 Meilin Mu
Meilin Mu Hongwei Gao
Hongwei Gao- School of Life Science, Ludong University, Yantai, China
The geometric structure of azido Pt(IV) compounds containing picoline was calculated by using density functional theory(DFT) at the LSDA/SDD level. The ESP distribution shows the possible reaction sites of the compounds. In addition, the frequency calculation results assigned the infrared spectra of these compounds, and specified important stretching and bending vibrations. The HOMO-LUMO energy gaps of these compounds are also calculated to explain the charge transfer of the molecules. The distribution of Mulliken charges and natural atomic charges of these atoms is also calculated. Natural bond orbital(NBO) analysis explains the intramolecular interactions and their electron density.
1 Introduction
Metal platinum therapy is usually a cancer treatment plan including cisplatin (1), carboplatin (2, 3) and oxaliplatin (4). Especially the successful medical treatment of cisplatin can be called a milestone in cancer chemotherapy. But the incidental properties of these three platinum drugs include poor drug resistance, side effects and targeting (5, 6). In order to solve the problems of these drugs, new drug designs have been implemented to obtain better therapeutic effects. One of the solutions involves introducing new groups into the system to become Pt(IV) to give the compound new stability and new functional groups (7, 8). Drug improvement based on cisplatin (9, 10) and carboplatin (11) are also promising directions to improve the properties of the parent compounds. In addition, a class of compounds can obtain active reactants through light-induced activation, which may be a new direction for targeted therapy. Photodynamic therapy(PDT) is a kind of light therapy that achieves therapeutic effects by producing highly reactive singlet oxygen 1O2 (12). However, the tumor affected by hypoxia is a barrier that is not easy to recover for oxygen-dependent PDT (13). Photoactivated chemotherapy (PACT), a light therapy that is not limited to oxygen, is considered a promising therapy. Some members of transition metals often become prodrugs of PACT (14, 15). The compounds suitable for this method are not limited by the oxygen concentration, and can form a reactive active substance after being irradiated (16).
As a new type of anti-tumor platinum drug, Pt(IV) compounds containing azido have excellent stability in the dark, and they involve the release of azido groups and the production of reactive Pt(II) reactants after being irradiated (17, 18). In addition after these compounds have been developed, they can obviously overcome the easy-to-reducible properties of diiodide Pt(IV) compounds (19). The introduction of new groups has become an effective way to improve the performance of azido-containing compounds. One of the transformation methods for cisplatin, namely the introduction of heterocyclic imines(especially pyridine), has been found to have significant damaging capabilities for cancer tissue (20, 21). In addition, the way cis-diammine-(pyridine)-chloroplatinum(II) (or cDPCP) binds to DNA and the way it inhibits transcription are different from drugs such as cisplatin (22). Such monofunctional compound provides an excellent idea for appropriately improving the pharmacological activity of drug such as cisplatin.
In previous studies, we have introduced and classified the mechanism of action and toxicity of azido-containing Pt(IV) complexes in detail (23). The addition of the aromatic group(py) can significantly increase the phototoxicity of the compound trans,trans,trans-[Pt(N3)2(OH)2(NH3)(py)] compared with the amine-based compound trans,trans,trans-[Pt(N3)2(OH)2(NH3)2] (24, 25). The single pyridine compound trans,trans,trans-[Pt(N3)2(OH)2(NH3)(py)] is non-selective to a variety of cisplatin cells. Moreover, the photoactivation process of trans,trans,trans-[Pt(N3)2(OH)2(NH3)(py)] and DFT/TDDFT calculations prove that its reaction involves the drop of azide ligand (26). Trans,trans,trans-[Pt(N3)2(OH)2(py)2](1) is a promising candidate, which can bind to DNA firmly after being activated, and can show much higher photocytotoxicity than cisplatin (27). Furthermore, compound 1 tends to have higher intracellular accumulation and photocytotoxicity compared with its monopyridine azido compound (28). The picoline analog trans,trans,trans-[Pt(N3)2(OH)2(R1)(R2)] of compound 1 also have excellent phototoxicity to cisplatin-resistant cells and other specific cell lines. They are the compounds 2(R1 = 2-picoline, R2=pyridine), 3(R1 = 3-picoline, R2=pyridine), 4(R1 = 4-picoline, R2=pyridine), 5(R1 = 3-picoline, R2 = 3-picoline) and 6(R1 = 4-picoline, R2 = 4-picoline) (29).
Generally speaking, we hope that Pt(IV) diazonium compounds are stable in the dark and have cytotoxic effects after being reduced to Pt(II) compounds. For Pt(IV) compounds, their stability is a very important criterion for evaluating whether a compound is excellent when it is not reduced to Pt(II). Therefore, our manuscript focuses more on the stability of tetravalent complexes. Although there have been many experimental studies on such compounds, there are few theoretical studies on these compounds (30). Previous studies have shown that long-range corrected functional, LC-ωPBE, exhibits good characteristics in DFT calculation of cisplatin geometry optimization and Pt ligand vibration. In addition, PBE0 and mPW1PW functional combined with the LanL2TZ(f) basis set of Pt can be used to calculate the IR and Raman spectra of cisplatin (31). Compared with the experimental data, the B3LYP/(aug-cc-pVDZ + GD3BJ) method can predict the vibrational spectra of carboplatin, oxaliplatin, nedaplatin and heptaplatin well (32). We have optimized the geometric structure of compound 1 and analyzed the vibration spectrum before (33). In addition, the HOMO-LUMO orbital, natural bond orbital (NBO) and charge distribution of the compounds are also analyzed.
2 Computational Details
In density functional theory, Local Spin Density Approximation (LSDA) will underestimate the band gap value, and it is not suitable for systems with rapidly changing electron densities. The Generalized Gradient Approximation (GGA) often includes the gradient correction as the LSDA correction (34, 35). Hybrid functionals can better describe systems with more serious electronic self-interactions. Hybrid functionals can adjust the ratio of HF exchange functionals and DFT exchange functionals by changing the linear combination coefficients, so that the calculated results are closer to the real results of the experiment.
In previous research, We screened some functional methods and basis sets in Gaussian 16 that are suitable for pyridine-containing azido Pt(IV) complexes (33). When calculating the geometric optimization data, the functionals with smaller deviations from the experimental data are CAM-B3LYP, PBE1PBE, LSDA, etc. But we found that the LSDA functional method has small errors with the experimental values on the key bond length data of Pt-N(N3), Pt-O(OH) and N(N3)-N(N3). In the same way, although LANL2DZ (36) also has a small deviation, the calculation results of the SDD basis set show that bond lengths [Pt-N(N3), Pt-O(OH) and N(N3)-N(N3)] fit well. Therefore, we choose LSDA functional and SDD basis set as our calculation method. All calculation results are obtained at the local spin-density approximation(LSDA) (37, 38) & Stuttgart/Dresden and D95 ECPs(SDD) (39, 40) level of the density functional theory (DFT) method in Gaussian 16 (41) software. Firstly, the molecular structure of the compounds was optimized to analyze the structural characteristics of the compounds. The optimized structure is used for frequency calculation, and their characteristic absorption peaks are assigned. Electrostatic potential (ESP) is considered to be a possible method to predict the location of the interaction reaction. The ESP distribution of the compounds is drawn by Multiwfn program (42) and Visual Molecular Dynamics 1.9.3 (VMD 1.9.3) program (43). In addition, electronic properties such as HOMO-LUMO energy, Mulliken charge distribution and natural bond orbital (NBO) and natural atomic charge are calculated and analyzed.
3 Results and Discussion
3.1 Cytotoxic Activity
For the structure of the selected compounds 1, 2, 3, 4, 5 and 6, the main difference lies in the different positions and different amounts of picoline. In this article, we will discuss the effect of substituents on the properties of the compounds. Previous studies have completed the toxicity testing of these compounds through experiments, and their toxicity data is shown in Table 1 (29). Except for compound 2 which exhibits moderate toxicity in the dark, other compounds have good stability in the dark. In addition, compound 2 also has poor phototoxicity (14.5μM in human ovarian cells), and other compounds have good phototoxicity IC50 values (3.1-7.2 μM in human ovarian cells). In addition, compared with compound 5 (10.4 μM), compounds 3 and 4 showed better phototoxicity(3.3 and 4.6 μM) for the cisplatin-resistant cell line (A2780cis). Compared with its analogs 4 and 5, compound 6 has good cytotoxicity among the selected compounds (44).
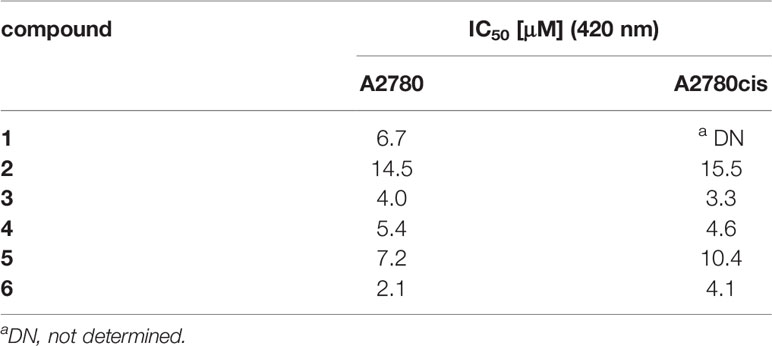
Table 1 Cytotoxicity of compounds 1, 2, 3, 4, 5 and 6 in A2780 and cisplatin-resistant cells(A2780cis) (29).
3.2 Molecular Geometry and ESP Analysis
The chemical structures and optimized molecular structures of the selected compounds 1, 2, 3, 4, 5 and 6 are shown in Figures 1 and 2. For the compounds, the leaving group N3 is usually the primary research object due to its importance in the first reaction stage, and the release rate of the N3 group is the reference feature for the rate of photoactivation (18, 45). The OH group is the key group when the compounds and nucleosides are combined, and the non-leaving group containing pyridine will significantly affect the photocytotoxicity of the compounds (24, 46). We have optimized their structure on the LSDA/SDD level, and the optimized structure data of the bond length and bond angle are summarized in Table 2. The date is used to evaluate the possible influence of the substituent effect on the stability and cytotoxicity of the compounds. We compared the calculated dates of complexes 1, 3, 4, 5 and 6 with the experimental values. The results show that their calculated bond length data is relatively close to the experimental data. In this part only, we have uniformly numbered the structures of the compounds for the convenience of research and discussion (Figure S1).
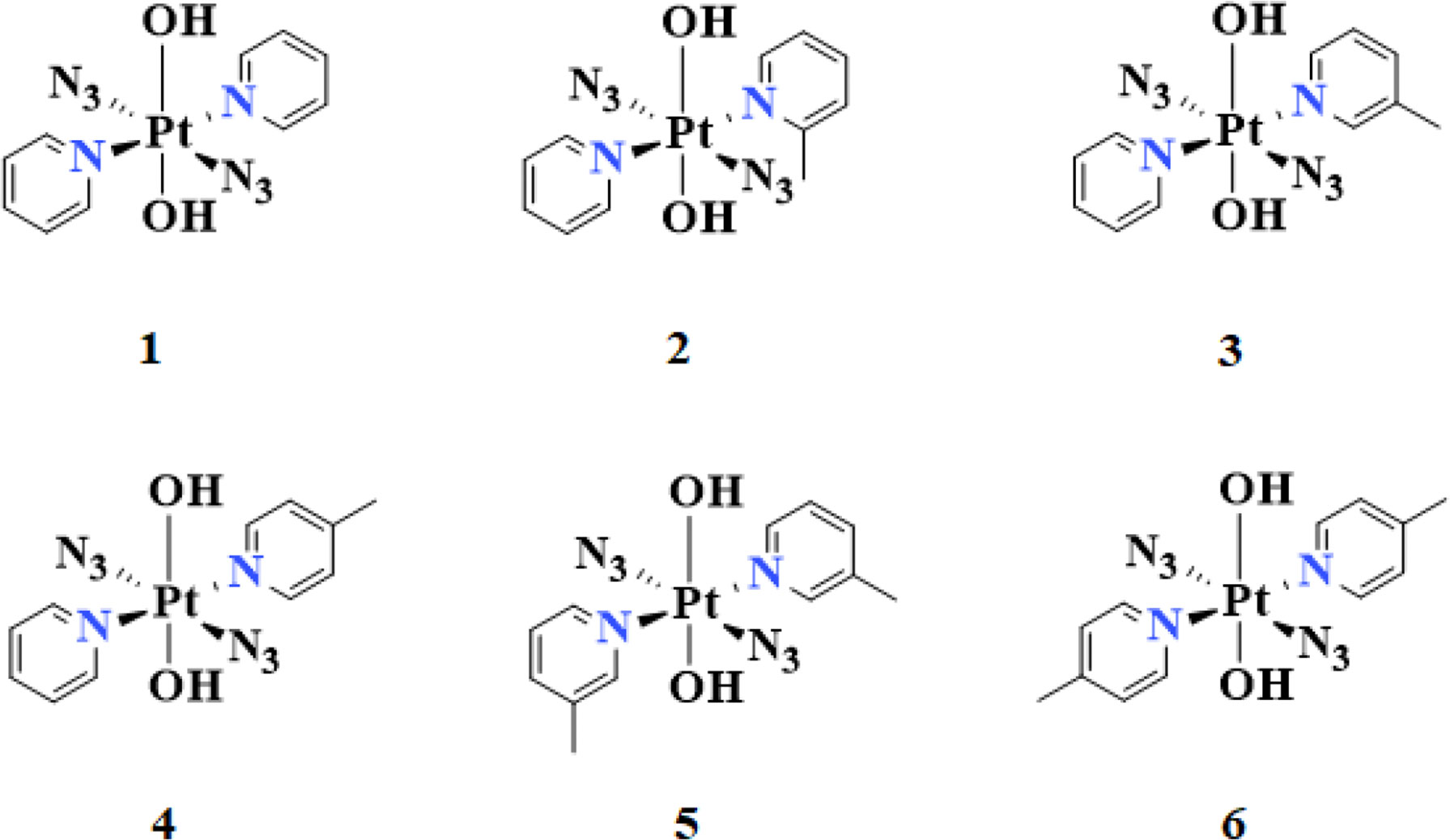
Figure 1 The calculated chemical structures of compounds: trans,trans,trans-[Pt(N3)2(OH)2(R1)(R2)][2(R1=2-picoline, R2=pyridine), 3(R1=3-picoline, R2=pyridine), 4(R1=4-picoline, R2=pyridine), 5(R1=3-picoline, R2=3-picoline) and 6(R1=4-picoline, R2=4-picoline)].
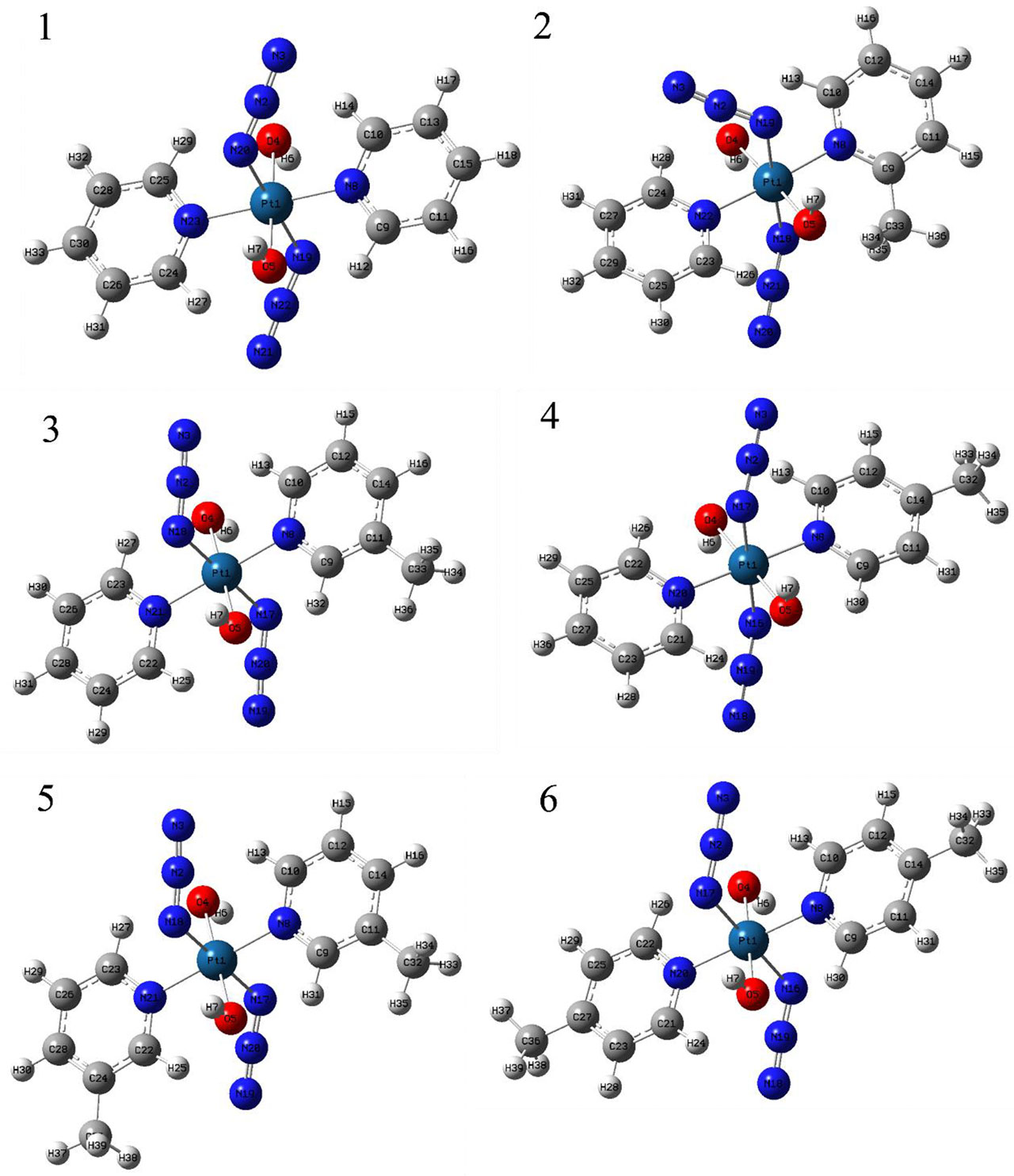
Figure 2 The optimized structure of compounds 1, 2, 3, 4, 5 and 6.
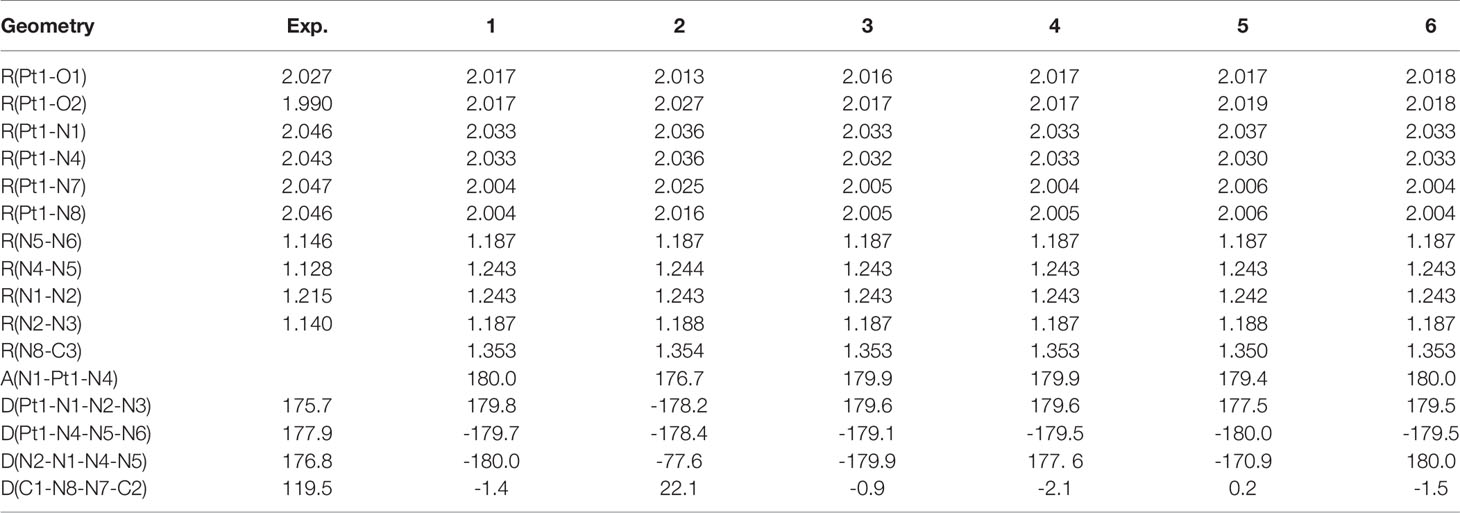
Table 2 Optimized bond length(in Å) and bond angle(in deg) of compounds 1, 2, 3, 4, 5 and 6.
The poorer cytotoxicity of compounds 2 and 4 than other compounds may be related to the bond length of the Pt-N bond. Previous studies have shown that after the introduction of 2-picoline, compound 2 has the fastest reduction rate among our selected compounds (29). The steric clash between the methyl group of 2-picoline and the axial hydroxyl ligand makes the compound 2 is easier to be restored (47). The data in Table 2 shows that the Pt-N3 bond lengths (Pt1-N1 and Pt1-N4) of compound 2 are 2.036 and 2.036 Å, respectively. The Pt-N3 bond lengths of compounds 1, 3, 4, 5 and 6 are 2.032-2.033 Å. Therefore, the results show that the binding ability between the Pt-N bonds between the Pt and the azido group in the compound 2 is weaker than that of the compounds 3, 4, and 6, which indicates that the azido group is easily released from the compound 2 after being irradiated by light. Because the photoreaction of the azido compound involves the reduction of Pt(IV) to Pt(II) and the drop of N3 after being irradiated. In addition, the Pt1-N1 and Pt1-N4 bond lengths of the compound 5 are 2.037 and 2.030 Å, which are the longest and shortest bond lengths respectively compared with other compounds. That is to say, the binding ability of the Pt atom and the N1 atom of the compound 5 is very weak, and the binding ability to the N4 atom is very strong compared with other compounds. Therefore, according to the previous report, the two azido groups of the compound 5 may be released one by one to participate in the photolysis pathway, and the second N3 group may be released after binding to the nucleotide (46).
It has been confirmed that the production of O2 in the photoproduct is likely to come from the hydroxyl groups of such compounds, and the decomposition of these hydroxyl groups after recombination is the key to the release of oxygen (46, 48). The results of the bond length between Pt atom and O atom show that the bond lengths of Pt-O1 and Pt-O2 of compound 2 are 2.013 and 2.027 Å, while the Pt-O bond lengths of the other compounds are relatively equal (2.017-2.019 Å). Therefore, the binding ability of the Pt-O2 bond of the compound 2 is weaker than other compounds, and the binding ability of the Pt-O1 of the compound 2 is less than other compounds.
An important event for the inactivation of cisplatin in cells is the replacement of amines, which affects the activity of the drug directly (49). The azido Pt(IV) compounds, trans,trans,trans-[Pt(N3)2(OH)2(NH3)2] & trans,trans,trans-[Pt(N3)2(OH)2(NH3)(py)], both have the dissociation of NH3 after being illuminated (24, 50). The pyridine group of the new compound 1 has not been observed to be released and has high cytotoxicity, so the addition of the pyridyl group can compensate for the loss of phototoxicity after the amino group is replaced (51). The bond lengths of Pt1-N7 and Pt1-N8 of the selected compound 2 are 2.025 and 2.016 Å, respectively, which are longer than the bond lengths of Pt-N(pyridine) bonds in other compounds (2.004-2.006 Å). In other words, the binding capacity of the compound 2 with Pt atoms is much smaller than that of other compounds. Therefore, according to previous studies, the stability of the Pt-N bond between the Pt atom and the pyridine group is directly related to the phototoxicity of the compounds. As shown in the data in Table 1, compound 2 has poor phototoxicity compared with other compounds, which is consistent with our calculation results.
We have compared compounds 2, 3 and 4 with different substitution positions, and the results show that meta-picoline compound 3 and para-picoline compound 4 are basically the same. For Ortho-picoline compound 2, although the bond length of Pt-O1 is smaller than that of compounds 3 and 4, the bond lengths of Pt-O2, Pt1-N1, Pt1-N4, Pt1-N7, and Pt1-N8 are all longer than compounds 3 and 4. Therefore, for single pyridine compounds with different substitution positions, the substituent effect is more significant on compound 2. And the bond length of compound 2 is mostly longer, which may be related to its poor phototoxicity and dark stability. For the effect of different numbers of picoline substituents, we have analyzed the bond lengths of compounds 1, 4 and 6, and the results show that their bond length data are not much different. The bond length analysis results of compounds 1, 3 and 5 show that the bond length data of compounds 1 and 3 are basically the same. The bond lengths of Pt1-N4 and N8-C3 of compound 5 are slightly smaller than that of compounds 1 and 3, and the bond lengths of Pt-O2, Pt1-N1, Pt1-N7 and Pt1-N8 are all longer. Moreover, the cytotoxicity of compound 5 is relatively small, so the number of substituents may also have a significant impact on cytotoxicity.
In addition, the two pyridine/picoline ligands of compound 2 have a large angular deflection (dihedral angle of 22.1°), which may be caused by the steric hindrance of compound 2. Among the six types of compounds, the bond lengths of Pt-N(N3), Pt-OH and Pt-N(pyridine) mentioned above are in the order of Pt-N(N3)>Pt-OH>Pt-N(pyridine). Therefore, the order of the binding ability between Pt and adjacent groups is the pyridine(non-leaving group)>OH>N3(leaving group) for the compounds we selected. The above analysis is consistent with the results shown by the mechanism of action of these compounds.
The surface electrostatic potential (ESP) can provide a reference for the interaction mode between molecules, and the interaction between molecules depends on the ESP extreme point of the binding site (52–57). The extreme values of the positive and negative potentials drawn with Multiwfn and VMD are shown in Figure 3. In the figure, the ESP value of the blue area is negative, and the red area is positive. The para-position of the N atom in pyridine/picoline in all compounds is the most vulnerable site to nucleophilic attack, and the maximum ESP value comes from compound 1 (32.80 kcal/mol). Compound 6 has a methyl substituent at the para position, which causes its extreme point to shift to the meta H position. The N3 and OH- regions of all compounds have one or more ESP minimums, which are the most vulnerable to proton attack. The lower ESP values are -39.89 and 39.90 kcal/mol for compounds 5 and 6.
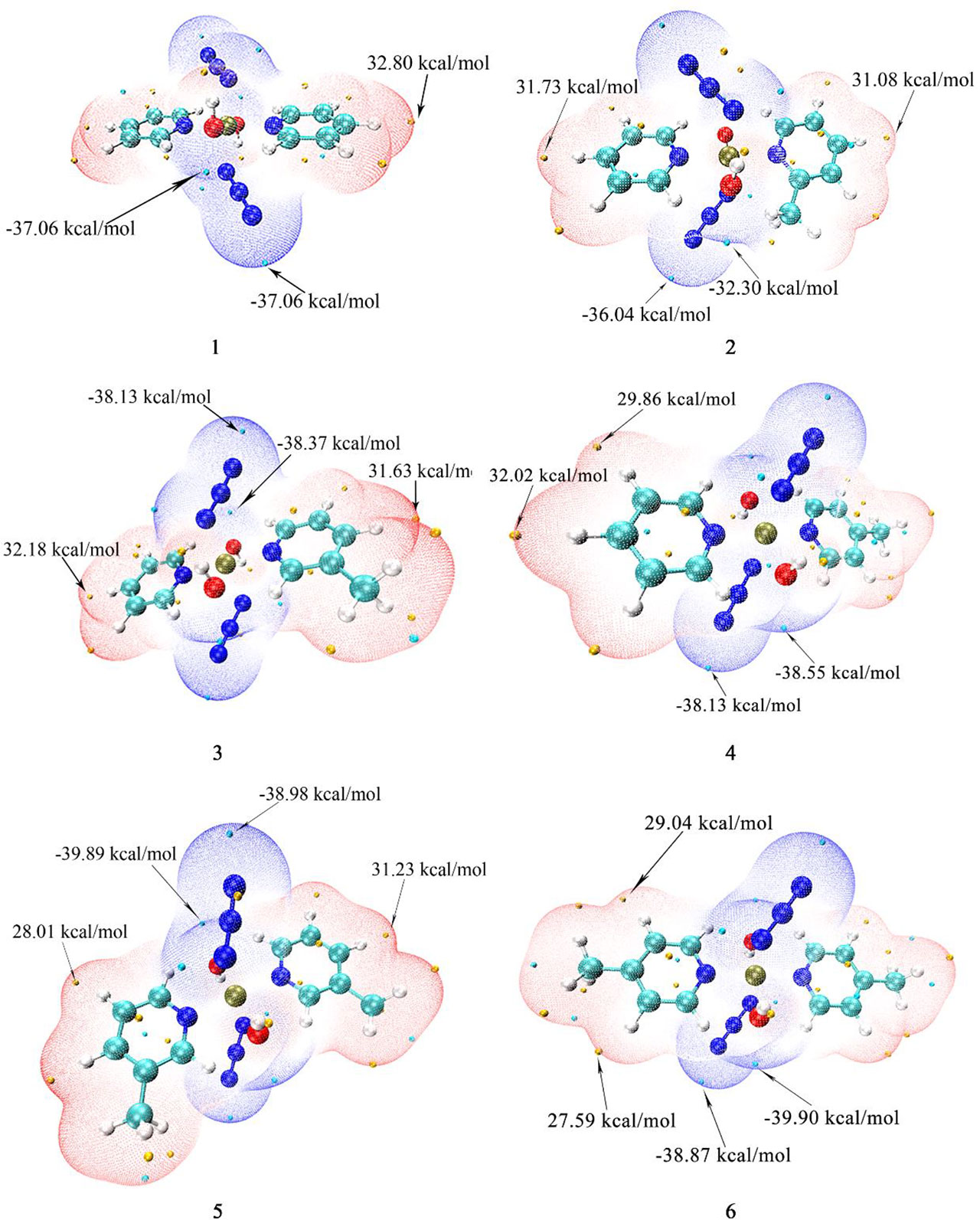
Figure 3 The calculated electrostatic potential of compounds 1, 2, 3, 4, 5 and 6.
For the mono-substituted compounds 2, 3 and 4 with different substitution positions, the minimum points of ESP are -36.04, -38.37 and -38.55 kcal/mol, respectively. So the para-compound 4 has the smallest extreme point for different substituent positions. For 3-picoline compounds 1, 3 and 5, their minimum ESP values are -37.06, -38.37, and -39.89 kcal/mol, respectively. For 4-picoline compounds 1, 4 and 6, their minimum ESP values are -37.06, -38.55 and -39.90 kcal/mol, respectively. Therefore, the ESP value results of the compounds with different numbers of substituents show that the minimum value of ESP decreases due to the increase of the number of substituents.
3.1 Vibrational Assignments
The optimized structure of the selected compounds has been shown in Figure 2. The infrared characteristic peaks of different compounds have been assigned, and Figure 4 has shown their infrared vibration spectra. The infrared vibration frequencies and corresponding vibration intensities of some important groups are summarized in Table 3. All vibrations are obtained by optimized calculations at the LSDA/SDD level. It is worth mentioning that in addition to the strong azido group’s stretching vibration intensity, the vibration modes of other groups also have high peak intensity. Among the selected vibration modes, stretching vibration and bending vibration are the most active vibration modes.
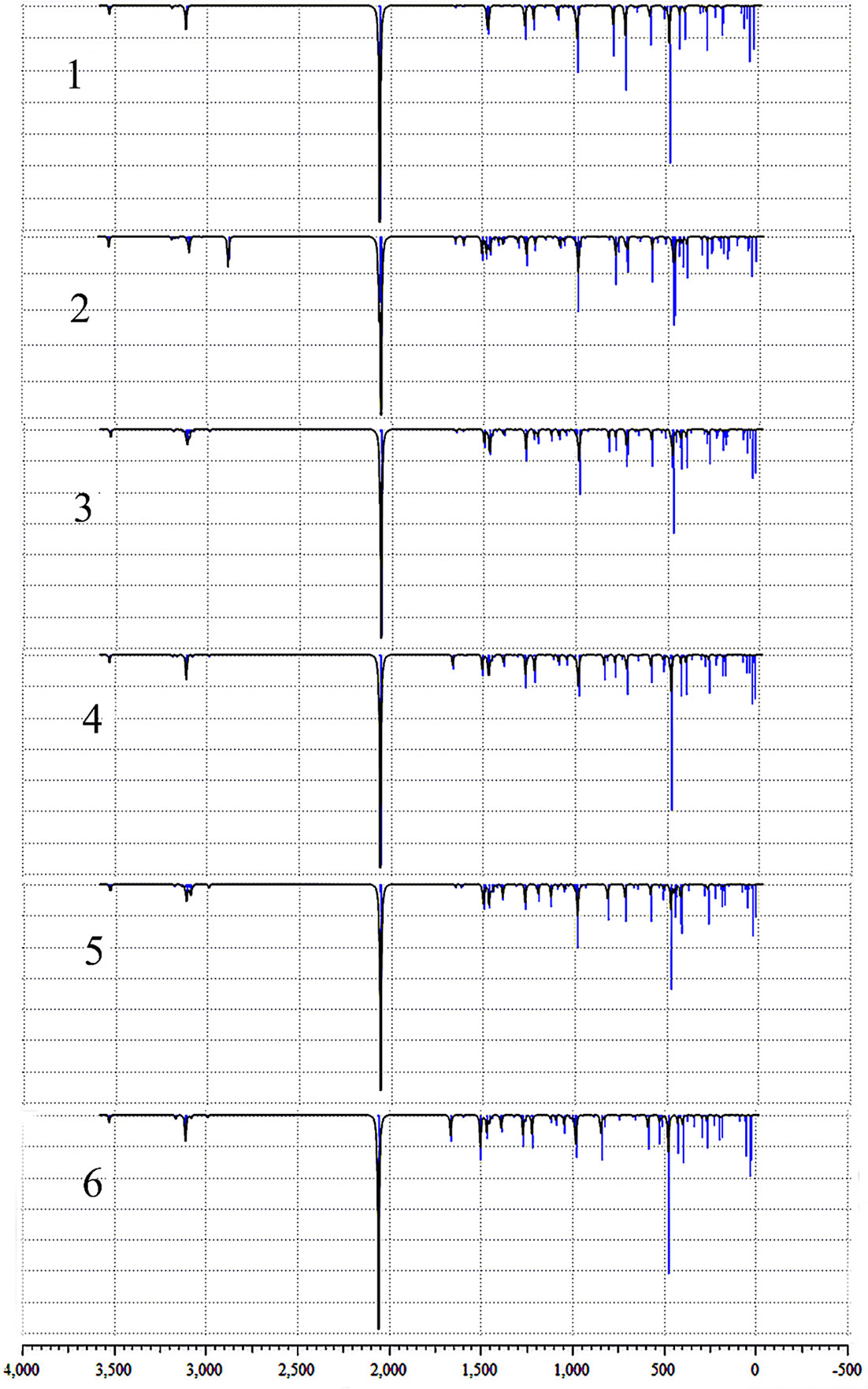
Figure 4 The calculated infrared spectra of the compounds 1, 2, 3, 4, 5 and 6.
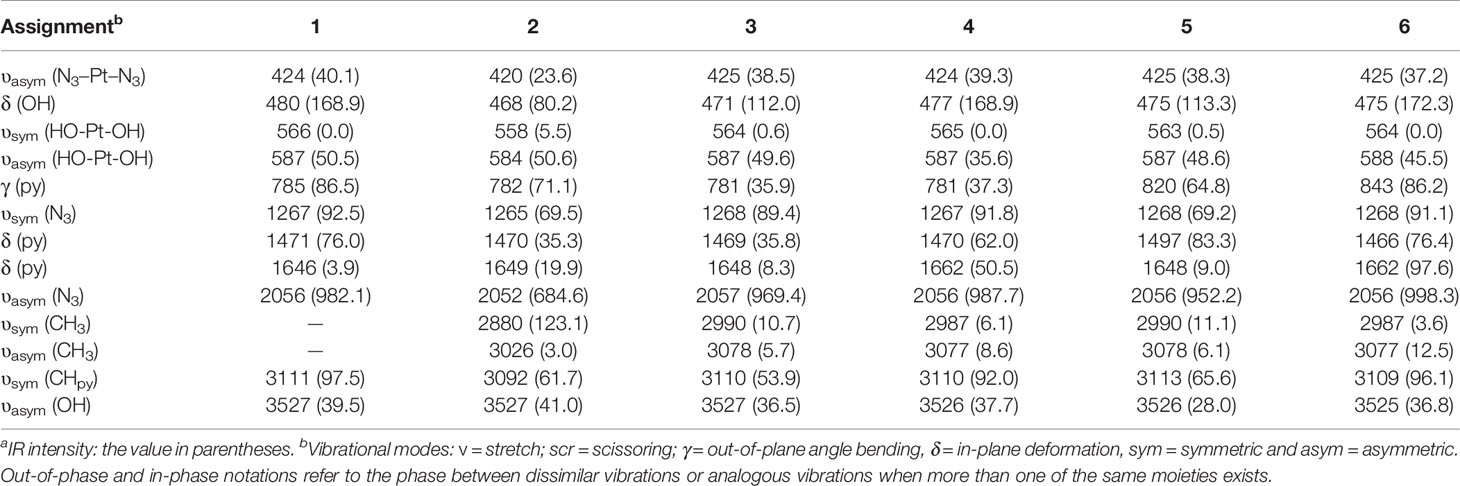
Table 3 Frequencies(cm-1) and infrared intensitiesa(km/mol) calculated for compounds 1, 2, 3, 4, 5 and 6.
3.3.1 Hydroxido Vibrations
In all the compounds, the asymmetric stretching vibration υasym(OH) of the hydroxyl group was observed at 3525-3527 cm-1, which is basically consistent with the previously measured OH stretching vibration (58, 59). In addition, OH in-plane deformation vibration δ(OH) was observed at 468-480 cm-1. After the pyridine group of compound 1 was replaced by picoline, the positions of the other complexes only shifted slightly (compounds 2 and 3 shifted around 10 cm-1, the compounds 4, 5 and 6 shifted around 5 cm-1). For the asymmetric stretching vibration υasym(HO-Pt-OH) between the metal Pt atom and the hydroxyl ligand, compound 2 was observed to appear at 584 cm-1, and the compounds 1, 3, 4, 5, 6 were observed at 587-588 cm-1. Therefore, in this vibration mode, the differently substituted picoline has little effect on the infrared absorption position. The symmetric stretching vibration υsym(HO-Pt-OH) between the metal Pt atom and the hydroxyl ligand is observed at 263-566 cm-1 (except for compound 2 at 558 cm-1). However, it is worth noting that the infrared absorption intensity of compounds 1, 2, 3, 4, 5, 6 in the υsym(HO-Pt-OH) vibration mode is not strong.
3.3.1 Azido Vibrations
The azido asymmetric stretching vibration υasym(N3) has the strongest absorption intensity peak in all vibration modes. Except for the slight shift of the wavenumber of compound 2 (2052 cm-1), the calculated wavenumbers of other compounds are all around 2056 cm-1. Another vibration mode of the azido group is symmetric stretching υsym(N3), and their absorption peaks are observed at 1265-1268 cm-1. In addition, the υasym(N3–Pt–N3) absorption peaks of compounds 1, 3, 4, 5, 6 were observed at 420-425 cm-1.
The azido asymmetric stretching vibration υasym(N3) has the strongest absorption intensity peak in all vibration modes. Except for the slight shift of the wavenumber of compound 2 (2052 cm-1), the calculated wavenumbers of other compounds are all around 2056 cm-1. Another vibration mode of the azido group is symmetric stretching υsym(N3), and their absorption peaks are observed at 1265-1268 cm-1. In addition, the υasym(N3–Pt–N3) absorption peaks of compounds 1, 3, 4, 5, 6 were observed at 420-425 cm-1.
3.3.3 Pyridine Vibrations
There have been previous studies on the vibrational spectroscopy of pyridine compounds bound to different metals (60, 61). There are many types of in-plane deformation vibration of pyridine groups δ(py), one of which is that the wavenumbers of compounds 1, 2, 3 and 5 are 1646-1649 cm-1. However, the corresponding vibration peaks of the two 4-picoline compounds 4 and 6 have a similar shift, and their peaks are observed at 1662 cm-1 and 1662 cm-1. Therefore, for compounds 1, 4 and 6 with different amounts of 4-picoline, the absorption wavenumber of these compounds increased after adding 4-picoline. For another δ(py) vibration, the wavenumbers of the other compounds are concentrated at 1466-1471 cm-1 except for the deviation of compound 5(1497 cm-1). The reason for the shift is that the methyl of the compound 5 is working, and the similar wavenumber is observed in the δ(3-pic) of the compound 3. For the out-of-plane angle bending vibration γ(py) of the pyridine group, the vibrations of compounds 1, 2, 3, 4, 5 are observed to be distributed at 781-785 cm-1, and the vibrations wavenumber of compounds 5 and 6 are respectively observed at 820 cm-1 and 843 cm-1. Therefore, for the influence of picoline substitution on the wavenumber of γ(py), the unsubstituted and monosubstituted compounds only have a small deviation, but the disubstituted compounds 5 and 6 have a larger deviation. For the wavenumber of γ(py) vibration, unsubstituted compound=mono-substituted compounds<di-substituted compounds, the wavenumber of double-substituted compounds has increased a lot. That is because the methyl group is added to the unsubstituted pyridine, which increases the wavenumber of the out-of-plane bending vibration of the compounds 5 and 6.
3.3.4 Methyl Vibrations and C-H Vibrations
For the compounds 2, 3, 4, 5 and 6, the introduction of picoline allowed the methyl characteristic absorption υsym(CH3) and υasym(CH3) to be observed. The wavenumber of the υsym(CH3) of compound 2 was observed at 2880 cm-1, and the wavenumbers of the other compounds (2987-2990 cm-1) shifted by about 100 cm-1. In addition, the infrared absorption intensity of the compound 2 is higher than other compounds for this vibration mode. For the methyl asymmetric stretching vibration υasym(CH3) on the pyridine group, the wavenumbers of compounds 2, 3, 4, 5, and 6 are observed at 3026, 3078,3077, 3078, and 3077 cm-1, respectively. The two C-H bonds connected to the N atom on the pyridine have a strong infrared absorption intensity in their symmetric stretching vibration υsym(CHpy). The absorption wavenumber of compound 2 is 3092 cm-1, while the compounds 1, 3, 4, 5, 6 have no significant shift (3109-3113 cm-1). The υ(CH3) vibration wavenumbers of compounds 2, 3, 4, 5, and 6 have been increased after the CH3 group forms a bond with pyridine. However, due to the steric hindrance of the compound 2, the vibration wavenumber of υ(CH3) of the compound 2 is smaller than that of compounds 1, 3, 4, 5, 6. Therefore, the vibrational spectral distribution of the ortho-picoline compound 2 is most affected by the substituent effect.
3.4 HOMO–LUMO Analysis
The highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) respectively represent the ability to provide electrons and the ability to obtain electrons (62). The energy gap between HOMO and LUMO determines the charge transfer effect in the molecule (63, 64). The energy gap of the compounds selected in this article is calculated using the LSDA/SDD level (Table 4, Figure 5). The energy gaps of compounds 1 and 3 are 2.13121 eV and 2.14264 eV, which are the two smaller energy gaps of all the compounds. The compound 2 has the largest energy gap value(2.23788 eV). The HOMO-LUMO transition means that the electron density of compound 2 is transferred from OH to N3, and compounds 1, 3, 4, 5 and 6 all have electron density transfer from N3 to py. It is only worth noting that the electron density transfer caused by the HOMO-LUMO transition of compounds 5 and 6 is not obvious, and their energy gap is second only to compound 2(2.16985 and 2.18618 eV).
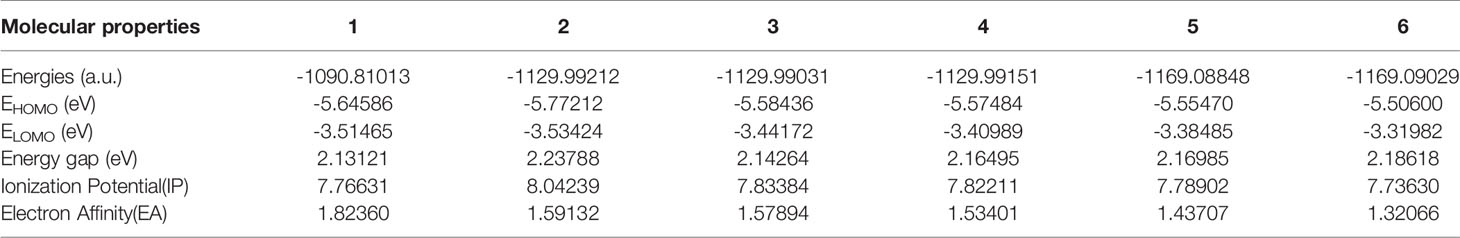
Table 4 HOMO–LUMO energy gaps and related molecular properties of the compounds 1, 2, 3, 4, 5 and 6.
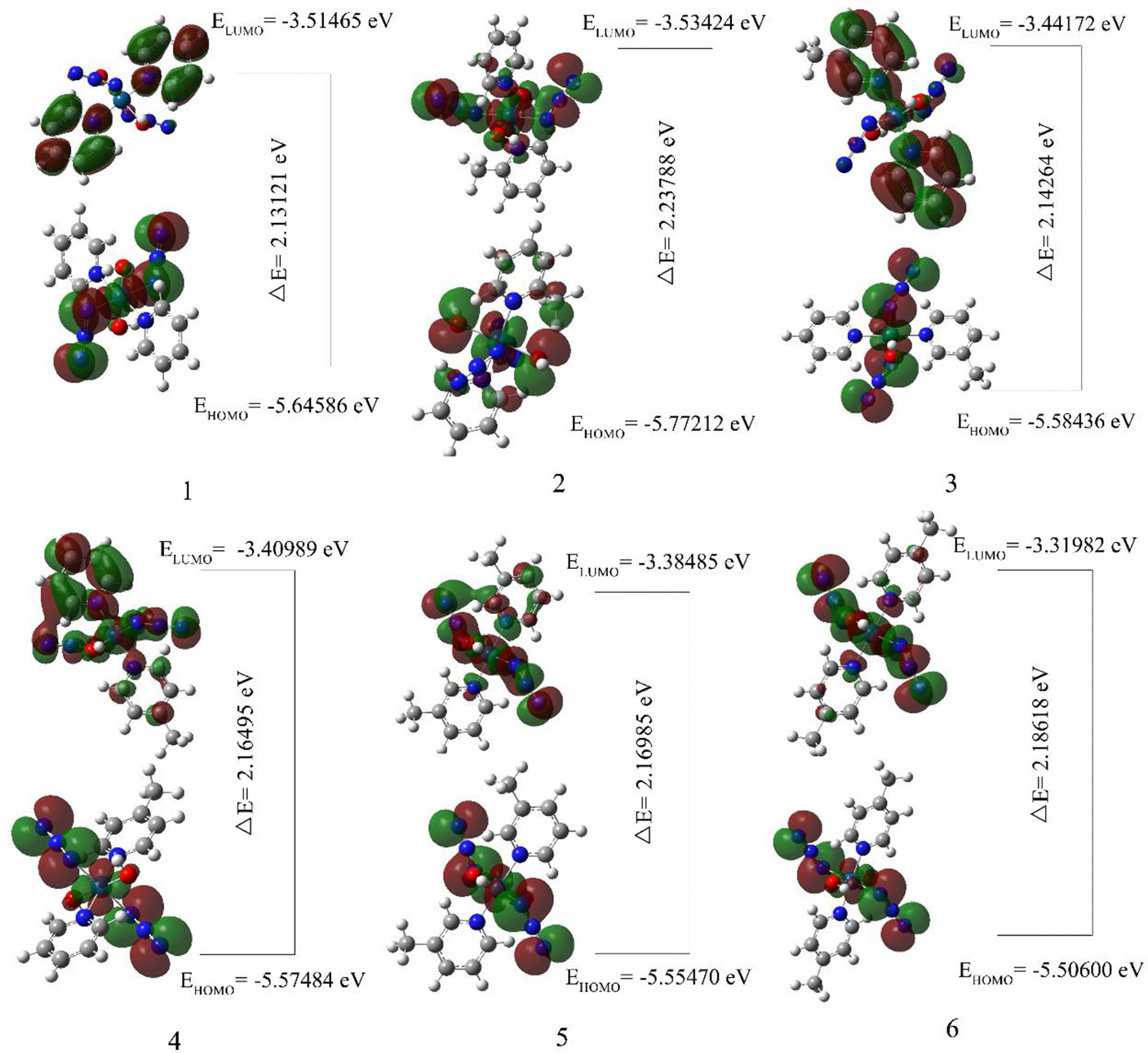
Figure 5 The calculated molecular orbital HOMO-LUMO of compounds 1, 2, 3, 4, 5 and 6.
The analysis of the mono-substituted compounds 2, 3 and 4 shows that the energy gap is 2>4>3. Therefore, the reactivity of these analogs is meta-compound>pair-compound>ortho-compound. The analysis of different numbers of substituents shows that the order of the energy gap of the compounds is 1>3>5. Di-3-picoline compound 5 has the highest reactivity, followed by mono-3-picoline compound 3, and non-picoline compound 1 is the smallest. Similarly, the order of reactivity of different numbers of 4-picoline compounds is also 1>4>6. Therefore, as the number of substituents in the compounds increases, the energy gap value of the compounds decreases.
DFT is not enough to satisfy Koopmans Theorem due to the difference between functionals, which results in the calculated vertical ionization potential (VIP) value not approximately equal to -E(HOMO) and vertical electron affinity (VEA) not approximately equal to -E(LUMO) (65). The electron affinity and ionization potential data in Table 4 are calculated using the LSDA functional method and the SDD basis set. The electron affinities corresponding to compounds 1, 2, 3, 4, 5 and 6 are 1.82360, 1.59132, 1.57894, 1.53401, 1.43707 and 1.32066 eV, and their ionization potentials are 7.76631, 8.04239, 7.83384, 7.82211, 7.778902 and 7.73630 eV respectively. For all the compounds, the electron affinity values of compounds 2, 3 and 4 are almost the same, which may be due to the fact that they are isomers. The value of the electron affinity of the compound 1 is the largest, which means that it has the greatest ability to gain an electron compared with other compounds. The electron affinity of compounds 5 and 6 indicates that they have poor electron-acquiring ability compared with other compounds. Therefore, the order of the ability to obtain electrons is unsubstituted compound>monosubstituted compounds>disubstituted compounds. The order of the ionization potential is 2>3>4>5>1>6. So compound 2 is the least easily ionized among all compounds. The compound 2 is easily reduced by obtaining an electron, which may be the reason for its large dark toxicity. It is worth noting that compound 6 has the largest phototoxic index (PI, PI is the ratio of cytotoxicity after irradiation to cytotoxicity in the dark) (29). The small IP and EA of compound 6 show that it neither loses nor gains electrons easily, and shows good stability in all compounds.
In different mono-substituent compounds 2, 3 and 4, the order of IP and EA values is the 2>3>4. Therefore, the ability of mono-substituted compounds to obtain electrons is ortho-compound>meta-compound>para-compound. The IP values of compounds with different numbers of 3-picoline substituents is 3>5>1, and the IP values of compounds with different numbers of 4-picoline substituents is 4>1>6. The EA values of different numbers of picoline substituents are 1>3>5 and 1>4>6, which indicates that the electron-acquiring ability of the compounds decreases after the number of their picoline substituents increases. The ability to be reduced after obtaining electrons is unsubstituted compound>monosubstituted compounds>disubstituted compounds.
3.5 Mulliken and Natural Population Analysis
Mulliken charges and natural atomic charges analysis is a good way to view the charge distribution between the atoms of a molecule. They can also indicate the positive and negative charges (electron donor and electron acceptor), dipole moment and polarizability of the selected compounds (66, 67). The distributions of Mulliken charges and natural atomic charges calculated using LSDA/SDD levels are shown in Table 5. The comparison of the charge distributions of the two methods is shown in Figure 6.
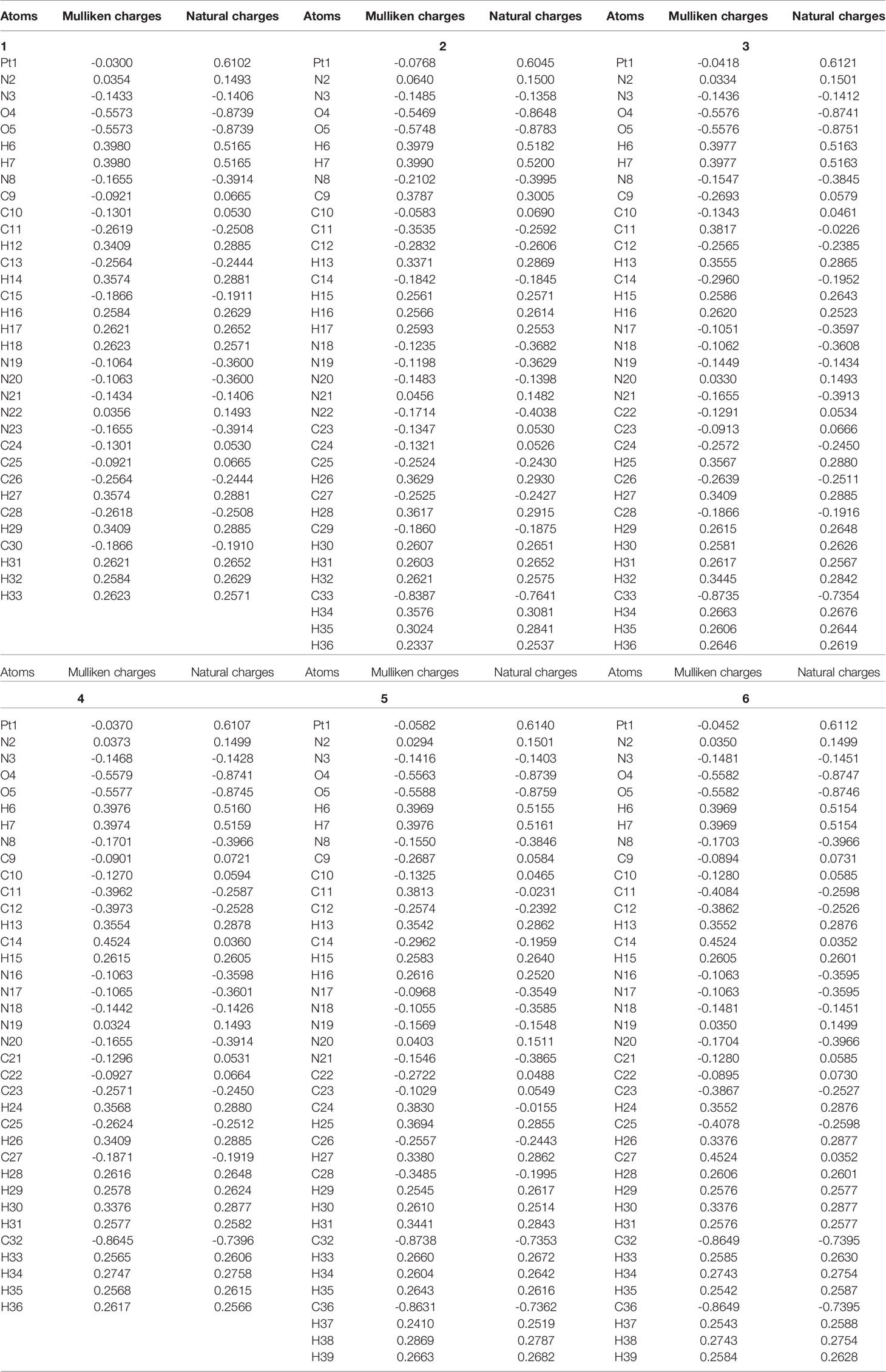
Table 5 The calculated Mulliken charges and natural atomic charges of compounds 1, 2, 3, 4, 5 and 6.
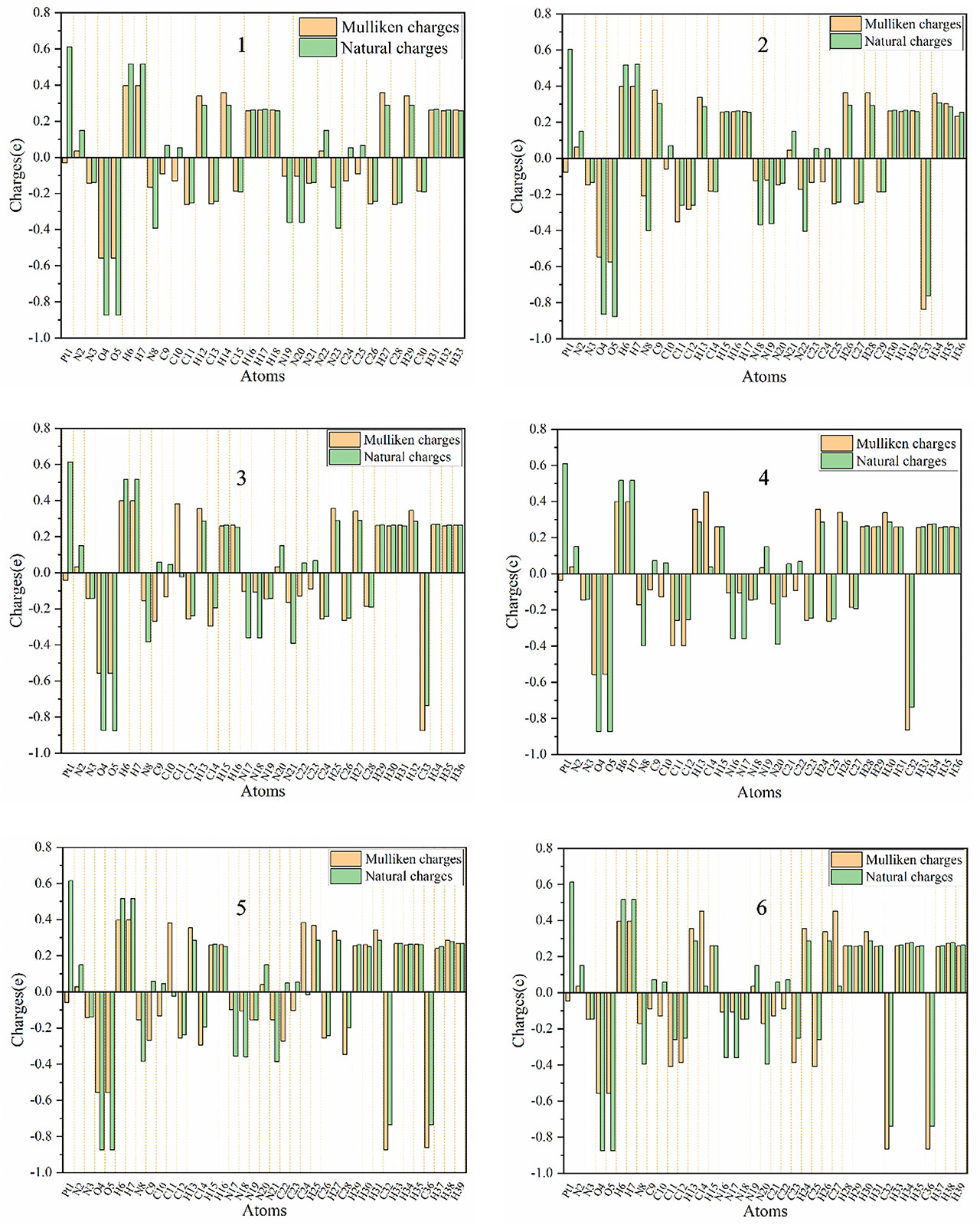
Figure 6 Distribution of calculated Mulliken charges and natural atomic charges for compounds 1, 2, 3, 4, 5 and 6.
The calculated natural charge shows that the Pt atoms of the selected compounds have positive charge, and the positive charge is the largest among all the atoms (0.6045-0.6140 e). Among all the compounds, the smallest Pt positive charge, the largest N(Pt-N3) negative charge and the larger R(Pt – NN3) show that compound 30 is easier to be reduced to Pt(IV) than other compounds (68) All H atoms are also positively charged (0.2514-0.5200 e). The O atoms of the hydroxyl groups of all compounds have negative charge (between -0.8648 e and -0.8783 e). Except for the N(N3) atoms in the middle of the azido group have positive charge, all other N atoms(the N(N3) atoms at both ends of the azido group, and the N(py) atom of pyridyl group connected to the Pt atom) have negative charge.
The C11, C26 and C28 (corresponding atoms of other compounds) of compound 1 all have negative charge, and its C9, C10, C24 and C25 (corresponding atoms of other compounds) all have positive charges. And the methyl C atoms of compounds 2, 3, 4, 5, and 6 are all negatively charged. And the CCH3 atoms of compounds 2, 3, 4, 5 and 6 all have negative charge. The C13 of the compound 1 has positive charge, while the other compounds have negative charge. The charge distribution of the para-C atoms of the py group is different between different compounds. The C15 of compound 1 (the corresponding para-C atoms of compounds 2, 3 and 5) have negative charge, while the corresponding atoms of compounds 4 and 6 have positive charge. The C30 of compounds 1, 2, 3, 4 and 5 all have negative charge, while the compound 6 has positive charge. Therefore, the main difference between them comes from the addition of a methyl group, which often results in the decrease of the negative charge of the C atom or the conversion of the C atom from negative to positive.
3.6 Natural Bond Orbital Analysis
Natural bond orbital(NBO) analysis explains the Lewis structure, bond type, hybridization type, and the strength of the orbital interaction between the electron donor(i) and the electron acceptor(j) (69). In this article, NBO analysis of selected compounds is performed at the LSDA/SDD level. The strength of the i-j interaction is estimated by the magnitude of the second-order perturbation interaction energy E(2), which is derived from the second-order perturbation method:
Where qi is the donor orbital occupancy, εi and are the diagonal elements orbital energies, and F(i,j) is the off-diagonal NBO Fock matrix element. The larger E(2) indicates the stronger the interaction, the greater the donation tendency between the electron donor and the electron donor, and the greater the degree of conjugation of the entire system. The delocalization of the electron density between the bonded/lone paired NBO orbital and the antibonded NBO orbital corresponds to a stable donor-acceptor interaction (70). The second-order perturbation stability energy E(2) (kcal/mol) of the compound 1 discussed in this article is listed in Table 6. The NBO information of other compounds is basically the same as that of the compound 1 (Table S1, Figure S2). The N(N3) bonding donor of compound 11 σ(Pt1-N19) to the anti-bonding acceptor σ*(Pt1-O4), σ*(Pt1-N8) and σ*(Pt1-N19) lead to large σ-σ* stability energy. Their stability energies are 114.62, 105.57 and 43.66 kcal/mol, respectively. The stability energy of the bond donor σ(Pt1-N19) to the anti-bond acceptor π*(N21-N22) is 39.22 kcal/mol. In addition, there is also a strong interaction between the bond donor σ(Pt1-N8) of N(py) and the anti-bond acceptor σ*(Pt1-O4)、σ*(Pt1-N8) and σ*(Pt1-N19). The E(2) are respectively 42.76, 16.27 and 36.74 kcal/mol. Another larger π-π* interaction stability energy related to the degree of conjugation occurs in pyridine. The bond donor π(C26-C30) to the anti-bonding acceptor π*(N23-C24) and π*(C25-C28), their interaction stability energies E(2) are 28.24 and 13.44 kcal/mol. The interaction energy between the bond donor π(N23-C24) and the anti-bond acceptor π*(C25-C28) is 17.11 kcal/mol. In addition, the magnitude of the interaction energy from the lone pair of electrons LP(N19) to π*(N21-N22) is 125.92 kcal/mol, and the large E(2) value shows the electron donation in the N3 group.
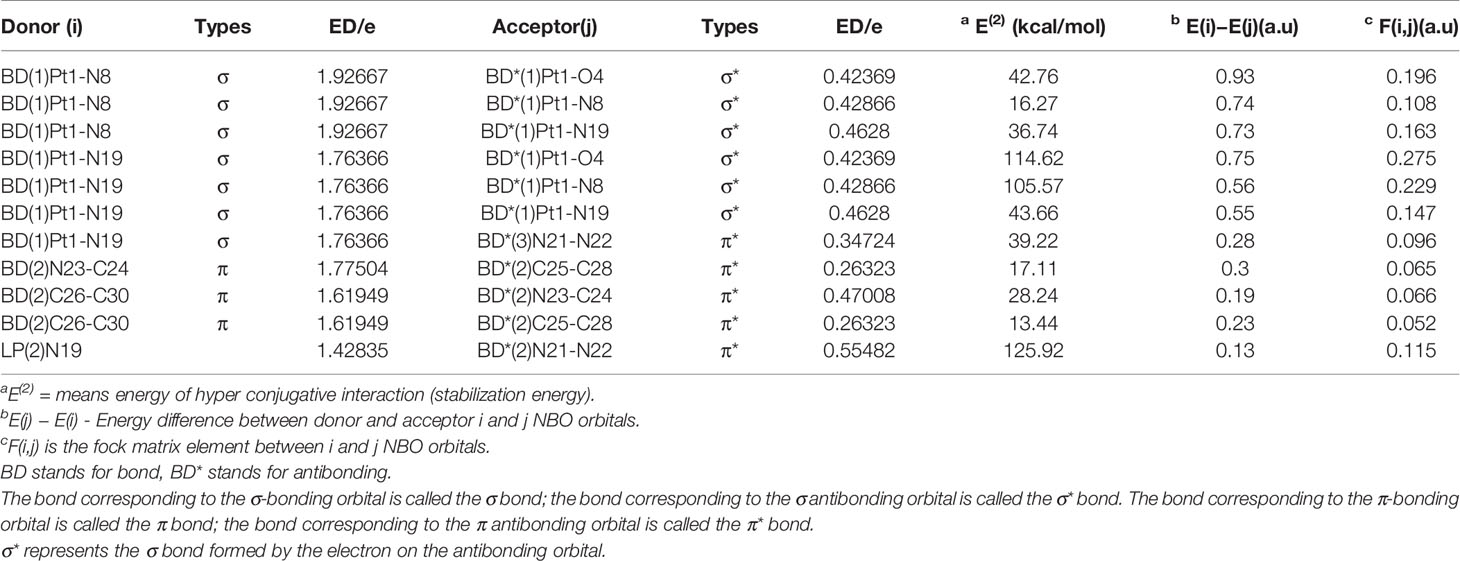
Table 6 Second-order perturbation theory analysis of Fock matrix in NBO basis for compound 1.
4 Conclusion
The azido picoline compound is a promising anti-tumor compound. In this work, for the first time, we have shown the structural information and infrared spectrum information of the azido picoline compounds. The predicted structural characteristics can be used as a theoretical basis for screening candidates in the future, and the infrared spectrum information accurately identifies the characteristic absorption peaks of the compounds. HOMO-LUMO, NBO analysis and the distribution of natural atomic charges are calculated to study the electronic properties of these compounds. In the later work, we will explore the interaction between these compounds and small biological molecules.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Author Contributions
MM performed the statistical analysis, organized the database and wrote the manuscript. HG contributed to conception and design of the study. All authors contributed to the article and approved the submitted version.
Funding
This work was financially supported by Natural Science Foundation of Shandong, China [Grant No. ZR2019MC004], the High-end Talent Team Construction Foundation [Grant No. 108-10000318], the Cooperation Project of University and Local Enterprise in Yantai of Shandong Province (2021XDRHXMXK23) and the High-end Full-time Innovative Talent Introduction Foundation "two-hundred plans" of Yantai.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2021.749178/full#supplementary-material
References
1. Rosenberg B, Vancamp L, Krigas T. Inhibition of Cell Division in Escherichia Coli by Electrolysis Products From a Platinum Electrode. Nature (1965) 205:698–9. doi: 10.1038/205698a0
2. Barnham KJ, Djuran MI, Murdoch P, Ranford JD, Sadler PJ. Ring-Opened Adducts of the Anticancer Drug Carboplatin With Sulfur Amino Acids. Inorganic Chem (1996) 35:1065–72. doi: 10.1021/ic950973d
3. Kelland L. The Resurgence of Platinum-Based Cancer Chemotherapy. Nat Rev Cancer (2007) 7:573–84. doi: 10.1038/nrc2167
4. Raymond E, Faivre S, Woynarowski JM, Chaney SG. Oxaliplatin: Mechanism of Action and Antineoplastic Activity. Semin Oncol (1998) p 4–12. Available at: https://europepmc.org/article/MED/9609103
5. Park SB, Lin CSY, Krishnan AV, Goldstein D, Friedlander ML, Kiernan MC. Long-Term Neuropathy After Oxaliplatin Treatment: Challenging the Dictum of Reversibility. The Oncologist (2019) 16(2011):708–16. doi: 10.1634/theoncologist.2010-0248
6. Kelland LR. The Molecular Basis of Cisplatin Sensitivity/Resistance. Eur J Cancer (1994) 30:725–7. doi: 10.1016/0959-8049(94)90281-X
7. Fei Chin C, Yuan Qiang Wong D, Jothibasu R, Han Ang W. Anticancer Platinum (IV) Prodrugs With Novel Modes of Activity. Curr Topics Med Chem (2011) 11:2602–12. doi: 10.2174/156802611798040778
8. Johnstone TC, Suntharalingam K, Lippard SJ. The Next Generation of Platinum Drugs: Targeted Pt(II) Agents, Nanoparticle Delivery, and Pt(IV) Prodrugs. Chem Rev (2016) 116:3436–86. doi: 10.1021/acs.chemrev.5b00597
9. Bera A, Gautam S, Raza MK, Kondaiah P, Chakravarty AR. Oxoplatin-B, a Cisplatin-Based Platinum(IV) Complex With Photoactive BODIPY for Mitochondria Specific “Chemo-PDT” Activity. J Inorganic Biochem (2021) 223:111526. doi: 10.1016/j.jinorgbio.2021.111526
10. Sun Y, Yin E, Tan Y, Yang T, Song D, Jin S, et al. Immunogenicity and Cytotoxicity of a Platinum(Iv) Complex Derived From Capsaicin. Dalton Trans (2021) 50:3516–22. doi: 10.1039/D0DT03470C
11. Deng Z, Li C, Chen S, Zhou Q, Xu Z, Wang Z, et al. An Intramolecular Photoswitch Can Significantly Promote Photoactivation of Pt(iv) Prodrugs. Chem Sci (2021) 12:6536–42. doi: 10.1039/D0SC06839J
12. Allison RR, Moghissi K. Photodynamic Therapy (PDT): PDT Mechanisms. Clin Endosc (2013) 46:24–9. doi: 10.5946/ce.2013.46.1.24
13. Wilson WR, Hay MP. Targeting Hypoxia in Cancer Therapy. Nat Rev Cancer (2011) 11:393–410. doi: 10.1038/nrc3064
14. Van Rixel V, Siewert B, Hopkins S, Askes S, Busemann A, Siegler M, et al. Green Light-Induced Apoptosis in Cancer Cells by a Tetrapyridyl Ruthenium Prodrug Offering Two Trans Coordination Sites. Chem Sci (2016) 7:4922–9. doi: 10.1039/C6SC00167J
15. Farrer NJ, Salassa L, Sadler PJ. Photoactivated Chemotherapy (PACT): The Potential of Excited-State D-Block Metals in Medicine. Dalton Trans (2009), 10690–701. doi: 10.1039/b917753a
16. Lameijer LN, Ernst D, Hopkins SL, Meijer MS, Askes SH, Le Dévédec SE, et al. A Red-Light-Activated Ruthenium-Caged NAMPT Inhibitor Remains Phototoxic in Hypoxic Cancer Cells. Angewandte Chemie (2017) 129:11707–11. doi: 10.1002/ange.201703890
17. Ronconi L, Sadler PJ. Photoreaction Pathways for the Anticancer Complex Trans,Trans,Trans-[Pt(N3)2(OH)2(Nh3)2]. Dalton Trans (2011) 40:262–8. doi: 10.1039/C0DT00546K
18. Wilson JJ, Lippard SJ. Synthetic Methods for the Preparation of Platinum Anticancer Complexes. Chem Rev (2013) 114:4470–95. doi: 10.1021/cr4004314
19. Shi H, Imberti C, Sadler PJ. Diazido Platinum(Iv) Complexes for Photoactivated Anticancer Chemotherapy. Inorganic Chem Front (2019) 6:1623–38. doi: 10.1039/C9QI00288J
20. Hollis LS, Amundsen AR, Stern EW. Chemical and Biological Properties of a New Series of Cis-Diammineplatinum(II) Antitumor Agents Containing Three Nitrogen Donors: Cis-[Pt(NH3)2(N-Donor)Cl]+. J Med Chem (1989) 32:128–36. doi: 10.1021/jm00121a024
21. Holford J, Sharp S, Murrer B, Abrams M, Kelland L. In Vitro Circumvention of Cisplatin Resistance by the Novel Sterically Hindered Platinum Complex AMD473. Br J Cancer (1998) 77:366–73. doi: 10.1038/bjc.1998.59
22. Wang D, Zhu G, Huang X, Lippard SJ. X-Ray Structure and Mechanism of RNA Polymerase II Stalled at an Antineoplastic Monofunctional Platinum-DNA Adduct. Proc Natl Acad Sci (2010) 107:9584–9. doi: 10.1073/pnas.1002565107
23. Mu M, Zhan J, Dai X, Gao H. Research Progress of Azido-Containing Pt(IV) Antitumor Compounds. Eur J Medicinal Chem (2022) 227:113927. doi: 10.1016/j.ejmech.2021.113927
24. Mackay FS, Woods JA, Heringova P, Kasparkova J, Pizarro AM, Moggach SA, et al. A Potent Cytotoxic Photoactivated Platinum Complex. Proc Natl Acad Sci USA (2007) 104:20743–8. doi: 10.1073/pnas.0707742105
25. Farrer NJ, Woods JA, Munk VP, Mackay FS, Sadler PJ. Photocytotoxic Trans-Diam(m)ine Platinum(IV) Diazido Complexes More Potent Than Their Cis Isomers. Chem Res Toxicol (2010) 23:413–21. doi: 10.1021/tx900372p
26. Westendorf AF, Woods JA, Korpis K, Farrer NJ, Salassa L, Robinson K, et al. Trans,trans,trans-[PtIV(N3)2(OH)2(py)(NH3)]: A Light-Activated Antitumor Platinum Complex That Kills Human Cancer Cells by an Apoptosis-Independent Mechanism. Mol Cancer Ther 11 (2012) 11(9):1894–904. doi: 10.1158/1535-7163.MCT-11-0959
27. Pracharova J, Zerzankova L, Stepankova J, Novakova O, Farrer NJ, Sadler PJ, et al. Interactions of DNA With a New Platinum(IV) Azide Dipyridine Complex Activated by UVA and Visible Light: Relationship to Toxicity in Tumor Cells. Chem Res Toxicol (2012) 25:1099–111. doi: 10.1021/tx300057y
28. Pizarro AM, McQuitty RJ, Mackay FS, Zhao Y, Woods JA, Sadler PJ. Cellular Accumulation, Lipophilicity and Photocytotoxicity of Diazido Platinum(IV) Anticancer Complexes. ChemMedChem (2014) 9:1169–75. doi: 10.1002/cmdc.201402066
29. Shaili E, Salassa L, Woods JA, Clarkson G, Sadler PJ, Farrer NJ. Platinum(iv) Dihydroxido Diazido N-(Heterocyclic)Imine Complexes Are Potently Photocytotoxic When Irradiated With Visible Light. Chem Sci (2019) 10:8610–7. doi: 10.1039/C9SC02644D
30. Malik M, Wysokiński R, Zierkiewicz W, Helios K, Michalska D. Raman and Infrared Spectroscopy, DFT Calculations, and Vibrational Assignment of the Anticancer Agent Picoplatin: Performance of Long-Range Corrected/Hybrid Functionals for a Platinum(II) Complex. J Phys Chem A (2014) 118:6922–34. doi: 10.1021/jp5056254
31. Malik M, Michalska D. Assessment of New DFT Methods for Predicting Vibrational Spectra and Structure of Cisplatin: Which Density Functional Should We Choose for Studying Platinum(II) Complexes? Spectrochimica Acta Part A: Mol Biomolecular Spectrosc (2014) 125:431–9. doi: 10.1016/j.saa.2014.01.107
32. Torres M, Khan S, Duplanty M, Lozano HC, Morris TJ, Nguyen T, et al. Raman and Infrared Studies of Platinum-Based Drugs: Cisplatin, Carboplatin, Oxaliplatin, Nedaplatin, and Heptaplatin. J Phys Chem A (2018) 122:6934–52. doi: 10.1021/acs.jpca.8b04023
33. Mu M, Gao H. Density Functional Study of Trans,Trans,Trans-[Pt(N3)2(OH)2(Py)2] on Molecular Structure and Vibrational Spectroscopy. Spectrochimica Acta Part A: Mol Biomolecular Spectrosc (2021) 261:120022. doi: 10.1016/j.saa.2021.120022
34. Perdew JP, Burke K, Ernzerhof M. Generalized Gradient Approximation Made Simple. Phys Rev Lett (1998) 77:3865–8. doi: 10.1103/PhysRevLett.77.3865
35. Becke AD. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J Chem Phys (1993) 98:5648–52. doi: 10.1063/1.464913
36. Hay PJ, Wadt WR. Ab Initio Effective Core Potentials for Molecular Calculations. Potentials for the Transition Metal Atoms Sc to Hg. J Chem Phys (1985) 82:270–0. doi: 10.1063/1.448799
37. Slater JC, Phillips JC. Quantum Theory of Molecules and Solids Vol. 4: The Self-Consistent Field for Molecules and Solids. Physics Today (1974) 27(1974):49–50. doi: 10.1063/1.3129035
38. Vosko SH, Wilk L, Nusair M. Accurate Spin-Dependent Electron Liquid Correlation Energies for Local Spin Density Calculations: A Critical Analysis. Can J Phys (1980) 58:1200–11. doi: 10.1139/p80-159
39. Fuentealba P, Stoll H, Szentpaly LV, Schwerdtfeger P, Preuss H. On the Reliability of Semi-Empirical Pseudopotentials: Simulation of Hartree-Fock and Dirac-Fock Results. J Phys B Atomic Mol Phys (1983) 16:L323. doi: 10.1088/0022-3700/16/11/001
40. Andrae D, H?U?Ermann U, Dolg M, Stoll H, Preu H. Energy-Adjustedab Initio Pseudopotentials for the Second and Third Row Transition Elements. Theoretica Chimica Acta (1990) 77:123–41. doi: 10.1007/BF01114537
41. Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, et al. Gaussian 16 Rev. C.01. Wallingford, CT (2016).
42. Lu T, Chen F. Multiwfn: A Multifunctional Wavefunction Analyzer. J Comput Chem (2012) 33:580–92. doi: 10.1002/jcc.22885
43. Humphrey W, Dalke A, Schulten K. VMD: Visual Molecular Dynamics. J Mol Graphics (1996) 14:33–8. doi: 10.1016/0263-7855(96)00018-5
44. Quiroga AG, Pérez JM, Alonso C, Navarro-Ranninger C, Farrell N. Novel Transplatinum(II) Complexes With [N2O2] Donor Sets. Cellular Pharmacology and Apoptosis Induction in Pam 212-Ras Cells. J Med Chem (2006) 49:224–31. doi: 10.1021/jm050804v
45. Shushakov AA, Pozdnyakov IP, Grivin VP, Plyusnin VF, Vasilchenko DB, Zadesenets AV, et al. Primary Photochemical Processes for Pt(iv) Diazido Complexes Prospective in Photodynamic Therapy of Tumors. Dalton Trans (2017) 46:9440–50. doi: 10.1039/C7DT01529A
46. Zhao Y, Farrer NJ, Li H, Butler JS, McQuitty RJ, Habtemariam A, et al. De Novo Generation of Singlet Oxygen and Ammine Ligands by Photoactivation of a Platinum Anticancer Complex. Angew Chem Int Ed Engl (2013) 52:13633–7. doi: 10.1002/anie.201307505
47. Battle AR, Choi R, Hibbs DE, Hambley TW. Platinum(IV) Analogues of AMD473 (Cis-[PtCl2(NH3)(2-Picoline)]): Preparative, Structural, and Electrochemical Studies. Inorganic Chem (2006) 45:6317–22. doi: 10.1021/ic060273g
48. Vernooij RR, Joshi T, Horbury MD, Graham B, Izgorodina EI, Stavros VG, et al. Spectroscopic Studies on Photoinduced Reactions of the Anticancer Prodrug, Trans,Trans,Trans-[Pt(N3)2(OH)2(Py)2]. Chem – A Eur J (2018) 24:5790–803. doi: 10.1002/chem.201705349
49. Lau JK-C, Deubel DV. Loss of Ammine From Platinum(II) Complexes: Implications for Cisplatin Inactivation, Storage, and Resistance. Chem – A Eur J (2005) 11:2849–55. doi: 10.1002/chem.200401053
50. Mackay FS, Woods JA, Moseley H, Ferguson J, Dawson A, Parsons S, et al. A Photoactivated Trans-Diammine Platinum Complex as Cytotoxic as Cisplatin. Chemistry (2006) 12:3155–61. doi: 10.1002/chem.200501601
51. Farrer NJ, Woods JA, Salassa L, Zhao Y, Robinson KS, Clarkson G, et al. A Potent Trans-Diimine Platinum Anticancer Complex Photoactivated by Visible Light. Angew Chem Int Ed Engl (2010) 49:8905–8. doi: 10.1002/anie.201003399
52. Lu T, Manzetti S. Wavefunction and Reactivity Study of Benzo[a]Pyrene Diol Epoxide and Its Enantiomeric Forms. Struct Chem (2014) 25:1521–33. doi: 10.1007/s11224-014-0430-6
53. Sun H, Cao B, Tian Q, Liu S, Du D, Xue Z, et al. A DFT Study on the Absorption Mechanism of Vinyl Chloride by Ionic Liquids. J Mol Liquids (2016) 215:496–502. doi: 10.1016/j.molliq.2016.01.026
54. Cao B, Du J, Cao Z, Sun X, Sun H, Fu H. DFT Study on the Dissolution Mechanisms of α-Cyclodextrin and Chitobiose in Ionic Liquid. Carbohydr Polymers (2017) 169:227–35. doi: 10.1016/j.carbpol.2017.04.012
55. Li H, Zhou P, Zhang J, Li D, Li X, Gao X. A Theoretical Guide for Screening Ionic Liquid Extractants Applied in the Separation of a Binary Alcohol-Ester Azeotrope Through a DFT Method. J Mol Liquids (2018) 251:51–60. doi: 10.1016/j.molliq.2017.12.049
56. Zhang Y, Wang H, Sun N, Chi R. Experimental and Computational Study on Mechanism of Dichromate Adsorption by Ionic Liquid-Bonded Silica Gel. Separation Purification Technol (2018) 205:84–93. doi: 10.1016/j.seppur.2018.04.084
57. Ma J, Zhu M, Yang X, Wang Y, Wang B. Different Cation-Anion Interaction Mechanisms of Diamino Protic Ionic Liquids: A Density Functional Theory Study. Chem Phys Lett (2021) 774:138615. doi: 10.1016/j.cplett.2021.138615
58. Hambley TW, Battle AR, Deacon GB, Lawrenz ET, Fallon GD, Gatehouse BM, et al. Modifying the Properties of Platinum(IV) Complexes in Order to Increase Biological Effectiveness. J Inorganic Biochem (1999) 77:3–12. doi: 10.1016/S0162-0134(99)00133-6
59. Johnstone TC, Lippard SJ. Reinterpretation of the Vibrational Spectroscopy of the Medicinal Bioinorganic Synthon C,C,T-[Pt(NH3)2Cl2(OH)2]. JBIC J Biol Inorganic Chem (2014) 19:667–74. doi: 10.1007/s00775-014-1109-6
60. Wu D-Y, Ren B, Jiang Y-X, Xu X, Tian Z-Q. Density Functional Study and Normal-Mode Analysis of the Bindings and Vibrational Frequency Shifts of the Pyridine–M (M = Cu, Ag, Au, Cu+, Ag+, Au+, and Pt) Complexes. J Phys Chem A (2002) 106:9042–52. doi: 10.1021/jp025970i
61. Kumar M, Srivastava M, Yadav RA. Vibrational Studies of Benzene, Pyridine, Pyridine-N-Oxide and Their Cations. Spectrochimica Acta Part A: Mol Biomolecular Spectrosc (2013) 111:242–51. doi: 10.1016/j.saa.2013.03.004
62. Fukui K. Role of Frontier Orbitals in Chemical Reactions. Science (1982) 218:747–54. doi: 10.1126/science.218.4574.747
63. Chaitanya K. Molecular Structure, Vibrational Spectroscopic (FT-IR, FT-Raman), UV–vis Spectra, First Order Hyperpolarizability, NBO Analysis, HOMO and LUMO Analysis, Thermodynamic Properties of Benzophenone 2,4-Dicarboxylic Acid by Ab Initio HF and Density Functional Method. Spectrochimica Acta Part A: Mol Biomolecular Spectrosc (2012) 86:159–73. doi: 10.1016/j.saa.2011.09.069
64. Nataraj A, Balachandran V, Karthick T, Karabacak M, Atac A, FT-Raman. FT-IR, UV Spectra and DFT and Ab Initio Calculations on Monomeric and Dimeric Structures of 3,5-Pyridinedicarboxylic Acid. J Mol Structure (2012) 1027:1–14. doi: 10.1016/j.molstruc.2012.05.048
65. Ranasinghe DS, Margraf JT, Perera A, Bartlett RJ. Vertical Valence Ionization Potential Benchmarks From Equation-of-Motion Coupled Cluster Theory and QTP Functionals. J Chem Phys (2019) 150:074108. doi: 10.1063/1.5084728
66. Khajehzadeh M, Sadeghi N. Molecular Structure, the Effect of Solvent on UV–vis and NMR, FT–IR and FT–Raman Spectra, NBO, Frontier Molecular Orbital Analysis of Mitomycin Anticancer Drug. J Mol Liquids (2018) 256:238–46. doi: 10.1016/j.molliq.2018.01.099
67. Khajehzadeh M, Rajabi M, Rahmaniasl S. Synthesis, Spectroscopic (UV–vis, FT-IR and NMR), Solubility in Various Solvents, X–ray, NBO, NLO and FMO Analysis of (L1) and [(L1)PdCl2] Complex: A Comprehensive Experimental and Computational Study. J Mol Structure (2019) 1175:139–51. doi: 10.1016/j.molstruc.2018.07.084
68. Zhao Y, Woods JA, Farrer NJ, Robinson KS, Pracharova J, Kasparkova J, et al. Diazido Mixed-Amine Platinum(IV) Anticancer Complexes Activatable by Visible-Light Form Novel DNA Adducts. Chem – A Eur J (2013) 19:9578–91. doi: 10.1002/chem.201300374
69. Glendening ED, Landis CR, Weinhold F. Natural Bond Orbital Methods. Wiley Interdiscip Rev Comput Mol Sci (2012) 2:1–42. doi: 10.1002/wcms.51
70. Sert Y, Sreenivasa S, Doğan H, Manojkumar KE, Suchetan PA, Ucun F. FT-IR, Laser-Raman Spectra and Quantum Chemical Calculations of Methyl 4-(Trifluoromethyl)-1H-Pyrrole-3-Carboxylate-A DFT Approach. Spectrochimica Acta Part A: Mol Biomolecular Spectrosc (2014) 127:122–30. doi: 10.1016/j.saa.2014.02.069
Keywords: DFT, IR spectroscopy, geometry optimization, HOMO and LUMO, natural bond orbital, Pt(IV)
Citation: Mu M and Gao H (2022) DFT Study on the Substituent Effect of Anticancer Picoline-Diazido-Pt(IV) Compounds. Front. Oncol. 11:749178. doi: 10.3389/fonc.2021.749178
Received: 30 July 2021; Accepted: 10 December 2021;
Published: 10 January 2022.
Edited by:
Carlos Alberto Manssour Fraga, Federal University of Rio de Janeiro, BrazilReviewed by:
Iti Gupta, Indian Institute of Technology Gandhinagar, IndiaTanmoy Chakraborty, Sharda University, India
Prabhat Ranjan, Manipal University Jaipur, India
Copyright © 2022 Mu and Gao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hongwei Gao, Z2FvaG9uZ3czNjlAbGR1LmVkdS5jbg==