch1 overexpression (48). This finding suggested that GASC1 promoted stemness in OSCC CSCs cells via Notch1 promoter demethylation (48). Therefore, the GASC1/Notch1 signaling axis could be a potential therapeutic target against CSCs of OSCC.
Targeting Hedgehog Signaling in Cancer Stem Cells of Esophageal Cancer
The Hedgehog (HH) signaling pathway plays a crucial role in growth and differentiation during embryonic development (58). However, abnormal activation of this pathway may also lead to cancer stemness along with stimulation of EMT, cancer metastasis and therapy resistance (59–61). Furthermore, activation of the Hedgehog pathway associated with distant metastases, advanced tumor stage in patients with esophageal cancers (both OSCC and OAC) (60, 62, 63). Although Hedgehog inhibitors have been extensively studied in clinical trials for different solid tumors, clinical trials on esophageal cancers are still limited (64). Vismodegib, also known as GDC-0449, is a small molecule inhibitor of Hedgehog signaling that blocks the interaction between the Ptch-receptors and their ligands (Figure 1, Table 1) (65). In addition, Vismodegib in combination with chemotherapy (FOLFOX) did not increase the survival of patients with gastroesophageal junction adenocarcinoma significantly (64).
Importantly, Vismodegib combined with neoadjuvant chemo-radiotherapy is under investigation in a clinical trial in Hedgehog activated OAC cells (49). Vismodegib treatment reduced the CSC pool derived in OSCC (OE21) and OAC (OE33) cells. Investigation of options for the suppression of the Hedgehog pathway may have additional importance, it has been suggested that neoadjuvant chemo-radiotherapy may activate the Hedgehog pathway, which in turn causes acquisition of more CSC features including the property of therapy resistance (49). For example, there is a subset of cancer cells with activated Hedgehog pathway prior to therapy that renders them able to survive chemo- and radiotherapy (66–69). By contrast, inhibiting the Hedgehog pathway resulted in a reduction of cells with CSC phenotype (CD44+/CD24−), inhibited sphere-forming capability and induced radio-sensitivity (70–72).
BMS-833923, an inhibitor of smoothened (SMO), another constituent of the Hedgehog pathway, combined with chemotherapy (FOLFOX) is currently under investigation in patients with metastatic esophageal carcinoma (73). SMO brings about the translocation of Gli protein into nucleus which results in the transcription of downstream target genes. Other SMO inhibitors such as Sonidegib and Taladegib are being explored currently against gastroesophageal adenocarcinomas (73, 74). In addition, activation of Hedgehog signaling could be inhibited by targeting transcription factor ATPase family AAA domain-containing protein 2 (ATAD2) (73). ATAD2, a member of the AAA + ATPase family, which is involved in various cancers by regulating cell proliferation, apoptosis, invasion and migration, and its overexpression is associated with poor prognosis of patients with cervical and gastric cancer (75, 76). High expression of ATAD2 has been identified in various types of tumors, including OSCC (75, 77). Interestingly, inhibition of ATAD2 resulted in subsequent inhibition of the Hedgehog signaling pathway, which was confirmed by reduced expression of Gli1, SMO, and Ptch11 in OSCC (50). On top of that, silencing of ATAD2 or inhibiting the Hedgehog signaling decreased the proliferation, invasion and migration abilities along with colony formation of CSCs in OSCC. Furthermore, increased apoptosis followed by the suppression of Hedgehog signaling was noted in CSCs derived from OSCC cells (50). Moreover, in vivo experiments in nude mice further validated the suppressive effect of siRNA mediated ATAD2 silencing on tumor growth (50). Thus, down-regulation of ATAD2 can certainly restrict the malignant phenotypes of OSCC cells through inhibition of the Hedgehog signaling pathway in CSCs derived from OSCC cells. These findings suggest that targeting the Hedgehog pathway via any of a number of mechanisms could be an effective approach to control CSCs in esophageal carcinomas.
Targeting Hippo Signaling of Esophageal Cells of Esophageal Cancer
The Hippo pathway has been implicated in the regulation of organ size, proliferation, and stem cell properties (78, 79). YAP1 plays a significant role in the maintenance of stemness of embryonic stem cells as well as contributing to the functions of CSCs (80–82).Therefore, deregulation of Hippo and activation of YAP1 in CSCs contributes many important properties of tumors, and thus, targeting YAP1 will be an effective strategy to target CSCs, thereby inhibiting tumor growth.
Several small-molecule inhibitors have been tested against the Hippo pathway in both OSCC and OAC cells (Figure 1, Table 1) (35, 80, 83–86). For example, a novel YAP inhibitor CA3 exhibited remarkable inhibitory activity on the transcriptional activity of YAP1/transcriptional enhanced associate domains (TEAD) (35). CA3 demonstrated strong inhibitory effects on the growth of OAC, especially on YAP1 overexpressing cancer cells both in vitro and in vivo (35). Most importantly, radio-resistant CSCs with aggressive phenotypes can be effectively suppressed by CA3 treatment. CA3inhibited proliferation, induced apoptosis and reduced tumor sphere formation of CSC (ALDH1+) cells derived from OSCC (35). Furthermore, CA3 in combination with 5-FU inhibited the growth of esophageal adenocarcinoma, especially in YAP1 overexpressing cancer cells (35). Taken together, these findings suggested that CA3 represents a new inhibitor of YAP1 and primarily targets YAP1 overexpressing and therapy-resistant CSCs generated from OAC.
Additionally, YAP1activity correlated with SOX9 expression in esophageal adenocarcinoma (35). SOX9 was found to be highly upregulated in various premalignant lesions and in tumor tissues and plays crucial roles in tumor development (83–85). The co-activator of Hippo pathway (YAP1) acts as a major determinant of CSC properties in non-transformed cells and as well as in OAC cells which directly upregulates the expression SOX9 (80). YAP1 regulates the transcription of SOX9 through a conserved TEAD binding site in the SOX9 promoter region. Exogenous expression of YAP1 or inhibition of its upstream negative regulators in vivo caused an increased SOX9 expression, which subsequently results in the acquisition of CSCs properties (80). On the other hand, shRNA-mediated knockdown of YAP1 or SOX9 in transformed cells inhibited CSC phenotypes in vitro and tumorigenicity in vivo (80). Furthermore, Verteporfin (VP), a small-molecule inhibitor of YAP1, significantly blocks CSCs (ALDH+ cells) properties in OAC cells overexpressingYAP1 (80). Thus, in the acquisition of CSC propertiesYAP1 driven SOX9 expression is critical, indicating that YAP1 inhibition might be an attractive option in targeting CSC population in esophageal cancer. For example, overexpression of YAP1 was positively associated with CDK6expression in radiation-resistant esophageal cancer tissues (both in OAC and OSCC) (86). CDK6 is a key regulator of the cell cycle. Induction of YAP1 expression in esophageal cancer cells up-regulated CDK6 expression, increased transcription, and consequently induced the resistance against radiotherapy (86). By blocking YAP1 and CDK6 with the YAP1 inhibitor CA3, and the CDK6 inhibitor LEE001 significantly suppressed esophageal cancer cell growth and CSC properties, particularly in radiation-resistant cells in both OAC and OSCC (86). The combination of LEE001 and CA3 exhibited the highest anti-tumor effects in radiation-resistant cells overexpressing YAP1 and CDK6 in both in vitro and in vivo by sensitizing resistant tumors to irradiation (86). Thus, it was implied that crosstalk between YAP1 and CDK6 seems to play a pivotal role in conferring radiation resistance and targeting both YAP1 and CDK6 could be a useful therapeutic strategy to treat both esophageal adenocarcinoma and squamous cell carcinoma.
Targeting Other Pathways in Esophageal Cancer Stem Cells
The pathways discussed above may act alone or in crosstalk with other pathways to induce stem cell properties in cancer cells or can even participate in driving therapy resistance upon interacting with other pathways (51). For example, the mTOR pathway is often activated in cancers and may generate therapy resistance followed by Hedgehog pathway inhibition (87, 88). The mTOR pathway along with Hedgehog and other pathways are associated with the maintenance of CSC phenotypes (89–93). Thus, interrupting mTOR with novel therapeutic could induce a reduction of stemness of cancer cells and sensitize them to the therapies. Metformin, an anti-diabetic agent, for instance, was found to significantly inhibit cell growth in both OSCC and OAC cells and sensitized them to 5-FU by targeting the mTOR signaling pathway in CSCs (80, 87–91). It increased the effectiveness of 5-FU against both OSCC and OAC cells and inhibited their growth in vitro and in a xenograft nude mouse model (51). Significant downregulation of mTOR pathway components including phospho-AKT, phospho-S6, phospho-70S6 was seen followed by metformin treatment, which are crucial to maintaining tumor cells’ growth. Furthermore, metformin treatment strongly decreased the expression of stem cell markers such as Jagged1, Shh, and YAP1 (51). Therefore, metformin-induced cell growth inhibition in vitro and in vivo in both OSCC and OAC cells by its ability to reduce the CSCs population as well as inhibition of the mTOR pathway. Furthermore, the synergistic effect of metformin with 5-FU was particularly of interest, because it would potentially provide an opportunity to treat both the CSCs and proliferating cell component at the same time, to effectively increase the sensitivity of chemo-radiation in patients with OSCC and OAC.
The JAK/STAT signaling pathway has been implicated in various physiological processes, and inhibition of this pathway could impede cancer cell growth and induce apoptosis in various cancers (94–96). Cyclooxygenase-2 (COX-2) together with JAK/STAT signaling has been found to be involved tumorigenesis. Specifically, the tumorigenesis pathway is associated with COX-2 upregulation (97, 98). Inhibition of COX-2 with nimesulide, a selective COX-2 inhibitor, results in suppression of the JAK/STAT signaling pathway, which subsequently inhibits the growth of Eca-109 human OSCC cells (52). Nimesulide induced apoptosis in Eca-109 cells by decreasing the expression of COX-2 and survivin and increasing caspase-3 expression (98). Also, nimesulide inhibited the JAK/STAT pathway by downregulating the phosphorylation of JAK2 and STAT3 (52). Inhibition of in vivo tumor growth of Eca-109 in xenotransplanted animals followed by a reduction inexpression of p-JAK2 and p-STAT3 were noted in Nimesulide treatment (52). Though Nimesulide could be used to inhibit JAK/STAT signaling pathway in OSCC cells, its effects on CSCs is yet to be evaluated. Thus, further studies are warranted to explore the effect of inhibition of JAK/STAT pathway in CSCs in esophageal cancers.
Epidermal growth factor receptors (EGFRs), a family of receptor kinases, are expressed in various cancers and contribute to a complex signaling cascade, which in turn controls growth, differentiation, adhesion, migration and survival of CSC and non-CSC cancer cells (53, 99). The wide range of roles for EGFRs in cancer progression makes them an attractive candidate for anti-cancer therapy. EGFRs are overexpressed in OSCC and play pivotal roles in the generation of stem-like cells via TGF-β (53). They induce EMT in CD44 overexpressing CSC cells derived from OSCC cells (53). CSCs (CD44+/CD24-) were significantly enriched in EPC2T and OKF6T cells (transformed keratinocyte cell lines) overexpressing EGFR, which could induce EMT by TGF-β1 in CSCs derived fromEPC2T and OKF6T cells (53). Interestingly, Erlotinib and Cetuximab (two EGFR inhibitors) significantly inhibited the enrichment of CSCs via inhibition of TGF-β1 mediated EMT (Table 1). Also, treatment with EGFR inhibitors resulted in increased expression of CD24 in the non-CSC population (CD44-/CD24+cells), indicating that EGFR inhibition could prompt differentiation in non-CSC populations as CD24 is a marker of keratinocyte differentiation (53). These results suggest that inhibition of EGFR may halt EMT by instigating differentiation in non-CSC populations, thereby suppressing enrichment of CSCs via inhibition of EMT. However, these EGFR inhibitors do not affect pre-existing CSCs. By contrast, some EGFR inhibitors suppress Zinc finger E-box binding proteins (ZEBs) and induce differentiation of CSCs in OSCC (53). These findings suggested that EGFR inhibition might suppress the expression of ZEBs and induce differentiation in a wider variety of cancers, thereby blocking EMT-mediated enrichment of CSCs.
NF-kβ, another prominent pathway, regulates various biological processes including apoptosis, proliferation, immune response, cell invasion, and cancer stem‐like cells (CSCs) (100). The key proteins in the NF‐κB pathway (e.g., p50, p52, and Rel) were overexpressed in patients with OSCC (101). In addition, the aberrant activation of the NF‐κB signaling pathway is a significant predictor for prognosis and recurrence of OSCC, which makes it a potential target in the treatment of patients with OSCC (102). A natural quinonemethide triterpenoid compound has been isolated from traditional Chinese herbals known as pristimerin, potently inhibited the growth of OSCC xenograft in nude mice (Table 1) (54). Pristimerin demonstrated its anti‐OSCC effects through the inhibition of NF‐κB pathway by suppressing tumor necrosis factor α (TNFα)‐induced Iκ Bα phosphorylation, p65 translocation, and the expression of NF‐κB‐ dependent genes (e.g., p50, p52, and Rel).Furthermore, pristimerin inhibited cell proliferation, migration, invasion, induced apoptosis, and eliminated cancer stem-like cells (CSCs) derived from OSCC cells (54). In addition, pristimerin exhibited a synergistic effect on OSCC when combined with 5‐FU (54). These results imply that pristimerin could increase chemo-sensitivity by suppressing the therapy-resistant CSC cell population in OSCCs.
Targeting MicroRNA Expression in Esophageal Cancer Stem Cells
MicroRNAs (miRNAs/miRs) are a class of small noncoding RNAs approximately 19–25 nucleotides in length, which regulate post-transcriptional gene expression by binding with their target mRNA transcripts (103, 104). Depending on the roles of their target genes, miRNAs can act either as tumor suppressors or oncogenes (105, 106). They are strongly involved in the formation of CSCs by regulating post transcriptional gene expressions in various cancer types (107). Altered expression of particular cancer-associated miRNAs causes significant changes in the level of potential oncogenic and anti-oncogenic proteins, which suggests miRNAs as useful therapeutic targets in cancer (108). Thus, miRNA mediated changes in gene expression in cancer has become a subject undergoing intense research nowadays.
MicroRNAs could act as molecular markers of cancer stem-like cells in esophageal cancer. Thereby, novel therapeutic strategies targeting miRNAs in CSCs have the potential to eradicate CSCs population, resulting in the improved clinical outcomes for patients with esophageal squamous cell carcinoma or adenocarcinoma (Table 2, Figure 2) (109–111, 116–118). For example, miRNA-203 is downregulated in cancer stem-like cells (Side population generated from OSCC (EC9706) cells) and expression of miR-203 was inversely associated with the expression of stem cell self-renewal factor Bmi-1 (109). Comparison of expression ofBmi-1 between SP and non-SP cells revealed that Bmi-1 was highly expressed in SP cells and its expression was significantly diminished during the differentiation from SP to non-SP cells (109, 110, 118). Therefore, miR-203 and Bmi-1 appear to play important roles in the generation of cancer stem-like cells in OSCC. In addition, lentiviral mediated expression of miR-203 resulted in decreased colony formation ability of SP cells, which was associated with the resistance to chemotherapy and responsible for tumorigenesis in nude mice (109). Since miR-203 and Bmi-1 were inversely expressed in SP cells, Bmi-1 might be a direct target of miR-203, thus therapeutics targeting miR-203 or Bmi-1could have the potential to eradicate CSCs in OSCC.

Table 2 MicroRNAs associated with functions of esophageal cancer stem cells.
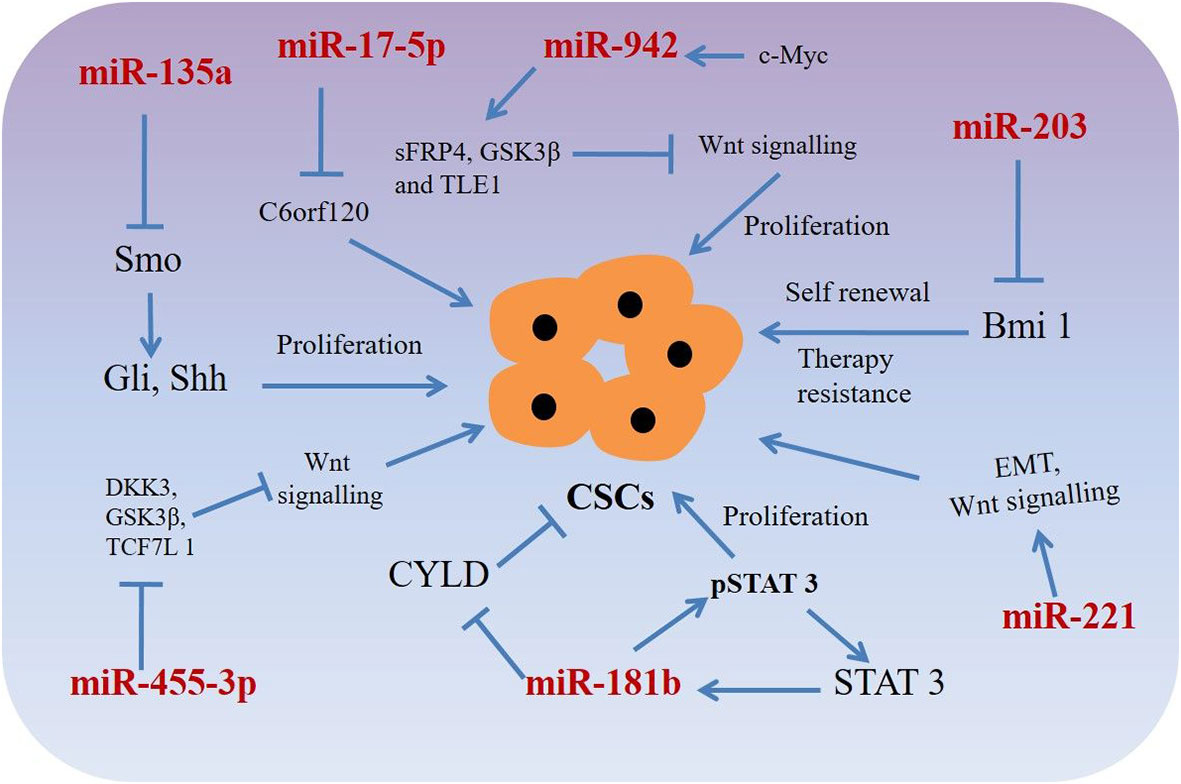
Figure 2 miRNAs targeting phenotypic markers in esophageal cancer stem cells (CSCs). miRNAs upregulate or downregulate the genes related to proliferation, sphere-formation, and therapy resistance.
Another miRNA, miR-181b in association with STAT3, plays a significant role in stem cell properties of esophageal squamous cell carcinoma stem-like cells (110). Isolating sphere-forming cells from OSCC cells (Eca109) exhibited proliferation and tumorigenicity characteristics of CSCs in a mouse xenograft model (110). The sphere-forming cells demonstrated cancer stem-like cell properties such as an enhanced population of CD44+/CD24- cells, increased stemness factors, mesenchymal marker expression, ATP-binding cassette (ABC) transporters and tumorigenicity in vivo when compared to that of parental cells (110). A mutual regulation between the signal transducer and activator transcription 3 (STAT3, a transcription factor) and miR-181b controls the sphere-forming cells’ proliferation and apoptosis resistance in esophageal cancer stem-like cells. STAT3 directly activated miR-181b transcription in a sphere-forming cells, which in turn potentiated p-STAT3 activity (110). Mechanistically, miR-181b binds with 3′-untranslated region (UTR) of cylindromatosis (CYLD) mRNA and regulates CYLD expression, which in turn regulates sphere-forming cells via modulating the STAT3/miR-181b loop in esophageal CSCs.
MicroRNAs such asmiR-135a may regulate biological behaviors of CSCs in OSCC through the Hedgehog signaling pathway by targeting its component SMO (111). Expressions of hedgehog pathway proteins such as SMO, Gli1, Shh, and Gli2 were happened to be increased and the expression of miR-135a was decreased in in esophageal CSCs of squamous cell carcinoma. However, exogenous overexpression of miR-135a or silencing of SMO decreased the expression of Gli1, Gli2, and Shh, resulting in reduced proliferation migration, invasion and increased apoptosis of CSCs derived from esophageal cancer cells (111). Interestingly, silencing of miR-135a was associated with increased carcinogenic capability of miR-135a in CSCs derived from OSCC (111). These results suggest that miR-135a mediated inhibition of CSCs derived from esophageal squamous cell carcinoma cells through suppression of the SMO/Hedgehog axis may act as a potential therapeutic option for patients with the carcinoma.
Another example of a miRNA promoting stem cell-like characteristics is miR-942, which in OSCCs causes activation of the Wnt/β-catenin signaling pathway (112). miR-942 was significantly upregulated in OSCC and was correlated with poor prognosis in patients with OSCC. Upregulation of miR-942 promoted cancer stem-like cell (CD90+ cells) traits in OSCC, whereas inhibition of miR-942 decreased tumor sphere formation and inhibited the expression of pluripotency-associated markers in the stem-like cells (112). Moreover, in vivo assays demonstrated thatmiR-942 overexpressing cells form larger tumors and display higher tumorigenesis capacity (112). miR-942 upregulates the Wnt/β-catenin signaling activity via directly targeting FRP4, GSK3β, and TLE1, which are prominent negative regulators of the Wnt/β-catenin signaling cascade (112). In addition, c-myc (a stem cell pluripotency-associated marker) directly binds to the miR-942 promoter and increased its expression, resulting in increased CSC mediated tumorigenesis (112). Considering the oncogenic role of miR-942 in OSCC, miR-942 might be an attractive therapeutic target for patients with OSCC.
Also, dysregulation of miR-455-3ppromoted chemoresistance and tumorigenesis of OSCC cells (113). Interestingly, treatment with a miR-455-3p antagomir significantly chemo-sensitized OSCC cells and decreased CD90+ and CD271 + cell populations (a CSC phenotype) through inhibition of various stemness-associated pathways including Wnt/β-catenin and TGF- β signaling (113). miR-455-3p targets several negative regulators e.g. DKK3, GSK3β, TCF7L 1, IGFBP4 etc. (Wnt/β-catenin pathway components) and Smurf2, NEDD4L, FKBP1A, BAMB I, etc. (TGF-β/Smad pathway components), resulting in inactivation of Wnt/β-catenin and TGF-β signaling in CSCs derived from OSCC cells (113). Association of miR-455-3p levels with chemoresistance and overall/relapse-free survival of patients with OSCC, indicating miR-455-3p antagonist could have potential as effective therapeutics for patients with OSCC. Another miRNA, miR-17 associated with the radio-resistant property of OAC cancer stem-like cells (114). An in vitro isogenic model using radio-resistantOE33 R cells derived from OE33 OAC cells demonstrated increased expression of CSC-associated markers and had enhanced tumorigenicity in vivo and increased holoclone forming capacity (114). Also, radio resistantOE33 R cells have increased ALDH activity. However, an in vitro study suggested that exogenous expression of miR-17-5p significantly sensitized radio-resistant cells to radiation therapy by repression of chromosome 6 open reading frame 120 (C6orf120) expression (114). This study sheds novel insights into the role of miR-17-5p as a potential prognostic biomarker in patients with esophageal adenocarcinomas.
Additionally, miR-221 is another miRNA upregulated in 5-FU resistant esophageal cancer cells (OAC) as well as in human OAC tissues (115). DKK2, a putative inhibitor of Wnt signaling was identified as a potential target for miR-221. Importantly, miR-221 knockdown in 5-FU resistant cells resulted in decreased cell proliferation, increased apoptosis, restored chemo-sensitivity, and led to inactivation of the stem cell pathway Wnt/β-catenin by activation of DKK2 activity (115). In addition, reduction of miR-221 expression resulted in alteration of EMT-associated genes e.g. E-cadherin and vimentin and slowed xenograft tumor growth in nude mice (115). Furthermore, a substantial dysregulation of Wnt/β-catenin signaling and chemoresistance target genes such as CDH1, CD44, MYC, and ABCG2 was reported as a result of miR-221 modulation in OAC (115). miR-221 may, therefore, could act as a prognostic marker and therapeutic target for patients with OAC.
Targeting Hypoxia-Related Pathways in Esophageal Cancer to Eliminate Cancer Stem Cells
Hypoxia, resulting from low oxygen concentration and nutrition deprivation, is a very common scenario in locally advanced solid tumors (119, 120). It regulates hypoxia-inducible factor (HIF) 1 and 2, which in turn can play critical roles in cancer metabolism, stem cell proliferation, maintaining aggressiveness and metastatic potential of both OSCC and OAC cells (Figure 3) (119, 120). Overexpression of HIFs also reduces radio-sensitivity (121, 122) and induces EMT in cancer cells (123, 124). On the other hand, inhibition of HIF1α resulted in suppression of tumorigenicity of OSCC cells in both in vitro and in vivo (125). At tissue levels, hypoxia and HIF1α are associated with therapy resistance and poor prognosis in patients with OSCC and OAC (126–129). Moreover, hypoxia regulates EMT and cancer stemness in various cancers by targeting Notch, Wnt/β-catenin, Hedgehog, PI3K/mTOR and unfolded protein response (UPR) pathways (130).
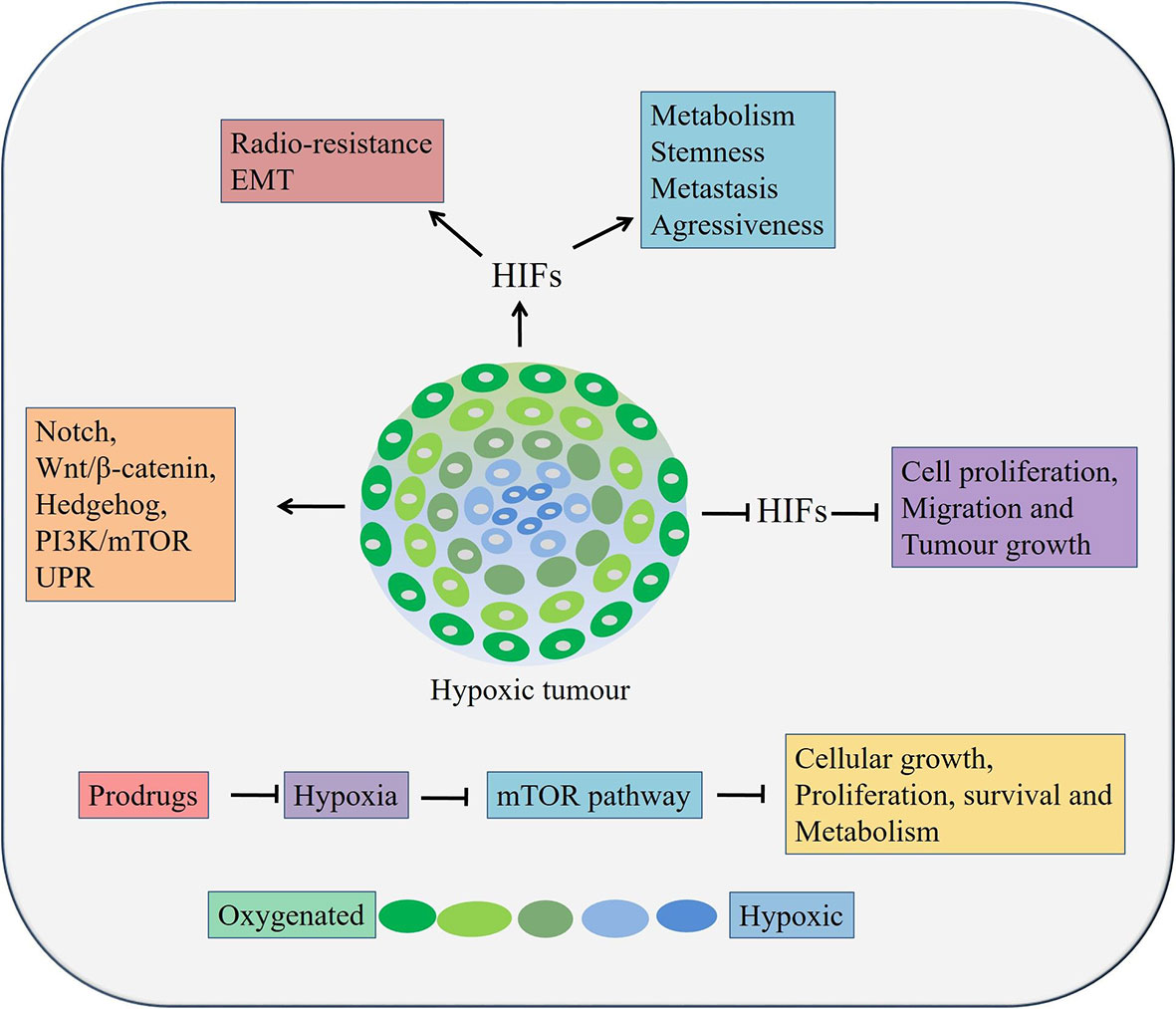
Figure 3 Role of hypoxia in esophageal cancers. Hypoxia can lead to cancer cell growth, metastasis, stemness, and therapy resistance through aberrant activation of pathways, inducing EMT processes etc. HIFs, Hypoxia inducible factors.
In esophageal cancer, a lower level of oxygen increases the CSC population, suggesting the need to target hypoxia in order to eradicate all tumor cells, especially the CSC population (131). It was reported that the expression of HIF‑1α and CSC‑related genes conditions were upregulated under hypoxic condition. A significant reduction of cell proliferation, migration and tumor growth was occurred followed by HIF‑1α knockdown in OSCC cells in vivo (131). In addition, knockdown of HIF‑1α also inhibited spheroid formation, inhibited expression of CSC‑related genes and Wnt/β‑catenin target genes, thereby decreased Wnt/β‑catenin activity CSCs derived from OSCC (131). Therefore, targeting hypoxia or its related factor and at the same time, inhibiting Wnt/β‑catenin might be an attractive option against patients with both OSCC and OAC. There are two main strategies targeting tumor hypoxia. Firstly, by applying bio-reductive prodrugs and secondly, inhibiting molecular targets associated with hypoxia using molecular inhibitors (132). A few prodrugs, for example, Tirapazamine, Apaziquone, TH-302, PR-104, Banoxantrone, and RH1 are effective in other solid cancers and are in clinical trials in minimizing tumor hypoxia (132). These prodrugs could be utilized against hypoxia in esophageal cancers. Interestingly, inhibition of the PI3K/mTOR pathway or a hypoxia may lead to activation of autophagy and could be used as an alternative therapeutic modality in esophageal cancers (130). The mTOR pathway negatively regulates autophagy in hypoxic conditions along with regulating cellular growth, proliferation, survival and metabolism (133). Thus, targeting the mTOR pathway mediated autophagy by Bafilomycin and Chloroquine could be useful against CSCs in both OAC and OSCC (73).
Finally, clinical trials targeting esophageal CSCs registered at https://clinicaltrials.gov/ were examined. To the best of our knowledge there is only a study using Fursultiamine, a nutrition supplement is undergoing a phase II clinical trial against OSCC patients in Taiwan in combination with concurrent chemo-radiation therapy to target CSCs (NCT02423811). Fursultiamine suppress OCT-4, SOX-2, NANOG expression and decreased ABCB1 and ABCG2 in tumor spheres. These findings encouraged the researchers to undertake a phase II trial to identify the effect of Fursultiamine combined with concurrent chemo-radiation therapy in ESCC patients. The outcome of the trial is not reported yet, however, they suggested that stem cell markers in clinical specimens collected before and after concurrent chemo-radiation therapy would be evaluated to identify whether Fursultiamine is effective against CSCs or not.
Concluding Remarks
Current conventional anticancer therapies are unable to eliminate CSCs. Therefore, relapse can occur, and CSCs can enable tumors to develop with further resistance to treatment and with more biological aggressiveness. In esophageal cancer, accumulating information has led to the hypothesis that the CSC population could be the seeds of carcinogenesis and are associated with therapy resistance and cancer recurrence. Thus, targeted therapy against CSCs could offer new options approaches to eliminate the malignant phenotypes of cancer without causing any harm to normal stem cells. In addition, careful analysis of a patient’s specific tumor may lead to a personalized approach, where both CSCs and the bulk tumor can potentially be eradicated. Eradicating both CSCs and bulk tumor should lead to a more promising outcome for patients with esophageal cancers. In some patients, conventional chemotherapy, surgical strategy along with targeted therapy will ultimately provide a more durable cure to this disease.
Author Contributions
PD drafted the manuscript. FI synthesized the concept and edited the manuscript. AL supervised the project and edited the manuscript. RS edited the concept and did the English proofreading. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin (2018) 68:394–424. doi: 10.3322/caac.21492
2. Global Burden of Disease Cancer Collaboration, Fitzmaurice C, Abate D, Abbasi N, Abbastabar H, Abd-Allah F, et al. Global, Regional, and National Cancer Incidence, Mortality, Years of Life Lost, Years Lived With Disability, and Disability-Adjusted Life-Years for 29 Cancer Groups, 1990 to 2017: A Systematic Analysis for the Global Burden of Disease Study. JAMA Oncol (2019) 5:1749–68. doi: 10.1001/jamaoncol.2019.2996
3. Codipilly DC, Qin Y, Dawsey SM, Kisiel J, Topazian M, Ahlquist D, et al. Screening for esophageal squamous cell carcinoma: recent advances. Gastrointest Endosc (2018) 88:413–26. doi: 10.1016/j.gie.2018.04.2352
4. Ajani JA, D’Amico TA, Almhanna K, Bentrem DJ, Besh S, Chao J, et al. National comprehensive cancer network. Esophageal and esophagogastric junction cancers, version 1.2015. J Natl Compr Canc Netw (2015) 13:194–227. doi: 10.6004/jnccn.2015.0028
5. van Hagen P, Hulshof MC, van Lanschot JJ, Steyerberg EW, van Berge Henegouwen MI, Wijnhoven BP, et al. Preoperative chemoradiotherapy for esophageal or junctional cancer. N Engl J Med (2012) 366:2074–84. doi: 10.1056/NEJMoa1112088
6. Das PK, Rakib MA, Khanam JA, Pillai S, Islam F. Novel therapeutics against breast cancer stem cells by targeting surface markers and signaling pathways. Curr Stem Cell Res Ther (2019) 14:669–82. doi: 10.2174/1574888X14666190628104721
7. Das PK, Islam F, Lam AK. The Roles of Cancer Stem Cells and Therapy Resistance in Colorectal Carcinoma. Cells (2020) 9:1392. doi: 10.3390/cells9061392
8. Islam F, Gopalan V, Lam AK. Identification of Cancer Stem Cells in Esophageal Adenocarcinoma. Methods Mol Biol (2018) 1756:165–76. doi: 10.1007/978-1-4939-7734-5_15
9. Islam F, Gopalan V, Smith RA, Lam AK. Translational potential of cancer stem cells: A review of the detection of cancer stem cells and their roles in cancer recurrence and cancer treatment. Exp Cell Res (2015) 335:135–47. doi: 10.1016/j.yexcr.2015.04.018
10. Islam F, Gopalan V, Wahab R, Smith RA, Lam AK. Cancer stem cells in oesophageal squamous cell carcinoma: Identification, prognostic and treatment perspectives. Crit Rev Oncol Hematol (2015) 96:9–19. doi: 10.1016/j.critrevonc.2015.04.007
11. Hoffmeyer K, Raggioli A, Rudloff S, Anton R, Hierholzer A, Del Valle I, et al. Wnt/β-catenin signaling regulates telomerase in stem cells and cancer cells. Science (2012) 336:1549–54. doi: 10.1126/science.1218370
12. Taniguchi H, Moriya C, Igarashi H, Saitoh A, Yamamoto H, Adachi Y, et al. Cancer stem cells in human gastrointestinal cancer. Cancer Sci (2016) 107:1556–62. doi: 10.1111/cas.13069
13. Wadhwa R, Wang X, Baladandayuthapani V, Liu B, Shiozaki H, Shimodaira Y, et al. Nuclear expression of Gli-1 is predictive of pathologic complete response to chemoradiation in trimodality treated oesophageal cancer patients. Br J Cancer (2017) 117:648–55. doi: 10.1038/bjc.2017.225
14. Wang Z, Chen J, Capobianco AJ. The Notch signaling pathway in esophageal adenocarcinoma. Cell Mol Biol (Noisy-le-grand) (2015) 61(6):24–32.
15. You Z, Xu D, Ji J, Guo W, Zhu W, He J. JAK/STAT signal pathway activation promotes progression and survival of human oesophageal squamous cell carcinoma. Clin Transl Oncol (2012) 14:143–9. doi: 10.1007/s12094-012-0774-6
16. Zanconato F, Cordenonsi M, Piccolo S. YAP/TAZ at the Roots of Cancer. Cancer Cell (2016) 29:783–803. doi: 10.1016/j.ccell.2016.05.005
17. He B, Yin B, Wang B, Xia Z, Chen C, Tang J. MicroRNAs in esophageal cancer (review). Mol Med Rep (2012) 6:459–65. doi: 10.3892/mmr.2012.975
18. Bao B, Azmi AS, Ali S, Ahmad A, Li Y, Banerjee S, et al. Thebiological kinship of hypoxia with CSC and EMT and their relationship withderegulated expression of miRNAs and tumor aggressiveness. Biochim Biophys Acta (2012) 1826:272–96. doi: 10.1016/j.bbcan.2012.04.008
19. Gu J, Wang Y, Wu X. MicroRNA in the pathogenesis and prognosis of esophageal cancer. Curr Pharm Des (2013) 19:1292–300. doi: 10.2174/138161213804805775
20. Pan Y, Ma S, Cao K, Zhou S, Zhao A, Li M, et al. Therapeuticapproaches targeting cancer stem cells. J Cancer Res Ther (2018) 14:1469–75. doi: 10.4103/jcrt.JCRT_976_17
21. Wang QE. DNA damage responses in cancer stem cells: Implications for cancer therapeutic strategies. World J Biol Chem (2015) 6:57–64. doi: 10.4331/wjbc.v6.i3.57
22. Das PK, Pillai S, Rakib MA, Khanam JA, Gopalan V, Lam AKY, et al. Plasticity of cancer stem cell: origin and role in disease progression and therapy resistance. Stem Cell Rev Rep (2020) 16:397–412. doi: 10.1007/s12015-019-09942-y
23. Singh AK, Arya RK, Maheshwari S, Singh A, Meena S, Pandey P, et al. Tumor heterogeneity and cancer stem cell paradigm: updates in concept,controversies and clinical relevance. Int J Cancer (2015) 136:1991–2000. doi: 10.1002/ijc.28804
24. Cohnheim J. Congenitales, quergestreiftesmuskelsarkomdernieren. Archivfürpathologische Anatomie und Physiologie und fürklinische Medicin (1875) 65:64–9. doi: 10.1007/BF01978936
25. Huntly BJ, Gilliland DG. Leukaemia stem cells and the evolution of cancer-stem-cell research. Nat Rev Cancer (2005) 5:311–21. doi: 10.1038/nrc1592
26. Islam F, Qiao B, Smith RA, Gopalan V, Lam AK. Cancer stem cell: fundamental experimental pathological concepts and updates. Exp Mol Pathol (2015) 98:184–91. doi: 10.1016/j.yexmp.2015.02.002
27. Rassouli FB, Matin MM, Saeinasab M. Cancer stem cells in human digestive tract malignancies. Tumour Biol (2016) 37:7–21. doi: 10.1007/s13277-015-4155-y
28. Harada K, Pool Pizzi M, Baba H, Shanbhag ND, Song S, Ajani JA. Cancer stem cells in esophageal cancer and response to therapy. Cancer (2018) 124:3962–4. doi: 10.1002/cncr.31697
29. Steinbichler TB, Dudás J, Skvortsov S, Ganswindt U, Riechelmann H, Skvortsova II. Therapy resistance mediated by cancer stem cells. Semin Cancer Biol (2018) 53:156–67. doi: 10.1016/j.semcancer.2018.11.006
30. Islam F, Gopalan V, Law S, Tang JC, Lam AK. FAM134B promotes esophageal squamous cell carcinoma in vitro and its correlations with clinicopathologic features. Hum Pathol (2019) 87:1–10. doi: 10.1016/j.humpath.2018.11.033
31. Islam F, Gopalan V, Lam AK. Detention and Identification of Cancer Stem Cells in Esophageal Squamous Cell Carcinoma. Methods Mol Biol (2020) 2129:177–91. doi: 10.1007/978-1-0716-0377-2_14
32. Lu C, Xu F, Gu J, Yuan Y, Zhao G, Yu X, et al. Clinical and biological significance of stem-like CD133(+)CXCR4(+) cells in esophageal squamous cell carcinoma. J Thorac Cardiovasc Surg (2015) 150:386–95. doi: 10.1016/j.jtcvs.2015.05.030
33. Wu Q, Wu Z, Bao C, Li W, He H, Sun Y, et al. Cancer stem cells in esophageal squamous cell cancer. Oncol Lett (2019) 18:5022–32. doi: 10.3892/ol.2019.10900
34. Zhao Y, Bao Q, Schwarz B, Zhao L, Mysliwietz J, Ellwart J, et al. Stem cell-like side populations in esophageal cancer: a source of chemotherapy resistance and metastases. Stem Cells Dev (2014) 23:180–92. doi: 10.1089/scd.2013.0103
35. Song S, Xie M, Scott AW, Jin J, Ma L, Dong X, et al. A Novel YAP1 Inhibitor Targets CSC-Enriched Radiation-Resistant Cells and Exerts Strong Antitumor Activity in Esophageal Adenocarcinoma. Mol Cancer Ther (2018) 17:443–54. doi: 10.1158/1535-7163.MCT-17-0560
36. Zhang X, Komaki R, Wang L, Fang B, Chang JY. Treatment of radioresistant stem-like esophageal cancer cells by an apoptotic gene-armed, telomerase-specific oncolytic adenovirus. Clin Cancer Res (2008) 14:2813–23. doi: 10.1158/1078-0432.CCR-07-1528
37. Hong L, Han Y, Zhang H, Li M, Gong T, Sun L, et al. The prognostic and chemotherapeutic value of miR-296 in esophageal squamous cell carcinoma. Ann Surg (2010) 251:1056–63. doi: 10.1097/SLA.0b013e3181dd4ea9
38. Hamano R, Miyata H, Yamasaki M, Kurokawa Y, Hara J, Moon JH, et al. Overexpression of miR-200c induces chemoresistance in esophageal cancers mediated through activation of the Aktsignaling pathway. Clin Cancer Res (2011) 17:3029–38. doi: 10.1158/1078-0432.CCR-10-2532
39. Wang D, Plukker JTM, Coppes RP. Cancer stem cells with increased metastatic potential as a therapeutic target for esophageal cancer. Semin Cancer Biol (2017) 44:60–6. doi: 10.1016/j.semcancer.2017.03.010
40. Zhou C, Fan N, Liu F, Fang N, Plum PS, Thieme R, et al. Linking Cancer Stem Cell Plasticity to Therapeutic Resistance-Mechanism and Novel Therapeutic Strategies in Esophageal Cancer. Cells (2020) 9:1481. doi: 10.3390/cells9061481
41. Long A, Giroux V, Whelan KA, Hamilton KE, Tétreault MP, Tanaka K, et al. WNT10A promotes an invasive and self-renewing phenotype in esophageal squamous cell carcinoma. Carcinogenesis (2015) 36:598–606. doi: 10.1093/carcin/bgv025
42. Wang Z, Da Silva TG, Jin K, Han X, Ranganathan P, Zhu X, et al. Notch signaling drives stemness and tumorigenicity of esophageal adenocarcinoma. Cancer Res (2014) 74:6364–74. doi: 10.1158/0008-5472.CAN-14-2051
43. Katoh M. Canonical and non-canonical WNT signaling in cancer stem cells and their niches: Cellular heterogeneity, omics reprogramming, targeted therapy and tumor plasticity (Review). Int J Oncol (2017) 5:1357–69. doi: 10.3892/ijo.2017.4129
44. Clevers H, Nusse R. Wnt/β-catenin signaling and disease. Cell (2012) 149:1192–205. doi: 10.1016/j.cell.2012.05.012
45. Han S, Gou Y, Jin D, Ma J, Chen M, Dong X. Effects of Icaritin on thephysiological activities of esophageal cancer stem cells. Biochem Biophys Res Commun (2018) 504:792–6. doi: 10.1016/j.bbrc.2018.08.060
46. Chen Q, Song S, Wei S, Liu B, Honjo S, Scott A, et al. ABT-263 induces apoptosis and synergizes with chemotherapy by targeting stemness pathways in esophageal cancer. Oncotarget (2015) 6:25883–96. doi: 10.18632/oncotarget.4540
47. Mao XM, Li H, Zhang XY, Zhou P, Fu QR, Chen QE, et al. Retinoic Acid Receptor α Knockdown Suppresses the Tumorigenicity of Esophageal Carcinoma via Wnt/β-catenin Pathway. Dig Dis Sci (2018) 63:3348–58. doi: 10.1007/s10620-018-5254-6
48. Jia R, Yang L, Yuan X, Kong J, Liu Y, Yin W, et al. GASC1 Promotes Stemness of Esophageal Squamous Cell Carcinoma via NOTCH1 Promoter Demethylation. J Oncol (2019) 2019:1621054. doi: 10.1155/2019/1621054
49. Lee SY, Jeong EK, Ju MK, Jeon HM, Kim MY, Kim CH, et al. Induction of metastasis, cancer stem cell phenotype, and oncogenic metabolism in cancer cells by ionizing radiation. Mol Cancer (2017) 16:10. doi: 10.1186/s12943-016-0577-4
50. Cattaneo M, Morozumi Y, Perazza D, Boussouar F, Jamshidikia M, Rousseaux S, et al. Lessons from yeast on emerging roles of the ATAD2 protein family in gene regulation and genome organization. Mol Cells (2014) 37:851–6. doi: 10.14348/molcells.2014.0258
51. Honjo S, Ajani JA, Scott AW, Chen Q, Skinner HD, Stroehlein J, et al. Metformin sensitizes chemotherapy by targeting cancer stem cells and the mTOR pathway in esophageal cancer. Int J Oncol (2014) 45:567–74. doi: 10.3892/ijo.2014.2450
52. Liu JR, Wu WJ, Liu SX, Zuo LF, Wang Y, Yang JZ, et al. Nimesulide inhibits the growth of human esophageal carcinoma cells by inactivating the JAK2/STAT3pathway. Pathol Res Pract (2015) 211(6):426–34. doi: 10.1016/j.prp.2015.01.007
53. Sato F, Kubota Y, Natsuizaka M, Maehara O, Hatanaka Y, Marukawa K, et al. EGFR inhibitors prevent induction of cancer stem-like cells in esophageal squamous cell carcinoma by suppressing epithelial-\mesenchymal transition. Cancer Biol Ther (2015) 16:933–40. doi: 10.1080/15384047.2015.1040959
54. Tu Y, Tan F, Zhou J, Pan J. Pristimerin targeting NF-κB pathway inhibitsproliferation, migration, and invasion in esophageal squamous cell carcinomacells. Cell Biochem Funct (2018) 36:228–40. doi: 10.1002/cbf.3335
55. Harb JG, Neviani P, Chyla BJ, Ellis JJ, Ferenchak GJ, Oaks JJ, et al. Bcl-xL anti-apoptotic network is dispensable for development and maintenance of CML but is required for disease progression where it represents a new therapeutic target. Leukemia (2013) 27:1996–2005. doi: 10.1038/leu.2013.151
56. Gronemeyer H, Gustafsson JA, Laudet V. Principles for modulation of the nuclear receptor superfamily. Nat Rev Drug Discovery (2004) 3:950–64. doi: 10.1038/nrd1551
57. Krishnamurthy N, Kurzrock R. Targeting the Wnt/beta-catenin pathway in cancer: Update on effectors and inhibitors. Cancer Treat Rev (2018) 62:50–60. doi: 10.1016/j.ctrv.2017.11.002
58. Briscoe J, Thérond PP. The mechanisms of Hedgehog signalling and its roles in development and disease. Nat Rev Mol Cell Biol (2013) 14:416–29. doi: 10.1038/nrm3598
59. Yu Z, Pestell TG, Lisanti MP, Pestell RG. Cancer stem cells. Int J Biochem Cell Biol (2012) 44:2144–51. doi: 10.1016/j.biocel.2012.08.022
60. Das PK, Zahan T, AbdurRakib M, Khanam JA, Pillai S, Islam F. Naturalcompounds targeting cancer stem cells: a promising resource for chemotherapy. Anticancer Agents Med Chem (2019) 19(15):1796–808. doi: 10.2174/1871520619666190704111714
61. Isohata N, Aoyagi K, Mabuchi T, Daiko H, Fukaya M, Ohta H, et al. Hedgehog and epithelial-mesenchymal transition signaling in normal and malignant epithelial cells of the esophagus. Int J Cancer (2009) 125:1212–21. doi: 10.1002/ijc.24400
62. Yang Z, Cui Y, Ni W, Kim S, Xuan Y. Gli1, a potential regulator of esophageal cancer stem cell, is identified as an independent adverse prognostic factor in esophageal squamous cell carcinoma. J Cancer Res Clin Oncol (2017) 143:243–54. doi: 10.1007/s00432-016-2273-6
63. Ma X, Sheng T, Zhang Y, Zhang X, He J, Huang S, et al. Hedgehog signaling is activated in subsets of esophageal cancers. Int J Cancer (2006) 118:139–48. doi: 10.1002/ijc.21295
65. Cohen DJ, Christos PJ, Sparano JA, Kindler HL, Catenacci DV, Bekaii-Saab TB, et al. A randomized phase II study of vismodegib (V), a hedgehog (HH) pathway inhibitor, combined with FOLFOX in patients (pts) with advanced gastric and gastroesophageal junction (GEJ) carcinoma: a New York Cancer Consortium led study (abstract). J Clin Oncol (2013) 31(suppl 4):(abstr 67). doi: 10.1200/jco.2013.31.4_suppl.67
66. Katoh Y, Katoh M. Hedgehog target genes: mechanisms of carcinogenesis induced by aberrant hedgehog signaling activation. Curr Mol Med (2009) 9:873–86. doi: 10.2174/156652409789105570
67. Du FY, Zhou QF, Sun WJ, Chen GL. Targeting cancer stem cells in drug discovery: Current state and future perspectives. World J Stem Cells (2019) 11:398–420. doi: 10.4252/wjsc.v11.i7.398
68. Takebe N, Miele L, Harris PJ, Jeong W, Bando H, Kahn M, et al. Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells:clinical update. Nat Rev Clin Oncol (2015) 12:445–64. doi: 10.1038/nrclinonc.2015.61
69. Sari IN, Phi LTH, Jun N, Wijaya YT, Lee S, Kwon HY. Hedgehog signaling in cancer: a prospective therapeutic target for eradicating cancer stem cells. Cells (2018) 7:208. doi: 10.3390/cells7110208
70. Pan S, Wu X, Jiang J, Gao W, Wan Y, Cheng D, et al. Discovery of NVP-LDE225, a potent and selective smoothened antagonist. ACS Med Chem Lett (2010) 1:130–4. doi: 10.1021/ml1000307
71. Dummer R, Guminski A, Gutzmer R, Dirix L, Lewis KD, Combemale P, et al. The 12-month analysis from basal cell carcinoma outcomes with LDE225 Treatment (BOLT): A phase II, randomized, double-blind study of sonidegib in patients with advanced basal cell carcinoma. J Am Acad Dermatol (2016) 75:113–125.e5. doi: 10.1016/j.jaad.2016.02.1226
72. D’Amato C, Rosa R, Marciano R, D’Amato V, Formisano L, Nappi L, et al. Inhibition of Hedgehog signalling by NVP-LDE225 (Erismodegib) interferes with growth and invasion of human renal cell carcinoma cells. Br J Cancer (2014) 111:1168–79. doi: 10.1038/bjc.2014.421
73. Rimkus TK, Carpenter RL, Qasem S, Chan M, Lo HW. Targeting the Sonic Hedgehog Signaling Pathway: Review of Smoothened and GLI Inhibitors. Cancers (Basel) (2016) 8:22. doi: 10.3390/cancers8020022
74. Li N, Yu Y, Wang B. Downregulation of AAA-domain-containing protein 2 restrains cancer stem cell properties in esophageal squamous cell carcinoma via blockade of the Hedgehog signaling pathway. Am J Physiol Cell Physiol (2020) 319:C93–C104. doi: 10.1152/ajpcell.00133.2019
75. Zheng L, Li T, Zhang Y, Guo Y, Yao J, Dou L, et al. Oncogene ATAD2 promotes cell proliferation, invasion and migration in cervical cancer. Oncol Rep (2015) 33:2337–44. doi: 10.3892/or.2015.3867
76. Zhang M, Zhang C, Du W, Yang X, Chen Z. ATAD2 is overexpressed in gastric cancer and serves as an independent poor prognostic biomarker. Clin Transl Oncol (2016) 18:776–81. doi: 10.1007/s12094-015-1430-8
77. Wu G, Lu X, Wang Y, He H, Meng X, Xia S, et al. Epigenetic high regulation of ATAD2 regulates the Hh pathway in human hepatocellular carcinoma. Int J Oncol (2014) 45:351–61. doi: 10.3892/ijo.2014.2416
78. Tumaneng K, Russell RC, Guan KL. Organ size control by Hippo and TOR pathways. Curr Biol (2012) 22:R368–379. doi: 10.1016/j.cub.2012.03.003
79. Tumaneng K, Schlegelmilch K, Russell RC, Yimlamai D, Basnet H, Mahadevan N, et al. YAP mediates crosstalk between the Hippo and PI(3)K–TOR pathways by suppressing PTEN via miR-29. Nat Cell Biol (2012) 14:1322–9. doi: 10.1038/ncb2615
80. Song S, Ajani JA, Honjo S, Maru DM, Chen Q, Scott AW, et al. Hippo coactivator YAP1 upregulates SOX9 and endows esophageal cancer cells with stem-like properties. Cancer Res (2014) 74:4170–82. doi: 10.1158/0008-5472.CAN-13-3569
81. Gregorieff A, Liu Y, Inanlou MR, Khomchuk Y, Wrana JL. Yap-dependent reprogramming of Lgr5(+) stem cells drives intestinal regeneration and cancer. Nature (2015) 526:715–8. doi: 10.1038/nature15382
82. Bora-Singhal N, Nguyen J, Schaal C, Perumal D, Singh S, Coppola D, et al. YAP1 regulates OCT4 activity and SOX2 expression to facilitate self-renewal and vascular mimicry of stem-like cells. Stem Cells (2015) 33:1705–18. doi: 10.1002/stem.1993
83. Aguilar-Medina M, Avendaño-Félix M, Lizárraga-Verdugo E, Bermúdez M, Romero-Quintana JG, Ramos-Payan R, et al. SOX9 stem-cellfactor: clinical and functional relevance in cancer. J Oncol (2019) 2019:6754040. doi: 10.1155/2019/6754040
84. Thomsen MK, Ambroisine L, Wynn S, Cheah KS, Foster CS, Fisher G, et al. Transatlantic Prostate Group. SOX9 elevation in the prostate promotes proliferation and cooperates with PTEN loss to drive tumor formation. Cancer Res (2010) 70:979–87. doi: 10.1158/0008-5472.CAN-09-2370
85. Matheu A, Collado M, Wise C, Manterola L, Cekaite L, Tye AJ, et al. Oncogenicity of the developmental transcription factor Sox9. Cancer Res (2012) 72:1301–15. doi: 10.1158/0008-5472.CAN-11-3660
86. Li F, Xu Y, Liu B, Singh PK, Zhao W, Jin J, et al. YAP1-Mediated CDK6 Activation ConfersRadiation Resistance in Esophageal Cancer-Rationale for the Combination of YAP1 and CDK4/6 Inhibitors in Esophageal Cancer. Clin Cancer Res (2019) 25:2264–77. doi: 10.1158/1078-0432.CCR-18-1029
87. Wang Y, Ding Q, Yen CJ, Xia W, Izzo JG, Lang JY, et al. The crosstalk of mTOR/S6K1 and Hedgehog pathways. Cancer Cell (2012) 21:374–87. doi: 10.1016/j.ccr.2011.12.028
88. Jiang JH, Pi J, Jin H, Cai JY. Oridonin-induced mitochondria-dependent apoptosis in esophageal cancer cells by inhibiting PI3K/AKT/mTOR and Ras/Raf pathways. J Cell Biochem (2019) 120(3):3736–46. doi: 10.1002/jcb.27654
89. Bao B, Wang Z, Ali S, Ahmad A, Azmi AS, Sarkar SH, et al. Metformin inhibits cell proliferation, migration and invasion by attenuating CSC function mediated by deregulating miRNAs in pancreatic cancer cells. Cancer Prev Res (Phila) (2012) 5:355–64. doi: 10.1158/1940-6207.CAPR-11-0299
90. Hirsch HA, Iliopoulos D, Tsichlis PN, Struhl K. Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer Res (2009) 69:7507–11. doi: 10.1158/0008-5472.CAN-09-2994
91. Iliopoulos D, Hirsch HA, Struhl K. Metformin decreases the dose of chemotherapy for prolonging tumor remission in mouse xenografts involvingmultiple cancer cell types. Cancer Res (2011) 71(9):3196–201. doi: 10.1158/0008-5472.CAN-10-3471
92. Bednar F, Simeone DM. Metformin and cancer stem cells: old drug, new targets. Cancer Prev Res (Phila) (2012) 5:351–4. doi: 10.1158/1940-6207.CAPR-12-0026
93. Vazquez-Martin A, Oliveras-Ferraros C, Cufí S, Del Barco S, Martin-Castillo B, Menendez JA. Metformin regulates breast cancer stem cell ontogeny by transcriptional regulation of the epithelial-mesenchymal transition (EMT) status. Cell Cycle (2010) 9:3807–14. doi: 10.4161/cc.9.18.13131
94. Buettner R, Mora LB, Jove R. Activated STAT signaling in human tumors provides novel molecular targets for therapeutic intervention. Clin Cancer Res (2002) 8(4):945–54.
95. Mora LB, Buettner R, Seigne J, Diaz J, Ahmad N, Garcia R, et al. Constitutive activation of Stat3 in human prostate tumors and cell lines: direct inhibition of Stat3 signaling induces apoptosis of prostate cancer cells. Cancer Res (2002) 62:6659–66.
96. Xiong H, Zhang ZG, Tian XQ, Sun DF, Liang QC, Zhang YJ, et al. Inhibition of JAK1, 2/STAT3 signaling induces apoptosis, cell cycle arrest, and reduces tumor cell invasion in colorectal cancer cells. Neoplasia (2008) 10:287–97. doi: 10.1593/neo.07971
97. Koon HW, Zhao D, Zhan Y, Rhee SH, Moyer MP, Pothoulakis C. Substance P stimulates cyclooxygenase-2 and prostaglandin E2 expression through JAK-STAT activation in human colonic epithelial cells. J Immunol (2006) 176:5050–9. doi: 10.4049/jimmunol.176.8.5050
98. Xuan YT, Guo Y, Zhu Y, Han H, Langenbach R, Dawn B, et al. Mechanism of cyclooxygenase-2 upregulation in late preconditioning. J Mol Cell Cardiol (2003) 35:525–37. doi: 10.1016/s0022-2828(03)00076-2
99. Mitchell RA, Luwor RB, Burgess AW. Epidermal growth factor receptor:structure-function informing the design of anticancer therapeutics. Exp Cell Res (2018) 371(1):1–19. doi: 10.1016/j.yexcr.2018.08.009
100. Soleimani A, Rahmani F, Ferns GA, Ryzhikov M, Avan A, Hassanian SM. Role of the NF-κB signaling pathway in the pathogenesis of colorectal cancer. Gene (2020) 726:144132. doi: 10.1016/j.gene.2019.144132
101. Kang MR, Kim MS, Kim SS, Ahn CH, Yoo NJ, Lee SH. NF-kappaB signalling proteins p50/p105, p52/p100, RelA, and IKKepsilon are over-expressed in oesophageal squamous cell carcinomas. Pathology (2009) 41:622–5. doi: 10.3109/00313020903257756
102. Peng Y, Zhou Y, Cheng L, Hu D, Zhou X, Wang Z, et al. The anti-esophageal cancer cell activity by a novel tyrosine/phosphoinositide kinase inhibitor PP121. Biochem Biophys Res Commun (2015) 465:137–44. doi: 10.1016/j.bbrc.2015.07.147
103. Yu SL, Chen HY, Chang GC, Chen CY, Chen HW, Singh S, et al. MicroRNA signature predicts survival and relapse in lung cancer. Cancer Cell (2008) 13:48–57. doi: 10.1016/j.ccr.2007.12.008
104. Das PK, Siddika MA, Asha SY, Aktar S, Rakib MA, Khanam JA, et al. MicroRNAs, a Promising Target for Breast Cancer Stem Cells. Mol Diagn Ther (2020) 24:69–83. doi: 10.1007/s40291-019-00439-5
105. Bertoli G, Cava C, Castiglioni I. MicroRNAs: New Biomarkers for Diagnosis, Prognosis, Therapy Prediction and Therapeutic Tools for Breast Cancer. Theranostics (2015) 5:1122–43. doi: 10.7150/thno.11543
106. Abdolvahabi Z, Nourbakhsh M, Hosseinkhani S, Hesari Z, Alipour M, Jafarzadeh M, et al. MicroRNA-590-3P suppresses cell survival and triggers breast cancer cell apoptosis via targeting sirtuin-1 and deacetylation of p53. J Cell Biochem (2018) 120:9356–68. doi: 10.1002/jcb.28211
107. Rahimi M, Sharifi-Zarchi A, Zarghami N, Geranpayeh L, Ebrahimi M, Alizadeh E. Down-Regulation of miR-200c and up-regulation of miR-30c target both stemness and metastasis genes in breast cancer. Cell J (2020) 21:467–78. doi: 10.22074/cellj.2020.6406
108. Carthew RW, Sontheimer EJ. Origins and mechanisms of miRNAs and siRNAs. Cell (2009) 136:642–55. doi: 10.1016/j.cell.2009.01.035
109. Yu X, Jiang X, Li H, Guo L, Jiang W, Lu SH. miR-203 inhibits the proliferation and self-renewal of esophageal cancer stem-like cells by suppressing stem renewal factor Bmi-1. Stem Cells Dev (2014) 23:576–85. doi: 10.1089/scd.2013.0308
110. Xu DD, Zhou PJ, Wang Y, Zhang L, Fu WY, Ruan BB, et al. Reciprocal activation between STAT3 and miR-181b regulates the proliferation of esophageal cancer stem-like cells via the CYLD pathway. Mol Cancer (2016) 15:40. doi: 10.1186/s12943-016-0521-7
111. Yang C, Zheng X, Ye K, Sun Y, Lu Y, Fan Q, et al. miR-135a inhibits the invasion and migration of esophageal cancer stem cells through the Hedgehog Signaling pathway by targeting Smo. Mol Ther Nucleic Acids (2020) 19:841–52. doi: 10.1016/j.omtn.2019.10.037
112. Ge C, Wu S, Wang W, Liu Z, Zhang J, Wang Z, et al. miR-942 promotes cancer stem cell-like traits in esophageal squamous cell carcinoma through activation of Wnt/β-catenin signalling pathway. Oncotarget (2015) 6:10964–77. doi: 10.18632/oncotarget.3696
113. Liu A, Zhu J, Wu G, Cao L, Tan Z, Zhang S, et al. Antagonizing miR-455-3p inhibits chemoresistance and aggressiveness in esophageal squamous cell carcinoma. Mol Cancer (2017) 16:106. doi: 10.1186/s12943-017-0669-9
114. Lynam-Lennon N, Heavey S, Sommerville G, Bibby BA, Ffrench B, Quinn J, et al. MicroRNA-17 is downregulated in esophageal adenocarcinoma cancer stem-like cells and promotes a radioresistant phenotype. Oncotarget (2017) 8:11400–13. doi: 10.18632/oncotarget.13940
115. Wang Y, Zhao Y, Herbst A, Kalinski T, Qin J, Wang X, et al. miR-221 Mediates Chemoresistance of Esophageal Adenocarcinoma by Direct Targeting of DKK2 Expression. Ann Surg (2016) 264:804–14. doi: 10.1097/SLA.0000000000001928
116. Sonkoly E, Wei T, Janson PC, Sääf A, Lundeberg L, Tengvall-Linder M, et al. MicroRNAs: novel regulators involved in the pathogenesis of psoriasis? PloS One (2007) 2:e610. doi: 10.1371/journal.pone.0000610
117. Feber A, Xi L, Luketich JD, Pennathur A, Landreneau RJ, Wu M, et al. MicroRNA expression profiles of esophageal cancer. J Thorac Cardiovasc Surg (2008) 135:255–60. doi: 10.1016/j.jtcvs.2007.08.055
118. Huang D, Gao Q, Guo L, Zhang C, Jiang W, Li H, et al. Isolation and identification of cancer stem-like cells in esophageal carcinoma cell lines. Stem Cells Dev (2009) 18:465–73. doi: 10.1089/scd.2008.0033
119. Carnero A, Lleonart M. The hypoxic microenvironment: A determinant of cancer stem cell evolution. Bioessays (2016) 38 Suppl 1:S65–74. doi: 10.1002/bies.201670911
120. Marie-Egyptienne DT, Lohse I, Hill RP. Cancer stem cells, the epithelial to mesenchymal transition (EMT) and radioresistance: potential role of hypoxia. Cancer Lett (2013) 341:63–72. doi: 10.1016/j.canlet.2012.11.019
121. Wang D, Qin Q, Jiang QJ, Wang DF. Bortezomib sensitizes esophageal squamous cancer cells to radiotherapy by suppressing the expression of HIF-1α and apoptosis proteins. J Xray Sci Technol (2016) 24:639–46. doi: 10.3233/XST-160571
122. Kato Y, Yashiro M, Fuyuhiro Y, Kashiwagi S, Matsuoka J, Hirakawa T, et al. Effects of acute and chronic hypoxia on the radiosensitivity of gastric and esophageal cancer cells. Anticancer Res (2011) 31:3369–75.
123. Wu X, Qiao B, Liu Q, Zhang W. Upregulation of extracellular matrix metalloproteinase inducer promotes hypoxia-induced epithelial-mesenchymal transition in esophageal cancer. Mol Med Rep (2015) 12:7419–24. doi: 10.3892/mmr.2015.4410
124. Jing SW, Wang YD, Kuroda M, Su JW, Sun GG, Liu Q, et al. HIF-1α contributes to hypoxia-induced invasion and metastasis of esophageal carcinoma via inhibiting E-cadherin and promoting MMP-2 expression. Acta Med Okayama (2012) 66:399–407. doi: 10.18926/AMO/48964
125. Zhu H, Feng Y, Zhang J, Zhou X, Hao B, Zhang G, et al. Inhibition of hypoxia inducible factor 1α expression suppresses the progression of esophageal squamous cell carcinoma. Cancer Biol Ther (2011) 11:981–7. doi: 10.4161/cbt.11.11.15707
126. Zhang HY, Wang ZQ, Li YY, Wang F, Zeng QR, Gao Y, et al. Transforming growth factor-β1-induced epithelial-mesenchymal transition in human esophageal squamous cell carcinoma via the PTEN/PI3K signaling pathway. Oncol Rep (2014) 32:2134–42. doi: 10.3892/or.2014.3453
127. Ping W, Sun W, Zu Y, Chen W, Fu X. Clinicopathological and prognostic significance of hypoxia-inducible factor-1α in esophageal squamous cell carcinoma: a meta-analysis. Tumour Biol (2014) 35:4401–9. doi: 10.1007/s13277-013-1579-0
128. Sohda M, Ishikawa H, Masuda N, Kato H, Miyazaki T, Nakajima M, et al. Pretreatment evaluation of combined HIF-1alpha, p53 and p21 expression is a useful and sensitive indicator of response to radiation and chemotherapy in esophageal cancer. Int J Cancer (2004) 110:838–44. doi: 10.1002/ijc.20215
129. Koukourakis MI, Giatromanolaki A, Skarlatos J, Corti L, Blandamura S, Piazza M, et al. Hypoxia inducible factor (HIF-1a and HIF-2a) expression in early esophageal cancer and response to photodynamic therapy and radiotherapy. Cancer Res (2001) 61:1830–2.
130. Wouters BG, Koritzinsky M. Hypoxia signalling through mTOR and the unfolded protein response in cancer. Nat Rev Cancer (2008) 8:851–64. doi: 10.1038/nrc2501
131. Lv Z, Liu RD, Chen XQ, Wang B, Li LF, Guo YS, et al. HIF−1α promotes the stemness of oesophageal squamous cell carcinoma by activating the Wnt/β−catenin pathway. Oncol Rep (2019) 42:726–34. doi: 10.3892/or.2019.7203
132. Wilson WR, Hay MP. Targeting hypoxia in cancer therapy. Nat Rev Cancer (2011) 11:393–410. doi: 10.1038/nrc3064
Keywords: esophageal cancer, esophageal cancer stem cells, cancer signaling, miRNAs, hypoxia, autophagy, therapeutic options
Citation: Das PK, Islam F, Smith RA and Lam AK (2021) Therapeutic Strategies Against Cancer Stem Cells in Esophageal Carcinomas. Front. Oncol. 10:598957. doi: 10.3389/fonc.2020.598957
Received: 26 August 2020; Accepted: 29 December 2020;
Published: 16 February 2021.
Edited by:
Hongjuan Cui, Southwest University, ChinaReviewed by:
Zhen Dong, Southwest University, ChinaShourong Wu, Chongqing University, China
Jun Mi, Shanghai Jiao Tong University, China
Copyright © 2021 Das, Islam, Smith and Lam. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Farhadul Islam, ZmFyaGFkX2JpbzgzQHJ1LmFjLmJk; Alfred K. Lam, YS5sYW1AZ3JpZmZpdGguZWR1LmF1