 Valeria Merz1,2*
Valeria Merz1,2* Marina Gaule1,3
Marina Gaule1,3 Camilla Zecchetto1,3Alessandro Cavaliere1,3Simona Casalino1,3
Camilla Zecchetto1,3Alessandro Cavaliere1,3Simona Casalino1,3 Camilla Pesoni1,3Serena Contarelli1Fabio Sabbadini1Monica Bertolini1Domenico Mangiameli1
Camilla Pesoni1,3Serena Contarelli1Fabio Sabbadini1Monica Bertolini1Domenico Mangiameli1 Michele Milella3
Michele Milella3 Vita Fedele1
Vita Fedele1 Davide Melisi1,3*
Davide Melisi1,3*- 1Digestive Molecular Clinical Oncology Research Unit, University of Verona, Verona, Italy
- 2Medical Oncology Unit, Santa Chiara Hospital, Trento, Italy
- 3Section of Medical Oncology, Università degli Studi di Verona, Verona, Italy
Mutations of the proto-oncogene KRAS are the most frequent gain-of-function alterations found in cancer. KRAS is mutated in about 30% of all human tumors, but it could reach more than 90% in certain cancer types such as pancreatic adenocarcinoma. Although historically considered to be undruggable, a particular KRAS mutation, the G12C variant, has recently emerged as an actionable alteration especially in non-small cell lung cancer (NSCLC). KRASG12C and pan-KRAS inhibitors are being tested in clinical trials and have recently shown promising activity. Due to the difficulties in direct targeting of KRAS, other approaches are being explored. The inhibition of target upstream activators or downstream effectors of KRAS pathway has shown to be moderately effective given the evidence of emerging mechanisms of resistance. Various synthetic lethal partners of KRAS have recently being identified and the inhibition of some of those might prove to be successful in the future. The study of escape mechanisms to KRAS inhibition could support the utility of combination strategies in overcoming intrinsic and adaptive resistance and enhancing clinical benefit of KRASG12C inhibitors. Considering the role of the microenvironment in influencing tumor initiation and promotion, the immune tumor niche of KRAS mutant tumors has been deeply explored and characterized for its unique immunosuppressive skewing. However, a number of aspects remains to be fully understood, and modulating this tumor niche might revert the immunoresistance of KRAS mutant tumors. Synergistic associations of KRASG12C and immune checkpoint inhibitors are being tested.
Introduction
Using an oversimplified description, cancer could be defined as a disease caused by the accumulation of alterations in genes coding for proteins involved in cell growth induction or control defined oncogenes or tumor suppressor genes, respectively. In the last half-century, the largest efforts made in the field of experimental targeted therapeutics have been mainly focusing towards the development of therapeutic agents capable of inhibiting oncogenes or restoring the function of tumor suppressor genes. The most important successes in cancer treatment have been, indeed, represented by experimental therapeutics able to effectively interfere with the product of some of the most relevant oncogenes in human cancers. In this regard, those human cancers for which the development of appropriate targeted therapeutics has been most frustrating are sustained in their proliferation by altered genes whose function is essential for the integration and the transduction of physiologic signals in normal cells.
KRAS has been the first oncogene identified in human cancer in 1982 (1). Mutations affecting members of the RAS family genes (KRAS, HRAS, NRAS) are the most frequent genetic alterations in human cancers accounting for about 27% of all tumors. KRAS mutations are involved in the pathogenesis of different epithelial cancer histotypes, including lung and colorectal cancer, but its role has been especially investigated in pancreatic ductal adenocarcinoma, which is considered the type of tumor mostly dependent on KRAS for its development, metastatic progression, and treatment resistance (2–5).
Because of its high incidence in different tumors and its role in cancer initiation and progression, many efforts have been made in finding effective treatments directly or indirectly targeting KRAS. However, due to the lack of accessible binding pockets and its complex downstream signaling, most of the efforts in targeting KRAS have failed, and mutated KRAS still remains an undruggable target.
Here, we describe and discuss the most recent efforts aimed to identify novel therapeutic approaches of mutated KRAS-driven tumors.
The RAS/MAPK Pathway
KRAS gene encodes for a small GTPase that in normal cells functions as a molecular switch between an active and an inactive state. In quiescent cells, KRAS is inactive and GDP-bound, while in cells receiving extracellular stimuli it is active and GTP-bound. KRAS in its active state leads to the activation of a number of different intracellular transduction signaling pathways, including MAPK and AKT pathways. The switching between inactive and active state is mediated by the guanine nucleotide exchange factors (GEFs) which allow GTP loading. Conversely, the inactive state is mediated by GTPase-activating proteins (GAPs) through GTP hydrolysis (6). GTP-bound RAS interacts and recruits RAF, promoting its accumulation at the plasma membrane and inducing its dimerization and activation of RAF kinases. Activated RAF phosphorylates MEK1 and MEK2 kinases, that consequently phosphorylate and activate ERK1 and ERK2 kinases. ERK1/2 translocate into the nucleus where they phosphorylate several transcription factors that regulate the expression of genes involved in proliferation and cancer progression. MAPK pathway is a linear cascade characterized by complex regulatory mechanisms and feedback loops controlling several kinases. The attempt of inhibiting MAPK pathway, in order to block proliferation signaling, generates cross-talk between different pathways and the activation of compensatory pathways such as the PI3K-AKT-mTORC1 signaling. Noteworthy, PI3K-AKT-mTORC1 pathway, unlike MAPK pathway, can also be activated independently from KRAS, by receptor tyrosine kinases (RTKs) or G-protein coupled receptors (GPCR) and integrin signaling (7).
KRAS mutated cancer cells carry mostly missense mutations causing single amino-acid substitutions in three hotspots, glycine12 (G12), glycine13 (G13), and glutamine61 (Q61). These mutations prevent GAPs from accessing GTP so that hydrolysis is blocked, resulting in a persistently activated GTP-bound state. KRAS activity becomes therefore independent from extracellular stimuli, resulting in overstimulation of downstream pathways and induction of signals for cell proliferation, migration, and metastasis (8). Interestingly, different KRAS mutations can reflect differences in signaling and oncogenic mechanisms, that can have a role in tailoring treatments. In an in vitro colorectal cancer study, phosphotyrosine proteomic profiles comparison between the two most frequent KRAS mutations, KRASG12D and KRASG13D, has been performed. KRASG12D mutation enhances membrane and adherens junction signaling, while KRASG13Dactivates signaling molecules such as MAPK kinases, non-receptor tyrosine kinases, and regulators of metabolic processes (9).
Direct Targeting of KRAS
KRAS mutations types and incidence vary among epithelial cancer histotypes. Whereas KRASG12C mutations are frequent in lung adenocarcinoma, they are rare in pancreatic ductal adenocarcinoma (PDAC). PDAC are enriched in KRASG12D, KRASG12V, and KRASG12R point mutations (10). These differences are crucial for the development of new potential therapeutic strategies.
Recently, encouraging results using direct KRASG12C inhibitors have been reported. KRAS mutation G12C is present in about 13% of lung cancer, 3% of colorectal cancer, and in a smaller percentage of other epithelial tumors (11). The mutant cysteine-12 is located next to a cryptic pocket (SWII) in GDP-KRAS. The proximity of this cryptic pocket (SWII) to cysteine-12 has driven the development of covalent inhibitors targeting SWII, getting an allosteric inhibition of cysteine-12. ARS-1620 was the first covalent inhibitor binding SWII pocket of KRASG12C-GDP complex developed (12). Starting from this pioneering milestone, many efforts have been made in order to improve potency, drug permeability, solubility, and oral bioavailability and create suitable drugs for clinical use. As a result of these efforts several drugs have been developed and tested in preclinical and clinical studies, including AMG 510 (or sotorasib) and MRTX849 (or adagrasib). Sotorasib was developed by Amgen. Improvements in drug potency have been achieved by taking advantage of an alternative orientation of His95, located in the switch II pocket. The alternative orientation creates a larger surface groove which guarantees an irreversible interaction between KRASG12C and its inhibitor. The almost complete inhibition of pERK observed upon sotorasib treatment confirmed its enhanced potency (13). Interestingly, while durable responses were obtained in immune-competent tumor-bearing murine models, this activity was not durable in immune-deficient models. Further studies demonstrated that sotorasib induces a pro-inflammatory microenvironment through the expression of chemokines, such as CXCL10 and CXCL11. These chemokines attract tumor-suppressive immune cells, including T cells, macrophages, and dendritic cells, leading to long-term anti-tumor T cells responses. This observation suggested that a more significant and prolonged tumor response could be induced by a combination therapy with immune checkpoint inhibitors (14).
The small molecule covalent inhibitor sotorasib is currently under active clinical development for the treatment of KRASG12C mutated tumors. Successful results have been recently reported in the phase 1 CodeBreaK100 trial, that investigated sotorasib in patients with advanced and pretreated solid tumors, mainly NSCLC and colorectal cancer, harboring a KRASG12C mutation (15). About one third of NSCLC patients responded to therapy. Disease control rate was remarkable in both NSCLC (88.1%) and colorectal cancer (73.8%). Median progression free survival was 6.3 months for NSCLC patients and 4.0 months for colorectal cancer patients. Some patients exhibited a rapid disease progression after an initial response, but a group of patients presented durable responses. The lower response rate in colorectal cancer patients (7.1%) suggests a different KRAS dependency across diverse tumor types harboring the same mutation. Sotorasib showed a good safety profile, with diarrhea, fatigue, and nausea as the most common adverse events, and no dose-limiting toxic effects have been observed. Combination treatments of sotorasib with immunotherapy are currently under clinical evaluation and invested of great expectations (NCT04303780, NCT04185883).
A different KRASG12C covalent inhibitor, named adagrasib, has been developed by Mirati Therapeutics Inc. Adagrasib also binds SWII pocket of GDP-KRAS, inhibiting KRAS pathway and inducing in turn a potent anti-tumor response, as demonstrated in different in vivo models. Nonetheless, resistance mechanisms emerged early through the activation of other pathways and activation of compensatory mechanisms, leading to transient or submaximal response to adagrasib. Indeed, high expression or activated mutations of RTKs can activate feedback mechanisms reactivating RAS and stimulating mTOR pathway. Similarly, the co-occurrence of alteration in genes involved in cell cycle regulation, such as CDKN2A and CDK4/6, can induce Rb phosphorylation and cell cycle transition. Based on these evidences, the combination of adagrasib with different drugs, such as EGFR, SHP2, and mTORC inhibitors have been tested in different in vivo murine models. The combination of adagrasib with afatinib, RMC-4550, and vistusertib respectively, obtained a stronger inhibition of ERK and S6 phosphorylation than did any single agent treatment with an improved anti-tumor activity. Moreover, the combination of adagrasib and palbociclib decreased Rb and E2F family target genes expression levels, reduced S6 phosphorylation level, and induced major tumor regression, especially in CDKN2A altered models (16). In the phase 1/2 multi-expansion cohort KRYSTAL-1 trial (NCT03785249), adagrasib has been evaluated in patients with advanced solid tumors harboring KRASG12C mutations, demonstrating an acceptable safety profile and promising clinical activity. In NSCLC patients previously treated with chemotherapy and anti-PD-1/PD-L1 therapy the disease control rate was 96% and objective response rate (ORR) was 45% (https://cm.eortc.org/cmPortal/Searchable/ENA2020/config/normal#!abstractdetails/0000902150). The only commonly reported (>2%) grade 3/4 adverse event was hyponatremia. Disease control was observed in 94% of colorectal patients. Confirmed partial responses (PRs) were observed in a patient with endometrial cancer and a patient with pancreatic cancer (https://cm.eortc.org/cmPortal/Searchable/ENA2020/config/normal#!abstractdetails/0000902140). The most commonly reported adverse events included diarrhea, nausea, fatigue, and vomiting. Other trials have been designed to evaluate the combination of adagrasib with other drugs, such as EGFR, SHP2, or PD1 inhibitors (KRISTAL-1, -2, -7). Among clinical trials conducted in patients with cancers harboring KRASG12C, ARS-3248/JNJ-74699157, LY3499446, and GDC-6036 are being investigated (NCT03114319, NCT04165031, NCT04449874).
PanKRAS inhibitors represent a different category of drugs that do not target a single KRAS isoform selectively but aim to inhibit a broader spectrum of targets. Among these molecules, we count BI 2852, which binds between switch I and switch II pocket and inhibits KRAS interactions with GEFs, GAPs, and its downstream effectors. BI 2852, indeed, effectively reduces pERK and pAKT levels, achieving antiproliferative effects on KRAS mutant cell lines. Because of the high conservation of the SI/II-pocket across RAS isoforms, this molecule can bind with similar affinity most of them (17). SOS1 inhibitors are also panKRAS inhibitors. These drugs, indeed, do not bind directly to KRAS but inhibit the interaction between KRAS: SOS1, preventing KRAS GTP loading and its switching in to the active state. The main representatives are BI 3406 and BI 1701963. BI 3406 has been proven to be active in in vitro and in vivo murine models, harboring KRAS G12 and G13 codon mutations, but not G12R mutation. SOS1 can be downregulated by ERK-mediated phosphorylation, representing an important negative feedback modulator of KRAS pathway. During treatment with MEK inhibitors, pERK levels reduction induces a decrease of SOS1 phosphorylation, resulting in RAS pathway activation. These observations suggest that inhibition at both levels represents a good strategy to efficiently block KRAS pathway and prevent escape. The combination treatment of SOS1 with MEK inhibitor achieved good results in vitro and in vivo murine models, with robust pathway inhibition and tumor regression (18). Based on these preliminary preclinical results, BI 1701963, the second representative of SOS1 inhibitors, is being tested, alone and in combination with MEK inhibitor trametinib, in a phase I clinical trial in cancer patients carrying pan-KRAS mutations (NCT04111458).
RAS direct targeting has also been investigated in several studies. The bacterial Ras/Rap1-specific endopeptidase (RRSP) represents a good candidate for RAS direct targeting therapy. RRSP induces a proteolytic cleavage of RAS proteins between residues Tyr-32- and Asp-33 in SWI pocket, this cleavage alters RAS SWI structure, blocking the interaction of RAS with GEFs and preventing the transition of RAS into the active state. It also prevents the interaction with RAF and the downstream signaling pathway (19). RRSP can disrupt both wild type and mutant RAS proteins (including KRAS, HRAS, and NRAS). This blockage allows the inhibition of signal transduction depending from various RAS mutations, overexpression of upstream receptor tyrosine kinases (RTKs), or amplification of wild-type RAS as it happens in head and neck squamous cell carcinoma, esophageal and gastric carcinoma, ovarian adenocarcinoma, and triple-negative breast cancer. To enable the migration of RRSP through the biological membrane, a chimeric toxin formed by RAS/Rap1 specific endopeptidase and the translocation machinery of diphtheria toxin has been developed. The use of this engineered chimeric toxin has achieved good results in vitro and in vivo murine models, especially in lung and colorectal tumor cell lines and in cells expressing high levels of HB EGF. In human cells HB EGF is highly represented and this could represent a limit for its clinical application, influencing the dose limiting toxicities (DLT). Further engineering steps could allow to overcome this possible limit, aiming to vehicle RRSP across tumoral membrane cells only and sparing normal human cells.
Indirect Targeting of KRAS Signaling
In the attempt of inhibiting KRAS signaling, different strategies intended to target upstream activators or downstream effectors of KRAS pathway have been developed (Figure 1). Because of the redundancy of the intracellular networks involved, the initial enthusiasm for the development of a single target therapy has been mitigated by evidence of emerging mechanisms of resistance. The combination of different drugs targeting different signal pathways could prevent or delay the development of resistance mechanisms, often, however, at cost of increased toxicities.
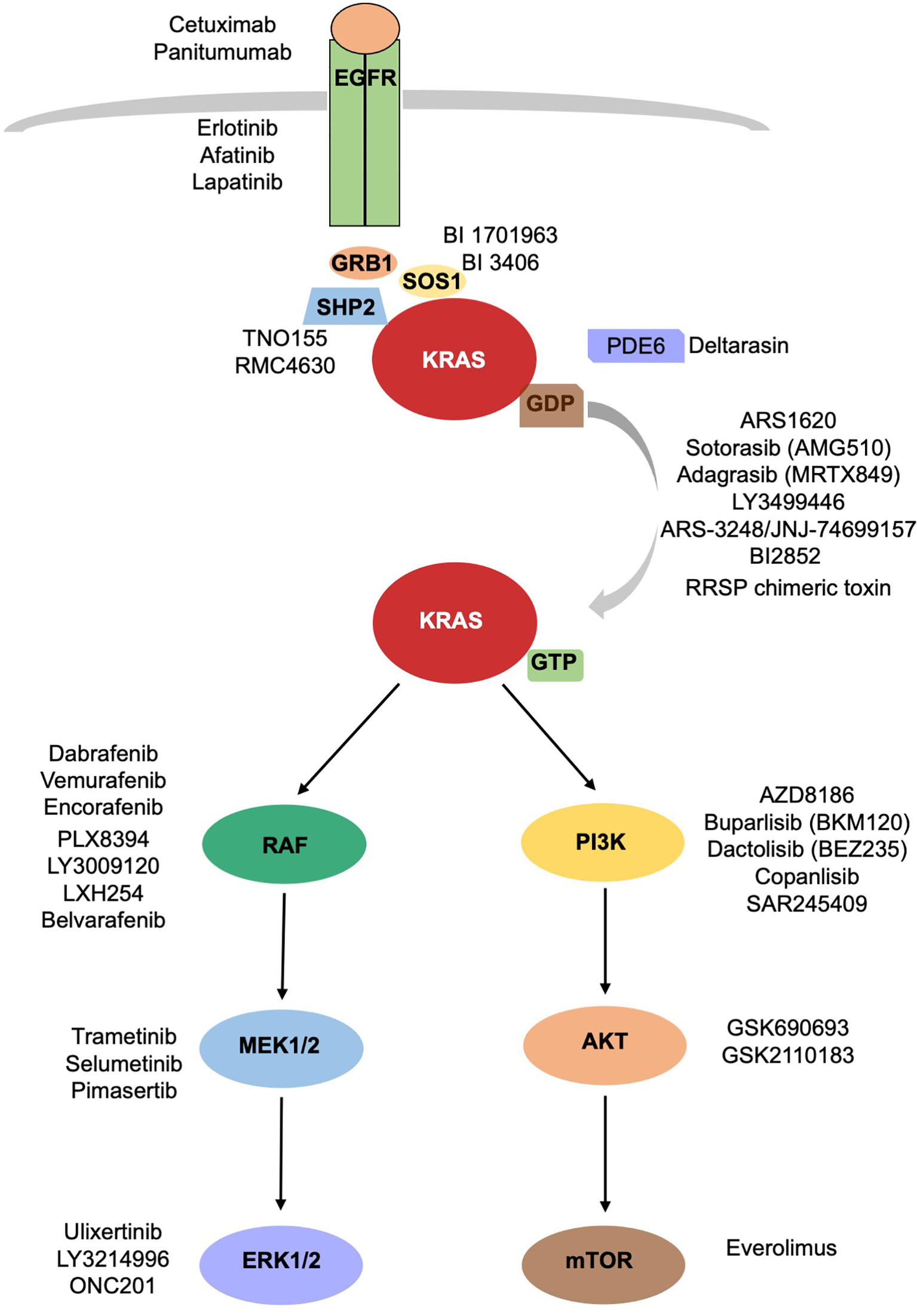
Figure 1 KRAS signaling cascade. Inhibitors of KRAS and upstream and downstream mediators of KRAS are reported.
Inhibiting KRAS Processing and Activation
Different critical steps are necessary for KRAS activation: nucleotide exchange, localization, processing, effector binding. The blockage of each of them could prevent KRAS activation.
- Nucleotide exchange: The previously described pan-KRAS inhibitors should belong to this category despite their name, since their function is linked to SOS1 (GEF) binding and not KRAS directly. Nucleotide exchange is also favored by SHP2, across their binding to GRB2 and SOS1. Different SHP2 inhibitors, including TNO155 and RMC-4630, directed versus non-receptor protein tyrosine phosphatase, are being clinically evaluated in combination therapies in advanced solid tumors (20, 21).
- Processing: prenylation–proteolysis–methylation.
Many efforts have been made to inhibit RAS farnesylation, aiming to prevent its membrane localization. These efforts led to the development of tipifarnib, a small molecule farnesyl transferase inhibitor (22). However, tipifarnib has been evaluated in HRAS mutated cancers only, since KRAS is prenylated by geranylgeranyl transferase and does not need farnesylation for membrane localization. Unfortunately, a dual inhibition of both transferases did not inhibit KRAS prenylation in human patients (23).
- Localization: KRAS splices into KRAS 4A and KRAS 4B. KRAS4B needs a chaperone, PDE6d, to translocate to the membrane surface. Deltarasin, a PDE6d inhibitor, prevents PDE6d from binding to KRAS, causing accumulation of KRAS 4B onto endomembranes (24). It is not clear yet if the cellular effects are due to KRAS inhibition or to other PDE6d effectors inhibition.
Targeting Downstream Mediators of Intracellular Signaling
Activated KRAS induces RAF proteins phosphorylation and dimerization with consequent activation of their kinases. There are three isoforms of RAF, represented by BRAF, RAF1/CRAF, and ARAF. Numerous studies conducted on BRAF, which is the best characterized isoform, have led to the development of therapies targeting V600E mutation specifically. These targeted therapies are widely used in melanoma and recently have been approved for the treatment of BRAF mutated NSCLC. Unfortunately, the application of BRAF inhibitors vemurafenib, dabrafenib, and encorafenib has failed in KRAS driven tumors, because of the paradoxical activation of ERK1/2. BRAF inhibitors, indeed, bind to BRAF and induce the heterodimerization BRAF/RAF1. The binding of BRAF inhibitors to BRAF mediates an allosteric activation of RAF1, with consequent MEK/ERK activation (25). Novel panRAF inhibitors, known as paradox breakers, have been developed to overcome this effect. Among these panRAF inhibitors we count PLX8394, which seems to have higher affinity with BRAF homodimers and BRAF/RAF1 heterodimers (26, 27), and LY3009120, which blocks the kinase activity of RAF dimers (28). Despite the promising data seen in preclinical studies (29, 30), LY3009120 failed to demonstrate efficacy in early clinical trials as monotherapy. A phase I study conducted in patients affected by RAS or BRAF mutated advanced tumors reported as best response stable disease in 8 of 51 patients (15%) with no complete or partial response achieved (31). LXH254 and belvarafenib are panRAF inhibitors also in active clinical development. A phase I clinical trial with LXH254 alone and in combination with an anti PD1 antibody is ongoing (NCT02607813). Belvarafenib has been tested in a phase I study including patients affected by advanced solid tumors harboring RAS or BRAF mutations, demonstrating good safety profile and antitumor activity (32). A study exploring its use in combination with anti-MEK agents is ongoing (NCT03284502).
MEK inhibitors, such as selumetinib, have been tested in KRAS mutated NSCLC as single agents and in combination with chemotherapy, without showing any clinical benefit (33). The same results have been seen with trametinib (34) and pimasertinib (35) in pancreatic cancer. This failure has been attributed partially to vertical compensation mechanisms of upstream elements, such as RTKs, BRAF, or KRAS, that lead to ERK reactivation, or alternative compensatory mechanisms as the activation of PI3K-AKT-mTORC1 pathway.
ERK inhibitors, such as ulixertinib/BVD523 and LY3214996, have been tested in phase I clinical trials (NCT02608229, NCT02857270). Ulixertinib has recently moved to phase II clinical development. The frequent reactivation of ERK observed during treatment with MEK or BRAF inhibitors calls for a better characterization of ERK inhibitors. This class of inhibitors is being studied accurately, to evaluate their employment in vertical combination (2, 36). Other strategies have been tested to target simultaneously different molecules aiming to vertical combination (NCT01229150, NCT02230553, NCT02258607, NCT04185883, NCT04330664) (Table 1).

Table 1 Clinical trials investigating combination treatments reported as “Drug 1” and “Drug 2” that target effectors of the same pathway (vertical combination). For each trial the study phase and the setting of patients is indicated.
The other pathway activated by KRAS is the PI3K-AKT-mTORC1 pathway, important for cellular proliferation, motility, and survival. Its persistent activation due to KRAS mutation contributes to cancer progression. Class I PI3K phosphorylates PIP2, which attracts AKT to plasma membrane and induces mTOR activation. Inhibition of PI3K pathway using AKT inhibitors failed in in vitro and in vivo trials, perhaps because of the activation of other signaling pathways. The compensatory mechanisms between the two pathways, MAPK and PI3K pathway, that emerge blocking one of them, led to the idea of blocking both pathways simultaneously. The combination of MEK inhibitors and AKT inhibitors showed promising results in pancreatic cancer in in vitro and in vivo studies (37). In a similar way, the combination of PI3K inhibitors with ERK inhibitors (38), MEK inhibitors (NCT01363232, NCT01337765, NCT01392521, NCT01390818) or RAF inhibitors are being tested with promising results in early clinical trials. However, at cost of higher toxicity associated to the double treatment (39).
Synthetic Lethal Partners of KRAS
Since direct inhibition of KRAS has been proven to be exceptionally challenging, one potential strategy to target tumors dependent upon this oncogene has been through the identification of its synthetic lethal partners. Synthetic lethal partners are genes that if mutated individually are compatible with viability, but the simultaneous perturbation of their expression or pharmacological inhibition of their products determines cell death (40). Synthetic lethality can be exploited in order to target tumor cells harboring undruggable mutations. An example of synthetic lethality is represented by the sensitivity of BRCA mutant cells to PARP inhibition (41). Synthetic lethal partners of KRAS could be downstream of its pathway or acting in parallel adaptative signaling. Targeting synthetic lethal partners should reduce the risk of adverse events because mutated cancer cells are more sensitive to this strategy compared to normal cells (42).
Among the different methods used to identify novel KRAS synthetic lethal partners, RNA interference has been the approach initially and more frequently applied (42). Unfortunately, evidence suggests that the reproducibility of this technology could be limited by the library quality and off target effects (43). More recently, CRISPR-Cas9 screening technology has been applied to loss-of-function genetic screening, enabling the complete knockout of target genes, that has been useful in identifying essential genes in KRAS mutant cancer cells (44). Results from different studies looking for synthetic lethal partners are scarcely overlapped and attempts to reproduce published KRAS synthetic lethal targets failed (45, 46). The novel CRISPR-Cas9 technology improves genetic perturbation but is not able alone to overcome limitations associated with different cellular and genetic contexts. Interestingly, a meta-analysis of published synthetic lethal screens highlighted that different studies’ results overlap at the pathway rather than at a gene level (47). Furthermore, these differences have been ascribed to changes in genetic context or cellular conditions.
Several studies have identified different putative KRAS synthetic lethal partners (Table 2). Inhibitors of the MAPK pathway showed greater sensitivity in KRAS mutated cancers compared to the wild-type ones (58). Probably because of their sole cytostatic effect, MEK inhibitors have proved no clinical efficacy as single agents in RAS mutant cancers (59). The combination of a MEK inhibitor with an inhibitor of the antiapoptotic BCL-XL led to increased apoptosis in many KRAS mutant cell lines from different histologies, and tumor regression in in vivo lung cancer mouse models (48). A phase 1b/2 trial is investigating the safety, pharmacokinetics, pharmacodynamics, and clinical activity of the combination of the MEK inhibitor trametinib and the BCL2-family inhibitor navitoclax (ABT-263) in patients with KRAS or NRAS mutated advanced solid tumors (NCT02079740). An interim analysis showed a good safety profile and initial signs of efficacy, in particular in gynecologic tumors. Evidence coming from other in vitro and in vivo studies demonstrated efficient induction of apoptosis in KRAS or BRAF mutant colorectal cancer cell lines treated with navitoclax in combination with the TORC1/2 inhibitor AZD8055 but not in the wild-type controls. Similar results were obtained in murine models (60). IGF1R and MEK inhibition resulted in growth inhibition of KRAS muted NSCLC cell lines and murine tumors (61). FGFR1 inhibition combined with the MEK inhibitor trametinib has shown to mediate cell death in KRAS-mutant lung cancer both in vitro and in vivo (49).
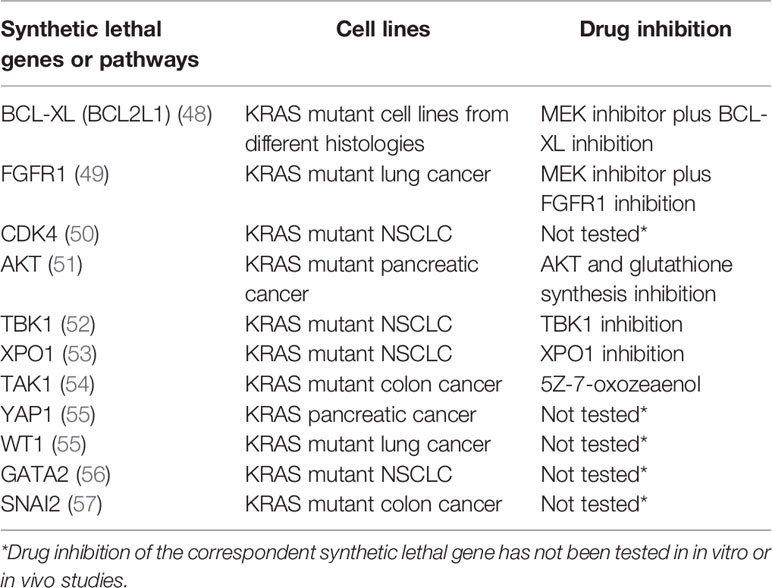
Table 2 RAS synthetic lethal partners and KRAS mutant cell lines in which they have been identified. Synthetic lethal gene inhibition is reported if tested.
CDK4 has been proposed as synthetic lethal partner in KRAS mutant NSCLC. This synthetic lethal interaction was observed only in lung cancer, not in colon or pancreatic cancer, pointing to a different tissue specific dependency of KRAS signaling (50). Targeting AKT and the glutathione antioxidant pathway mimicking Nrf2 ablation inhibited pancreatic adenocarcinoma tumors ex vivo and in vivo (51). NF-κB pathway has a central role in KRAS mutated cancers (62, 63). RAL-GEF family is one of the effectors of KRAS and mediates the activation of NF-κB, contributing to oncogenesis (64). NF-κB inhibition in a mouse model of lung adenocarcinoma expressing KRASG12D and lacking p53 has been demonstrated to reduce tumor development (65).
Another putative synthetic lethal partner of oncogenic KRAS is represented by the IκB kinase (IKK)–related kinase Tank-binding kinase-1 (TBK1). TBK1 is activated by RalB, a small GTPase downstream of KRAS belonging to the Ral signaling pathway, and Sec5, a component of the exocyst (66). TBK1 regulates an autocrine CCL5 and IL-6 signaling, inducing carcinogenesis in KRAS mutated cancer (52). Furthermore, activated TBK1 promotes NF-κB signaling through BCL-XL and the c-Rel protooncogene. Inhibiting TBK1 induces cell death in KRAS driven NSCLC adenocarcinoma murine models.
The JAK-STAT signaling pathway has a recognized role in pancreatic cancer development. In KRAS driven pancreatic cancer models, inhibiting JAK1/2 and TBK1 with momelotinib showed preclinical efficacy in vitro and in vivo (52). Nevertheless, to date it has not exhibited signs of activity in human pancreatic cancer (67).
XPO1 has also been proposed as synthetic lethal partner of KRAS. XPO1, overexpressed in many types of human cancers, is an export receptor in charge of the nuclear-cytoplasmic transport of many proteins. XPO1 has been proposed as a therapeutic target in several tumors including KRAS-mutant lung cancer. The effect of XPO1 inhibition consists in the accumulation of nuclear IκBα and consequent suppression of NFκB activity. Studies conducted on KRAS-mutant NSCLC cells showed that inhibition of the nuclear export XPO1 leads to a synthetic lethal interaction with oncogenic KRAS (53).
TAK1 has been suggested as mutant KRAS synthetic lethal target in colon cancer (54). In APC/KRAS mutant cells, KRAS mediates TAK1 activation and enhances Wnt activity by stimulating BMP-7 secretion and BMP signaling. TAK1 inhibition prompted apoptosis in KRAS-dependent colon cancer cells. However, TAK1 dependency may not be restricted to colon cancer, and approaches in targeting TAK1 have shown activity in other KRAS dependent tumors as well (68–70).
The transcription factor YAP1 is sustained by TAK1 and mediates KRAS independent growth (71, 72). YAP1 has been shown to overcome KRAS blockade to prompt pancreatic cancer growth in murine models (55). Representing a central hub in resistance to RAF and MEK inhibition, targeting YAP1 could represent a combination therapy in KRAS mutated cancers (73).
Loss of the transcription factor Wilms tumor 1 (WT1) has been correlated with decreased proliferation and increased cell senescence in KRAS driven cancer cell lines (55). However, WT1 remains not druggable to date (74).
The transcription factor GATA2 has been identified as a synthetic lethal target in RAS pathway mutant NSCLC models. However, GATA2 itself remains undruggable (56).
Deficiency of the DNA repair machinery has been described in KRAS mutant cells (47). BRCA1 is a strong synthetic lethal partner of PARP inhibition. PARP inhibition has also been proposed as a putative effective strategy in KRAS mutant cells.
RAS signaling is a known mediator of epithelial-mesenchymal transition (EMT). Thus, EMT regulators could represent therapeutic targets in KRAS driven tumors (75). The SNAI2 gene encoding SNAIL, a transcription factor and regulator of EMT, has been identified as a KRAS synthetic lethal target in colorectal cancer cell lines (57).
Direct targeting of KRAS has been approached and the need for targeting synthetic lethal partners could be questioned. However, synthetic lethal partner inhibitors could be used in the future in combination with direct inhibition in order to overcome possible escape mechanisms.
Targeting Metabolic Reprogramming in KRAS Mutant Cancers
Studies conducted using murine pancreatic cancer models have shown that KRASG12D stimulates the expression of glucose transporter 1 (GLUT1) and glycolytic enzymes and conveys glucose intermediates into the hexosamine biosynthesis pathway (HBP) and non-oxidative pentose phosphate pathway (PPP). The inhibition of the HBP gene (Gfpt1) or non-oxidative PPP genes (Rpia or Rpe) suppresses the KRAS dependent tumor growth (76). It has been shown that an increase in glucose uptake through enhanced GLUT1 expression is dependent on KRAS and BRAF mutation in colorectal cancer cell lines and sustained their survival (77). Furthermore, glucose deprivation with a glycolysis inhibitor suppressed tumor growth. Mutated KRAS determines higher 18F-fluorodeoxyglucose accumulation possibly by upregulation of GLUT1 (78). A retrospective study reported a significantly higher 18F-fluorodeoxyglucose accumulation detected with positron emission tomography in KRAS mutant colorectal cancer patients compared with wild-type ones (79). High levels of vitamin C have been found to selectively kill colorectal cancer cells harboring KRAS or BRAF mutations. The increased uptake of the oxidized form of vitamin C through GLUT1 causes oxidative stress and cell death only in KRAS or BRAF mutant cells (80).
Cancer cells are characterized by increased anabolic metabolism, which requires the use of the amino acid glutamine. It has been demonstrated that oncogenic KRAS mediates the reprogramming of glutamine metabolism in pancreatic adenocarcinoma cells by modifying the transcription of metabolic enzymes in a noncanonical pathway of glutamine (81). However, the tissue of origin and the microenvironment can impact on metabolic features. For example, pancreatic cells do not depend on the branched-chain amino acid (BCAA) processing enzymes Bcat1 and Bcat2, which enables BCAAs to be utilized as a nitrogen source, contrary to NSCLC (82). In KRAS mutated colorectal cancer cells the pentose phosphate pathway has been demonstrated to be essential for the growth in aerobic conditions and glutamine conversion into α-ketoglutarate and alanine aminotransferase for KRAS induced anchorage-independent growth (83). In KRAS driven lung cancer mouse models mitochondrial metabolism and mitochondrial reactive oxygen species generation, which is allowed by glutamine conversion into α-ketoglutarate, are essential for KRAS induced tumorigenicity (83). Models obtaining the suppression of KRAS led to reveal potential KRAS independent escape mechanisms. In KRAS G12D mouse model of pancreatic cancer surviving cells responsible for tumor relapse rely on oxidative phosphorylation, making the combined inhibition of the KRAS pathway and mitochondrial respiration a possible therapeutic strategy (84).
Autophagy is a mechanism characterized by degradation of intracellular components. It is stimulated by oxidative stress, nutrient shortage, and protein damage through inhibition of the AMPK and mTOR pathways and the activation of the unfolded protein response system (85). Pancreatic adenocarcinoma tumors show raised autophagy, whose inhibition demonstrated to reduce tumor growth (86). However, the role of KRAS in autophagy remains controversial. In a study conducted in different cancer cell lines, KRAS mutation was not correlated with the dependance to autophagy (87). The use of hydroxychloroquine, that inhibits autophagy preventing lysosome acidification, failed to show therapeutic activity in pancreatic cancer patients (88). However, several studies are ongoing to investigate hydroxychloroquine in combination with chemotherapy in pancreatic cancer (NCT04524702, NCT04132505). The deficiency of atg7, an essential autophagy gene, in KRASG12D mutated NSCLC mouse models determined the accumulation of dysfunctional mitochondria and inhibited cancer growth (89).
RAS proteins have been demonstrated to enhance macropinocytosis, a process by which extracellular fluid and extracellular proteins are internalized through vesicles. Macropinocytosis inhibition with amiloride blocked the growth of KRAS mutated pancreatic cancer xenografts (90).
The metabolism of fatty acids has been correlated with KRAS mutation in NSCLC. It has been shown that KRAS regulates lipid homeostasis and Acyl-coenzyme A synthetase, an enzyme involved in fatty acid metabolism, essential for mutant KRAS lung cancer tumorigenesis in vivo (91). Furthermore, KRAS has been reported to promote lipogenesis through the induction of fatty acid synthase in lung cancer (92).
In KRAS/p53 mutant lung cancer mouse models the inhibition of HSP90 combined with rapamycin was shown to promote endoplasmic reticulum stress and mitochondrial damage and tumor regression (93).
Putative Escape Pathways to KRAS Inhibition
Although a clinically relevant strategy for effectively targeting KRAS in all of its mutated status seems still far to be developed, potential mechanisms of resistance for KRAS inhibition have been already explored in several preclinical models.
KRASG12C inhibitors bind specifically to inactive GDP-bound form of KRAS. Thus, the potency of KRASG12C inhibition is reduced by increased RTK activity, that promotes cycling of KRASG12C to its active GTP-bound form, hindering KRASG12C drug inhibition (94). Furthermore, the suppression of nucleotide exchange activity downstream of tyrosine kinases enhances KRASG12C inhibition, suggesting possible combination strategies.
By targeting KRASG12C with ARS-1620, the phosphorylation of multiple RTKs was augmented in different ways across diverse KRASG12C mutant models (95). Synergistic effects of RTK inhibitors combined with KRAS blockade may vary across different tumor cell types (96).
KRASG12C inhibitors induce growth inhibition mainly by targeting MAPK/ERK pathway. The redundancy of parallel growth factor signals can bypass KRAS blockade, underlying intrinsic resistance to KRASG12C inhibitors (96). However, combining this strategy with the inhibition of SHP2, a phosphatase that mediates signaling of different RTKs to KRAS, blocked the feedback reactivation and enhanced efficacy of KRASG12C inhibition in vitro and in vivo, also in models refractory to KRASG12C inhibition alone (16). This encouraging preclinical evidence led to move to an early-phase clinical trial investigating combination therapies aimed to simultaneously targeting KRASG12C and SHP2 (NCT04330664, NCT04185883). Another central node stimulated by RTK is represented by SOS1, a guanine nucleotide exchange factor activating KRAS (97). The SOS1 inhibitor BAY-293 can synergize with the KRASG12C inhibitor ARS-853 reducing cell proliferation (98).
Noteworthy, the scenario of KRAS mutated cancer is extremely heterogeneous and complex. The dependency on KRAS signaling varies across different KRAS mutant cancer types and could reflect the variability in the tumor response, representing a possible mechanism of intrinsic resistance (75, 99). KRASG12C colorectal cancer cells have been shown to have higher basal EGFR activity compared to NSCLC cells, leading to higher phospho-ERK rebound and thus resistance to KRASG12C blockade (100). This finding is consistent with clinical results, in which activity of sotorasib seems to be lower in colorectal cancer patients. Thus, combining KRASG12C inhibition with EGFR inhibition could represent an effective treatment strategy. Indeed, in KRAS mutant cancer cells KRASG12C inhibition with ARS-853 was increased by the combination with EGFR inhibitors (94).
Other adaptive resistance mechanisms for KRASG12C inhibition involved reactivation of MAPK pathway and failed PI3K–AKT pathway inactivation (96). The combination of the KRASG12C inhibitor ARS1620 with PI3K inhibition has demonstrated to be effective in vitro and in vivo in different models resistant to single-agent KRASG12C inhibitor. Also a strategy of blocking PI3K effectors, such as AKT and mTOR, together with KRASG12C, proved to be effective in preclinical studies (94, 101).
Activation of RTK signaling in KRASG12C mutant cancers could limit the KRASG12C therapeutic inhibition both by increasing regulation of GTPase activity and promoting KRAS independent ERK and mTOR/S6 pathway activation (16). The combination of the mTOR inhibitor vistusertib with the KRASG12C inhibitor MRTX849 also improved antitumor activity in vitro.
mTOR and IGF1R could also play a central role in KRAS inhibition resistance. The addition of mTOR and IGF1R to the KRASG12C inhibitor ARS1620 improved efficacy in KRASG12C mutant lung cancer in in vitro and in vivo mouse models.
Another mechanism proposed for the adaptive resistance to KRASG12C inhibitors is represented by feedback reactivation of wild-type RAS (95). In KRASG12C models, an adaptive RAS pathway reactivation after a rapid KRASG12C inhibition with ARS-1620 and AMG-510 is driven by activation of wild-type RAS (NRAS or HRAS) mediated by RTKs and is not inhibited by KRASG12C inhibitors.
In response to KRASG12C inhibitors, proliferation of cancer cells can be resumed through the production of new KRASG12C (102). The distribution of newly synthetized KRASG12C between the active and inactive state, which is the only conformation bound by KRASG12C inhibitors, modulated the divergent response. Cells producing new KRASG12C, which is converted to the active and drug insensitive state, are able to escape KRASG12C inhibition.
Another possible mechanism responsible for resistance to KRASG12C inhibitors is represented by the presence of additional KRAS genetic alterations that can potentiate nucleotide exchange or impair inherent GTPase activity (94). Furthermore, the resistance to KRASG12C inhibitors could be cause by the presence of a heterogeneous spectrum of KRAS mutations in the same patient (103).
Moreover, aurora kinase A (AURKA) was shown to promote drug inhibition escape by interacting with KRASG12C and c-Raf (102). In KRASG12C mutant cancer models a synergic effect was demonstrated with the KRASG12C inhibitor ARS-1620 and the AURKA inhibitor alisertib (102).
In an inducible KRASG12D pancreatic cancer mouse model, the amplification and overexpression of the transcriptional coactivator Yap1 has been demonstrated to be a potential KRAS independent bypass mechanism (55). In this study, indeed, after KRAS extinction and complete tumor regression in all mice, about two thirds of them relapsed. At least three possible resistance mechanisms have been identified. In about half of the relapsed tumors, a KRAS transgene amplification has been found, meaning that genomic alteration on target itself could bypass target blockade. Another possible mechanism leading to tumor relapse is represented by the compensatory activation of other key growth pathways. According to this, previous findings showed that expression of receptor tyrosine kinases bypasses the KRAS dependency (75). Furthermore, a novel mechanism of resistance to KRAS inhibition through a Yap1-mediated transcriptional program has been proposed. Although Yap1 is not sufficient for driving de novo pancreatic cancer development, it can drive tumor recurrence in inducible KRASG12D pancreatic cancer models (104).
Since increased cell proliferation and antiapoptotic signaling could represent a possible mechanism of resistance to KRASG12C inhibitors, their combination with chemotherapy, that inhibits cell proliferation, could boost responses and deter resistance. There are also evidences showing a synergistic effect of cell cycle inhibitors like palbociclib in combination with KRASG12C inhibitors (14). Indeed, genetic alterations in CDKN2A, CDK4, or CCND1 can be found in up to 20% of KRAS mutated NSCLC cancers (105).
Overall, these data support the utility of combination therapies in overcoming intrinsic and adaptive resistance and enhancing clinical benefit of KRASG12C inhibitors.
KRAS Reprogramming of Tumor Microenvironment and Potential Implication for Immunotherapeutic Approaches
The development and progression of tumors depend not only on oncogenic mutations but also on the interaction with the surrounding microenvironment, which creates a nurturing niche for cancer cells. KRAS mutant tumors are typically characterized by an immunosuppressive state (106). KRAS signaling induces in tumor cells the expression of immunomodulatory factors and inflammatory cytokines, with subsequent recruitment of neutrophils and myeloid-derived suppressor cells (MDSCs), creating an immunosuppressive tumor microenvironment. KRASG12D was shown to induce ELR CXC chemokines in human embryonic kidney cells (107). Large production of chemokines was observed also in KRAS mutant pancreatic cell lines (108). In murine lung cancer models KRASG12D demonstrated to stimulate CXCL1, 2, and 5, leading to neutrophils and macrophages infiltration (109). A tumor growth promoting role for CXCL2 and CXCL5 was also found in KRAS mutated pancreatic cancer cell lines (110).
The binding of CXCL3 with CXCR2 and the production of GM-CSF induce the accumulation of MDSCs. In colorectal cancer models KRASG12D has shown to downregulate the expression of interferon regulatory factor 2 (IRF2), which in turn suppresses CXCL3 expression, resulting in high expression of CXCL3 and promoting migration of myeloid-derived suppressor cells to the tumor microenvironment (111). Responsiveness to anti-PD-1 therapy was increased in colorectal cancers with higher IRF2 expression. The tumor microenvironment is populated by other myeloid cells, such as alternatively activated immune suppressive M2 macrophages, and lymphoid cells, including CD4+FoxP3+ T regulatory (Treg) cells, CD19+IL-10+ B regulatory (Breg) cells, and interleukin (IL)-17-producing T helper (Th)17 cells (112, 113).
IL-6 expression has been correlated with KRAS mutated signaling and seems to play a central role in shaping the immune milieu. In pancreatic cancer models IL-6 signaling was accompanied by an infiltration of myeloid cells and lymphocytes (114).
Upregulation of IL-10 transcription through MEK/ERK/AP-1 pathway was shown in KRAS mutant colorectal cancer cells and its secretion was required for the conversion of CD4+ T cell to CD4+FoxP3+ Treg cells (113). High IL-10 levels were associated with a worse prognosis in patients with KRAS mutated cancers (106).
The capacity of TGF-β in regulating the immune system and inhibiting inflammation is acknowledged since many years (115). Either RAS downstream MAPK and PI3K pathways seem to contribute to TGF-β production (116). In KRAS mutated colorectal cancer lines TGF-β secretion was required for Treg cell differentiation as mediated via the MEK/ERK/AP-1 pathway (117). In a lung cancer mouse model, it has been demonstrated that IL-10 and TGFβ secreted by KRAS mutated cancer cells, induce the conversion of CD4+ CD25- T-cells into FOXP3+/CTLA4+/CD122+ T regulatory cells (Tregs) (117). In immune-excluded colorectal cancer models the inhibition of TGF-β promoted anti-tumorigenic immune infiltration, restoring sensitivity to PD-L1/PD-1 blockade (118). Considering that pancreatic cancer is a poorly immunogenic, “cold” tumor, novel approaches targeting the microenvironment have been explored. Signals of activity using TGF-β-inhibitor galunisertib in combination with gemcitabine have been showed in advanced pancreatic cancer patients (119). Moreover, conventional therapy is able to shape the immune landscape in KRAS mutant tumors. It has been demonstrated that mutant KRAS pancreatic cancer cell lines treated with chemotherapy activate MAPK and NF-κB pathways, inducing the secretion of inflammatory cytokines able to enhance monocyte differentiation towards MDSCs and thus counteracting therapy response (120). Other mechanisms have also been proposed. High circulating IL-8 levels have been suggested to be a potential predictive biomarker of resistance to nanoliposomal irinotecan (nal-IRI) in gemcitabine-refractory patients with pancreatic cancer (121). Nal-IRI has been developed to exploit tumor-associated macrophages (TAMs) for accumulation and conversion into its active metabolite. IL-8 has shown an increased mobilization of immature CD11b+Gr-1+ myeloid cells, thus, it has been hypothesized that high IL-8 levels and low TAMs activity could be correlated with lack of nal-IRI activity (122).
Mutated KRAS has a central role in pancreatic cancer development and growth through regulation of T cell cytokines in the microenvironment, therefore shaping the metabolic cancer cell landscape (123). The presence of T cells in the microenvironment is of crucial importance considering their therapeutic potential with immune checkpoints inhibitors. TH1 cells are generally associated with response to immunotherapy and promote CD8+ T cell infiltration (124). TH2 cells prevent tumor rejection and promote tumor growth (125). In addition to promoting macrophage M2 polarization, IL-4, which is abundantly produced by TH2 cells, has been recently demonstrated to stimulate tumor cell proliferation through KRAS in pancreatic cancer. Mutant KRAS in cancer cells stimulates cytokine receptor expression such as such as IL4R, IL2Rγ, and IL13Rα1 that, in turn, facilitate the Jak1-Stat6-cMyc pathway activation by IL-4 and IL-13. cMyc, which is activated by Stat6, is required for metabolic reprogramming and drives glycolysis.
GM-CSF can exert both immune suppression and stimulation and the balance could be dependent on its levels (106). KRASG12D is responsible for GM-CSF transcription through MAPK and PI3K pathways in pancreatic cancer cells (126). The correlation between reduced overall survival and high levels of GM-CGF observed in pancreatic cancer patients is probably due to the ability of GM-CSF to cause MDSC differentiation and inhibition of T cell proliferation (120).
Although IL-10 and TGF-β can induce a shifting of macrophages towards the alternative activated immunosuppressive M2 state, a clear correlation between their secretion by KRAS mutated cancer and macrophage polarization has not been established (106). In pancreatic cancer both M1 and M2 macrophage phenotypes have been hypothesized to play an important role in tumor initiation and progression and growth (127). In advanced pancreatic cancer macrophages represent the most abundant immune cell population, playing mainly an immunosuppressive role (128). The correlation of macrophages with prognosis in lung cancer patients remains controversial (129). Mechanisms of macrophage recruitment in KRAS mutant lung cancer are not well defined, but it has been hypothesized a role for CXCR2 signaling (130).
Also, a crosstalk between cancer-associated fibroblasts (CAFs) and KRAS mutant cancer cells has been shown. In a KRASG12D mutant lung cancer and CT26 colon cancer mouse models, the depletion of fibroblast activation protein (FAP), expressed by CAFs, was demonstrated to inhibit tumor cell proliferation through accumulation of collagen and decrease of myofibroblast content and blood vessel density (131). In pancreatic cancer cells KRAS activates Hedgehog pathway, which is involved in the generation and maintenance of the typical dense tumor stroma (132). In a pancreatic cancer mouse model, mutant KRAS induced the expression of Sonic hedgehog, which in turn activated the transcription factor GLI1. GLI1 regulates IL-6 expression in fibroblasts by binding its promoter and IL-6/STAT3 axis is involved in pancreatic carcinogenesis (133).
Pancreatic stellate cells are essential in disease progression and are the most represented cell type of tumor stroma (134). TGF-β and many other factors secreted by pancreatic cancer cells contribute to the activation of stellate cells which, in turn, produce and release several other growth factors and cytokines (106). Pancreatic stellate cells and mutant KRAS cancer cells have a synergistic effect on the immune microenvironment.
The composition of the immune population and its crosstalk with KRAS altered tumor cells have a central role not only in determining tumor onset and progression but also in sensitivity to immunotherapeutic drugs (135). A study reported that oncogenic RAS signaling can upregulate PD-L1 expression on tumor cells through a mechanism of increased PD-L1 mRNA stability (136). Indeed, KRAS-induced MEK signaling promotes the inhibition of tristetetrapolin, a negative regulator of PD-1 expression. In human lung and colorectal tumors, RAS pathway activation has been correlated with elevated expression of PD-L1. It has been reported that PD-1 and PD-L1 expression is more frequent in KRAS mutated NSCLC (137). Some studies have already shown a clinical relevance of the combination of MEK inhibitors with immunotherapy (138, 139). An ongoing phase 1b/2 trial is testing the activity of the treatment with MEK inhibitor binimetinib in combination with nivolumab or nivolumab plus ipilimumab in pretreated patients with microsatellite stable metastatic colorectal cancer harboring a RAS mutation (NCT03271047).
Differently from other TKIs, novel KRASG12C inhibitors are specifically selective for the mutation variant of KRAS and should not have any effects on the immune cells directly. Thus, KRAS inhibition in cancer cells can shift the balance from an immunosuppressive state to a microenvironment favoring effective antitumor activity and can sensitize tumors to checkpoint inhibitor therapy.
The predictive role of KRAS status to immune checkpoint inhibitors in NSCLC is controversial. Although KRAS status has never been included as stratification factor in clinical trials with immune checkpoint inhibitors for NSCLC, a subgroup analysis of the CheckMate 057 trial revealed that patients with tumors harboring a KRAS mutation had a greater clinical benefit with nivolumab compared to docetaxel (140). A meta-analysis conducted on five prospective randomized trials has revealed that (141) KRAS mutation is associated with a better outcome in patients treated with PD-1/PD-L1 inhibitors in second-line setting (142). However, the study failed to prove that KRAS status is an independent predictive factor for treatment. The retrospective IMMUNOTARGET registry confirmed a greater benefit from immune checkpoint inhibitors in patients with KRAS mutated NSCLC compared to those with EGFR mutant tumors (143). Another retrospective study found similar activity of immunotherapeutic agents in KRAS mutated compared to KRAS wild-type lung cancer patients (141). The mutation variants KRASG12V, KRASG12D, and KRASG13C have been associated with higher tumor expression of PD-L1 compared with other variants in NSCLC.
Interestingly, some evidence supports the hypothesis that STK11/LKB1 co-mutation in KRAS mutated NSCLC could represent a negative predictive factor for immunotherapy (144). LKB1 loss is involved in the suppression of stimulator of interferon genes (STING), determining a decreased expression of type I interferon genes and chemokines that facilitate T-cell recruitment (145). STING activation has been associated with response to immunotherapy and is stimulated by chemotherapy (146). A subgroup of STK11 and p53 co-mutated NSCLC is characterized by high STING- and immune-related gene expression. KRAS mutated tumors with co-occurring CDKN2A/B mutations have a scarce immune infiltrate and low PD-L1 expression, resulting in resistance to anti-PD-1 therapies (147). Another group of KRAS mutant NSCLC presents p53 co-mutation and they also have high PD-L1 expression, high T-cell infiltration and, thus, enhanced response to immunotherapy. For the resistance to anti-PD-1 observed in this latter group, a mechanism involving STAT signaling has been proposed (148). In KRAS/p53 mutant murine lung cancer models neurotrophic receptor tyrosine kinase 1 (NTRK1) has been found to be upregulated after treatment with PD-1 inhibitors and to regulate JAK/STAT signaling, promoting PD-L1 expression and CD8+ T cell exhaustion in the microenvironment.
p21-activated kinase 4 (PAK4) is a serine/threonine kinase acting downstream of RAS signaling. PAK4 overexpression has been found in tumor biopsies of anti-PD-1 non-responders and was correlated with low T cell and dendritic cell infiltration across different cancer types, with a strong negative correlation in pancreatic cancer (https://doi.org/10.1038/s43018-019-0003-0). The genetic knockout of PAK4 augmented tumor infiltration by T cells and natural killer cells and pharmacological inhibition of PAK4 synergized with PD-1 blockade immunotherapy in melanoma mouse models, suggesting the possibility of enhancing the efficacy of immunotherapy also in KRAS mutant tumors.
Therefore, combining KRAS inhibition with immune checkpoint blockade has a strong biological rationale and could open the way to therapeutic options, reversing the innately immunoresistant phenotype of some RAS mutant cancers.
A recent study has suggested that the novel KRASG12C inhibitor sotorasib (AMG 510) can potentiate immune rejection when combined with anti-PD-1 immune checkpoint inhibitor (14). On one side, sotorasib promotes tumor regression by blocking growth and proliferation pathways, on the other side, it induces a change in the expression of immunomodulating factors in cancer cells, such as increased production of T-cell chemoattractants CXCL10 and CXCL11. The combination of sotorasib with anti-PD-1 determined complete regression in nine out of ten CT26 KRAS mutated colon carcinoma mice, which is one of the most immune-responsive mouse tumor models, and induced T cell memory. The immunological memory was demonstrated by the fact that the growth of isogenic KRAS G12D tumors in treated mice was impaired.
The phase 1b trial CodeBreakTM 101 testing the combination of sotorasib with anti-PD-1 is ongoing in patients with a KRASG12C advanced solid tumors (NCT04185883).
Further investigation about the synergistic association of KRASG12C and immune checkpoint blockade is warranted. It has to be explored if this combination will be effective only in tumors that are already moderately sensitive to immunotherapy or even in those intrinsically resistant to immune checkpoint inhibition.
Conclusions
Although KRAS is the most mutated oncogene in human cancer, it has considered to be undruggable because of its structural biology. Recently, exciting data of activity have been reported with KRASG12C inhibitors in early-phase clinical trials, raising a growing interest for KRAS inhibition, especially in lung cancer.
Different strategies are being explored in order to overcome resistance mechanisms and enhance the efficacy of KRAS inhibition, for example targeting synthetic lethal partners of KRAS. There is a hope that in the next future it will be achievable to block other mutation variants of KRAS other than G12C, making possible to exploit this approach also in other KRAS mutant tumors. Combinations of KRAS inhibitors and immune checkpoint inhibitors are being tested, since they showed a synergistic effect in a preclinical setting. Considering the immunosuppressive microenvironment characterizing KRAS mutant cancers, results from clinical trials utilizing this mechanism are anxiously awaited.
Many improvements have been made in targeting the oncogene KRAS, that was previously thought impossible to block, paving the way for a novel clinical field of research that will probably lead to new horizons in the future clinical practice.
Author Contributions
VM and DaM designed the review. VM and MG wrote the manuscript. VM, MG, CZ, AC, SCa, CP, SCo, FS, MB, DoM, MM contributed to literature revision. VF and DaM contributed to the final revision. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by Associazione Italiana per la Ricerca sul Cancro (AIRC) through the Investigator Grant n°23719 and 5x1000 Grant n°12182, by the Italian Ministry of Health through the Ricerca Finalizzata 2016 GR-2016-02361134 grant and by the patient associations “Nastro Viola” and “Voglio il Massimo” through their donations to DaM.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Tsuchida N, Ryder T, Ohtsubo E. Nucleotide sequence of the oncogene encoding the p21 transforming protein of Kirsten murine sarcoma virus. Science (1982) 217(4563):937–9. doi: 10.1126/science.6287573
2. Ryan MB, Corcoran RB. Therapeutic strategies to target RAS-mutant cancers. Nat Rev Clin Oncol (2018) 15(11):709–20. doi: 10.1038/s41571-018-0105-0
3. Melisi D, Budillon A. Pancreatic cancer: between bench and bedside. Curr Drug Targets (2012) 13(6):729–30. doi: 10.2174/138945012800564130
4. Melisi D, Calvetti L, Frizziero M, Tortora G. Pancreatic cancer: systemic combination therapies for a heterogeneous disease. Curr Pharm Des (2014) 20(42):6660–9. doi: 10.2174/1381612820666140826154327
5. Tamburrino A, Piro G, Carbone C, Tortora G, Melisi D. Mechanisms of resistance to chemotherapeutic and anti-angiogenic drugs as novel targets for pancreatic cancer therapy. Front Pharmacol (2013) 4:56. doi: 10.3389/fphar.2013.00056
6. Cox AD, Der CJ. Ras history: The saga continues. Small GTPases (2010) 1(1):2–27. doi: 10.4161/sgtp.1.1.12178
7. Holderfield M. Efforts to Develop KRAS Inhibitors. Cold Spring Harbor Perspect Med (2018) 8(7). doi: 10.1101/cshperspect.a031864
8. Scheffzek K, Ahmadian MR, Kabsch W, Wiesmuller L, Lautwein A, Schmitz F, et al. The Ras-RasGAP complex: structural basis for GTPase activation and its loss in oncogenic Ras mutants. Science (1997) 277(5324):333–8. doi: 10.1126/science.277.5324.333
9. Tahir R, Renuse S, Udainiya S, Madugundu AK, Cutler JA, Nirujogi RS, et al. Mutation-Specific and Common Phosphotyrosine Signatures of KRAS G12D and G13D Alleles. J Proteome Res (2020) 20(1):670–83. doi: 10.1021/acs.jproteome.0c00587
10. Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ. Drugging the undruggable RAS: Mission possible? Nat Rev Drug Discovery (2014) 13(11):828–51. doi: 10.1038/nrd4389
11. Consortium APG. AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discovery (2017) 7(8):818–31. doi: 10.1158/2159-8290.CD-17-0151
12. Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature (2013) 503(7477):548–51. doi: 10.1038/nature12796
13. Lanman BA, Allen JR, Allen JG, Amegadzie AK, Ashton KS, Booker SK, et al. Discovery of a Covalent Inhibitor of KRAS(G12C) (AMG 510) for the Treatment of Solid Tumors. J Med Chem (2020) 63(1):52–65. doi: 10.1039/d0md00096e
14. Canon J, Rex K, Saiki AY, Mohr C, Cooke K, Bagal D, et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature (2019) 575(7781):217–23. doi: 10.1038/s41586-019-1694-1
15. Hong DS, Fakih MG, Strickler JH, Desai J, Durm GA, Shapiro GI, et al. KRAS(G12C) Inhibition with Sotorasib in Advanced Solid Tumors. N Engl J Med (2020) 383(13):1207–17. doi: 10.1056/NEJMoa1917239
16. Hallin J, Engstrom LD, Hargis L, Calinisan A, Aranda R, Briere DM, et al. The KRAS(G12C) Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discovery (2020) 10(1):54–71. doi: 10.1158/2159-8290.CD-19-1167
17. Kessler D, Gmachl M, Mantoulidis A, Martin LJ, Zoephel A, Mayer M, et al. Drugging an undruggable pocket on KRAS. Proc Natl Acad Sci USA (2019) 116(32):15823–9. doi: 10.1073/pnas.1904529116
18. Hofmann MH, Gmachl M, Ramharter J, Savarese F, Gerlach D, Marszalek JR, et al. BI-3406, a potent and selective SOS1::KRAS interaction inhibitor, is effective in KRAS-driven cancers through combined MEK inhibition. Cancer Discovery (2020) 10.1158/2159-8290. doi: 10.1158/2159-8290.CD-20-0142
19. Biancucci M, Minasov G, Banerjee A, Herrera A, Woida PJ, Kieffer MB, et al. The bacterial Ras/Rap1 site-specific endopeptidase RRSP cleaves Ras through an atypical mechanism to disrupt Ras-ERK signaling. Sci Signaling (2018) 11(550). doi: 10.1126/scisignal.aat8335
20. Chen YN, LaMarche MJ, Chan HM, Fekkes P, Garcia-Fortanet J, Acker MG, et al. Allosteric inhibition of SHP2 phosphatase inhibits cancers driven by receptor tyrosine kinases. Nature (2016) 535(7610):148–52. doi: 10.1038/nature18621
21. Chen X, Zou F, Hu Z, Du G, Yu P, Wang W, et al. PCC0208023, a potent SHP2 allosteric inhibitor, imparts an antitumor effect against KRAS mutant colorectal cancer. Toxicol Appl Pharmacol (2020) 398:115019. doi: 10.1016/j.taap.2020.115019
22. Untch BR, Dos Anjos V, Garcia-Rendueles MER, Knauf JA, Krishnamoorthy GP, Saqcena M, et al. Tipifarnib Inhibits HRAS-Driven Dedifferentiated Thyroid Cancers. Cancer Res (2018) 78(16):4642–57. doi: 10.1158/0008-5472.CAN-17-1925
23. Lobell RB, Liu D, Buser CA, Davide JP, DePuy E, Hamilton K, et al. Preclinical and clinical pharmacodynamic assessment of L-778,123, a dual inhibitor of farnesyl:protein transferase and geranylgeranyl:protein transferase type-I. Mol Cancer Ther (2002) 1(9):747–58. doi: 10.1021/acs.chemrev.8b00201
24. Zimmermann G, Papke B, Ismail S, Vartak N, Chandra A, Hoffmann M, et al. Small molecule inhibition of the KRAS-PDEdelta interaction impairs oncogenic KRAS signalling. Nature (2013) 497(7451):638–42. doi: 10.1038/nature12205
25. Lito P, Rosen N, Solit DB. Tumor adaptation and resistance to RAF inhibitors. Nat Med (2013) 19(11):1401–9. doi: 10.1038/nm.3392
26. Zhang C, Spevak W, Zhang Y, Burton EA, Ma Y, Habets G, et al. RAF inhibitors that evade paradoxical MAPK pathway activation. Nature (2015) 526(7574):583–6. doi: 10.1038/nature14982
27. Yao Z, Gao Y, Su W, Yaeger R, Tao J, Na N, et al. RAF inhibitor PLX8394 selectively disrupts BRAF dimers and RAS-independent BRAF-mutant-driven signaling. Nat Med (2019) 25(2):284–91. doi: 10.1038/s41591-018-0274-5
28. Peng SB, Henry JR, Kaufman MD, Lu WP, Smith BD, Vogeti S, et al. Inhibition of RAF Isoforms and Active Dimers by LY3009120 Leads to Anti-tumor Activities in RAS or BRAF Mutant Cancers. Cancer Cell (2015) 28(3):384–98. doi: 10.1016/j.ccell.2015.08.002
29. Vakana E, Pratt S, Blosser W, Dowless M, Simpson N, Yuan XJ, et al. LY3009120, a panRAF inhibitor, has significant anti-tumor activity in BRAF and KRAS mutant preclinical models of colorectal cancer. Oncotarget (2017) 8(6):9251–66. doi: 10.18632/oncotarget.14002
30. Henry JR, Kaufman MD, Peng SB, Ahn YM, Caldwell TM, Vogeti L, et al. Discovery of 1-(3,3-dimethylbutyl)-3-(2-fluoro-4-methyl-5-(7-methyl-2-(methylamino)pyrido[2,3- d]pyrimidin-6-yl)phenyl)urea (LY3009120) as a pan-RAF inhibitor with minimal paradoxical activation and activity against BRAF or RAS mutant tumor cells. J Med Chem (2015) 58(10):4165–79. doi: 10.1021/acs.jmedchem.5b00067
31. Sullivan RJ, Hollebecque A, Flaherty KT, Shapiro GI, Rodon Ahnert J, Millward MJ, et al. A Phase I Study of LY3009120, a Pan-RAF Inhibitor, in Patients with Advanced or Metastatic Cancer. Mol Cancer Ther (2020) 19(2):460–7. doi: 10.1158/1535-7163.MCT-19-0681
32. Kim TW, Lee J, Shin SJ, Kim J-S, Kim YJ, Han HS, et al. Belvarafenib, a novel pan-RAF inhibitor, in solid tumor patients harboring BRAF, KRAS, or NRAS mutations: Phase I study. J Clin Oncol (2019) 37(15_suppl):3000–. doi: 10.1200/JCO.2019.37.15_suppl.3000
33. Janne PA, van den Heuvel MM, Barlesi F, Cobo M, Mazieres J, Crino L, et al. Selumetinib Plus Docetaxel Compared With Docetaxel Alone and Progression-Free Survival in Patients With KRAS-Mutant Advanced Non-Small Cell Lung Cancer: The SELECT-1 Randomized Clinical Trial. JAMA (2017) 317(18):1844–53. doi: 10.1001/jama.2017.3438
34. Infante JR, Somer BG, Park JO, Li CP, Scheulen ME, Kasubhai SM, et al. A randomised, double-blind, placebo-controlled trial of trametinib, an oral MEK inhibitor, in combination with gemcitabine for patients with untreated metastatic adenocarcinoma of the pancreas. Eur J Cancer (2014) 50(12):2072–81. doi: 10.1016/j.ejca.2014.04.024
35. Van Cutsem E, Hidalgo M, Canon JL, Macarulla T, Bazin I, Poddubskaya E, et al. Phase I/II trial of pimasertib plus gemcitabine in patients with metastatic pancreatic cancer. Int J Cancer (2018) 143(8):2053–64. doi: 10.1002/ijc.31603
36. Morris EJ, Jha S, Restaino CR, Dayananth P, Zhu H, Cooper A, et al. Discovery of a novel ERK inhibitor with activity in models of acquired resistance to BRAF and MEK inhibitors. Cancer Discovery (2013) 3(7):742–50. doi: 10.1158/2159-8290.CD-13-0070
37. Collisson EA, Trejo CL, Silva JM, Gu S, Korkola JE, Heiser LM, et al. A central role for RAF–>MEK–>ERK signaling in the genesis of pancreatic ductal adenocarcinoma. Cancer Discovery (2012) 2(8):685–93. doi: 10.1158/2159-8290.CD-11-0347
38. Hayes TK, Neel NF, Hu C, Gautam P, Chenard M, Long B, et al. Long-Term ERK Inhibition in KRAS-Mutant Pancreatic Cancer Is Associated with MYC Degradation and Senescence-like Growth Suppression. Cancer Cell (2016) 29(1):75–89. doi: 10.1016/j.ccell.2015.11.011
39. Shimizu T, Tolcher AW, Papadopoulos KP, Beeram M, Rasco DW, Smith LS, et al. The clinical effect of the dual-targeting strategy involving PI3K/AKT/mTOR and RAS/MEK/ERK pathways in patients with advanced cancer. Clin Cancer Res (2012) 18(8):2316–25. doi: 10.1158/1078-0432.CCR-11-2381
40. Kaelin WG Jr. The concept of synthetic lethality in the context of anticancer therapy. Nat Rev Cancer (2005) 5(9):689–98. doi: 10.1038/nrc1691
41. Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature (2005) 434(7035):913–7. doi: 10.1038/nature03443
42. Aguirre AJ, Hahn WC. Synthetic Lethal Vulnerabilities in KRAS-Mutant Cancers. Cold Spring Harbor Perspect Med (2018) 8(8). doi: 10.1101/cshperspect.a031518
43. Mullard A. Synthetic lethality screens point the way to new cancer drug targets. Nat Rev Drug Discovery (2017) 16(10):736. doi: 10.1038/nrd.2017.189
44. Koike-Yusa H, Li Y, Tan EP, Velasco-Herrera Mdel C, Yusa K. Genome-wide recessive genetic screening in mammalian cells with a lentiviral CRISPR-guide RNA library. Nat Biotechnol (2014) 32(3):267–73. doi: 10.1038/nbt.2800
45. Downward J. RAS Synthetic Lethal Screens Revisited: Still Seeking the Elusive Prize? Clin Cancer Res (2015) 21(8):1802–9. doi: 10.1158/1078-0432.CCR-14-2180
46. Babij C, Zhang Y, Kurzeja RJ, Munzli A, Shehabeldin A, Fernando M, et al. STK33 kinase activity is nonessential in KRAS-dependent cancer cells. Cancer Res (2011) 71(17):5818–26. doi: 10.1158/0008-5472.CAN-11-0778
47. Ku AA, Hu HM, Zhao X, Shah KN, Kongara S, Wu D, et al. Integration of multiple biological contexts reveals principles of synthetic lethality that affect reproducibility. Nat Commun (2020) 11(1):2375. doi: 10.1038/s41467-020-16078-y
48. Corcoran RB, Cheng KA, Hata AN, Faber AC, Ebi H, Coffee EM, et al. Synthetic lethal interaction of combined BCL-XL and MEK inhibition promotes tumor regressions in KRAS mutant cancer models. Cancer Cell (2013) 23(1):121–8. doi: 10.1016/j.ccr.2012.11.007
49. Manchado E, Weissmueller S, Morris JPT, Chen CC, Wullenkord R, Lujambio A, et al. A combinatorial strategy for treating KRAS-mutant lung cancer. Nature (2016) 534(7609):647–51. doi: 10.1038/nature18600
50. Puyol M, Martin A, Dubus P, Mulero F, Pizcueta P, Khan G, et al. A synthetic lethal interaction between K-Ras oncogenes and Cdk4 unveils a therapeutic strategy for non-small cell lung carcinoma. Cancer Cell (2010) 18(1):63–73. doi: 10.1016/j.ccr.2010.05.025
51. Chio IIC, Jafarnejad SM, Ponz-Sarvise M, Park Y, Rivera K, Palm W, et al. NRF2 Promotes Tumor Maintenance by Modulating mRNA Translation in Pancreatic Cancer. Cell (2016) 166(4):963–76. doi: 10.1016/j.cell.2016.06.056
52. Zhu Z, Aref AR, Cohoon TJ, Barbie TU, Imamura Y, Yang S, et al. Inhibition of KRAS-driven tumorigenicity by interruption of an autocrine cytokine circuit. Cancer Discovery (2014) 4(4):452–65. doi: 10.1158/2159-8290.CD-13-0646
53. Kim J, McMillan E, Kim HS, Venkateswaran N, Makkar G, Rodriguez-Canales J, et al. XPO1-dependent nuclear export is a druggable vulnerability in KRAS-mutant lung cancer. Nature (2016) 538(7623):114–7. doi: 10.1038/nature19771
54. Singh A, Sweeney MF, Yu M, Burger A, Greninger P, Benes C, et al. TAK1 inhibition promotes apoptosis in KRAS-dependent colon cancers. Cell (2012) 148(4):639–50. doi: 10.1016/j.cell.2011.12.033
55. Kapoor A, Yao W, Ying H, Hua S, Liewen A, Wang Q, et al. Yap1 activation enables bypass of oncogenic Kras addiction in pancreatic cancer. Cell (2014) 158(1):185–97. doi: 10.1016/j.cell.2014.06.003
56. Kumar MS, Hancock DC, Molina-Arcas M, Steckel M, East P, Diefenbacher M, et al. The GATA2 transcriptional network is requisite for RAS oncogene-driven non-small cell lung cancer. Cell (2012) 149(3):642–55. doi: 10.1016/j.cell.2012.02.059
57. Wang Y, Ngo VN, Marani M, Yang Y, Wright G, Staudt LM, et al. Critical role for transcriptional repressor Snail2 in transformation by oncogenic RAS in colorectal carcinoma cells. Oncogene (2010) 29(33):4658–70. doi: 10.1038/onc.2010.218
58. Garnett MJ, Edelman EJ, Heidorn SJ, Greenman CD, Dastur A, Lau KW, et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature (2012) 483(7391):570–5. doi: 10.1038/nature11005
59. Adjei AA, Cohen RB, Franklin W, Morris C, Wilson D, Molina JR, et al. Phase I Pharmacokinetic and Pharmacodynamic Study of the Oral, Small-Molecule Mitogen-Activated Protein Kinase Kinase 1/2 Inhibitor AZD6244 (ARRY-142886) in Patients With Advanced Cancers. J Clin Oncol (2008) 26(13):2139–46. doi: 10.1200/JCO.2007.14.4956
60. Faber AC, Coffee EM, Costa C, Dastur A, Ebi H, Hata AN, et al. mTOR inhibition specifically sensitizes colorectal cancers with KRAS or BRAF mutations to BCL-2/BCL-XL inhibition by suppressing MCL-1. Cancer Discovery (2014) 4(1):42–52. doi: 10.1158/2159-8290.CD-13-0315
61. Molina-Arcas M, Hancock DC, Sheridan C, Kumar MS, Downward J. Coordinate direct input of both KRAS and IGF1 receptor to activation of PI3 kinase in KRAS-mutant lung cancer. Cancer Discovery (2013) 3(5):548–63. doi: 10.1158/2159-8290.CD-12-0446
62. Carbone C, Melisi D. NF-kappaB as a target for pancreatic cancer therapy. Expert Opin Ther Targets (2012) 16 Suppl 2:S1–10. doi: 10.1517/14728222.2011.645806
63. Melisi D, Chiao PJ. NF-kappa B as a target for cancer therapy. Expert Opin Ther Targets (2007) 11(2):133–44. doi: 10.1517/14728222.11.2.133
64. Hamad NM, Elconin JH, Karnoub AE, Bai W, Rich JN, Abraham RT, et al. Distinct requirements for Ras oncogenesis in human versus mouse cells. Genes Dev (2002) 16(16):2045–57. doi: 10.1101/gad.993902
65. Meylan E, Dooley AL, Feldser DM, Shen L, Turk E, Ouyang C, et al. Requirement for NF-kappaB signalling in a mouse model of lung adenocarcinoma. Nature (2009) 462(7269):104–7. doi: 10.1038/nature08462
66. Chien Y, Kim S, Bumeister R, Loo YM, Kwon SW, Johnson CL, et al. RalB GTPase-mediated activation of the IkappaB family kinase TBK1 couples innate immune signaling to tumor cell survival. Cell (2006) 127(1):157–70. doi: 10.1016/j.cell.2006.08.034
67. Ng K, Hendifar A, Starodub A, Chaves J, Yang Y, Koh B, et al. Phase 1 dose-escalation study of momelotinib, a Janus kinase 1/2 inhibitor, combined with gemcitabine and nab-paclitaxel in patients with previously untreated metastatic pancreatic ductal adenocarcinoma. Invest New Drugs (2019) 37(1):159–65. doi: 10.1007/s10637-018-0650-5
68. Melisi D, Xia Q, Paradiso G, Ling J, Moccia T, Carbone C, et al. Modulation of pancreatic cancer chemoresistance by inhibition of TAK1. J Natl Cancer Inst (2011) 103(15):1190–204. doi: 10.1093/jnci/djr243
69. Santoro R, Carbone C, Piro G, Chiao PJ, Melisi D. TAK-ing aim at chemoresistance: The emerging role of MAP3K7 as a target for cancer therapy. Drug Resist Updat (2017) 33-35:36–42. doi: 10.1016/j.drup.2017.10.004
70. Santoro R, Zanotto M, Simionato F, Zecchetto C, Merz V, Cavallini C, et al. Modulating TAK1 expression inhibits YAP and TAZ oncogenic functions in pancreatic cancer. Mol Cancer Ther (2019) 11:142–57. doi: 10.1158/1535-7163.MCT-19-0270
71. Santoro R, Zanotto M, Simionato F, Zecchetto C, Merz V, Cavallini C, et al. Modulating TAK1 Expression Inhibits YAP and TAZ Oncogenic Functions in Pancreatic Cancer. Mol Cancer Ther (2020) 19(1):247–57. doi: 10.1158/1535-7163.MCT-19-0270
72. Shao DD, Xue W, Krall EB, Bhutkar A, Piccioni F, Wang X, et al. KRAS and YAP1 converge to regulate EMT and tumor survival. Cell (2014) 158(1):171–84. doi: 10.1016/j.cell.2014.06.004
73. Lin L, Sabnis AJ, Chan E, Olivas V, Cade L, Pazarentzos E, et al. The Hippo effector YAP promotes resistance to RAF- and MEK-targeted cancer therapies. Nat Genet (2015) 47(3):250–6. doi: 10.1038/ng.3218
74. Vicent S, Chen R, Sayles LC, Lin C, Walker RG, Gillespie AK, et al. Wilms tumor 1 (WT1) regulates KRAS-driven oncogenesis and senescence in mouse and human models. J Clin Invest (2010) 120(11):3940–52. doi: 10.1172/JCI44165
75. Singh A, Greninger P, Rhodes D, Koopman L, Violette S, Bardeesy N, et al. A gene expression signature associated with “K-Ras addiction” reveals regulators of EMT and tumor cell survival. Cancer Cell (2009) 15(6):489–500. doi: 10.1016/j.ccr.2009.03.022
76. Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E, et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell (2012) 149(3):656–70. doi: 10.1016/j.cell.2012.01.058
77. Yun J, Rago C, Cheong I, Pagliarini R, Angenendt P, Rajagopalan H, et al. Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science (2009) 325(5947):1555–9. doi: 10.1126/science.1174229
78. Iwamoto M, Kawada K, Nakamoto Y, Itatani Y, Inamoto S, Toda K, et al. Regulation of 18F-FDG accumulation in colorectal cancer cells with mutated KRAS. J Nucl Med Off Publication Soc Nucl Med (2014) 55(12):2038–44. doi: 10.2967/jnumed.114.142927
79. Kawada K, Nakamoto Y, Kawada M, Hida K, Matsumoto T, Murakami T, et al. Relationship between 18F-fluorodeoxyglucose accumulation and KRAS/BRAF mutations in colorectal cancer. Clin Cancer Res (2012) 18(6):1696–703. doi: 10.1158/1078-0432.CCR-11-1909
80. Yun J, Mullarky E, Lu C, Bosch KN, Kavalier A, Rivera K, et al. Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Science (2015) 350(6266):1931–6.
81. Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature (2013) 496(7443):101–5. doi: 10.1038/nature12040
82. Mayers JR, Torrence ME, Danai LV, Papagiannakopoulos T, Davidson SM, Bauer MR, et al. Tissue of origin dictates branched-chain amino acid metabolism in mutant Kras-driven cancers. Science (2016) 353(6304):1161–5. doi: 10.1126/science.aaf5171
83. Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M, et al. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci USA (2010) 107(19):8788–93. doi: 10.1073/pnas.1003428107
84. Viale A, Pettazzoni P, Lyssiotis CA, Ying H, Sanchez N, Marchesini M, et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature (2014) 514(7524):628–32. doi: 10.1038/nature13611
85. Rabinowitz JD, White E. Autophagy and metabolism. Science (2010) 330(6009):1344–8. doi: 10.1126/science.1193497
86. Yang A, Rajeshkumar NV, Wang X, Yabuuchi S, Alexander BM, Chu GC, et al. Autophagy is critical for pancreatic tumor growth and progression in tumors with p53 alterations. Cancer Discovery (2014) 4(8):905–13. doi: 10.1158/2159-8290.CD-14-0362
87. Eng CH, Wang Z, Tkach D, Toral-Barza L, Ugwonali S, Liu S, et al. Macroautophagy is dispensable for growth of KRAS mutant tumors and chloroquine efficacy. Proc Natl Acad Sci USA (2016) 113(1):182–7. doi: 10.1073/pnas.1515617113
88. Wolpin BM, Rubinson DA, Wang X, Chan JA, Cleary JM, Enzinger PC, et al. Phase II and pharmacodynamic study of autophagy inhibition using hydroxychloroquine in patients with metastatic pancreatic adenocarcinoma. Oncol (2014) 19(6):637–8. doi: 10.1634/theoncologist.2014-0086
89. Guo JY, Karsli-Uzunbas G, Mathew R, Aisner SC, Kamphorst JJ, Strohecker AM, et al. Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes Dev (2013) 27(13):1447–61. doi: 10.1101/gad.219642.113
90. Commisso C, Davidson SM, Soydaner-Azeloglu RG, Parker SJ, Kamphorst JJ, Hackett S, et al. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature (2013) 497(7451):633–7. doi: 10.1038/nature12138
91. Padanad MS, Konstantinidou G, Venkateswaran N, Melegari M, Rindhe S, Mitsche M, et al. Fatty Acid Oxidation Mediated by Acyl-CoA Synthetase Long Chain 3 Is Required for Mutant KRAS Lung Tumorigenesis. Cell Rep (2016) 16(6):1614–28. doi: 10.1016/j.celrep.2016.07.009
92. Gouw AM, Eberlin LS, Margulis K, Sullivan DK, Toal GG, Tong L, et al. Oncogene KRAS activates fatty acid synthase, resulting in specific ERK and lipid signatures associated with lung adenocarcinoma. Proc Natl Acad Sci U.S.A. (2017) 114(17):4300–5. doi: 10.1073/pnas.1617709114
93. De Raedt T, Walton Z, Yecies JL, Li D, Chen Y, Malone CF, et al. Exploiting cancer cell vulnerabilities to develop a combination therapy for ras-driven tumors. Cancer Cell (2011) 20(3):400–13. doi: 10.1016/j.ccr.2011.08.014
94. Lito P, Solomon M, Li LS, Hansen R, Rosen N. Allele-specific inhibitors inactivate mutant KRAS G12C by a trapping mechanism. Science (2016) 351(6273):604–8. doi: 10.1126/science.aad6204
95. Ryan MB, Fece de la Cruz F, Phat S, Myers DT, Wong E, Shahzade HA, et al. Vertical Pathway Inhibition Overcomes Adaptive Feedback Resistance to KRAS(G12C) Inhibition. Clin Cancer Res (2020) 26(7):1633–43. doi: 10.1158/1078-0432.CCR-19-3523
96. Misale S, Fatherree JP, Cortez E, Li C, Bilton S, Timonina D, et al. KRAS G12C NSCLC Models Are Sensitive to Direct Targeting of KRAS in Combination with PI3K Inhibition. Clin Cancer Res (2019) 25(2):796–807. doi: 10.1158/1078-0432.CCR-18-0368
97. Boriack-Sjodin PA, Margarit SM, Bar-Sagi D, Kuriyan J. The structural basis of the activation of Ras by Sos. Nature (1998) 394(6691):337–43. doi: 10.1038/28548
98. Hillig RC, Sautier B, Schroeder J, Moosmayer D, Hilpmann A, Stegmann CM, et al. Discovery of potent SOS1 inhibitors that block RAS activation via disruption of the RAS-SOS1 interaction. Proc Natl Acad Sci USA (2019) 116(7):2551–60. doi: 10.1073/pnas.1812963116
99. Vasan N, Baselga J, Hyman DM. A view on drug resistance in cancer. Nature (2019) 575(7782):299–309. doi: 10.1038/s41586-019-1730-1
100. Amodio V, Yaeger R, Arcella P, Cancelliere C, Lamba S, Lorenzato A, et al. EGFR Blockade Reverts Resistance to KRAS(G12C) Inhibition in Colorectal Cancer. Cancer Discovery (2020) 10(8):1129–39. doi: 10.1158/2159-8290.CD-20-0187
101. Molina-Arcas M, Moore C, Rana S, van Maldegem F, Mugarza E, Romero-Clavijo P, et al. Development of combination therapies to maximize the impact of KRAS-G12C inhibitors in lung cancer. Sci Trans Med (2019) 11(510). doi: 10.1126/scitranslmed.aaw7999
102. Xue JY, Zhao Y, Aronowitz J, Mai TT, Vides A, Qeriqi B, et al. Rapid non-uniform adaptation to conformation-specific KRAS(G12C) inhibition. Nature (2020) 577(7790):421–5. doi: 10.1038/s41586-019-1884-x
103. Kordiak J, Szemraj J, Grabska-Kobylecka I, Bialasiewicz P, Braun M, Kordek R, et al. Intratumor heterogeneity and tissue distribution of KRAS mutation in non-small cell lung cancer: implications for detection of mutated KRAS oncogene in exhaled breath condensate. J Cancer Res Clin Oncol (2019) 145(1):241–51. doi: 10.1007/s00432-018-2779-1
104. George NM, Day CE, Boerner BP, Johnson RL, Sarvetnick NE. Hippo signaling regulates pancreas development through inactivation of Yap. Mol Cell Biol (2012) 32(24):5116–28. doi: 10.1128/MCB.01034-12
105. Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med (2017) 23(6):703–13. doi: 10.1038/nm.4333
106. Cullis J, Das S, Bar-Sagi D. Kras and Tumor Immunity: Friend or Foe? Cold Spring Harbor Perspect Med (2018) 8(9). doi: 10.1101/cshperspect.a031849
107. O’Hayer KM, Brady DC, Counter CM. ELR+ CXC chemokines and oncogenic Ras-mediated tumorigenesis. Carcinogenesis (2009) 30(11):1841–7. doi: 10.1093/carcin/bgp198
108. Purohit A, Varney M, Rachagani S, Ouellette MM, Batra SK, Singh RK. CXCR2 signaling regulates KRAS(G(1)(2)D)-induced autocrine growth of pancreatic cancer. Oncotarget (2016) 7(6):7280–96. doi: 10.18632/oncotarget.6906
109. Ji H, Houghton AM, Mariani TJ, Perera S, Kim CB, Padera R, et al. K-ras activation generates an inflammatory response in lung tumors. Oncogene (2006) 25(14):2105–12. doi: 10.1038/sj.onc.1209237
110. Li A, King J, Moro A, Sugi MD, Dawson DW, Kaplan J, et al. Overexpression of CXCL5 is associated with poor survival in patients with pancreatic cancer. Am J Pathol (2011) 178(3):1340–9. doi: 10.1016/j.ajpath.2010.11.058
111. Liao W, Overman MJ, Boutin AT, Shang X, Zhao D, Dey P, et al. KRAS-IRF2 Axis Drives Immune Suppression and Immune Therapy Resistance in Colorectal Cancer. Cancer Cell (2019) 35(4):559–72 e7. doi: 10.1016/j.ccell.2019.02.008
112. Almand B, Clark JI, Nikitina E, van Beynen J, English NR, Knight SC, et al. Increased production of immature myeloid cells in cancer patients: a mechanism of immunosuppression in cancer. J Immunol (2001) 166(1):678–89. doi: 10.4049/jimmunol.166.1.678
113. Kusmartsev S, Gabrilovich DI. Effect of tumor-derived cytokines and growth factors on differentiation and immune suppressive features of myeloid cells in cancer. Cancer Metastasis Rev (2006) 25(3):323–31. doi: 10.1007/s10555-006-9002-6
114. Lesina M, Kurkowski MU, Ludes K, Rose-John S, Treiber M, Kloppel G, et al. Stat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell (2011) 19(4):456–69. doi: 10.1016/j.ccr.2011.03.009
115. Shull MM, Ormsby I, Kier AB, Pawlowski S, Diebold RJ, Yin M, et al. Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature (1992) 359(6397):693–9. doi: 10.1038/359693a0
116. Tsubaki M, Yamazoe Y, Yanae M, Satou T, Itoh T, Kaneko J, et al. Blockade of the Ras/MEK/ERK and Ras/PI3K/Akt pathways by statins reduces the expression of bFGF, HGF, and TGF-beta as angiogenic factors in mouse osteosarcoma. Cytokine (2011) 54(1):100–7. doi: 10.1016/j.cyto.2011.01.005
117. Zdanov S, Mandapathil M, Abu Eid R, Adamson-Fadeyi S, Wilson W, Qian J, et al. Mutant KRAS Conversion of Conventional T Cells into Regulatory T Cells. Cancer Immunol Res (2016) 4(4):354–65. doi: 10.1158/2326-6066.CIR-15-0241
118. Tauriello DVF, Palomo-Ponce S, Stork D, Berenguer-Llergo A, Badia-Ramentol J, Iglesias M, et al. TGFbeta drives immune evasion in genetically reconstituted colon cancer metastasis. Nature (2018) 554(7693):538–43. doi: 10.1038/nature25492
119. Melisi D, Garcia-Carbonero R, Macarulla T, Pezet D, Deplanque G, Fuchs M, et al. Galunisertib plus gemcitabine vs. gemcitabine for first-line treatment of patients with unresectable pancreatic cancer. Br J Cancer (2018) 119(10):1208–14. doi: 10.1038/s41416-018-0246-z
120. Takeuchi S, Baghdadi M, Tsuchikawa T, Wada H, Nakamura T, Abe H, et al. Chemotherapy-Derived Inflammatory Responses Accelerate the Formation of Immunosuppressive Myeloid Cells in the Tissue Microenvironment of Human Pancreatic Cancer. Cancer Res (2015) 75(13):2629–40. doi: 10.1158/0008-5472.CAN-14-2921
121. Merz V, Zecchetto C, Santoro R, Simionato F, Sabbadini F, Mangiameli D, et al. Plasma IL8 Is a Biomarker for TAK1 Activation and Predicts Resistance to Nanoliposomal Irinotecan in Patients with Gemcitabine-Refractory Pancreatic Cancer. Clin Cancer Res (2020) 26(17):4661–9. doi: 10.1158/1078-0432.CCR-20-0395
122. Asfaha S, Dubeykovskiy AN, Tomita H, Yang X, Stokes S, Shibata W, et al. Mice that express human interleukin-8 have increased mobilization of immature myeloid cells, which exacerbates inflammation and accelerates colon carcinogenesis. Gastroenterology (2013) 144(1):155–66. doi: 10.1053/j.gastro.2012.09.057
123. Dey P, Li J, Zhang J, Chaurasiya S, Strom A, Wang H, et al. Oncogenic KRAS-Driven Metabolic Reprogramming in Pancreatic Cancer Cells Utilizes Cytokines from the Tumor Microenvironment. Cancer Discovery (2020) 10(4):608–25. doi: 10.1158/2159-8290
124. Ubaldi V, Gatta L, Pace L, Doria G, Pioli C. CTLA-4 engagement inhibits Th2 but not Th1 cell polarisation. Clin Dev Immunol (2003) 10(1):13–7. doi: 10.1080/10446670310001598519
125. De Monte L, Reni M, Tassi E, Clavenna D, Papa I, Recalde H, et al. Intratumor T helper type 2 cell infiltrate correlates with cancer-associated fibroblast thymic stromal lymphopoietin production and reduced survival in pancreatic cancer. J Exp Med (2011) 208(3):469–78. doi: 10.1084/jem.20101876
126. Pylayeva-Gupta Y, Lee KE, Hajdu CH, Miller G, Bar-Sagi D. Oncogenic Kras-induced GM-CSF production promotes the development of pancreatic neoplasia. Cancer Cell (2012) 21(6):836–47. doi: 10.1016/j.ccr.2012.04.024
127. Liou GY, Doppler H, Necela B, Edenfield B, Zhang L, Dawson DW, et al. Mutant KRAS-induced expression of ICAM-1 in pancreatic acinar cells causes attraction of macrophages to expedite the formation of precancerous lesions. Cancer Discovery (2015) 5(1):52–63. doi: 10.1158/2159-8290.CD-14-0474
128. Mielgo A, Schmid MC. Impact of tumour associated macrophages in pancreatic cancer. BMB Rep (2013) 46(3):131–8. doi: 10.5483/bmbrep.2013.46.3.036
129. Busch SE, Hanke ML, Kargl J, Metz HE, MacPherson D, Houghton AM. Lung Cancer Subtypes Generate Unique Immune Responses. J Immunol (2016) 197(11):4493–503. doi: 10.4049/jimmunol.1600576
130. Cortez-Retamozo V, Etzrodt M, Newton A, Rauch PJ, Chudnovskiy A, Berger C, et al. Origins of tumor-associated macrophages and neutrophils. Proc Natl Acad Sci U S A (2012) 109(7):2491–6. doi: 10.1073/pnas.1113744109
131. Santos AM, Jung J, Aziz N, Kissil JL, Pure E. Targeting fibroblast activation protein inhibits tumor stromagenesis and growth in mice. J Clin Invest (2009) 119(12):3613–25. doi: 10.1172/JCI38988
132. Ji Z, Mei FC, Xie J, Cheng X. Oncogenic KRAS activates hedgehog signaling pathway in pancreatic cancer cells. J Biol Chem (2007) 282(19):14048–55. doi: 10.1074/jbc.M611089200
133. Mills LD, Zhang Y, Marler RJ, Herreros-Villanueva M, Zhang L, Almada LL, et al. Loss of the transcription factor GLI1 identifies a signaling network in the tumor microenvironment mediating KRAS oncogene-induced transformation. J Biol Chem (2013) 288(17):11786–94. doi: 10.1074/jbc.M112.438846
134. Omary MB, Lugea A, Lowe AW, Pandol SJ. The pancreatic stellate cell: a star on the rise in pancreatic diseases. J Clin Invest (2007) 117(1):50–9. doi: 10.1172/JCI30082
135. Pauken KE, Nelson CE, Martinov T, Spanier JA, Heffernan JR, Sahli NL, et al. Cutting edge: identification of autoreactive CD4+ and CD8+ T cell subsets resistant to PD-1 pathway blockade. J Immunol (2015) 194(8):3551–5. doi: 10.4049/jimmunol.1402262
136. Coelho MA, de Carne Trecesson S, Rana S, Zecchin D, Moore C, Molina-Arcas M, et al. Oncogenic RAS Signaling Promotes Tumor Immunoresistance by Stabilizing PD-L1 mRNA. Immunity (2017) 47(6):1083–99.e6. doi: 10.1016/j.immuni.2017.11.016
137. Chen N, Fang W, Lin Z, Peng P, Wang J, Zhan J, et al. KRAS mutation-induced upregulation of PD-L1 mediates immune escape in human lung adenocarcinoma. Cancer Immunol Immunother CII (2017) 66(9):1175–87. doi: 10.1007/s00262-017-2005-z
138. Ebert PJR, Cheung J, Yang Y, McNamara E, Hong R, Moskalenko M, et al. MAP Kinase Inhibition Promotes T Cell and Anti-tumor Activity in Combination with PD-L1 Checkpoint Blockade. Immunity (2016) 44(3):609–21. doi: 10.1016/j.immuni.2016.01.024
139. Poon E, Mullins S, Watkins A, Williams GS, Koopmann JO, Di Genova G, et al. The MEK inhibitor selumetinib complements CTLA-4 blockade by reprogramming the tumor immune microenvironment. J Immunother Cancer (2017) 5(1):63. doi: 10.1186/s40425-017-0268-8
140. Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N Engl J Med (2015) 373(17):1627–39. doi: 10.1056/NEJMoa1507643
141. Jeanson A, Tomasini P, Souquet-Bressand M, Brandone N, Boucekine M, Grangeon M, et al. Efficacy of Immune Checkpoint Inhibitors in KRAS-Mutant Non-Small Cell Lung Cancer (NSCLC). J Thorac Oncol (2019) 14(6):1095–101. doi: 10.1016/j.jtho.2019.01.011
142. Lee CK, Man J, Lord S, Cooper W, Links M, Gebski V, et al. Clinical and Molecular Characteristics Associated With Survival Among Patients Treated With Checkpoint Inhibitors for Advanced Non-Small Cell Lung Carcinoma: A Systematic Review and Meta-analysis. JAMA Oncol (2018) 4(2):210–6. doi: 10.1001/jamaoncol.2017.4427
143. Mazieres J, Drilon A, Lusque A, Mhanna L, Cortot AB, Mezquita L, et al. Immune checkpoint inhibitors for patients with advanced lung cancer and oncogenic driver alterations: results from the IMMUNOTARGET registry. Ann Oncol (2019) 30(8):1321–8. doi: 10.1093/annonc/mdz167
144. Skoulidis F, Goldberg ME, Greenawalt DM, Hellmann MD, Awad MM, Gainor JF, et al. STK11/LKB1 Mutations and PD-1 Inhibitor Resistance in KRAS-Mutant Lung Adenocarcinoma. Cancer Discovery (2018) 8(7):822–35. doi: 10.1158/2159-8290.CD-18-0099
145. Kitajima S, Ivanova E, Guo S, Yoshida R, Campisi M, Sundararaman SK, et al. Suppression of STING Associated with LKB1 Loss in KRAS-Driven Lung Cancer. Cancer Discovery (2019) 9(1):34–45. doi: 10.1158/2159-8290.CD-18-0689
146. Della Corte CM, Sen T, Gay CM, Ramkumar K, Diao L, Cardnell RJ, et al. STING Pathway Expression Identifies NSCLC With an Immune-Responsive Phenotype. J Thorac Oncol (2020) 15(5):777–91. doi: 10.1016/j.jtho.2020.01.009
147. Skoulidis F, Byers LA, Diao L, Papadimitrakopoulou VA, Tong P, Izzo J, et al. Co-occurring genomic alterations define major subsets of KRAS-mutant lung adenocarcinoma with distinct biology, immune profiles, and therapeutic vulnerabilities. Cancer Discovery (2015) 5(8):860–77. doi: 10.1158/2159-8290.CD-14-1236
Keywords: KRAS, NSCLC, pancreatic cancer, colon cancer, G12C mutation
Citation: Merz V, Gaule M, Zecchetto C, Cavaliere A, Casalino S, Pesoni C, Contarelli S, Sabbadini F, Bertolini M, Mangiameli D, Milella M, Fedele V and Melisi D (2021) Targeting KRAS: The Elephant in the Room of Epithelial Cancers. Front. Oncol. 11:638360. doi: 10.3389/fonc.2021.638360
Received: 06 December 2020; Accepted: 27 January 2021;
Published: 11 March 2021.
Edited by:
Floriana Morgillo, Second University of Naples, ItalyReviewed by:
Hong-Quan Duong, Hanoi University of Public Health, VietnamCarminia Maria Della Corte, Second University of Naples, Italy
Copyright © 2021 Merz, Gaule, Zecchetto, Cavaliere, Casalino, Pesoni, Contarelli, Sabbadini, Bertolini, Mangiameli, Milella, Fedele and Melisi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Valeria Merz, dmFsZXJpYW1lcnpAZ21haWwuY29t; Davide Melisi, ZGF2aWRlLm1lbGlzaUB1bml2ci5pdA==