 Danila Comandini1†Fabio Catalano1†Massimiliano Grassi1
Danila Comandini1†Fabio Catalano1†Massimiliano Grassi1 Guido Pesola2Rossella Bertulli3Antonio Guadagno4Bruno Spina4
Guido Pesola2Rossella Bertulli3Antonio Guadagno4Bruno Spina4 Matteo Mascherini5Franco De Cian5,6
Matteo Mascherini5Franco De Cian5,6 Federico Pistoia7
Federico Pistoia7 Sara Elena Rebuzzi1,8*
Sara Elena Rebuzzi1,8*- 1Medical Oncology Unit 1, IRCCS Ospedale Policlinico San Martino, Genova, Italy
- 2Clinic of Medical Oncology, Oncology Institute of Southern Switzerland, Bellinzona, Switzerland
- 3Adult Mesenchymal Tumor Medical Oncology Unit, Fondazione IRCCS Istituto Nazionale Tumori, Milan, Italy
- 4Anatomic Pathology Unit, IRCCS Ospedale Policlinico San Martino, Genova, Italy
- 5Surgical Clinic Unit 1, IRCCS Ospedale Policlinico San Martino, Genova, Italy
- 6Department of Surgical Sciences and Integrated Diagnostics (DISC), University of Genova, Genova, Italy
- 7Department of Health Sciences (DISSAL), University of Genova, Genova, Italy
- 8Department of Internal Medicine and Medical Specialties (Di.M.I.), University of Genova, Genova, Italy
Inflammatory myofibroblastic tumor (IMT) is a very rare subtype of sarcoma, which frequently harbor chromosomal rearrangements, including anaplastic lymphoma kinase (ALK) rearrangements (almost 50% of the IMTs) and other kinase fusions such as ROS1. ROS1 fusions are present in about 10% of IMT, almost half of the ALK-negative IMT patients. Apart from radical surgery for resectable tumors, there is no standard-of-care therapy for advanced IMTs. Nonetheless, the use of tyrosine kinase inhibitors has shown promising efficacy in IMT patients with targetable genomic alterations. We report the case of a 24-year-old patient with chemotherapy-refractory metastatic IMT harboring ROS1 kinase fusion, who experienced a significant clinical and pathological response to crizotinib. This clinical case highlights the need to assess all patients with unresectable IMTs for chromosomal abnormalities and gene mutations and address them to targeted agents as well as clinical trials.
Introduction
Inflammatory myofibroblastic tumor (IMT), also known as inflammatory pseudotumor, is a rare mesenchymal soft tissue tumor characterized by myofibroblastic spindle-cells and a prominent inflammatory infiltrate in an extracellular myxoid/collagenous stroma (1, 2). IMT predominantly affects children and young adults, primarily during the first two decades of life, and occurs mostly in soft tissues and viscera (3, 4). The most common sites of origin are retroperitoneum, abdominopelvic region and lungs (5). Differential diagnosis includes other spindle cell neoplasms such as desmoid fibromatosis, myofibroblastic sarcoma, leiomyosarcoma, gastrointestinal stromal tumor, follicular dendritic cell sarcoma, dedifferentiated liposarcoma and nodular fasciitis (4).
IMT shows a tendency for local recurrence with low risk of distant metastasis and radical surgery is the mainstay of treatment (1, 3, 5). For unresectable or metastatic disease there is no standard-of-care, although many systemic chemotherapeutic regimens have been investigated and showed activity in several trials and case reports (6–14). An overall response rate (ORR) of about 50–60% after first- or second-line treatment was reported in studies on different types of chemotherapy, including anthracycline-based and vinorelbine/vinblastine-based regimens (15, 16).
Almost 50% of the IMTs harbors rearrangements involving the anaplastic lymphoma kinase (ALK), which seem to be associated with localized tumors and better prognosis compared to ALK-negative IMT (17). A substantial proportion of IMTs displays other kinase fusions such as ROS1, PDGFRβ, NTRK3, RET and FN1–IGF1R (18). ROS1 is a receptor tyrosine kinase structurally similar to ALK and ROS1 fusions, mainly YWHAE–ROS1 and TFG-ROS1, are present in about 10% of IMT, almost half of the ALK-negative IMT patients (19, 20).
Given the rarity of IMT, few clinical trials and case series are available on targeted therapy in ALK-positive IMT (21–34). Nonetheless, crizotinib is the only FDA-approved targeted therapy for advanced unresectable ALK-positive IMTs based on the findings of the CREATE study (34).
Moreover, very little data are currently available on the prognosis of ALK-negative IMTs and the role of ROS1 as a driver and potential target in IMT has been documented only in two single cases (17–20).
Here we reported the clinical case of an advanced IMT patient harboring a ROS1 rearrangement who extraordinarily responds to crizotinib (Figure 1).
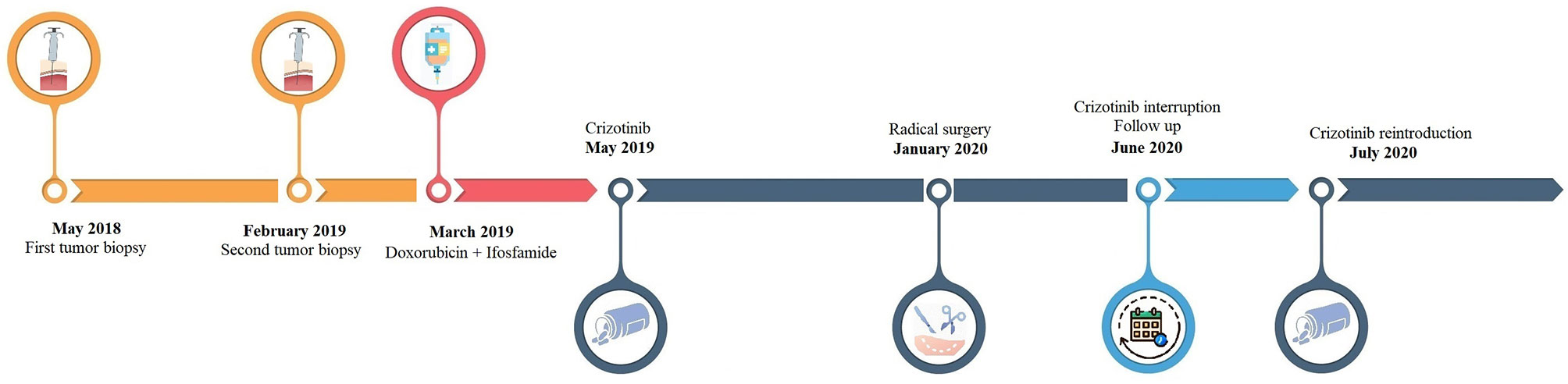
Figure 1 Timeline of patient’s clinical history.
Case Presentation
In April 2018 a 23-year-old Caucasian man presented to the general practitioner for the appearance of a rapidly increasing palpable mass in the proximal part of the left thigh in the last month. His clinal history was negative, as well as his oncological family history. He worked as a plumber and did not report any traumatism. He was a light smoker (less than 5 cigarettes per day for 3 years) and denied the consumption of alcohol or drugs. The clinical examination revealed only a solid lesion of 6 cm of the upper third of the left thigh which was hard, fixed on the deep plane and not painful. A magnetic resonance imaging (MRI) scan was performed, showing an enhancing solid lesion of 50 × 45 × 60 mm localized between the rectus femoris and the tensor fasciae latae muscles, associated with enlargement of homolateral inguinal lymph nodes, radiologically suspected for reactive lymphadenopathies, but not clinically palpable for the presence of the main lesion in the proximal part of the thigh.
In May 2018 a biopsy of the mass was performed, revealing an initial diagnosis of nodular fasciitis, and consequently the patient was addressed to follow up. In consideration of the clinical dimensional increase of the mass, a new MRI was performed in July 2018 confirming the increase of the left thigh lesion (54 × 64 × 75 mm). A surgical approach was, therefore, planned but the pre-operative blood tests reported an important elevation of serum transaminases values (GOT = 145 U/L, GPT = 440 U/L, GOT/GPT = 0.32). According to the suspicion of liver metastases, a staging Total Body (TB) computed tomography (CT) scan was performed in November 2018, but no secondary lesions were shown. According to the hepatological consultation, the transaminase elevation was not amenable to a viral or autoimmune disease, but a paraneoplastic syndrome.
The radical surgery was, therefore, planned in February 2019 but the pre-operative 18F-FDG-PET revealed, in addition to the main tight lesion (SUV max 23), the presence of two paravertebral lesions (SUV max 21), suspected for metastases, and a minimal uptake of the homolateral inguinal lymph nodes (SUV max 2.4), questioning the initial diagnosis of nodular fasciitis.
In consideration of the different uptake compared with the main lesion, the inguinal lymph nodes were considered reactive lymphadenopathies.
A new biopsy was performed in February 2019 with the diagnosis of myofibroblastic sarcoma. Considering the complexity of the case, in agreement with the patient, a second opinion from an expert pathologist was asked by the first hospital where the patient was followed.
The TB CT scan of March 2019 showed the left thigh mass of 170 × 135 mm, a similar mass in the context of the left iliopsoas (40 × 25 mm) and gluteus muscles (19 × 21 mm), other two tumoral nodules in the dorsal muscles next to D11 vertebral body (37 × 30 mm) and L5 vertebral body (40 × 30 mm) (Figures 2A, B and 3A). The reactive inguinal lymphadenopathies were not viewable.
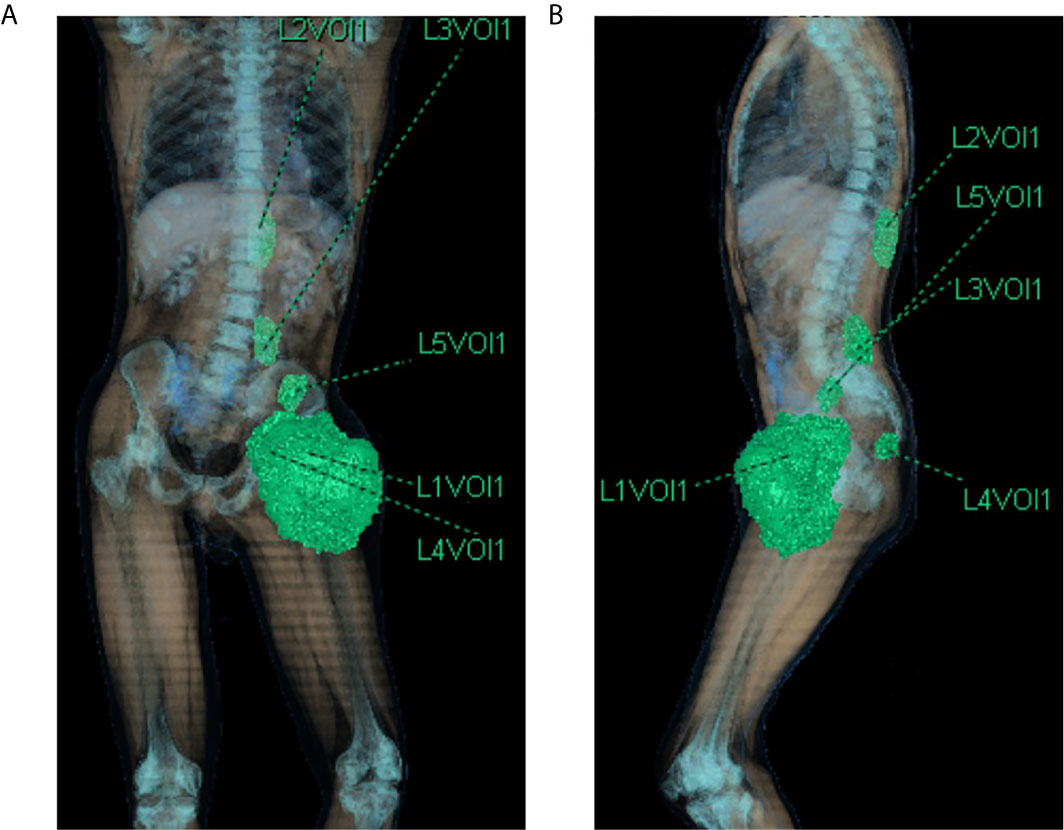
Figure 2 CT scan 3d-reconstruction of the baseline tumor lesions (A) coronal plane; (B) sagittal plane). L1Vol1 = left thigh mass; L2Vol1 = lesion of the left dorsal muscles next to D11 vertebral body; L3Vol1 = lesion of the left dorsal muscles next to L5 vertebral body; L4Vol1 = lesion of the left gluteus muscles; L5Vol1 = lesion of the left iliopsoas.
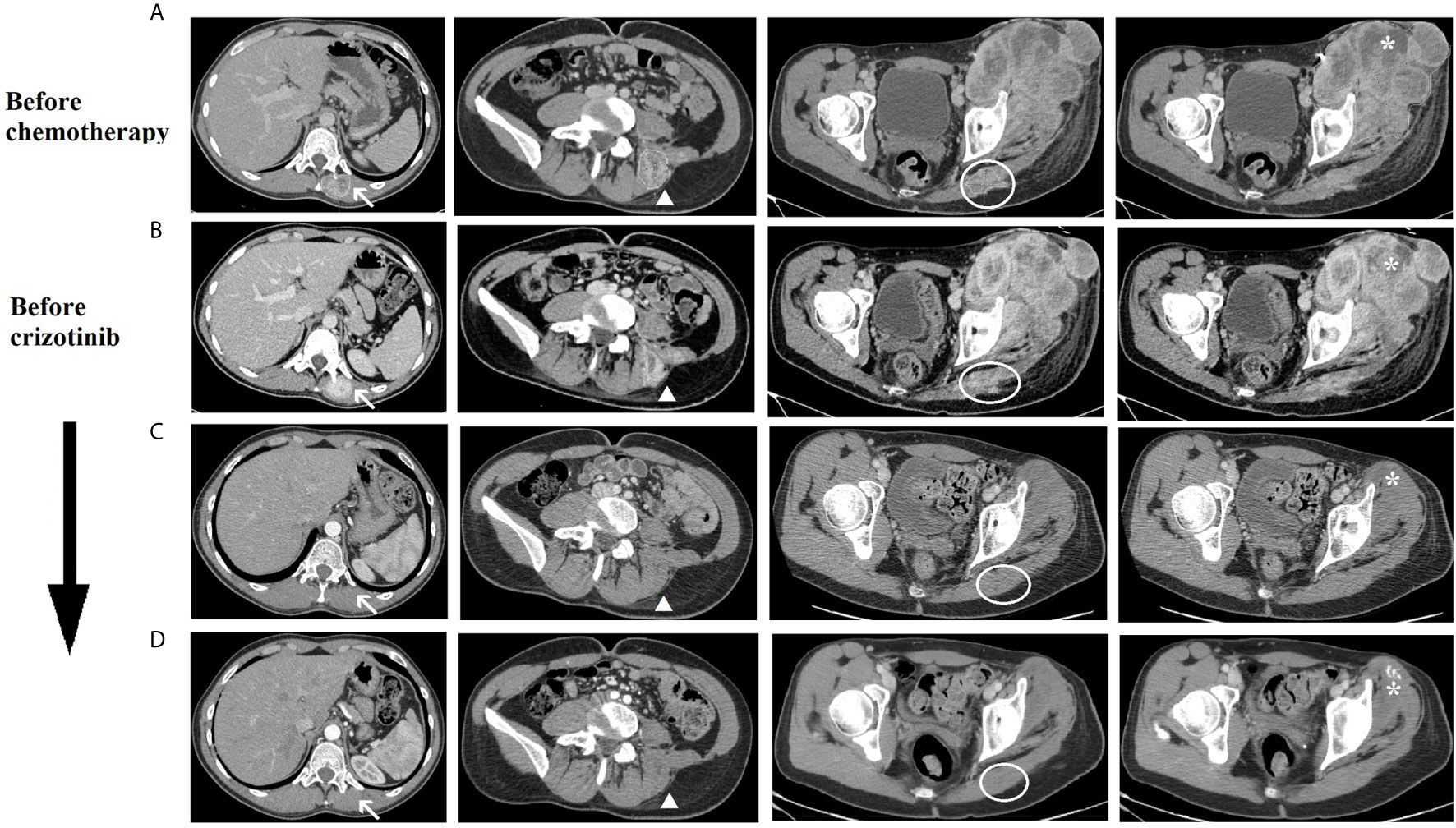
Figure 3 CT scan imaging of the tumoral lesions during patient’s treatment history. Evolution during treatment of the tumoral lesions: lesions of the left dorsal muscles next to D11 vertebral body (arrow) and L5 vertebral body (arrowhead), lesion of the left gluteus (circle) and left thigh mass (asterisk). (A) before chemotherapy (March 2019), (B) after second cycle of chemotherapy and before crizotinib (May 2019), (C) after 4 months of crizotinib (September 2019), (D) after 7 months of crizotinib (December 2019).
In the same month, in consideration of the rapid progression of the primary lesion (Figure 4A) and the final diagnosis of sarcoma, the start of standard chemotherapy with doxorubicin and ifosfamide was indicated. Only at this point, the patient presented to our cancer department for the start of the chemotherapy. The patient generally did not tolerate the chemotherapy due to the onset of severe hematological toxicity (grade 4 neutropenia according to Common Terminology Criteria for Adverse Events (CTCAE) v5.0) but a normalization of serum transaminases values (GOT 12 U/L and GPT 16 U/L) was observed. The physical examination showed a dimensional stability of the main tight lesion. In April 2019 the patient started complaining of dorsal pain and we, therefore, opted for palliative radiotherapy of the dorsal lesions (20 Gy in two fractions).
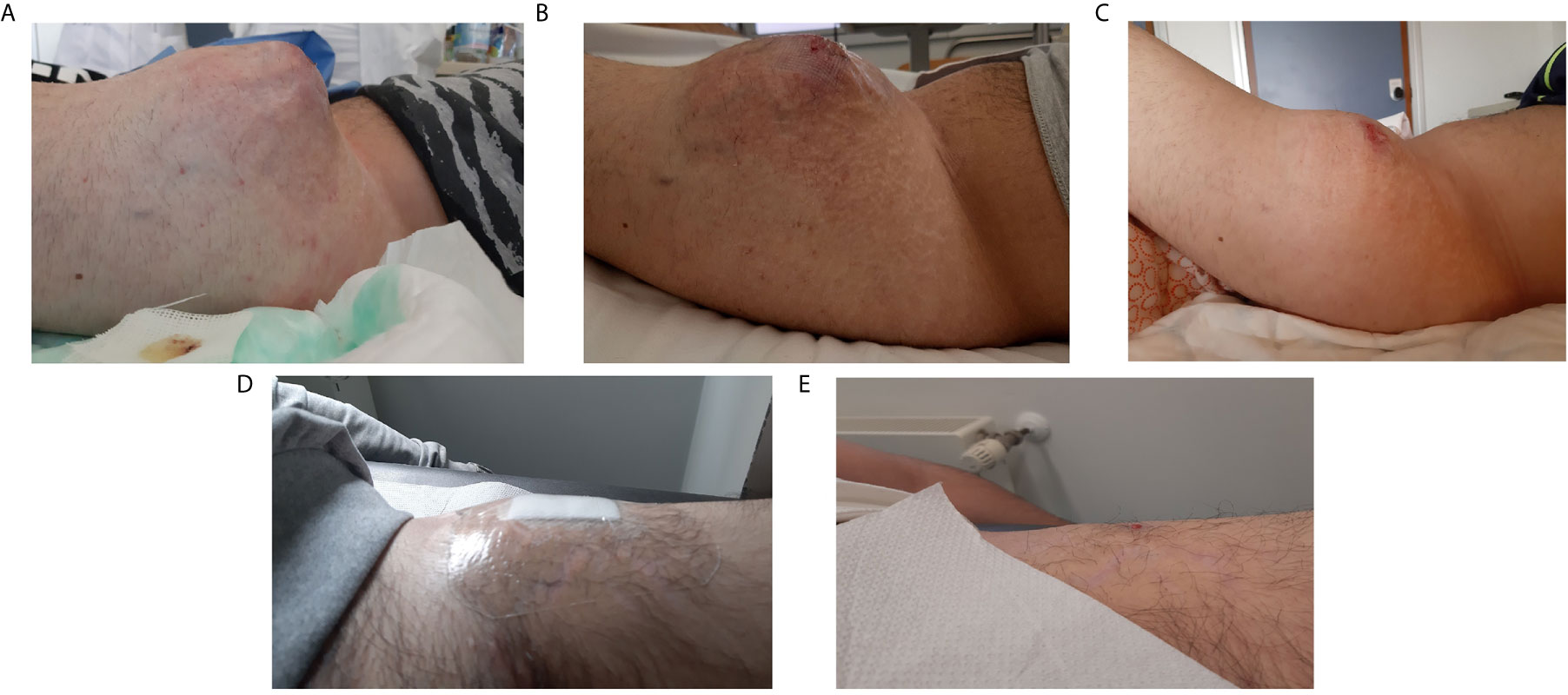
Figure 4 Clinical presentation of the left thigh mass during patient’s treatment history. (A) before chemotherapy (March 2019), (B) after second cycle of chemotherapy and before crizotinib (April 2019), (C) after 7 days of crizotinib, (D) after 3 months of therapy (August 2019), (E) after 7 months of therapy (December 2019).
Between the second and the third cycle of chemotherapy, the final histological diagnosis of IMT was provided by the second opinion of the external expert pathologist. Immunohistochemistry (IHC) for the assessment of ALK was performed but the result was negative. A next-generation sequencing (NGS) of the tumoral tissue was subsequently performed and revealed the presence of the YWHAE1-ROS1 fusion (images not available).
According to the absence of clinical and radiological response to chemotherapy (Figures 3B and 4B), the presence of this rare targetable mutation and the patient’s preference to stop chemotherapy, the patient started crizotinib (250 mg orally, twice daily) as off-label therapy in May 2019. After only 7 days of treatment, we observed a significant dimensional decrease of the left thigh mass (Figure 4C). Despite the good clinical response, the patient developed visual disturbances (blurred vision), which is a common adverse event reported with crizotinib (34). In consideration of the mild grade of the adverse event and the rapid clinical benefit, the treatment with crizotinib was continued with no dose reduction. The visual symptoms spontaneously resolved after the first month of therapy.
In August 2019, after 3 months of therapy, an outstanding clinical response was observed (Figure 4D). The CT scan of September 2019, also, showed a significant dimensional decrease of the left thigh mass (40 × 30 mm) and a complete regression of the lesions of the iliopsoas, gluteus and dorsal muscles (Figure 3C).
The patient experienced also a significant clinical benefit with the disappearance of dorsal pain and the functional recovery of the left leg with the resumption of normal daily activities.
In December 2019, after 7 months of therapy, an additional minimal shrinkage of the tumor mass was observed (38 × 30 mm) (Figure 3D). In January 2020, after 8 months of treatment, in consideration of the outstanding clinical and radiological response and the patient’s preference to remove all the visible disease, the patient underwent wide excision of the residual tight lesion (Figure 4E).
The histological examination showed a large tumor bed composed of necrotic area, sclerotic fibrous tissue and a mixed chronic inflammatory infiltrate with abundant foamy histiocytes. Within the tumor bed, two residual foci of neoplasia were observed (both about 5 mm in diameter). The neoplastic foci displayed spindle-shaped cells embedding in a loose myxoid stroma (Figures 5A–D) (TNM: ypT1, according to UICC—VIII ed. 2017).
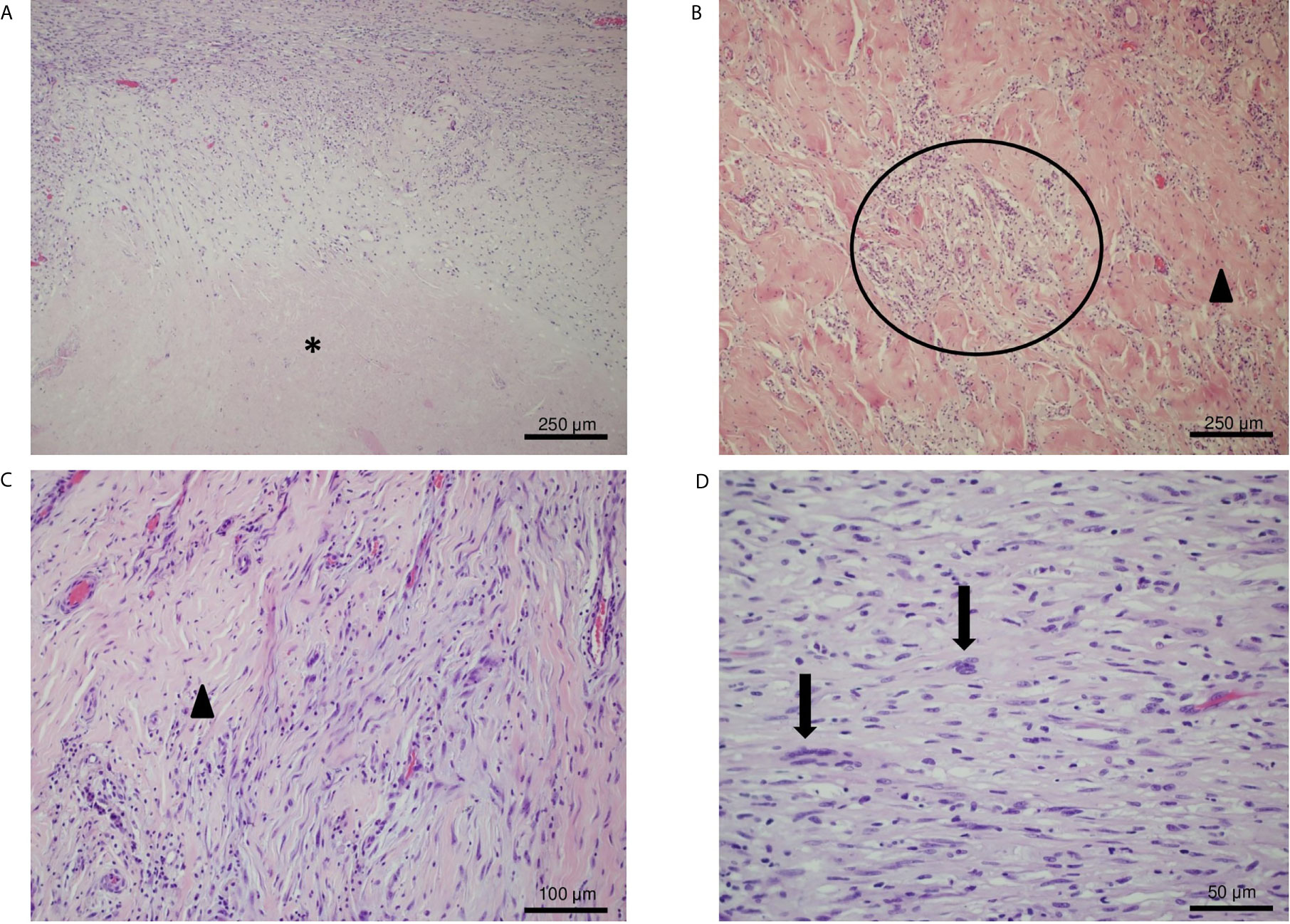
Figure 5 IHC analysis of the resected tumor. (A) Hematoxylin and eosin (H&E) stain, original magnification 10×: necrotic area (asterisk) and sclerotic tissue with residual neoplastic spindle cells. (B, C) H&E stain, original magnification 20×: foamy histiocytes (circle) and a vascularized fibrotic area of connective tissue (arrowhead) mark the tumor bed. (D) H&E stain, original magnification 40×: atypical spindled neoplastic cells set in a loose collagenous matrix with a mild inflammatory infiltrate. Neoplastic cells show hyperchromatic and enlarged nuclei (arrows).
In consideration of the initial advanced stage of the disease, crizotinib was resumed in February 2020, two weeks after radical surgery. In consideration of the absence of scientific evidence on the efficacy of crizotinib in the “adjuvant” setting and the patient’s preference to stop any oncological treatment, crizotinib was continued until June 2020 when the patient reached the completion of a full year of treatment. Moreover, the patient agreed with this clinical decision of treatment discontinuation also to reduce the hospital accesses during the SARS-CoV-2 pandemia.
In July 2020, the first radiological assessment off-therapy (2 months) the TB CT scan and the subsequent MRI (Figure 6A) showed a multifocal local recurrence in the left thigh without evidence of new distant metastasis. Therefore, in August 2020 we decided, in agreement with the patient, to resume the treatment with crizotinib. After 3 months of therapy (November 2020) the TB CT scan and the MRI (Figure 6B) showed a complete regression of the disease.
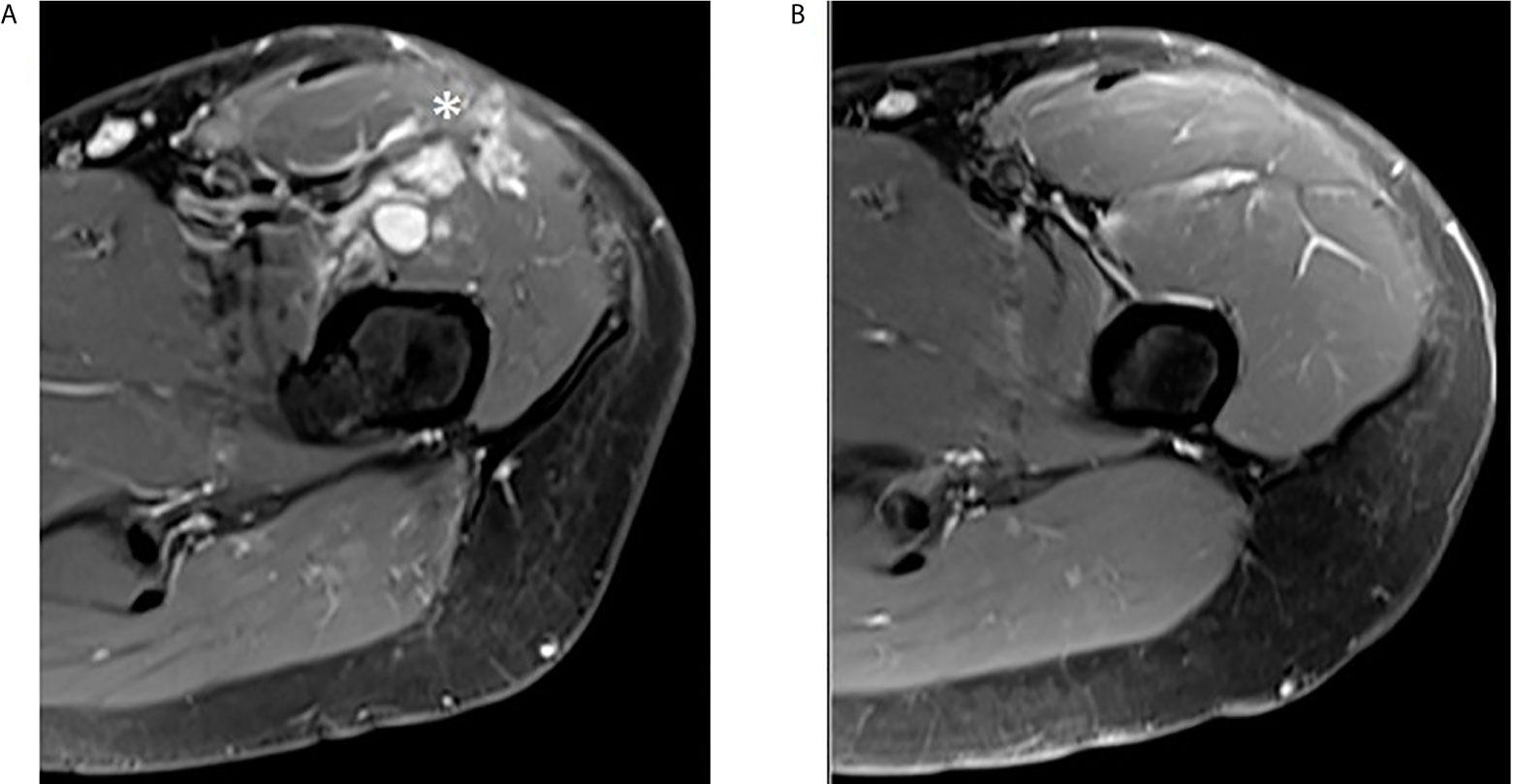
Figure 6 Complete regression of multifocal local recurrence in the left thigh. (A) Post-contrast MR T1 fat saturated axial image of upper left thigh shows some irregular enhancing nodules in the anterior compartment (asterisk). (B) The same sequence obtained three months later in the same area reveals complete nodules disappearance.
In April 2021 the patient is still on treatment with crizotinib, in good clinical conditions and without symptoms or clinical or radiological evidence of disease.
Discussion
The histopathological diagnosis of rare tumors, especially sarcomas, is one of the most difficult tasks to get to, but also one of the most important ones as it may change their multidisciplinary approach. We know from the literature that the initial histological diagnosis often needs to be reviewed by an expert pathologist because in about 15% of cases major discordances are found (35).
In our case, we got to the final diagnosis only after the expert pathology revision and after almost one year from the initial biopsy. IMT is indeed an ultra-rare sarcoma whose diagnosis can be very hard, both for clinicians and pathologists, also considering the multiple differential diagnosis (4).
When the clinical and morphological aspects are compatible with the IMT diagnosis, the immunohistochemical assessment of ALK-staining is paramount for diagnosis as its positivity is observed in 50–60% of IMT cases. In case of a positive result (ALK-positive by IHC), the experts suggest performing FISH analysis for the evaluation of ALK fusions (17, 36). In case of negative IHC result, FISH analysis for the detection of ROS1 and NTRK3 should be performed considering that new target therapies are recently available (20). NGS may be more appealing in this scenario because can also detect other gene fusions characteristic for IMT such as PDGFR, RET, FN1–IGF1R and other rarer mutations, that may be possible targetable mutations in ALK/ROS1-negative IMTs (18, 20, 37, 38). The molecular assessment of IMTs is, therefore, crucial to choose the best target treatment (15, 16).
In 2010 the first case of ALK-positive IMT responding to crizotinib was reported by Butrynski et al., as compared with no response observed in an ALK-negative IMT patient receiving the same treatment (21). Other case series and phase I/II trials reported positive activity and efficacy results of crizotinib in ALK-positive IMTs, which are increased when compared with ALK-negative IMTs (22, 23, 30, 34).
The CREATE trial was the most important study assessing the activity and safety of crizotinib in advanced or inoperable IMTs with and without ALK alterations (34). ALK-positive patients (n = 12) reported an objective response rate (ORR) of 50%, including 17% complete responses, with a disease control rate (DCR) of 100%. On the other hand, ALK-negative patients (n = 7) reported lower ORR and DCR (14 and 86% respectively). Also, survival rates were higher in ALK-positive IMT patients: progression-free survival at 1-year and 2-year (1-yr/2-yr PFS) was of 73.3 and 48.9% in ALK-positive patients compared to 53.6 and 35.7% in ALK-negative ones.
These data suggested the use of crizotinib only in the ALK-positive population, but it is important to notice that the mutations in the ALK-negative group were not reported and the number of patients was very small.
The first case of a ROS1-positive pulmonary IMT pediatric patient responding to crizotinib was reported by Lovly et al. (20). Similarly, a long-lasting remission with crizotinib was observed in another ROS1-rearranged pulmonary pediatric IMT reported by Mai et al. (39).
In consideration of these promising results and those observed in ROS-1-positive non-small cell lung cancer patients, we decided to propose crizotinib to our patient. This clinical case is the first case reported in the literature on an adult patient with a ROS1-rearranged IMT of soft tissues who experienced an outstanding clinical and radiological response to crizotinib.
However, this clinical case is not interesting only for the rarity of the tumor and the exceptional treatment response, but also for the different clinical points in the decision-making of such a rare tumor. Considering the evident limitation due to the absence of literature data, many clinical concerns were raised during the clinical management of the patient. Every diagnostic and therapeutic choice has been taken considering these limits and was previously discussed with the patient.
After the excellent clinical response to crizotinib, we opted for wide excision after 8 months of therapy for the following different reasons. Neoadjuvant or adjuvant therapies for IMT patients have been reported in few clinical cases and seemed a feasible approach (40–42). The only data on crizotinib treatment in ALK-negative patients came from the phase II trial CREATE: in these patients the median duration of response was 7.6 months (34), so that we could not exclude a possible progression of disease in a short time continuing the targeted therapy. Moreover, this decision was guided by the patient’s desire to undergo surgery because of his fear of the local disease that impacted his quality of life.
In the after-surgery scenario, the main problem was to decide whether to resume the treatment with crizotinib even in the absence of evident disease or not. In the literature, very few reports have been written on neoadjuvant or adjuvant use of targeted therapy or chemotherapy in IMTs (40–42). Nevertheless, the impressive response to crizotinib observed in our patient and the initial metastatic disease led us to continue therapy after surgery, even after an almost complete pathological response.
The key point then was to determine for how long this therapy should be continued. A recent retrospective study published by Trahair et al. reported eight cases of ALK-positive IMT children and adolescents treated with crizotinib and surgery with different stages of disease (42). The median duration of treatment with crizotinib, either in the adjuvant or neoadjuvant setting, was 1 year with most patients still disease-free at a median follow up of three years (42). This study revealed that different strategies were all justified and, after treatment discontinuation, most patients maintained the response achieved. These data, even if weak, in addition to the patient’s preference, lead us to discontinue treatment after the completion of one year of treatment.
At disease progression, after therapy discontinuation, we decided to resume crizotinib because, as reported in the literature, new responses are still possible and, moreover, in this rare disease other effective therapies are still not approved and difficult to get the access (25–29, 32, 33, 42–45).
In fact, in ALK-positive IMT patients, after progression to crizotinib, different ALK inhibitors, such as ceritinib, alectinib and lorlatinib, have been investigated reporting promising responses (25–29, 32, 33, 42–45).
Also in ROS1-rearranged patients, especially those with NSCLC, other ROS1 inhibitors (lorlatinib, entrectinib, repotrectinib) have been investigated and may be a possible future therapeutic option in ROS1-positive disease, including IMT (46–50).
In this scenario, molecular assessment is recommended in ALK-negative IMT patients to identify potentially actionable mutations for which new target therapies are available (36). Further clinical trials and expanded access programs are, therefore, needed to address rare tumors, like IMT, to specific and effective treatments.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Ethics Statement
Written informed consent was obtained from the patient for the publication of any potentially identifiable images or data included in this article.
Author Contributions
DC, FCa, and SER: conception, design, writing and review of the manuscript. FCa, MG, and GP: draft of the manuscript and analysis of the patient’s data. RB, MM, FDC, and FP: clinical management of the patient and provision of patient’s data. AG and BS: elaboration and analysis of the pathology. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Coffin CM, Watterson J, Priest JR, Dehner LP. Extrapulmonary Inflammatory Myofibroblastic Tumor (Inflammatory Pseudotumor): A Clinicopathologic and Immunohistochemical Study of 84 Cases. Am J Surg Pathol (1995) 19(8):859–72. doi: 10.1097/00000478-199508000-00001
2. Pettinato G, Manivel JC, De Rosa N, Dehner LP. Inflammatory Myofibroblastic Tumor (Plasma Cell Granuloma). Clinicopathologic Study of 20 Cases With Immunohistochemical and Ultrastructural Observations. Am J Clin Pathol (1990) 94(5):538–46. doi: 10.1093/ajcp/94.5.538
3. Meis JM, Enzinger FM. Inflammatory Fibrosarcoma of the Mesentery and Retroperitoneum: A Tumor Closely Simulating Inflammatory Pseudotumor. Am J Surg Pathol (1991) 15(12):1146–56. doi: 10.1097/00000478-199112000-00005
4. Jo VY, Fletcher CD. WHO Classification of Soft Tissue Tumours: An Update Based on the 2013 (4th) Edition. Pathology (2014) 46(2):95–104. doi: 10.1097/PAT.0000000000000050
5. Gleason BC, Hornick JL. Inflammatory Myofibroblastic Tumours: Where Are We Now? J Clin Pathol (2008) 61(4):428–37. doi: 10.1136/jcp.2007.049387
6. Maruyama Y, Fukushima T, Gomi D, Kobayashi T, Sekiguchi N, Sakamoto A, et al. Relapsed and Unresectable Inflammatory Myofibroblastic Tumor Responded to Chemotherapy: A Case Report and Review of the Literature. Mol Clin Oncol (2017) 7(4):521–4. doi: 10.3892/mco.2017.1383
7. Favini F, Resti AG, Collini P, Casanova M, Meazza C, Trecate G, et al. Inflammatory Myofibroblastic Tumor of the Conjunctiva: Response to Chemotherapy With Low-Dose Methotrexate and Vinorelbine. Pediatr Blood Cancer (2010) 54(3):483–5. doi: 10.1002/pbc.22342
8. Tao YL, Wang ZJ, Han JG, Wei P. Inflammatory Myofibroblastic Tumor Successfully Treated With Chemotherapy and Nonsteroidals: A Case Report. World J Gastroenterol (2012) 18(47):7100–3. doi: 10.3748/wjg.v18.i47.7100
9. Navinan MR, Liyanage I, Herath S, Yudhishdran J, Shivanthan C, Beneragama D, et al. Inoperable Inflammatory Myofibroblastic Tumour of the Para-Nasal Sinuses and Orbit With Recurrence Responding to Methotrexate and Prednisolone: A Case Report. BMC Res Notes (2015) 8:27. doi: 10.1186/s13104-015-0993-3
10. Casali PG, Abecassis N, Aro HT, Bauer S, Biagini R, Bielack S, et al. Soft Tissue and Visceral Sarcomas: ESMO-EURACAN Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up. Ann Oncol (2018) 29(Suppl 4):iv51–67. doi: 10.1093/annonc/mdy096
11. Kube S, Vokuhl C, Dantonello T, Scheer M, Hallmen E, Feuchtgruber S, et al. Inflammatory Myofibroblastic Tumors—A Retrospective Analysis of the Cooperative Weichteilsarkom Studiengruppe. Pediatr Blood Cancer (2018) 65(6):e27012. doi: 10.1002/pbc.27012
12. Kusunoki-Nakamoto F, Matsukawa T, Tanaka M, Miyagawa T, Yamamoto T, Shimizu J, et al. Successful Treatment of an Unresectable Inflammatory Myofibroblastic Tumor of the Frontal Bone Using a Cyclooxygenase-2 Inhibitor and Methotrexate. Intern Med (2013) 52(5):623–8. doi: 10.2169/internalmedicine.52.8785
13. Kubo N, Harada T, Anai S, Otsubo K, Yoneshima Y, Ijichi K, et al. Carboplatin Plus Paclitaxel in the Successful Treatment of Advanced Inflammatory Myofibroblastic Tumor. Intern Med (2012) 51(17):2399–401. doi: 10.2169/internalmedicine.51.7599
14. Chen M, Zhang L, Cao G, Zhu W, Chen X. Fang Q. Partial Response to Chemotherapy in a Patient With Retroperitoneal Inflammatory Myofibroblastic Tumor. Mol Clin Oncol (2016) 5(4):463–6. doi: 10.3892/mco.2016.967
15. Casanova M, Brennan B, Alaggio R, Kelsey A, Orbach D, van Noesel MM, et al. Inflammatory Myofibroblastic Tumor: The Experience of the European Pediatric Soft Tissue Sarcoma Study Group (EpSSG). Eur J Cancer (2020) 127:123–9. doi: 10.1016/j.ejca.2019.12.021
16. Baldi GG, Brahmi M, Lo Vullo S, Cojocaru E, Mir O, Casanova M, et al. The Activity of Chemotherapy in Inflammatory Myofibroblastic Tumors: A Multicenter, European Retrospective Case Series Analysis. Oncologist (2020) 25(11):e1777–84. doi: 10.1634/theoncologist.2020-0352
17. Antonescu CR, Suurmeijer AJH, Zhang L, Sung YS, Jungbluth AA, Travis WD, et al. Molecular Characterization of Inflammatory Myofibroblastic Tumors With Frequent ALK and ROS1 Gene Fusions and Rare Novel RET Rearrangement. Am J Surg Pathol (2015) 39(7):957–67. doi: 10.1097/PAS.0000000000000404
18. Yamamoto H, Yoshida A, Taguchi K, Kohashi K, Hatanaka Y, Yamashita A, et al. Alk, ROS1 and NTRK3 Gene Rearrangements in Inflammatory Myofibroblastic Tumours. Histopathology (2016) 69(1):72–83. doi: 10.1111/his.12910
19. Davies KD, Doebele RC. Molecular Pathways: ROS1 Fusion Proteins in Cancer. Clin Cancer Res (2013) 19(15):4040–5. doi: 10.1158/1078-0432.CCR-12-2851
20. Lovly CM, Gupta A, Lipson D, Otto G, Brennan T, Chung CT, et al. Inflammatory Myofibroblastic Tumors Harbor Multiple Potentially Actionable Kinase Fusions. Cancer Discov (2014) 4(8):889–95. doi: 10.1158/2159-8290.CD-14-0377
21. Butrynski JE, D’Adamo DR, Hornick JL, Dal Cin P, Antonescu CR, Jhanwar SC, et al. Crizotinib in ALK-Rearranged Inflammatory Myofibroblastic Tumor. N Engl J Med (2010) 363(18):1727–33. doi: 10.1056/NEJMoa1007056
22. Mossé YP, Lim MS, Voss SD, Wilner K, Ruffner K, Laliberte J, et al. Safety and Activity of Crizotinib for Paediatric Patients With Refractory Solid Tumours or Anaplastic Large-Cell Lymphoma: A Children’s Oncology Group Phase 1 Consortium Study. Lancet Oncol (2013) 14(6):472–80. doi: 10.1016/S1470-2045(13)70095-0
23. Gambacorti-Passerini C, Orlov S, Zhang L, Braiteh F, Huang H, Esaki T, et al. Long-Term Effects of Crizotinib in ALK-Positive Tumors (Excluding NSCLC): A Phase 1b Open-Label Study. Am J Hematol (2018) 93(5):607–14. doi: 10.1002/ajh.25043
24. Brivio E, Zwaan CM. ALK Inhibition in Two Emblematic Cases of Pediatric Inflammatory Myofibroblastic Tumor: Efficacy and Side Effects. Pediatr Blood Cancer (2019) 66(5):e27645. doi: 10.1002/pbc.27645
25. Parker BM, Parker JV, Lymperopoulos A. A Case Report: Pharmacology and Resistance Patterns of Three Generations of ALK Inhibitors in Metastatic Inflammatory Myofibroblastic Sarcoma. J Oncol Pharm Pract (2019) 25(5):1226–30. doi: 10.1177/1078155218781944
26. Honda K, Kadowaki S, Kato K, Hanai N, Hasegawa Y, Yatabe Y, et al. Durable Response to the ALK Inhibitor Alectinib in Inflammatory Myofibroblastic Tumor of the Head and Neck With a Novel SQSTM1–ALK Fusion: A Case Report. Invest New Drugs (2019) 37(4):791–5. doi: 10.1007/s10637-019-00742-2
27. Nishio M, Murakami H, Horiike A, Takahashi T, Hirai F, Suenaga N, et al. Phase I Study of Ceritinib (LDK378) in Japanese Patients With Advanced, Anaplastic Lymphoma Kinase-Rearranged Non-Small-Cell Lung Cancer or Other Tumors. J Thorac Oncol (2015) 10(7):1058–66. doi: 10.1097/JTO.0000000000000566
28. Mansfield AS, Murphy SJ, Harris FR, Robinson SI, Marks RS, Johnson SH, et al. Chromoplectic TPM3-ALK Rearrangement in a Patient With Inflammatory Myofibroblastic Tumor Who Responded to Ceritinib After Progression on Crizotinib. Ann Oncol (2016) 27(11):2111–7. doi: 10.1093/annonc/mdw405
29. Ono A, Murakami H, Serizawa M, Wakuda K, Kenmotsu H, Naito T, et al. Drastic Initial Response and Subsequent Response to Two ALK Inhibitors in a Patient With a Highly Aggressive ALK-Rearranged Inflammatory Myofibroblastic Tumor Arising in the Pleural Cavity. Lung Cancer (2016) 99:151–4. doi: 10.1016/j.lungcan.2016.07.002
30. Mossé YP, Voss SD, Lim MS, Rolland D, Minard CG, Fox E, et al. Targeting ALK With Crizotinib in Pediatric Anaplastic Large Cell Lymphoma and Inflammatory Myofibroblastic Tumor: A Children’s Oncology Group Study. J Clin Oncol (2017) 35(28):3215–21. doi: 10.1200/JCO.2017.73.4830
31. Yuan C, Ma MJ, Parker JV, Mekhail TM. Metastatic Anaplastic Lymphoma Kinase-1 (ALK-1)-Rearranged Inflammatory Myofibroblastic Sarcoma to the Brain With Leptomeningeal Involvement: Favorable Response to Serial ALK Inhibitors: A Case Report. Am J Case Rep (2017) 18:799–804. doi: 10.12659/AJCR.903698
32. Saiki M, Ohyanagi F, Ariyasu R, Koyama J, Sonoda T, Nishikawa S, et al. Dramatic Response to Alectinib in Inflammatory Myofibroblastic Tumor With Anaplastic Lymphoma Kinase Fusion Gene. Jpn J Clin Oncol (2017) 47(12):1189–92. doi: 10.1093/jjco/hyx133
33. Rao N, Iwenofu H, Tang B, Woyach J, Liebner DA. Inflammatory Myofibroblastic Tumor Driven by Novel NUMA1-ALK Fusion Responds to ALK Inhibition. JNCCN J Natl Compr Cancer Netw (2018) 16(2):115–21. doi: 10.6004/jnccn.2017.7031
34. Schöffski P, Sufliarsky J, Gelderblom H, Blay JY, Strauss SJ, Stacchiotti S, et al. Crizotinib in Patients With Advanced, Inoperable Inflammatory Myofibroblastic Tumours With and Without Anaplastic Lymphoma Kinase Gene Alterations (European Organisation for Research and Treatment of Cancer 90101 CREATE): A Multicentre, Single-Drug, Prosp. Lancet Respir Med (2018) 6(6):431–41. doi: 10.1016/S2213-2600(18)30116-4
35. Perrier L, Rascle P, Ray-Coquard I, Bui Nuguyen B, Morelle M, Ranchere Vince D, et al. Cost of Discordant Diagnoses in Sarcoma, Gist, and Desmoid Tumors in France: Results From the RREPS (Reseau De Reference En Pathologie Des Sarcomes) Network. Value Heal (2014) 17(3):95–6. doi: 10.1016/j.jval.2014.03.556
36. Chang JC, Zhang L, Drilon AE, Chi P, Alaggio R, Borsu L, et al. Expanding the Molecular Characterization of Thoracic Inflammatory Myofibroblastic Tumors Beyond ALK Gene Rearrangements. J Thorac Oncol (2019) 14(5):825–34. doi: 10.1016/j.jtho.2018.12.003
37. Hornick JL, Sholl LM, Dal Cin P, Childress MA, Lovly CM. Expression of ROS1 Predicts ROS1 Gene Rearrangement in Inflammatory Myofibroblastic Tumors. Mod Pathol (2015) 28(5):732–9. doi: 10.1038/modpathol.2014.165
38. Piarulli G, Puls F, Wängberg B, Fagman H, Hansson M, Nilsson J, et al. Gene Fusion Involving the Insulin-Like Growth Factor 1 Receptor in an ALK-Negative Inflammatory Myofibroblastic Tumour. Histopathology (2019) 74(7):1098–102. doi: 10.1111/his.13839
39. Mai S, Xiong G, Diao D, Wang W, Zhou Y, Cai R. Case Report: Crizotinib Is Effective in a Patient With ROS1-Rearranged Pulmonary Inflammatory Myofibroblastic Tumor. Lung Cancer (2019) 128:101–4. doi: 10.1016/j.lungcan.2018.12.016
40. Nagumo Y, Maejima A, Toyoshima Y, Komiyama M, Yonemori K, Yoshida A, et al. Neoadjuvant Crizotinib in ALK-Rearranged Inflammatory Myofibroblastic Tumor of the Urinary Bladder: A Case Report. Int J Surg Case Rep (2018) 48:1–4. doi: 10.1016/j.ijscr.2018.04.027
41. Bertocchini A, Lo Zupone C, Callea F, Gennari F, Serra A, Monti L, et al. Unresectable Multifocal Omental and Peritoneal Inflammatory Myofibroblastic Tumor in a Child: Revisiting the Role of Adjuvant Therapy. J Pediatr Surg (2011) 46(4):e17–21. doi: 10.1016/j.jpedsurg.2011.01.007
42. Trahair T, Gifford AJ, Fordham A, Mayoh C, Fadia M, Lukeis R, et al. Crizotinib and Surgery for Long-Term Disease Control in Children and Adolescents With ALK-Positive Inflammatory Myofibroblastic Tumors. JCO Precis Oncol (2019) 3:PO.18.00297. doi: 10.1200/PO.18.00297
43. Alan O, Kuzhan O, Koca S, Telli TA, Basoglu T, Ercelep O, et al. How Long Should We Continue Crizotinib in ALK Translocation-Positive Inflammatory Myofibroblastic Tumors? Long-Term Complete Response With Crizotinib and Review of the Literature. J Oncol Pharm Pract (2020) 26(4):1011–8. doi: 10.1177/1078155219879757
44. Theilen TM, Soerensen J, Bochennek K, Becker M, Schwabe D, Rolle U, et al. Crizotinib in ALK + Inflammatory Myofibroblastic Tumors—Current Experience and Future Perspectives. Pediatr Blood Cancer (2018) 65(4). doi: 10.1002/pbc.26920
45. Li Y, Chen X, Qu Y, Fan JM, Li Y, Peng H, et al. Zhang HB. Partial Response to Ceritinib in a Patient With Abdominal Inflammatory Myofibroblastic Tumor Carrying a TFG-ROS1 Fusion. J Natl Compr Canc Netw (2019) 17(12):1459–62. doi: 10.6004/jnccn.2019.7360
46. Drilon A, Siena S, Ou SI, Patel M, Ahn MJ, Lee J, et al. Safety and Antitumor Activity of the Multitargeted Pan-TRK, ROS1, and ALK Inhibitor Entrectinib: Combined Results From Two Phase I Trials (ALKA-372-001 and STARTRK-1). Cancer Discov (2017) 7(4):400–9. doi: 10.1158/2159-8290.CD-16-1237
47. Yun MR, Kim DH, Kim S-Y, Joo HS, Lee YW, Choi HM, et al. Repotrectinib Exhibits Potent Antitumor Activity in Treatment-Naïve and Solvent-Front–Mutant ROS1-Rearranged Non–Small Cell Lung Cancer. Clin Cancer Res (2020) 26(13):3287–95. doi: 10.1158/1078-0432.ccr-19-2777
48. Shaw AT, Felip E, Bauer TM, Besse B, Navarro A, Postel-Vinay S, et al. Lorlatinib in Non-Small-Cell Lung Cancer With ALK or ROS1 Rearrangement: An International, Multicentre, Open-Label, Single-Arm First-in-Man Phase 1 Trial. Lancet Oncol (2017) 18(12):1590–9. doi: 10.1016/S1470-2045(17)30680-0
49. Ambati SR, Slotkin EK, Chow-Maneval E, Basu EM. Entrectinib in Two Pediatric Patients With Inflammatory Myofibroblastic Tumors Harboring ROS1 or ALK Gene Fusions. JCO Precis Oncol (2018). doi: 10.1200/po.18.00095
Keywords: inflammatory myofibroblastic tumor, inflammatory pseudotumor, sarcoma, ROS1, crizotinib, retreatment, target therapy
Citation: Comandini D, Catalano F, Grassi M, Pesola G, Bertulli R, Guadagno A, Spina B, Mascherini M, De Cian F, Pistoia F and Rebuzzi SE (2021) Outstanding Response in a Patient With ROS1-Rearranged Inflammatory Myofibroblastic Tumor of Soft Tissues Treated With Crizotinib: Case Report. Front. Oncol. 11:658327. doi: 10.3389/fonc.2021.658327
Received: 17 March 2021; Accepted: 26 April 2021;
Published: 15 June 2021.
Edited by:
Massimo Fantini, Precision Biologics, Inc., United StatesReviewed by:
Victor C. Kok, Asia University, TaiwanVladislav Korobeynikov, Columbia University Irving Medical Center, United States
Copyright © 2021 Comandini, Catalano, Grassi, Pesola, Bertulli, Guadagno, Spina, Mascherini, De Cian, Pistoia and Rebuzzi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sara Elena Rebuzzi, c2FyYWVsZW5hODlAaG90bWFpbC5pdA==; orcid.org/0000-0003-0546-6304
†These authors have contributed equally to this work