 Giulia Pinton1*Zhuo Wang2Cecilia Balzano1
Giulia Pinton1*Zhuo Wang2Cecilia Balzano1 Sara Missaglia3
Sara Missaglia3 Daniela Tavian3Renzo Boldorini4Dean A. Fennell5Martin Griffin2
Daniela Tavian3Renzo Boldorini4Dean A. Fennell5Martin Griffin2 Laura Moro1*
Laura Moro1*- 1Department of Pharmaceutical Sciences, University of Piemonte Orientale (UPO), Novara, Italy
- 2School of Life and Health Sciences, Aston University, Birmingham, United Kingdom
- 3Laboratory of Cellular Biochemistry and Molecular Biology, Centro di Ricerca in Biochimica E Nutrizione dello Sport (CRIBENS), Catholic University of the Sacred Heart, Milan, Italy
- 4Department of Health Science, University of Piemonte Orientale (UPO), Novara, Italy
- 5Leicester Cancer Research Centre, University of Leicester, Leicester, United Kingdom
Malignant pleural mesothelioma is an aggressive cancer, heterogeneous in its presentation and behaviour. Despite an increasing knowledge about molecular markers and their diagnostic and prognostic value, they are not used as much as they might be for treatment allocation. It has been recently reported that mesothelioma cells that lack BAP1 (BRCA1 Associated Protein) are sensitive to inhibition of the EZH2 (Enhancer of Zeste Homolog 2) histone methyltransferase. Since we observed strong H3K27me3 (histone H3 lysine 27 trimetylation) immunoreactivity in BAP1 wild-type mesothelioma biopsies, we decided to characterize in vitro the response/resistance of BAP1 wild-type mesothelioma cells to the EZH2 selective inhibitor, EPZ-6438. Here we demonstrate that BAP1 wild-type mesothelioma cells were rendered sensitive to EPZ-6438 upon SIRT1 (Sirtuin 1) silencing/inhibition or when cultured as multicellular spheroids, in which SIRT1 expression was lower compared to cells grown in monolayers. Notably, treatment of spheroids with EPZ-6438 abolished H3K27me3 and induced the expression of CDKN2A (Cyclin-Dependent Kinase Inhibitor 2A), causing cell growth arrest. EPZ-6438 treatment also resulted in a rapid and sustained induction of the genes encoding HIF2α (Hypoxia Inducible Factor 2α), TG2 (Transglutaminase 2) and IL-6 (Interleukin 6). Loss of CDKN2 is a common event in mesothelioma. CDKN2A silencing in combination with EPZ-6438 treatment induced apoptotic death in mesothelioma spheroids. In a CDKN2A wild-type setting apoptosis was induced by combining EPZ-6438 with 1-155, a TG2 selective and irreversible inhibitor. In conclusion, our data suggests that the expression of CDKN2A predicts cell fate in response to EZH2 inhibition and could potentially stratify tumors likely to undergo apoptosis.
Introduction
Malignant pleural mesothelioma (MPM) is a lethal cancer that originates from the mesothelial cells aligning the pleura (1). The causal association of MPM with asbestos exposure is well established and supported by epidemiological and toxicological studies (2, 3). Although asbestos has been banned in numerous countries, it continues to be used worldwide, and a rise in the MPM global incidence is predicted (4).
The most commonly used chemotherapy drugs for treating MPM include pemetrexed with cisplatin or carboplatin (5). The high number of non-responders to chemotherapy, as well as the frequent recurrences of the disease (6, 7), suggests the presence of drug-resistant clones within the tumor. Given that MPM develops over many years and its growth rate is in most cases quite low, it is highly likely that it has profound heterogeneity, which makes it challenging to eradicate (8).
MPM intra- and inter-tumor heterogeneity manifests with a morphological spectrum, ranging from epithelioid to sarcomatoid tumors, with the biphasic subtype containing a combination of both components (9). Besides histological diversity, an increasing number of publications highlight the importance of genetic and epigenetic intra-tumor heterogeneity for MPM therapeutic resistance (10–13). Genomic interrogation has revealed extensive interpatient heterogeneity with frequent tumor suppressor inactivation being a dominant feature of the mutation landscape mediated by multiple mechanisms, which include single nucleotide variation, copy number losses, gene fusions and splicing alterations (14, 15). Loss or mutation of BAP1 (BRCA1 Associated Protein 1) coding gene is recurrently identified in MPM and translates into nuclear negativity for BAP1 expression (16). Levine’s group has reported that BAP1 inactivation leads to upregulation and dependence on the chromatin modifying Polycomb Repressive Complex 2 (PRC2), comprising the methyltransferase EZH2 (Enhancer of Zeste Homolog 2), which trimethylates lysine 27 on histone H3 (H3K27me3) (17). Besides transcriptional regulation, post-translational modifications of EZH2, including phosphorylation (18), acetylation (19), ubiquitination (20), sumoylation (21) and GlcNAcylation (22), have also been found to be important for its expression and silencing function on target genes.
Another common genetic alteration in MPM is the homozygous deletion of the 9p21 locus, within a cluster of genes that includes CDKN2A (cyclin-dependent kinase inhibitor 2A) encoding p16ink4a (10, 23, 24). Heterozygous deletion of CDKN2A is also commonly observed, sometimes at higher levels than homozygous deletion, however, few reports have addressed its role in MPM (25–27). Furthermore, a discrepancy between p16ink4a protein expression and gene deletion suggests that epigenetic mechanisms play a role in its regulation (28). Indeed, H3K27me3-dependent repression of CDKN2A transcription is a common feature of many tumors (29, 30).
H3K27 methylation can also be influenced by hypoxia (31). We have described that HIF-2α (Hypoxia Inducible Factor 2α) induces the expression of the H3K27 demethylase KDM6B and reduces the H3K27me3 repressive mark in MPM cells exposed to chronic hypoxia (32). Furthermore, we have described that HIF-2α-mediates the induction of TGM2 and the increase of TG2 (Transglutaminase 2) activity (33). TG2 is a multifunctional enzyme that exhibits crosslinking, GTPase, cell adhesion, protein disulfide isomerase, kinase, and scaffold activities (34). TG2 expression and downstream IL-6 (Interleukin-6) production have been profoundly correlated with primary tumor growth, peritoneal spreading, distant metastasis and resistance to standard cytotoxic agents (35, 36). We have demonstrated that TG2 inhibition or TGM2 silencing causes, under hypoxic conditions, a significant reduction of MPM cell viability (33).
Here we describe strong H3K27me3 immunoreactivity in tumor tissue samples obtained from patients diagnosed with BAP1 wild-type MPM. Moreover, we report that the treatment of MPM multicellular spheroids with an EZH2 selective inhibitor induces CDKN2A expression and cell cycle arrest. Finally, we describe the involvement of p16ink4a and TG2 in the control of MPM apoptotic cell death in response to EZH2 inhibition.
Materials and Methods
Reagents and Antibodies
The polyclonal antibodies specific for histone H3 trimethyl lysine 27 (H3K27me3), histone H3, SIRT1, KDM6B and the monoclonal antibodies specific for Poly (ADP-ribose) polymerase 1 (PARP1), acetyl-lysine, HIF2α, BAP1 and α-tubulin were purchased from Santa Cruz Biotechnology (Santa Cruz CA, USA). Polyclonal antibody anti-p16ink4a was from Cell Signaling Technology (Leiden, The Netherlands). Polyclonal antibody specific for EZH2 was from Active Motif (La Hulpe, Belgium). Anti-mouse and anti-rabbit IgG peroxidase or FITC conjugated antibodies and chemical reagents were from Sigma-Aldrich (St. Louis, MO, USA). ECL, nitrocellulose membranes and protein assay kit were from Bio-Rad (Hercules, CA, USA). Culture media, sera, antibiotics, LipofectAMINE transfection reagent and the monoclonal antibody specific for TG2 (TG100) were from Thermo Fisher (Waltham, MA, USA). EX-527 and the EZH2-selective inhibitor, EPZ-6438, were from Selleckchem (Houston, TX, USA). The highly selective TG2 inhibitor, 1-155 (cell-permeable) was designed and synthesized as previously documented (37, 38).
Immunohistochemistry on Formalin-Fixed Paraffin-Embedded Tissue Specimens
BAP1 and histone H3 trimethyl lysine 27 (H3K27me3) expression were evaluated by immunohistochemistry (IHC) in eight biphasic MPM human biopsies using a BenchMark standard automated immunostainer (Ventana Medical System, Tucson, AZ, USA). Specific primary antibodies against the anti-human BAP1 and the polyclonal antibodies specific for H3K27me3 were used. BAP1 and H3K27me3 were considered positive when a weak-to-strong nuclear positivity was shown. Negative controls were obtained by replacing the primary antibody by PBS. Non-neoplastic cells, such as vascular endothelium or inflammatory cells, were considered as internal positive controls. Slides were counterstained with hematoxylin.
Cell Cultures and Transfection
The biphasic MPM derived MSTO-211H cell lines was obtained from the Istituto Scientifico Tumori (IST) Cell-bank, Genoa, Italy; the epithelioid MPM BR95 cell line was kindly provided by Prof. Osella D. (University of Piemonte Orientale, Alessandria, Italy). Cells were grown in standard conditions in RPMI medium supplemented with 10% FBS, 100 μg/ml streptomycin and 10 μg/ml penicillin at 37°C in a humidified environment containing 5% CO2. Mycoplasma infection was excluded by the use of Mycoplasma PlusTM PCR Primer Set kit from Stratagene (La Jolla, CA, USA). Cells grown to 80% confluence in tissue culture dishes were transiently transfected with negative control or specific siRNAs from Qiagen (Hilden, Germany) using LipofectAMINE reagent as described by the manufacturer. To obtain cell number and viability information following treatments, cell were trypsinized and stained with Trypan blue; the number of cells considered viable (unstained cells) was counted in a Bürker chamber within 5 min after staining.
Multicellular Spheroids
Multicellular spheroids were generated in non-adsorbent round-bottomed 96-well plates, as previously described (31). The 96-well plates were coated with a 1:24 dilution of polyHEMA (120 mg/ml) in 95% ethanol and dried at 37°C for 24 h. Before use, plates were sterilized by UV light for 30 min. For generation of multicellular spheroids, 1 × 104 cells were added into each well of polyHEMA-coated 96-well plate and placed in a 37°C humidified incubator with 5% CO2. Every 24 h, 50% of supernatant was replaced with fresh medium ± 10 μM EPZ-6438 and/or 5 μM 1-155 inhibitors. For cell dissemination, spheroids treated for 48 h with or without EPZ-6438, were transferred to flat-bottomed culture dishes and incubated in complete medium for additional 24 h. Migration area was observed under the microscope.
Immunofluorescence Staining
Cell spheroids were let to adhere for 1/2 h to poly-L-lysine coated glass slides and then fixed in 4% paraformaldehyde, permeabilized with 0.5% Triton X-100 in PBS and blocked in 3% BSA/PBS 10% FBS. The primary antibody (mouse anti-H3K27me3 1:100) was incubated for 2 h at 4°C. The fluorescent secondary antibody (rabbit anti-mouse IgG antibody conjugated with fluorescein isothiocyanate (FITC); 1:100) was incubated for 1 h at 4°C. Fluorescent images were captured using a Leica MB5000B microscope equipped with a DFC480 R2 digital camera and a Leica Application Suite (LAS) software.
Cell Cycle Analysis
For cell cycle/apoptosis analysis, 5 × 105 cells were silenced for SIRT1 and treated or not with EPZ-6438 for 48 h at 37°C in a 5% CO2 atmosphere. After incubation, detached and suspended cells were harvested in complete RPMI and centrifuged at 500×g for 10 min. Pellets were washed with PBS, pH 7.4, in ice-cold 75% ethanol at 4°C, treated with 100 mg/ml RNAse A for 1 h at 37°C, stained with 25 μg/ml propidium iodide and finally analyzed by using a Bio-Rad S3e Cell Sorter (Hercules, CA, USA) and the Modfit software (Verity Software House, Topsham, ME, USA).
Cell Lysis and Immunoblot
Cells were extracted with 1% NP-40 lysis buffer (50 mM Tris–HCl pH 8.5 containing 1% NP-40, 150 mM NaCl, 10 mM EDTA, 10 mM NaF, 10 mM Na4P2O7 and 0.4 mM Na3VO4) with freshly added protease inhibitors (10 μg/ml leupeptin, 4 μg/ml pepstatin and 0.1 Unit/ml aprotinin). Lysates were centrifuged at 13.000×g for 10 min at 4°C and the supernatants were collected and assayed for protein concentration with the Bradford assay method (Bio-Rad). Histones were acid extracted from nuclei with 0.4 N HCl and precipitated with trichloroacetic acid (TCA), followed by washing with ice-cold acetone containing 0.006% HCl, and then with pure ice-cold acetone. The resulting pellets were air-dried, dissolved in a minimal volume of sterile distilled water and the protein concentration was determined.
Proteins were separated by SDS-PAGE under reducing conditions. Following SDS-PAGE, proteins were transferred to nitrocellulose, reacted with specific antibodies and then detected with peroxidase-conjugate secondary antibodies and chemioluminescent ECL reagent. Digital images were taken with the Bio-Rad ChemiDoc™ Touch Imaging System and quantified using Bio-Rad Image Lab 5.2.1.
RNA Isolation and Real-Time PCR
Total RNA was extracted using the guanidinium thiocyanate method. Starting from equal amounts of RNA, cDNA used as template for amplification in the real-time PCR (5 µg), was synthesized by the reverse transcription reaction using RevertAid Minus First Strand cDNA Synthesis Kit from Fermentas-Thermo Scientific (Burlington, ON, Canada), using random hexamers as primers, according to the manufacturer’s instructions. Some 20 ng of cDNA were used to perform RT-PCR amplification of mRNA. The real-time reverse transcription-PCR was performed using the double-stranded DNA-binding dye SYBR Green PCR Master Mix (Fermentas-Thermo Scientific) on an ABI GeneAmp 7000 Sequence Detection System machine, as described by the manufacturer. The instrument, for each gene tested, obtained graphical Cycle threshold (Ct) values automatically. Triplicate reactions were performed for each marker and the melting curves were constructed using Dissociation Curves Software (Applied Biosystems, Foster City, CA, USA), to ensure that only a single product was amplified.
Statistical Analysis
Statistical evaluation of the differential analysis was performed by one-way ANOVA and Student’s t-test.
Results
BAP1 Wild-Type Mesotheliomas Exhibit H3K27me3 Immunoreactivity
BAP1 is inactive in 40–60% of MPM patients (10, 14). Recent evidence suggesting the potential efficacy of EZH2 inhibition in preclinical models of BAP1 loss led to a phase II clinical trial evaluating the EZH2 inhibitor Tazemetostat (EPZ-6438) in MPM patients with inactive BAP1 (NCT02860286).
Here, we examined the H3K27me3 status in biopsies obtained at diagnosis from a cohort of BAP1 positive, biphasic MPM, using immunohistochemistry (IHC). All tissues examined showed strong BAP1 and H3K27me3 nuclear staining in the majority of tumor cells (3+ in >67%) (Figures 1A–D and Figures S1A–F). A minority of cells displayed a reduced H3K27me3 nuclear staining, however, further and deeper investigation is needed in order to associate significant differences in intensity with the epithelioid or the sarcomatoid component of the tumor. Strong immunoreactivity for H3K27me3 was also observed in the non-neoplastic infiltrating inflammatory cells (Figures S1E, F).
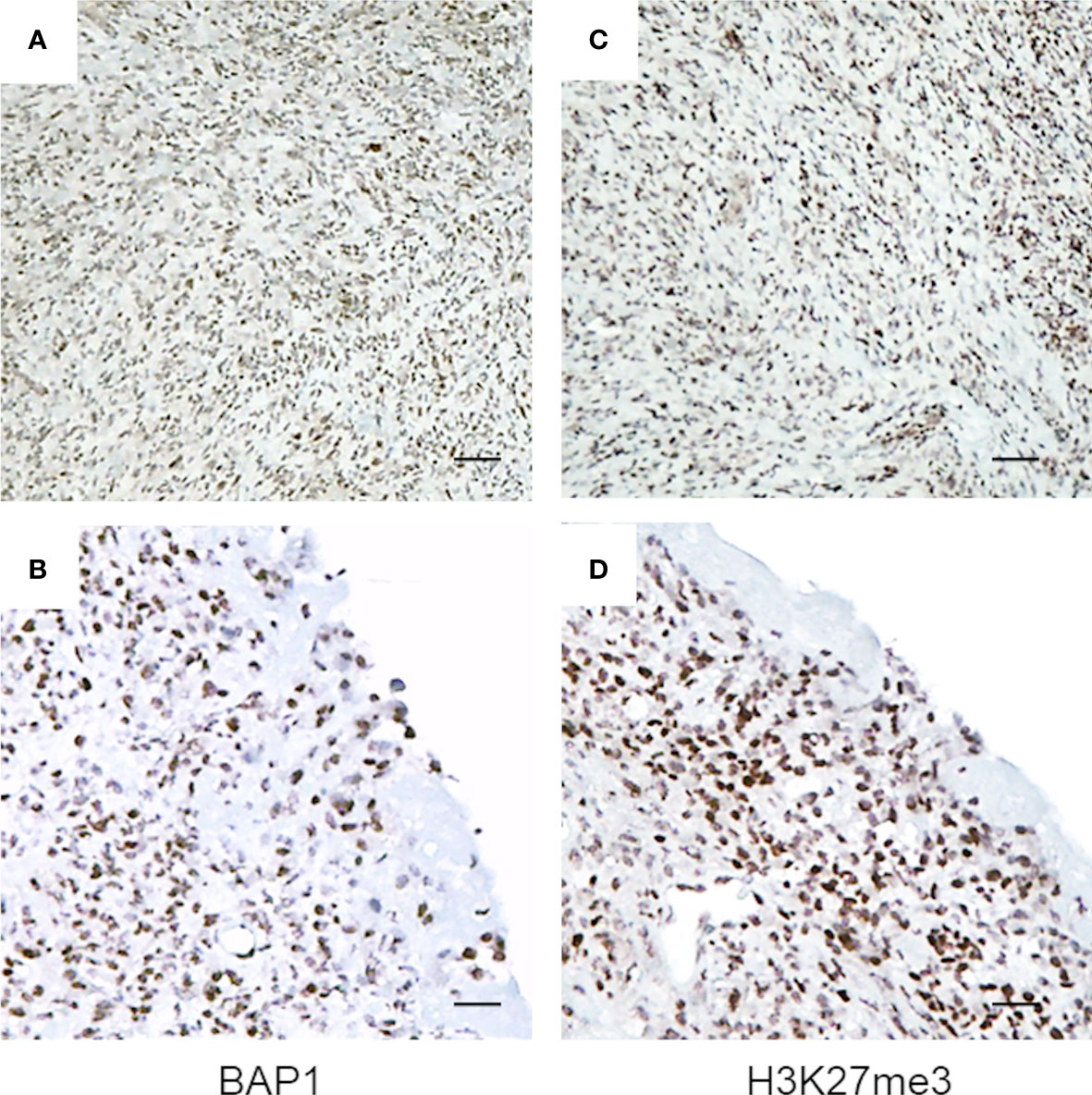
Figure 1 BAP1 wild-type MPM tumor biopsies exhibit high immunoreactivity for H3K27me3. Representative images of immunostaining. Byphasic MPM tissue samples were stained for BAP1 (A, B) and H3K27me3 (C, D). Images were captured at magnification ×40 (A, C) and ×200 (B, D) using light microscopy. Scale bar = 100 μM.
Inhibition of EZH2 Activity Exerts Anti-Proliferative Effect in BAP1 Wild-Type MPM Spheroids
Based on IHC results, we decided to use the MSTO-211H cell line, established from the pleural effusion of a patient with biphasic mesothelioma, as a model to better characterize in vitro the response of BAP1 wild-type MPM to EPZ-6438, a potent and selective inhibitor of the H3K27 methyltransferase EZH2.
We first report, as described by LaFave et al. (17), that inhibition of the EZH2 methyltransferase activity, by EPZ-6438, did not affect MSTO-211H cell viability, when cultured as two-dimensional (2D) monolayer (Figures 2A, a, b, B). Differently, we observed that EPZ-6438 treatment significantly reduced the size and modified the architecture of MSTO-211H multicellular spheroids (MCS), as evidenced by bright field and pseudo-color images (Figures 2A, c–f), and reduced cell dissemination after re-adhesion of MCS to flat-bottomed plates (Figures 2A, g, h). Cell counts confirmed that the reduction in size of EPZ-6438 treated MCS was due to a lower number of cells and not to an increase in cell packing density (Figure 2B). Immunofluorescence images in Figures 2A(i, j) show that H3K27me3 was spatially restricted to the external proliferating region of the spheroids and was completely abolished by EPZ-6438 treatment. In agreement, we documented high expression of the H3K27 demethylase KDM6B in the internal hypoxic zone of the spheroids, enriched in HIF2α expression (Figure S2) (31). Western-blot analysis confirmed an increase in H3K27 methylation in MSTO-211H MCS versus 2D culture that was completely abolished by EPZ-6438 treatment (Figure 2C).
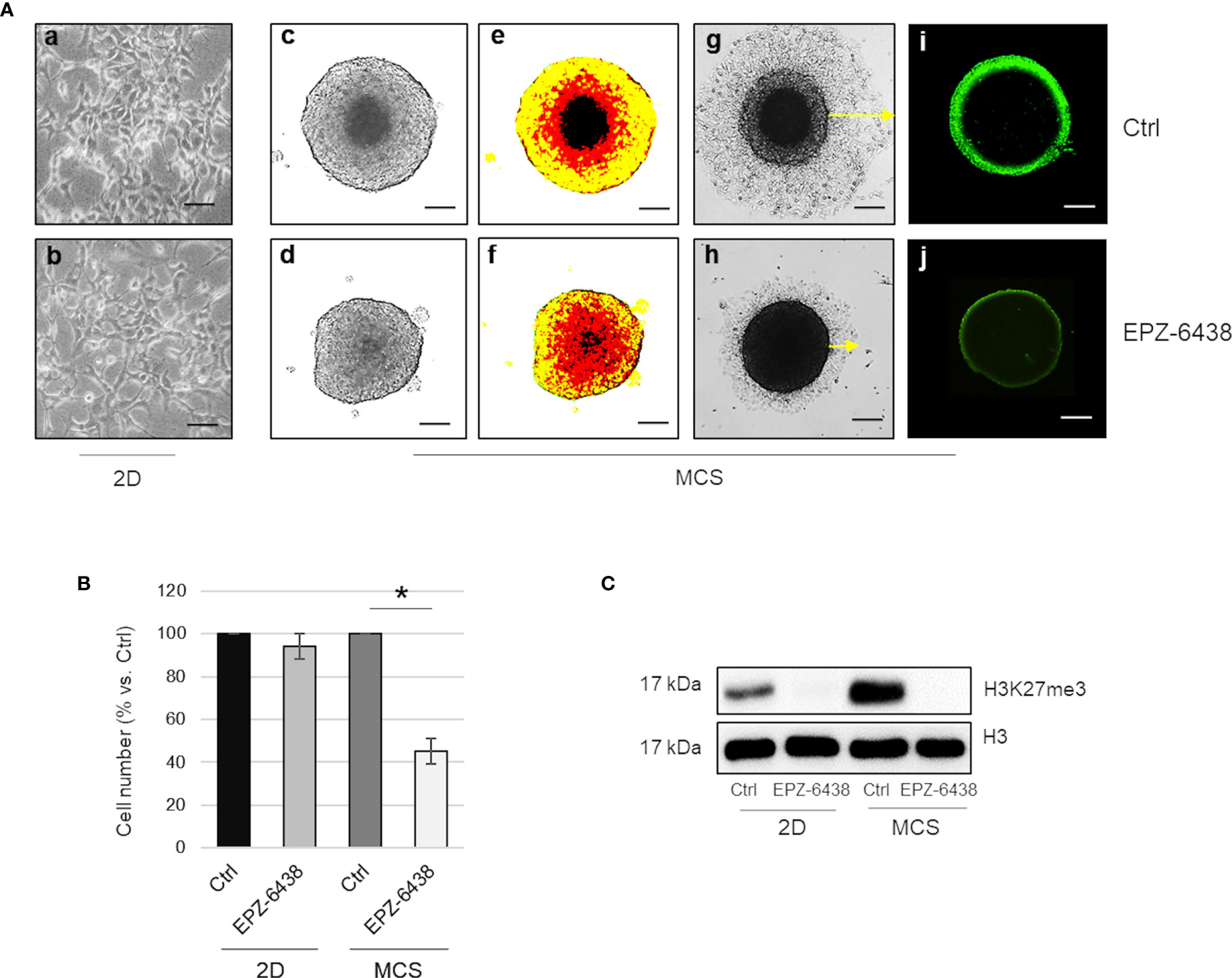
Figure 2 EZH2 inhibition displays an antiproliferative effect in BAP1 wild-type MPM MCS. (A) Representative phase contrast images (×40 magnification) of MSTO-211H cultured as monolayer (a, b) or MCS (1 of 12) (c, d) with relative pseudo-color images (e, f). Representative phase contrast images of dissemination of MSTO-211H MCS (one of 12) after 24 h re-adhesion to flat-bottomed plates, post 48 h treatment in the presence or absence of EPZ-6438 (g, h). H3K27me3 immunofluorescence image of MSTO-211H MCS treated for 48 h, with or without EPZ-6438 (i, j). Scale bar = 100 μM. (B) Bar graph shows the percentage of viability of MSTO-211H cells cultured as monolayer or MCS ± 48 h treatment with EPZ-6438. Each bar represents mean of three independent experiments ± s.d., *p ≤0.05. (C) Representative Western blot analysis of H3K27me3 in MSTO-211H cultured in 2D or as MCS ± 48 h treatment with EPZ-6438. histone H3 was used as loading control.
SIRT1 Inhibition Confers Sensitivity to EZH2 Inhibition in BAP1 Wild-Type MPM Cells
It has been reported that depletion of the NAD-dependent deacetylase SIRT1 increases EZH2 protein acetylation, stability and its repressive effect on target genes (19). In our previous studies, we demonstrated lactate-mediated SIRT1 downregulation, in MPM cells cultured as MCS compared to monolayer (39). Here, along with SIRT1 downregulation, we observed increased expression of EZH2 in MSTO-211H MCS (Figure 3A). To evaluate the regulatory role of SIRT1 on EZH2 expression and activity in BAP1 wild-type MPM, we treated MSTO-211H cells in 2D with the SIRT1 selective inhibitor, EX-527, alone or in combination with EPZ-6438. In MSTO-211H cells treated with EX-527, we demonstrated the expression of EZH2 protein significantly increased, whereas EZH2 mRNA levels remained stable (Figure 3B). In this experimental condition, we demonstrated, by immunoprecipitation experiments, an increase in EZH2 acetylation (Figure 3C). In accordance with increased EZH2 protein expression and acetylation, we observed increased H3K27me3 level in EX-257 treated cells (Figure 3B). The combined treatment of EPZ-6438 and EX-527 resulted in a significant reduction in the MSTO-211H growth rate (Figure 3D). H3K27 demethylation was confirmed by Western blot analysis (Figure 3E).
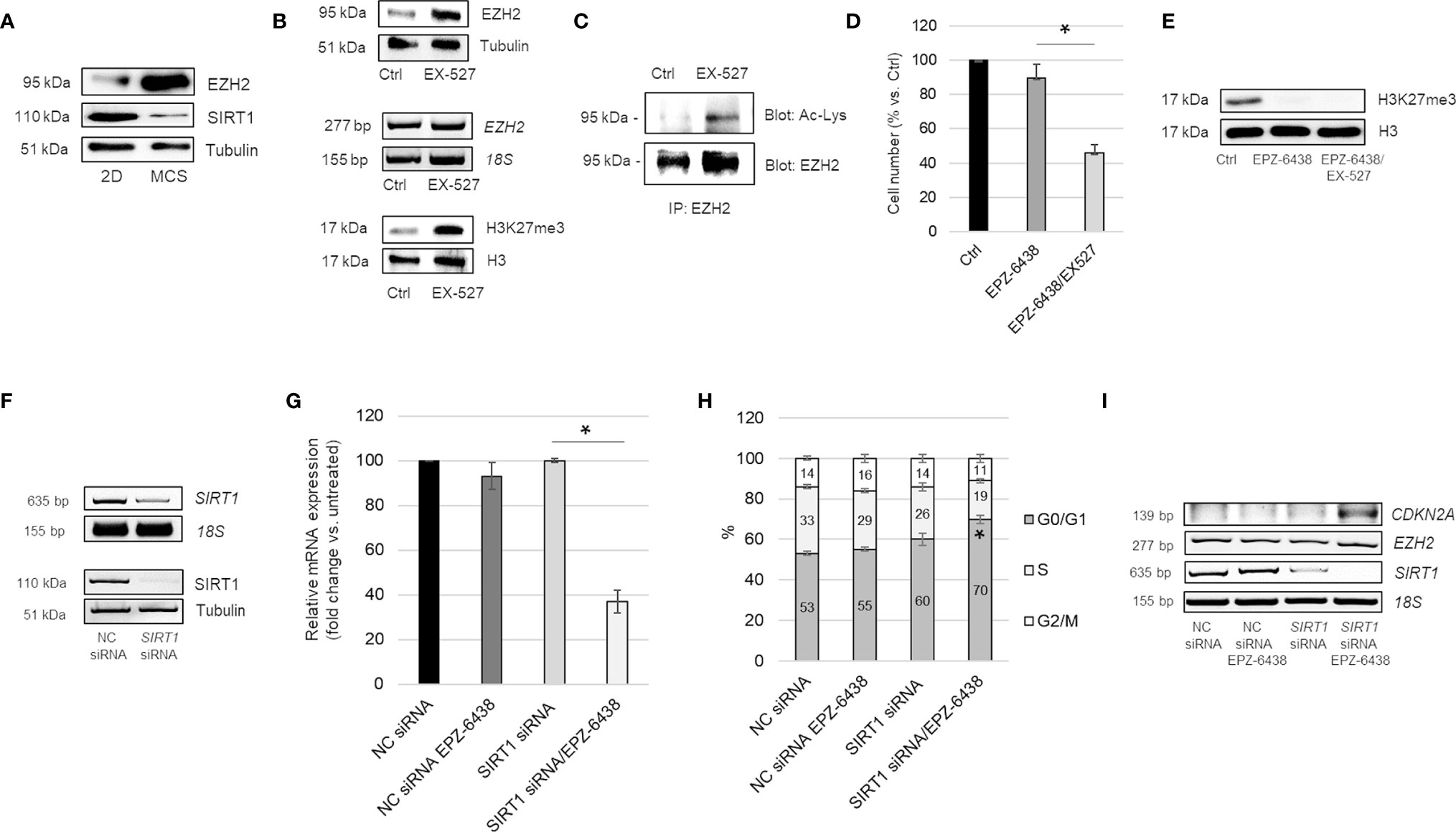
Figure 3 SIRT1 inhibition increases EZH2 expression, acetylation and activity. (A) Representative Western blot analysis of EZH2 and SIRT1 in MSTO-211H cells cultured 48 h as monolayer or as MCS. Tubulin was used as loading control. (B) Representative Western blot analysis and RT-PCR analysis of EZH2 expression and of H3K27 trimethylation (H3K27me3) in MSTO-211H cells cultured as monolayer ± 48 h EX527 treatment. Tubulin and histone H3 were used as loading control and 18S rRNA as housekeeping gene. (C) Immunoprecipitation of EZH2 from lysates of MSTO-211H cells ± EX527 48 h treatment. Lysine acetylation and EZH2 were detected by Western blot analysis with the respective antibodies (AcLys and EZH2). (D) Bar graph shows the percentage of viability of MSTO-211H cells cultured as monolayer ± 48 h treatment with EPZ-6438 or EPZ-6438/EX527 combination. Each bar represents mean of three independent experiments ± s.d., *p ≤ 0.05. (E) Representative Western blot analysis of H3K27me3 in MSTO-211H cells treated 48 h with EPZ-6438 or EPZ-6438/EX527 combination. Histone H3 was used as loading control. (F) Representative Western blot analysis and RT-PCR analysis of SIRT1 expression in MSTO-211H cells transfected with negative control (NC) or SIRT1 specific siRNAs (SIRT1 siRNA). Tubulin was used as loading control and 18S rRNA as housekeeping gene. (G) Bar graph shows the percentage of viability of MSTO-211H cells transfected with negative control (NC) or SIRT1 specific siRNAs (SIRT1 siRNA) after 48 h treatment ± EPZ-6438. Each bar represents mean of three independent experiments ± s.d., *p ≤ 0.05. (H) Bar graph representing the percentage of cells in G0/G1, S and G2 phase of MSTO-211H cells transfected with negative control (NC) or SIRT1 specific siRNAs (SIRT1 siRNA) and treated or not, 48 h, with EPZ-6438. Bars represent the means of three measurements ± s.d., *p < 0.05. (I) Representative quantitative RT-PCR analysis of CDKN2A, EZH2 and SIRT1 in MSTO-211H cells transfected with negative control (NC) or SIRT1 specific siRNAs (SIRT1 siRNA) and treated 48 h ± EPZ-6438. 18S rRNA was used as housekeeping gene.
To confirm the role of SIRT1 in cell response to EPZ-6438 treatment, we transfected MSTO-211H cells with specific siRNAs and evaluated the effect on cell proliferation. SIRT1 silencing was confirmed by RT-PCR and Western blot analyses (Figure 3F). Comparable EPZ-6438 anti-proliferative effect, accompanied by a G1 phase cell cycle arrest, was observed in SIRT1 silenced cells (Figures 3G, H). We observed that EPZ-6438 treatment induced the expression of CDKN2A, encoding for the cell cycle inhibitor p16ink4a, in SIRT1 silenced cells cultured as monolayer (Figure 3I), but not in cells transfected with negative control siRNA.
EZH2 Inhibition Mediates Strong Induction of CDKN2A in MPM Spheroids
Notably, in EPZ-6438 treated MSTO-211H MCS, we observed a strong induction of CDKN2A within 24 h, with a gradual increase at 48 and 72 h (Figures 4A, B). Comparable results were observed in the BAP1/CDKN2A wild-type BR95 MPM cells, cultured as MCS (Figure S3).
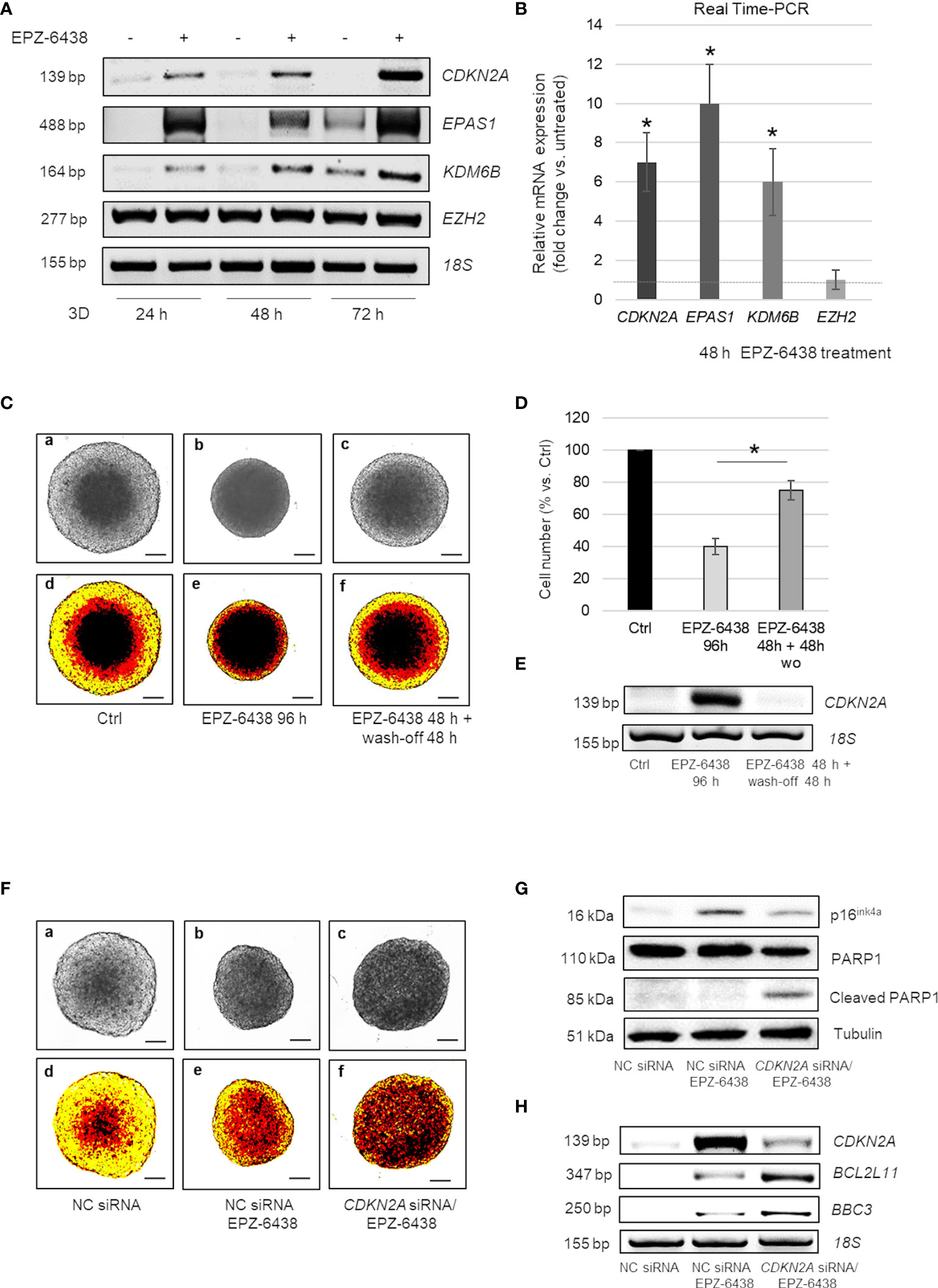
Figure 4 EZH2 inhibition induces cell cycle arrest in CDKN2A+ or apoptosis in CDKN2A- MPM MCS (A) Representative RT-PCR analysis of CDKN2A, EPAS1, KDM6B and EZH2 in MSTO-211H MCS treated or not 24, 48 and 72 h with EPZ-6438. (B) CDKN2A, EPAS1, KDM6B and EZH2 mRNA expression (fold increase over Ctrl) after 48 h treatment with EPZ-6438 of MSTO-211H MCS confirmed by Real time PCR (qPCR). 18S rRNA was used as housekeeping gene. (C) Representative phase contrast images (×40 magnification) (a–c) and relative pseudo-color images (d–f) of MSTO-211H MCS treated ± EPZ-6438 for 96 h or treated for 48 h with EPZ-6438 and, after wash-off, grown for additional 48 h in normal medium. (D) Bar graph shows the percentage of viability of MSTO-211H MCS treated 96 h with EPZ-6438 or treated for 48 h with EPZ-6438 and, after wash-off, grown for additional 48 h in normal medium. Each bar represents mean of three independent experiments ± s.d., *p ≤0.05. (E) Representative quantitative RT-PCR analysis of CDKN2A in MSTO-211H MCS treated as described above. 18S rRNA was used as housekeeping gene. (F) Representative phase contrast images (×40 magnification) (one of 12) (a–c) and relative pseudo-color images (d–f) of MSTO-211H MCS transfected with negative control (NC) siRNAs or CDKN2A specific siRNAs (CDKN2A siRNA) and treated ± EPZ-6438, 48 h. Scale bar = 100 μM. (G) Representative Western blot analysis of p16ink4a, PARP1 and cleaved PARP1 in MSTO-211H MCS transfected with negative control (NC) siRNAs or CDKN2A specific siRNAs (CDKN2A siRNA) and treated with or without EPZ-6438 for 48 h. Tubulin was used as loading control. (H) Representative RT-PCR analyses of CDKN2A, BCL2L11 and BBC3 in MSTO-211H MCS transfected with negative control (NC) siRNAs or CDKN2A specific siRNAs (CDKN2A siRNA) and treated, 48 h, with EPZ-6438. 18S rRNA was used as housekeeping gene.
Moreover, here we show that treatment with EPZ-6438 strongly induced the expression of genes encoding HIF2α and KDM6B, starting from 24 h of treatment (Figures 4A, B). In untreated MCS the expression of these genes increased starting from 72 h of culture, as we had previously described (31).
To determine whether EZH2 inhibition induced a temporary cell cycle arrest or a permanent exit from cycling, MSTO-211H MCS were treated 48 h with EPZ-6438, followed by its wash-off, and continued growth in normal culture medium for an additional 48 h. Interestingly, the cell growth and architecture of MSTO-211H MCS (Figures 4C, a–f, D) along with CDKN2A expression (Figure 4E) returned back to control levels after 48 h of EPZ-6438 removal.
EZH2 Inhibition Induces Apoptosis in CDKN2A Silenced MPM Cells
As homozygous and heterozygous deletions of the 9p21 locus, encompassing CDKN2A, are frequent in MPM, we hypothesized that p16ink4a induction was responsible for the arrest in cell growth observed in MCS treated with EPZ-6438 and that cells deleted for this gene could be insensitive to EZH2 inhibition. We next generated MCS from CDKN2A silenced MSTO-211H cells to counteract its induced expression in response to EPZ-6438 treatment. Different to our hypothesis, EPZ-6438 treatment induced apoptosis in CDKN2A silenced MSTO-211H MCS, as demonstrated by bright field (Figures 4F, a–c) and pseudo-color microscope images (Figures 4F, d–f), PARP1 cleavage (Figure 4H), BCL2L11 (encoding BIM) and BBC3 (encoding PUMA) induction (Figure 4F). CDKN2A silencing was confirmed by Western blot (Figure 4G) and RT-PCR (Figure 4H) analysis.
Dual Inhibition of EZH2 and TG2 Exhibits Proapoptotic Synergy in CDKN2A Wild-Type MPM Cells
Looking for a potential druggable target in CDKN2A expressing MPM MCS, we tested whether EZH2 inhibition, by inducing rapid HIF2 expression, could also lead to increased TG2 expression. We have recently reported that HIF2 controls TGM2 expression and that TG2 activity is essential for MPM cell survival under prolonged hypoxic condition. As shown in Figures 5A, C, EPZ-6438 induced the expression of EPAS1,TGM2 and its target IL-6 at 48 h treatment in MSTO-211H and in BR95 MCS (Figures S3C, D).
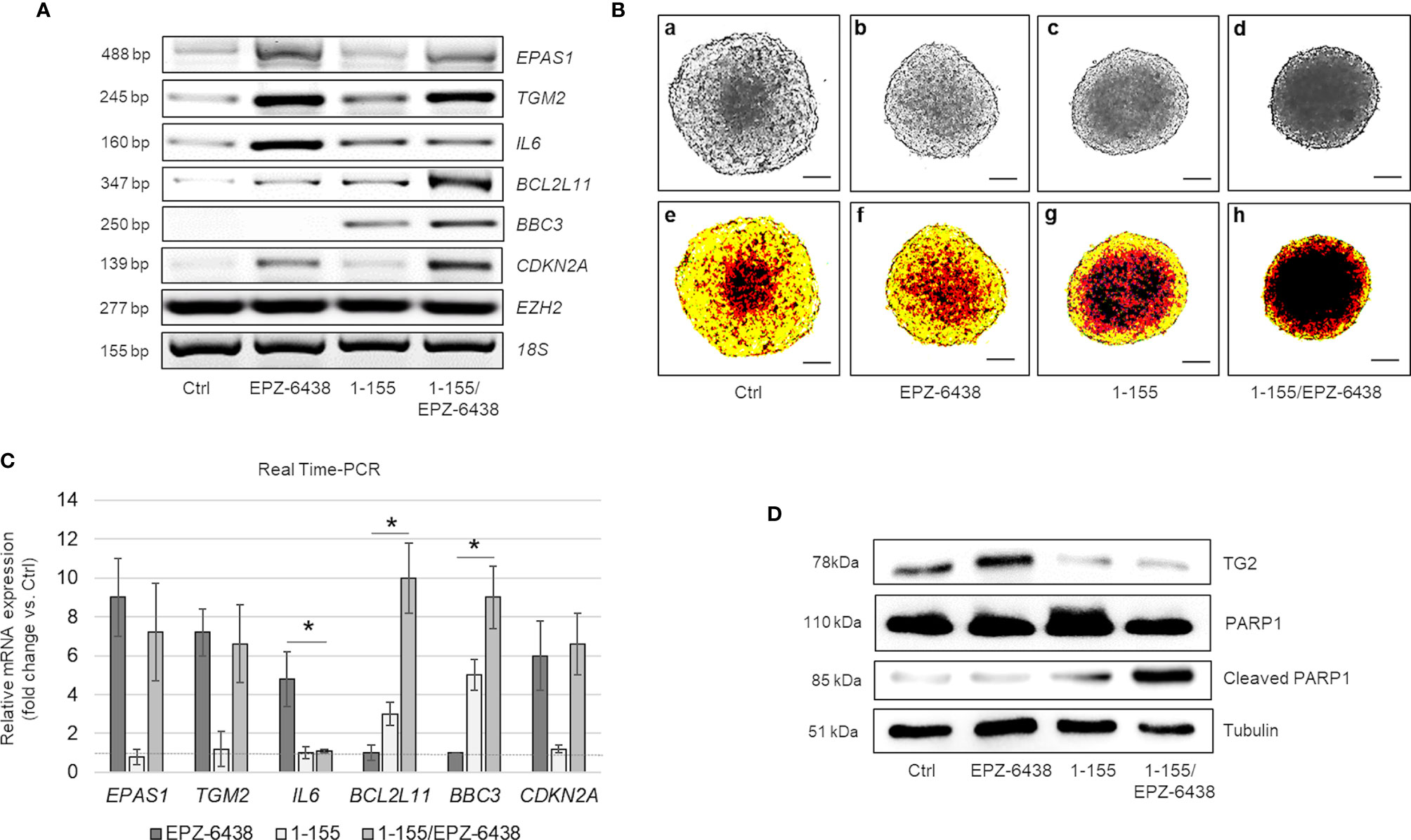
Figure 5 Dual inhibition of TG2 and EZH2 exhibits pro-apoptotic synergy in MPM MCS. (A) Representative RT-PCR of EPAS1, TGM2, IL6, BCL2L11, BBC3, CDKN2A, and EZH2 expression in MSTO-211H MCS untreated or treated 48 h with EPZ-6438 and 1-155 as single agents or in combination. 18S rRNA was used as housekeeping gene. (B) Representative phase contrast images (×40 magnification) (one of 12) (a–d) and relative pseudo-color images (e–h) of MSTO-211H MCS untreated or treated 48 h with EPZ-6438 and 1-155 as single agents or in combination. Scale bar = 100 μM. (C) Real Time PCR (qPCR) analysis of EPAS1, TGM2, IL6, BCL2L11, BBC3 and CDKN2A expression in MSTO-211H MCS untreated or treated 48 h with EPZ-6438 and 1-155 as single agents or in combination. 18S rRNA was used as housekeeping gene. Each bar represents mean of three independent experiments ± s.d., *p ≤ 0.05. (D) Representative Western blot analysis of TG2, PARP1 and cleaved PARP1 in MSTO-211H MCS untreated or treated 48 h with EPZ-6438 and 1-155 as single agents or in combination. Tubulin was used as loading control.
In order to test the importance of TG2 induction in preventing apoptosis, we treated MSTO-211H cells, cultured as MCS, with EPZ-6438 and the cell permeable TG2 selective inhibitor, 1-155, as single agents or in combination, for 48 h. As documented by bright field (Figures 5B, a–d) and pseudo-color microscope images (Figures 5B, e–h) the two drugs synergized in reducing MCS size. By Western-blot analysis, we observed a slow induction of TG2 expression in response to EPZ-6438 treatment and, as expected, accelerated breakdown in response to 1-155 treatment (Figure 5D). The EPZ-6438/1-155 combined treatment induced apoptosis as documented by increased PARP1 cleavage (Figure 5D), BCL2L11 and BBC3 expression, as documented by RT-PCR (Figure 5A) and Real Time PCR (Figure 5C), 1-155 treatment did not modify the EPZ-6438 induced expression of CDKN2A, EPAS1 and TGM2, but significantly reduced the expression of the gene encoding IL-6 (Figures 5A, C).
Discussion
MPM is known to show intrinsic therapy resistances and is so far incurable. Therefore, establishment of new therapies for this disease could be a valuable addition to current treatment options.
The high number of non-responders to chemotherapy (6), as well as the frequent recurrences of the disease (7), suggests a high degree of genetic heterogeneity within individual tumors. In concert with genetic variation, dynamic regulation of the epigenetic state may have important consequences for tumor plasticity and biology. Recent work has implicated mutation and/or dysregulated expression of histone lysine methyltransferases (KMTs) and demethylases (KDMs) in cancer (40).
Among KMTs, EZH2 has been reported to be overexpressed in various cancers, including MPM, and particularly to be associated with aggressiveness and poor prognosis. Pharmacological inhibition of EZH2 by 3-Deazanplanocin A (DZNep), an S-adenosylhomocysteine (SAH) hydrolase inhibitor, has been shown to effectively inhibit the growth of MPM cells (41, 42). A phase II clinical trial (NCT02860286) of Tazemetostat (EPZ-6438), a potent and selective EZH2 inhibitor, in MPM with inactive BAP1 has shown promising results (43).
However, BAP1 inactivation and H3K27me3 positivity in tissues may be sufficient, but not essential to predict response to EZH2 inhibition. Here we uncovered an antiproliferative activity of EPZ-6438 in BAP1 wild-type MPM cells, when cultured in vitro as three-dimensional (3D) multicellular spheroids, but not when grown in conventional two-dimensional (2D) monolayers.
Consistent evidence suggests that preclinical models based on standard 2D culture of cancer cells largely fail to predict drug efficacy, because they do not recapitulate the three-dimensional architecture, heterogeneity and complexity of human tumors (44). Multicellular tumor spheroids formed from established tumor cell lines, approximating physiologic conditions, may contribute to bridging this gap.
We show by immunofluorescence that EPZ-6438 treatment nearly abolished H3K27 trimethylation, which was spatially restricted to only the external oxygenated region of the spheroid. In agreement with this, we have previously demonstrated that expression of the H3K27 demethylase KDM6B was mostly restricted to the internal hypoxic core of MPM MCS, enriched in HIF2α expression (31).
It has been described that, under metabolic or hypoxic stress, changes in NAD+/NADH are key events that initiate the cellular adaptation processes. The NAD+ level and NAD+/NADH ratio decrease during hypoxia and this is associated with SIRT1 downregulation and increased acetylation (45). We observed that SIRT1 silencing or inhibition, with EX-527, resulted in increased EZH2 protein acetylation and stability. In accordance with increased EZH2 protein expression, we observed increased H3K27me3 levels. It has been described that acetylated EZH2 exerts a gain-of-function in cells and promotes lung cancer progression (19). Interestingly, the observed highest levels of global H3K27me3 in MPM cells might indicate a higher degree of addiction towards EZH2 activity and explain increased sensitivity to its inhibition (Figures 6A, B).
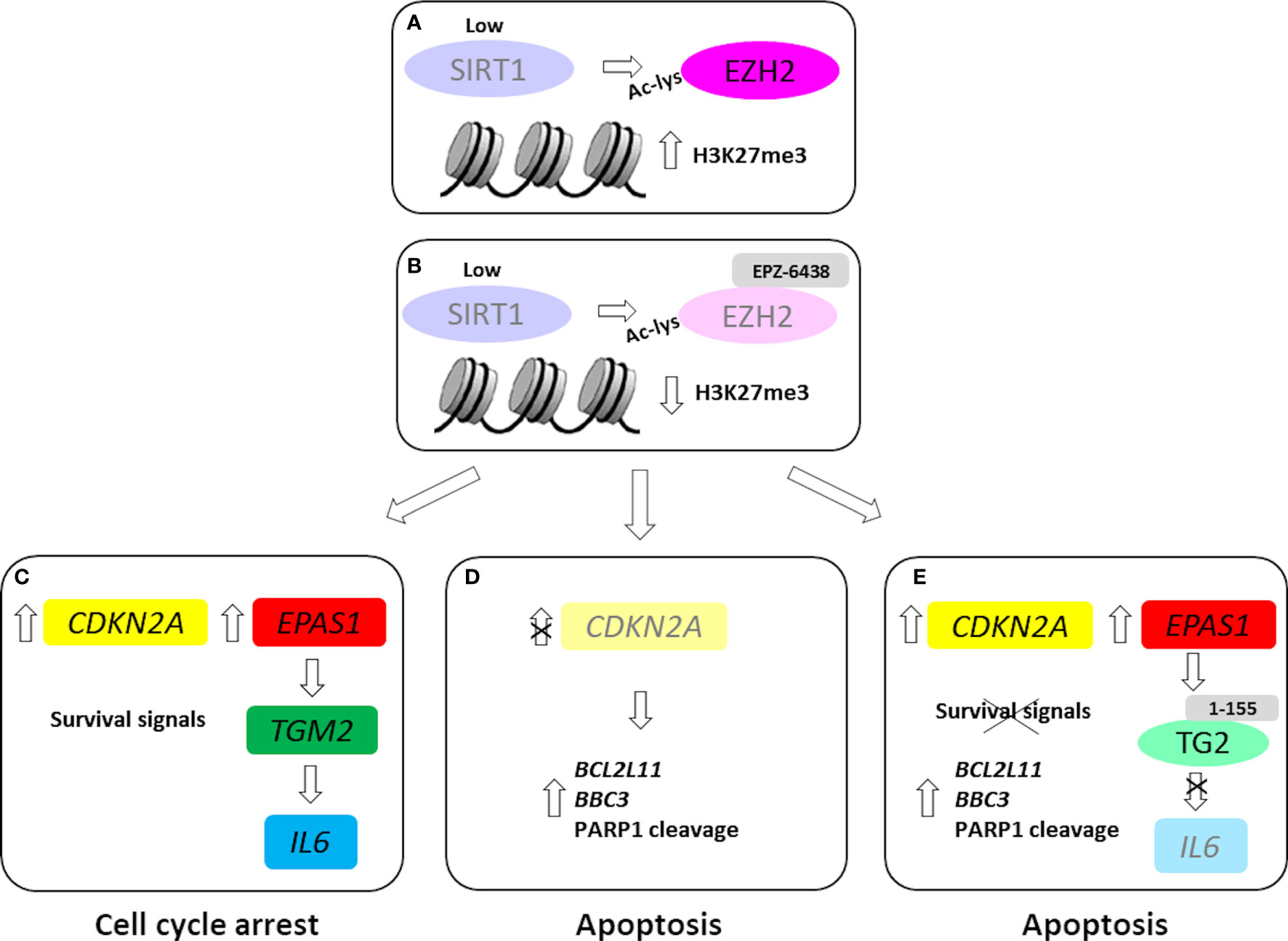
Figure 6 EZH2 inhibition in wild-type and CDKN2A silenced MPM MCS. In our MPM model, SIRT1 inhibition/silencing induced EZH2 acetylation and stability (A). EZH2 inhibition, by EPZ-6438 treatment (B), induced the expression CDKN2A, causing cell growth arrest. Furthermore, EPZ-6438 treatment upregulated EPAS1 and downstream TGM2 and IL6 (C). EPZ-6438 treatment induced apoptosis in CDKN2A silenced MPM MCS (D). Combined treatment with EPZ-6438 and 1-155, a selective TG2 inhibitor, abolished IL6 induction and led to apoptotic death (E). Light colors were used to indicate low or null levels of gene expression or protein activity.
In low SIRT1 conditions, EPZ-6438 treatment significantly reduced MPM cell proliferation, arresting cells in G0/G1 phase. We observed that EPZ-6438 treatment significantly reduced the size and modified the architecture of MPM MCS in which SIRT1 was expressed at lower and EZH2 at higher levels, compared to the cells cultured in 2D. Notably, in EPZ-6438 treated MCS, we observed a strong induction of CDKN2A, encoding for the cell cycle inhibitor p16ink4a. p16ink4a exerts tumor suppressive function by inhibiting the activities of the cyclin D-dependent kinases, CDK4 and CDK6, and preventing the retinoblastoma protein (Rb) phosphorylation and dissociation from the transcription factor E2F1. This biochemical pathway, essential for the transition from G1 to S phase, is frequently disrupted in tumor cells, by either deletions or inactivating mutations. Furthermore, the literature supports that H3K27me3 regulates CDKN2A expression and that EZH2 repression of CDKN2A is a critical control point to promote transformation (46).
It has been described that several stress stimuli may activate p16ink4a expression to orchestrate cell cycle exit and senescence response. Cellular senescence is a potent tumor suppressor mechanism, acting in coordination with the immune system to clear potentially malignant cells from the tissues. However, senescence and cancer might be considered related endpoints of accumulating cellular damage (24).
CDKN2A induction and cell cycle arrest were reverted upon EPZ-6438 removal, indicating growth inhibition and not cytotoxicity in cells exposed to the treatment. Indeed, our results show that MPM cells in which CDKN2A/p16ink4a was induced were more resistant to apoptosis than CDKN2A silenced cells (Figure 6C).
Several studies have since demonstrated the reversibility of full-featured senescence, indicating that pre-senescent or early senescent cells are poised to re-enter the cell cycle. Based on the massive epigenetic remodeling underlying the state switch, senescence has also been linked to enhanced plasticity and reprogramming (47). Our data strongly suggests that EPZ-6438 treatment not only suppressed MPM cell proliferation, but also induced pro-survival pathways, which protected cancer cells from apoptosis. EZH2 inhibition increased the expression of TG2 coding gene, via HIF2α (31), which in turn increased the expression of the IL-6 gene coding for the pro-inflammatory cytokine, IL-6. IL-6 has emerged as a mediator of pivotal processes in MPM, such as cell proliferation and chemoresistance (48). In MPM MCS expressing CDKN2A, blocking TG2 activity, by the selective inhibitor 1-155, abrogated the EPZ-6438-mediated increase in IL-6 expression and induced apoptosis (Figure 6E). While, in CDKN2A silenced MPM MCS, EPZ-6438 treatment induced apoptosis (Figure 6D), probably by promoting the pro-apoptotic activity of E2F1 (49). However, the mechanistic difference between wild-type and CDKN2A silenced MCS response to EPZ-6438 treatment needs further and deeper investigation.
Our data support the view that treatment approaches based on inducing p16ink4a and related G0/G1 growth arrest, in tumor cells that lack p16ink4a, can induce tumor stabilization, but may also enhance pro-survival signals and thereby confer resistance to chemotherapy treatments. Deletion of CDKN2A on chromosome 9p21 is a common molecular alteration in MPM. However, fluorescent in situ hybridization (FISH) on MPM tumor tissues has revealed that the CDKN2A homozygous deletion cannot be detected in all cells. Indeed, the status of the CDKN2A gene is highly variable, with no loss, hemizygous and homozygous losses within the same tumor. This detection of non-homogeneous deletions of CDKN2A suggests that besides the polyclonal origin, several genetic subclones might also exist within one tumor (8).
In conclusion, we demonstrated that, in cells with no loss or hemizygous deletion, such as the MSTO-211H MPM cells, CDKN2A expression can be epigenetically modulated. Translating our findings into the clinical context, we could suggest that patients with homozygous deletion for CDKN2A should respond to EZH2 inhibition (apoptosis). Conversely, in MPMs with wild-type CDKN2A, a cell growth arrested phenotype would be anticipated. Apoptosis could be augmented in a CDKN2A wild-type setting by TG2 inhibition. In conclusion, these findings open up possibilities for refining patient stratification and/or potentiation of EZH2 inhibitors.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
MPM tissue samples were collected in accordance with Ospedale Maggiore della Carità (Novara, Italy) approval and with patients’ informed consent.
Author Contributions
Conception and Design: GP and LM. Data Acquisition: GP, ZW, CB, SM, DT, and RB. Analysis of Data: GP, ZW, RB, MG, DF, and LM. Writing: GP, LM, MG, and DF. All authors contributed to the article and approved the submitted version.
Funding
The authors acknowledge the financial support of project HERMES (HEreditary Risk in MESothelioma) and Università del Piemonte Orientale (Bando ricerca locale 2019).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Authors gratefully acknowledge Dr. Priscilla Bernardi for technical assistance.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2021.678447/full#supplementary-material
References
1. Jaurand MC, Fleury-Feith J. Pathogenesis of Malignant Pleural Mesothelioma. Respirology (2005) 10:2–8. doi: 10.1111/j.1440-1843.2005.00694.x
2. Yap TA, Aerts JG, Popat S, Fennell DA. Novel Insights Into Mesothelioma Biology and Implications for Therapy. Nat Rev Cancer (2017) 17:475–88. doi: 10.1038/nrc.2017.42
3. Frank AL, Joshi T. The Global Spread of Asbestos. Ann Glob Health (2014) 80:257–62. doi: 10.1016/j.aogh.2014.09.016
4. Stayner L, Welch LS, Lemen R. The Worldwide Pandemic of Asbestos-Related Diseases. Annu Rev Public Health (2013) 34:205–16. doi: 10.1146/annurev-publhealth-031811-124704
5. Vogelzang NJ, Rusthoven JJ, Symanowski J, Denham C, Kaukel E, Ruffie P, et al. Phase III Study of Pemetrexed in Combination With Cisplatin Versus Cisplatin Alone in Patients With Malignant Pleural Mesothelioma. J Clin Oncol (2003) 21:2636–44. doi: 10.1200/JCO.2003.11.136
6. Politi L, Borzellino G. Second Surgery for Recurrence of Malignant Pleural Mesothelioma After Extrapleural Pneumonectomy. Ann Thorac Surg (2010) 89:207–10. doi: 10.1016/j.athoracsur.2009.09.028
7. Baldini EH, Richards WG, Gill RR, Goodman BM, Winfrey OK, Eisen HM, et al. Updated Patterns of Failure After Multimodality Therapy for Malignant Pleural Mesothelioma. J Thorac Cardiovasc Surg (2015) 149:1374–81. doi: 10.1016/j.jtcvs.2014.10.128
8. Oehl K, Vrugt B, Opitz I, Meerang M. Heterogeneity in Malignant Pleural Mesothelioma. Int J Mol Sci (2018) 30:19. doi: 10.3390/ijms19061603
9. Travis WD, Brambilla E, Burke AP, Marx A, Nicholson AG. World Health Organization Classification of Tumours of the Lung, Pleura, Thymus and Heart. 4th. Lyon-France: IARC Press (2015).
10. Guo G, Chmielecki J, Goparaju C, Heguy A, Dolgalev I, Carbone M, et al. Whole-Exome Sequencing Reveals Frequent Genetic Alterations in bap1, nf2, cdkn2a, and Cul1 in Malignant Pleural Mesothelioma. Cancer Res (2015) 75:264–9. doi: 10.1158/0008-5472.CAN-14-1008
11. Blum Y, Meiller C, Quetel L, Elarouci N, Ayadi M, Tashtanbaeva D, et al. Dissecting Heterogeneity in Malignant Pleural Mesothelioma Through Histo-Molecular Gradients for Clinical Applications. Nat Commun (2019) 10:1333. doi: 10.1038/s41467-019-09307-6
12. Kato S, Tomson BN, Buys TP, Elkin SK, Carter JL, Kurzrock R. Genomic Landscape of Malignant Mesotheliomas. Mol Cancer Ther (2016) 15:2498–507. doi: 10.1158/1535-7163.MCT-16-0229
13. Sage AP, Martinez VD, Minatel BC, Pewarchuk ME, Marshall EA, MacAulay GM, et al. Genomics and Epigenetics of Malignant Mesothelioma. High Throughput (2018) 7:20. doi: 10.3390/ht7030020
14. Yap TA, Aerts JG, Popat S, Fennell DA. Novel Insight Into Mesothelioma Biology and Implications for Therapy. Nat Rev Cancer (2017) 17:475–88. doi: 10.1038/nrc.2017.42
15. Lo Iacono M, Monica V, Righi L, Grosso F, Libener R, Vatrano S, et al. Targeted Next-Generation Sequencing of Cancer Genes in Advanced Stage Malignant Pleural Mesothelioma: A Retrospective Study. J Thorac Oncol (2015) 10:492–9. doi: 10.1097/JTO.0000000000000436
16. Bueno R, Stawiski EW, Goldstein LD, Durinck S, De Rienzo A, Modrusan Z, et al. Comprehensive Genomic Analysis of Malignant Pleural Mesothelioma Identifies Recurrent Mutations, Gene Fusions and Splicing Alterations. Nat Genet (2016) 48:407–16. doi: 10.1038/ng.3520
17. LaFave LM, Béguelin W, Koche R, Teater M, Spitzer B, Chramiec A, et al. Loss of BAP1 Function Leads to EZH2-Dependent Transformation. Nat Med (2015) 21:1344–9. doi: 10.1038/nm.3947
18. Cha TL, Zhou BP, Xia W, Wu Y, Yang CC, Chen CT, et al. Akt-Mediated Phosphorylation of EZH2 Suppresses Methylation of Lysine 27 in Histone H3. Science (2005) 310:306–10. doi: 10.1126/science.1118947
19. Wan J, Zhan J, Li S, Ma J, Xu W, Liu C, et al. PCAF-Primed EZH2 Acetylation Regulates Its Stability and Promotes Lung Adenocarcinoma Progression. Nucleic Acids Res (2015) 43:3591–604. doi: 10.1093/nar/gkv238
20. Yu YL, Chou RH, Shyu WC, Hsieh SC, Wu CS, Chiang SY, et al. Smurf2-mediated Degradation of EZH2 Enhances Neuron Differentiation and Improves Functional Recovery After Ischaemic Stroke. EMBO Mol Med (2013) 5:531–47. doi: 10.1002/emmm.201201783
21. Riising EM, Boggio R, Chiocca S, Helin K, Pasini D. The Polycomb Repressive Complex 2 Is a Potential Target of SUMO Modifications. PLoS One (2008) 3:e2704. doi: 10.1371/journal.pone.0002704
22. Chu CS, Lo PW, Yeh YH, Hsu PH, Peng SH, Teng YC, et al. O-GlcNAcylation Regulates EZH2 Protein Stability and Function. Proc Natl Acad Sci U S A (2014) 111:1355–60. doi: 10.1073/pnas.1323226111
23. Cheng JQ, Jhanwar SC, Klein WM, Bell DW, Lee WC, Altomare DA, et al. p16 Alterations and Deletion Mapping of 9p21-p22 in Malignant Mesothelioma. Cancer Res (1994) 54:5547–51.
24. Kettunen E, Savukoski S, Salmenkivi K, Böhling T, Vanhala E, Kuosma E, et al. CDKN2A Copy Number and p16 Expression in Malignant Pleural Mesothelioma in Relation to Asbestos Exposure. BMC Cancer (2019) 19:507. doi: 10.1186/s12885-019-5652-y
25. López-Ríos F, Chuai S, Flores R, Shimizu S, Ohno T, Wakahara K, et al. Global Gene Expression Profiling of Pleural Mesotheliomas: Overexpression of Aurora Kinases and P16/CDKN2A Deletion as Prognostic Factors and Critical Evaluation of Microarray-Based Prognostic Prediction. Cancer Res (2006) 66:2970–9. doi: 10.1158/0008-5472.CAN-05-3907
26. Dacic S, Kothmaier H, Land S, Shuai Y, Halbwedl I, Morbini P, et al. Prognostic Significance of p16/cdkn2a Loss in Pleural Malignant Mesotheliomas. Virchows Arch (2008) 453:627–35. doi: 10.1007/s00428-008-0689-3
27. Ivanov SV, Miller J, Lucito R, Tang C, Ivanova AV, Pei J, et al. Genomic Events Associated With Progression of Pleural Malignant Mesothelioma. Int J Cancer (2009) 124:589–99. doi: 10.1002/ijc.23949
28. Wong L, Zhou J, Anderson D, Kratzke RA. Inactivation of p16INK4a Expression in Malignant Mesothelioma by Methylation. Lung Cancer (2002) 38:131–6. doi: 10.1016/S0169-5002(02)00178-2
29. Burchfield JS, Li Q, Wang HY, Wang RF. JMJD3 as an Epigenetic Regulator in Development and Disease. Int J Biochem Cell Biol (2015) 67:148–57. doi: 10.1016/j.biocel.2015.07.006
30. Hojfeldt W, Agger K, Helin K. Histone Lysine Demethylases as Targets for Anticancer Therapy. Nat Rev Drug Discov (2013) 12:917–30. doi: 10.1038/nrd4154
31. Johnson AB, Denko N, Barton MC. Hypoxia Induces a Novel Signature of Chromatin Modifications and Global Repression of Transcription. Mutat Res (2008) 640:174–9. doi: 10.1016/j.mrfmmm.2008.01.001
32. Manente AG, Pinton G, Zonca S, Tavian D Habib T, Jithesh PV, et al. KDM6B Histone Demethylase Is an Epigenetic Regulator of Estrogen Receptor β Expression in Human Pleural Mesothelioma. Epigenomics (2016) 8:1227–38. doi: 10.2217/epi-2016-0025
33. Zonca S, Pinton G, Wang Z, Soluri MF, Tavian D, Griffin M, et al. Tissue Transglutaminase (TG2) Enables Survival of Human Malignant Pleural Mesothelioma Cells in Hypoxia. Cell Death Dis (2017) 8:e2592. doi: 10.1038/cddis.2017.30
34. Lorand L, Graham RM. Transglutaminases: Crosslinking Enzymes With Pleiotropic Functions. Nat Rev Mol Cell Biol (2003) 4:140–56. doi: 10.1038/nrm1014
35. Oh K, Moon HG, Lee DS, Yoo YB. Tissue Transglutaminase-Interleukin-6 Axis Facilitates Peritoneal Tumor Spreading and Metastasis of Human Ovarian Cancer Cells. Lab Anim Res (2015) 31:188–97. doi: 10.5625/lar.2015.31.4.188
36. Oh K, Lee OY, Park Y, Seo MW, Lee DS. Il-1β Induces IL-6 Production and Increases Invasiveness and Estrogen-Independent Growth in a TG2-dependent Manner in Human Breast Cancer Cells. BMC Cancer (2016) 16:724. doi: 10.1186/s12885-016-2746-7
37. Badarau E, Wang Z, Rathbone DL, Costanzi A, Thbault T, Murdoch CE, et al. Development of Potent and Selective Tissue Tranglutaminase Inhibitors: Their Effect on TG2 Function and Application in Pathological Conditions. Chem Biol (2015) 22:1347–61. doi: 10.1016/j.chembiol.2015.08.013
38. Griffin M, Mongeot M, Collighen R, Saint RE, Jones RA, Coutts IG, et al. Synthesis of Potent Water-Soluble Tissue Transglutaminase Inhibitors. Bioorg Med Chem Lett (2008) 18:5559–62. doi: 10.1016/j.bmcl.2008.09.006
39. Manente AG, Pinton G, Zonca S, Cilli M, Rinaldi M, Daga A, et al. Intracellular Lactate-Mediated Induction of Estrogen Receptor Beta (Erβ) in Biphasic Malignant Pleural Mesothelioma Cells. Oncotarget (2015) 6:25121–34. doi: 10.18632/oncotarget.4486
40. McGrath J, Trojer P. Targeting Histone Lysine Methylation in Cancer. Pharmacol Ther (2015) 150:1–22. doi: 10.1016/j.pharmthera.2015.01.002
41. Comet I, Riising EM, Leblanc B, Helin K. Maintaining Cell Identity: PRC2-mediated Regulation of Transcription and Cancer. Nat Rev Cancer (2016) 16:803–10. doi: 10.1038/nrc.2016.83
42. Kemp CD, Rao M, Xi S, Inchauste S, Mani H, Fetsch P, et al. Polycomb Repressor Complex-2 Is a Novel Target for Mesothelioma Therapy. Clin Cancer Res (2012) 18:77–90. doi: 10.1158/1078-0432.CCR-11-0962
43. Zauderer MG, Szlosarek P, Le Moulec S, Popat S, Taylor P, Planchard D, et al. Phase 2, Multicenter Study of the EZH2 Inhibitor Tazemetostat as Monotherapy in Adults With Relapsed or Refractory (R/R) Malignant Mesothelioma (MM) With BAP1 Inactivation. J Clinic Oncol (2018) 36:8515. doi: 10.1200/JCO.2018.36.15_suppl.8515
44. Sant S, Johnston PA. The Production of 3D Tumor Spheroids for Cancer Drug Discovery. Drug Discov Today Technol (2017) 23:27–36. doi: 10.1016/j.ddtec.2017.03.002
45. Lim JH, Lee YM, Chun YS, Chen J, Kim JE, Park JW. Sirtuin 1 Modulates Cellular Responses to Hypoxia by Deacetylating Hypoxia-Inducible Factor1alpha. Mol Cell (2010) 38:864–78. doi: 10.1016/j.molcel.2010.05.023
46. Bracken AP, Kleine-Kohlbrecher D, Dietrich N, Pasini D, Gargiulo G, Beekman C, et al. The Polycomb Group Proteins Bind Throughout the INK4A-ARF Locus and Are Disassociated in Senescent Cells. Genes Dev (2007) 21:525–30. doi: 10.1101/gad.415507
47. Soyoung L, Clemens AS. The Dynamic Nature of Senescence in Cancer. Nat Cell Biol (2019) 21:94–101. doi: 10.1038/s41556-018-0249-2
48. Abdul Rahim SN, Ho GY, Coward JI. The Role of Interleukin-6 in Malignant Mesothelioma. Transl Lung Cancer Res (2015) 4:55–66. doi: 10.3978/j.issn.2218-6751.2014.07.01
Keywords: malignant pleural mesothelioma, EZH2 inhibitor, CDKN2A/p16ink4a, TG2, multicellular spheroids
Citation: Pinton G, Wang Z, Balzano C, Missaglia S, Tavian D, Boldorini R, Fennell DA, Griffin M and Moro L (2021) CDKN2A Determines Mesothelioma Cell Fate to EZH2 Inhibition. Front. Oncol. 11:678447. doi: 10.3389/fonc.2021.678447
Received: 09 March 2021; Accepted: 14 June 2021;
Published: 01 July 2021.
Edited by:
Soldano Ferrone, Massachusetts General Hospital and Harvard Medical School, United StatesReviewed by:
Yogesh Chawla, Mayo Clinic, United StatesMin Hee Kang, Texas Tech University Health Sciences Center, United States
SrinivasVinod Saladi, Massachusetts Eye & Ear Infirmary and Harvard Medical School, United States
Copyright © 2021 Pinton, Wang, Balzano, Missaglia, Tavian, Boldorini, Fennell, Griffin and Moro. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Laura Moro, bGF1cmEubW9yb0B1bml1cG8uaXQ=; Giulia Pinton, Z2l1bGlhLnBpbnRvbkB1bml1cG8uaXQ=