 Shantel Angstadt
Shantel Angstadt Qingfeng Zhu
Qingfeng Zhu Elizabeth M. Jaffee2
Elizabeth M. Jaffee2 Douglas N. Robinson
Douglas N. Robinson- 1Department of Pathology Johns Hopkins University School of Medicine, Baltimore, MD, United States
- 2Department of Oncology, Johns Hopkins University School of Medicine, Baltimore, MD, United States
- 3Department of Cell Biology, Johns Hopkins University School of Medicine, Baltimore, MD, United States
Pancreatic ductal adenocarcinoma (PDAC) remains one of the deadliest cancers due to low therapeutic response rates and poor prognoses. Majority of patients present with symptoms post metastatic spread, which contributes to its overall lethality as the 4th leading cause of cancer-related deaths. Therapeutic approaches thus far target only one or two of the cancer specific hallmarks, such as high proliferation rate, apoptotic evasion, or immune evasion. Recent genomic discoveries reveal that genetic heterogeneity, early micrometastases, and an immunosuppressive tumor microenvironment contribute to the inefficacy of current standard treatments and specific molecular-targeted therapies. To effectively combat cancers like PDAC, we need an innovative approach that can simultaneously impact the multiple hallmarks driving cancer progression. Here, we present the mechanical properties generated by the cell’s cortical cytoskeleton, with a spotlight on PDAC, as an ideal therapeutic target that can concurrently attack multiple systems driving cancer. We start with an introduction to cancer cell mechanics and PDAC followed by a compilation of studies connecting the cortical cytoskeleton and mechanical properties to proliferation, metastasis, immune cell interactions, cancer cell stemness, and/or metabolism. We further elaborate on the implications of these findings in disease progression, therapeutic resistance, and clinical relapse. Manipulation of the cancer cell’s mechanical system has already been shown to prevent metastasis in preclinical models, but it has greater potential for target exploration since it is a foundational property of the cell that regulates various oncogenic behaviors.
Introduction
Cell Mechanics and Cancer
Cell mechanics refers to the cell’s physical properties and the mechanisms of force detection, force production, and load bearing to generate cell shape and behavior. More broadly, cell mechanics can encompass the application of solid and fluid concepts from physics and engineering to cells and the larger structures they compose (1, 2). To perform essential functions, such as tissue development, cell division, apoptosis, and migration, cells use internal and external stimuli to drive cell shape change and other highly mechanical processes. Morphogenesis results from the rearrangement of the cell’s underlying cytoskeleton, which changes the physical properties of the cell. Cytoskeletal rearrangement and the resulting cell shape change require active force generation initiated by the integration of chemical and mechanical (mechanochemical) signals. Understanding processes involving cell shape modification will lend new insights for the treatment of pathological states resulting from dysfunctions in cell division, apoptosis, and migration, i.e. cancer transformation and metastasis (3, 4).
The cortical cytoskeleton is responsible for cell shape change and is primarily composed of actin filaments, crosslinking proteins, and nonmuscle myosin II (NMII) filaments. The various regulators of these proteins are also vital contributors to morphogenesis since they control the spatiotemporal assembly and disassembly of cytoskeletal filaments. Actin filaments’ semi-flexible property, in addition to actin crosslinkers, allow for the interconnected network of filaments to form the cortical cytoskeleton and propagate mechanical stresses around the entire cell. The molecular properties and resulting function of the three components culminate into the mechanical properties of cell behavior. For example, cooperative interactions between NMII and crosslinking proteins allow these proteins to accumulate at sites of stress along the cell cortex. Overall cell shape change is managed through the integration of actin filament turnover, actin crosslinking, and NMII contractility and cooperativity (5–9).
The molecular binding affinities of the structural proteins with each other allow the cytoskeleton to maintain a fixed structure and resist deformation on short time scales, behaving elastically like a solid material. Likewise, these same binding affinities and regulatory mechanisms allow for protein disassociation and cytoskeletal rearrangement lending the cells viscous behavior, like a liquid, on longer timescales. Therefore, the cell is defined mechanically as a viscoelastic material often represented by both elastic spring and viscous damper components in mathematical models. Modeling can help predict cell behaviors, which is an invaluable tool for understanding cell processes such as cytokinesis and motility (8, 10, 11).
A major focus of the field has been to characterize mechanical properties that regulate cell and tissue function, especially in disease states. The ultimate goal is to use mechanical properties to generate new perspectives for various diseases and corresponding prognoses and treatments. Researchers have proposed a structure-property-function-disease paradigm in investigating the mechanics of cancer transformation and progression. For example, investigating the signaling effectors leading to physical properties of cells, how these physical properties lend function, and how the dysfunction at any part (signaling molecules, physical properties, resulting function) leads to diseased states. But cell mechanics is a complex and multiscale field: properties at the molecular, cellular, and tissue levels must each be characterized as well as integrated with the dynamic feedback systems. In the specific case of tumor tissue, the actin cytoskeleton at the molecular level generates the mechanical properties of individual cells, and the tissue level mechanical properties (i.e., stiffness) of the tumor feedback into individual cell’s mechanics resulting in altered function (differentiation, proliferation, invasion). Although complex, an understanding of tumor cell mechanics and the metastatic process opens an entirely new field for prognoses and therapeutic targeting (1, 2, 7, 8, 12).
Moreover, mechanical microenvironments in tumors are transformed along with the individual tumor cells. For example, the extracellular matrix (ECM) of the tumor is often stiffer than the surrounding healthy tissue. Tumor cells have been shown to have more invasive phenotype on stiffer substrates and this phenotype can be reversed with substrate tension relaxation. There is evidence that tumor cells soften in response to this stiffening ECM. Additionally, tumor tissues experience increased fluid pressures due to angiogenesis and tissue restructuring. The altered mechanical states of the tumor tissue initiate mechanosensing in individual tumor cells. Mechanosensing pathways have been shown to drive proliferation, survival, invasion, stemness, and therapy resistance. Therefore, mechanical stimuli and their effects are multiscale: tissue-level, cell-to-cell interaction, cell-to-matrix interactions, and biochemical reactions (13).
In addition to the ECM altering tumor cell function through mechanosensing, tumor cells have the ability to remodel the ECM and the polarity of cancer-associated fibroblasts and different immune cells. For example, another focus of cancer cell mechanics has been traction forces and polarity generated by tumor cell contractility during adhesion and migration. Understanding cell-generated traction forces is necessary because these stresses help restructure the ECM and push cells forward during migration. A correlation exists between traction forces, contractility, and metastatic potential. For example, metastatic cells across three cancer types exerted greater traction forces in response to matrix stiffness compared to their non-metastatic counterparts (14–16). These highly metastatic cells with larger traction forces lead the way for collective migration by restructuring collagen fiber alignment into tracks that others cells could more easily follow. Cell polarity also plays a role in mesenchymal modes of cell migration where the direction and persistence of migration is dictated by the alignment of cell and matrix-remodeling polarity (17, 18). Additionally, cytoskeletal forces are physically transmitted to the nucleus through the Linker of Nucleoskeleton and Cytoskeleton (LINC) complex. For example, substrate stiffness can lead to alterations in nuclear stress and shape, resulting in changes of gene expression, nuclear stiffness, and the cell’s differentiation state (19–21).
Furthermore, the metastatic cascade is a physical and mechanically-driven process. Metastasis is both the process and result of cancer cells migrating and colonizing in a location other than the primary tumor site. First, tumor cells must break adhesion complexes with their surrounding cells and migrate through tumor stroma. This migration involves deformation to squeeze through ECM pores, push and pull ECM fibers, and degrade fibers. Second, the tumor cell must invade through a basement membrane and vasculature wall for intravasation, which requires continued matrix degradation and shape deformation. Third, tumor cells in circulation must resist shear forces inflicted by blood flow and adhere to the vessel wall for extravasation. Extravasation involves another deformation process followed by migration/invasion into the new tissue site. Finally, tumor cells must initiate proliferation to colonize the metastatic tumor (22).
Cell deformability is essential for the metastatic cascade and is dependent on the cell’s viscoelasticity. Another major focus within cancer cell mechanics is on the viscoelastic differences between normal, transformed, and metastatic cells. The aim is to characterize the relationship between deformability and metastatic potential. Using optical stretching and atomic force microscopy, several studies across different cancer types revealed that cell deformability increases with metastatic potential. Interestingly, cells can actively resist externally-imposed deformation by polymerizing actin and recruiting specific cytoskeletal proteins to increase tension. It is the dynamic ability of being able to deform and simultaneously resist deformation that allows tumor cells to undergo the metastatic cascade. Therefore, a method to prevent metastasis for solid tumors would be to stiffen and decrease the cell shape change ability (8, 13, 23–27).
Cell and tissue deformation mechanics during cancer transformation and progression is a relatively young field in cancer biology and is working on uncovering a different system of therapeutic targets to address metastasis and patient mortality. Cell deformability and morphogenesis are foundational components of signaling networks, division, adhesion, migration, invasion, and metastatic potential. The shape and rigidity of a cell is due to the cytoskeleton and its molecular components. Previous reports have shown altered cytoskeletal structure, regulation pathways, and extracellular matrix structures in various cancer types and stages. Additionally, metrics of mechanical properties correlate with disease state and metastatic potential. Targeting the cytoskeleton has already been shown to alter mechanical properties and metastasis in preclinical models and is being investigated as an indicator for disease stage and prognoses (26–32). The major challenge in deformation mechanics is that the cell cannot be fully characterized by the static mechanical properties of solids and fluids. Cells are dynamic systems that react and respond to internal and external stimuli. Therefore, their subcellular and physical properties are constantly changing (33). We have already seen the potential of altering the cell’s physical properties to prevent cancer progression. Therefore, a critical need exists to fully integrate the field of cell mechanics in cancer therapeutic and prognostic development.
PDAC and PDAC-Specific Cortical Mechanics
Pancreatic ductal adenocarcinoma (PDAC) is the most common type of pancreatic cancer. Pancreatic cancer incidence continues to increase across the country in conjunction with it having one of the lowest 5-year relative survival rates around 10%. Patients typically present with symptoms post metastatic spread, which contributes to its overall lethality as the 4th leading cause of cancer-related deaths (34, 35). Advancements in therapy and precision medicine have helped to increase the low 5-year survival rate, but recent discoveries have also uncovered how little we understand PDAC transformation and progression. Our lack of knowledge for the PDAC-specific pathogenesis of transformation and metastasis limits our ability to innovate more effective treatments.
PDAC forms from precursor lesions and has historically presented as a genetic disease, gradually progressing through a sequence of acquired mutations. Four driver genes have been linked to each stage of transformation (KRAS, TP53, SMAD4, and CDKN2A), but recurrent somatic mutations (SNV, indel, scNA) and germline mutations (in DNA damage repair genes) have also been found. PDAC is characterized by desmoplastic reaction due to interactions between cancer, vasculature, pancreatic stellate, and inflammatory cells. Over half of all cases are diagnosed post-metastatic spread, and the most common sites for metastasis are stomach, lung, colorectum, esophagus, gall bladder, liver, and common bile duct (2, 36, 37).
The standard treatment is surgical resection and/or two chemotherapeutic agents, gemcitabine and FOLFIRINOX (the combination of oxaliplatin, irinotecan, fluorouracil, and leucovorin) depending on disease severity and stage, but these treatments are least effective post metastatic spread (38, 39). Novel whole genome sequencing and bioinformatic techniques on primary and metastatic tumor clonal populations have revealed that not all PDAC tumors progress through the gradual sequence of transformation steps. Additionally, some PDAC tumors result from a more prolonged precursor lesion stage and show micrometastasis early in the tumor formation process (5, 40–44). Altogether, recent genomic discoveries reveal that genetic heterogeneity and early micrometastases within PDAC progression result in the inefficacy of current standard treatments and specific molecular targeted therapies.
The PDAC tumor microenvironment (TME) is a major contributor to inefficacious treatments, especially for immunotherapies. The PDAC TME is characterized by dense stroma and a small cell content composed of tumor cells, cancer-associated fibroblasts, muscle fibroblasts, pancreatic stellate cells, and infiltrated immune cells. Tumors are typically 70-90% stroma, mostly deposited by fibroblasts and stellate cells, which causes increased intratumoral pressure, poor vascularization, and hypoxia. Therefore, drugs (chemotherapy, immunotherapy, molecular targeted therapies) delivered systemically cannot penetrate throughout the tumor (45–48). Additionally, the infiltrated immune cells generate a tumor promoting and immunosuppressive environment. For example, the immune cells of the TME are predominately regulatory T cells (Tregs), macrophages, and myeloid-derived suppressor cells, which all work together and impede cytotoxic T cells from infiltrating, identifying, and killing tumor cells (49–53). Preclinical and clinical studies targeting the stroma and immunosuppressive pathways have had contradictory results and revealed the complexity and labyrinth-like network of interactions and pathways of the PDAC TME (54). Though progress has been made, it is essential to continue exploring and uncovering new avenues for therapeutic approaches because our current methods generate low clinical response rates. Since cancer transformation and progression are intensive mechanical processes, the molecular machinery and system responsible for cortical mechanics has the potential to be the next avenue of therapeutic approaches for PDAC.
Mechanical states of cells are defined by their underlying cytoskeletal structural and contractile machinery, which governs morphology and morphogenesis. Morphogenesis is an essential part of tumor formation and progression (i.e. proliferation, differentiation, polarization, migration, invasion), Figure 1. In general, tumor cells alter their cytoskeletal machinery to be more deformable and responsive to their changing environment. In fact, deformability has been correlated to metastatic potential and aggressiveness in many different cancer types (33, 55–57). Therefore, it is no surprise that the mechanical landscape of PDAC cells is significantly altered, which makes cortical mechanics an opportune field to explore for PDAC prognostic and therapeutic purposes (26).
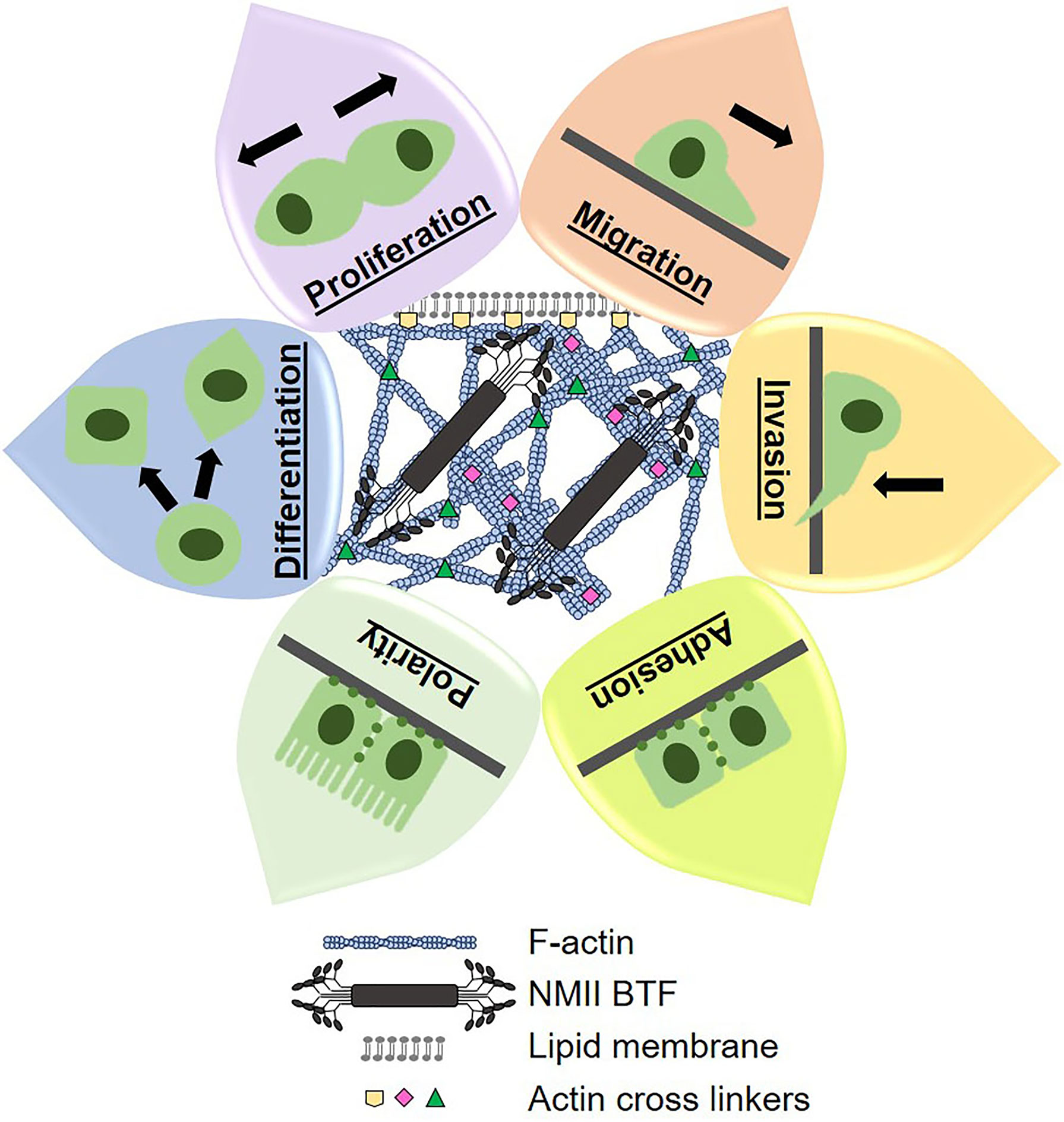
Figure 1 Cytoskeletal dynamics control cortical mechanics, morphogenesis, and cell behavior. The cytoskeletal components of filamentous actin (F-actin), actin crosslinking proteins, and non-muscle myosin II bipolar thick filaments (NMII BTF) dynamically assemble, polymerize, and depolymerize to generate whole cell mechanical properties and cell shape. Mechanical properties and cell shape change underlie the cell behaviors that we observe and measure. Therefore, we can manipulate cell behaviors by altering cortical mechanics and cytoskeletal dynamics, which lends great therapeutic potential.
Specifically, the four cytoskeletal proteins non-muscle myosin IIA (NMIIA), NMIIC, α-actinin 4, and filamin B have increased expression in PDAC patient tissues as compared to normal pancreatic ductal epithelium. The NMII paralogs are responsible for contractile forces in the cytoskeleton, and α-actinin 4 and filamin B are actin crosslinkers. In response to external stresses, each of these proteins accumulates to the site of stress to generate forces and maintain membrane-cortex integrity. We have defined this accumulation in response to physical stress as a mechanoresponse. Additionally, NMIIC is responsible for the formation of traverse actin arcs in single cells and for cortical actin belts in PDAC spheroids. Traverse actin arcs are actin bundles generated by NMII contractility and propagate toward the rear end of migrating cells as a necessary structural element driving migration. Cortical actin belts between epithelial cells generate the epithelial boundary that forms apical sides of tissues. Activating NMIIC assembly into the cytoskeleton using the small molecule 4-Hydroxyacetophenone (4-HAP) decreased in vitro dissemination from PDAC spheroids and in vivo metastasis. Specifically, 4-HAP induced cortical actin belt formation and slowed down retrograde flow of transverse actin arcs. Furthermore, 4-HAP increased cell cortical tension by activating the assembly of NMIIB and NMIIC, which also decreased migration and invasion in vitro. Therefore, inducing NMIIB and NMIIC assembly is a therapeutic strategy to reduce cell mobility and metastasis overall (26, 58).
Research from UCLA indicated stiffness and invasion are differentially regulated by actin and NMII proteins in accordance with disease stage in PDAC (59). The study used MIA PaCa-2 and PANC-1 cell lines derived from primary tumors and Hs766T cells derived from a metastatic site in the lymph node. Hs766T cells had higher stiffness than Panc-1 cells and were slower to round up. Furthermore, the Hs766T cells did not require NMII for invasion. Interestingly, Hs766T cells required actin filament nucleators, Arp2/3 and formin, to maintain cell stiffness and invasion. The dependence on actin filament nucleation suggests that actin polymerization is a major driving force for invasion and mechanotype (stiffness) in this metastasis-derived cell line. Invasion was not dependent on expression or activity levels in any of the cell lines. A characteristic hallmark of Arp2/3-dependent invasion is longer protrusions at the leading edge of the invading cell, which the Hs766T cell line exhibited more than the Panc-1 cells (59). This work further elucidates the altered mechanical landscape in PDAC progression and identifies potential targets specifically for metastatic PDAC cells.
Another interesting finding from an immunotherapy clinical trial implicated the NMII regulator, myosin phosphatase targeting subunit 1 (MYPT1), in PDAC. MYPT1 was originally identified through its being targeted by the immune system in a clinical trial for a cytokine-secreting whole tumor cell vaccine. An antibody response against MYPT1 in patients treated with the tumor cell vaccine correlated to a positive treatment outcome of greater than 3-years of disease-free survival. In addition to MYPT1’s elicited antibody response, its expression is highly upregulated in PDAC patient samples and in established PDAC cell lines. The function of MYPT1 overexpression and its implication in cortical mechanics has yet to be characterized in PDAC tumor cells, but its discovery through this immunotherapy clinical trial is a harbinger for the interconnectedness of the individual tumor cell cytoskeleton, immune cells of the tumor microenvironment, and therapeutic responses (60–62).
In summary, PDAC’s mechanical landscape comprising various cytoskeletal and regulatory proteins is significantly altered during transformation and the metastatic cascade. The targetability of this altered mechanical landscape using the small molecule 4-HAP demonstrated our ability to prevent metastasis, but there is much more that needs to be explored to uncover other potential therapeutics. Moreover, cortical mechanics can be used to target more than proliferation, invasion, and metastasis. While the number of studies in PDAC are still limited, when combined with research in other cancer types (12, 27), the implications and potential of targeting cell mechanics are becoming increasingly apparent via connections to cell stemness, differentiation, immune cell modulation, and metabolic reprogramming. The following section will provide evidence in other cancer types for such connections and illuminate cortical mechanics as a foundational property of the cell that can tie the various drivers of cancer together for more efficacious therapeutic targeting. In combination, the studies presented in this section on PDAC and the studies presented in the following section further highlight the potential of PDAC's altered mechanical landscape as being a revolutionary field for PDAC-specific therapeutic development.
Implications for Disease Severity and Clinical Outcome
Proliferation, Metastatic Potential, Disease Progression
Numerous reports have centered the molecular determinants of cortical mechanics in cell proliferation, migration, invasion, metastasis, and overall disease prognoses. For example, the cytoskeletal scaffolding protein anillin is extensively implicated in cancer progression and patient survival of several cancer types (e.g., breast, pancreatic, colorectal, lung, gastric, liver). Generally, anillin is upregulated during cancer transformation and invasion and is associated with both positive and poor prognoses clinically depending on cell localization (63). Anillin binds to both actin filaments and NMII in the cytoskeleton and facilitates NMII localization and actomyosin contractility at the cortex of dividing cells (64, 65). Regarding migration, anillin depletion in breast cancer cells decreased in vitro migration and in vivo metastasis (66). Additionally, anillin regulates assembly of adherens and tight junctions in epithelial cells to establish the epithelial barrier. The apical actomyosin cytoskeleton physically interacts with the adhesion complexes and transfers tensile forces between cells of the epithelial barrier. Therefore, anillin being implicated in various adenocarcinoma’s disease severity and its impact on the metastatic cascade is intuitive (67–70). Additionally, the actin cross-linker and mechanoresponsive protein, filamin B, has increased expression in pancreatic cancer primary tumors and has been correlated with reduced patient survival through the analysis of publicly available data on the Oncomine and UALCAN databases (71, 72). Furthermore, filamin B is positively regulated by the pancreatic cancer-associated transcription factor MYB, which is suggested to be a potential biomarker of PDAC aggression (73, 74).
The NMII contractile protein paralogs have extensively been implicated in cancer cell behaviors and disease progression. Singh et al. found that NMIIA suppressed tumor formation, metastasis, and regulated immune cell infiltration in in vitro and in vivo melanoma models. NMIIA knockdown in B16F10 cells enhanced migration and invasion in transwell-based assays. Furthermore, subcutaneous and intravenous xenograft mouse models showed enhanced tumor cell proliferation, metastasis, and inflammatory cell infiltration in response to NMIIA knockdown (75). Specifically, they saw an increase in several oncogenes, ERK signaling, and endothelial cells in tumor sections, which suggests NMIIA is a tumor suppressor in melanoma cells. Picariello etal. (76) found that NMIIA regulates glioblastoma proliferation and invasion depending on the mechanical environment. Glioblastoma commonly invades into the surrounding brain tissue, which results in its overall aggressiveness and lethality. NMII activity was suspected to be a potential target for glioblastoma since the invasive ability of the tumor cells is dependent on NMII function (76).
Interestingly, this study discovered complete NMIIA knockout regulated cell proliferation and motility differentially depending on the stiffness of the substrate in the cells’ environment. On softer substrates, NMIIA depletion led to ERK1/2 activation, resulting in higher proliferation rates. On stiffer substrates, NMIIA depletion led to NFκB activation impacting cell survival and cell stemness. NFκB activation in response to NMIIA depletion was also observed in triple negative breast cancer cells and keratinocytes. Overall, in vivo experiments revealed NMIIA depletion in a mouse model led to reduced invasion, but larger tumors which hastened the overall lethality of the disease. The significant findings from this study show that NMIIA is a downstream effector that can be targeted to prevent invasion, but is also an up/mid-stream signaling component that responds to the mechanical environment and impacts disease progression. This is further complicated by the fact that each of the NMII paralogs can serve different functions within the cell (76).
The Nguyen-Ngoc et al. study found that NMIIA and NMIIB suppress breast epithelial proliferation. Using transgenic mice, they developed mammary organoids where 50-75% of the cells were NMIIA and NMIIB null, resulting in a mosaic tissue of IIA and IIB expression. Additionally, they used organoids from transgenic mice that had ubiquitous deletion of NMIIA and NMIIB expression and found that the mosaic NMIIA/IIB organoids had increased proliferation compared to the ubiquitously deleted NMIIA/IIB organoids. Furthermore, simple stimulation via fibroblast growth factor signaling induced hyperplasia. These results were also confirmed in an in vivo model. Overall, this study demonstrated NMIIA and NMIIB’s suppressive regulation of proliferation in breast epithelium (77).
The Kapoor et al. study elucidated the RhoA-ROCK-NMII pathway regulation of two distinct modes of invasion. The first being mesenchymal invasion where single cells have a spindle-like morphology and use adhesion- and matrix metalloproteinase (MMP)- dependent migration to move though tissue. The second mode uses amoeboid migration where single cells have a rounded morphology and use adhesion- and MMP-independent motility to squeeze through ECM pores. They demonstrated ovarian cancer cell lines with resistance to cisplatin used mesenchymal invasion, whereas cells resistant to paclitaxel or both drugs used amoeboid invasion. In both modes, signaling through the RhoA-ROCK2-NMII regulated invasion and, more specifically, NMIIA and NMIIB function mediated both nuclear squeezing and MMP-9 activity. Conclusively, NMIIA and NMIIB regulated both modes of invasion in ovarian cancer cells, demonstrating NMII’s role in invasion and the metastatic cascade (78).
Phosphorylation of the NMIIA heavy chain regulates breast cancer cell ability to degrade ECM and invade. NMIIA heavy chain phosphorylation regulates the myosin heavy chain’s ability to assemble into the bipolar myosin II filaments that are functional in the cytoskeleton. Phosphorylation of the heavy chain on the Ser-1943 residue promotes myosin disassembly and increased EGF-stimulated lamellipodia formation of breast cancer cells. Additionally, Ser-1943 phosphorylation is required for in vitro matrix degradation and increased invadopodia function, as well as increased in vivo metastasis. Therefore, NMIIA function regulates breast cancer cell invasion and metastasis (79). NMIIA-mediated cortical mechanics has also been implicated in colorectal cancer. Using tissue microarrays of patient tumor samples, NMIIA heavy chain overexpression positively correlated with disease progression and poor survival of patients. Overexpression of NMIIA heavy chain increased proliferation, invasion, and metastasis in both in vitro and in vivo models. At the molecular level, NMIIA heavy chain overexpression increased phosphorylation levels of both ERK and AKT, which subsequently reversed with NMIIA heavy chain knockdown (80). Altogether, the studies of the NMII paralogs in various cancer types realize NMII-modulated cortical mechanics as a foundational property of cancer cell shape control and function.
Various reports have implicated MYPT1, the NMII assembly regulator found overexpressed in PDAC (62), in the formation and prognoses of different cancers. For example, MYPT1 knockdown in HeLa cells resulted in nuclear fragmentation, nuclear compartment breakdown and genome instability (81). MYPT1 knockdown also increased histone methylation levels via the methyl transferase PRMT5, which is associated with transformation in hepatocellular carcinoma (82). Overexpression of the micro-RNA molecule microRNA-30d that targets MYPT1 knockdown predicted aggressive disease in prostate cancer (83). Finally, low copy number of the MYPT1 gene in colorectal cancer predicted poor clinical outcome for oxaliplatin treatment (84).
In gastric cancer, overexpression of MYPT1 presents as a tumor suppressor. MYPT1 expression in normal tissue was compared to patients’ cancer tissue and overall patient survival. MYPT1 is decreased in gastric tumors, which also correlated with poor patient survival. In vitro, MYPT1 overexpression inhibited proliferation, migration, and invasion. MYPT1 functions to negatively regulate NMII activity in the cytoskeleton. Therefore, gastric tumor cells decreased a myosin inhibitor to promote disease progression, ultimately increasing NMII activity and yielding NMII activity as oncogenic (85). MYPT1 was also discovered in prostate cancer as a biomarker of disease progression. Gene expression profiles of tumor cells had not adequately predicted patient outcome. Therefore, researchers investigated the TME to identify potential prognostic biomarkers using tumor microarrays of patient tumor samples paired with Aperio Imagescope software analysis. They then evaluated biomarker association to biochemical recurrence and time to biochemical recurrence. Overall, MYPT1 positively correlated with disease progression (86).
The reports mentioned thus far have focused on individual cytoskeletal elements and regulators, but there are also many reports characterizing general mechanical properties. For example, mechanical properties of human ovarian and breast cancer cell lines predicted the invasive ability of these cells. Microfluidic devices were developed to perform quantitative deformability cytometry and allowed measurement of physical phenotypes such as the cell’s elastic modulus, cell fluidity, entry time, maximum strain, and cell size. Using prediction models paired with the known phenotypes of individual cell lines, analysis indicated that the elastic modulus correlated the most with invasive ability, but the additional parameters of fluidity, entry time, and size improved the model’s predictive accuracy. Altogether, this study demonstrated the value of mechanical characteristics as biomarkers of invasion and potential targets for therapeutic intervention (87).
An in vitro study using osteosarcoma cells investigated the relationship between mechanical properties and metastatic potential. Researchers used a low metastatic parental line and a corresponding high metastatic line. Overall, highly metastatic cells spread less and exerted weaker forces than the line with low metastatic ability. The weaker forces of the highly metastatic cells contradict the increased traction forces of metastatic cells in other cancer types (14–16), but osteosarcoma differs in that it has mesenchymal origin and does not undergo epithelial-to-mesenchymal transition (EMT). Therefore, osteosarcoma cells do not experience similar polarity and differentiation trends as other adenocarcinomas, which exemplifies differences in mechanical properties according to differentiation and the need to characterize mechanics specific to each cancer type (88). Finally, cell and tissue stiffness are determinants in metastatic organotropism (89–92). For example, breast cancer cells subcategorized by their cytoskeletal and biophysical properties had specific metastatic preferences due to cytoskeletal adaptation ability and their corresponding gene expression patterns (93).
In summary, mechanical properties of individual cells and tissues dictate cell behaviors and overall disease outcomes, but we see a contradiction of effect for specific cytoskeletal proteins like the NMII paralogs (76–83). The contradiction of NMII function in different cancer types reveals the complexity of the cytoskeletal system and our lack of understanding of the cortical cytoskeletal role in cell function, further revealing the need for investigation and integration of cortical mechanics in the different cancer types. Yet, each of these studies points toward the cell’s mechanical landscape as a foundational system that can tie the various drivers of cancer together for more efficacious therapeutic targeting, further highlighting the role of PDAC’s altered mechanical landscape.
Immune Cell Interaction and Immunotherapy Resistance
Cytoskeletal forces of both the TME immune cells and tumor cells regulate immune cell infiltration into tumors, immune cell polarity, the molecular interactions between cells, and the resulting anti- or pro-tumor immune response. Traction force microscopy has been used to measure Jurkat T cell force exertion during T cell receptor (TCR) activation. TCR activation is central to any adaptive immune response and mediates both antigen-specificity and cytotoxic activity against targeted cells. For the anti-tumor response, TCR activation in CD4+ T cells enables identification of neoantigens on tumor cells vs. normal cells and the fatal interaction of CD8+ T cells recognizing the tumor cells. Previous work had indicated that primary CD4+ T cells exerted traction forces in response to CD3 or CD28 stimulation, which is required for TCR activation and a T cell response. The cytoskeletal forces generated in the T cell are mediated by actin polymerization and NMII contractility (94–104).
A study of Jurkat T cells demonstrated that T cells spread more uniformly and exhibited larger and longer TCR signaling responses on stiffer substrates as compared to softer substrates. The differential responses of T cells based on substrate stiffness have major implications for the interactions between tumor cells and T cells and the regulation of cell stiffness and cortical tension of the individual tumor cells (105). The exact molecular mechanisms of T cell response to substrate stiffness are already being elucidated. For example, cytotoxic CD8+ T cells were able to kill the bulk of tumor cells, but not the undifferentiated cells with self-renewal capabilities. The undifferentiated cells are referred to as tumor repopulating cells, a subset of cancer stem cells that can be dormancy competent, and were immediately characterized as inducing PD-1 expression in CD8+ T cells and as being softer than the differentiated tumor cells (106–110).
Further investigation revealed cell softness prevented formation of the perforin pore in the targeted tumor cell. Perforin is released from activated CD8+ T cells to form a pore on the tumor cells and allow granzymes from the T cell to enter the targeted cell for apoptotic induction. NMIIA heavy chain is required for perforin pore formation because tumor cells with NMIIA heavy chain knocked down were unable to generate the actomyosin-mediated forces at the cell membrane and failed to form perforin pores. Pharmacologically increasing the stiffness of tumor repopulating cells allowed perforin pore formation and T cell-induced apoptosis in tumor cells using both in vitro and in vivo models (111). Cell softness also prevented immune synapse formation and target-induced apoptosis of natural killer cells, in addition to cytotoxic T cells. Human NK cells more effectively secreted granzymes A and B, FasL, granulysin, and IFNγ on stiffer substrates than softer substrates (112), a trend that was similarly observed in the cytotoxic T cells. Moreover, a study in cervical and colorectal cancer cell lines revealed NMII paralog-specific activity and localization induced MHCI and CD59 uptake via clathrin-mediated endocytosis (113).
The previous studies focused on T cell and natural killer cell interactions with tumor cells. Work has shown that the mechanical properties of tumor cells also impact the immune cells’ ability to enter and incorporate with the tumor. Specifically, knockdown of the NMIIA heavy chain in melanoma cells regulated immune cell infiltration of the TME. Using subcutaneous tumor formation and intravenous lung metastasis models, melanoma tumors with NMIIA heavy chain knocked down had increased recruitment of leukocytes (CD45+) and macrophages (F4/80+). The exact polarity and function of these cells was not further investigated, but we know immune cells in general greatly impact tumor growth, metastatic ability, and therapeutic response (75).
In summary, cortical mechanics of both tumor cells and the cells of the tumor microenvironment coordinate to facilitate immune evasion, detection, and therapeutic responses. The studies presented here indicate that targeting cell softness could enhance the tumor response to immunotherapies that induce cytotoxic T cell killing. This is pertinent to PDAC as it has repeatedly shown low response rates to anti-PD-1/PD-L1 immunotherapy. Cancer cell stemness adds to the story since mechanisms of T cell evasion have been elucidated in the tumor repopulating cells. These findings imply that we can target cortical mechanics to induce cancer cell differentiation and simultaneously prevent immune evasion. The next section will further elucidate the connection between the mechanical properties of cancer cell stemness, differentiation state, and their impact on immune cell interactions.
Cancer Cell Stemness, Differentiation, and Disease Relapse
The case for cancer stem cells (CSC) has faced criticism due to the complexity of differentiation states and cell plasticity/adaptability, which hinder our ability to fully understand these cell populations. CSCs are characterized by self-renewal, resistance to stress, dormancy, and evading cell death (114). Clinical data from cancer patients with disease recurrence or relapse revealed the metastatic tumor cells and circulating tumor cells within these patients came from cancer cells that persisted after treatment of the primary tumor. Therefore, CSCs are suspected of being the therapy-resistant cells that are responsible for disease recurrence (115).
Molecular and genetic characterization of CSCs have further revealed subcategories such as dormancy-competent CSCs, dormancy-incompetent CSCs, and cancer-repopulating cells. Dormant cells are simply defined as cells that exit a highly proliferative state. Dormant cells are able to evade standard chemotherapies that kill highly proliferative cells due to the quiescent-like state (116, 117). Additionally, CSCs can maintain short- and long-term dormant states, which helps to explain clinical relapse after just months or years of remission (118–120). Moreover, dormancy-competent cells are able to resist immunological targeting directly and indirectly through immunosuppression. The less proliferative dormant cells are not capable of generating neoantigens like highly proliferative tumor cells, which ultimately leads to immune evasion. Dormant cells have also displayed immunosuppressive mechanisms of inhibition for T cell activation via overexpression of B7 homolog 1, cytotoxic T cell-induced apoptosis via methylation of suppressor of cytokine signaling 1, and antigen inhibition via decreased human leukocyte antigen expression (121–124).
We are emphasizing cancer stem cells, and the subsequent dormant and differentiated cells, in this review because interesting mechanical data has revealed a new avenue of characterization and targetability for these therapy-resistant cells. For example, research with breast, ovarian, lung, bladder, and prostate cancer cell lines revealed that dormancy competent cells entered states of dormancy and survival in response to high matrix stiffness, whereas dormancy-incompetent cells rapidly died. Both proliferative and metabolic activity were inhibited, and chemoresistance was increased in these dormant cells (125–132, 145). Ongoing research is developing ways to induce cancer cells to exit dormancy by using biomaterials. Using an agarose-silica gel-based method, breast cancer cells were able to enter dormant states and then exit by immediately regaining proliferative and migratory capabilities that were lost in the dormant state (134). Uncovering the mechanism of matrix stiffness regulating dormancy will provide new insight on how to effectively target dormant cancer cells and prevent clinical relapse. Additionally, research has focused on uncovering ways to identify cancer stem cells from the bulk tumor cells. Interestingly, cell stiffness or cell softness is a unique marker of cancer stem cells. Specifically, CSCs are significantly softer across various cancer types, and this property is mediated through stem cell factor signaling pathways (135–137).
Interestingly, NMII activity regulates the self-renewal capability in human pluripotent stem cells and mouse embryonic stem cells. For example, NMII inhibition via blebbistatin and RNA knockdown increased cell viability and the expression of self-renewal regulators Oct3/4 and Nanog (138). Additionally, NMIIA expression in mouse embryonic stem cells maintained E-cadherin-mediated cell adhesions. Furthermore, NMII assembly regulation via its phosphorylation sites is intertwined with EMT and migration. When EMT is induced via TGF-β stimulation in mouse epithelial cells, there was a stark increase in NMIIA Ser-1916 phosphorylation, which increased the invasive behavior of these cells. In mesenchymal stem cells, phosphorylation of NMIIA at Ser-1943 resulted in random migration on soft substrates, but dephosphorylation and subsequent assembly of NMIIA at Ser-1943 resulted when these same cells were placed on stiff substrates (139). Altogether, these results reveal the molecular determinants of cortical mechanics to actively regulate stemness.
Focusing on differentiation state, the Singh et al. study in melanoma revealed NMIIA knockdown regulated EMT. Specifically, NMIIA heavy chain knockdown increased the mesenchymal markers slug and twist and the epithelial marker E-cadherin. The final effect of the altered EMT markers is inconclusive, but demonstrates the clear connection between myosin II expression and differentiation (75). The Wang et al. study revealed NMIIA heavy chain overexpression induced EMT through upregulation of mesenchymal markers fibronectin, N-cadherin, and MMP9, and downregulation of epithelial markers ZO-1 and β-catenin. NMIIA heavy chain-mediated EMT was required for the observed aggressive phenotype (80).
Returning to the case of anillin (63, 66), this scaffolding protein impacts differentiation and stemness in addition to proliferation, migration, and invasion. Depletion of anillin in two mesenchymal-type breast cancer cell lines decreased stem cell properties. Similarly, increased anillin expression in an epithelial cell line increased stemness properties (66, 67). Interestingly, pluripotent cells of mouse embryos, Drosophila testes, and zebrafish retina all have higher anillin expression, while senescent human fibroblasts and cervical cancer cells have decreased anillin expression (133, 140–144). This correlation in stem-like properties is interconnected with differentiation state. For example, a decrease of anillin in lung and breast cancer cells resulted in mesenchymal-to-epithelial transition. The exact mechanisms and transcription factors responsible for connecting anillin to stemness and plasticity are currently being elucidated (63, 66).
Collectively, the studies of this section reveal the intertwined relationship between cell stemness, differentiation, and mechanical state. Additional reports exemplify the axis of regulation between matrix stiffness, cell stemness/differentiation, and NMII expression/function (146–150). Most importantly, cancer stem cells evade T cell-induced apoptosis by reducing cell stiffness and subsequently preventing perforin pore formation. Overall, these studies elucidate how we can use cytoskeletal components to identify and target cancer cell stemness and differentiation, which contribute to cancer progression, therapeutic resistance, and clinical relapse. This is relevant to PDAC because we do not have a clear consensus for cancer stem cell identification. Using PDAC cortical mechanics, we can potentially identify and target stem cells to eradicate therapeutic resistance and disease relapse caused by CSCs.
Targeting Cytoskeletal and Metabolic Connections
One of the hallmarks of cancer cells is the metabolic reprogramming undergone to allow the cancer cells to survive in densely populated tissues with limited nutrients. The Warburg effect is a well-characterized metabolic shift in which cancer cells increase glucose uptake and glycolytic rates to increase energy production. More recent discoveries uncovered the heterogeneity of metabolic reprogramming in cancer cells to also include increases in oxidative phosphorylation and the use of alternative carbon sources (i.e., glutamine, fatty acids, and serine). Various studies have revealed a metabolic regulation system between cancer cells, immune cells, and stroma and have demonstrated the targetability of these metabolic shifts in preventing disease progression (151–158). Surprisingly, studies have also revealed regulation and correlation between metabolic shifts and cytoskeletal proteins in cancer.
For example, proteomic analysis of breast cancer cells treated with doxorubicin, a chemotherapeutic agent, identified connections between the transcription of several metabolic enzymes, actin, and α-actinin. Ongoing work aims to use these proteins as treatment targets for breast cancer cells (159). Breast epithelial cells’ EMT via TGF-β induction can be mitigated through inhibition of phosphocholine anabolism, which subsequently changed the increased actin stress fiber formation that is associated with EMT (160). Another breast cancer study demonstrated the association of actin binding proteins with proteasome activity (161). Ezrin, a scaffolding protein that links the cytoskeleton to the plasma membrane, regulates osteosarcoma tumor progression and metastasis through alterations in lactate production and ATP-dependent oxygen consumption (162). A limitation of the aforementioned studies is the lack of a clear mechanistic pathway, but there are reports which implicate various mechanisms of regulation between metabolism and cytoskeletal components. For example, the pro-metastatic actin-binding protein Fascin that is overexpressed in lung cancer directly increases the transcription and activity of glycolytic enzymes phosphofructokinase 1 and 2 through the YAP1 transcription factor (163). Fructose-bisphosphate aldolase A (ALDOA), a glycolytic enzyme, directly interacts with actin and regulates the polymerization of actin filaments, which is crucial for migration and invasion. Furthermore, ALDOA has increased expression in renal, liver, and lung cancer cells and correlated with disease aggression and prognoses (164–167). Targeting ALDOA to prevent its interaction with actin reduced actin stress fiber content, proliferation, migration, ATP synthesis and survival in cancer cells (168, 169).
In conclusion, cytoskeletal composition and assembly level directly and indirectly regulate different metabolic pathways and can be used to reveal mechanisms and potential targets in cancer progression. Research has revealed upregulated cytoskeletal components in PDAC (26, 62). Work investigating the connection between these proteins and metabolism has yet to be published, but PDAC’s metabolic reprogramming is well documented and known to drive disease progression (Li et al., 2019). Therefore, there is a need for investigations of altered PDAC-associated metabolic pathways in connection with the upregulated cytoskeletal components.
Future Outlook and Clinical Perspectives
The latest technology and research have revealed cancer to not only be a disease of genetics, but also a disease of epigenetics, immunology, and metabolism. Yet, therapeutic approaches thus far have targeted only one or two of the cancer-specific hallmarks, such as high proliferation rate, apoptotic evasion, immune evasion, etc. Unfortunately, current therapies and molecular targeting have not been a huge success for all cancers due to various reasons, including pathway redundancies and circumvention, low levels of immune cell invasion, cell plasticity, and metabolic reprogramming. For these reasons, cancers such as PDAC continue to have low clinical response rates and poor prognoses. To effectively combat cancers like PDAC, we need an innovative approach that can simultaneously impact the multiple systems driving cancer progression. Traditional combination therapies are an option, but may be limited due to only targeting a couple systems at a time, the overall drug toxicity, and the tendency of cancer cells to forget their initial disease drivers. Therefore, an ideal therapeutic approach may be to target multiple systems concurrently. The cancer cell’s mechanical system is a prospective field for target exploration, seeing as it is a foundational property of the cells and has already been tied to various systems driving the disease.
Cortical mechanics refers to the physical properties and capabilities of a cell given by the underlying cytoskeleton and the molecular properties of its various components. For example, actin polymerization and NMII contraction are largely responsible for force generation, but actin crosslinkers and mechanoresponsiveness of these proteins contribute to the load-bearing capability of cells. Ultimately, these properties of force generation and load-bearing drive cell morphogenesis and culminates into cell behaviors such as proliferation, migration, polarity, differentiation, and invasion. Therefore, these proteins are a major focus within the realm of cortical mechanics and have been implicated throughout various cancer types and studies. Thus far, cytoskeletal components and their regulation are heavily involved in invasive and metastatic potential, disease prognoses, cell fate and polarity, immune cell interactions of the tumor microenvironment, and metabolic regulation. Additionally, preclinical targeting of the cytoskeleton, specifically NMIIC assembly, has already been shown to prevent metastasis in PDAC and colorectal cancer in vivo models (26, 27).
Major challenges in the field of cancer cortical mechanics would need to be addressed to use this knowledge to its full potential. The first is the full elucidation of the mechanoresponse system regulating cell morphogenesis and the subsequent cell behaviors. Work thus far has focused on specific proteins in various cancer types at various stages with no indication of changes in the remaining cytoskeletal components. Therefore, there is confusing and contradicting conclusions regarding these specific proteins. For example, NMII has been characterized as both a tumor suppressor and promotor, depending on the specific paralog, cancer type, and methods used. Additionally, studies focus on expression levels of proteins with simplified binary descriptions of high vs. low. Unfortunately, many of the cytoskeletal components are filamentous and/or need to be assembled to be functional, but also must maintain a free pool of subunits in order to facilitate rapid remodeling in response to various mechanical and signaling inputs. Therefore, expression levels do not inform us of the assembled or functional fractions of these components in the cytoskeleton vs. the cytoplasm. Moreover, a concept of an optimal setpoint for the function of these proteins is becoming increasingly fundamental, which is exemplified through NMII being able to drive cancer progression, while at the same time, being tumor suppressive in other cancer types. Collectively, simply targeting these systems using inhibitors is not the correct strategy. As we see in PDAC and colorectal cancer models, the strategy of pushing the system towards over-assembly, i.e., beyond the “optimal setpoint”, may block cancer progression, including metastasis, without inhibiting the protein’s tumor suppressive functions (26, 58).
Another major challenge is the siloed nature of many studies. The studies presented here have focused on cortical mechanics and just a few aspects of a cancer cell phenotypes, such as proliferation/migration, metabolism, and/or immune cell infiltration. For example, the melanoma study characterized proliferation, metastasis, and immune cell infiltration, but did not characterize the functionality or role of the immune cells in the tumor (75). We will need to understand the full integration of these concepts in order to draw conclusions on the impacts of any manipulation on the system. Finally, we need better characterization and identification of cancer stem cells within each cancer type. Undifferentiated cancer cells will remain a clinical problem due to their ability to resist various therapies, maintain migratory and proliferative capabilities, and further differentiate into tumor repopulating cells. Majority of cancer patient deaths are due to metastases, and we know metastasis results from persistent cancer cells that are able to micro- and macro-metastasize prior, during, and after therapies. Therefore, we need to develop methods to target the population of cancer stem cells in addition to the bulk tumor cells to fully address the disease long-term. Some cancers, such as PDAC, lack consensus on how to best identify these populations, creating yet another gap in our understanding. Without this understanding, we will not be able to treat clinical relapse.
In summary, aggressive cancers require innovative therapeutic approaches that can concurrently target the multiple systems driving disease progression to be effective. We propose that the field of cancer cortical mechanics is a prospective area for targetability since it encompasses foundational properties of cells that interact with multiple systems (proliferation, migration, invasion, differentiation, metabolism, immune evasion) driving disease progression, Figure 2. Work has already uncovered PDAC’s altered mechanical landscape at the molecular level and revealed targeting NMIIC assembly can prevent metastasis, but we need further integration of this work with other fields such as metabolism, cancer stem cells, and immune cell infiltration/interaction. The integration of these systems will provide an understanding for therapeutic development that will be applicable to numerous cancer types in addition to PDAC.
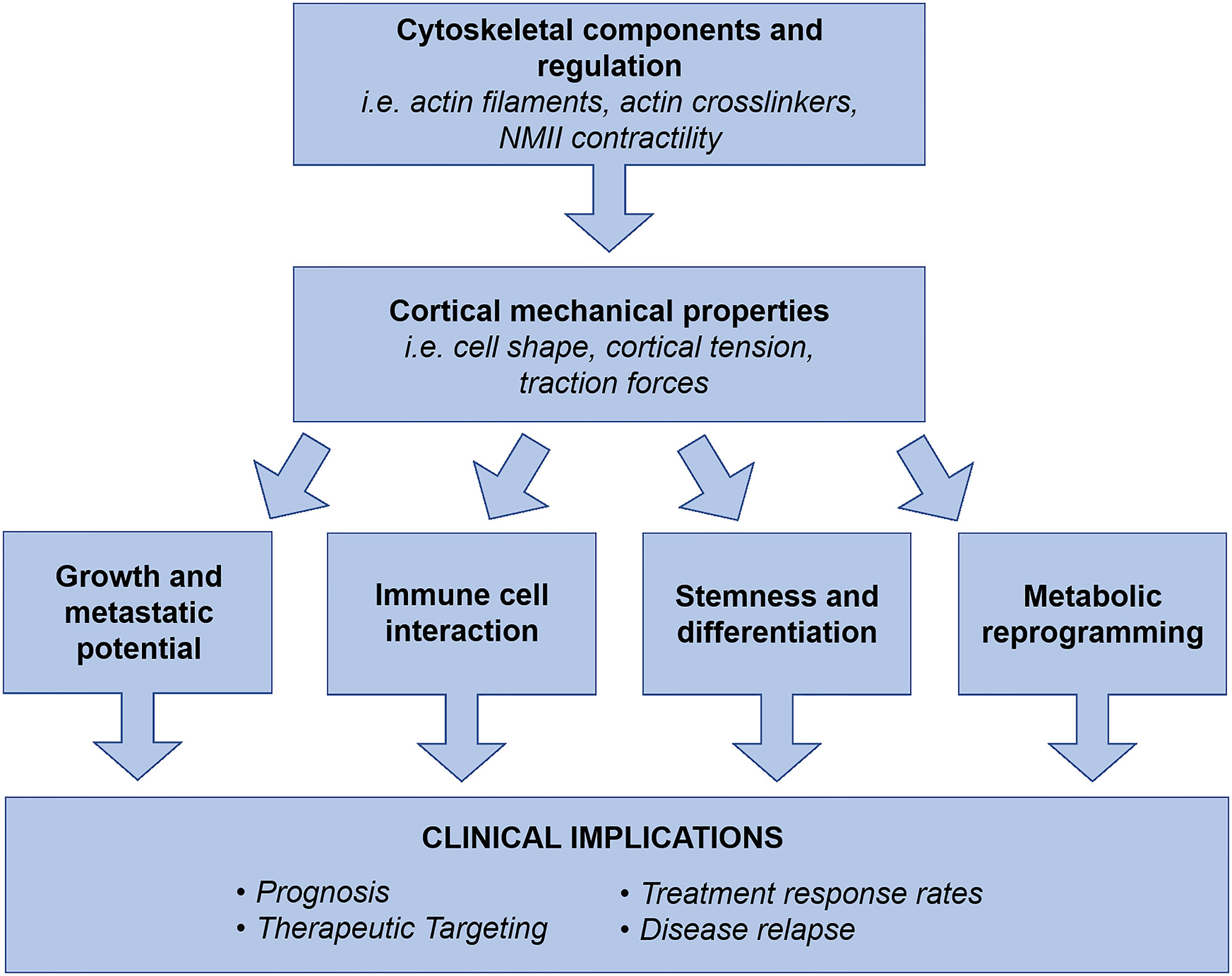
Figure 2 Cell cortical mechanics and its molecular drivers generate cell shape and morphogenesis, which collectively result in observed cell behaviors. The mechanical landscape and properties of cancer cells are significantly altered to drive metastasis and disease progression. Collectively, cytoskeletal components, regulators, and their resulting physical properties have been revealed as regulators of cancer cell growth and metastatic potential, TME interactions, stemness and differentiation, and metabolic reprogramming. Therefore, we propose the field of cancer cortical mechanics as a prospective area for exploration in disease prognosis, therapeutic targeting, elimination of therapeutic resistance, and prevention of clinical relapse. As a foundational system of cell behavior, cortical mechanics has the potential to concurrently address multiple drivers of disease progression, which is an ideal strategy for treating aggressive and unresponsive cancer types such as PDAC.
Author Contributions
SA: Conceptualization, writing (original draft), writing (review and editing), figure visualization and generation. QZ: Conceptualization, writing (review and editing). EJ: Writing (review and editing), funding acquisition, supervision. DR: Writing (review and editing), funding acquisition, supervision. RA: Writing (review and editing), funding acquisition, supervision. All authors contributed to the article and approved the submitted version.
Funding
Our research is supported by the NIH (National Institute of General Medical Sciences Grant R01 GM66817) and the Lustgarten Foundation for Pancreatic Cancer (Project 90094641).
Conflict of Interest
RA reports grants and personal fees from Bristol Myers Squibb, and personal fees from AstraZeneca and Merck SD. DR is currently exploring formation of a startup company. A patent has been granted on the use of 4-HAP to treat disease, and DR is an inventor on this patent. EJ reports grants from Bristol Myers Squibb and Lustgarten, as well as grants from Abmeta and Lustgarten, and personal fees from Genocea, Achilles, DragonFly, CSTONE, Candel Therapeutics, and Parker Institute.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank the members of the Robinson, Anders, and Jaffee labs for many helpful discussions, reviews, and edits.
References
1. Moeendarbary E, Harris AR. Cell Mechanics: Principles, Practices, and Prospects. Wiley Interdiscip Rev Syst Biol Med (2014) 6(5):371–88. doi: 10.1002/wsbm.1275
2. Chugh P, Paluch EK. The Actin Cortex at a Glance. J Cell Sci (2018) 131(14):jcs186254. doi: 10.1242/jcs.186254
3. Basoli F, Giannitelli SM, Gori M, Mozetic P, Bonfanti A, Trombetta M, et al. Biomechanical Characterization at the Cell Scale: Present and Prospects. Front Physiol (2018) 9:1449. doi: 10.3389/fphys.2018.01449
4. Harris MJ, Wirtz D, Wu PH. Dissecting Cellular Mechanics: Implications for Aging, Cancer, and Immunity. Sem Cell Dev Bio (2019) 93:16–25. doi: 10.1016/j.semcdb.2018.10.008
5. Luo T, Mohan K, Srivastava V, Ren Y, Iglesias PA, Robinson DN. Understanding the Cooperative Interaction Between Myosin II and Actin Cross-Linkers Mediated by Actin Filaments During Mechanosensation. Biophys J (2012) 102(2):238–47. doi: 10.1016/j.bpj.2011.12.020
6. Luo T, Mohan K, Iglesias PA, Robinson DN. Molecular Mechanisms of Cellular Mechanosensing. Nat Mater (2013) 12:1064–71. doi: 10.1038/nmat3772
7. Schiffhauer ES, Luo T, Mohan K, Srivastava V, Qian X, Griffis ER, et al. Mechanoaccumulative Elements of the Mammalian Actin Cytoskeleton. Curr Biol (2016) 26(11):1473–9. doi: 10.1016/j.cub.2016.04.007
8. Schiffhauer ES, Robinson DN. Mechanochemical Signaling Directs Cell-Shape Change. Biophys J (2017) 112(2):207–14. doi: 10.1016/j.bpj.2016.12.015
9. Pegoraro AF, Janmey P, Weitz DA. Mechanical Properties of the Cytoskeleton and Cells. Cold Spring Harb Perspect Biol (2017) 9(11):a022038. doi: 10.1101/cshperspect.a022038
10. Reinhart-King CA. Cancer Cell Mechanics. In: Janmey P, editor. Physical Sciences and Engineering Advances in Life Sciences and Oncology. Science Policy Reports. Cham:Springer (2012). Available at: https://doi.org/10.1007/978-3-319-17930-8_4.
11. Robinson DN, Kee YS, Luo T, Surcel A. Understanding How Dividing Cells Change Shape. In: Comprehensive Biophysics, vol. 7. Amsterdam: Elsevier Inc (2012). p. 48–72. Available at: https://doi.org/10.1016/B978-0-12-374920-8.00705-0.
12. Parajón E, Surcel A, Robinson DN. The Mechanobiome: A Goldmine for Cancer Therapeutics. Am J Physiol Cell Physiol (2021) 320:C306–23. doi: 10.1152/ajpcell.00409.2020
13. Spill F, Bakal C, Mak M. Mechanical and Systems Biology of Cancer. Comput Struct Biotechnol J (2018) 16:237–45. doi: 10.1016/j.csbj.2018.07.002
14. Kraning-Rush CM, Califano JP, Reinhart-King CA. Cellular Traction Stresses Increase With Increasing Metastatic Potential. PloS One (2012) 7(2):e32572. doi: 10.1371/journal.pone.0032572
15. Kopanska KS, Alcheikh Y, Staneva R, Vignjevic D, Betz T. Tensile Forces Originating From Cancer Spheroids Facilitate Tumor Invasion. PloS One (2016) 11(6):e0156442. doi: 10.1371/journal.pone.0156442
16. Wang H, Abhilash AS, Chen CS, Wells RG, Shenoy VB. Long-Range Force Transmission in Fibrous Matrices Enabled by Tension-Driven Alignment of Fibers. Biophys J (2014) 107(11):2592–603. doi: 10.1016/j.bpj.2014.09.044
17. Arjonen A, Kaukonen R, Ivaska J. Filopodia and Adhesion in Cancer Cell Motility. Cell Adh Migr (2011) 5(5):421–30. doi: 10.4161/cam.5.5.17723
18. Malandrino A, Trepat X, Kamm RD, Mak M. Dynamic Filopodial Forces Induce Accumulation, Damage, and Plastic Remodeling of 3D Extracellular Matrices. PloS Comput Biol (2019) 15(4):e1006684. doi: 10.1371/journal.pcbi.1006684
19. Wang N, Tytell J, Ingber D. Mechanotransduction at a Distance: Mechanically Coupling the Extracellular Matrix With the Nucleus. Nat Rev Mol Cell Biol (2009) 10:75–82. doi: 10.1038/nrm2594
20. Swift J, Ivanovska IL, Buxboim A, Harada T, Dingal PC, Pinter J, et al. Nuclear Lamin-A Scales With Tissue Stiffness and Enhances Matrix-Directed Differentiation. Sci (2013) 341(6149):1240104. doi: 10.1126/science.1240104
21. Denais CM, Gilbert RM, Isermann P, McGregor AL, Lindert M, Weigelin B, et al. Nuclear Envelope Rupture and Repair During Cancer Cell Migration. Sci (2016) 352(6283):353–8. doi: 10.1126/science.aad7297
22. Welch DR, Hurst DR. Defining the Hallmarks of Metastasis. Cancer Res (2019) 79(12):3011–27. doi: 10.1158/0008-5472.CAN-19-0458
23. Ujihara Y, Ono D, Nishitsuji K, Ito M, Sugita S, Nakamura M. B16 Melanoma Cancer Cells With Higher Metastatic Potential Are More Deformable at a Whole-Cell Level. Cell Mol Bioeng (2021) 14(4):309–20. doi: 10.1007/s12195-021-00677-ww
24. Nel I, Morawetz EW, Tschodu D, Käs JA, Aktas B. The Mechanical Fingerprint of Circulating Tumor Cells (CTCs) in Breast Cancer Patients. Cancers (2021) 13(5):1119. doi: 10.3390/cancers13051119
25. Luo D, Liu N, Chen Y, Peng Y, Yue T, Cao S, et al. Microfluidic Assessment of Drug Effects on Physical Properties of Androgen Sensitive and Non-Sensitive Prostate Cancer Cells. Micromachines (2021) 12(5):532. doi: 10.3390/mi12050532
26. Surcel A, Schiffhauer ES, Thomas DG, Zhu Q, DiNapoli K, Herbig M, et al. Targeting Mechanoresponsive Proteins in Pancreatic Cancer: 4-Hydroxyacetophenone Blocks Dissemination and Invasion by Activating MYH14. Cancer Res (2019) 79(18):4665–78. doi: 10.1158/0008-5472.CAN-18-3131
27. Bryan DS, Stack M, Krystofiak K, Cichoń U, Thomas DG, Surcel A, et al. 4-Hydroxyacetophenone Modulates the Actomyosin Cytoskeleton to Reduce Metastasis. Proc Natl Acad Sci USA (2020) 117(36):22423–9. doi: 10.1073/pnas.2014639117
28. Rangamani P, Fardin MA, Xiong Y, Lipshtat A, Rossier O, Sheetz MP, et al. Signaling Network Triggers and Membrane Physical Properties Control the Actin Cytoskeleton-Driven Isotropic Phase of Cell Spreading. Biophys J (2011) 100(4):845–57. doi: 10.1016/j.bpj.2010.12.3732
29. Meyers J, Craig J, Odde DJ. Potential for Control of Signaling Pathways via Cell Size and Shape. Curr Biol (2006) 16(17):1685–93. doi: 10.1016/j.cub.2006.07.056
30. Gevari MT, Aydemir G, Gharib G, Kutlu O, Uvet H, Ghorbani M, et al. Local Carpet Bombardment of Immobilized Cancer Cells With Hydrodynamic Cavitation. IEEE Access IEEE (2021) 9:14983–91. doi: 10.1109/ACCESS.2021.3052893ff.hal-03151148
31. Ron A, Azeloglu EU, Calizo RC, Hu M, Bhattacharya S, Chen Y, et al. Cell Shape Information Is Transduced Through Tension-Independent Mechanisms. Nat Commun (2017) 8(1):2145. doi: 10.1038/s41467-017-02218-45
32. Spill F, Andasari V, Mak M, Kamm RD, Zaman MH. Effects of 3D Geometries on Cellular Gradient Sensing and Polarization. Phys Biol (2016) 13(3):36008. doi: 10.1088/1478-3975/13/3/036008
33. Suresh S. Biomechanics and Biophysics of Cancer Cells. Acta Biomater (2007) 3:413–38. doi: 10.1016/j.actbio.2007.04.002
34. Mauro LA, Herman JM, Jaffee EM, Laheru DA. Chapter 81: Carcinoma of the Pancreas. In: Niederhuber JE, Armitage JO, Dorshow JH, Kastan MB, Tepper JE, editors. Abeloff’s Clinical Oncology, 5th. Philadelphia, Pa: Elsevier (2014).
35. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer Statistic. CA Cancer J Clin (2021) 71(1):7–33. doi: 10.3322/caac.21654
36. Yamakawa K, Ye J, Nakano-Narusawa Y, Matsuda Y. Pathological Changes in Pancreatic Carcinogenesis: A Review. Cancers (Basel) (2021) 13(4):686. doi: 10.3390/cancers13040686
37. Cancer Genome Atlas Research Network. Electronic Address:QW5kcmV3X0FndWlycmVARGZjaS5IYXJ2YXJkLkVkdQ==; Cancer Genome Atlas Research Network. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Can Cell (2017) 32(2):185–203.e13. doi: 10.1016/j.ccell.2017.07.007
38. Vaccaro V, Sperduti I, Milella M. FOLFIRINOX Versus Gemcitabine for Metastatic Pancreatic Cancer. N Engl J Med (2011) 365(8):768–9. doi: 10.1056/NEJMc1107627
39. Sarantis P, Koustas E, Papadimitropoulou A, Papavassiliou AG, Karamouzis MV. Pancreatic Ductal Adenocarcinoma: Treatment Hurdles, Tumor Microenvironment and Immunotherapy. World J Gastrointest Oncol (2020) 12(2):173–81. doi: 10.4251/wjgo.v12.i2.173
40. Le Large TYS, Bijlsma MF, Kazemier G, van Laarhoven HWM, Giovannetti E, Jimenez CR. Key Biological Processes Driving Metastatic Spread of Pancreatic Cancer as Identified by Multi-Omics Studies. Semin Cancer Biol (2017) 44:153–69. doi: 10.1016/j.semcancer.2017.03.008
41. Rhim AD, Mirek ET, Aiello NM, Maitra A, Bailey JM, McAllister F, et al. EMT and Dissemination Precede Pancreatic Tumor Formation. Cell (2012) 148:349–61. doi: 10.1016/j.cell.2011.11.025
42. Notta F, Chan-Seng-Yue M, Lemire M, Li Y, Wilson GW, Connor AA, et al. A Renewed Model of Pancreatic Cancer Evolution Based on Genomic Rearrangement Pattern. Nature (2016) 538(7625):378–82. doi: 10.1038/nature19823
43. Hayashi A, Fan J, Chen R, Ho Y, Makohon-Moore AP, Lecomte N, et al. A Unifying Paradigm for Transcriptional Heterogeneity and Squamous Features in Pancreatic Ductal Adenocarcinoma. Nat Can (2020) 1:59–74. doi: 10.1038/s43018-019-0010-1
44. Yarchoan M, Hopkins A, Jaffee EM. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N Engl J Med (2017) 377(25):2500–1. doi: 10.1056/NEJMc1713444
45. Uzunparmak B, Sahin IH. Pancreatic Cancer Microenvironment: A Current Dilemma. Clin Transl Med (2019) 8(1):2. doi: 10.1186/s40169-019-0221-1
46. Erkan M, Hausmann S, Michalski CW, Fingerle AA, Dobritz M, Kleeff J, et al. The Role of Stroma in Pancreatic Cancer: Diagnostic and Therapeutic Implications. Nat Rev Gastroenterol Hepatol (2012) 9:454–67. doi: 10.1038/nrgastro.2012.115
47. Karagiannis GS, Poutahidis T, Erdman SE, Kirsch R, Riddell RH, Diamandis EP. Cancer-Associated Fibroblasts Drive the Progression of Metastasis Through Both Paracrine and Mechanical Pressure on Cancer Tissue. Mol Cancer Res (2012) 10:1403–18. doi: 10.1158/1541-7786.MCR-12-0307
48. Binenbaum Y, Na’ara S, Gil Z. Gemcitabine Resistance in Pancreatic Ductal Adenocarcinoma. Drug Resist Update (2015) 23:55–68. doi: 10.1016/j.drup.2015.10.002
49. Rhim AD, Oberstein PE, Thomas DH, Mirek ET, Palermo CF, Sastra SA, et al. Stromal Elements Act to Restrain, Rather Than Support, Pancreatic Ductal Adenocarcinoma. Can Cell (2014) 25:735–47. doi: 10.1016/j.ccr.2014.04.021
50. Liu Q, Li Y, Niu Z, Zong Y, Wang M, Yao L, et al. Atorvastatin (Lipitor) Attenuates the Effects of Aspirin on Pancreatic Cancerogenesis and the Chemotherapeutic Efficacy of Gemcitabine on Pancreatic Cancer by Promoting M2 Polarized Tumor Associated Macrophages. J Exp Clin Cancer Res (2016) 35:33. doi: 10.1186/s13046-016-0304-4
51. Dijkgraaf EM, Heusinkveld M, Tummers B, Vogelpoel LT, Goedemans R, Jha V, et al. Chemotherapy Alters Monocyte Differentiation to Favor Generation of Cancer-Supporting M2 Macrophages in the Tumor Microenvironment. Cancer Res (2013) 73:2480–92. doi: 10.1158/0008-5472.CAN-12-3542
52. Karakhanova S, Link J, Heinrich M, Shevchenko I, Yang Y, Hassenpflug M, et al. Characterization of Myeloid Leukocytes and Soluble Mediators in Pancreatic Cancer: Importance of Myeloid-Derived Suppressor Cells. Oncoimmunology (2015) 4:e998519. doi: 10.1080/2162402X.2014.998519
53. Young K, Hughes DJ, Cunningham D, Starling N. Immunotherapy and Pancreatic Cancer: Unique Challenges and Potential Opportunities. Ther Adv Med Oncol (2018) 10:1758835918816281. doi: 10.1177/17588359188162811
54. Ho WJ, Jaffee EM, Zheng L. The Tumour Microenvironment in Pancreatic Cancer - Clinical Challenges and Opportunities. Nat Rev Clin Oncol (2020) 17(9):527–40. doi: 10.1038/s41571-020-0363-5
55. Staunton JR, Doss BL, Lindsay S, Ros R. Correlating Confocal Microscopy and Atomic Force Indentation Reveals Metastatic Cancer Cells Stiffen During Invasion Into Collagen I Matrices. Sci Rep (2016) 6:19686. doi: 10.1038/srep19686
56. Kim TH, Gill NK, Nyberg KD, Nguyen AV, Hohlbauch SV, Geisse NA, et al. Cancer Cells Become Less Deformable and More Invasive With Activation of Beta-Adrenergic Signaling. J Cell Sci (2016) 129:4563–75. doi: 10.1242/jcs.194803
57. Nguyen AV, Nyberg KD, Scott MB, Welsh AM, Nguyen AH, Wu N, et al. Stiffness of Pancreatic Cancer Cells Is Associated With Increased Invasive Potential. Integr Biol (Camb) (2016) 8:1232–45. doi: 10.1039/c6ib00135a
58. Surcel A, Ng WP, West-Foyle H, Zhu Q, Ren Y, Avery LB, et al. Pharmacological Activation of Myosin II Paralogs to Correct Cell Mechanics Defects. Proc Natl Acad Sci USA (2015) 112(5):1428–33. doi: 10.1073/pnas.1412592112
59. Nguyen AV, Trompetto B, Tan XHM, Scott MB, Hu KHH, Deeds E, et al. Differential Contributions of Actin and Myosin to the Physical Phenotypes and Invasion of Pancreatic Cancer Cells. Cell Mol Bioeng (2019) 13(1):27–44. doi: 10.1007/s12195-019-00603-1
60. Eric L, Yeo CJ, Lillemoe KD, Biedrzycki B, Kobrin B, Herman J, et al. A Lethally Irradiated Allogeneic Granulocyte-Macrophage Colony Stimulating Factor-Secreting Tumor Vaccine for Pancreatic Adenocarcinoma. Ann Surg (2011) 253:328–35. doi: 10.1097/sla.0b013e3181fd271c
61. Johnson BA, Yarchoan M, Lee V, Laheru DA, Jaffee EM. Strategies for Increasing Pancreatic Tumor Immunogenicity. Clin Can Res (2017) 23:1656–69. doi: 10.1158/1078-0432.ccr-16-2318
62. Jhaveri DT, Kim MS, Thompson ED, Huang L, Sharma R, Klein AP, et al. Using Quantitative Seroproteomics to Identify Antibody Biomarkers in Pancreatic Cancer. Can Immun Res (2016) 4:225–33. doi: 10.1158/2326-6066.cir-15-0200-t
63. Naydenov NG, Koblinski JE, Ivanov AI. Anillin is an Emerging Regulator of Tumorigenesis, Acting as a Cortical Cytoskeletal Scaffold and a Nuclear Modulator of Cancer Cell Differentiation. Cell Mol Life Sci (2021) 78(2):621–33. doi: 10.1007/s00018-020-03605-9
64. Straight AF, Field CM, Mitchison TJ. Anillin Binds Nonmuscle Myosin II and Regulates the Contractile Ring. Mol Biol Cell (2005) 16:193–201. doi: 10.1091/mbc.e04-08-0758
65. Zhao WM, Fang G. Anillin is a Substrate of Anaphase-Promoting Complex/Cyclosome (APC/C) That Controls Spatial Contractility of Myosin During Late Cytokinesis. J Biol Chem (2005) 280:33516–24. doi: 10.1074/jbc.m504657200
66. Wang D, Naydenov NG, Dozmorov MG, Koblinski JE, Ivanov AI. Anillin Regulates Breast Cancer Cell Migration, Growth, and Metastasis by Non-Canonical Mechanisms Involving Control of Cell Stemness and Differentiation. Breast Cancer Res (2020) 22:3. doi: 10.1186/s13058-019-1241-x
67. Wang D, Chadha GK, Feygin A, Ivanov AI. F-Actin Binding Protein, Anillin, Regulates Integrity of Intercellular Junctions in Human Epithelial Cells. Cell Mol Life Sci (2015) 72:3185–200. doi: 10.1007/s00018-015-1890-6
68. Budnar S, Husain KB, Gomez GA, Naghibosadat M, Varma A, Verma S, et al. Anillin Promotes Cell Contractility by Cyclic Resetting of RhoA Residence Kinetics. Dev Cell (2019) 49:894–906. doi: 10.1016/j.devcel.2019.04.031
69. Arnold TR, Shawky JH, Stephenson RE, Dinshaw KM, Higashi T, Huq F, et al. Anillin Regulates Epithelial Cell Mechanics by Structuring the Medial-Apical Actomyosin Network. eLife (2019) 8:e39065. doi: 10.7554/elife.39065
70. Reyes CC, Jin M, Breznau EB, Espino R, Delgado-Gonzalo R, Goryachev AB, et al. Anillin Regulates Cell-Cell Junction Integrity by Organizing Junctional Accumulation of Rho-GTP and Actomyosin. Curr Biol (2014) 24:1263–70. doi: 10.1016/j.cub.2014.04.021
71. Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D, et al. ONCOMINE: A Cancer Microarray Database and Integrated Data-Mining Platform. Neoplasia (2004) 6:1–6. doi: 10.1016/s1476-5586(04)80047-2
72. Chandrashekar DS, Bashel B, Balasubramanya SAH, Creighton CJ, Ponce-Rodriguez I, Chakravarthi B, et al. UALCAN: A Portal for Facilitating Tumor Subgroup Gene Expression and Survival Analyses. Neoplasia (2017) 19:649–58. doi: 10.1016/j.neo.2017.05.002
73. Srivastava SK, Bhardwaj A, Arora S, Singh S, Azim S, Tyagi N, et al. MYB Is a Novel Regulator of Pancreatic Tumour Growth and Metastasis. Br J Cancer (2015) 113:1694–703. doi: 10.1038/bjc.2015.400
74. Zubair H, Patel GK, Khan MA, Azim S, Zubair A, Singh S, et al. Proteomic Analysis of MYB-Regulated Secretome Identifies Functional Pathways and Biomarkers: Potential Pathobiological and Clinical Implications. J Proteome Res (2020) 19(2):794–804. doi: 10.1021/acs.jproteome.9b00641
75. Singh SK, Sinha S, Padhan J, Jangde N, Ray R, Rai V. MYH9 Suppresses Melanoma Tumorigenesis, Metastasis and Regulates Tumor Microenvironment. Med Oncol (2020) 37(10):88. doi: 10.1007/s12032-020-01413-6
76. Picariello HS, Kenchappa RS, Rai V, Crish JF, Dovas A, Pogoda K, et al. Myosin IIA Suppresses Glioblastoma Development in a Mechanically Sensitive Manner. Proc Natl Acad Sci USA (2019) 116(31):15550–9. doi: 10.1073/pnas.1902847116
77. Nguyen-Ngoc KV, Silvestri VL, Georgess D, Fairchild AN, Ewald AJ. Mosaic Loss of non-Muscle Myosin IIA and IIB Is Sufficient to Induce Mammary Epithelial Proliferation. J Cell Sci (2017) 130(19):3213–21. doi: 10.1242/jcs.208546
78. Kapoor A, Barai A, Thakur B, Das A, Patwardhan SR, Monteiro M, et al. Soft Drug-Resistant Ovarian Cancer Cells Migrate via Two Distinct Mechanisms Utilizing Myosin II-Based Contractility. Biochim Biophys Acta Mol Cell Res (2018) 1865(2):392–405. doi: 10.1016/j.bbamcr.2017.11.012
79. Norwood Toro LE, Wang Y, Condeelis JS, Jones JG, Backer JM, Bresnick AR. Myosin-IIA Heavy Chain Phosphorylation on S1943 Regulates Tumor Metastasis. Exp Cell Res (2018) 370(2):273–82. doi: 10.1016/j.yexcr.2018.06.028
80. Wang B, Qi X, Liu J, Zhou R, Lin C, Shangguan J, et al. MYH9 Promotes Growth and Metastasis via Activation of MAPK/AKT Signaling in Colorectal Cancer. J Cancer (2019) 10(4):874–84. doi: 10.7150/jca.27635
81. Takaki T, Montagner M, Serres MP, Russell M, Collinson L, Szuhai K, et al. Actomyosin Drives Cancer Cell Nuclear Dysmorphia and Threatens Genome Stability. Nat Commun (2017) 8:16013. doi: 10.1038/ncomms16013
82. Sipos A, Iván J, Bécsi B, Darula Z, Tamás I, Horváth D, et al. Myosin Phosphatase and RhoA-Activated Kinase Modulate Arginine Methylation by the Regulation of Protein Arginine Methyltransferase 5 in Hepatocellular Carcinoma Cells. Sci Rep (2017) 7:40590. doi: 10.1038/srep40590
83. Lin Z, Chen G, Zhang Y, He H, Liang Y, Ye J, et al. MicroRNA-30d Promotes Angiogenesis and Tumor Growth via MYPT1/c-JUN/VEGFA Pathway and Predicts Aggressive Outcome in Prostate Cancer. Mol Cancer (2017) 16:48. doi: 10.1186/s12943-017-0615-x
84. Zhang C, Li A, Li H, Peng K, Wei Q, Lin M, et al. PPP1R12A Copy Number Is Associated With Clinical Outcomes of Stage III CRC Receiving Oxaliplatin-Based Chemotherapy. Mediators Inflammation (2015) 2015:417184. doi: 10.1155/2015/417184
85. Wang F, Sun Y. Overexpression of Myosin Phosphatase Target Subunit 1 (MYPT1) Inhibits Tumor Progression and Metastasis of Gastric Cancer. Med Sci Monit (2018) 24:2508–17. doi: 10.12659/msm.906852
86. Jia Z, Zhu J, Zhuo Y, Li R, Qu H, Wang S, et al. Offsetting Expression Profiles of Prognostic Markers in Prostate Tumor vs. Its Microenvironment. Front Oncol (2019) 9:539. doi: 10.3389/fonc.2019.00539
87. Nyberg KD, Bruce SL, Nguyen AV, Chan CK, Gill NK, Kim T, et al. Predicting Cancer Cell Invasion by Single-Cell Physical Phenotyping. Integr Biol (Camb) (2018) 10(4):218–31. doi: 10.1039/c7ib00222j
88. Holenstein CN, Horvath A, Schär B, Shoenenberger AD, Bollhalder M, Goedecke N, et al. The Relationship Between Metastatic Potential and In Vitro Mechanical Properties of Osteosarcoma Cells. Mol Biol Cell (2019) 30(7):887–98. doi: 10.1091/mbc.E18-08-0545
89. Lambert AW, Pattabirama DR, Weinberg RA. Emerging Biological Principles of Metastasis. Cell (2017) 168:670–91. doi: 10.1016/j.cell.2016.11.037
90. Nguyen DX, Bos PD, Massague J. Metastasis: From Dissemination to Organ-Specific Colonization. Nat Rev Cancer (2009) 9:274–84. doi: 10.1038/nrc2622
91. Yates LR, Knappskog S, Wedge D, Farmery JHR, Gonzalez S, Martincorena I, et al. Genomic Evolution of Breast Cancer Metastasis and Relapse. Cancer Cell (2017) 32:169–184 e167. doi: 10.1016/j.ccell.2017.07.005
92. Hess KR, Varadhachary GR, Taylor SH, Wei W, Raber MN, Lenzi R, et al. Metastatic Patterns in Adenocarcinoma. Cancer (2006) 106:1624–33. doi: 10.1002/cncr.21778
93. Tang K, Xin Y, Li K, Chen X, Tan Y. Cell Cytoskeleton and Stiffness Are Mechanical Indicators of Organotropism in Breast Cancer. Biol (Basel) (2021) 10(4):259. doi: 10.3390/biology10040259
94. Kim ST, Takeuchi K, Sun ZY, Touma M, Castro CE, Fahmy A, et al. The Alpha Beta T Cell Receptor Is an Anisotropic Mechanosensor. J Biol Chem (2009) 284:31028–37. doi: 10.1074/jbc.M109.052712
95. Li YC, Chen BM, Wu PC, Cheng TL, Kao LS, Tao MH, et al. Cutting Edge: Mechanical Forces Acting on T Cells Immobilized via the TCR Complex can Trigger TCR Signaling. J Immunol (2010) 184(11):5959–63. doi: 10.4049/jimmunol.0900775
96. Husson J, Chemin K, Bohineust A, Hivroz C, Henry N. Force Generation Upon T Cell Receptor Engagement. PloS One (2011) 6(5):e19680. doi: 10.1371/journal.pone.0019680
97. Bashour KT, Gondarenko A, Chen H, Shen K, Liu X, Huse M, et al. CD28 and CD3 Have Complementary Roles in T-Cell Traction Forces. Proc Natl Acad Sci USA (2014) 111(6):2241–6. doi: 10.1073/pnas.1315606111
98. Yu Y, Smoligovets AA, Groves JT. Modulation of T Cell Signaling by the Actin Cytoskeleton. J Cell Sci (2013) 126(Pt 5):1049–58. doi: 10.1242/jcs.098210
99. Hammer JA 3rd, Burkhardt JK. Controversy and Consensus Regarding Myosin II Function at the Immunological Synapse. Curr Opin Immunol (2013) 25(3):300–6. doi: 10.1016/j.coi.2013.03.010
100. Yi J, Wu XS, Crites T, Hammer JA 3rd. Actin Retrograde Flow and Actomyosin II Arc Contraction Drive Receptor Cluster Dynamics at the Immunological Synapse in Jurkat T Cells. Mol Biol Cell (2012) 23(5):834–52. doi: 10.1091/mbc.E11-08-0731
101. Kumari S, Vardhana S, Cammer M, Curado S, Santos L, Sheetz MP, et al. T Lymphocyte Myosin IIA Is Required for Maturation of the Immunological Synapse. Front Immunol (2012) 3:230. doi: 10.3389/fimmu.2012.00230
102. Jacobelli J, Chmura SA, Buxton DB, Davis MM, Krummel MF. A Single Class II Myosin Modulates T Cell Motility and Stopping, But Not Synapse Formation. Nat Immunol (2004) 5(5):531–8. doi: 10.1038/ni1065
103. Comrie WA, Burkhardt JK. Action and Traction: Cytoskeletal Control of Receptor Triggering at the Immunological Synapse. Front Immunol (2016) 7:68. doi: 10.3389/fimmu.2016.00068
104. Kumari S, Curado S, Mayya V, Dustin ML. T Cell Antigen Receptor Activation and Actin Cytoskeleton Remodeling. Biochim Biophys Acta (2014) 1838(2):546–56. doi: 10.1016/j.bbamem.2013.05.004
105. Hui KL, Balagopalan L, Samelson LE, Upadhyaya A. Cytoskeletal Forces During Signaling Activation in Jurkat T-Cells. Mol Biol Cell (2015) 26(4):685–95. doi: 10.1091/mbc.E14-03-0830
106. Liu Y, Liang X, Dong W, Fang Y, Lv J, Zhang T, et al. Tumor-Repopulating Cells Induce PD-1 Expression in CD8(+) T Cells by Transferring Kynurenine and AhR Activation. Can Cell (2018) 33:480–94. doi: 10.1016/j.ccell.2018.02.005
107. Liu J, Tan Y, Zhang H, Zhang Y, Xu P, Chen J, et al. Soft Fibrin Gels Promote Selection and Growth of Tumorigenic Cells. Nat Mater (2012) 11:734–41. doi: 10.1038/nmat3361
108. Liu X, Liang X, Yin J, Lv K, Tang J, Ma T, et al. Blockade of IDO-Kynurenine-AhR Metabolic Circuitry Abrogates IFN-γ-Induced Immunologic Dormancy of Tumor-Repopulating Cells. Nat Commun (2017) 8:15207. doi: 10.1038/ncomms15207
109. Müller-Hermelink N, Braumüller H, Pichler B, Wieder T, Mailhammer R, Schaak K, et al. TNFR1 Signaling and IFN-γ Signaling Determine Whether T Cells Induce Tumor Dormancy or Promote Multistage Carcinogenesis. Can Cell (2008) 13:507–18. doi: 10.1016/j.ccr.2008.04.001
110. Li Y, Luo S, Ma R, Liu J, Xu P, Zhang H, et al. Upregulation of Cytosolic Phosphoenolpyruvate Carboxykinase Is a Critical Metabolic Event in Melanoma Cells That Repopulate Tumors. Cancer Res (2015) 75:1191–6. doi: 10.1158/0008-5472.CAN-14-2615
111. Liu Y, Zhang T, Zhang H, Li J, Zhou N, Fiskesund R, et al. Cell Softness Prevents Cytolytic T-Cell Killing of Tumor-Repopulating Cells. Cancer Res (2021) 81(2):476–88. doi: 10.1158/0008-5472.CAN-20-2569
112. Friedman D, Simmonds P, Hale A, Bere L, Hodson NW, White MRH, et al. Natural Killer Cell Immune Synapse Formation and Cytotoxicity Are Controlled by Tension of the Target Interface. J Cell Sci (2021) 134(7):jcs258570. doi: 10.1242/jcs.258570
113. Wayt J, Cartagena-Rivera A, Dutta D, Donaldson JG, Waterman CM. Myosin II Isoforms Promote Internalization of Spatially Distinct Clathrin-Independent Endocytosis Cargoes Through Modulation of Cortical Tension Downstream of ROCK2. Mol Biol Cell (2021) 32(3):226–36. doi: 10.1091/mbc.E20-07-0480
114. Plaks V, Kong N, Werb Z. The Cancer Stem Cell Niche: How Essential Is the Niche in Regulating Stemness of Tumor Cells? Cell Stem Cell (2015) 16(3):225–38. doi: 10.1016/j.stem.2015.02.015
115. Gao X, Zhang M, Tang Y, Liang X. Cancer Cell Dormancy: Mechanisms and Implications of Cancer Recurrence and Metastasis. OncoTargets Ther (2017) 10:5219–28. doi: 10.2147/OTT.S140854
116. Naumov GN, Townson JL, MacDonald IC, Wilson SM, Bramwell VHC, Groom AC, et al. Ineffectiveness of Doxorubicin Treatment on Solitary Dormant Mammary Carcinoma Cells or Late-Developing Metastases. Breast Cancer Res Treat (2003) 82(3):199– 206. doi: 10.1023/B:BREA.0000004377.12288.3c
117. Ghajar CM, Peinado H, Mori H, Matie M, Evason KJ, Brazier H, et al. The Perivascular Niche Regulates Breast Tumour Dormancy. Nat Cell Biol (2013) 15(7):807–817. doi: 10.1038/ncb2767
118. Talukdar S, Bhoopathi P, Emdad L, Das S, Sarkar D, Fisher PB. Dormancy and Cancer Stem Cells: An Enigma for Cancer Therapeutic Targeting. Adv Cancer Res (2019) 141:43–84. doi: 10.1016/bs.acr.2018.12.002
119. Gordon RR, Nelson PS. Cellular Senescence and Cancer Chemotherapy Resistance. Drug Resist Update (2012) 15(1-2):123–31. doi: 10.1016/j.drup.2012.01.002
120. Görner K, Bachmann J, Holzhauer C, Kirchner R, Raba K, Fiscer JC, et al. Genetic Analysis of Circulating Tumor Cells in Pancreatic Cancer Patients: A Pilot Study. Genomics (2015) 106(1):7–14. doi: 10.1016/j.ygeno.2015.02.003
121. Quesnel B. Tumor Dormancy and Immunoescape. APMIS (2008) 116(7-8):685–94. doi: 10.1111/j.1600-0463.2008.01163.x
122. Dunn GP, Old LJ, Schreiber RD. The Three Es of Cancer Immunoediting. Annu Rev Immunol (2004) 22:329–60. doi: 10.1146/annurev.immunol.22.012703.104803
123. Ge W, Ma X, Li X, Wang Y, Li C, Meng H, et al. B7-H1 Up-Regulation on Dendritic-Like Leukemia Cells Suppresses T Cell Immune Function Through Modulation of IL-10/IL-12 Production and Generation of Treg Cells. Leuk Res (2009) 33(7):948–57. doi: 10.1016/j.leukres.2009.01.007
124. Teng MW, Swann JB, Koebel CM, Schreiber RD, Smyth MJ. Immune-Mediated Dormancy: An Equilibrium With Cancer. J Leukoc Biol (2008) 84(4):988–93. doi: 10.1189/jlb.1107774
125. Kim RS, Avivar-Valderas A, Estrada Y, Bragado P, Sosa MS, Aguirre-Ghiso JA, et al. Dormancy Signatures and Metastasis in Estrogen Receptor Positive and Negative Breast Cancer. PloS One (2012) 7(4):e35569. doi: 10.1371/journal.pone.0035569
126. Tibbitt MW, Anseth KS. Hydrogels as Extracellular Matrix Mimics for 3D Cell Culture. Biotechnol Bioeng (2009) 103(4):655– 663. doi: 10.1002/bit.22361
127. Keeratichamroen S, Lirdprapamongkol K, Svasti J. Mechanism of ECM-Induced Dormancy and Chemoresistance in A549 Human Lung Carcinoma Cells. Oncol Rep (2018) 39(4):1765– 1774. doi: 10.3892/or.2018.6258
128. Pradhan S, Slater JH. Tunable Hydrogels for Controlling Phenotypic Cancer Cell States to Model Breast Cancer Dormancy and Reactivation. Biomat (2019) 215:119177. doi: 10.1016/j.biomaterials.2019.04.022
129. Ovadia EM, Pradhan L, Sawicki LA, Cowart JE, Huber RE, Polson SW, et al. Understanding ER+ Breast Cancer Dormancy Using Bioinspired Synthetic Matrices for Long-Term 3D Culture and Insights Into Late Recurrence. Adv Biosyst (2020) 4(9):2000119. doi: 10.1002/adbi.202000119
130. Lam H-M, Vessella RL, Morrissey C. The Role of the Microenvironment-Dormant Prostate Disseminated Tumor Cells in the Bone Marrow. Drug Discovery Today Technol (2014) 11:41– 47. doi: 10.1016/j.ddtec.2014.02.002
131. Reynolds DS, Bougher KM, Letendre JH, Fitzgerald SF, Gisladottir UO, Grinstaff MW, et al. Mechanical Confinement via a PEG/collagen Interpenetrating Network Inhibits Behavior Characteristic of Malignant Cells in the Triple Negative Breast Cancer Cell Line MDA.MB.231. Acta Biomater (2018) 77:85– 95. doi: 10.1016/j.actbio.2018.07.032
132. Lam T, Aguirre-Ghiso JA, Geller MA, Aksan A, Azarin SM. Immobilization Rapidly Selects for Chemoresistant Ovarian Cancer Cells With Enhanced Ability to Enter Dormancy. Biotechnol Bioeng (2020) 117(10):3066– 3080. doi: 10.1002/bit.27479
133. Hurst RE, Kyker KD, Bonner RB, Bowditch RD, Hemstreet GP III. Matrix-Dependent Plasticity of the Malignant Phenotype of Bladder Cancer Cells. Anticancer Res (2003) 23(4):3119– 3128.
134. Preciado J, Lam T, Azarin SM, Lou E, Aksan A. Induction of Dormancy by Confinement: An Agarose-Silica Biomaterial for Isolating and Analyzing Dormant Cancer Cells. J BioMed Mater Res B Appl Biomater (2021) 109(12):2117–30. doi: 10.1002/jbm.b.34859
135. Lv J, Liu Y, Cheng F, Li J, Zhou Y, Zhang T, et al. Cell Softness Regulates Tumorigenicity and Stemness of Cancer Cells. EMBO J (2021) 40(2):e106123. doi: 10.15252/embj.2020106123
136. Lv J, Liu Y, Huang B. Mechanical Softness: A True Stemness Feature for Cancer Cells. Mol Cell Oncol (2021) 8(2):1882285. doi: 10.1080/23723556.2021.1882285
137. Xiao W, Gao Z, Duan Y, Yuan W, Ke Y. Notch Signaling Plays a Crucial Role in Cancer Stem-Like Cells Maintaining Stemness and Mediating Chemotaxis in Renal Cell Carcinoma. J Exp Clin Cancer Res (2017) 36(1):41. doi: 10.1186/s13046-017-0507-3
138. Walker A, Su H, Conti MA, Harb N, Adelstein RS, Sato N. Non-Muscle Myosin II Regulates Survival Threshold of Pluripotent Stem Cells. Nat Commun (2010) 1:71. doi: 10.1038/ncomms1074
139. Conti MA, Even-Ram S, Liu C, Yamada KM, Adelstein RS. Defects in Cell Adhesion and the Visceral Endoderm Following Ablation of Nonmuscle Myosin Heavy Chain II-A in Mice. J Biol Chem (2004) 279:41263–6. doi: 10.1074/jbc.C400352200
140. Hesse M, Raulf A, Pilz GA, Haberlandt C, Klein AM, Jabs R, et al. Direct Visualization of Cell Division Using High-Resolution Imaging of M-Phase of the Cell Cycle. Nat Commun (2012) 3:1076. doi: 10.1038/ncomms2089
141. Tulina N, Matunis E. Control of Stem Cell Self-Renewal in Drosophila Spermatogenesis by JAK-STAT Signaling. Science (2001) 294:2546–9. doi: 10.1126/science.1066700
142. Paolini A, Duchemin AL, Albadri S, Patzel E, Bornhorst D, Gonzalez Avalos P, et al. Asymmetric Inheritance of the Apical Domain and Self-Renewal of Retinal Ganglion Cell Progenitors Depend on Anillin Function. Development (2015) 142:832–9. doi: 10.1242/dev.118612
143. Johung K, Goodwin EC, DiMaio D. Human Papillomavirus E7 Repression in Cervical Carcinoma Cells Initiates a Transcriptional Cascade Driven by the Retinoblastoma Family, Resulting in Senescence. J Virol (2007) 81:2102–16. doi: 10.1128/jvi.02348-06
144. Marthandan S, Baumgart M, Priebe S, Groth M, Schaer J, Kaether C, et al. Conserved Senescence Associated Genes and Pathways in Primary Human Fibroblasts Detected by RNA-Seq. PloS One (2016) 11:e0154531. doi: 10.1371/journal.pone.0154531
145. Cepero Malo M, Duchemin AL, Guglielmi L, Patzel E, Sel S, Auffarth GU, et al. The Zebrafish anillin-eGFP Reporter Marks Late Dividing Retinal Precursors and Stem Cells Entering Neuronal Lineages. PloS One (2017) 12:e0170356. doi: 10.1371/journal.pone.0170356
146. Beach JR, Hussey GS, Miller TE, Chaudhury A, Patel P, Monslow J, et al. Myosin II Isoform Switching Mediates Invasiveness After TGF-Beta-Induced Epithelial-Mesenchymal Transition. Proc Natl Acad Sci USA (2011) 108(44):17991–6. doi: 10.1073/pnas.1106499108
147. Dulyaninova NG, House RP, Betapudi V, Bresnick AR. Myosin-IIA Heavy-Chain Phosphorylation Regulates the Motility of MDA-MB-231 Carcinoma Cells. Mol Biol Cell (2007) 18:3144–55. doi: 10.1091/mbc.E06-11-1056
148. Dulyaninova NG, Bresnick AR. The Heavy Chain Has Its Day: Regulation of Myosin-II Assembly. Bioarchitecture (2013) 3(4):77–85. doi: 10.4161/bioa.26133
149. Raab M, Swift J, Dingal PC, Shah P, Shin JW, Discher DE. Crawling From Soft to Stiff Matrix Polarizes the Cytoskeleton and Phosphoregulates Myosin-II Heavy Chain. J Cell Biol (2012) 199:669–83. doi: 10.1083/jcb.201205056
150. Pecci A, Ma X, Savoia A, Adelstein RS. MYH9: Structure, Functions and Role of Non-Muscle Myosin IIA in Human Disease. Gene (2018) 664:152–67. doi: 10.1016/j.gene.2018.04.048
151. Gentric G, Mieulet V, Mechta-Grigoriou F. Heterogeneity in Cancer Metabolism: New Concepts in an Old Field. Antioxid Redox Signal (2017) 26(9):462–85. doi: 10.1089/ars.2016.6750
152. Shao W, Hwang J, Liu C, Mukhopadhyay D, Zhoa S, Shen M, et al. Serum Lipoprotein-Derived Fatty Acids Regulate Hypoxia-Inducible Factor. J Biol Chem (2020) 295(52):18284–300. doi: 10.1074/jbc.RA120.015238
153. Esquejo RM, Roqueta-Rivera M, Shao W, Phelan PE, Seneviratne U, Am Ende CW, et al. Dipyridamole Inhibits Lipogenic Gene Expression by Retaining SCAP-SREBP in the Endoplasmic Reticulum. Cell Chem Biol (2021) 28(2):169–179.e7. doi: 10.1016/j.chembiol.2020.10.003
154. Williamson T, de Abreu MC, Trembath DG, Brayton C, Kang B, Mendes TB, et al. Mebendazole Disrupts Stromal Desmoplasia and Tumorigenesis in Two Models of Pancreatic Cancer. Oncotarget (2021) 12(14):1326–38. doi: 10.18632/oncotarget.28014
155. Wang LB, Karpova A, Gritsenko MA, Kyle JE, Cao S, Li Y, et al. Proteogenomic and Metabolomic Characterization of Human Glioblastoma. Cancer Cell (2021) 39(4):509–528.e20. doi: 10.1016/j.ccell.2021.01.006
156. Yamashita AS, da Costa Rosa M, Borodovsky A, Festuccia WT, Chan T, Riggins GJ. Demethylation and Epigenetic Modification With 5-Azacytidine Reduces IDH1 Mutant Glioma Growth in Combination With Temozolomide. Neuro Oncol (2019) 21(2):189–200. doi: 10.1093/neuonc/noy146
157. Lopez-Bujanda ZA, Obradovic A, Nirschl TR, Crowley L, Macedo R, Papachristodoulou A, et al. TGM4: An Immunogenic Prostate-Restricted Antigen. J Immunother Cancer (2021) 9(6):e001649. doi: 10.1136/jitc-2020-001649
158. Manukian G, Kivolowitz C, DeAngelis T, Shastri AA, Savage JE, Camphausen K, et al. Caloric Restriction Impairs Regulatory T Cells Within the Tumor Microenvironment After Radiation and Primes Effector T Cells. Int J Radiat Oncol Biol Phys (2021) 110(5):1341–9. doi: 10.1016/j.ijrobp.2021.02.029
159. Ahmed H, Ajat M, Mahmood RI, Mansor R, Razak ISA, Al-Obaidi JR, et al. LC-MS/MS Proteomic Study of MCF-7 Cell Treated With Dox and Dox-Loaded Calcium Carbonate Nanoparticles Revealed Changes in Proteins Related to Glycolysis, Actin Signalling, and Energy Metabolism. Biol (Basel) (2021) 10(9):909. doi: 10.3390/biology10090909
160. Hua W, Kostidis S, Mayboroda O, Giera M, Hornsveld M, Ten Dijke P. Metabolic Reprogramming of Mammary Epithelial Cells During TGF-β-Induced Epithelial-To-Mesenchymal Transition. Metabolites (2021) 11(9):626. doi: 10.3390/metabo11090626
161. Sereda EE, Kolegova ES, Kakurina GV, Sidenko EA, Korshunov DA, Kondakova IV. Relationship Estimation of Cell Mobility Proteins Level With Processes of Proteolysis and Lymphogenic Metastasis in Breast Cancer. Dokl Biochem Biophys (2021) 499(1):211–4. doi: 10.1134/S1607672921040153
162. Ren L, Hong SH, Chen QR, Briggs J, Cassavaugh J, Srinivasan S, et al. Dysregulation of Ezrin Phosphorylation Prevents Metastasis and Alters Cellular Metabolism in Osteosarcoma. Cancer Res (2012) 72(4):1001–12. doi: 10.1158/0008-5472.CAN-11-0210
163. Lin S, Li Y, Wang D, Huang C, Marino D, Bollt O, et al. Fascin Promotes Lung Cancer Growth and Metastasis by Enhancing Glycolysis and PFKFB3 Expression. Cancer Lett (2021) 518:230–42. doi: 10.1016/j.canlet.2021.07.025
164. Na N, Li H, Xu CF, Miao B, Hong LQ, Huang ZY, et al. High Expression of Aldolase a Predicts Poor Survival in Patients With Clear-Cell Renal Cell Carcinoma. Ther Clin Risk Manag (2017) 13:279–85. doi: 10.2147/Tcrm.S123199
165. Du S, Guan ZZ, Hao LH, Song Y, Wang L, Gong LL, et al. Fructose-Bisphosphate Aldolase A Is a Potential Metastasis-Associated Marker of Lung Squamous Cell Carcinoma and Promotes Lung Cell Tumorigenesis and Migration. PloS One (2014) 9(1):e85804. doi: 10.1371/journal.pone.0085804
166. Li JT, Wang YP, Yin M, Lei QY. Metabolism Remodeling in Pancreatic Ductal Adenocarcinoma. Cell Stress (2019) 3(12):361–8. doi: 10.15698/cst2019.12.205
167. Li X, Jiang FX, Ge Z, Chen B, Yu J, Xin MJ, et al. Fructose-Bisphosphate Aldolase A Regulates Hypoxic Adaptation in Hepatocellular Carcinoma and Involved With Tumor Malignancy. Dig Dis Sci (2019) 64(11):3215–27. doi: 10.1007/s10620-019-05642-2
168. Sun J, He D, Fu Y, Zhang R, Guo H, Wang Z, et al. A Novel lncRNA ARST Represses Glioma Progression by Inhibiting ALDOA-Mediated Actin Cytoskeleton Integrity. J Exp Clin Cancer Res (2021) 40(1):187. doi: 10.1186/s13046-021-01977-9
Keywords: PDAC, cytoskeleton, cortical mechanics, cell shape, clinical implications
Citation: Angstadt S, Zhu Q, Jaffee EM, Robinson DN and Anders RA (2022) Pancreatic Ductal Adenocarcinoma Cortical Mechanics and Clinical Implications. Front. Oncol. 12:809179. doi: 10.3389/fonc.2022.809179
Received: 04 November 2021; Accepted: 05 January 2022;
Published: 31 January 2022.
Edited by:
Liang Qiao, Westmead Institute for Medical Research, AustraliaReviewed by:
Saravanakumar Marimuthu, University of Nebraska Medical Center, United StatesAlessandro Poli, IFOM—The FIRC Institute of Molecular Oncology, Italy
Copyright © 2022 Angstadt, Zhu, Jaffee, Robinson and Anders. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Douglas N. Robinson, ZG5yQGpobWkuZWR1; Robert A. Anders, cmFuZGVyNTRAamhtaS5lZHU=