 Daniele Fanale1‡
Daniele Fanale1‡ Alessia Fiorino1‡
Alessia Fiorino1‡ Lorena Incorvaia2‡
Lorena Incorvaia2‡ Alessandra Dimino1
Alessandra Dimino1 Clarissa Filorizzo1
Clarissa Filorizzo1 Marco Bono1Daniela Cancelliere1
Marco Bono1Daniela Cancelliere1 Valentina Calò1
Valentina Calò1 Chiara Brando1Lidia Rita Corsini1Roberta Sciacchitano1Luigi Magrin1Alessia Pivetti1Erika Pedone1Giorgio Madonia1Alessandra Cucinella1
Chiara Brando1Lidia Rita Corsini1Roberta Sciacchitano1Luigi Magrin1Alessia Pivetti1Erika Pedone1Giorgio Madonia1Alessandra Cucinella1 Giuseppe Badalamenti1§
Giuseppe Badalamenti1§ Antonio Russo1*†§
Antonio Russo1*†§ Viviana Bazan2*§
Viviana Bazan2*§- 1Department of Surgical, Oncological and Oral Sciences, Section of Medical Oncology, University of Palermo, Palermo, Italy
- 2Department of Biomedicine, Neuroscience and Advanced Diagnostics (Bi.N.D.), Section of Medical Oncology, University of Palermo, Palermo, Italy
About 10–20% of breast/ovarian (BC/OC) cancer patients undergoing germline BRCA1/2 genetic testing have been shown to harbor Variants of Uncertain Significance (VUSs). Since little is known about the prevalence of germline BRCA1/2 VUS in Southern Italy, our study aimed at describing the spectrum of these variants detected in BC/OC patients in order to improve the identification of potentially high-risk BRCA variants helpful in patient clinical management. Eight hundred and seventy-four BC or OC patients, enrolled from October 2016 to December 2020 at the “Sicilian Regional Center for the Prevention, Diagnosis and Treatment of Rare and Heredo-Familial Tumors” of University Hospital Policlinico “P. Giaccone” of Palermo, were genetically tested for germline BRCA1/2 variants through Next-Generation Sequencing analysis. The mutational screening showed that 639 (73.1%) out of 874 patients were BRCA-w.t., whereas 67 (7.7%) were carriers of germline BRCA1/2 VUSs, and 168 (19.2%) harbored germline BRCA1/2 pathogenic/likely pathogenic variants. Our analysis revealed the presence of 59 different VUSs detected in 67 patients, 46 of which were affected by BC and 21 by OC. Twenty-one (35.6%) out of 59 variants were located on BRCA1 gene, whereas 38 (64.4%) on BRCA2. We detected six alterations in BRCA1 and two in BRCA2 with unclear interpretation of clinical significance. Familial anamnesis of a patient harboring the BRCA1-c.3367G>T suggests for this variant a potential of pathogenicity, therefore it should be carefully investigated. Understanding clinical significance of germline BRCA1/2 VUS could improve, in future, the identification of potentially high-risk variants useful for clinical management of BC or OC patients and family members.
Introduction
Since the discovery of BRCA1 and BRCA2 genes, genetic testing requests have been steadily increasing. Inherited Pathogenic Variants (PVs) or Likely Pathogenic Variants (LPVs) detected in these major susceptibility genes have been shown to be involved in the Hereditary Breast and Ovarian Cancer syndrome (HBOC) (1–4). However, these sequence variants confer in carriers an increased lifetime risk also to develop other tumors such as pancreatic carcinoma (5, 6), prostate cancer (7–9), and melanoma (10).
As regards the meaning of variants, the Italian Association of Medical Oncology (AIOM) adopts the classification criteria proposed by the Evidence-based Network for the Interpretation of Germline Mutant Alleles (ENIGMA) consortium (https://enigmaconsortium.org/), according to the International Agency for Research on Cancer (IARC) recommendations (11), considering a classification system of variants into five classes: Benign (class I), Likely Benign (class II), Variant of Uncertain Significance (VUS, class III), Likely Pathogenic (class IV), and Pathogenic (class V) (12). Thanks to Next-Generation Sequencing (NGS) technologies, novel variants defined as VUSs have been shown to be harbored by 10–20% of patients undergoing BRCA1/2 genetic screening (13). A VUS is a nucleotide sequence alteration with unknown consequences on the possible loss of function of the gene product or on the potential risk of causing disease. Consequently, the clinical significance remains unclear leading to a difficult clinical management by the oncologist and not easy explanation to the patient, since a VUS exhibits a probability of being reclassified as pathogenic between 5 and 94.9% (14).
Most of VUSs could have no effect on the BRCA1/2 tumor suppressor function. Some of these variants behave as low-penetrance gene mutations and should not be managed as highly penetrant alterations (15). However, many of these may be crucial in the inheritance of high-risk neoplasms, and therefore, their identification in family members could be essential. As consequence, in case of VUS identification in the genome of a proband, the clinical decisions concerning the risk reduction and prevention strategies depend on family history and other risk factors, pending a reclassification of the variant. Generally, it has been demonstrated that most of VUS will be subsequently reclassified as class I benign variants (16, 17). The aim of the BRCA Challenge project and the BRCA Exchange database is to minimize differences in clinical interpretations of VUS between different laboratories, both worldwide and in national territory (18–20). Nowadays, in silico and experimental approaches are adopted for the classification of new and unclear sequence variants in order to improve the clinical management of BRCA1/2 VUS carriers. Data regarding family history of cancer are integrated as co-segregation with disease and co-occurrence with known PVs/LPVs into computational models in order to assess the probability that a VUS is a cause of disease (21).
To date, the knowledge about the prevalence of BRCA1/2 VUS in BC or OC patients belonging to some regions of Southern Italy such as Sicily is poor. Based on a Breast and Ovarian Cancer BRCA System database harvested at the University Hospital Policlinico “P. Giaccone” of Palermo, the aim of this retrospective investigation was to describe the typology and gene location of germline variants of unknown significance detected in BRCA1 and BRCA2 coding sequences and splicing sites of BC or OC patients in order to investigate the prevalence and spectrum of these inherited genetic variants observed in Southern Italy. Furthermore, the analysis of all molecular and clinical data of BC or OC patients could favor the identification of potentially high-risk susceptibility variants distributed in Sicilian population, contributing to the future reclassification of these variants with unclear clinical significance. Therefore, this work may provide information which, in the future, could be helpful in the clinical management of BC/OC patients harboring VUSs.
Patients and Methods
Study Population
A retrospective collection of clinical and molecular data from 874 unrelated BC or OC patients was carried out from October 2016 to December 2020 at the “Sicilian Regional Center for the Prevention, Diagnosis and Treatment of Rare and Heredo-Familial Tumors” of the Section of Medical Oncology of University Hospital Policlinico “P. Giaccone” of Palermo. Eight hundred seventy-four probands, 531 of which were with BC and 343 with OC, have been subjected to BRCA genetic testing according to specific susceptibility criteria based on a strong family and personal history of BC and/or OC. BC and/or OC patients with at least two other family members affected by HBOC-associated tumors are considered individuals with a strong family history. The personal and family anamnesis of patients was acquired during oncogenetic counseling performed by a multidisciplinary group of specialists which included an oncologist, a geneticist, and a psychologist. All patients have provided a signed informed consent. All information regarding personal and familial history of cancer, family geographical origin, age at diagnosis, histological tumor subtype, molecular phenotype and disease stages (I–IV) was anonymously recorded. The study (Protocol “G-Land 2017”) was approved by the ethical committee (Comitato Etico Palermo 1; approval number: 0103-2017) of the University-affiliated Hospital A.O.U.P. ‘P. Giaccone’ of Palermo. Data concerning the histological type and cancer diagnosis were provided by medical pathology reports in diagnostic core biopsies or tumor resections.
All patients were screened for germline BRCA1/2 genetic testing based on probability rate of carrying BRCA1/2 variants calculated by means of BRCAPRO genetic risk prediction model (22, 23) and according to the criteria established by guidelines of the Italian Association of Medical Oncology (AIOM) (24).
The genetic analysis result has been considered informative when a PV/LPV was identified or non-informative if no PV/LPV was detected, but it was not excluded that it could be present, or if a VUS, belonging to class III, was found (15, 25). A genetic testing will be positive in case of identification of inherited PVs/LPVs, but negative only in case of lack of identification of a known PV/LPV in a family member.
Sample Collection and Next-Generation Sequencing Analysis
Peripheral blood samples were harvested at diagnosis from BC or OC patients through a vacutainer syringe containing EDTA. Genomic DNA was isolated using the DNeasy® Blood Kit (QIAGEN, Hilden, Germany). After the extraction phase, DNA has been quantified by Qubit®3.0 fluorometer (Thermofisher Scientific, Waltham, MA, USA) and its quality has been assessed through the use of 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA).
The genetic analysis for BRCA1/2 was performed as previously described (4, 26).
Sequencing analysis was performed using Ion 520 Chip (Thermofisher Scientific, Waltham, MA, USA) and Ion Torrent S5 (Thermofisher Scientific, Waltham, MA, USA) NGS platform. Obtained data were analyzed using both Amplicon Suite (SmartSeq s.r.l.) and Ion Reporter Software v.5.12 (Thermofisher Scientific, Waltham, MA, USA). NGS data analysis was performed with the standardization of sequencing coverage depth in order to minimize the probability of false positive and negative results in clinical practice, considering a minimum coverage of 500× to each sample.
Sanger Sequencing Analysis
Sanger sequencing analysis was used to confirm the BRCA1/2 VUS identified by NGS analysis. We used the SeqStudio analyzer (Thermofisher Scientific, Waltham, MA, USA) and BigDye Therminator 3.1 Cycle Sequencing Kit (Life Technologies, Carlsbad, CA, USA), according to manufacturer’s protocols, as previously described (26).
Genetic Variant Classification
Identified BRCA variants have been classified according to the criteria developed by the Evidence-based Network for the Interpretation of Germline Mutant Alleles (ENIGMA) consortium (https://enigmaconsortium.org/) and IARC recommendations (11), using a system of classification in five classes: benign (class I), likely benign (class II), variant of uncertain significance (VUS, class III), likely pathogenic (class IV), and pathogenic (class V). After variant meaning understanding, available databases, such as ClinVar, VarSome and Priors V2.0 Software packages, were our weapon to perform an in silico analysis in order to investigate the molecular and clinical meaning of an identified variant with unclear properties. Algorithms developed to predict the effect of missense changes on protein structure and function (PolyPhen-2, SIFT, Align-GVGD) have been used.
The localization of the variants on BRCA1 and BRCA2 genes was obtained and graphically represented using the informatic tool Mutation Mapper-cBioPortal for Cancer Genomics (27, 28). The identified BRCA1/2 VUS was named according to the systematic nomenclature of the recommendations for the description of sequence variants established by the Human Genome Variation Society (HGVS) with authorization by the HGVS, Human Variome Project (HVP), and the Human Genome Organization (HUGO) (29).
Results
Detection of Germline BRCA1/2 VUS in BC or OC Patients
Eight hundred seventy-four patients with BC or OC, enrolled from October 2016 to December 2020 at our institute, who met the criteria concerning personal and family history of cancer recommended by the AIOM national guidelines, were genetically tested for germline BRCA1/2 variants. Among recruited 874 probands, 531 were BC patients and 343 were OC women.
The mutational screening of the examined study cohort showed that 639 (73.1%) out of 874 patients harbored germline BRCA1/2 benign/likely benign variants (BRCA-w.t.), whereas 67 (7.7%) probands were carriers of germline BRCA1/2 VUS (class III), and 168 (19.2%) subjects carried a germline BRCA1/2 PV/LPV (BRCA-positive) (Supplementary Figure 1).
The genetic analysis revealed the presence of 59 different VUS detected in 67 patients, 46 of which had BC (68.7%) and 21 were women affected by OC (31.3%). The median age at diagnosis of analyzed BC or OC patients was 45 years (range 22–75 years) and 51 years (range 28–78 years), respectively. In particular, most of BC patients had an invasive ductal carcinoma (31/46 probands; 67.4%), a luminal A molecular phenotype (15/46; 32.6%), and more than one relative with BRCA-related tumor (22/46; 47.8%). Furthermore, 18 (39%) out 46 probands harbored early onset BCs (before age 40 year), 16 of which were hormone-dependent tumors and 2 triple-negative BCs (Table 1). On the other side, most of OC patients had monolateral ovarian carcinoma with high-grade serous carcinoma (HGSC) histological subtype (16/21 women; 76.2%) (Table 1).
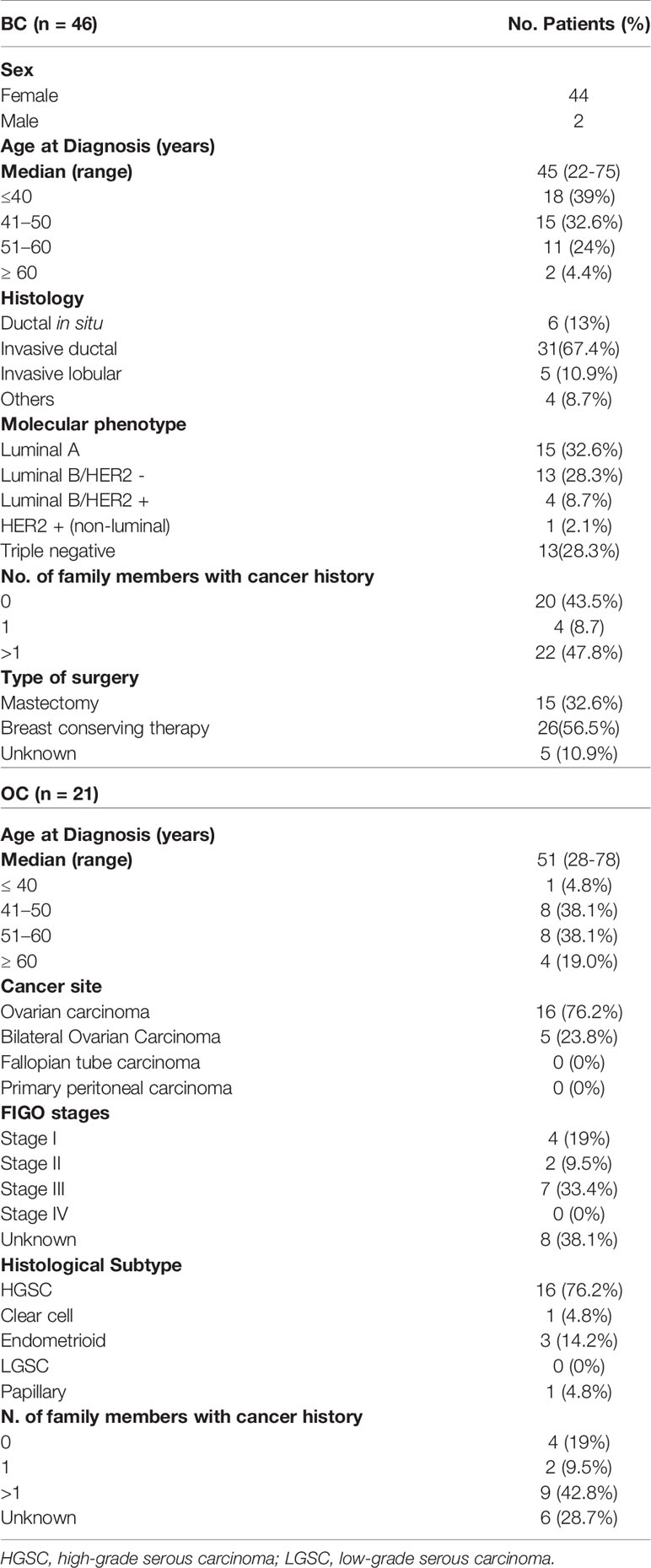
Table 1 Clinico-pathological features of germline BRCA1/2 VUS carriers affected by BC or OC.
Considering a distinction for pathology, among the 46 BC patients, 18 different variants have been identified in BRCA1 gene and 33 in BRCA2 gene. Among the 21 OC women, eight harbored BRCA1 VUS and 13 BRCA2 VUS.
Twenty-five (37.3%) out 67 probands, 17 of which were affected by BC and eight by OC, harbored VUS in BRCA1 gene; 37 (55.2%) subjects, 24 of which were with BC and 13 with OC in BRCA2 gene, whereas four (6.0%) BC women carried simultaneously two VUS in BRCA2, and one (1.5%) BC patient had VUS both in BRCA1 and BRCA2. Twenty-one (35.6%) out of 59 different variants were located on BRCA1 gene, whereas 38 (64.4%) have been identified on BRCA2 gene (Tables 2 and 3).
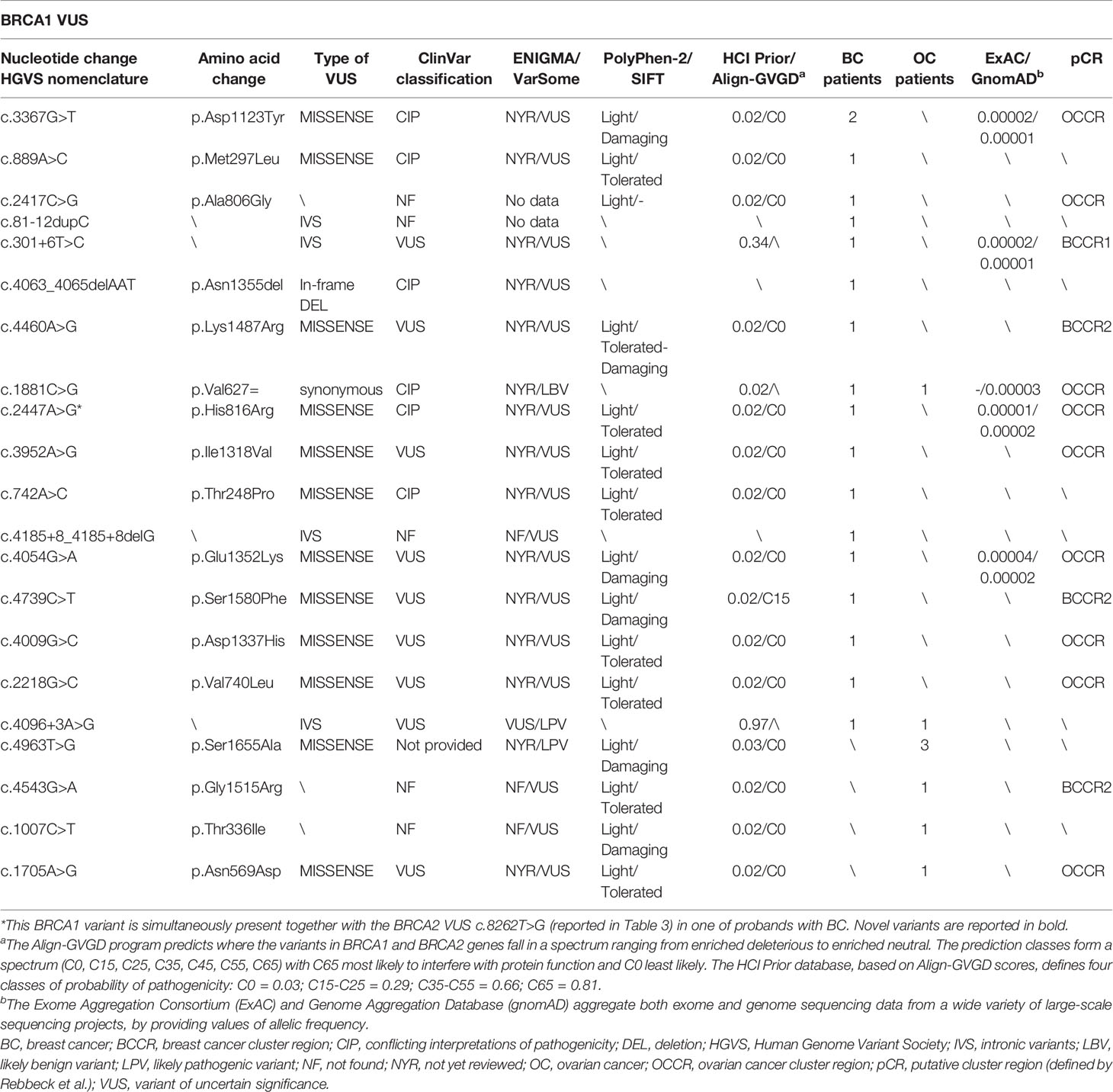
Table 2 BRCA1 gene variants of unclear significance harboured by patients with breast and ovarian cancers.
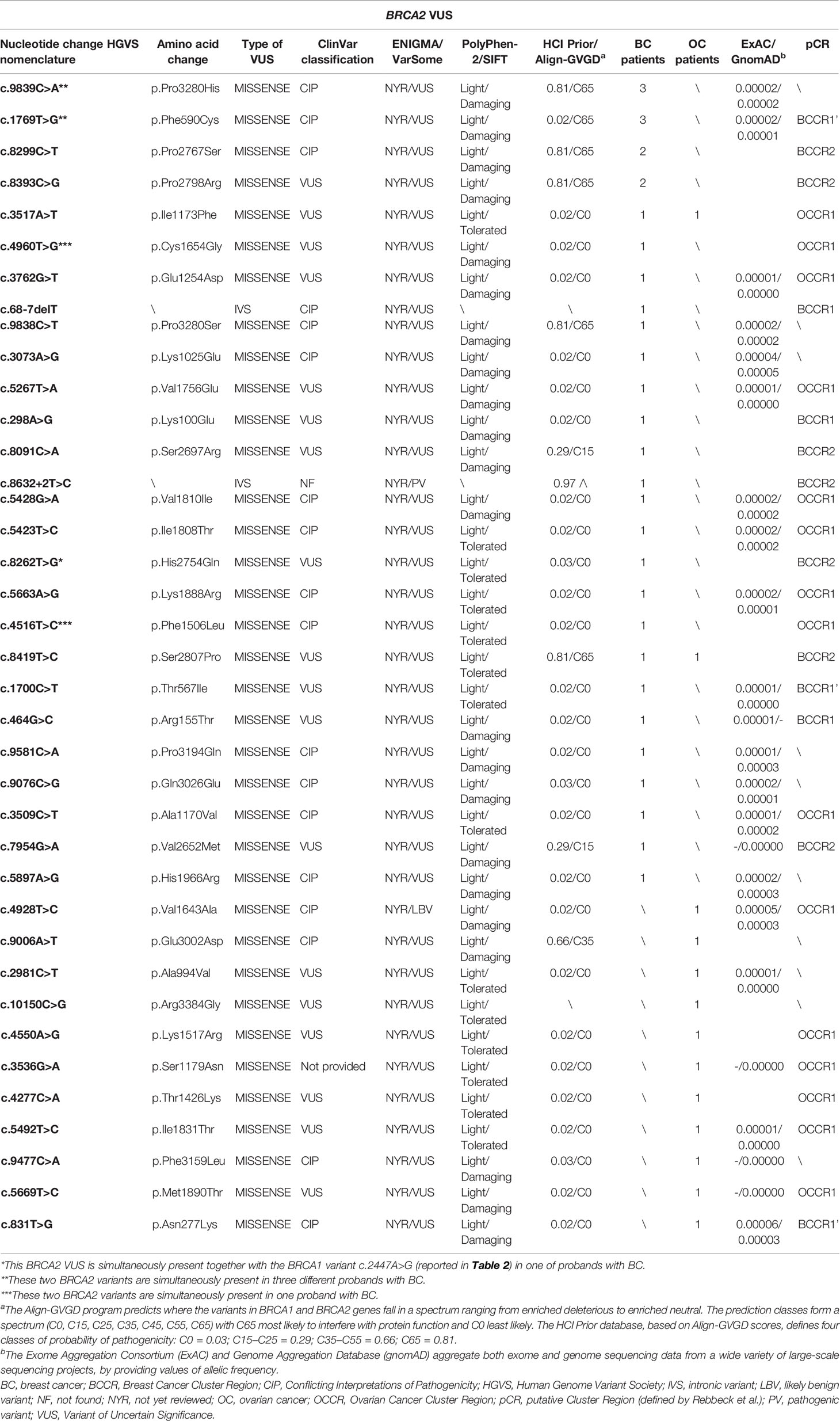
Table 3 BRCA2 gene variants of unclear significance harbored by patients with breast and ovarian cancers.
Several studies showed a strong correlation between specific BRCA1/2 variants and changes in BC/OC relative risk, by identifying specific putative Breast Cancer Cluster Regions (BCCRs) and Ovarian Cancer Cluster Regions (OCCRs) located on the coding DNA sequences of BRCA1/2 genes (30–34). As regards the gene location of BRCA1 variants detected in our study cohort, most of them (14/21) were mainly located within the hypothetical cluster region present in the BRCA1 protein structure which includes the exon 11 (nucleotides: 861–4,218; codons: 248–1,366), with a greater distribution inside the serine cluster domain (SCD). No variant was observed at BRCT domain near the C-terminus (Figure 1). A correlation between the variant localization in the putative BCCRs and OCCRs of BRCA1 and type of tumor was observed only in three BC and two OC patients (Table 2).
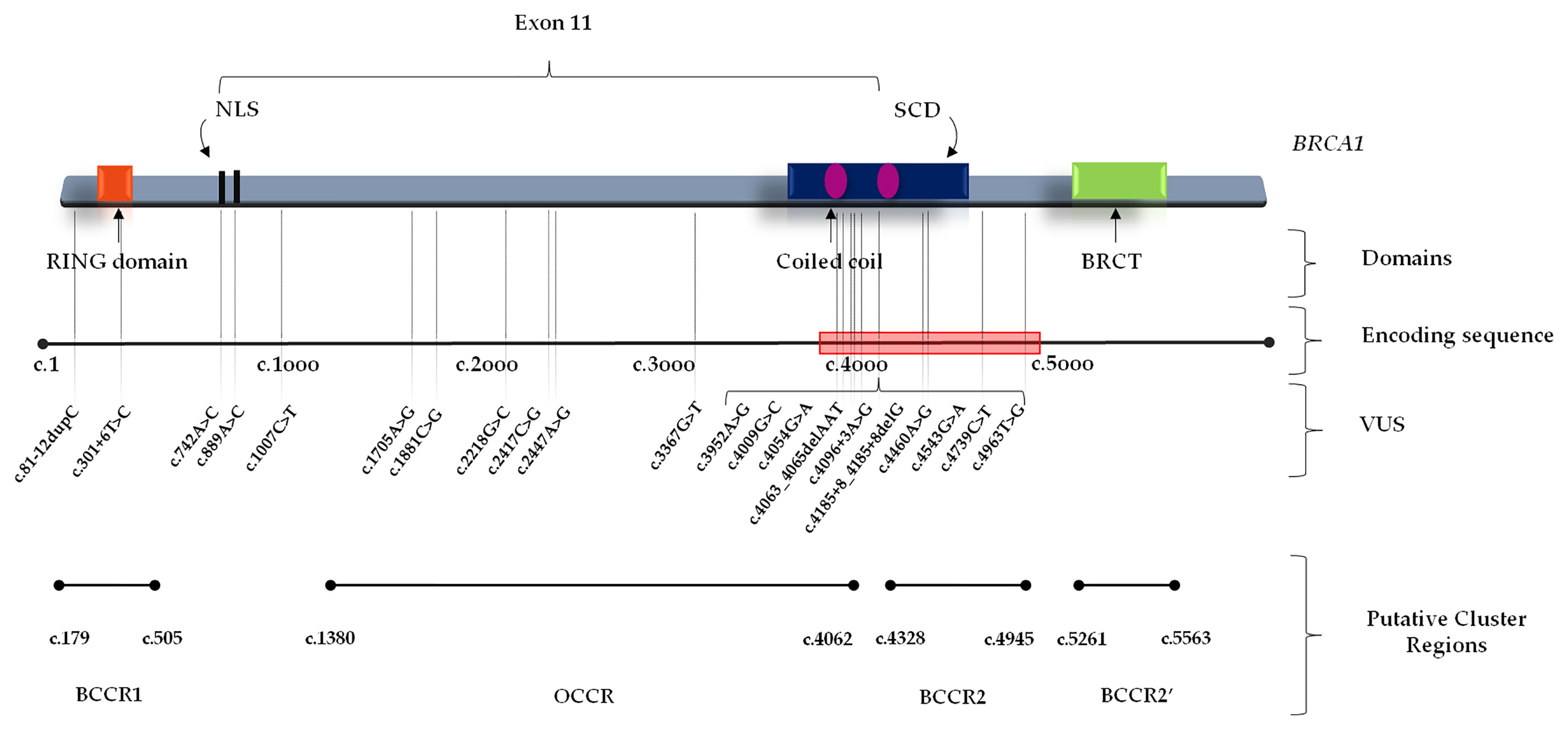
Figure 1 BRCA1 functional domains and gene location of BRCA1 Variants of Uncertain Significance in BC and/or OC patients. BCCR, Breast Cancer Cluster Region; BRCT, BRCA1 C-terminus domain; NLS, nuclear localization sequence; OCCR, Ovarian Cancer Cluster Region; SCD, serine cluster domain; VUS, Variant of Uncertain Significance.
Conversely, BRCA2 variants were distributed along the entire gene sequence, but most of them were mainly localized inside three putative cluster regions present in the BRCA2 protein structure which includes the BRC repeats (located within the exon 11), DNA binding helical domain, and OB fold domain near the C-terminus. Specifically, 17 BRCA2 variants were located at the BRC repeats, included inside exon 11 (nucleotides: 3,301–6,125; codons: 1,025–1,966), whereas seven were detected in the DNA binding helical domain (nucleotides: 8,182–8,862; codons: 2,652–2,878), and, finally, five variants were observed at the C-terminus, within the OB fold domain (nucleotides: 9,705–10,378; codons: 3,159–3,384) (Figure 2). A correlation between the variant localization in the BCCRs and OCCRs of BRCA2 and type of tumor was observed in 16 BC and seven OC patients (Table 3).
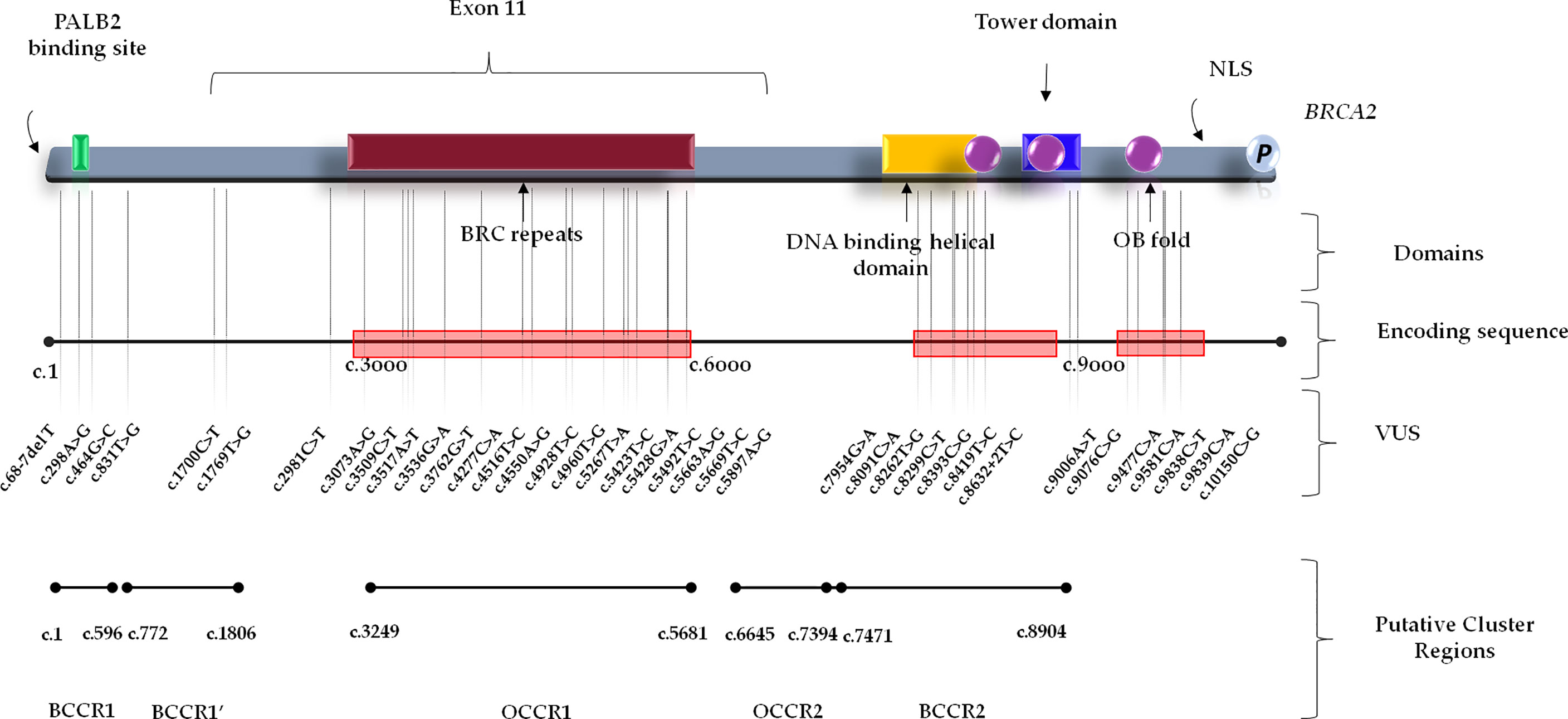
Figure 2 BRCA2 functional domains and gene location of BRCA2 Variants of Uncertain Significance in BC and/or OC patients. BCCR, breast cancer cluster region; NLS, nuclear localization sequence; OB, oligonucleotide binding; OCCR, Ovarian Cancer Cluster Region; VUS, Variant of Uncertain Significance.
Online available databases, such as ClinVar, ENIGMA, and VarSome, have been used to perform an in silico analysis to investigate the molecular and clinical meaning of an identified variants of unclear significance. Twenty-seven (45.7%) out of 59 observed alterations were defined as VUS in the major databases, whereas 24/59 (40.7%) were categorized as variants in a condition of “Conflicting Interpretations of Pathogenicity” (CIP) on ClinVar database. Furthermore, 3/59 (5.1%) variants have been defined as “not provided” on ClinVar. Among these, the BRCA1-c.4963T>G (p.Ser1655Ala) variant identified in three OC women, instead, was reported as Likely Pathogenic on VarSome database. This type of missense variant could be considered disease-causing. Finally, 5/59 (8.5%) variants have been unreported on most common databases and never described until now (Tables 2 and 3).
Based on the available information and online databases, after a deep study of personal and familial anamnesis of patients, we described the most representative variants detected in our study cohort.
Interestingly, the co-presence of two different BRCA2 variants (CIP) named c.9839C>A and c.1769T>G has been observed in three BC probands. The c.9839C>A variant, located in coding exon 26 of the BRCA2 gene at nucleotide position 9839, has been shown to cause a change in a poorly conserved region of the encoded protein sequence, involving the substitution of a proline amino acid residue with a histidine at codon 3280 (P3280H) (35). Some experimental lines of evidence supported by functional assays reported that this alteration is to be considered as likely benign due to its high frequency in the population and neutral effect on protein function (35, 36), whereas in silico tools predicted a damaging effect which could be disease-causing as reported on ClinVar database. For these reasons, the clinical significance of this variant remains yet unclear. The other c.1769T>G alteration causes a nucleotide substitution which involves the replacement of a phenylalanine with a cysteine at codon 590 (F590C) in a known functional domain. Based on current evidence reporting discordant findings, this variant still has an unclear significance (37, 38).
In addition, other two BRCA2 missense variants, named c.4960T>G and c.4516T>C, have been shown to be simultaneously present in one proband affected by BC. The c.4960T>G alteration involves the change of a thymine with guanine at nucleotide position 4,960 in coding exon 10 of the BRCA2 gene, resulting in substitution of a cysteine with a glycine at codon 1654, two amino acids with largely different physicochemical properties. The other sequence variant c.4516T>C, instead, determines minor physicochemical changes through the replacement of phenylalanine with leucine at codon 1,506.
Other two sequence variants named c.2447A>G and c.8262T>G (39) located in BRCA1 and BRCA2, respectively, were simultaneously detected in one BC patient. Until today, in silico analyses and population frequency data for both alterations have not shown evidence sufficient to associate them with a pathogenicity condition, therefore their clinical significance remains yet unclear.
Furthermore, three OC women have been shown to be carriers of a BRCA1 sequence variant named c.4963T>G (40), whose interpretation on Clinvar database results to be “not provided”.
The BRCA1 variant named c.3367G>T, which involves the change of aspartic acid with tyrosine (p.Asp1123Tyr) in an unknown functional domain of protein, has been identified in two probands affected by hormone-sensitive BC with early onset (37 and 39 years, respectively). Evidence from some in silico studies and low allele frequency observed in large population cohorts supported the hypothesis that this alteration is deleterious (41). Based on currently available data, this alteration is considered to have an unclear clinical significance. However, the case of one of two BC women who were carriers of this variant is interesting. Her family members were offered genetic testing because of the strong family history of BRCA-related tumors. Our genetic investigation showed that maternal uncle and grandmother, affected by prostate cancer and OC respectively, were carriers of same variant, whereas patient’s mother and sister were unaffected carriers (Figure 3).
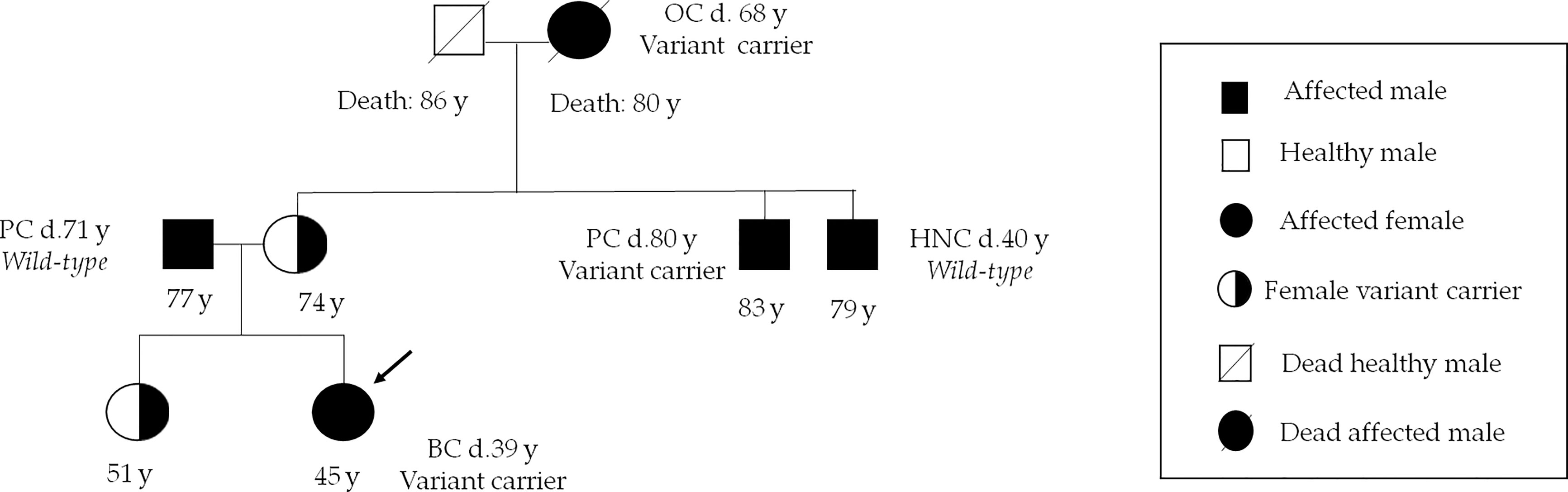
Figure 3 Family pedigree of the c.3367G>T carrier patient. Index case is indicated by an arrow. Numbers under symbols show the age of the patient and relatives. Instead, the age at diagnosis for the affected individuals is indicated by letter “d”. BC, breast cancer; HNC, head and neck cancer; OC, ovarian cancer; PC, prostate cancer.
Finally, interestingly, 10 variants of unclear significance have been observed in patients who already harbored a BRCA1 or BRCA2 PV (Supplementary Table 1).
Description and In Silico Analysis of Variants of Unclear Clinical Significance
Though most of the above described variants are known, we detected six alterations in BRCA1 and two in BRCA2, whose interpretation of clinical significance has not reported from data published in literature or main reference databases (Tables 2 and 3). We have defined as “not found” the variants not reported on the Clinvar, while those reported in the same database but without an interpretation of clinical significance have been defined as “not provided”.
Three variants, one of which was in BRCA1 (c.4963T>G) and two in BRCA2 (c.3536G>A and c.8632+2T>C), were reported as “not provided” on the Clinvar database.
Controversial is the case of BRCA1 variant named c.4963T>G (p.Ser1655Ala), which has been identified in three OC women. This alteration is defined as “not provided” on Clinvar, whereas it is reported as LPV on VarSome database. Literature data reported that the BRCT domain of BRCA1 is a key binding domain for phosphorylated serine (pSer) (42). Batenburg et al. (43) assessed if S1655 residue might mediate the interaction between BRCA1 and CSB (Cockayne syndrome complementation group B protein), a member of the SWI2/SNF2 superfamily, promoting a DNA repair mechanism via homologous recombination. These findings demonstrated that S1655A abolishes the interaction of BRCA1–BRCT with CSB, suggesting that the BRCA1–BRCT complex binds probably to pSer on CSB (43).
The BRCA2 variant named c.3536G>A (p.Ser1179Asn) has been identified in one 69 years old OC woman. The substitution of a serine with an asparagine at position 1,179 could be considered a novel alteration. The serine residue is located within the BRCA2 region involved in the interaction with RAD51 protein (44, 45). Both asparagine and serine are hydrophilic amino acids and have similar size, but the significance of this alteration is controversial. This BRCA2 variant of unclear significance has been simultaneously detected in the same patient together with BRCA1 PV named c.514delC.
The BRCA2 variant named c.8632+2T>C has been detected in a hormone-sensitive BC patient. This is an intronic variant localized in the splicing regions (46). This variant is actually classified as PV on VarSome, but it was not found on Clinvar. Its meaning is still not well defined.
Three BRCA1 variants named c.2417C>G and c.81-12dupC, never described before, and c.4185+8_4185+8delG, unreported on ClinVar, but described as VUS on VarSome, have been identified in three out of 46 BC patients, respectively.
Our genetic investigation has allowed identifying two novel frameshift variants, c.81-12dupC and c.4185+8_4185+8delG. Literature data showed that the small frameshift deletion c.4185+8_4185+8delG may lead to a possible alteration damaging the BRCA1 protein (47).
Furthermore, we have detected, in two women respectively, two germline BRCA1 variants, named c.4543G>A and c.1007C>T, never described before and never reported both on ClinVar and VarSome databases. The c.1007C>T alteration has been considered as “damaging” on SIFT database (48) and has been simultaneously detected in the same patient together with the BRCA1 PV named c.984_985insC. Further analysis could clarify, in future, the role of this variant.
Discussion
The frequency of identified VUS worldwide is different and strongly depends on the number of performed genetic testing as well as ancestral origin of the examined population. Literature data reported that VUS prevalence rate reaches 21% for patients of African-American ethnicity, and approximately the 5–6% for individuals of European ancestry (49, 50).
Recently, some studies showed how the prevalence of germline genetic variants and gene-specific risk estimates could change based on family history of cancer or tumor molecular subtype, but also according to other factors such as race, ancestry, and geographic location (51–53).
About 1,500 VUS as well as numerous BRCA1/2 PVs/LPVs are available in several publicly accessible databases. However, no public databases currently give the possibility to annotate cumulative evidence finalized to reclassify VUS (54).
To understand the clinical meaning of currently identified VUS may be accelerated by enabling increased sharing of information, which results complicated ethical issues (19).
Gene expression studies and in silico analysis predicting the impact of the amino acid change on protein folding, by testing the effect of a VUS on functions of a protein have been performed (55, 56).
In our retrospective study, we showed that prevalence of BRCA1/2 VUS in our population cohort was 7.7%, a concordant value with data reported in literature (15). Some VUS have been shown to be simultaneously present in some enrolled BC and/or OC patients, whereas other variants without an interpretation of clinical significance have been reported and described for the first time.
The collected data about familial anamnesis of a patient harboring the BRCA1-c.3367G>T variant suggested that this alteration should be further considered carefully because other affected relatives have been shown to be carriers of this variant.
Our results showed that there is a high heterogeneity of VUS among individuals of our population cohort and only a very few variants are shared between BC or OC patients. The lack of a specific territorial prevalence and heterogeneous distribution of VUS could be attributed to the genetic heterogeneity of the people belonging to regions of Southern Italy and their historical background due to the coexistence of different civilizations and critical geographical position of Sicily at the centre of Mediterranean Sea, crossroads of several ethnicities (57).
This study, through the use of prediction tools and databases, was aimed at describing the BRCA1/2 variants of unclear significance detected in the Sicilian population, which could be useful, in future, for improving the identification of novel disease-causing variants in BRCA1 and BRCA2 genes, allowing their eventual re-classification in potentially high-risk BRCA variants eligible for clinical purposes.
To understand whether these variants may potentially belong to class IV/V, an accurate assessment of the proband’s family history should be carried out, offering genetic testing to all consenting family members. However, many variants often cannot be investigated within a family due to the poor knowledge about cancer family history by proband.
In general, the classification of unclear significance variants results to be difficult due to the lack of functional evidence, insufficiency of population-based statistical evidence, and different evaluation approaches by scientists and clinicians (14). Since VUSs are mostly synonymous or missense alterations which involve the substitution of amino acid residues with similar physicochemical properties, or in-frame insertions/deletions, their effect on protein function is more complicated to unveil than nonsense variants (58, 14). Therefore, further experimental evidence is requested for enhancing the number of in vitro assays, given their complexity and consequent shortage. In addition, in some cases, suitable statistical assessments may be hindered by presence of slightly more frequent variants detected in population subsets or several pathological conditions (18). Finally, if, on one hand, the researchers take into consideration the VUS, polymorphisms, and novel variants because of their potential impact on the biochemical processes (14), even without clinical purposes, on the other hand, instead, the medical geneticists prefer to assess only variants with a well-defined clinical significance which may be helpful in the patient’s clinical management. These different approaches may hamper the studies regarding the clinical interpretation of variants with unclear significance, causing loss of information potentially useful for patient care (59). Another issue is represented by lack of universal standardization method for VUS among different diagnostic laboratories (60).
Nowadays, further in vitro functional and in silico analysis based on the use of updated databases and predictive algorithms are needed to allow a reclassification of alterations of unclear clinical significance in potentially high-risk variants (58). For this reason, today the development of in vitro assays to improve the VUS classification is the main objective of clinical research.
Surely, advances in molecular biology, such as the use of multi-gene panels, exome sequencing, and/or RNA-seq, are increasing the amount of data in the field of research on these unknown variants with unclear significance (61).
Further linkage analyses will be able to provide additional information helpful to geneticists for the understanding of inherited variants of unclear clinical significance associated with BC and/or OC.
Data Availability Statement
The datasets presented in this article are not readily available because of ethical and privacy reasons. Requests to access the datasets should be directed to YW50b25pby5ydXNzb0B1c2EubmV0 and dml2aWFuYS5iYXphbkB1bmlwYS5pdA==.
Ethics Statement
The studies involving human participants were reviewed and approved by the ethical committee (Comitato Etico Palermo 1; approval number: 0103-2017) of the University-affiliated Hospital A.O.U.P. ‘P. Giaccone’ of Palermo. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
Conceptualization: DF, AF, LI, GB, AR, and VB; genetic counseling: DF, VC, CF, AD, RS, and LM; sample collection and gene testing: AF, MB, DC, and AP; data curation and analysis: DF, AF, CB, CF, AD, GM, AC, EP, and LC; writing: DF and AF; Project administration: AR, GB, and VB. All authors contributed to the article and approved the submitted version.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
All authors thank Dr. Chiara Drago for the language English revision.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2021.682445/full#supplementary-material
Supplementary Figure 1 | Number of genetically tested patients with BC or OC (Oct 2016–Dec 2020). Total number of analyzed patients is divided into carriers of BRCA w.t. (green), BRCA VUS (yellow), and BRCA PVs/LPVs (red). Patients harboring benign/likely benign variants (BVs/LBVs) are considered carriers of BRCA w.t.
References
1. Futreal P, Liu Q, Shattuck-Eidens D, Cochran C, Harshman K, Tavtigian S, et al. BRCA1 Mutations in Primary Breast and Ovarian Carcinomas. Science (1994) 266:120–2. doi: 10.1126/science.7939630
2. Wooster R, Bignell G, Lancaster J, Swift S, Seal S, Mangion J, et al. Identification of the Breast Cancer Susceptibility Gene BRCA2. Nature (1995) 378:789–92. doi: 10.1038/378789a0
3. Fanale D, Incorvaia L, Filorizzo C, Bono M, Fiorino A, Calò V, et al. Detection of Germline Mutations in a Cohort of 139 Patients With Bilateral Breast Cancer by Multi-Gene Panel Testing: Impact of Pathogenic Variants in Other Genes Beyond BRCA1/2. Cancers (2020b) 12:2415. doi: 10.3390/cancers12092415
4. Incorvaia L, Fanale D, Bono M, Calò V, Fiorino A, Brando C, et al. BRCA1/2 Pathogenic Variants in Triple-Negative Versus Luminal-Like Breast Cancers: Genotype–Phenotype Correlation in a Cohort of 531 Patients. Ther Adv Med Oncol (2020b) 12:1–19. doi: 10.1177/1758835920975326
5. Shindo K, Yu J, Suenaga M, Fesharakizadeh S, Cho C, Macgregor-Das A, et al. Deleterious Germline Mutations in Patients With Apparently Sporadic Pancreatic Adenocarcinoma. J Clin Oncol (2017) 35:3382–90. doi: 10.1200/JCO.2017.72.3502
6. Hu C, Hart SN, Polley EC, Gnanaolivu R, Shimelis H, Lee KY, et al. Association Between Inherited Germline Mutations in Cancer Predisposition Genes and Risk of Pancreatic Cancer. JAMA (2018) 319:2401. doi: 10.1001/jama.2018.6228
7. Agalliu I, Gern R, Leanza S, Burk RD. Associations of High-Grade Prostate Cancer With BRCA1 and BRCA2 Founder Mutations. Clin Cancer Res (2009) 15:1112–20. doi: 10.1158/1078-0432.CCR-08-1822
8. Leongamornlert D, Mahmud N, Tymrakiewicz M, Saunders E, Dadaev T, Castro E, et al. Germline BRCA1 Mutations Increase Prostate Cancer Risk. Br J Cancer (2012) 106:1697–701. doi: 10.1038/bjc.2012.146
9. Pilarski R. The Role of BRCA Testing in Hereditary Pancreatic and Prostate Cancer Families. Am Soc Clin Oncol Educ Book (2019) 39:79–86. doi: 10.1200/EDBK_238977
10. Di Lorenzo S, Fanale D, Corradino B, Caló V, Rinaldi G, Bazan V, et al. Absence of germlineCDKN2Amutation in Sicilian Patients With Familial Malignant Melanoma: Could it be a Population-Specific Genetic Signature? Cancer Biol Ther (2015) 17:83–90. doi: 10.1080/15384047.2015.1108494
11. Plon SE, Eccles DM, Easton D, Foulkes WD, Genuardi M, Greenblatt MS, et al. Sequence Variant Classification and Reporting: Recommendations for Improving the Interpretation of Cancer Susceptibility Genetic Test Results. Hum Mutat (2008) 29:1282–91. doi: 10.1002/humu.20880
12. Landrum MJ, Lee JM, Riley GR, Jang W, Rubinstein WS, Church DM, et al. ClinVar: Public Archive of Relationships Among Sequence Variation and Human Phenotype. Nucleic Acids Res (2014) 42:D980–5. doi: 10.1093/nar/gkt1113
13. Eccles BK, Copson E, Maishman T, Abraham JE, Eccles DM. Understanding of BRCA VUS Genetic Results by Breast Cancer Specialists. BMC Cancer (2015) 15:936. doi: 10.1186/s12885-015-1934-1
14. Federici G, Soddu S. Variants of Uncertain Significance in the Era of High-Throughput Genome Sequencing: A Lesson From Breast and Ovary Cancers. J Exp Clin Cancer Res (2020) 39:46. doi: 10.1186/s13046-020-01554-6
15. Chern J-Y, Lee SS, Frey MK, Lee J, Blank SV. The Influence of BRCA Variants of Unknown Significance on Cancer Risk Management Decision-Making. J Gynecol Oncol (2019) 30:60. doi: 10.3802/jgo.2019.30.e60
16. Spurdle AB, Whiley PJ, Thompson B, Feng B, Healey S, Brown MA, et al. BRCA1 R1699Q Variant Displaying Ambiguous Functional Abrogation Confers Intermediate Breast and Ovarian Cancer Risk. J Med Genet (2012) 49:525–32. doi: 10.1136/jmedgenet-2012-101037
17. Makhnoon S, Shirts BH, Bowen DJ. Patients’ Perspectives of Variants of Uncertain Significance and Strategies for Uncertainty Management. J Genet Couns (2019) 28:313–25. doi: 10.1002/jgc4.1075
18. Moghadasi S, Eccles DM, Devilee P, Vreeswijk MPG, Van Asperen CJ. Classification and Clinical Management of Variants of Uncertain Significance in High Penetrance Cancer Predisposition Genes. Hum Mutat (2016) 37:331–6. doi: 10.1002/humu.22956
19. Eng C, Cline MS, Liao RG, Parsons MT, Paten B, Alquaddoomi F, et al. BRCA Challenge: BRCA Exchange as a Global Resource for Variants in BRCA1 and BRCA2. PloS Genet (2018) 14:e1007752. doi: 10.1371/journal.pgen.1007752
20. Lee J-S, Oh S, Park SK, Lee M-H, Lee JW, Kim S-W, et al. Reclassification of BRCA1 and BRCA2 Variants of Uncertain Significance: A Multifactorial Analysis of Multicentre Prospective Cohort. J Med Genet (2018) 55:794–802. doi: 10.1136/jmedgenet-2018-105565
21. Lindor NM, Guidugli L, Wang X, Vallée MP, Monteiro ANA, Tavtigian S, et al. A Review of a Multifactorial Probability-Based Model for Classification of BRCA1 and BRCA2 Variants of Uncertain Significance (VUS). Hum Mutat (2012) 33:8–21. doi: 10.1002/humu.21627
22. Fischer C, Kuchenbäcker K, Engel C, Zachariae S, Rhiem K, Meindl A, et al. Evaluating the Performance of the Breast Cancer Genetic Risk Models BOADICEA, Ibis, BRCAPRO and Claus for Predicting BRCA1/2 Mutation Carrier Probabilities: A Study Based on 7352 Families From the German Hereditary Breast and Ovarian Cancer Consortium. J Med Genet (2013) 50:360–7. doi: 10.1136/jmedgenet-2012-101415
23. Mazzola E, Blackford A, Parmigiani G, Biswas S. Recent Enhancements to the Genetic Risk Prediction Model BRCAPRO. Cancer Inf (2015) 14s2. CIN.S17292:147-57. doi: 10.4137/CIN.S17292
24. Pujol P, Barberis M, Beer P, Friedman E, Piulats JM, Capoluongo ED, et al. Clinical Practice Guidelines for BRCA1 and BRCA2 Genetic Testing. Eur J Cancer (2021) 146:30–47. doi: 10.1016/j.ejca.2020.12.023
25. Choi MC. Clinical Significance of Variants of Unknown Significances in BRCA Genes. J Gynecol Oncol (2019) 30:80. doi: 10.3802/jgo.2019.30.e80
26. Incorvaia L, Fanale D, Badalamenti G, Bono M, Calò V, Cancelliere D, et al. Hereditary Breast and Ovarian Cancer in Families From Southern Italy (Sicily)—Prevalence and Geographic Distribution of Pathogenic Variants in BRCA1/2 Genes. Cancers (2020a) 12:1158. doi: 10.3390/cancers12051158
27. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The Cbio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data: Figure 1. Cancer Discovery (2012) 2:401–4. doi: 10.1158/2159-8290.CD-12-0095
28. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci Signaling (2013) 6:pl1–1. doi: 10.1126/scisignal.2004088
29. Den Dunnen JT, Dalgleish R, Maglott DR, Hart RK, Greenblatt MS, Mcgowan-Jordan J, et al. Hgvs Recommendations for the Description of Sequence Variants: 2016 Update. Hum Mutat (2016) 37:564–9. doi: 10.1002/humu.22981
30. Thompson D, Easton D. Variation in Cancer Risks, by Mutation Position, in BRCA2 Mutation Carriers. Am J Hum Genet (2001) 68:410–9. doi: 10.1086/318181
31. Lubinski J, Phelan CM, Ghadirian P, Lynch HT, Garber J, Weber B, et al. Cancer Variation Associated With the Position of the Mutation in the BRCA2 Gene. Familial Cancer (2002) 3:1–10. doi: 10.1023/B:FAME.0000026816.32400.45
32. Rebbeck TR, Mitra N, Wan F, Sinilnikova OM, Healey S, Mcguffog L, et al. Association of Type and Locationof BRCA 1 and BRCA2 Mutations With Risk of Breast and Ovarian Cancer. JAMA (2015) 313:1347. doi: 10.1001/jama.2014.5985
33. Kuchenbaecker KB, Hopper JL, Barnes DR, Phillips K-A, Mooij TM, Roos-Blom M-J, et al. Risks of Breast, Ovarian, and Contralateral Breast Cancer for BRCA1 and BRCA2 Mutation Carriers. JAMA (2017) 317:2402. doi: 10.1001/jama.2017.7112
34. Teixeira N, van der Hout A, Oosterwijk JC, Vos JR, Devilee P, Van Engelen K, et al. The Association Between Cancer Family History and Ovarian Cancer Risk in BRCA1/2 Mutation Carriers: Can it be Explained by the Mutation Position? Eur J Hum Genet (2018) 26:848–57. doi: 10.1038/s41431-018-0111-9
35. Guidugli L, Carreira A, Caputo SM, Ehlen A, Galli A, Monteiro ANA, et al. Functional Assays for Analysis of Variants of Uncertain Significance in BRCA2. Hum Mutat (2014) 35:151–64. doi: 10.1002/humu.22478
36. Hucl T, Rago C, Gallmeier E, Brody JR, Gorospe M, Kern SE. A Syngeneic Variance Library for Functional Annotation of Human Variation: Application to BRCA2. Cancer Res (2008) 68:5023–30. doi: 10.1158/0008-5472.CAN-07-6189
37. Azzollini J, Scuvera G, Bruno E, Pasanisi P, Zaffaroni D, Calvello M, et al. Mutation Detection Rates Associated With Specific Selection Criteria for BRCA1/2 Testing in 1854 High-Risk Families: A Monocentric Italian Study. Eur J Internal Med (2016) 32:65–71. doi: 10.1016/j.ejim.2016.03.010
38. Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of Protein-Coding Genetic Variation in 60,706 Humans. Nature (2016) 536:285–91. doi: 10.1038/nature19057
39. Zuntini R, Ferrari S, Bonora E, Buscherini F, Bertonazzi B, Grippa M, et al. Dealing With Brca1/2 Unclassified Variants in a Cancer Genetics Clinic: Does Cosegregation Analysis Help? Front Genet (2018) 9:378. doi: 10.3389/fgene.2018.00378
40. Findlay GM, Daza RM, Martin B, Zhang MD, Leith AP, Gasperini M, et al. Accurate Classification of BRCA1 Variants With Saturation Genome Editing. Nature (2018) 562:217–22. doi: 10.1038/s41586-018-0461-z
41. Seymour IJ, Casadei S, Zampiga V, Rosato S, Danesi R, Scarpi E, et al. Results of a Population-Based Screening for Hereditary Breast Cancer in a Region of North-Central Italy: Contribution of BRCA1/2 Germ-Line Mutations. Breast Cancer Res Treat (2007) 112:343–9. doi: 10.1007/s10549-007-9846-7
42. Yu X. The BRCT Domain Is a Phospho-Protein Binding Domain. Science (2003) 302:639–42. doi: 10.1126/science.1088753
43. Batenburg NL, Walker JR, Coulombe Y, Sherker A, Masson J-Y, Zhu X-D. CSB Interacts With BRCA1 in Late S/G2 to Promote MRN- and CtIP-Mediated DNA End Resection. Nucleic Acids Res (2019) 47:10678–92. doi: 10.1093/nar/gkz784
44. Wong AKC, Pero R, Ormonde PA, Tavtigian SV, Bartel PL. Rad51 Interacts With the Evolutionarily Conserved BRC Motifs in the Human Breast Cancer Susceptibility Gene BRCA2. J Biol Chem (1997) 272:31941–4. doi: 10.1074/jbc.272.51.31941
45. Chen C-F, Chen P-L, Zhong Q, Sharp ZD, Lee W-H. Expression of BRC Repeats in Breast Cancer Cells Disrupts the BRCA2-Rad51 Complex and Leads to Radiation Hypersensitivity and Loss of G2/M Checkpoint Control. J Biol Chem (1999) 274:32931–5. doi: 10.1074/jbc.274.46.32931
46. Esteban Cardeñosa E, Bolufer Gilabert P, De Juan Jimenez I, Palanca Suela S, Barragán González E, Chirivella González I, et al. Broad BRCA1 and BRCA2 Mutational Spectrum and High Incidence of Recurrent and Novel Mutations in the Eastern Spain Population. Breast Cancer Res Treat (2009) 121:257–60. doi: 10.1007/s10549-009-0680-y
47. Bouwman P, van der Gulden H, van der Heijden I, Drost R, Klijn CN, Prasetyanti P, et al. A High-Throughput Functional Complementation Assay for Classification of BRCA1 Missense Variants. Cancer Discovery (2013) 3:1142–55. doi: 10.1158/2159-8290.CD-13-0094
48. Tang H, Thomas PD. Tools for Predicting the Functional Impact of Nonsynonymous Genetic Variation. Genetics (2016) 203:635–47. doi: 10.1534/genetics.116.190033
49. Ready K, Gutierrez-Barrera AM, Amos C, Meric-Bernstam F, Lu K, Hortobagyi G, et al. Cancer Risk Management Decisions of Women With BRCA1 or BRCA2 Variants of Uncertain Significance. Breast J (2011) 17:210–2. doi: 10.1111/j.1524-4741.2010.01055.x
50. Lindor NM, Goldgar DE, Tavtigian SV, Plon SE, Couch FJ. Brca1/2 Sequence Variants of Uncertain Significance: A Primer for Providers to Assist in Discussions and in Medical Management. Oncol (2013) 18:518–24. doi: 10.1634/theoncologist.2012-0452
51. Manchanda R, Loggenberg K, Sanderson S, Burnell M, Wardle J, Gessler S, et al. Population Testing for Cancer Predisposing BRCA1/BRCA2 Mutations in the Ashkenazi-Jewish Community: A Randomized Controlled Trial. JNCI: J Natl Cancer Institute (2015) 107:379. doi: 10.1093/jnci/dju379
52. Akbari MR, Gojska N, Narod S. Coming of Age in Canada: A Study of Population-Based Genetic Testing for Breast and Ovarian Cancer. Curr Oncol (2017) 24:282–3. doi: 10.3747/co.24.3828
53. Rowley SM, Mascarenhas L, Devereux L, Li N, Amarasinghe KC, Zethoven M, et al. Population-Based Genetic Testing of Asymptomatic Women for Breast and Ovarian Cancer Susceptibility. Genet Med (2018) 21:913–22. doi: 10.1038/s41436-018-0277-0
54. Easton DF, Deffenbaugh AM, Pruss D, Frye C, Wenstrup RJ, Allen-Brady K, et al. A Systematic Genetic Assessment of 1,433 Sequence Variants of Unknown Clinical Significance in the BRCA1 and BRCA2 Breast Cancer–Predisposition Genes. Am J Hum Genet (2007) 81:873–83. doi: 10.1086/521032
55. Millot GA, Carvalho MA, Caputo SM, Vreeswijk MPG, Brown MA, Webb M, et al. A Guide for Functional Analysis of BRCA1 Variants of Uncertain Significance. Hum Mutat (2012) 33:1526–37. doi: 10.1002/humu.22150
56. Ernst C, Hahnen E, Engel C, Nothnagel M, Weber J, Schmutzler RK, et al. Performance of In Silico Prediction Tools for the Classification of Rare BRCA1/2 Missense Variants in Clinical Diagnostics. BMC Med Genomics (2018) 11:35. doi: 10.1186/s12920-018-0353-y
57. Fanale D, Incorvaia L, Bono M, Calò V, Cancelliere D, Fiorino A, et al. 247p Population-based Testing for Hereditary Breast and Ovarian Cancer in a Cohort of 1,346 Patients From Southern Italy (Sicily): When Historical Background Affects Genetics. Ann Oncol (2020a) 31:S338–9. doi: 10.1016/j.annonc.2020.08.368
58. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med (2015) 17:405–23. doi: 10.1038/gim.2015.30
59. Macarthur DG, Manolio TA, Dimmock DP, Rehm HL, Shendure J, Abecasis GR, et al. Guidelines for Investigating Causality of Sequence Variants in Human Disease. Nature (2014) 508:469–76. doi: 10.1038/nature13127
60. Makhnoon S, Shirts BH, Bowen DJ, Fullerton SM. Hereditary Cancer Gene Panel Test Reports: Wide Heterogeneity Suggests Need for Standardization. Genet Med (2018) 20:1438–45. doi: 10.1038/gim.2018.23
Keywords: BRCA1, BRCA2, breast cancer, genetic testing, ovarian cancer, variants of uncertain significance (VUS)
Citation: Fanale D, Fiorino A, Incorvaia L, Dimino A, Filorizzo C, Bono M, Cancelliere D, Calò V, Brando C, Corsini LR, Sciacchitano R, Magrin L, Pivetti A, Pedone E, Madonia G, Cucinella A, Badalamenti G, Russo A and Bazan V (2021) Prevalence and Spectrum of Germline BRCA1 and BRCA2 Variants of Uncertain Significance in Breast/Ovarian Cancer: Mysterious Signals From the Genome. Front. Oncol. 11:682445. doi: 10.3389/fonc.2021.682445
Received: 18 March 2021; Accepted: 25 May 2021;
Published: 11 June 2021.
Edited by:
Daniela Turchetti, University of Bologna, ItalyReviewed by:
Andrea Tinelli, Moscow Institute of Physics and Technology, RussiaAlessandra Brambati, Memorial Sloan Kettering Cancer Center, United States
Concetta Santonocito, Catholic University of the Sacred Heart, Italy
George Wiggins, University of Otago, Christchurch, New Zealand
Copyright © 2021 Fanale, Fiorino, Incorvaia, Dimino, Filorizzo, Bono, Cancelliere, Calò, Brando, Corsini, Sciacchitano, Magrin, Pivetti, Pedone, Madonia, Cucinella, Badalamenti, Russo and Bazan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Viviana Bazan, dml2aWFuYS5iYXphbkB1bmlwYS5pdA==; Antonio Russo, YW50b25pby5ydXNzb0B1c2EubmV0
†ORCID ID: Antonio Russo, orcid.org/0000-0002-4370-2008
‡These authors have contributed equally to this work
§These authors share last authorship