 Maria Valeria Freire1
Maria Valeria Freire1 Marie Martin1
Marie Martin1 Romain Thissen1
Romain Thissen1 Cédric Van Marcke2,3Karin Segers1Edith Sépulchre1
Cédric Van Marcke2,3Karin Segers1Edith Sépulchre1 Natacha Leroi1Céline Lété1Corinne Fasquelle1Jean Radermacher4Yeter Gokburun5Joelle Collignon6Anne Sacré7
Natacha Leroi1Céline Lété1Corinne Fasquelle1Jean Radermacher4Yeter Gokburun5Joelle Collignon6Anne Sacré7 Claire Josse6
Claire Josse6 Leonor Palmeira1
Leonor Palmeira1 Vincent Bours1*
Vincent Bours1*- 1Department of Human Genetics, GIGA Research Center – University of Liège and Centre Hospitalier Universitaire (CHU) Liège, Liège, Belgium
- 2Institute for Experimental and Clinical Research (Institut de Recherche Expérimentale et Clinique (IREC), Pôle Molecular Imaging, Radiotherapy and Oncology (MIRO)), Université Catholique de Louvain (UCLouvain), Brussels, Belgium
- 3Department of Medical Oncology, Institut Roi Albert II, Cliniques Universitaires Saint‐Luc, Brussels, Belgium
- 4Department of Pathology, Institut de Pathologie et de Génétique, Charleroi, Belgium
- 5Department of Gastroenterology, Centre Hospitalier Régional Sambre et Meuse, Namur, Belgium
- 6Department of Medical Oncology, GIGA Research Center – University of Liège and Centre Hospitalier Universitaire (CHU) Liège, Liège, Belgium
- 7Onco-Hematology Department, Centre Hospitalier Régional (CHR) Verviers, Verviers, Belgium
Objective: The link between BRCA1 and homologous recombination deficiency (HRD) in cancer has gained importance with the emergence of new targeted cancer treatments, while the available data on the role of the gene in colorectal cancer (CRC) remain contradictory. The aim of this case series was to elucidate the role of known pathogenic BRCA1 variants in the development of early-onset CRC.
Design: Patients were evaluated using targeted next generation sequencing, exome sequencing and chromosomal microarray analysis of the paired germline and tumor samples. These results were used to calculate the HRD score and the frequency of mutational signatures in the tumors.
Results: Three patients with metastatic CRC were heterozygous for a previously known BRCA1 nonsense variant. All tumors showed remarkably high HRD scores, and the HRD-related signature 3 had the second highest contribution to the somatic pattern of variant accumulation in the samples (23% in 1 and 2, and 13% in sample 3).
Conclusions: A BRCA1 germline pathogenic variant can be involved in CRC development through HRD. Thus, BRCA1 testing should be considered in young patients with a personal history of microsatellite stable CRC as this could further allow a personalized treatment approach.
Introduction
BRCA1 is a tumor suppressor gene encoding a large protein that coordinates several cellular pathways including DNA repair, transcriptional regulation, cell-cycle control, centrosome duplication, and apoptosis (1). Pathogenic germline variants in BRCA1 gene have been associated with familial risk of breast and ovarian cancers (OMIM: 604370) (2, 3). As early as in 1994, it was observed that women with a history of breast, endometrial, or ovarian cancer presented a statistically significant although small risk for subsequent colorectal cancer (CRC), suggesting the existence of common etiologic factors for the development of these tumors (4).
Data concerning young patients with BRCA1 variants that develop CRC have been scarce. Germline pathogenic variants in BRCA1 gene have not been causally linked to an increased risk of familial colorectal cancer, but the reports on the subject are contradictory (5–9). Indeed, patients carrying a germline BRCA1 variant can develop a sporadic tumor, independently of BRCA1 loss of function, highlighting the need to demonstrate the causal role of the variant in the cancer development (10).
The aim of this case series was to gain insight into the role of known pathogenic BRCA1 variants in the development of early-onset CRC.
Case Description
Three patients were diagnosed in 2020 and 2021 with aggressive early-onset CRC. The demographic, familial, clinical, histopathological, and molecular characteristics, as well as the treatment regimens of these patients are presented in Table 1.
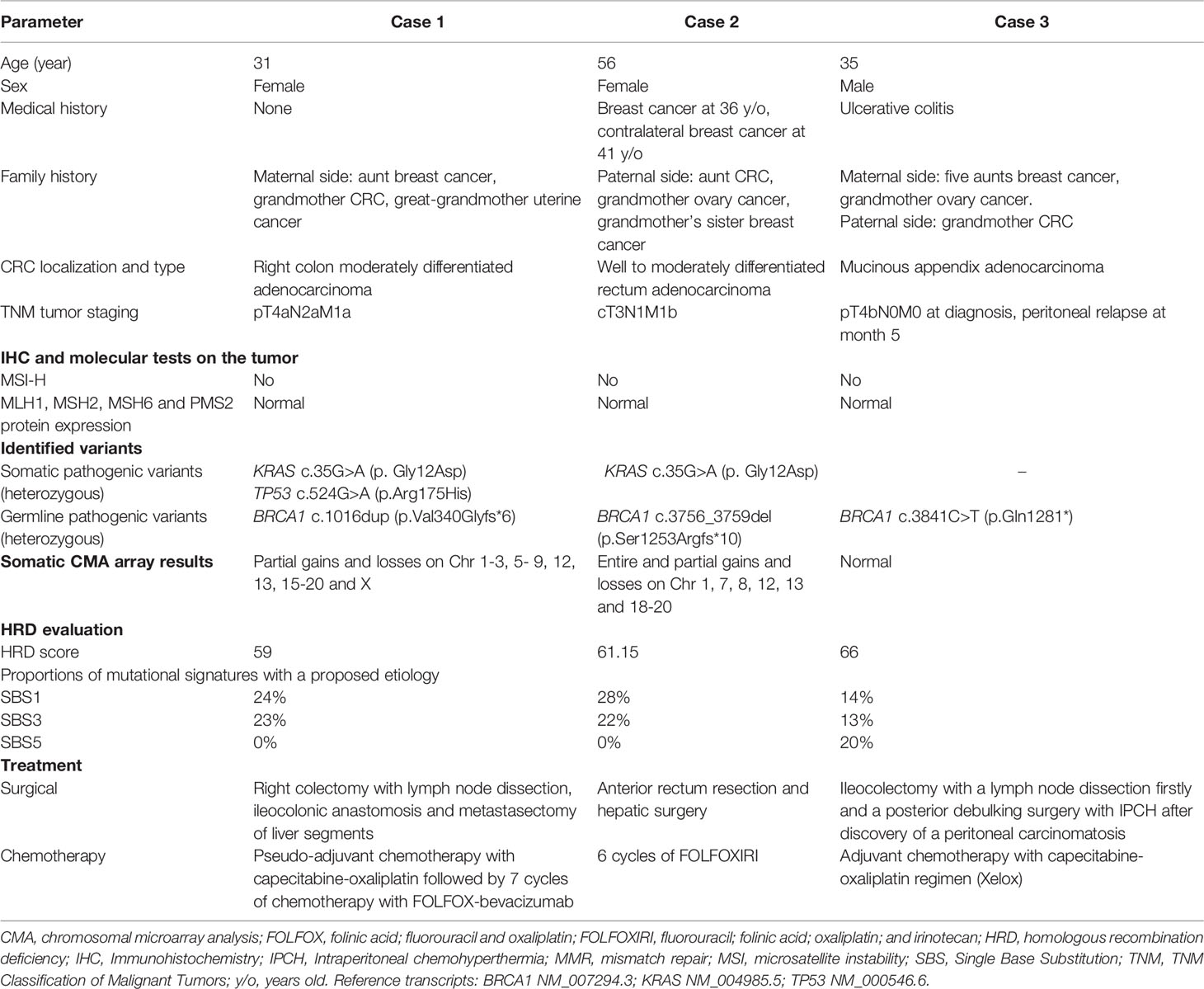
Table 1 Patient characteristics.
The first case was referred to oncogenetic consultation due to the young age of presentation of an aggressive disease without evidence of Lynch syndrome (no mismatch repair deficiency or microsatellite instability) and history of a BRCA1 pathogenic variant in the family. Given the age at the diagnosis of CRC, genes associated with familial polyposis (NTHL1, RNF43, SMAD4, BMPR1A), CRC (POLE, POLD1) and Li-Fraumeni syndrome (TP53) were analyzed. However, the patient only carried the heterozygous BRCA1 pathogenic variant NM_007294.3(BRCA1_v001):c.1016dup (p.Val340Glyfs*6) identified in her maternal aunt.
To further evaluate the disease, targeted next-generation sequencing (NGS) and a high-resolution (180K) chromosomal microarray analysis (CMA) were performed on the DNA extracted from the tumor (estimated proportion of tumor cells in the sample - 50%). After sequencing, the familial pathogenic variant BRCA1 c.1016dup was identified at an allele frequency (AF) of 70%, suggesting a loss of heterozygosity at the BRCA1 locus. Further analysis revealed a somatic variant of TP53 NM_000546.6(TP53):c.524G>A (p.Arg175His) at an AF of 40%. The CMA showed multiple rearrangements indicating genomic instability (chromosomal partial gains and losses on chromosomes 1-3, 5- 9, 12, 13, 15-20 and X).
The personal and family history of cancer in case 2 already led in 2011 to the identification of the pathogenic BRCA1 germline variant NM_007294.3(BRCA1_v001):c.3756_3759del (p.Ser1253Argfs*10). Taking this information into account, a CMA and NGS of the tumor DNA (estimated tumor infiltration – 30%) were performed, identifying the known germline BRCA1 variant with an AF of 35% and an additional NM_004985.5(KRAS_v001):c.35G>A variant with an AF of 23%. The CMA results were monosomies 18 and 19, trisomies 1q, 7, 8, 12, 13 and 20, partial chromosomal losses in the 1p region and partial chromosomal gains in the 1p region.
In case 3, CRC was diagnosed from a surgical specimen obtained after an appendectomy with the subsequent identification of a tumor-like lesion with low-grade dysplasia at the base of the cecum. Considering that the patient’s mother carried a BRCA1 germline variant, the patient DNA was tested, confirming the presence of the heterozygous BRCA1 pathogenic variant NM_007294.3(BRCA1_v001):c.3841C>T (p.Gln1281*). Subsequently, BRCA1 sequencing and CMA array on tumor DNA (sample estimated tumor infiltration – 20%) showed the BRCA1 c.3841C>T family variant with an AF of 43%, while the CMA was normal.
The three variants are predicted to cause truncation of the translation in exon 10 (out of a total of 23) which will result in a severely shortened or absent protein due to nonsense-mediated decay of the mRNA. BRCA1 protein truncations downstream of this position have been described as pathogenic (11, 12). BRCA1 c.1016dup and BRCA1 c.3841C>T variants were absent in 251174 control chromosomes in gnomAD, whereas BRCA1 c.3756_3759del was present at an AF of 1.267e-05. BRCA1 c.1016dupA has been reported in the literature as a founder variant in Norway and Canada (13, 14) and also in multiple individuals affected with hereditary breast and ovarian cancer syndrome in other populations (15–18). Case 2 four-nucleotide deletion was widely reported in the literature in Polish and French-Canadian gynecological cancer patients (19, 20). The BRCA1 variant present in case 3 has been reported as a France, Belgium, and Holland founder variant (21). ClinVar submitters including an expert panel (ENIGMA) cite the three variants as pathogenic. These data indicate that the three variants are highly likely to be associated with high breast and ovarian cancer risk.
Homologous recombination deficiency (HRD) evaluation can be performed using HRD score, an aggregate score of loss of heterozygosity (LOH), telomeric-allelic imbalance (TAI) and large-scale state transitions (LST). To confirm the HRD score in the CRC samples we used an alternative method of HRD detection by investigating single base substitution (SBS) signatures.
To assess homologous recombination deficiency (HRD) in CRC samples, a paired germline and tumoral DNA exome sequencing using Twist Comprehensive Exome Panel and Twist Human RefSeq Panel (according to the manufacturer’s instructions) from all three patients was performed. We used Sequenza (22) to detect and quantify copy number variation and estimate tumor cellularity and ploidy. These results were used as an input to calculate the HRD score with a threshold of positivity ≥33 (23). Mutational signatures in the samples were analyzed Using MutationalPatterns R package (24) and COSMIC v2 signatures (25), taking only the somatic variants into account.
Through Sequenza, the estimated tumor cellularity was of 95% in the case 3 sample, while this value was lower for cases 1 and 2 – 22% and 27%, respectively. All three samples showed remarkably high HRD scores (59, 61.15 and 66, respectively), while no somatic copy number alteration was identified in PALB2, BRCA1 and BRCA2.
The three most frequent SBS signatures with a proposed etiology in the samples were SBS1, 3 and 5 (see Figure 1). Signature 3 was the second most frequent signature with a contribution to 23% of the somatic pattern of variant accumulation in samples 1 and 2, and 13% in sample 3. While signature 1 and 5 reflect clock-like accumulation of somatic variants, signature 3 has been directly related to HRD (25).
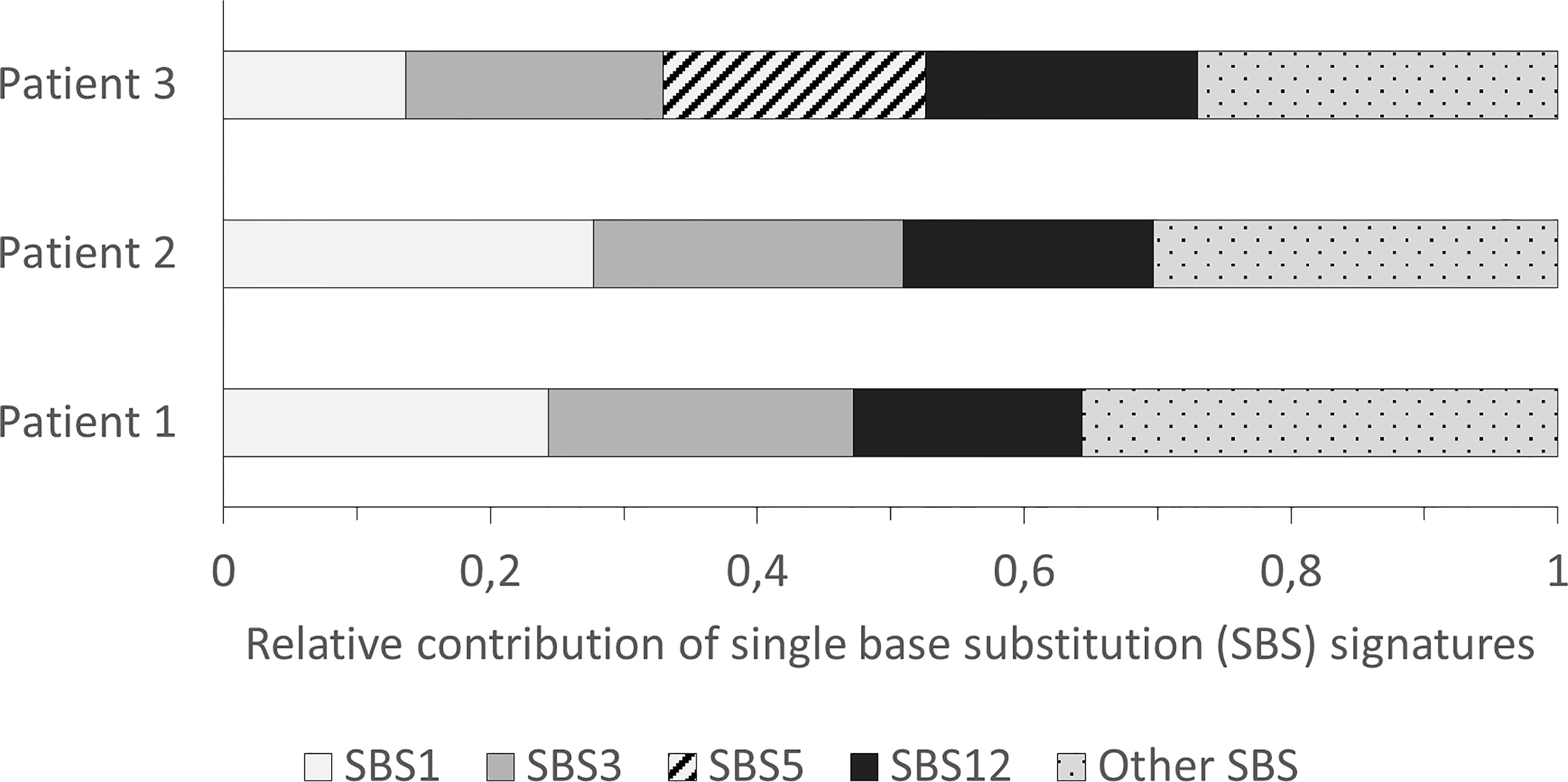
Figure 1 Single base substitution (SBS) signatures identified in the patients with a relative contribution ≥ 15% of the global pattern.
Discussion
The existing data linking germline pathogenic variants in the BRCA1 gene to an increased risk of CRC are scarce. Two large studies reported that BRCA1 variants conferred approximatively a fivefold increased risk for CRC, especially in young patients from high-risk families (6, 26). Out of three recent meta-analyses, one of them found an increased risk of colorectal cancer associated with BRCA1 variants (odds ratio = 1.49, 95% CI = 1.19 to 1.85, P < 0.001) (8), while the other two did not identify any increase in CRC risk among patients carrying a BRCA1 variant (7, 9). A study evaluating a cohort of BRCA1 or BRCA2 pathogenic variant carriers mostly of Ashkenazy ancestry concluded that they may be prone to developing anal carcinoma and left-sided mucinous histology CRC (27). One single publication reported a young male patient with a BRCA1 germinal variant who presented with rectal adenocarcinoma and showed an excellent response to oxaliplatin-containing neoadjuvant therapy (28). These data thus remain contradictory and do not allow to recommend to screen for CRC in BRCA1 variants heterozygotes, or to consider BRCA1 pathogenic variants as a factor predisposing to familial CRC.
Given the frequency of CRC and of BRCA1 variant heterozygotes in European populations (29), co-occurrence may be incidental rather than indicative of a causal relationship, as suggested previously (30). However, a few lines of evidence indicate that co-occurrence might be relevant.
Recently, a large report investigated the frequencies of various cancers, including CRCs, in 6902 men with BRCA variants (31). The probability for developing a CRC was, according to this report, two times lower in men with BRCA2 variants than in BRCA1 variant heterozygotes. As it seems unlikely that BRCA2 variants had a protective role against CRCs, these data could indicate a slightly but significantly increased risk of these cancers in men with BRCA1 variants.
In our samples, we did not evaluate BRCA1 protein expression. Although we describe patients with aggressive metastatic cancer, the presence of low levels of BRCA1 protein had a worse prognosis even in early-stage CRC (32).
In our study, we not only confirmed that the BRCA1 germline variants were still present in the tumor (with evidence of positive selection in case 1), but we also demonstrate scars of HRD in the three tumors. Indeed, the presence of germline variants in HRD-associated genes alone is not sufficient to predict clinically relevant HRD. We highlighted the presence of specific mutational signatures (COSMIC signature 3) (33) and genomic instability characteristics (LOH, TAI and LST) (34–36), reflecting significant HRD, comparable with that observed in ovarian cancers with a BRCA1 or BRCA2 pathogenic variants. Interestingly, the initial somatic NGS analysis of cases 2 and 3 was not conclusive, possibly because of low tumor infiltration, but it could also be indicative of an epigenetic event leading to loss of BRCA1 function and demonstrates the role of HRD testing even in cases where the mechanism driving HRD is not fully elucidated. Taken together, these observations indicate that germline BRCA1 variants may, in a small proportion of variant carriers, play a driver role in CRC development or progression and that these patients might thus benefit from a treatment with poly (ADP-ribose) polymerase-inhibitors (PARPi). Indeed, clinical trials clearly demonstrated the efficacy of platinum-based chemotherapy and PARPi to treat BRCA mutated and/or HRD positive cancers inside the spectrum of BRCA-related cancers (37). Further evidence demonstrating that some CRC could be linked to BRCA deficiencies could open new perspectives for treatment with PARPi of these rare aggressive tumors.
The small number of patients and the bias in recruitment are the main limitations of our study, precluding to justify any specific surveillance or screening program in the absence of a personal or family history.
In conclusion, our data indicate that a BRCA1 germline pathogenic variant can be involved in CRC development through HRD. Thus, BRCA1 testing should be considered in young patients with a personal history of microsatellite stable CRC. This could further allow a personalized treatment approach with a PARPi.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Ethics Statement
The “Comité d’Ethique Hospitalo-facultaire Universitaire de Liège” (CHU/University of Liège) approved the study. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author Contributions
Conceptualization: VB. Data curation: MF, MM, and VB. Formal analysis: MF. Funding acquisition: VB. Investigation: MF, MM, RT, CM, KS, ES, NL, CL, and CF. Methodology: MF, MM, RT, CM, CJ, and LP. Project administration: VB. Resources: VB. Supervision: CJ, LP, and VB. Validation: JR, YG, JC, and AS. Writing-original draft: MF and VB. Writing-review and editing: MF, MM, RT, CM, KS, ES, NL, CL, JR, YG, JC, AS, CJ, LP, VB, and CF. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by a Télévie fellowship (MV.F., grant N° 7451419F), a grant from the CHU Liége (N° 981481200) and the WALGEMED grant (Région Wallonne, grant N° 1710180).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The CHU of Liége is member of the GENTURIS (GENetic TUmor RISk) European Reference Network).
References
1. Wu W, Koike A, Takeshita T, Ohta T. The Ubiquitin E3 Ligase Activity of BRCA1 and Its Biological Functions. Cell Div (2008) 3:1. doi: 10.1186/1747-1028-3-1
2. Miki Y, Swensen J, Shattuck-Eidens D, Futreal PA, Harshman K, Tavtigian S, et al. A Strong Candidate for the Breast and Ovarian Cancer Susceptibility Gene Brca1. Science (1994) 266(5182):66–71. doi: 10.1126/science.7545954
3. Mavaddat N, Peock S, Frost D, Ellis S, Platte R, Fineberg E, et al. Cancer Risks for BRCA1 and BRCA2 Mutation Carriers: Results From Prospective Analysis of EMBRACE. J Natl Cancer Inst (2013) 105(11):812–22. doi: 10.1093/jnci/djt095
4. Schoen RE, Weissfeld JL, Kuller LH. Are Women With Breast, Endometrial, or Ovarian Cancer at Increased Risk for Colorectal Cancer? Am J Gastroenterol (1994) 89(6):835–42.
5. Mersch J, Jackson M, Park M, Nebgen D, Peterson SK, Singletary C, et al. Cancers Associated With BRCA1 and BRCA2 Mutations Other Than Breast and Ovarian. Cancer (2015) 121(2):269–75. doi: 10.1002/cncr.29041
6. Phelan CM, Iqbal J, Lynch HT, Lubinski J, Gronwald J, Moller P, et al. Incidence of Colorectal Cancer in BRCA1 and BRCA2 Mutation Carriers: Results From a Follow-Up Study. Br J Cancer (2014) 110(2):530–4. doi: 10.1038/bjc.2013.741
7. Cullinane CM, Creavin B, O’Connell EP, Kelly L, O’Sullivan MJ, Corrigan MA, et al. Risk of Colorectal Cancer Associated With BRCA1 and/or BRCA2 Mutation Carriers: Systematic Review and Meta-Analysis. BJS (British J Surgery) (2020) 107(8):951–9. doi: 10.1002/bjs.11603
8. Oh M, McBride A, Yun S, Bhattacharjee S, Slack M, Martin JR, et al. BRCA1 and BRCA2 Gene Mutations and Colorectal Cancer Risk: Systematic Review and Meta-Analysis. J Natl Cancer Inst (2018) 110(11):1178–89. doi: 10.1093/jnci/djy148
9. Lee Y-C, Lee Y-L, Li C-Y. BRCA Genes and Related Cancers: A Meta-Analysis From Epidemiological Cohort Studies. Medicina (2021) 57(9):905. doi: 10.3390/medicina57090905
10. Curtit E, Benhamo V, Gruel N, Popova T, Manie E, Cottu P, et al. First Description of a Sporadic Breast Cancer in a Woman With BRCA1 Germline Mutation. Oncotarget (2015) 6(34):35616–24. doi: 10.18632/oncotarget.5348
11. Song H, Cicek MS, Dicks E, Harrington P, Ramus SJ, Cunningham JM, et al. The Contribution of Deleterious Germline Mutations in BRCA1, BRCA2 and the Mismatch Repair Genes to Ovarian Cancer in the Population. Hum Mol Genet (2014) 23(17):4703–9. doi: 10.1093/hmg/ddu172
12. de Juan Jiménez I, García Casado Z, Palanca Suela S, Esteban Cardeñosa E, López Guerrero JA, Segura Huerta Á, et al. Novel and Recurrent BRCA1/BRCA2 Mutations in Early Onset and Familial Breast and Ovarian Cancer Detected in the Program of Genetic Counseling in Cancer of Valencian Community (Eastern Spain). Relationship of Family Phenotypes With Mutation Prevalence. Familial Cancer (2013) 12(4):767–77. doi: 10.1007/s10689-013-9622-2
13. Simard J, Tonin P, Durocher F, Morgan K, Rommens J, Gingras S, et al. Common Origins of BRCA1 Mutations in Canadian Breast and Ovarian Cancer Families. Nat Genet (1994) 8(4):392–8. doi: 10.1038/ng1294-392
14. Dørum A, Heimdal K, Hovig E, Inganäs M, Møller P. Penetrances of BRCA1 1675dela and 1135insa With Respect to Breast Cancer and Ovarian Cancer. Am J Hum Genet (1999) 65(3):671–9. doi: 10.1086/302530
15. Foretova L, Machackova E, Navratilova M, Pavlu H, Hruba M, Lukesova M, et al. BRCA1 and BRCA2 Mutations in Women With Familial or Early-Onset Breast/Ovarian Cancer in the Czech Republic. Hum Mutat (2004) 23(4):397–8. doi: 10.1002/humu.9226
16. Machackova E, Foretova L, Lukesova M, Vasickova P, Navratilova M, Coene I, et al. Spectrum and Characterisation of BRCA1 and BRCA2 Deleterious Mutations in High-Risk Czech Patients With Breast and/or Ovarian Cancer. BMC Cancer (2008) 8:140. doi: 10.1186/1471-2407-8-140
17. Laraqui A, Uhrhammer N, Lahlou-Amine I, EL Rhaffouli H, El Baghdadi J, Dehayni M, et al. Mutation Screening of the BRCA1 Gene in Early Onset and Familial Breast/Ovarian Cancer in Moroccan Population. Int J Med Sci (2012) 10(1):60–7. doi: 10.7150/ijms.5014
18. Janavičius R. Founder BRCA1/2 Mutations in the Europe: Implications for Hereditary Breast-Ovarian Cancer Prevention and Control. EPMA J (2010) 1(3):397–412. doi: 10.1007/s13167-010-0037-y
19. Behl S, Hamel N, de Ladurantaye M, Lepage S, Lapointe R, Mes-Masson A-M, et al. Founder BRCA1/BRCA2/PALB2 Pathogenic Variants in French-Canadian Breast Cancer Cases and Controls. Sci Rep (2020) 10:6491. doi: 10.1038/s41598-020-63100-w
20. Łukomska A, Menkiszak J, Gronwald J, Tomiczek-Szwiec J, Szwiec M, Jasiówka M, et al. Recurrent Mutations in BRCA1, BRCA2, RAD51C, PALB2 and CHEK2 in Polish Patients With Ovarian Cancer. Cancers (Basel) (2021) 13(4):849. doi: 10.3390/cancers13040849
21. Caputo S, Benboudjema L, Sinilnikova O, Rouleau E, Béroud C, Lidereau R. Description and Analysis of Genetic Variants in French Hereditary Breast and Ovarian Cancer Families Recorded in the UMD-BRCA1/BRCA2 Databases. Nucleic Acids Res (2012) 40(Database issue):D992–1002. doi: 10.1093/nar/gkr1160
22. Favero F, Joshi T, Marquard AM, Birkbak NJ, Krzystanek M, Li Q, et al. Sequenza: Allele-Specific Copy Number and Mutation Profiles From Tumor Sequencing Data. Ann Oncol (2015) 26(1):64–70. doi: 10.1093/annonc/mdu479
23. Coleman RL, Fleming GF, Brady MF, Swisher EM, Steffensen KD, Friedlander M, et al. Veliparib With First-Line Chemotherapy and as Maintenance Therapy in Ovarian Cancer. New Engl J Med (2019) 381:2403–15. doi: 10.1056/NEJMoa1909707
24. Blokzijl F, Janssen R, van Boxtel R, Cuppen E. MutationalPatterns: Comprehensive Genome-Wide Analysis of Mutational Processes. Genome Med (2018) 10(1):33. doi: 10.1186/s13073-018-0539-0
25. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behjati S, Biankin AV, et al. Signatures of Mutational Processes in Human Cancer. Nature (2013) 500(7463):415–21. doi: 10.1038/nature12477
26. Sopik V, Phelan C, Cybulski C, Narod SA. BRCA1 and BRCA2 Mutations and the Risk for Colorectal Cancer. Clin Genet (2015) 87(5):411–8. doi: 10.1111/cge.12497
27. Grinshpun A, Halpern N, Granit RZ, Hubert A, Hamburger T, Laitman Y, et al. Phenotypic Characteristics of Colorectal Cancer in BRCA1/2 Mutation Carriers. Eur J Hum Genet (2018) 26(3):382–6. doi: 10.1038/s41431-017-0067-1
28. Soyano AE, Baldeo C, Kasi PM. BRCA Mutation and Its Association With Colorectal Cancer. Clin Colorectal Cancer (2018) 17(4):e647–50. doi: 10.1016/j.clcc.2018.06.006
29. Maxwell KN, Domchek SM, Nathanson KL, Robson ME. Population Frequency of Germline BRCA1/2 Mutations. JCO (2016) 34(34):4183–5. doi: 10.1200/JCO.2016.67.0554
30. Evans DG, Clancy T, Hill J, Tischkowitz M. Is There Really an Increased Risk of Early Colorectal Cancer in Women With BRCA1 Pathogenic Mutations? Clin Genet (2016) 89(3):399–9. doi: 10.1111/cge.12687
31. Silvestri V, Leslie G, Barnes DR, CIMBA Group, Agnarsson BA, Aittomäki K, et al. Characterization of the Cancer Spectrum in Men With Germline BRCA1 and BRCA2 Pathogenic Variants: Results From the Consortium of Investigators of Modifiers of BRCA1/2 (CIMBA). JAMA Oncol (2020) 6(8):1218–30. doi: 10.1001/jamaoncol.2020.2134
32. Du C, Peng Y, He Y, Chen G, Chen H. Low Levels of BRCA1 Protein Expression Predict a Worse Prognosis in Stage I–II Colon Cancer. Int J Biol Markers (2021) 36(1):47–53. doi: 10.1177/1724600820986572
33. Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Tian Ng AW, Wu Y, et al. The Repertoire of Mutational Signatures in Human Cancer. Nature (2020) 578(7793):94–101. doi: 10.1038/s41586-020-1943-3
34. Abkevich V, Timms KM, Hennessy BT, Potter J, Carey MS, Meyer LA, et al. Patterns of Genomic Loss of Heterozygosity Predict Homologous Recombination Repair Defects in Epithelial Ovarian Cancer. Br J Cancer (2012) 107(10):1776–82. doi: 10.1038/bjc.2012.451
35. Birkbak NJ, Wang ZC, Kim J-Y, Eklund AC, Li Q, Tian R, et al. Telomeric Allelic Imbalance Indicates Defective DNA Repair and Sensitivity to DNA Damaging Agents. Cancer Discov (2012) 2(4):366–75. doi: 10.1158/2159-8290.CD-11-0206
36. Popova T, Manié E, Rieunier G, Caux-Moncoutier V, Tirapo C, Dubois T, et al. Ploidy and Large-Scale Genomic Instability Consistently Identify Basal-Like Breast Carcinomas With BRCA1/2 Inactivation. Cancer Res (2012) 72(21):5454–62. doi: 10.1158/0008-5472.CAN-12-1470
Keywords: colorectal (colon) cancer, BRCA1, homologous recombination deficiency (HRD), exome sequencing (ES), case report
Citation: Freire MV, Martin M, Thissen R, Van Marcke C, Segers K, Sépulchre E, Leroi N, Lété C, Fasquelle C, Radermacher J, Gokburun Y, Collignon J, Sacré A, Josse C, Palmeira L and Bours V (2022) Case Report Series: Aggressive HR Deficient Colorectal Cancers Related to BRCA1 Pathogenic Germline Variants. Front. Oncol. 12:835581. doi: 10.3389/fonc.2022.835581
Received: 14 December 2021; Accepted: 01 February 2022;
Published: 24 February 2022.
Edited by:
Walter Hernán Pavicic, Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), ArgentinaReviewed by:
Moom Roosan, Chapman University, United StatesNatalija Dedić Plavetić University Hospital Centre Zagreb, Croatia
Copyright © 2022 Freire, Martin, Thissen, Van Marcke, Segers, Sépulchre, Leroi, Lété, Fasquelle, Radermacher, Gokburun, Collignon, Sacré, Josse, Palmeira and Bours. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vincent Bours, dmJvdXJzQHVsaWVnZS5iZQ==