 David Bakhshinyan
David Bakhshinyan Neil Savage1
Neil Savage1 Sabra Khalid Salim
Sabra Khalid Salim Sheila K. Singh
Sheila K. Singh- 1Department of Biochemistry and Biomedical Sciences, Faculty of Health Sciences, McMaster University, Hamilton, ON, Canada
- 2Department of Surgery, Faculty of Health Sciences, McMaster University, Hamilton, ON, Canada
During embryonic development, radial glial precursor cells give rise to neural lineages, and a small proportion persist in the adult mammalian brain to contribute to long-term neuroplasticity. Neural stem cells (NSCs) reside in two neurogenic niches of the adult brain, the hippocampus and the subventricular zone (SVZ). NSCs in the SVZ are endowed with the defining stem cell properties of self-renewal and multipotent differentiation, which are maintained by intrinsic cellular programs, and extrinsic cellular and niche-specific interactions. In glioblastoma, the most aggressive primary malignant brain cancer, a subpopulation of cells termed glioblastoma stem cells (GSCs) exhibit similar stem-like properties. While there is an extensive overlap between NSCs and GSCs in function, distinct genetic profiles, transcriptional programs, and external environmental cues influence their divergent behavior. This review highlights the similarities and differences between GSCs and SVZ NSCs in terms of their gene expression, regulatory molecular pathways, niche organization, metabolic programs, and current therapies designed to exploit these differences.
Introduction
Glioblastoma [GBM, International Classification of Diseases for Oncology (ICD-O) code 9440/3] is the most common and aggressive primary CNS malignancy in adults. The short median survival of 9–18 months in patients with GBM has been attributed to the highly invasive nature of the disease with rapid cell infiltration, frequent relapses, and therapy resistance (1–5). Anatomically, GBMs arise predominantly in the cerebral cortex (40%), followed by temporal lobe (29%), the parietal lobe (14%), deeper brain structures (14%), and the occipital lobe (3%) (6). The current therapy for GBM, consisting of maximal surgical resection followed by radiation and temozolomide (TMZ), a cytotoxic chemotherapy (7), has yielded minimal survival benefit with a vast majority of GBM patients presenting with tumor recurrence. Recently, the addition of tumor-treating fields (TTFs) to the standard chemoradiotherapy regimen has extended survival of patients from 16 to 20.9 months (8).
Limited therapeutic options, poor survival, and the universally fatal nature of the disease have fueled research efforts to uncover novel molecular vulnerabilities within GBM. However, despite the best efforts, the discovery of novel effective treatments remains elusive. GBM tumors exhibit a large degree of intra- and inter-tumoral heterogeneity, which frequently renders majority of targeted therapies ineffective (9). In an attempt to deconvolute this heterogeneity, an increasing body of scientific work has been established to identify the cell-of-origin in GBM to shed light on the hierarchical organization of GBM tumors and identify vulnerabilities to target the tumor at its roots. Historically, two major candidate cells of origin of GBMs have been proposed, neural stem cells (NSCs) and oligodendrocyte precursor cells (OPCs). The supporting evidence and shortcomings of the two hypotheses have been recently reviewed in detail by Fan et al. (10). The initial identification of a subpopulation of GBM cells with multilineage potency, increased self-renewal ability, proliferation, and migration, termed glioma stem cells (GSCs) (11–13) has provided correlative evidence for the possibility of GBMs arising from transformed neural stem cells (NSCs). Through analysis of patient samples and genetically engineered mouse models of GBM, several studies have subsequently provided molecular evidence suggesting that GBM arises from migration of mutated, astrocyte-like NSCs from the subventricular zone (SVZ) (11–15). In this review, we describe the intrinsic and extrinsic regulations of SVZ NSCs and GSCs including molecular pathways, microenvironment, and metabolic activity to further evaluate how the differences can be exploited in the next generation of targeted therapies for GBM.
Subventricular Zone Neural Stem Cells in Adult Neurogenesis
Neural stem and progenitor cells are a specialized population of multipotent cells that contribute to lifelong neural plasticity. During embryonic neurogenesis, NSCs are spatiotemporally regulated to generate multiple neural populations including neurons and glial cells (16). Beyond development, a small pool of NSCs are maintained and become spatially restricted to two neurogenic niches in the brain; the dentate gyrus of the hippocampus known as the subgranular zone (SGZ), or the ventricular-subventricular zone (SVZ) (17). NSCs were long believed to be a retained pool of self-renewing stem cells as suggested by long-term expansion and retention of differentiation potential by neurosphere culturing (17). Much of our current understanding of the human SVZ has been derived from studies in other mammals, namely mice. While mice display robust SVZ neurogenesis, humans have shown an increased preponderance of SGZ or hippocampal neurogenesis (18). Nonetheless, studies in other mammals provide deep insight into comparable NSC regulation and differentiation, highlighting significant complexity and heterogeneity in the adult brain. The SVZ is the largest germinal center in the adult human brain found on the walls of the lateral ventricles (19). SVZ-NSCs, also known as B1 cells, are displaced and surrounded by bi- or multi-ciliated ependymal cells to form a pinwheel-like structure, in which the NSC apical surface contacts the cerebrospinal fluid (CSF) and some the ventricle, while the basal process terminates vascular vessels and the extravascular basal lamina (17, 20). The morphology of B1 cells is reminiscent of radial glia in the embryonic ventricular zone from which they are hypothesized to originate (20, 21). These NSCs, which express GFAP and CD133 at quiescence, can become activated, express Nestin and EGFR, and become highly proliferative (22). Activation of these NSCs ultimately gives rise to EGFR+ transient amplifying cells, which in turn differentiate into progenitors and finally, neuroblasts. These cells follow a specialized migratory route known as the rostral migratory stream to the olfactory bulb in which they disperse radially and differentiate into GABAergic interneurons, or form corpus callosum oligodendrocytes (23, 24). Purification and subsequent single-cell transcriptomics have revealed that SVZ-NSCs exhibit a phenotypic continuum between quiescence and activation suggesting a high degree of transcriptional dynamics (21, 25, 26). NSCs present a heterogeneous profile of multiple activation states in the adult SVZ niche regulated by various molecular programs affected by both intrinsic and extrinsic programs (21, 23).
Intrinsic Regulation
Adult NSC self-renewal and multipotency have been proposed to be regulated by various transcriptional factors. One such factor is the orphan nuclear receptor TLX, which has been shown to be an essential transcriptional regulator of NSC maintenance and proliferation in the adult brain (27). Transcriptional regulation has also been demonstrated to be controlled by arsenite-resistance protein 2, a critical activator of the Sox family of DNA binding proteins, particularly Sox2 (28). TLX has also been suggested to regulate NSC maintenance by repression of cell-cycle inhibitory factors and recruitment of a host of tumor suppressor genes including Bmi1 (29), p53 (30), and the PTEN pathway (31) which regulate stem cell maintenance (27). Adult SVZ NSCs have also been shown to be regulated by basic helix-loop-helix (bHLH) transcription factors, which inhibit differentiation and maintain stemness. BHLH genes, particularly of the Hes family have also been implicated as Notch signaling effectors, which inhibit neuronal differentiation, and maintain NSCs by inducing quiescence (17, 32, 33). Beyond transcriptional regulators, other nuclear receptors such as estrogen receptors (34), thyroid hormone receptors (35), and peroxisome proliferator activated receptor-gamma (36), have been shown to regulate NSC proliferation and differentiation (17, 32).
Cell-intrinsic regulation is also maintained through epigenetic modification and chromatin remodeling. Epigenetic control has been demonstrated to be regulated by the aforementioned polycomb repressor Bmi1 by methylation of the histone tail H3K27 to promote self-renewal (37). SVZ-NSC differentiation is alternatively regulated by methylation of the histone tail H3K4 by the TrxG family of proteins (38). The balance between self-renewal and differentiation is subsequently mediated by switches from a polycomb-repressor driven chromatin remodeling to that of the TrxG family (39, 40). Epigenetic regulation has also been shown to work through histone acetyltransferase (HATs) and deacetylases (HDACs) in NSCs (41). HDACs promote the silencing of key neurogenic transcription and cell-cycle factors in a comparatively more dynamic fashion relative to the polycomb family of epigenetic regulators to tightly regulate fate specification, differentiation, and cell-cycle exit (32, 37).
Epigenetic mechanisms in SVZ-NSCs are also regulated by a network of miRNAs and non-coding RNAs, which play an additional regulatory role in adult neurogenesis. Many members of the small RNA family have been implicated in modulating neuronal differentiation by binding to the RE1-silencing transcription factor (REST), a crucial regulator of neuronal gene expression (42). Together, small non-coding RNAs fine tune epigenetic programs to regulate cell states of SVZ-NSCs.
Extrinsic Regulation
NSC intrinsic programs are also regulated by signals from the neurogenic niche (37, 43). The NSC niche is an extensive microenvironment that hosts cell-cell and cell-microenvironment interactions (44). Here, NSC proliferation and fate determination is facilitated by various cell-extrinsic molecular signals.
The extracellular matrix is a critical component of the SVZ niche that has been identified as a regulator of NSC proliferation (Figure 1A). It is composed of vessel basal lamina rich in laminin, collagen-I, and other molecules including metalloproteinases, brevican, tenascin-C, growth factors, and proteoglycans (44, 45). Unlike embryonic development, the adult SVZ also consists of a unique extravascular component consisting of ECM aggregates near the ventricular surface known as fractones (46). Fractones play an important role in facilitating the binding of growth-factors, cytokines, and chemokines from the circulating CSF to fractone-associated heparan sulfate proteoglycans (HSPGs) (47). Fractones then present these molecules to their cognate receptors on NSCs. Some of these molecules include fibroblast growth factor-2 (FGF-2) (48) and bone morphogenetic proteins (BMP) to influence NSC proliferation (49).
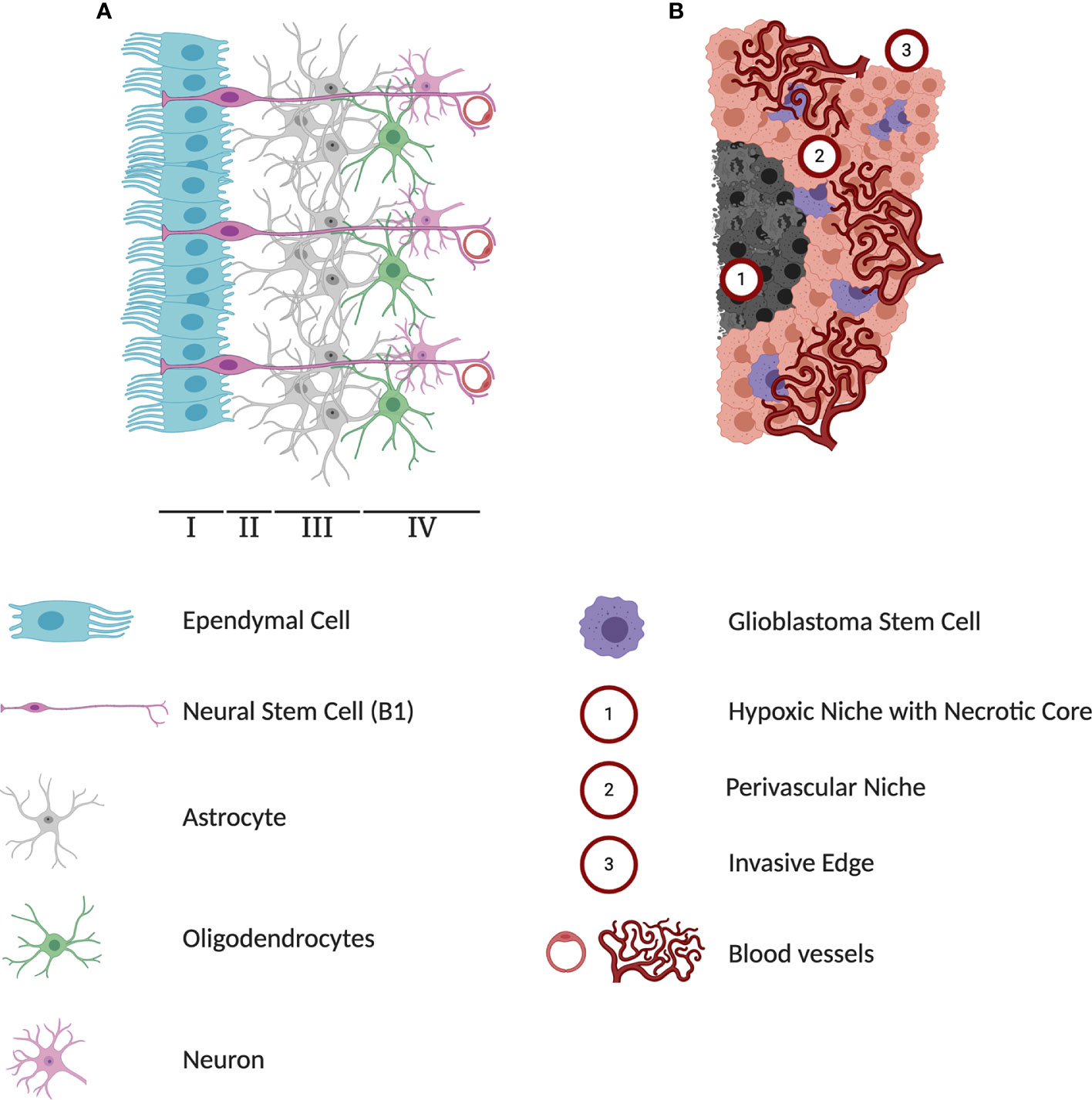
Figure 1 Differences in cytoarchitectures surrounding subventricular zone (SVZ) neural stem cells (NSCs) and glioblastoma stem cells (GSCs). (A) A schematic representation of adult human SVZ. Human SVZ is composed of four distinct layers. The superficial ependymal monolayer, Lamina I, is in contact with ventricular lumen. The second layer, Lamina II is a vastly acellular layer formed by the neuroblast depletion, containing ependymal expansions and numerous astrocyte processes. Lamina III, is a region known as astrocytic ribbon, containing densely packed astrocytes. Lastly, Lamina IV is a transitional zone rich in oligodendrocytes and myelinated neurons. The NSC niche is an extensive microenvironment that hosts cell-cell and cell-microenvironment interactions that contribute extensively to the extrinsic regulation of NSC proliferation and self-renewal. (B) A schematic representation of three major microenvironments within glioblastoma (GBM) tumors. The hypoxia region formed in the course of tumor growth lacks any blood and oxygen supply and has been implicated in playing a protective role against chemoradiotherapy. Perivascular niches exist along capillaries or arterioles where endothelial cells come into direct contact with glioblastoma cells. In addition to producing high levels of pro-angiogenic factors driving tumor vascularization, cells in the perivascular contribute to activation of pathways regulating self-renewal and proliferation of GSCs. As a highly invasive tumor, GBMs can infiltrate into healthy brain tissue and limit the effectiveness of surgical interventions.
Molecular signals in the ECM regulating adult NSC activity can originate from multiple cell types. In assessing the cellular composition of the SVZ microenvironment, endothelial, pericyte and vascular cells, as well as immature and mature lineages of NSCs are found (50, 51). Most significant are the ependymal cells which uniquely line the ventricular surface in a pinwheel formation around single NSCs (20, 52). Ependymal cells secrete local signaling factors into the circulating CSF, which include noggin, a BMP signaling inhibitor, to activate adult human NSCs and promote fate commitment (53). Endothelial cells in the SVZ also secrete factors including vascular endothelial growth factor (VEGF) and neurotrophin-3 which promote self-renewal and quiescence, respectively (54). NSCs and their immediate progeny also self-regulate through autocrine and paracrine mechanisms (55, 56). This regulation is particularly controlled by diffusible factors transmitted through gap junctions such as the neurotransmitter GABA which modulates quiescence (57), and cytokines such as IL-1β and IL-6 which promote NSC differentiation (58). Cell-cell interactions of adult NSCs are also observed in the SVZ microenvironment through signaling molecules such as ephrin B2 and Jagged1 on endothelial cells which promote quiescence and cell cycle suppression (59). Direct cell-cell interactions have also been demonstrated, such as that with endothelial cells through α6β1 integrins which modulate NSC proliferation (60). Thus, multiple cell types contribute to the signaling milieu of the SVZ niche.
Extrinsic factors that affect adult NSC regulation can also be derived from the cerebrospinal fluid (CSF) and blood-derived systemic signals. NSCs have direct access to the CSF which provides a rich supply of various additional mitogens such as PDGF, and morphogens including the Wnt ligands which particularly promote proliferation and self-renewal of adult NSCs via canonical Wnt signaling (61). Peripheral circulating morphogens have also been implicated in modulating mouse NSC behavior such as GDF11 which induces vascular remodeling leading to NSC proliferation (62, 63).
NSC multipotency and stemness is also modified by endogenous and niche-derived metabolic factors. Adult NSCs are known to rely on aerobic glycolysis prior to differentiation. Changes in metabolic activity affects adult NSC differentiation and cell-fate commitment, particularly by activation of mitochondrial respiration and reactive oxygen species production (64). Oxygen tension or hypoxia in the microenvironment also stimulates proliferation within the SVZ and migration into the hypoxic region (65). Metabolomic analyses of NSCs has also revealed that lipid metabolism can induce changes in NSC state. Adult NSCs have been shown to require lipogenesis for proliferation to ensure quiescence (66, 67). Extrinsic insulin/insulin-like growth factor signaling has also been shown to stimulate NSC reactivation and proliferation through regulation of CDK4 activity (68, 69).
While other niche-mediated cues such as regional identity (70) and positional information (71) modulate adult NSC activity, it is the combination of molecular stimuli, cytoarchitecture, and structural components of the SVZ niche that continually regulate NSC state and function.
Cancer Stem Cell Hypothesis and Glioblastoma
The cancer stem cell (CSC) hypothesis has been used as a framework describe and provide explanation for the high degree of molecular heterogeneity, cellular plasticity, and the molecular divergence of recurrent GBM. CSCs were observed to share many of the similar properties to the healthy stem cells including multipotent differentiation and self-renewal (72), low frequency and low proliferative rate (73–76), ability to regulate the surrounding microenvironment (77), strict re-regulation of proliferation and cell death, and reliance of similar molecular pathways (78). The initial evidence of cancer stem cell-driven tumorigenesis came through studies involving serial re-transplantation of a specific subpopulation of leukemic cells in immunodeficient mice (74, 79). Since the early 2000s, CSCs have successfully been identified in numerous solid tumors including breast cancer (80), colorectal cancer (81, 82), and brain cancers including GBM (12) in which they are specifically termed GBM stem cells (GSCs). GSCs have demonstrated chemo- (83, 84) and radiotherapy (85) resistant, while contributing to invasion (86), angiogenesis (87) and tumor recurrence (87). Comparison of underlying molecular mechanisms within GSCs to those in NSCs will allow for development of selective therapies to target the rare cell population responsible for tumor initiation, propagation, and evasion of current therapies.
While the precise identification of the GBM cell of origin remains elusive, two major hypotheses have been explored over the years. In one theory, GBM arises from a transformation events in differentiated astrocytes, while others have suggested that a GBM pathogenesis begins with a transformed NSC [Comprehensive review by Fan et al. (10)]. Previously, astrocyte progenitor cells were believed to be the sole proliferating cells in the adult brain (88) and were hypothesized to drive GBM tumorigenesis due to extensive expression of the marker GFAP in both healthy astrocytes and glioma samples (89). This would require a fully committed astrocytes to acquire mutations, de-differentiate and become tumorigenic. Other attempts to identify the cell-of-origin in GBM using lineage tracing experiments in mouse models have suggested oligodendrocyte precursor cells (OPCs) (90, 91). The similarity in expression levels of PDGFRα and NG2 in OPCs and GBM provided further support to the notion of OPC-derived GSC (92–94), and in a study by Hide et al., the authors have proposed a model where a transformation of both OPCs and NSCs is required for generation of GSCs (90).
Early mouse models exploring effects of genetic alterations in either NSCs or differentiated astrocytes have failed to provide definitive resolution to the cell-of-origin question. Some reports suggested that overexpression of Ras and Akt signaling in neural progenitor cells but not in more differentiated astrocytes was sufficient to induce formation of GBM-like lesions (95). On the other hand, other groups have provided evidence that genetic alterations in either population is sufficient to induce GBM formation (96). Mounting evidence of the hierarchical organization of GBM tumors and the upregulation of developmental pathways in GSCs became the principal evidence for the notion of transformation of NSCs from SVZ as the initial stage of gliomagenesis. In addition to evident functional overlap, similarities in expression patterns of a number of genes including CD133 (97, 98), Sox10 (99), Nestin (100, 101), Musashi (101, 102), GFAP (103), and Olig1/2 (104, 105) highlight shared molecular programs between NSCs and GSCs. Through deep sequencing of isocitrate dehydrogenase wild-type GBM patient samples and normal SVZ tissue, researchers observed similar expression of driver mutations in both the SVZ and patient matched-tumor tissue (15). Intriguingly, multiple studies have reported shorter survival of GBM patients in cases where tumors were in contact with SVZ (106–109). The comprehensive profiling and understanding of GBM cell of origin may pave the way for identification of prognostic markers along with targeted preventative and curative therapies.
Intrinsic Deregulations of Glioma Stem Cells Compared to Neural Stem Cells
Like all cancers, GBM exhibits behavioral hallmarks that distinguish it from healthy tissue (110). Compared to NSCs, GSCs are self-sufficient in providing growth signals, resistant to growth inhibition, evade programmed cell death, have limitless replicative potential, sustain angiogenesis, and invade surrounding tissue (Figure 2). Markers of interest to explain these phenotypes have been extensively studied and while some have been exploited in clinical settings, no single one is responsible for GBM’s relentless growth.
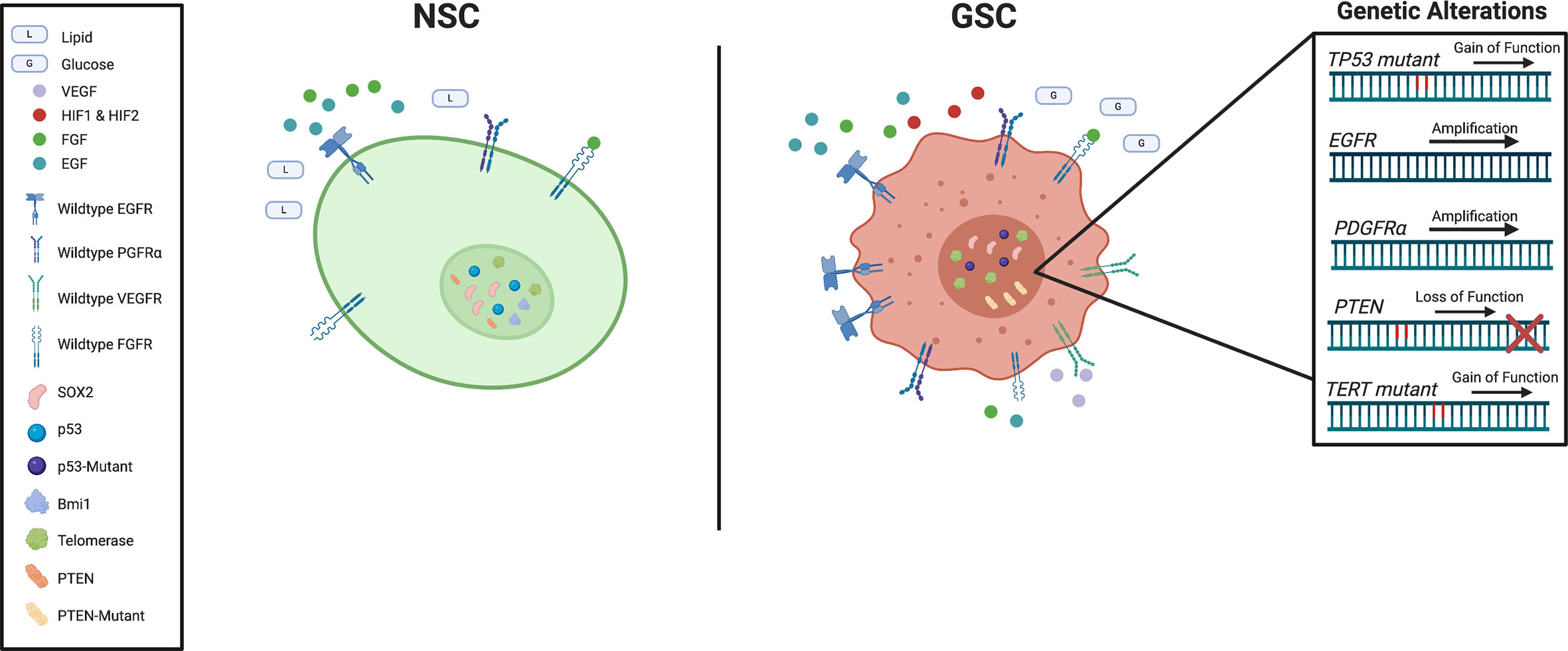
Figure 2 Schematic representation of key genetic and signaling difference between subventricular zone (SVZ) neural stem cells (NSCs) and glioblastoma stem cells (GSCs).
Aberrant Growth Signals
Epidermal growth factor (EGF), is a critical regulator in the proliferation of normal NSCs in mice (111). Aberrant EGF signaling prevents mouse NSC differentiation while increasing proliferative capacity and invasiveness, properties that closely resemble those of high-grade gliomas (112, 113). As epidermal growth factor receptor (EGFR) amplification is one of the most frequent mutations in GBM patients (114), and has been implicated in human gliomagenesis (115), its targeting in the clinical setting has been extensively investigate (116). Even in the absence of EGF, aberrant behavior of the EGFR pathway maintains stemness properties and promotes self-sufficient growth in tumors human (117).
Along with EGF, fibroblast growth factors (FGF) play an important role in the regulation of stemness in GSCs in vitro (118, 119). The FGF superfamily consists of 22 genes with various isoforms (120). Of particular interest is FGF-2, which does not follow conventional secretion (121) and was found to increase proliferation of NSCs in rat SVZs (122). The low molecular weight isoform of FGF-2 can be excreted and internalized for autocrine or paracrine signaling via fibroblast growth factor receptors (FGFRs), or be translocated directly to the cytoplasm and nucleus (123). The transcription factor ZEB1, which has previously been implicated in regulation of glioma stemness (124), has been found to regulate FGFR1 expression (125), suggesting that FGFRs could also be associated with GSCs. Indeed, FGFR1 was found to be preferentially expressed on GSCs, and regulated stem cell transcription factors SOX2, OLIG2, and ZEB1 to promote GBM growth in vivo (126). While FGF is a large and cumbersome family to investigate, further inquiries into their functions and potential redundancies may offer insights into preferential novel therapeutic vulnerabilities of GSCs compared to NSCs.
Platelet-derived growth factor (PDGF) is another important regulator of NSCs in the SVZ. When neural stem cells were identified to express PDGF-receptor-alpha (PDGFRA) in the adult mouse SVZ, supplemental PDGF alone was sufficient to induce hyperplasia with some features of GBM (127). These receptors show functional significance by promoting evasion of radiotherapy (128). Amplification of PDGFRA is one of the most common mutations in GBM and is most commonly associated with the proneural subtype (129). As PDGFRs are important in the healthy development of the normal central nervous system (130), its increased activity has led to important relationships between subclasses of GBM (131). While these receptors provide another intriguing avenue to potentially eradicate GBM, selective inhibition to avoid targeting of surrounding healthy tissue requires further research and clinical investigation.
Insensitive to Growth Inhibition
As one of the most studied tumor suppressors in cancer, TP53 has been shown to regulate a wide range of cellular functions of various cancers including GBM. As a key stem cell maintenance regulator, TP53 is expressed in proliferating cells within the SVZ and has been implicated in controlling cell division and differentiation (132). The key regulatory role of TP53 is not limited to development as it has been shown to regulate proliferation and self-renewal of NSCs in adult mice (133, 134). While deletion mutations in TP53 are predominant in several cancers, in the context of GBM, mutations in TP53 are often gain-of-function, resulting in a wider range of downstream effect (135). Mutations in TP53 may cause healthy NSCs to prematurely migrate out of the SVZ effectively seeding the brain with pre-cancerous stem cells (136, 137).
Another common loss of tumor suppressor in GBM is phosphatase and tensin homolog (PTEN). The role of PTEN in the regulation of mouse NSCs in SVZ is extensively reviewed in Li et al. (138). Together with TP53, PTEN also regulates the self-renewal and differentiation of both NSCs and GSCs (139). Loss of PTEN was observed to represses GSC differentiation (140), and similarly promote NSC conversion to a GSC-like phenotype (141). Unsurprisingly, mutational status of PTEN positively correlates with a worse overall prognosis for GBM patients (142).
Evasion of Programmed Cell Death
The PI3K/AKT/mTOR intracellular pathway is a critical pathway for cell cycle regulation, proliferation, and it is directly antagonized by PTEN (143). The pathway exerts direct influences on cell quiescence, proliferation, longevity by acting predominately through phosphorylation and subsequent activation of AKT/mTOR driving downstream effects (144). Although the PI3K/AKT/mTOR pathway is found throughout the body, its role in promoting growth and proliferation and preventing differentiation in adult NSCs (145) make it an important area of research in GBM and GSCs (146). In addition to being a key pathway in preventing GSC differentiation (147), PI3K/AKT/mTOR further contributes to GBM growth by blocking apoptosis signaling (146).
Signal transducer and activator of transcription-3 (STAT3) is a transcription factor whose activity is directly regulated by PI3K/AKT/mTOR (148). A variety of cytokines, growth factors and interferons converge to regulate STAT3, but its influence on an array of pathways within normal and cancerous stem cells is well documented as it is highly conserved (149). Most importantly, STAT3 plays a major role in maintaining stemness and promoting tumor survival and invasion while suppressing anti-tumor immunity (150). Several studies have shown that reducing levels of STAT3 can lead to a reduction of CD133 and other stemness markers while increasing the propensity for apoptosis and differentiation (151, 152). Inhibition of STAT3 in recurrent GBM has also been shown to reduce levels of BCL-XL and survivin, leading to caspase-3 activation and apoptosis in GSCs (153).
Limitless Replicative Potential
Healthy replication of cells requires proper activity of telomerase enzymes to ensure the end of chromosomes do not shorten or fuse with other chromosomes (154). Telomerase activity becomes restricted to the SVZ as mammals age (155) but proper maintenance allows NSCs to remain present into adulthood (156). Likewise, increased activity of telomerase leads to replicative immortality within GSCs and is one of the most frequent mutations in GBM (157, 158). The most common gain-of-function mutation of telomerase is located in the promoter region of TERT (159) and is predictive of shorter survival times (160).
Sustained Angiogenesis
Angiogenesis is a tightly controlled pathways in normal tissue and is initiated in response to injury (161). However, several of the receptors discussed before that are upregulated in GSCs are also involved in angiogenic pathways. EGFR, PDGFR, FGFR, and VEGFR have all been shown be involved in angiogenesis with GBM cells and GSCs themselves being major producers of the signaling molecules and their respective receptors (162–167).
Similar, to the intrinsic regulation seen in NSCs, GSCs themselves can directly promote their own survival by modulating the microenvironment. In a study by Takahashi et al., mice engrafted with OCT3/4 overexpressing GBM cells were observed to have larger tumors and increased number of blood vessels (168). Furthermore, tumor-conditioned media accelerated capillary formation in vitro and elevated mRNA levels of VEGF in OCT3/4-overexpressing cells providing additional evidence of tumor cell contributing to angiogenesis (168). GSCs have also been shown to directly secrete VEGF-A in extracellular vesicles (169). Along with their influence on surrounding cells, when GSCs asymmetrically divide to self-renew, the differentiated daughter cell is capable of forming blood vessel structures (170).
Increased Invasiveness
Although GBM cells rarely metastasize to other organs, they do demonstrate a highly invasive growth pattern. Once a GBM is established, the infiltrative edge presents a challenge for surgical resection as the edge is enriched with chemoradioresistant GSCs, meaning the remaining cells are poised to drive tumor relapse upon removal of therapeutic pressures (171, 172). The invasive nature of GBM cells is further exuberated by surrounding non-malignant cells, such as astrocytes secreting cytokines and chemokines (173). Additionally, GBM tumors can degrade the extracellular matrix via metalloproteinases (174) and cathepsins (175) to modulate their cell structure for efficient cell movement (176).
Altered Cellular Metabolism
Compared to the rest of the body, the brain naturally has a higher dependence on glucose as a source of energy, consuming 60% of our daily intake (177). In normal cells, catabolism via glycolysis/oxidative phosphorylation and anabolism via gluconeogenesis pathways achieve a glucose homeostasis. However, a phenomenon known as the Warburg effect describing the preferential usage of anaerobic glycolysis in CSCs, even in the presence of sufficient oxygen, heightens the dependency of differentiated GBM cells on glucose (178). However, in a side by side comparison, GSCs consumed less glucose and produced less lactate while maintaining higher ATP levels than their differentiated counterparts (179). GSCs are therefore thought to rely mainly on oxidative phosphorylation, however, if challenged, are capable of using other metabolic pathways (179).
Metabolic flexibility and plasticity of cellular states influence each other. In GBM cells, functional p53 leads to increased glutaminase 2 (GLS2) under stress which increases oxidative metabolism and ATP generation, by catalyzing the conversion of glutamine to glutamate and increasing α-ketoglutarate (α-KG) levels (180). This metabolic shift, known as glutaminolysis, is also observed in freshly resected GSCs (181). Glutaminolysis produces precursors for macromolecules (nucleic acids, amino acids, fatty acids), regulates redox homeostasis (via NADH, NADPH, and ROS levels) and contributes to immunosuppression by glutamate production to ensure pro-tumor survival (182). As an abundant non-essential amino acid, glutamine is transported through the blood and capable of crossing the blood-brain barrier, making it a particularly useful energy source by tumors (183).
Glutamate can be converted to α-KG by either glutamate dehydrogenase 1 (GDH1) or transaminases such as glutamate pyruvate transaminase 2 (GPT2) and glutamate oxaloacetate transaminase 2 (GOT2) (184). Conversions fluctuate according to nitrogen and carbon availability. In addition to a metabolite, α-KG behaves as a cofactor (along with oxygen) in the activity of α-ketoglutarate-dependent hydroxylases. These are non-heme, iron-containing enzymes that catalyze a wide range of oxygenation reactions including biosynthesis (ex. collagen and L-carnitine), post-translational modifications (ex. protein hydroxylation), epigenetic regulations (ex. histone and DNA demethylation), as well as sensors of energy metabolism. So far majority of these processes in GBM have been restricted to isocitrate dehydrogenase mutants of GBM, however, collagen prolyl hydroxylases were found to induce metastasis of breast cancer (185) by mechanistically stabilizing HIF-1α in chemoresistance (186). While the importance of HIF-1α in the conversion of GSCs in different tumor niches has been mentioned above, only recently was it reported that collagen-prolyl hydroxylases promote proliferation and invasion in GBM. Interestingly, mouse embryonic stem cells were found to maintain high αKG/succinate ratios via glucose and glutamine catabolism that promoted histone/DNA demethylation and maintenance of pluripotency (187). By altering intracellular αKG/succinate ratios, multiple chromatin modifications such as H3K27me3 and ten-eleven translocation-dependent (26) DNA demethylation were shown to regulate genes associated with pluripotency (187). In addition to glucose metabolism, GSCs were observed to have higher expression of genes involved in iron trafficking and metabolism when compared to healthy astroglial and neural progenitor cells, presenting an opportunity for targeted therapeutic intervention (188).
The Extrinsic Glioma Stem Cell Microenvironment
Unlike the NSC microenvironment in the SVZ, the GBM microenvironment is defined by three unique regions, the hypoxic-necrotic core, the perivascular niche, and the invasive edge, each with distinct contribution to the tumor progression (Figure 1B) (189). Each niche influences and activates different cellular programs in GSCs to express distinct markers and transcriptional profiles. This plasticity allows cells to change states and adapt to stressors as needed. The interconnected relationship between GBM and their environment maintains stemness and contributes to heterogeneity which is why emphasis to target the tumor microenvironment has gained traction is recent years, and why more advanced in vitro experimental methods such as 3D-culture methods and cerebral organoids are becoming more prevalent (190, 191).
Hypoxia and necrosis are defining features of GBM, caused by the tumor’s exceeding growth requirements on available blood flow to supply oxygen and nutrients. This subsequent lack of oxygen protects cells from irradiation, the most effective treatment modality against GBM, by limiting the amount of molecules capable of turning into cytotoxic free radicals (192). Restricted blood flow also limits the delivery of chemotherapies such as temozolomide to the tumor cells (193). In both cases, the hypoxic environment forces tumor cells into a quiescent state, where the lack of cell division prevents cytotoxic DNA damage induced by chemo-radiotherapies (194). Effects of hypoxia on stemness and tumor survival are largely mediated through hypoxia-inducible factors (HIF-1 and HIF-2), which upregulate signaling pathways including Klf4, Sox2, Oct4, CD133, and VEGF (195, 196). Cell death in the center of the hypoxic region leads to formation of the necrotic core and contributes to the release of pro-inflammatory signals, IL-1β, IL-6, and IL-8, into the surrounding microenvironment. This signaling in turn, contributes to the conversion of tissue-associated macrophages and neutrophils into immune-suppressive and angiogenesis-promoting cells, allowing for continued GBM progression and expansion (197–200). Similar to NSC metabolism, hypoxia forces a metabolic shift in GBM toward aerobic glycolysis and fatty acid metabolism rather than oxidative phosphorylation. Together, the hypoxic niche plays key regulatory roles leading to heterogeneity and cancer progression.
Interestingly, hypoxia can influence GBM cells to transdifferentiate into endothelial-like cells (201) which contribute to feedback loops of the second major tumor environment-the perivascular niche (202, 203). This niche most closely resembles the SVZ where NSCs reside (20, 50, 60). Perivascular niches exist along capillaries or arterioles where endothelial cells come into direct contact with GSCs (204). GSCs in the perivascular niche in turn remodel the microenvironment by producing high levels of pro-angiogenic factors, such as VEGF, that drive endothelial cell proliferation, survival, migration, and blood vessel permeability. This is critical for angiogenesis as GBM is one of the most vascularized human tumors and requires a supply of nutrients for tumor progression. The perivascular niche thus regulates stemness and induces pathways enriching for GSCs, namely nitric oxide and NOTCH (205), TGF-β (206, 207), as well as sonic hedgehog signaling pathways (208). Other perivascular cell populations in this niche, such as tumor-associated macrophages (TAMs) secrete chemokines to promote GSC growth and expansion.
The infiltrating (or “invasive”) edge is the third and final major GBM niche. As a highly invasive tumor, GBMs can infiltrate into healthy brain tissue and limit the effectiveness of maximal surgical resection. To circumvent and eradicate infiltrative GSCs, patients receive whole-brain radiation therapy. However, GBM cells, and most notably GSCs, have been shown to adapt and resist the applied environmental stress of irradiation (85). Once exposed to radiation, cells undergo a process known as the proneural-mesenchymal transition, similar to the metastatic cascade known as the epithelial-mesenchymal transition (EMT). In this process, cells lose cell polarity, cell-cell adhesions, and alter their cytoskeletal organization for migration. GSCs are observed to invade along white matter tracts of the human brain through a NOTCH1-Sox2 mediated feedback loop (209). Mesenchymal GSCs are regulated by STAT3, N-cadherin, NF-κB, and integrins (210–215). These phenotypes exhibited within the infiltrative niche are also influenced by the hypoxic and perivascular niches.
Leveraging Differences Between Neural Stem Cells and Glioblastoma Stem Cells for Development of Novel Target Therapies
While understanding the similarities between GSCs and NSCs is instrumental for contextualization of gliomagenesis and underlying mechanisms driving GBM progression and therapy resistance, it is leveraging the differences between two cell populations that may offer avenues for generation of novel targeted therapies. Unlike other solid tumors, brain tumors present a unique set of challenges for development of new treatment options. First, the blood-brain-barrier (BBB), which normally protects the brain from harmful toxins, can also hinder the access of targeted therapies against GBM. Although BBB permeability can be theoretically increased through chemical modification of small molecule-based inhibitors, such approach does not expand to more precise modalities including antibody-drug conjugates (ADCs) and adoptive cell transfer therapies. In both mouse models (216, 217) and patient studies, locoregional delivery of the therapeutic offers distinct advantages. In addition to expanding the range of possible treatment modalities, while minimizing systemic toxicities, the locoregional delivery route allows for direct targeting of potential source of GSCs residing in SVZ at the border of lateral ventricle.
One of the most widespread strategies in targeting GSCs is through identification and subsequent development of targeted therapies against cell surface markers. Several different targeting approaches have been investigated in the recent years including antibody-drug conjugates (ADCs) and chimeric antigen receptor T cells (216, 218). And while researchers were able to demonstrate efficacy in mouse models, it is likely that additional combinatorial strategies will be needed to yield complete tumor clearance. Anti-angiogenic therapies have become an attractive modality to prevent tumor progression by cutting off the tumor’s supply of nutrients and oxygen (219). Bevacizumab and other anti-angiogenic therapies showed great promise, but repeated failures show the adaptability of tumors to overcome single agent therapies (220). Reducing the invasion of GBMs has been tested to reduce overall tumor progression, but further therapies would be required to fully eradicate the tumor (221). The use of ibrutinib, and FDA-approved drug to treat lymphoma and leukemia, was shown to suppress the BMX-STAT3 axis in GSCs making them vulnerable to radiation therapy (222). This signaling axis was previously shown to maintain self-renewal in GSCs (223) and mitigate apoptosis (224). Additionally, because BMX is not expressed in neural progenitor cells, ibrutinib may be a selective and beneficial therapy for GBM patients (222).
While there is biological overlap between NSCs and GSCs, promising research is exposing differences and vulnerabilities of each, presenting an avenue for novel therapeutic interventions. Research on telomerase activity in a variety of tumors has resulted in development of distinct therapeutic approaches including small-molecule inhibitors, plant-derived compounds, gene therapy, and immunotherapy. Although they remain to be tested in the clinical setting, several of these therapies have demonstrated promising efficacy in mouse models of GBM (225–228). More recently, CRISPR/Cas9 technology has been tested pre-clinically as a modality to repair mutations in cancers to induce cell cycle arrest with few off-target effects in GBM (229). Moving forward, it will be vital to further interrogate the therapeutic window of telomerase activity modulating treatments by comparing their effects on GSCs and SVZ NSCs. The proximity of SVZ NSCs to the lateral ventricles allows for intracerebroventricular (ICV) delivery of potential therapeutic interventions, bypassing the challenges presented by the BBB while increasing penetrance and distribution. Delivery of cell and gene therapies intracranially and intracerebroventricularly has been tested in both human clinical trials and mouse models of GBM and was shown to reduce tumor progression and invasion (230–232). It is important to note, that due to the extensive intra- and inter-tumoral heterogeneity between GBM tumors, it is unlikely that a single intervention will be sufficient to eradicate the tumor or control its progression, requiring more research into combinatorial approaches in both mouse models and clinical trials. Further research profiling the mechanisms by which SVZ NSCs and the surrounding microenvironment contribute to the chemoradiotherapy evasion of GBM is needed to identify therapies that will synergize with the current SoC. For example, in several in vitro and in vivo pre-clinical studies, inhibiting CXCL12/CXCR4 signaling in the mouse SVZ promoted radiosensitization and reduced GBM tumor cell proliferation (233, 234). Finally, in the past few years, the difference in the metabolic flexibility between GSCs and NSCs has become more apparent and is now being extensively investigated for the therapeutic potential.
Concluding Remarks
The aggressive growth characteristic, resistance to therapies and poor clinical outcome have been attributed to the extensive intra- and intertumoral heterogeneity within GBM tumors. Over the years, the observed similarities between GSCs and NSCs of the SVZ, have led to the hypothesis of a transformed NSCs cell presiding at the apex of GBM cytoarchitecture. Although the recent findings have corroborated this hypothesis, it has become evident that that understanding both similarities and differences between GSCs and the healthy NSCs of the SVZ is essential in the search for novel targeted therapies. The comparison of similarities can allow for improved understanding of the molecular mechanisms driving GBM formation, while the comparison of the differences can allow in identifying unique molecular vulnerabilities for development of targeted therapies with a large therapeutic index.
Author Contributions
All authors (DB, NS, SSa, CV, SSi) contributed to article writing and editing. Figures were generated with BioRender.com. All authors contributed to the article and approved the submitted version.
Funding
NS is funded by Mitacs Accelerate Fellowship, SSa is generously supported by CIHR CGS-M award and SSi holds Terri Fox Program Project Grant.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med (2008) 359(5):492–507. doi: 10.1056/NEJMra0708126
2. Witthayanuwat S, Pesee M, Supaadirek C, Supakalin N, Thamronganantasakul K, Krusun S. Survival Analysis of Glioblastoma Multiforme. Asian Pac J Cancer Prev (2018) 19(9):2613–7. doi: 10.22034/APJCP.2018.19.9.2613
3. Poon MTC, Sudlow CLM, Figueroa JD, Brennan PM. Longer-term (>/= 2 years) survival in patients with glioblastoma in population-based studies pre- and post-2005: a systematic review and meta-analysis. Sci Rep (2020) 10(1):11622. doi: 10.1038/s41598-020-68011-4
4. Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, et al. The somatic genomic landscape of glioblastoma. Cell (2013) 155(2):462–77. doi: 10.1016/j.cell.2013.09.034
5. Thakkar JP, Dolecek TA, Horbinski C, Ostrom QT, Lightner DD, Barnholtz-Sloan JS, et al. Epidemiologic and molecular prognostic review of glioblastoma. Cancer Epidemiol Biomarkers Prev (2014) 23(10):1985–96. doi: 10.1158/1055-9965.EPI-14-0275
6. Onizuka H, Masui K, Komori T. Diffuse gliomas to date and beyond 2016 WHO Classification of Tumours of the Central Nervous System. Int J Clin Oncol (2020) 25(6):997–1003. doi: 10.1007/s10147-020-01695-w
7. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med (2005) 352(10):987–96. doi: 10.1056/NEJMoa043330
8. Stupp R, Taillibert S, Kanner A, Read W, Steinberg D, Lhermitte B, et al. Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs Maintenance Temozolomide Alone on Survival in Patients With Glioblastoma: A Randomized Clinical Trial. JAMA (2017) 318(23):2306–16. doi: 10.1001/jama.2017.18718
9. Qazi MA, Vora, Venugopal C, Sidhu SS, Moffat J, Swanton C, et al. Intratumoral heterogeneity: pathways to treatment resistance and relapse in human glioblastoma. Ann Oncol (2017) 28(7):1448–56. doi: 10.1093/annonc/mdx169
10. Fan X, Xiong Y, Wang Y. A reignited debate over the cell(s) of origin for glioblastoma and its clinical implications. Front Med (2019) 13(5):531–9. doi: 10.1007/s11684-019-0700-1
11. Galli R, Binda E, Orfanelli U, Cipelletti B, Gritti A, De Vitis S, et al. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res (2004) 64(19):7011–21. doi: 10.1158/0008-5472.CAN-04-1364
12. Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, et al. Identification of human brain tumour initiating cells. Nature (2004) 432(7015):396–401. doi: 10.1038/nature03128
13. Kroonen J, Nassen J, Boulanger YG, Provenzano F, Capraro V, Bours V, et al. Human glioblastoma-initiating cells invade specifically the subventricular zones and olfactory bulbs of mice after striatal injection. Int J Cancer (2011) 129(3):574–85. doi: 10.1002/ijc.25709
14. Sanai N, Tramontin AD, Quinones-Hinojosa A, Barbaro NM, Gupta N, Kunwar S, et al. Unique astrocyte ribbon in adult human brain contains neural stem cells but lacks chain migration. Nature (2004) 427(6976):740–4. doi: 10.1038/nature02301
15. Lee JH, Lee JE, Kahng JY, Kim SH, Park JS, Yoon SJ, et al. Human glioblastoma arises from subventricular zone cells with low-level driver mutations. Nature (2018) 560(7717):243–7. doi: 10.1038/s41586-018-0389-3
16. Urban N, Guillemot F. Neurogenesis in the embryonic and adult brain: same regulators, different roles. Front Cell Neurosci (2014) 8:396. doi: 10.3389/fncel.2014.00396
17. Bond AM, Ming GL, Song H. Adult Mammalian Neural Stem Cells and Neurogenesis: Five Decades Later. Cell Stem Cell (2015) 17(4):385–95. doi: 10.1016/j.stem.2015.09.003
18. Lazarov O, Marr RA. Of mice and men: neurogenesis, cognition and Alzheimer’s disease. Front Aging Neurosci (2013) 5:43. doi: 10.3389/fnagi.2013.00043
19. van Strien ME, van den Berge SA, Hol EM. Migrating neuroblasts in the adult human brain: a stream reduced to a trickle. Cell Res (2011) 21(11):1523–5. doi: 10.1038/cr.2011.101
20. Mirzadeh Z, Merkle FT, Soriano-Navarro M, Garcia-Verdugo JM, Alvarez-Buylla A. Neural stem cells confer unique pinwheel architecture to the ventricular surface in neurogenic regions of the adult brain. Cell Stem Cell (2008) 3(3):265–78. doi: 10.1016/j.stem.2008.07.004
21. Katsimpardi L, Lledo PM. Regulation of neurogenesis in the adult and aging brain. Curr Opin Neurobiol (2018) 53:131–8. doi: 10.1016/j.conb.2018.07.006
22. Codega P, Silva-Vargas V, Paul A, Maldonado-Soto AR, Deleo AM, Pastrana E, et al. Prospective identification and purification of quiescent adult neural stem cells from their in vivo niche. Neuron (2014) 82(3):545–59. doi: 10.1016/j.neuron.2014.02.039
23. Obernier K, Tong CK, Alvarez-Buylla A. Restricted nature of adult neural stem cells: re-evaluation of their potential for brain repair. Front Neurosci (2014) 8:162. doi: 10.3389/fnins.2014.00162
24. Ming GL, Song H. Adult neurogenesis in the mammalian brain: significant answers and significant questions. Neuron (2011) 70(4):687–702. doi: 10.1016/j.neuron.2011.05.001
25. Llorens-Bobadilla E, Zhao S, Baser A, Saiz-Castro G, Zwadlo K, Martin-Villalba A. Single-Cell Transcriptomics Reveals a Population of Dormant Neural Stem Cells that Become Activated upon Brain Injury. Cell Stem Cell (2015) 17(3):329–40. doi: 10.1016/j.stem.2015.07.002
26. Dulken BW, Leeman DS, Boutet SC, Hebestreit K, Brunet A. Single-Cell Transcriptomic Analysis Defines Heterogeneity and Transcriptional Dynamics in the Adult Neural Stem Cell Lineage. Cell Rep (2017) 18(3):777–90. doi: 10.1016/j.celrep.2016.12.060
27. Hsieh J. Orchestrating transcriptional control of adult neurogenesis. Genes Dev (2012) 26(10):1010–21. doi: 10.1101/gad.187336.112
28. Andreu-Agullo C, Maurin T, Thompson CB, Lai EC. Ars2 maintains neural stem-cell identity through direct transcriptional activation of Sox2. Nature (2011) 481(7380):195–8. doi: 10.1038/nature10712
29. Liu HK, Wang Y, Belz T, Bock D, Takacs A, Radlwimmer B, et al. The nuclear receptor tailless induces long-term neural stem cell expansion and brain tumor initiation. Genes Dev (2010) 24(7):683–95. doi: 10.1101/gad.560310
30. Zou Y, Niu W, Qin S, Downes M, Burns DK, Zhang CL. The nuclear receptor TLX is required for gliomagenesis within the adult neurogenic niche. Mol Cell Biol (2012) 32(23):4811–20. doi: 10.1128/MCB.01122-12
31. Liu HK, Belz T, Bock D, Takacs A, Wu H, Lichter P, et al. The nuclear receptor tailless is required for neurogenesis in the adult subventricular zone. Genes Dev (2008) 22(18):2473–8. doi: 10.1101/gad.479308
32. Shi Y, Sun G, Zhao C, Stewart R. Neural stem cell self-renewal. Crit Rev Oncol Hematol (2008) 65(1):43–53. doi: 10.1016/j.critrevonc.2007.06.004
33. Ohtsuka T, Ishibashi M, Gradwohl G, Nakanishi S, Guillemot F, Kageyama R. Hes1 and Hes5 as notch effectors in mammalian neuronal differentiation. EMBO J (1999) 18(8):2196–207. doi: 10.1093/emboj/18.8.2196
34. Brannvall K, Korhonen L, Lindholm D. Estrogen-receptor-dependent regulation of neural stem cell proliferation and differentiation. Mol Cell Neurosci (2002) 21(3):512–20. doi: 10.1006/mcne.2002.1194
35. Lopez-Juarez A, Remaud S, Hassani Z, Jolivet, Pierre Simons J, Sontag T, et al. Thyroid hormone signaling acts as a neurogenic switch by repressing Sox2 in the adult neural stem cell niche. Cell Stem Cell (2012) 10(5):531–43. doi: 10.1016/j.stem.2012.04.008
36. Di Giacomo E, Benedetti E, Cristiano L, Antonosante A, d’Angelo M, Fidoamore A, et al. Roles of PPAR transcription factors in the energetic metabolic switch occurring during adult neurogenesis. Cell Cycle (2017) 16(1):59–72. doi: 10.1080/15384101.2016.1252881
37. Ma DK, Marchetto MC, Guo JU, Ming GL, Gage FH, Song H. Epigenetic choreographers of neurogenesis in the adult mammalian brain. Nat Neurosci (2010) 13(11):1338–44. doi: 10.1038/nn.2672
38. Lim DA, Huang YC, Swigut T, Mirick AL, Garcia-Verdugo JM, Wysocka J, et al. Chromatin remodelling factor Mll1 is essential for neurogenesis from postnatal neural stem cells. Nature (2009) 458(7237):529–33. doi: 10.1038/nature07726
39. Grossniklaus U, Paro R. Transcriptional silencing by polycomb-group proteins. Cold Spring Harb Perspect Biol (2014) 6(11):a019331. doi: 10.1101/cshperspect.a019331
40. Murao N, Noguchi H, Nakashima K. Epigenetic regulation of neural stem cell property from embryo to adult. Neuroepigenetics (2016) 5(5):1–10. doi: 10.1016/j.nepig.2016.01.001
41. Koreman E, Sun X, Lu QR. Chromatin remodeling and epigenetic regulation of oligodendrocyte myelination and myelin repair. Mol Cell Neurosci (2018) 87:18–26. doi: 10.1016/j.mcn.2017.11.010
42. Gao Z, Ure K, Ding, Nashaat M, Yuan L, Ma J, et al. The master negative regulator REST/NRSF controls adult neurogenesis by restraining the neurogenic program in quiescent stem cells. J Neurosci (2011) 31(26):9772–86. doi: 10.1523/JNEUROSCI.1604-11.2011
43. Cheng LC, Pastrana E, Tavazoie M, Doetsch F. miR-124 regulates adult neurogenesis in the subventricular zone stem cell niche. Nat Neurosci (2009) 12(4):399–408. doi: 10.1038/nn.2294
44. Capilla-Gonzalez V, Bonsu JM, Redmond KJ, Garcia-Verdugo JM, Quinones-Hinojosa A. Implications of irradiating the subventricular zone stem cell niche. Stem Cell Res (2016) 16(2):387–96. doi: 10.1016/j.scr.2016.02.031
45. Kazanis I, ffrench-Constant C. Extracellular matrix and the neural stem cell niche. Dev Neurobiol (2011) 71(11):1006–17. doi: 10.1002/dneu.20970
46. McClenahan FK, Sharma H, Shan X, Eyermann C, Colognato H. Dystroglycan Suppresses Notch to Regulate Stem Cell Niche Structure and Function in the Developing Postnatal Subventricular Zone. Dev Cell (2016) 38(5):548–66. doi: 10.1016/j.devcel.2016.07.017
47. Kerever A, Yamada T, Suzuki Y, Mercier F, Arikawa-Hirasawa E. Fractone aging in the subventricular zone of the lateral ventricle. J Chem Neuroanat (2015) 66–67:52–60. doi: 10.1016/j.jchemneu.2015.06.001
48. Kerever A, Schnack J, Vellinga D, Ichikawa N, Moon C, Arikawa-Hirasawa E, et al. Novel extracellular matrix structures in the neural stem cell niche capture the neurogenic factor fibroblast growth factor 2 from the extracellular milieu. Stem Cells (2007) 25(9):2146–57. doi: 10.1634/stemcells.2007-0082
49. Mercier F. Fractones: extracellular matrix niche controlling stem cell fate and growth factor activity in the brain in health and disease. Cell Mol Life Sci (2016) 73(24):4661–74. doi: 10.1007/s00018-016-2314-y
50. Tavazoie M, Van der Veken L, Silva-Vargas V, Louissaint M, Colonna L, Zaidi B, et al. A specialized vascular niche for adult neural stem cells. Cell Stem Cell (2008) 3(3):279–88. doi: 10.1016/j.stem.2008.07.025
51. Ma DK, Ming GL, Song H. Glial influences on neural stem cell development: cellular niches for adult neurogenesis. Curr Opin Neurobiol (2005) 15(5):514–20. doi: 10.1016/j.conb.2005.08.003
52. Quinones-Hinojosa A, Sanai N, Soriano-Navarro M, Gonzalez-Perez O, Mirzadeh Z, Gil-Perotin S, et al. Cellular composition and cytoarchitecture of the adult human subventricular zone: a niche of neural stem cells. J Comp Neurol (2006) 494(3):415–34. doi: 10.1002/cne.20798
53. Lim DA, Tramontin AD, Trevejo JM, Herrera DG, Garcia-Verdugo JM, Alvarez-Buylla A. Noggin antagonizes BMP signaling to create a niche for adult neurogenesis. Neuron (2000) 28(3):713–26. doi: 10.1016/S0896-6273(00)00148-3
54. Delgado AC, Ferron SR, Vicente D, Porlan E, Perez-Villalba A, Trujillo CM, et al. Endothelial NT-3 delivered by vasculature and CSF promotes quiescence of subependymal neural stem cells through nitric oxide induction. Neuron (2014) 83(3):572–85. doi: 10.1016/j.neuron.2014.06.015
55. Rhee YH, Yi SH, Kim JY, Chang MY, Jo AY, Kim J, et al. Neural stem cells secrete factors facilitating brain regeneration upon constitutive Raf-Erk activation. Sci Rep (2016) 6:32025. doi: 10.1038/srep32025
56. Nguyen L, Malgrange B, Breuskin I, Bettendorff L, Moonen G, Belachew S, et al. Autocrine/paracrine activation of the GABA(A) receptor inhibits the proliferation of neurogenic polysialylated neural cell adhesion molecule-positive (PSA-NCAM+) precursor cells from postnatal striatum. J Neurosci (2003) 23(8):3278–94. doi: 10.1523/JNEUROSCI.23-08-03278.2003
57. Liu X, Wang Q, Haydar TF, Bordey A. Nonsynaptic GABA signaling in postnatal subventricular zone controls proliferation of GFAP-expressing progenitors. Nat Neurosci (2005) 8(9):1179–87. doi: 10.1038/nn1522
58. Barkho BZ, Song H, Aimone JB, Smrt RD, Kuwabara T, Nakashima K, et al. Identification of astrocyte-expressed factors that modulate neural stem/progenitor cell differentiation. Stem Cells Dev (2006) 15(3):407–21. doi: 10.1089/scd.2006.15.407
59. Ottone C, Krusche B, Whitby A, Clements M, Quadrato G, Pitulescu ME, et al. Direct cell-cell contact with the vascular niche maintains quiescent neural stem cells. Nat Cell Biol (2014) 16(11):1045–56. doi: 10.1038/ncb3045
60. Shen Q, Wang Y, Kokovay E, Lin G, Chuang SM, Goderie SK, et al. Adult SVZ stem cells lie in a vascular niche: a quantitative analysis of niche cell-cell interactions. Cell Stem Cell (2008) 3(3):289–300. doi: 10.1016/j.stem.2008.07.026
61. Faigle R, Song H. Signaling mechanisms regulating adult neural stem cells and neurogenesis. Biochim Biophys Acta (2013) 1830(2):2435–48. doi: 10.1016/j.bbagen.2012.09.002
62. Katsimpardi L, Litterman NK, Schein PA, Miller CM, Loffredo FS, Wojtkiewicz GR, et al. Vascular and neurogenic rejuvenation of the aging mouse brain by young systemic factors. Science (2014) 344(6184):630–4. doi: 10.1126/science.1251141
63. Villeda SA, Luo J, Mosher KI, Zou B, Britschgi M, Bieri G, et al. The ageing systemic milieu negatively regulates neurogenesis and cognitive function. Nature (2011) 477(7362):90–4. doi: 10.1038/nature10357
64. Gothie JD, Sebillot A, Luongo C, Legendre M, Nguyen Van C, Le Blay K, et al. Adult neural stem cell fate is determined by thyroid hormone activation of mitochondrial metabolism. Mol Metab (2017) 6(11):1551–61. doi: 10.1016/j.molmet.2017.08.003
65. Candelario KM, Shuttleworth CW, Cunningham LA. Neural stem/progenitor cells display a low requirement for oxidative metabolism independent of hypoxia inducible factor-1alpha expression. J Neurochem (2013) 125(3):420–9. doi: 10.1111/jnc.12204
66. Knobloch M, Braun SM, Zurkirchen L, von Schoultz C, Zamboni N, Arauzo-Bravo MJ, et al. Metabolic control of adult neural stem cell activity by Fasn-dependent lipogenesis. Nature (2013) 493(7431):226–30. doi: 10.1038/nature11689
67. Stoll EA, Makin R, Sweet IR, Trevelyan AJ, Miwa S, Horner PJ, et al. Neural Stem Cells in the Adult Subventricular Zone Oxidize Fatty Acids to Produce Energy and Support Neurogenic Activity. Stem Cells (2015) 33(7):2306–19. doi: 10.1002/stem.2042
68. Li S, Koe CT, Tay ST, Tan ALK, Zhang S, Zhang Y, et al. An intrinsic mechanism controls reactivation of neural stem cells by spindle matrix proteins. Nat Commun (2017) 8(1):122. doi: 10.1038/s41467-017-00172-9
69. Chirivella L, Kirstein M, Ferron SR, Domingo-Muelas A, Durupt FC, Acosta-Umanzor C, et al. Cyclin-Dependent Kinase 4 Regulates Adult Neural Stem Cell Proliferation and Differentiation in Response to Insulin. Stem Cells (2017) 35(12):2403–16. doi: 10.1002/stem.2694
70. Alvarez-Buylla A, Kohwi M, Nguyen TM, Merkle FT. The heterogeneity of adult neural stem cells and the emerging complexity of their niche. Cold Spring Harb Symp Quant Biol (2008) 73:357–65. doi: 10.1101/sqb.2008.73.019
71. Ihrie RA, Shah JK, Harwell CC, Levine JH, Guinto CD, Lezameta M, et al. Persistent sonic hedgehog signaling in adult brain determines neural stem cell positional identity. Neuron (2011) 71(2):250–62. doi: 10.1016/j.neuron.2011.05.018
72. Galderisi U, Cipollaro M, Giordano A. Stem cells and brain cancer. Cell Death Differ (2006) 13(1):5–11. doi: 10.1038/sj.cdd.4401757
73. Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med (1997) 3(7):730–7. doi: 10.1038/nm0797-730
74. Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature (1994) 367(6464):645–8. doi: 10.1038/367645a0
75. Uckun FM, Sather H, Reaman G, Shuster J, Land V, Trigg M, et al. Leukemic cell growth in SCID mice as a predictor of relapse in high-risk B-lineage acute lymphoblastic leukemia. Blood (1995) 85(4):873–8. doi: 10.1182/blood.V85.4.873.bloodjournal854873
76. Papaccio F, Paino F, Regad T, Papaccio G, Desiderio V, Tirino V. Concise Review: Cancer Cells, Cancer Stem Cells, and Mesenchymal Stem Cells: Influence in Cancer Development. Stem Cells Transl Med (2017) 6(12):2115–25. doi: 10.1002/sctm.17-0138
77. Marusyk A, Polyak K. Tumor heterogeneity: causes and consequences. Biochim Biophys Acta (2010) 1805(1):105–17. doi: 10.1016/j.bbcan.2009.11.002
78. Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature (2001) 414(6859):105–11. doi: 10.1038/35102167
79. Hope KJ, Jin L, Dick JE. Acute myeloid leukemia originates from a hierarchy of leukemic stem cell classes that differ in self-renewal capacity. Nat Immunol (2004) 5(7):738–43. doi: 10.1038/ni1080
80. Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA (2003) 100(7):3983–8. doi: 10.1073/pnas.0530291100
81. O’Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature (2007) 445(7123):106–10. doi: 10.1038/nature05372
82. Ricci-Vitiani L, Lombardi DG, Pilozzi E, Biffoni M, Todaro M, Peschle C, et al. Identification and expansion of human colon-cancer-initiating cells. Nature (2007) 445(7123):111–5. doi: 10.1038/nature05384
83. Liu G, Yuan X, Zeng Z, Tunici, Ng H, Abdulkadir IR, et al. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol Cancer (2006) 5:67. doi: 10.1186/1476-4598-5-67
84. Chen J, Li Y, Yu TS, McKay RM, Burns DK, Kernie SG, et al. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature (2012) 488(7412):522–6. doi: 10.1038/nature11287
85. Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature (2006) 444(7120):756–60. doi: 10.1038/nature05236
86. Wakimoto H, Kesari S, Farrell CJ, Curry WT Jr, Zaupa C, Aghi M, et al. Human glioblastoma-derived cancer stem cells: establishment of invasive glioma models and treatment with oncolytic herpes simplex virus vectors. Cancer Res (2009) 69(8):3472–81. doi: 10.1158/0008-5472.CAN-08-3886
87. Cheng L, Huang Z, Zhou W, Wu Q, Donnola S, Liu JK, et al. Glioblastoma stem cells generate vascular pericytes to support vessel function and tumor growth. Cell (2013) 153(1):139–52. doi: 10.1016/j.cell.2013.02.021
88. Cavanagh JB. The proliferation of astrocytes around a needle wound in the rat brain. J Anat (1970) 106(Pt 3):471–87.
89. Jones TR, Bigner SH, Schold SC Jr, Eng LF, Bigner DD. Anaplastic human gliomas grown in athymic mice. Morphology and glial fibrillary acidic protein expression. Am J Pathol (1981) 105(3):316–27.
90. Hide T, Takezaki T, Nakatani Y, Nakamura H, Kuratsu J, Kondo T. Combination of a ptgs2 inhibitor and an epidermal growth factor receptor-signaling inhibitor prevents tumorigenesis of oligodendrocyte lineage-derived glioma-initiating cells. Stem Cells (2011) 29(4):590–9. doi: 10.1002/stem.618
91. Lindberg N, Kastemar M, Olofsson T, Smits A, Uhrbom L. Oligodendrocyte progenitor cells can act as cell of origin for experimental glioma. Oncogene (2009) 28(23):2266–75. doi: 10.1038/onc.2009.76
92. Cancer Genome Atlas Research, N. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature (2008) 455(7216):1061–8. doi: 10.1038/nature07385
93. Shoshan Y, Nishiyama A, Chang A, Mork S, Barnett GH, Cowell JK, et al. Expression of oligodendrocyte progenitor cell antigens by gliomas: implications for the histogenesis of brain tumors. Proc Natl Acad Sci USA (1999) 96(18):10361–6. doi: 10.1073/pnas.96.18.10361
94. Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt, et al. An integrated genomic analysis of human glioblastoma multiforme. Science (2008) 321(5897):1807–12. doi: 10.1126/science.1164382
95. Holland EC, Celestino J, Dai C, Schaefer L, Sawaya RE, Fuller GN. Combined activation of Ras and Akt in neural progenitors induces glioblastoma formation in mice. Nat Genet (2000) 25(1):55–7. doi: 10.1038/75596
96. Bachoo RM, Maher EA, Ligon KL, Sharpless NE, Chan SS, You MJ, et al. Epidermal growth factor receptor and Ink4a/Arf: convergent mechanisms governing terminal differentiation and transformation along the neural stem cell to astrocyte axis. Cancer Cell (2002) 1(3):269–77. doi: 10.1016/S1535-6108(02)00046-6
97. Wu B, Sun C, Feng F, Ge M, Xia L. Do relevant markers of cancer stem cells CD133 and Nestin indicate a poor prognosis in glioma patients? A systematic review and meta-analysis. J Exp Clin Cancer Res (2015) 34:44. doi: 10.1186/s13046-015-0163-4
98. Pallini R, Ricci-Vitiani L, Banna GL, Signore M, Lombardi D, Todaro M, et al. Cancer stem cell analysis and clinical outcome in patients with glioblastoma multiforme. Clin Cancer Res (2008) 14(24):8205–12. doi: 10.1158/1078-0432.CCR-08-0644
99. Feng W, Liu S, Zhu R, Li B, Zhu Z, Yang J, et al. SOX10 induced Nestin expression regulates cancer stem cell properties of TNBC cells. Biochem Biophys Res Commun (2017) 485(2):522–8. doi: 10.1016/j.bbrc.2017.02.014
100. Shih AH, Holland EC. Notch signaling enhances nestin expression in gliomas. Neoplasia (2006) 8(12):1072–82. doi: 10.1593/neo.06526
101. Strojnik T, Rosland GV, Sakariassen PO, Kavalar R, Lah T. Neural stem cell markers, nestin and musashi proteins, in the progression of human glioma: correlation of nestin with prognosis of patient survival. Surg Neurol (2007) 68(2):133–43. discussion 143-4. doi: 10.1016/j.surneu.2006.10.050
102. Toda M, Iizuka Y, Yu W, Imai T, Ikeda E, Yoshida K, et al. Expression of the neural RNA-binding protein Musashi1 in human gliomas. Glia (2001) 34(1):1–7. doi: 10.1002/glia.1034
103. Kimura T, Budka H, Soler-Federsppiel S. An immunocytochemical comparison of the glia-associated proteins glial fibrillary acidic protein (GFAP) and S-100 protein (S100P) in human brain tumors. Clin Neuropathol (1986) 5(1):21–7.
104. Bouvier C, Bartoli C, Aguirre-Cruz L, Virard I, Colin C, Fernandez C, et al. Shared oligodendrocyte lineage gene expression in gliomas and oligodendrocyte progenitor cells. J Neurosurg (2003) 99(2):344–50. doi: 10.3171/jns.2003.99.2.0344
105. Riemenschneider MJ, Koy TH, Reifenberger G. Expression of oligodendrocyte lineage genes in oligodendroglial and astrocytic gliomas. Acta Neuropathol (2004) 107(3):277–82. doi: 10.1007/s00401-003-0809-8
106. Lim DA, Cha S, Mayo MC, Chen MH, Keles E, VandenBerg S, et al. Relationship of glioblastoma multiforme to neural stem cell regions predicts invasive and multifocal tumor phenotype. Neuro Oncol (2007) 9(4):424–9. doi: 10.1215/15228517-2007-023
107. Pope WB, Sayre J, Perlina A, Villablanca JP, Mischel PS, Cloughesy TF. MR imaging correlates of survival in patients with high-grade gliomas. AJNR Am J Neuroradiol (2005) 26(10):2466–74.
108. Kappadakunnel M, Eskin A, Dong J, Nelson SF, Mischel PS, Liau LM, et al. Stem cell associated gene expression in glioblastoma multiforme: relationship to survival and the subventricular zone. J Neurooncol (2010) 96(3):359–67. doi: 10.1007/s11060-009-9983-4
109. Chaichana KL, McGirt MJ, Frazier J, Attenello F, Guerrero-Cazares H, Quinones-Hinojosa A. Relationship of glioblastoma multiforme to the lateral ventricles predicts survival following tumor resection. J Neurooncol (2008) 89(2):219–24. doi: 10.1007/s11060-008-9609-2
110. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
111. Rietze RL, Reynolds BA. Neural stem cell isolation and characterization. Methods Enzymol (2006) 419:3–23. doi: 10.1016/S0076-6879(06)19001-1
112. Reynolds BA, Tetzlaff W, Weiss S. A multipotent EGF-responsive striatal embryonic progenitor cell produces neurons and astrocytes. J Neurosci (1992) 12(11):4565–74. doi: 10.1523/JNEUROSCI.12-11-04565.1992
113. Doetsch F, Petreanu L, Caille I, Garcia-Verdugo JM, Alvarez-Buylla A. EGF converts transit-amplifying neurogenic precursors in the adult brain into multipotent stem cells. Neuron (2002) 36(6):1021–34. doi: 10.1016/S0896-6273(02)01133-9
114. Neftel C, Laffy J, Filbin MG, Hara T, Shore ME, Rahme GJ, et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell (2019) 178(4):835–49.e21. doi: 10.1016/j.cell.2019.06.024
115. Wechsler-Reya R, Scott MP. The developmental biology of brain tumors. Annu Rev Neurosci (2001) 24:385–428. doi: 10.1146/annurev.neuro.24.1.385
116. Westphal M, Maire CL, Lamszus K. EGFR as a Target for Glioblastoma Treatment: An Unfulfilled Promise. CNS Drugs (2017) 31(9):723–35. doi: 10.1007/s40263-017-0456-6
117. Liffers K, Lamszus K, Schulte A. EGFR Amplification and Glioblastoma Stem-Like Cells. Stem Cells Int (2015) 2015:427518. doi: 10.1155/2015/427518
118. Lee JG, Kay EP. FGF-2-mediated signal transduction during endothelial mesenchymal transformation in corneal endothelial cells. Exp Eye Res (2006) 83(6):1309–16. doi: 10.1016/j.exer.2006.04.007
119. Lathia JD, Hitomi M, Gallagher J, Gadani SP, Adkins J, Vasanji A, et al. Distribution of CD133 reveals glioma stem cells self-renew through symmetric and asymmetric cell divisions. Cell Death Dis (2011) 2:e200. doi: 10.1038/cddis.2011.80
120. Itoh N, Ornitz DM. Evolution of the Fgf and Fgfr gene families. Trends Genet (2004) 20(11):563–9. doi: 10.1016/j.tig.2004.08.007
121. Sorensen V, Nilsen T, Wiedlocha A. Functional diversity of FGF-2 isoforms by intracellular sorting. Bioessays (2006) 28(5):504–14. doi: 10.1002/bies.20405
122. Frinchi M, Bonomo A, Trovato-Salinaro A, Condorelli DF, Fuxe K, Spampinato MG, et al. Fibroblast growth factor-2 and its receptor expression in proliferating precursor cells of the subventricular zone in the adult rat brain. Neurosci Lett (2008) 447(1):20–5. doi: 10.1016/j.neulet.2008.09.059
123. Powers CJ, McLeskey SW, Wellstein A. Fibroblast growth factors, their receptors and signaling. Endocr Relat Cancer (2000) 7(3):165–97. doi: 10.1677/erc.0.0070165
124. Edwards LA, Li A, Berel D, Madany M, Kim NH, Liu M, et al. ZEB1 regulates glioma stemness through LIF repression. Sci Rep (2017) 7(1):69. doi: 10.1038/s41598-017-00106-x
125. Kowalski-Chauvel A, Gouaze-Andersson V, Baricault L, Martin E, Delmas C, Toulas C, et al. Alpha6-Integrin Regulates FGFR1 Expression through the ZEB1/YAP1 Transcription Complex in Glioblastoma Stem Cells Resulting in Enhanced Proliferation and Stemness. Cancers (Basel) (2019) 11(3):406–20. doi: 10.3390/cancers11030406
126. Jimenez-Pascual A, Hale JS, Kordowski A, Pugh J, Silver DJ, Bayik D, et al. ADAMDEC1 Maintains a Growth Factor Signaling Loop in Cancer Stem Cells. Cancer Discov (2019) 9(11):1574–89. doi: 10.1158/2159-8290.CD-18-1308
127. Jackson EL, Garcia-Verdugo JM, Gil-Perotin S, Roy M, Quinones-Hinojosa A, VandenBerg S, et al. PDGFR alpha-positive B cells are neural stem cells in the adult SVZ that form glioma-like growths in response to increased PDGF signaling. Neuron (2006) 51(2):187–99. doi: 10.1016/j.neuron.2006.06.012
128. Alexandru O, Sevastre AS, Castro J, Artene SA, Tache DE, Purcaru OS, et al. Platelet-Derived Growth Factor Receptor and Ionizing Radiation in High Grade Glioma Cell Lines. Int J Mol Sci (2019) 20(19):4663–79. doi: 10.3390/ijms20194663
129. Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell (2010) 17(1):98–110. doi: 10.1016/j.ccr.2009.12.020
130. Funa K, Sasahara M. The roles of PDGF in development and during neurogenesis in the normal and diseased nervous system. J Neuroimmune Pharmacol (2014) 9(2):168–81. doi: 10.1007/s11481-013-9479-z
131. Brennan C, Momota H, Hambardzumyan D, Ozawa T, Tandon A, Pedraza A, et al. Glioblastoma subclasses can be defined by activity among signal transduction pathways and associated genomic alterations. PLoS One (2009) 4(11):e7752. doi: 10.1371/journal.pone.0007752
132. van Lookeren Campagne M, Gill R. Tumor-suppressor p53 is expressed in proliferating and newly formed neurons of the embryonic and postnatal rat brain: comparison with expression of the cell cycle regulators p21Waf1/Cip1, p27Kip1, p57Kip2, p16Ink4a, cyclin G1, and the proto-oncogene Bax. J Comp Neurol (1998) 397(2):181–98. doi: 10.1002/(SICI)1096-9861(19980727)397:2<181::AID-CNE3>3.0.CO;2-X
133. Gil-Perotin S, Haines JD, Kaur J, Marin-Husstege M, Spinetta MJ, Kim KH, et al. Roles of p53 and p27(Kip1) in the regulation of neurogenesis in the murine adult subventricular zone. Eur J Neurosci (2011) 34(7):1040–52. doi: 10.1111/j.1460-9568.2011.07836.x
134. Gil-Perotin S, Marin-Husstege M, Li J, Soriano-Navarro M, Zindy F, Roussel MF, et al. Loss of p53 induces changes in the behavior of subventricular zone cells: implication for the genesis of glial tumors. J Neurosci (2006) 26(4):1107–16. doi: 10.1523/JNEUROSCI.3970-05.2006
135. Zhang Y, Dube C, Gibert M Jr, Cruickshanks N, Wang B, Coughlan M, et al. The p53 Pathway in Glioblastoma. Cancers (Basel) (2018) 10(9):297–314. doi: 10.3390/cancers10090297
136. Flores ER, Sengupta S, Miller JB, Newman JJ, Bronson R, Crowley D, et al. Tumor predisposition in mice mutant for p63 and p73: evidence for broader tumor suppressor functions for the p53 family. Cancer Cell (2005) 7(4):363–73. doi: 10.1016/j.ccr.2005.02.019
137. Muller PA, Vousden KH, Norman JC. p53 and its mutants in tumor cell migration and invasion. J Cell Biol (2011) 192(2):209–18. doi: 10.1083/jcb.201009059
138. Li L, Liu F, Ross AH. PTEN regulation of neural development and CNS stem cells. J Cell Biochem (2003) 88(1):24–8. doi: 10.1002/jcb.10312
139. Zheng H, Ying H, Yan H, Kimmelman AC, Hiller DJ, Chen AJ, et al. p53 and Pten control neural and glioma stem/progenitor cell renewal and differentiation. Nature (2008) 455(7216):1129–33. doi: 10.1038/nature07443
140. Gont A, Hanson JE, Lavictoire SJ, Parolin DA, Daneshmand M, Restall IJ, et al. PTEN loss represses glioblastoma tumor initiating cell differentiation via inactivation of Lgl1. Oncotarget (2013) 4(8):1266–79. doi: 10.18632/oncotarget.1164
141. Duan S, Yuan G, Liu X, Ren R, Li J, Zhang W, et al. PTEN deficiency reprogrammes human neural stem cells towards a glioblastoma stem cell-like phenotype. Nat Commun (2015) 6:10068. doi: 10.1038/ncomms10068
142. Han F, Hu R, Yang H, Liu J, Sui J, Xiang X, et al. PTEN gene mutations correlate to poor prognosis in glioma patients: a meta-analysis. Onco Targets Ther (2016) 9:3485–92. doi: 10.2147/OTT.S99942
143. Papa A, Pandolfi PP. The PTEN(-)PI3K Axis in Cancer. Biomolecules (2019) 9(4):153–63. doi: 10.3390/biom9040153
144. Xu F, Na L, Li Y, Chen L. Roles of the PI3K/AKT/mTOR signalling pathways in neurodegenerative diseases and tumours. Cell Biosci (2020) 10:54. doi: 10.1186/s13578-020-00416-0
145. Peltier J, O’Neill A, Schaffer DV. PI3K/Akt and CREB regulate adult neural hippocampal progenitor proliferation and differentiation. Dev Neurobiol (2007) 67(10):1348–61. doi: 10.1002/dneu.20506
146. Langhans J, Schneele L, Trenkler N, von Bandemer H, Nonnenmacher L, Karpel-Massler G, et al. The effects of PI3K-mediated signalling on glioblastoma cell behaviour. Oncogenesis (2017) 6(11):398. doi: 10.1038/s41389-017-0004-8
147. Ojeda L, Gao J, Hooten KG, Wang E, Thonhoff JR, Dunn TJ, et al. Critical role of PI3K/Akt/GSK3beta in motoneuron specification from human neural stem cells in response to FGF2 and EGF. PLoS One (2011) 6(8):e23414. doi: 10.1371/journal.pone.0023414
148. Vogt PK, Hart JR. PI3K and STAT3: a new alliance. Cancer Discov (2011) 1(6):481–6. doi: 10.1158/2159-8290.CD-11-0218
149. Galoczova M, Coates P, Vojtesek B. STAT3, stem cells, cancer stem cells and p63. Cell Mol Biol Lett (2018) 23:12. doi: 10.1186/s11658-018-0078-0
150. Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer (2009) 9(11):798–809. doi: 10.1038/nrc2734
151. Sherry MM, Reeves A, Wu JK, Cochran BH. STAT3 is required for proliferation and maintenance of multipotency in glioblastoma stem cells. Stem Cells (2009) 27(10):2383–92. doi: 10.1002/stem.185
152. Li L, Cheung SH, Evans EL, Shaw PE. Modulation of gene expression and tumor cell growth by redox modification of STAT3. Cancer Res (2010) 70(20):8222–32. doi: 10.1158/0008-5472.CAN-10-0894
153. Ashizawa T, Miyata H, Iizuka A, Komiyama M, Oshita C, Kume A, et al. Effect of the STAT3 inhibitor STX-0119 on the proliferation of cancer stem-like cells derived from recurrent glioblastoma. Int J Oncol (2013) 43(1):219–27. doi: 10.3892/ijo.2013.1916
154. Shay JW, Wright WE. Telomeres and telomerase: three decades of progress. Nat Rev Genet (2019) 20(5):299–309. doi: 10.1038/s41576-019-0099-1
155. Caporaso GL, Lim DA, Alvarez-Buylla A, Chao MV. Telomerase activity in the subventricular zone of adult mice. Mol Cell Neurosci (2003) 23(4):693–702. doi: 10.1016/S1044-7431(03)00103-9
156. Ferron SR, Marques-Torrejon MA, Mira H, Flores I, Taylor K, Blasco MA, et al. Telomere shortening in neural stem cells disrupts neuronal differentiation and neuritogenesis. J Neurosci (2009) 29(46):14394–407. doi: 10.1523/JNEUROSCI.3836-09.2009
157. Farooqi A, Yang J, Sharin V, Ezhilarasan R, Danussi C, Alvarez C, et al. Identification of patient-derived glioblastoma stem cell (GSC) lines with the alternative lengthening of telomeres phenotype. Acta Neuropathol Commun (2019) 7(1):76. doi: 10.1186/s40478-019-0732-4
158. Nonoguchi N, Ohta T, Oh JE, Kim YH, Kleihues P, Ohgaki H. TERT promoter mutations in primary and secondary glioblastomas. Acta Neuropathol (2013) 126(6):931–7. doi: 10.1007/s00401-013-1163-0
159. Vinagre J, Almeida A, Populo H, Batista R, Lyra J, Pinto V, et al. Frequency of TERT promoter mutations in human cancers. Nat Commun (2013) 4:2185. doi: 10.1038/ncomms3185
160. Killela PJ, Reitman ZJ, Jiao Y, Bettegowda C, Agrawal N, Diaz LA Jr, et al TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc Natl Acad Sci USA (2013) 110(15):6021–6. doi: 10.1073/pnas.1303607110
161. Demidova-Rice TN, Durham JT, Herman IM. Wound Healing Angiogenesis: Innovations and Challenges in Acute and Chronic Wound Healing. Adv Wound Care (New Rochelle) (2012) 1(1):17–22. doi: 10.1089/wound.2011.0308
162. Koch S, Claesson-Welsh L. Signal transduction by vascular endothelial growth factor receptors. Cold Spring Harb Perspect Med (2012) 2(7):a006502. doi: 10.1101/cshperspect.a006502
163. Minder P, Zajac E, Quigley JP, Deryugina EI. EGFR regulates the development and microarchitecture of intratumoral angiogenic vasculature capable of sustaining cancer cell intravasation. Neoplasia (2015) 17(8):634–49. doi: 10.1016/j.neo.2015.08.002
164. Guo P, Hu B, Gu W, Xu L, Wang D, Huang HJ, et al. Platelet-derived growth factor-B enhances glioma angiogenesis by stimulating vascular endothelial growth factor expression in tumor endothelia and by promoting pericyte recruitment. Am J Pathol (2003) 162(4):1083–93. doi: 10.1016/S0002-9440(10)63905-3
165. Roy H, Bhardwaj S, Yla-Herttuala S. Biology of vascular endothelial growth factors. FEBS Lett (2006) 580(12):2879–87. doi: 10.1016/j.febslet.2006.03.087
166. Bruna A, Darken RS, Rojo F, Ocana A, Penuelas S, Arias A, et al. High TGFbeta-Smad activity confers poor prognosis in glioma patients and promotes cell proliferation depending on the methylation of the PDGF-B gene. Cancer Cell (2007) 11(2):147–60. doi: 10.1016/j.ccr.2006.11.023
167. Staberg M, Rasmussen RD, Michaelsen SR, Pedersen H, Jensen KE, Villingshoj M, et al. Targeting glioma stem-like cell survival and chemoresistance through inhibition of lysine-specific histone demethylase KDM2B. Mol Oncol (2018) 12(3):406–20. doi: 10.1002/1878-0261.12174
168. Hosokawa Y, Takahashi H, Inoue A, Kawabe Y, Funahashi Y, Kameda K, et al. Oct-3/4 modulates the drug-resistant phenotype of glioblastoma cells through expression of ATP binding cassette transporter G2. Biochim Biophys Acta (2015) 1850(6):1197–205. doi: 10.1016/j.bbagen.2015.01.017
169. Treps L, Perret R, Edmond S, Ricard D, Gavard J. Glioblastoma stem-like cells secrete the pro-angiogenic VEGF-A factor in extracellular vesicles. J Extracell Vesicles (2017) 6(1):1359479. doi: 10.1080/20013078.2017.1359479
170. Jhaveri N, Chen TC, Hofman FM. Tumor vasculature and glioma stem cells: Contributions to glioma progression. Cancer Lett (2016) 380(2):545–51. doi: 10.1016/j.canlet.2014.12.028
171. Minata M, Audia A, Shi J, Lu S, Bernstock J, Pavlyukov MS, et al. Phenotypic Plasticity of Invasive Edge Glioma Stem-like Cells in Response to Ionizing Radiation. Cell Rep (2019) 26(7):1893–905.e7. doi: 10.1016/j.celrep.2019.01.076
172. Guerra-Rebollo M, Garrido C, Sanchez-Cid L, Soler-Botija C, Meca-Cortes O, Rubio N, et al. Targeting of replicating CD133 and OCT4/SOX2 expressing glioma stem cells selects a cell population that reinitiates tumors upon release of therapeutic pressure. Sci Rep (2019) 9(1):9549. doi: 10.1038/s41598-019-46014-0
173. Rath BH, Fair JM, Jamal M, Camphausen K, Tofilon PJ. Astrocytes enhance the invasion potential of glioblastoma stem-like cells. PLoS One (2013) 8(1):e54752. doi: 10.1371/journal.pone.0054752
174. Wang C, Tong X, Jiang X, Yang F. Effect of matrix metalloproteinase-mediated matrix degradation on glioblastoma cell behavior in 3D PEG-based hydrogels. J BioMed Mater Res A (2017) 105(3):770–8. doi: 10.1002/jbm.a.35947
175. Koh SP, Wickremesekera AC, Brasch HD, Marsh R, Tan ST, Itinteang T. Expression of Cathepsins B, D, and G in Isocitrate Dehydrogenase-Wildtype Glioblastoma. Front Surg (2017) 4:28. doi: 10.3389/fsurg.2017.00028
176. Mair DB, Ames HM, Li R. Mechanisms of invasion and motility of high-grade gliomas in the brain. Mol Biol Cell (2018) 29(21):2509–15. doi: 10.1091/mbc.E18-02-0123
177. Erbsloh F, Bernsmeier A, Hillesheim H. [The glucose consumption of the brain & its dependence on the liver]. Arch Psychiatr Nervenkr Z Gesamte Neurol Psychiatr (1958) 196(6):611–26. doi: 10.1007/BF00344388
178. Marin-Valencia I, Yang C, Mashimo T, Cho S, Baek H, Yang XL, et al. Analysis of tumor metabolism reveals mitochondrial glucose oxidation in genetically diverse human glioblastomas in the mouse brain in vivo. Cell Metab (2012) 15(6):827–37. doi: 10.1016/j.cmet.2012.05.001
179. Vlashi E, Lagadec C, Vergnes L, Matsutani T, Masui K, Poulou M, et al. Metabolic state of glioma stem cells and nontumorigenic cells. Proc Natl Acad Sci USA (2011) 108(38):16062–7. doi: 10.1073/pnas.1106704108
180. Hu W, Zhang C, Wu R, Sun Y, Levine A, Feng Z. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc Natl Acad Sci USA (2010) 107(16):7455–60. doi: 10.1073/pnas.1001006107
181. Michelakis ED, Sutendra G, Dromparis P, Webster L, Haromy A, Niven E, et al. Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med (2010) 2(31):31ra34. doi: 10.1126/scitranslmed.3000677
182. Kesarwani P, Kant S, Prabhu A, Chinnaiyan P. The interplay between metabolic remodeling and immune regulation in glioblastoma. Neuro Oncol (2017) 19(10):1308–15. doi: 10.1093/neuonc/nox079
183. Oizel K, Chauvin C, Oliver L, Gratas C, Geraldo F, Jarry U, et al. Efficient Mitochondrial Glutamine Targeting Prevails Over Glioblastoma Metabolic Plasticity. Clin Cancer Res (2017) 23(20):6292–304. doi: 10.1158/1078-0432.CCR-16-3102
184. Jin L, Li D, Alesi GN, Fan J, Kang HB, Lu Z, et al. Glutamate dehydrogenase 1 signals through antioxidant glutathione peroxidase 1 to regulate redox homeostasis and tumor growth. Cancer Cell (2015) 27(2):257–70. doi: 10.1016/j.ccell.2014.12.006
185. Gilkes DM, Chaturvedi, Bajpai S, Wong CC, Wei H, Pitcairn S, et al. Collagen prolyl hydroxylases are essential for breast cancer metastasis. Cancer Res (2013) 73(11):3285–96. doi: 10.1158/0008-5472.CAN-12-3963
186. Xiong G, Stewart RL, Chen J, Gao T, Scott TL, Samayoa LM, et al. Collagen prolyl 4-hydroxylase 1 is essential for HIF-1alpha stabilization and TNBC chemoresistance. Nat Commun (2018) 9(1):4456. doi: 10.1038/s41467-018-06893-9
187. Carey BW, Finley LW, Cross JR, Allis CD, Thompson CB. Intracellular alpha-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature (2015) 518(7539):413–6. doi: 10.1038/nature13981
188. Schonberg DL, Miller TE, Wu Q, Flavahan WA, Das NK, Hale JS, et al. Preferential Iron Trafficking Characterizes Glioblastoma Stem-like Cells. Cancer Cell (2015) 28(4):441–55. doi: 10.1016/j.ccell.2015.09.002
189. Hambardzumyan D, Bergers G. Glioblastoma: Defining Tumor Niches. Trends Cancer (2015) 1(4):252–65. doi: 10.1016/j.trecan.2015.10.009
190. Linkous A, Balamatsias D, Snuderl M, Edwards L, Miyaguchi K, Milner T, et al. Modeling Patient-Derived Glioblastoma with Cerebral Organoids. Cell Rep (2019) 26(12):3203–11.e5. doi: 10.1016/j.celrep.2019.02.063
191. Jacob F, Salinas RD, Zhang DY, Nguyen PTT, Schnoll JG, Wong SZH, et al. A Patient-Derived Glioblastoma Organoid Model and Biobank Recapitulates Inter- and Intra-tumoral Heterogeneity. Cell (2020) 180(1):188–204.e22. doi: 10.1016/j.cell.2019.11.036
192. Chaplin DJ, Durand RE, Olive PL. Acute hypoxia in tumors: implications for modifiers of radiation effects. Int J Radiat Oncol Biol Phys (1986) 12(8):1279–82. doi: 10.1016/0360-3016(86)90153-7
193. Uribe D, Torres A, Rocha JD, Niechi I, Oyarzun C, Sobrevia L, et al. Multidrug resistance in glioblastoma stem-like cells: Role of the hypoxic microenvironment and adenosine signaling. Mol Aspects Med (2017) 55:140–51. doi: 10.1016/j.mam.2017.01.009
194. Jing X, Yang F, Shao C, Wei K, Xie M, Shen H, et al. Role of hypoxia in cancer therapy by regulating the tumor microenvironment. Mol Cancer (2019) 18(1):157. doi: 10.1186/s12943-019-1089-9
195. Mathieu J, Zhang Z, Zhou W, Wang AJ, Heddleston JM, Pinna CM, et al. HIF induces human embryonic stem cell markers in cancer cells. Cancer Res (2011) 71(13):4640–52. doi: 10.1158/0008-5472.CAN-10-3320
196. Kaur B, Khwaja FW, Severson EA, Matheny SL, Brat DJ, Van Meir EG. Hypoxia and the hypoxia-inducible-factor pathway in glioma growth and angiogenesis. Neuro Oncol (2005) 7(2):134–53. doi: 10.1215/S1152851704001115
197. Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell (2010) 141(1):39–51. doi: 10.1016/j.cell.2010.03.014
198. Yeung YT, McDonald KL, Grewal T, Munoz L. Interleukins in glioblastoma pathophysiology: implications for therapy. Br J Pharmacol (2013) 168(3):591–606. doi: 10.1111/bph.12008
199. Ruffell B, Coussens LM. Macrophages and therapeutic resistance in cancer. Cancer Cell (2015) 27(4):462–72. doi: 10.1016/j.ccell.2015.02.015
200. Rivera LB, Bergers G. Intertwined regulation of angiogenesis and immunity by myeloid cells. Trends Immunol (2015) 36(4):240–9. doi: 10.1016/j.it.2015.02.005
201. Soda Y, Marumoto T, Friedmann-Morvinski D, Soda M, Liu F, Michiue H, et al. Transdifferentiation of glioblastoma cells into vascular endothelial cells. Proc Natl Acad Sci USA (2011) 108(11):4274–80. doi: 10.1073/pnas.1016030108
202. Mei X, Chen YS, Chen FR, Xi SY, Chen ZP. Glioblastoma stem cell differentiation into endothelial cells evidenced through live-cell imaging. Neuro Oncol (2017) 19(8):1109–18. doi: 10.1093/neuonc/nox016
203. Calabrese C, Poppleton H, Kocak M, Hogg TL, Fuller C, Hamner B, et al. A perivascular niche for brain tumor stem cells. Cancer Cell (2007) 11(1):69–82. doi: 10.1016/j.ccr.2006.11.020
204. Chen Z, Feng X, Herting CJ, Garcia VA, Nie K, Pong WW, et al. Cellular and Molecular Identity of Tumor-Associated Macrophages in Glioblastoma. Cancer Res (2017) 77(9):2266–78. doi: 10.1158/0008-5472.CAN-16-2310
205. Charles N, Ozawa T, Squatrito M, Bleau AM, Brennan CW, Hambardzumyan D, et al. Perivascular nitric oxide activates notch signaling and promotes stem-like character in PDGF-induced glioma cells. Cell Stem Cell (2010) 6(2):141–52. doi: 10.1016/j.stem.2010.01.001
206. Penuelas S, Anido J, Prieto-Sanchez RM, Folch G, Barba I, Cuartas I, et al. TGF-beta increases glioma-initiating cell self-renewal through the induction of LIF in human glioblastoma. Cancer Cell (2009) 15(4):315–27. doi: 10.1016/j.ccr.2009.02.011
207. Ikushima H, Todo T, Ino Y, Takahashi M, Miyazawa K, Miyazono K. Autocrine TGF-beta signaling maintains tumorigenicity of glioma-initiating cells through Sry-related HMG-box factors. Cell Stem Cell (2009) 5(5):504–14. doi: 10.1016/j.stem.2009.08.018
208. Clement V, Sanchez P, de Tribolet N, Radovanovic I, Ruiz i Altaba A. HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr Biol (2007) 17(2):165–72. doi: 10.1016/j.cub.2006.11.033
209. Wang J, Xu SL, Duan JJ, Yi L, Guo YF, Shi Y, et al. Invasion of white matter tracts by glioma stem cells is regulated by a NOTCH1-SOX2 positive-feedback loop. Nat Neurosci (2019) 22(1):91–105. doi: 10.1038/s41593-018-0285-z
210. Hossain A, Gumin J, Gao F, Figueroa J, Shinojima N, Takezaki T, et al. Mesenchymal Stem Cells Isolated From Human Gliomas Increase Proliferation and Maintain Stemness of Glioma Stem Cells Through the IL-6/gp130/STAT3 Pathway. Stem Cells (2015) 33(8):2400–15. doi: 10.1002/stem.2053
211. Asano K, Duntsch CD, Zhou Q, Weimar JD, Bordelon D, Robertson JH, et al. Correlation of N-cadherin expression in high grade gliomas with tissue invasion. J Neurooncol (2004) 70(1):3–15. doi: 10.1023/B:NEON.0000040811.14908.f2
212. Rinkenbaugh AL, Cogswell PC, Calamini B, Dunn DE, Persson AI, Weiss WA, et al. IKK/NF-kappaB signaling contributes to glioblastoma stem cell maintenance. Oncotarget (2016) 7(43):69173–87. doi: 10.18632/oncotarget.12507
213. Lathia JD, Gallagher J, Heddleston JM, Wang J, Eyler CE, Macswords J, et al. Integrin alpha 6 regulates glioblastoma stem cells. Cell Stem Cell (2010) 6(5):421–32. doi: 10.1016/j.stem.2010.02.018
214. Carrasco-Garcia E, Auzmendi-Iriarte J, Matheu A. Integrin alpha7: a novel promising target in glioblastoma stem cells. Stem Cell Invest (2018) 5:2. doi: 10.21037/sci.2017.12.05
215. Nakada M, Nambu E, Furuyama N, Yoshida Y, Takino T, Hayashi Y, et al. Integrin alpha3 is overexpressed in glioma stem-like cells and promotes invasion. Br J Cancer (2013) 108(12):2516–24. doi: 10.1038/bjc.2013.218
216. Wang D, Starr R, Chang WC, Aguilar B, Alizadeh D, Wright SL, et al. Chlorotoxin-directed CAR T cells for specific and effective targeting of glioblastoma. Sci Transl Med (2020) 12(533):eaaw2672. doi: 10.1126/scitranslmed.aaw2672
217. Theruvath J, Sotillo E, Mount CW, Graef CM, Delaidelli A, Heitzeneder S, et al. Locoregionally administered B7-H3-targeted CAR T cells for treatment of atypical teratoid/rhabdoid tumors. Nat Med (2020) 26(5):712–9. doi: 10.1038/s41591-020-0821-8
218. Vora P, Venugopal C, Salim SK, Tatari N, Bakhshinyan D, Singh M, et al. The Rational Development of CD133-Targeting Immunotherapies for Glioblastoma. Cell Stem Cell (2020) 26(6):832–44.e6. doi: 10.1016/j.stem.2020.04.008
219. Batchelor TT, Reardon DA, de Groot JF, Wick W, Weller M. Antiangiogenic therapy for glioblastoma: current status and future prospects. Clin Cancer Res (2014) 20(22):5612–9. doi: 10.1158/1078-0432.CCR-14-0834
220. Li Y, Ali S, Clarke J, Cha S. Bevacizumab in Recurrent Glioma: Patterns of Treatment Failure and Implications. Brain Tumor Res Treat (2017) 5(1):1–9. doi: 10.14791/btrt.2017.5.1.1
221. Marin-Ramos NI, Thein TZ, Cho HY, Swenson SD, Wang W, Schonthal AH, et al. NEO212 Inhibits Migration and Invasion of Glioma Stem Cells. Mol Cancer Ther (2018) 17(3):625–37. doi: 10.1158/1535-7163.MCT-17-0591
222. Shi L, Tashiro S. Estimation of the effects of medical diagnostic radiation exposure based on DNA damage. J Radiat Res (2018) 59(suppl_2):ii121–9. doi: 10.1093/jrr/rry006
223. Guryanova OA, Wu Q, Cheng L, Lathia JD, Huang Z, Yang J, et al. Nonreceptor tyrosine kinase BMX maintains self-renewal and tumorigenic potential of glioblastoma stem cells by activating STAT3. Cancer Cell (2011) 19(4):498–511. doi: 10.1016/j.ccr.2011.03.004
224. Fox JL, Storey A. BMX Negatively Regulates BAK Function, Thereby Increasing Apoptotic Resistance to Chemotherapeutic Drugs. Cancer Res (2015) 75(7):1345–55. doi: 10.1158/0008-5472.CAN-14-1340
225. Mocellin S, Pooley KA, Nitti D. Telomerase and the search for the end of cancer. Trends Mol Med (2013) 19(2):125–33. doi: 10.1016/j.molmed.2012.11.006
226. Mizukoshi E, Kaneko S. Telomerase-Targeted Cancer Immunotherapy. Int J Mol Sci (2019) 20(8):1823–38. doi: 10.3390/ijms20081823
227. Fan HC, Chen CM, Chi CS, Tsai JD, Chiang KL, Chang YK, et al. Targeting Telomerase and ATRX/DAXX Inducing Tumor Senescence and Apoptosis in the Malignant Glioma. Int J Mol Sci (2019) 20(1):200–20. doi: 10.3390/ijms20010200
228. Lin PC, Lin SZ, Chen YL, Chang JS, Ho LI, Liu PY, et al. Butylidenephthalide suppresses human telomerase reverse transcriptase (TERT) in human glioblastomas. Ann Surg Oncol (2011) 18(12):3514–27. doi: 10.1245/s10434-011-1644-0
229. Wen L, Zhao C, Song J, Ma L, Ruan J, Xia X, et al. CRISPR/Cas9-Mediated TERT Disruption in Cancer Cells. Int J Mol Sci (2020) 21(2):653–64. doi: 10.3390/ijms21020653
230. Meijer DH, Maguire CA, LeRoy SG, Sena-Esteves M. Controlling brain tumor growth by intraventricular administration of an AAV vector encoding IFN-beta. Cancer Gene Ther (2009) 16(8):664–71. doi: 10.1038/cgt.2009.8
231. Panciani PP, Fontanella M, Tamagno I, Battaglia L, Garbossa D, Inghirami G, et al. Stem cells based therapy in high grade glioma: why the intraventricular route should be preferred? J Neurosurg Sci (2012) 56(3):221–9.
232. Brown CE, Aguilar B, Starr R, Yang X, Chang WC, Weng L, et al. Optimization of IL13Ralpha2-Targeted Chimeric Antigen Receptor T Cells for Improved Anti-tumor Efficacy against Glioblastoma. Mol Ther (2018) 26(1):31–44. doi: 10.1016/j.ymthe.2017.10.002
233. Goffart N, Kroonen J, Di Valentin E, Dedobbeleer M, Denne A, Martinive P, et al. Adult mouse subventricular zones stimulate glioblastoma stem cells specific invasion through CXCL12/CXCR4 signaling. Neuro Oncol (2015) 17(1):81–94. doi: 10.1093/neuonc/nou144
234. Gravina GL, Mancini A, Colapietro A, Vitale F, Vetuschi A, Pompili S, et al. The novel CXCR4 antagonist, PRX177561, reduces tumor cell proliferation and accelerates cancer stem cell differentiation in glioblastoma preclinical models. Tumour Biol (2017) 39(6):1010428317695528. doi: 10.1177/1010428317695528
Keywords: glioblastoma stem cells, neural stem cells, neurogenic niche, tumor microenvironment, tumor metabolism
Citation: Bakhshinyan D, Savage N, Salim SK, Venugopal C and Singh SK (2021) The Strange Case of Jekyll and Hyde: Parallels Between Neural Stem Cells and Glioblastoma-Initiating Cells. Front. Oncol. 10:603738. doi: 10.3389/fonc.2020.603738
Received: 07 September 2020; Accepted: 24 November 2020;
Published: 08 January 2021.
Edited by:
Natanael Zarco, Mayo Clinic, United StatesReviewed by:
Justin Lathia, Case Western Reserve University, United StatesJoanne C. Conover, University of Connecticut, United States
Copyright © 2021 Bakhshinyan, Savage, Salim, Venugopal and Singh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sheila K. Singh, c3NpbmdoQG1jbWFzdGVyLmNh