 Christian Vogeley
Christian Vogeley Katharina M. Rolfes†
Katharina M. Rolfes† Thomas Haarmann-Stemmann
Thomas Haarmann-Stemmann- IUF - Leibniz-Research Institute for Environmental Medicine, Düsseldorf, Germany
Cutaneous squamous cell carcinoma (SCC) is one of the most frequent malignancies in humans and academia as well as public authorities expect a further increase of its incidence in the next years. The major risk factor for the development of SCC of the general population is the repeated and unprotected exposure to ultraviolet (UV) radiation. Another important risk factor, in particular with regards to occupational settings, is the chronic exposure to polycyclic aromatic hydrocarbons (PAH) which are formed during incomplete combustion of organic material and thus can be found in coal tar, creosote, bitumen and related working materials. Importantly, both exposomal factors unleash their carcinogenic potential, at least to some extent, by activating the aryl hydrocarbon receptor (AHR). The AHR is a ligand-dependent transcription factor and key regulator in xenobiotic metabolism and immunity. The AHR is expressed in all cutaneous cell-types investigated so far and maintains skin integrity. We and others have reported that in response to a chronic exposure to environmental stressors, in particular UV radiation and PAHs, an activation of AHR and downstream signaling pathways critically contributes to the development of SCC. Here, we summarize the current knowledge about AHR’s role in skin carcinogenesis and focus on its impact on defense mechanisms, such as DNA repair, apoptosis and anti-tumor immune responses. In addition, we discuss the possible consequences of a simultaneous exposure to different AHR-stimulating environmental factors for the development of cutaneous SCC.
Introduction
Non-melanoma skin cancers, in particular basal cell carcinoma and SCC, are among the most frequent malignancies in humans (1–3). Cutaneous SCC primarily develop on sun-exposed areas of the body. Accordingly, a chronic exposure to artificial (tanning beds) or solar UVB radiation and the associated accumulation of damaged keratinocytes is the most important risk factor for SCC (1–3). Due to the continuously growing number of elderly individuals in the general population as well as the unbroken popularity of tanned skin among younger generations, the incidence of SCC is predicted to further increase (1–3). This trend might be exacerbated by environmental, occupational and life style-related exposure to carcinogenic chemicals, especially combustion-derived PAHs, alone or in combination with UV exposure (3). In addition, the climate change and associated global weather shifts may have an impact on human health and will probably increase the incidence of skin cancers and other malignancies (4, 5). Because SCC is not only a growing medical problem but also a substantial economic burden to health care systems (3, 6), there is an urgent need for the development of novel preventive and therapeutic measures. In this context, the AHR, a ligand-activated transcription factor and key regulator in xenobiotic metabolism and immunity, seems to be a promising molecular target. This notion is strengthened by the outcome of a two-stage genome-wide association study identifying the AHR as a novel susceptibility locus for cutaneous SCC in humans (7).
In this review article, we focus on the critical functions of AHR for DNA damage-dependent processes and immune responses which may contribute to the development of SCC in chronically UV- and/or PAH-exposed skin. Please note that while we are focusing on the mentioned aspects other functions of the AHR system might fall short, which may be also relevant for the process of skin cancer development.
AHR in Xenobiotic Metabolism and Chemical Carcinogenesis
The multistage model of carcinogenesis, defining the process of tumor development as a strict sequence of initiation, promotion and progression, was established more than 70 years ago (8, 9). In these studies, the researchers induced skin tumorigenesis in mice by applying 7,12-dimethylbenz[a]anthracene (DMBA) as tumor-initiator and phorbol ester-containing croton oil as tumor-promoting agent. Sequencing of the tumor DNA as well as further mechanistic studies provided evidence that PAHs initiate carcinogenesis by inducing mutations primarily in the Ha-Ras oncogene and that this process requires a metabolic conversion of the per se non-toxic chemicals to highly reactive metabolites, a process which is primarily carried out by AHR-regulated cytochrome P450 (CYP) monooxygenases (10–12).
AHR Ligands and Signaling Pathways
The AHR belongs to the basic helix-loop-helix Per-ARNT-Sim superfamily of transcription factors whose members translate developmental, physiological and environmental signals into biochemical processes and cell biological responses (13). Within this protein superfamily, the AHR is the only member which is activated by binding of small molecular weight compounds (ligands). AHR ligands can be divided into exogenous and endogenous compounds (14–17). The list of exogenous AHR ligands encompasses environmental pollutants, such as PAHs and dioxins, plant- and microbiota-derived indoles and polyphenols, and pharmaceutical drugs. Indole derivatives, such as indolo[3,2-b]carbazole, 6-formylindolo[3,2-b]carbazole (FICZ) and 2-(1’H-Indole-3’-carbonyl)-thiazole-4-carboxylic acid methyl ester as well as tryptophan metabolites, such as kynurenic acid and xanthurenic acid, are considered as relevant endogenous agonists of AHR (14–17).
In its inactive state, the AHR is trapped in a cytosolic multiprotein complex, composed of two heat-shock protein 90 (HSP90) molecules, the AHR-interacting protein, the co-chaperone p23 and the soluble tyrosine kinase c-Src (18) (Figure 1). Upon ligand binding, the cytosolic multiprotein complex dissociates, the AHR translocates into the nucleus, and dimerizes with its binding partner AHR nuclear translocator (ARNT). This heterodimer binds to xenobiotic-responsive elements (XRE) in the enhancer region of target genes to induce their expression (14–16). Typical AHR target genes encode for xenobiotic-metabolizing enzymes, such as CYP1A1, CYP1A2 and CYP1B1 (14–16). Another XRE-regulated gene codes for the AHR repressor, an AHR-related protein that lacks a transactivation domain and represses AHR signaling by competing for ARNT- and XRE-binding (19). Next to this so-called canonical AHR signaling pathway, the dissociation of the multiprotein complex leads to the release of c-Src, which subsequently may activate the epidermal growth factor receptor (EGFR) and downstream mitogen-activated protein kinase signal transduction (20–22) (Figure 1). Furthermore, AHR interacts with other transcription factors, including nuclear factor-κB (NF-κB) (23, 24), hypoxia-inducible factor-1α (25, 26), estrogen receptor-α (27, 28), and nuclear factor erythroid 2-related factor 2 (29, 30). These non-canonical functions may explain the frequently observed tissue- and cell-specific effects of AHR signaling and probably contribute to the pathogenesis of inflammatory and malignant diseases.
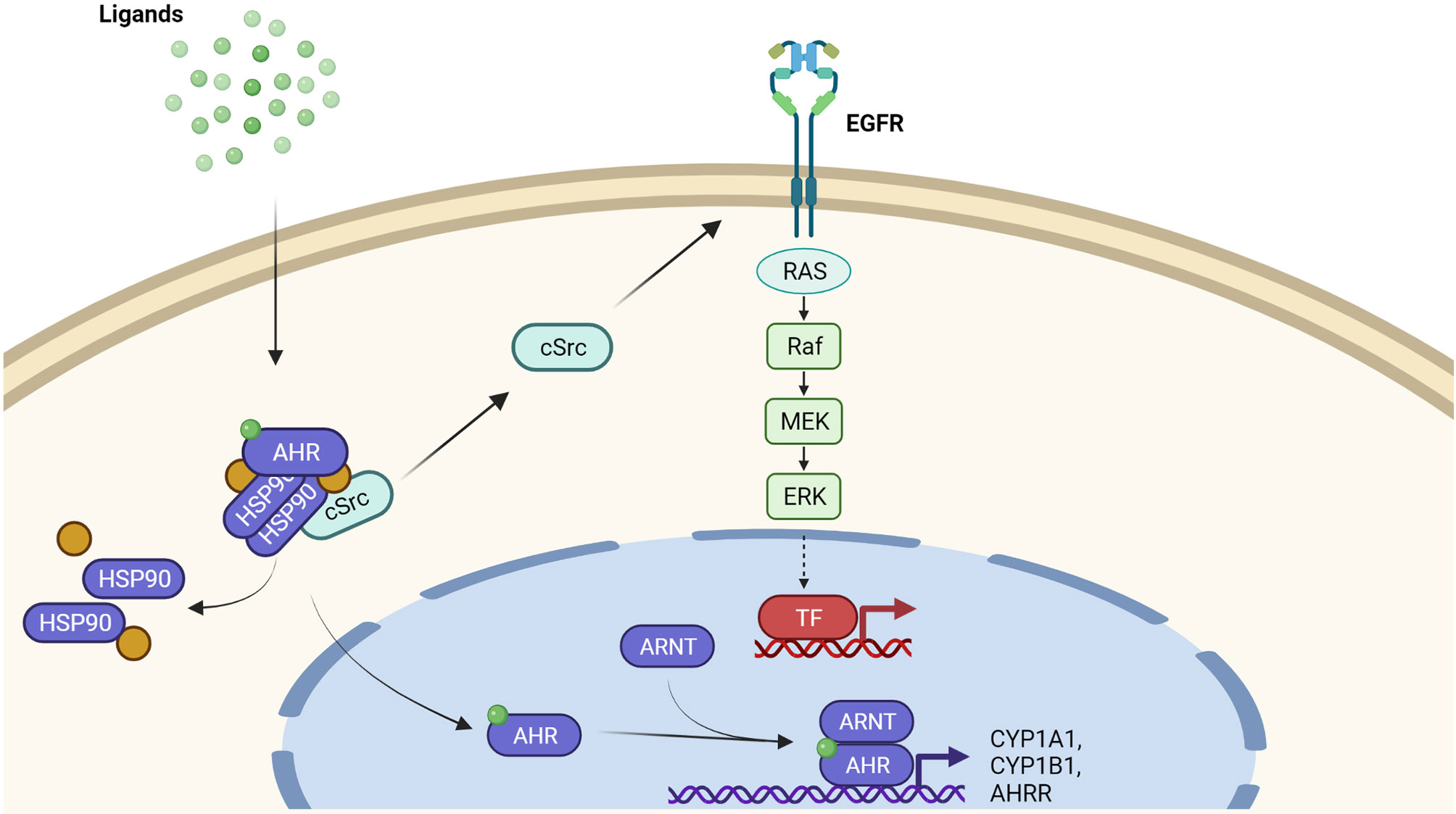
Figure 1 AHR-dependent signaling pathways. Inactive AHR is part of a cytosolic multiprotein. complex consisting of a HSP90 dimer, other chaperone molecules and tyrosine kinase c-Src. Upon ligand binding, the multiprotein complex dissociates and AHR translocates into the nucleus. Nuclear AHR heterodimerizes with ARNT and binds to the enhancer region of target genes, for instance encoding CYP1A1, CYP1B1, and AHRR, to enhance their expression. In addition to this canonical pathway, the AHR ligand-driven release of c-Src from the cytosolic multiportion complex may stimulate EGFR and downstream mitogen-activated protein kinases, resulting in the transcriptional induction of another set of genes.
AHR and Metabolic Activation of Polycyclic Aromatic Hydrocarbons
Exposure to environmental, occupational and life-style related organic pollutants is considered to be involved in the onset of cutaneous SCC (3). Especially a long-lasting occupational exposure to PAHs present in soot and various working materials, such as coal tar, bitumen and petroleum, may facilitate skin carcinogenesis (3, 31). In addition, the elevated risk of smokers to develop cutaneous SCC is largely attributed to the PAH fraction present in tobacco smoke (32–34). The genotoxic potential of PAHs is primarily unleashed by their activation through AHR-dependent CYP1 isoforms (10–12) (Figure 2). For instance, CYP1A1 and microsomal epoxide hydrolase 1 (EPHX1) sequentially metabolize benzo[a]pyrene (B[a]P) to B[a]P-7,8-dihydrodiol-9,10-epoxide (BPDE), a highly carcinogenic compound which forms bulky DNA adducts by binding to guanine at the N2 position (10–12). Since CYP1A1 expression is regulated by the AHR, AHR-deficient mice as well as mice bearing an epidermis-specific ARNT-deletion were resistant towards B[a]P induced skin carcinogenesis (35, 36). In contrast to B[a]P, DMBA is metabolized by CYP1A1, CYP1B1 and EPHX1. Whereas CYP1A1 leads to the detoxification of DMBA, the CYP1B1-mediated oxidation results in an accumulation of highly carcinogenic DMBA-3,4-diol-1,2-epoxide (37, 38). CYP1-specific alterations in the detoxification and metabolic activation was also reported for other PAHs, such as dibenzo[a,l]pyrene (39) and dibenzo[def,p]chrysene (40). Thus, the carcinogenic potential of PAHs is determined by the CYP1 isoform predominantly expressed in the exposed cell population. Interestingly, the expression of CYP1B1 is not exclusively regulated by AHR (41–43) and, accordingly, AHR-deficient mice still express sufficient amounts of CYP1B1 to toxify DMBA and initiate skin carcinogenesis (44). Another carcinogenesis study revealed that AHR-deficiency protects mice against the skin carcinogenicity of PAH-rich airborne particulate matter (PM) (45).
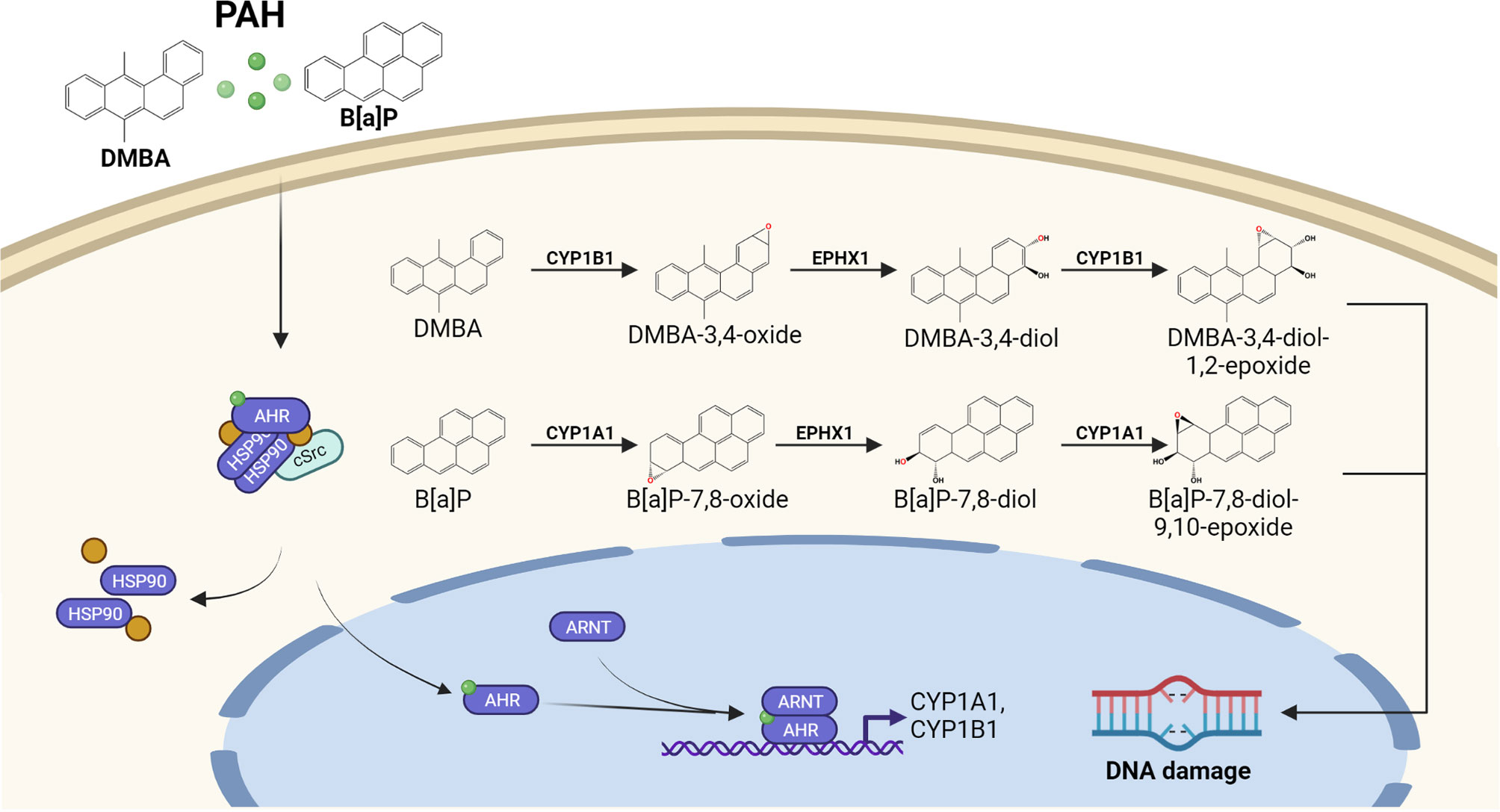
Figure 2 AHR-dependent metabolic activation of PAHs by CYP1 isoforms. Lipophilic PAHs can easily pass the plasma membrane and activate the canonical AHR signaling pathway. The resulting induction of CYP1 isoform expression and enzyme activity accelerates the oxidative metabolism of the invading chemicals. In case a proper detoxification of the resulting reactive PAH metabolites by the conjugating enzyme system (not shown) fails, reactive PAH-diol epoxides may covalently bind to the DNA and form highly mutagenic bulky DNA adducts.
At this point, we should mention that the CYP1-derived reactive PAH metabolites are efficiently detoxified through conjugation to glutathione, glucuronic acid or other hydrophilic molecules by phase 2 enzymes (11). However, in case the capacity of the conjugating enzyme system is exhausted, relevant amounts of reactive phase 1 metabolites may react with the DNA. Depending on the efficacy of other defense mechanisms, such as DNA repair and apoptosis, these DNA lesions may give rise to mutations (11, 46, 47).
The same is true for oxidative DNA damage that may occur during PAH metabolism. Specifically, CYP1-derived PAH dihydrodiols may serve as substrate for aldo-keto reductases (AKR), a family of cytosolic NADPH-dependent oxidoreductases (48). Several AKR1 isoforms, including AKR1C3, are capable of converting PAH dihydrodiols to the respective catechols which in the presence of oxygen can undergo redox-cycling. This results in the generation of reactive oxygen species (ROS), such as hydrogen peroxide and superoxide, which may contribute to skin carcinogenesis by oxidatively damaging DNA and other macromolecules (49, 50). In addition, AKR1C3 reduces prostaglandin (PG) D2 to 9α,11β-PGF2, a process which may fuel type 2 T helper (Th2) cell-related inflammatory responses in the skin (51, 52). Noteworthy, this AKR1C3-catalyzed reaction reduces the spontaneous dehydration of PGD2 to 15Δ-PGJ2, an eicosanoid that acts anti-inflammatory by inducing peroxisome proliferator-activated receptor-γ signaling and inhibiting pro-inflammatory NF-κB signaling pathways (51). Importantly, AKR1C3 is highly expressed in human SCC (53) and, moreover, we found that a PAH exposure of human keratinocytes results in an AHR-dependent upregulation of this enzyme (22). Taken together, these findings suggest that an AHR/AKR1C3-dependent modulation of PGD2 metabolism may foster the growth and apoptosis-resistance of initiated keratinocytes and SCC cells.
AHR and PAH-Induced Immune Reactions
An epicutaneous sensitization with PAHs, more precisely AHR/CYP1-derived reactive PAH metabolites, results in the development of an early inflammatory response which is responsible for contact hypersensitivity of the skin (54–56). This acute inflammation is triggered by antigen-specific CD8+ cytotoxic T cells and CD4+ type 1 T helper (Th1) cells which may prevent skin tumor development by secreting respective cytokines, such as interleukin (IL)-2 and interferon-γ (54–56). In its extent, this response is controlled by Th2 cells and immunosuppressive FoxP3+ regulatory T cells (Tregs). The induction of keratinocyte apoptosis by cytotoxic T cells leads to the release of further mediators which may stimulate other immune cells to infiltrate the inflamed skin. In case the damage is not resolved, chronic inflammatory condition facilitates the growth and progression of skin tumors (57). In experimental carcinogenesis studies, the application of phorbol ester fosters the expansion of IL-17-producing T cells which further promote tumor growth (58). Recently, various studies reported that by inhibiting the function of immunosuppressive Tregs (59–61) and promoting the polarization of Th2 and Th17 cells (Figure 3), an exposure to PAHs or PAH-rich particulate matter facilitates the worsening of chronic inflammatory diseases, including asthma and atopic dermatitis, in an AHR-dependent manner (61–67). Given that Th2 as well as Th17 responses are well recognized for their tumor-promoting capabilities (57), it is tempting to speculate that a chronic exposure of the skin to genotoxic PAH or PAH-rich materials may not only initiate the development of tumors but also promote their growth by creating an inflammatory micromilieu.
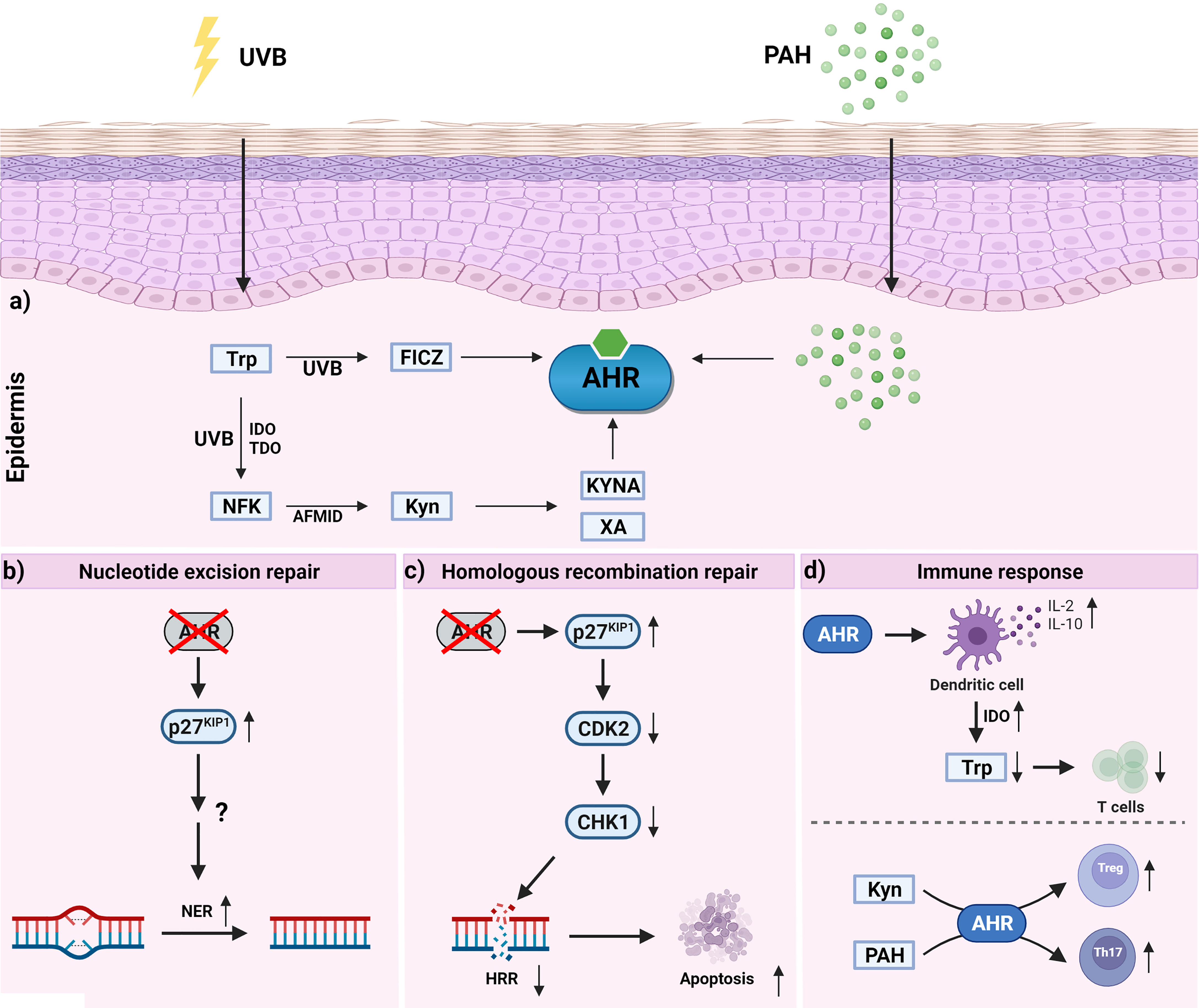
Figure 3 Activation of epidermal AHR signaling and AHR-dependent effects which may contribute to skin carcinogenesis. a) UVB irradiation of epidermal cells results in the formation of tryptophan photoproducts, such as FICZ and NFK. While FICZ is a high-affinity ligand for AHR, NFK is considered as a pro-ligand. Specifically, aryl formamidase converts NFK to kynurenine (kyn) which in subsequent metabolic reactions is metabolized to the AHR agonists kynurenic acid (KYNA) and xanthurenic acid (XA). b) Inhibition of AHR was shown to increase p27KIP1 protein content which improves NER of UVB radiation-induced DNA photoproducts by a yet enigmatic mechanism. c) Inhibition of AHR was shown to increase p27KIP1 protein content resulting in an inhibition of CDK2 and a time-dependent decline of the CHK1 level. The reduction of CHK1 is associated with a reduced performance of the HRR system and an elevated rate of keratinocyte apoptosis. d) AHR activation affects T cell polarization in a ligand-dependent manner. The upregulation of IL-2, IL-10 and IDO in DC may lead to an expansion of Tregs and inhibit the proliferation of effector T cells via tryptophan depletion. In contrast to the pro-ligand KYN, PAHs seem to interfere with differentiation and function of immunosuppressive Tregs.
Ultraviolet Radiation and Skin Photocarcinogenesis
Ultraviolet (UV) radiation is part of the electromagnetic spectrum of sunlight and can be subdivided into UVA, UVB and UVC radiation (68, 69). The latter (100 nm – 280 nm) is almost completely absorbed by the stratospheric ozone layer and thus does not reach our skin in sufficient amounts to cause biological effects. High-energy UVB photons (280 nm – 315 nm) are nearly completely absorbed by the DNA and other macromolecules of epidermal cells, i.e. keratinocytes and melanocytes. In contrast, UVA radiation (315 nm – 400 nm) can penetrate deep into our skin and even reach dermal fibroblasts (68, 69). UVA radiation induces oxidative stress and associated macromolecular damage by excitation of endogenous chromophores, such riboflavin and protoporphyrin IX (70). Importantly, the vast majority of skin cancers, i.e. basal cell carcinoma, SCC and malignant melanoma, originate from epidermal cells. Hence, in the context of skin photocarcinogenesis, UVB radiation can be considered as the most dangerous part of the UV spectrum.
DNA Damage, DNA Repair, and Apoptosis
Skin photocarcinogenesis is a multistep process, involving initiating and promoting events (71, 72). These include DNA damage and failure of appropriate cell rescue (DNA repair) or cell death (apoptosis) responses, the suppression of anti-tumor immune responses, and the clonal expansion of malignant cells (71–74). To provoke effects at a cellular level, UVB radiation needs to be absorbed by chromophores to convert its physical into chemical energy. The most important chromophore for UVB radiation is the DNA (75). In addition, other cellular components, in particular aromatic amino acids, such as tryptophan (76), can absorb UVB photons and contribute to the generation of the UVB stress response in the epidermal compartment (21, 77). The DNA damage-dependent part of this response is initiated by the UVB radiation-induced formation of two photoproducts between adjacent pyrimidine bases: cyclobutene pyrimidine dimers (CPD) and pyrimidine (6–4) pyrimidone photoproducts (75, 78, 79). Although both DNA photoproducts are highly mutagenic, CPDs and the resulting signature mutations, in particular C>T and CC>TT transitions, are considered as being mainly responsible for skin photocarcinogenesis (78, 80, 81). Numerous of these signature mutations are present in the p53 gene, a tumor suppressor gene that is inactivated or compromised by respective mutations in nearly 100% of UV radiation-associated skin cancers (78, 79). In placental mammals, UVB radiation-induced DNA photoproducts (as well as bulky PAH-DNA adducts) are removed by nucleotide excision repair (NER) which consists of four steps: Damage recognition, incision, gap filling and ligation (46, 82). NER is divided into two distinct sub-pathways that differ in their way of damage recognition: Transcription-coupled repair (TCR), which quickly removes DNA adducts in actively transcribed genes, and global genome repair (GGR), which removes DNA lesions in the entire genome. In case of TCR, a stalled RNA polymerase II which then is recognized by Cockayne Syndrome (CS) A and CSB proteins serves as damage sensor. In GGR, a complex of xeroderma pigmentosum (XP) C, centrin-2, RAD23B and DNA damage-binding proteins recognizes the DNA damage. DNA unwinding is performed by the DNA helicases XPB and XPD, which are part of the general transcription factor IIH (TFIIH). Excision of the DNA damage is performed by the endonucleases XPG and XPF-ERCC1 and, subsequently, the gap of 25 to 30 nucleotides is filled and sealed by DNA polymerases and DNA ligases, respectively (46, 82).
In case NER fails, the remaining DNA photoproducts will cause DNA double-strand breaks (DSB), which have been denominated as lethal DNA lesions (83). In fact, DSBs are not a direct consequence of UVB irradiation but occur when CPD-positive cells enter mitosis. During S phase, these helix-distorting DNA lesions cause a collapse of the replication fork leading to breakage (enzymatic cleavage) of both DNA strands (84–86). Subsequently, the DNA damage response, amongst others encompassing the activation of ataxia telangiectasia mutated (ATM) kinase and downstream checkpoint kinase 1 (CHK1), is induced to halt the cell-cycle and initiate homologous recombination repair (HRR) (87, 88), which is primarily in charge of fixing replication fork-associated DSBs (89). When HRR fails, apoptosis is initiated by ATM (or related ATR) kinase in both p53-dependent and p53–independent manners (47). Importantly, an elevation of keratinocyte apoptosis may effectively restrain UVB radiation-induced skin carcinogenesis (90–93).
UV Radiation-Induced Immunosuppression
As indicated above, UV radiation suppresses the immune system in an antigen-specific manner by inducing Tregs thereby promoting skin carcinogenesis (73, 74). Using mouse models of contact hypersensitivity, the induction of DNA photoproducts, especially CPDs, has been identified to be the major molecular trigger for the UV radiation-induced suppression of the immune system (94, 95). This result has been confirmed in human volunteers treated with UVB light alone or in combination with photolyase-containing liposomes and photo-reactivating light exposure (96). The Tregs induced by UVB irradiation are CD4+ and CD25+, express FoxP3 and secrete IL-10. Although bearing lymph node-homing receptors, Tregs may switch to skin-homing receptors upon contact with epidermal Langerhans cells, migrate into the skin and suppress cancer cell-killing effector T cells (74, 97). Given that an enforced removal of UVB radiation-induced DNA photoproducts does not completely abrogate immunosuppression (94), other chromophores might also be involved. Besides trans-urocanic acid which upon UVB irradiation may isomerize to immunosuppressive cis-urocanic acid in the stratum corneum (74), tryptophan is another candidate compound. Indeed, as discussed below in more detail (see AHR and Immunosuppression), AHR has been identified to contribute to the suppression of immune responses in UVB-irradiated mice (97) suggesting an involvement of tryptophan photoproducts. FICZ, however, has been reported to enforce the generation of Th17 cells and thereby exacerbate experimental autoimmunity in mice (98, 99).
Role of AHR in Skin Photocarcinogenesis
AHR Activation by UVB Irradiation
In the epidermis, UVB rays are absorbed by the aromatic amino acid tryptophan, resulting in the formation of photoproducts, such as FICZ and 1-(1H-indol-3-yl)-9H-pyrido(3,4-b)indole, which serve as high-affinity AHR ligands (21, 76, 100–102). FICZ is detectable in human skin in vivo (103), and FICZ metabolites, i.e. sulfoconjugates of hydroxylated FICZ molecules, are present in human urine samples. Accordingly, an exposure to UVB radiation enhances cutaneous and hepatic CYP1 enzyme activities in rodents (104, 105), and induces the expression of AHR target genes in the skin of human volunteers (106, 107). FICZ is a very good substrate for CYP1 isoforms and their induction by FICZ-stimulated AHR signaling thus ensures a transient activation of the AHR system in response to acute UVB exposure (108, 109). Even though experimental evidence is lacking, it is tempting to speculate that epidermal AHR activity is also fueled by N-formylkynurenine (NFK), another tryptophan photoproduct formed in UVB-irradiated cells (110–112). Subsequently, arylformamidase (kynurenine formamidase) may convert NFK to kynurenine which is further metabolized to endogenous AHR ligands, such as kynurenic acid and xanthurenic acid (113, 114). As discussed later on, this process bypasses the first and rate limiting step of tryptophan catabolism, i.e. the tryptophan 2,3-dioxygenase (TDO)- or indoleamine 2,3-dioxygenase (IDO)-mediated oxidation of tryptophan to NFK, and thus may be relevant for a modulation of UVB radiation-induced immunosuppressive effects by the AHR system. Other genes whose expression is upregulated via non-canonical AHR signaling pathways in UVB-irradiated keratinocytes and human skin ex vivo and which might be relevant concerning skin carcinogenesis encode for cyclooxygenase-2 and matrix metalloproeinase-1 (21, 106).
Under chronic UVB irradiation, overactivation of AHR signaling pathways may have detrimental consequences (Figure 3). In fact, in a photocarcinogenesis study AHR-deficient SKH-1 hairless mice developed approximately 50% less cutaneous SCC than their AHR-proficient littermates, providing evidence that AHR signaling critically contributes to UVB radiation-induced skin carcinogenesis (115). Further analyses of the skin lesions did not reveal any obvious genotype-specific differences in tumor histology/biochemistry (115), indicating that, in the context of photocarcinogenesis, AHR activity mainly affects the tumor initiation phase. However, given that UVB irradiation enhances the expression of inflammatory mediators (e.g. chemokine (C-X-C motif) ligand 5, cyclooxygenase-2) in murine and human skin in an AHR-dependent manner (106, 116), and that a transgenic overexpression of a constitutively active AHR in mice is associated with inflammatory skin lesions (117), it seems to be likely that cutaneous AHR signaling also exhibits tumor-promoting effects.
AHR and Nucleotide Excision Repair
Given that skin photocarcinogenesis depends on the formation and repair of UVB-induced DNA photoproducts, in particular CPDs, our group has elucidated whether AHR activation affects CPD removal via NER. In fact, chemical inhibition and genetic targeting of AHR in human epidermal keratinocytes accelerates CPD removal at early time points (4 hrs) after UVB exposure (115). Treatment of keratinocytes with a pan-caspase inhibitor and subsequent CPD quantification excluded an early clearance of CPD-positive cells through apoptosis. Transient RNAi experiments in which the expression of either XPC, the damage recognition factor of GGR, or CSB, an initiator of TCR, was silenced, revealed that AHR attenuated CPD repair by specifically repressing the GGR sub-pathway (115) (Figure 3). The clinical relevance of GGR is illustrated by the fact that patients with GGR-inactivating mutations, but not patients suffering from TCR-deficiency, have a 1,000-fold increased risk to develop cutaneous SCC (118, 119). Further RNAi-based mechanistic studies revealed that AHR inhibits GGR by activating EGFR and downstream PI3K/AKT signal transduction, resulting in the phosphorylation and subsequent proteasomal degradation of the cyclin-dependent kinase (CDK) inhibitor and tumor suppressor protein p27KIP1 (115, 120). Accordingly, AHR inhibition results in a stabilization of the p27KIP1 protein level in keratinocytes in vitro and mouse skin in vivo (115, 121, 122). This stabilizing effect of AHR inhibition on p27KIP1 is not restricted to epidermal keratinocytes but also present in DAYO medulloblastoma (123) and A549 lung adenocarcinoma cells (124). Ectopic overexpression of p27KIP1 accelerates CPD repair in UVB radiation-exposed keratinocytes, whereas chemical inhibitors targeting CDK7, mimicked high levels of p27KIP1, i.e. inhibited CPD repair. CDK7 is the catalytically active subunit of the CDK-activating kinase, a component of the general transcription factor and NER complex TFIIH (125). In fact, a chemical inhibition of CDK7 has been previously shown to specifically stimulate GGR activity (126). Elevated p27KIP1 protein levels were present in the skin of AHR-deficient mice and associated with a faster removal of UVB radiation-induced CPDs (115). These data indicate that by repressing the repair of UVB radiation-induced DNA photoproducts, AHR may critically contribute to skin photocarcinogenesis. Recently, this concept was challenged by a study reporting that an activation of AHR signaling by keratinocyte growth factor-2 (KGF2) stimulates CPD clearance as early as one hour after UVB exposure (127). However, it is not clear whether this effect depended on alterations in NER activity or apoptosis. Given that peptide growth factors do probably not bind to the ligand-binding site of the AHR protein, the mode of AHR activation by KGF2 remains quite enigmatic. KGF2 serves as a ligand for the fibroblast growth factor receptor-2, which acts mitogenic and thereby may reduce apoptotic cell death (128). Hence, it is tempting to speculate that a cross-talk between the receptor tyrosine kinase and AHR signaling may be causative for the described discrepancy in UVB radiation-induced keratinocyte apoptosis.
AHR, Homologues Recombination Repair and Apoptosis
As outlined above, an abrogation of AHR signaling accelerates the removal of mutagenic CPDs through the NER sub-pathway GGR and thus should decrease UVB radiation-induced keratinocyte apoptosis (129). However, it has been previously shown that AHR serves an anti-apoptotic function in UVB-irradiated keratinocytes and mouse skin (115, 121). Interestingly, this anti-apoptotic effect also seems to depend on the AHR-mediated reduction of the p27KIP1 protein level. Accordingly, an inhibition of AHR enhances the apoptosis susceptibility of UVB-irradiated keratinocytes by upregulating p27KIP1 levels (121). Subsequently, p27KIP1 inhibits the activity of its substrate CDK2 and thereby abolishes the downstream phosphorylation of the retinoblastoma protein (RB). RB phosphorylation is necessary to activate E2F1, which controls the expression of CHK1 (121), a stress kinase critically involved in initiating cell-cycle arrest upon DNA damage (88). In UVB-irradiated keratinocytes, DNA double-strand breaks (DSBs) mainly occur when CPD-positive cells start to divide (84–86). Results from Comet assays and γH2AX quantification indicated that the enhanced apoptosis susceptibility of AHR-compromised keratinocytes is indeed due to an elevated formation of DSBs (115). Accordingly, at later time points after UVB exposure (18 h), AHR-compromised keratinocytes exhibited an elevated amount of DSBs as compared to respective control cells. Given that CHK1 is also essential for the initiation of HRR (87), AHR-compromised keratinocytes, exhibiting reduced CHK1 levels, are prone to DSB-induced apoptosis (115). These data indicate that AHR is a positive regulator of the HRR (Figure 3) and the associated fixation of DSBs and confirm previously published observations in 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD)-treated Chinese hamster ovary cells (130, 131). However, in contrast to the described anti-apoptotic role of cutaneous AHR in the context of UVB irradiation, it has been reported that in the absence of DNA damage AHR activation may sensitize keratinocytes to cytokine-/death receptor-induced apoptosis (132).
AHR and Immunosuppression
UVB radiation-induced immunosuppression largely depends on the occurrence of DNA photoproducts (see UV Radiation-Induced Immunosuppression). An inhibition of both GGR and apoptotic clearance probably results in an accumulation of CPDs and thus may represent one mechanism through which active AHR maintains the UVB radiation-induced suppression of the immune system. Studies on a mouse model of contact hypersensitivity confirmed this hypothesis by demonstrating that a chemical or genetic inhibition of AHR attenuates the UV radiation-induced expansion of Tregs and associated immunosuppressive effects (97). Further mechanistic studies using 4-n-nonylphenol to induce AHR-dependent immunosuppression, however, showed that AHR activation switches antigen-presenting dendritic cells (DC) from a stimulatory into a regulatory phenotype thereby leading to an induction of Tregs independently from DNA damage (97, 133). The underlying mechanism involves an AHR-dependent induction of IL-2 (133, 134) which subsequently induces IL-10 while repressing the expression of the negative regulatory protein B7-H4, a co-inhibitory molecule of the B7 family (133). In addition, AHR activation by 4-n-nonylphenol induced the expression of IDO in bone marrow-derived DC, thus confirming a previous study reporting that AHR is required for proper IDO induction (135). The enforced degradation of tryptophan and the associated formation of tryptophan metabolites may inhibit T cell proliferation and thereby suppresses antitumor immune responses (114, 136, 137). In fact, the derived tryptophan metabolites may activate AHR to generate regulatory DC which foster the expansion of Tregs while inhibiting T cell polarization towards Th17 cells (114). Noteworthy, kynurenine was found to induce the generation of immunosuppressive Tregs in mice and certain tumor entities in patients in an AHR-dependent manner (136, 138–141). Along the same line, activation of AHR by its prototypic ligand TCDD promotes the expansion of CD4+ CD25+ and FoxP3+ Tregs to suppress experimental autoimmune disease (encephalomyelitis, uveoretinitis) (99, 142, 143). This, however, stands in stark contrast to an activation of AHR signaling by either FICZ (see UV Radiation-Induced Immunosuppression) or airborne PAHs and PAH-rich PM (see AHR and PAH-Induced Immune Reactions) which stimulate the generation of Th17 cells and exacerbate autoimmune disorders. This discrepancy clearly points to a ligand-specific effect of the AHR system on fate and function of T lymphocytes. Interestingly, results from a study conducted by the Kerkvliet laboratory suggest that this ligand-specific effect is due to differences in the metabolic half-life of the respective AHR ligand and associated dose-dependent effects (144). However, in the context of UVB irradiation, a very interesting facet is that an absorption of UVB rays by tryptophan results in the formation of NFK (110–112), a photochemical reaction which bypasses the first and so-called rate-limiting IDO/TDO-catalyzed step of tryptophan degradation. At biologically relevant doses of UVB radiation and simulated sunlight, approximately 20% of the free tryptophan contained in cell culture medium is converted into the AHR pro-ligand NFK (112). In general, tryptophan is highly susceptible to many oxidizing agents and NFK is one of its major oxidation products (145). Although probably less relevant for the therapy of cutaneous SCC, this observation provides a potential mechanism through which other epidermal cancers, in particular advanced melanomas, may overcome a pharmacological inhibition of IDO/TDO. In fact, a phase III trial (ECHO-301) testing pembrolizumab, an antibody targeting the immune checkpoint protein PD-L1, in combination with the IDO blocker epacadostat revealed that the co-treatment has no benefit for patients suffering from advanced melanoma as compared to the patients treated with pembrolizumab alone (114, 146). The failure of IDO1 inhibitors for melanoma therapy might be related to an enhanced TDO activity or an elevated expression of IL-4-induced gene 1, another tryptophan-metabolizing enzyme that induces immunosuppression by producing AHR ligands (147, 148). Nevertheless, it is tempting to speculate that the UV radiation-induced formation of NFK in the skin and its further catabolism to kynurenic acid and other AHR-agonistic metabolites may, at least partially, contribute to the expansion of Tregs and the suppression of appropriate anti-tumor immune responses. As indicated by studies on patients with lung cancer or oral SCC, the activated AHR may not only enhance the expression of IDO but also attenuate the response to immune checkpoint inhibition by inducing PD-L1 (141, 149). In addition, tumor repopulating melanoma cells may produce kynurenine and associated AHR ligands to activate AHR and induce the expression of the PD-L1 receptor PD-1 in CD8+ T cells (150). Hence, a combination of small molecules inhibiting AHR activity with PD-1 checkpoint inhibitors might be a suitable approach to combat immunotherapy-resistant tumors (151).
Although experimental evidence is yet lacking, it is conceivable that at least in advanced stages of cutaneous SCC, the AHR may play a comparable role in modulating tumor immunity.
Co-Carcinogenicity of UV Radiation and PAHs?
Given that environmentally ubiquitous PAHs as well as UV radiation are capable of modulating AHR activity in keratinocytes and other epidermal cell populations, a simultaneous exposure to these factors may cause co-carcinogenic effects. However, as specified below, the data published so far on this topic produce a heterogenous and sometimes even contradictory picture. For instance, some early carcinogenesis studies reported an increased skin tumor formation upon UV irradiation of coal tar-treated mice (152, 153), whereas in another study on albino mice alternately exposed to UV radiation and PAHs, namely 3-methylcholanthrene, DMBA and B[a]P, no additive skin cancer formation was observed (154). A major factor that may contribute to this discrepancy is the pronounced sensitizing property of various PAHs toward UVA radiation. The resulting generation of ROS may on the one hand facilitate carcinogenesis by inducing oxidative DNA damage and inhibiting the function of DNA repair enzymes and other proteins (155–162). On the other hand, strong or longer lasting phototoxic stress may cause ROS-mediated cytotoxicity and necrotic cell death not only in normal epidermal cells but also in initiated keratinocytes and malignant cells. Interestingly, several UVB radiation-induced photoproducts of tryptophan, including NFK and FICZ, have been identified as potent UVA photosensitizers (110, 163, 164), and a combinatorial treatment with FICZ and UVA radiation was proposed as a novel therapeutic approach for skin cancer (165). Notably, other investigators regard the oxidative stress resulting from the FICZ/UVA exposure and the associated inhibition of DNA repair enzymes as a pro-carcinogenic event (166). In general, this process, i.e. the application of a photosensitizing agent and its subsequent irradiation with light of a certain wavelength, has been successfully implemented into the clinical routine for the treatment of certain solid tumors (167), including melanoma and non-melanoma skin cancers (168), and is known as photodynamic therapy. Other studies have shown that UV irradiation enhances the skin permeation rates of simultaneously applied PAHs (161, 169, 170) and may affect their metabolic activation (105, 155, 171, 172). UVB radiation was proven to sensitize epidermal keratinocytes to PAH-DNA adduct formation and subsequent mutagenesis (105, 171). The laboratory of David Bickers applied the Goeckerman regimen, i.e. a sequential treatment of the skin with PAH-rich crude coal tar and UVB radiation, to neonatal rats and observed an enhanced metabolic activation of B[a]P and associated DNA adduct formation in subsequent ex vivo experiments (105). An enhanced amount of PAH metabolites and markers for DNA damage were also observed in the blood and urine of psoriasis patients that underwent the Goeckerman regimen (161). Interestingly, Bickers and co-workers were able to show that the opposite application sequence, i.e. UVB exposure first followed by coal tar treatment, did not cause any significant differences in metabolic activation and BPDE-DNA adduct formation as compared to the samples of the coal tar-only treated animals (105). In contrast, Nair and colleagues reported that a treatment of HaCaT keratinocytes with either UVB radiation, photooxidized tryptophan or FICZ prior to B[a]P application significantly enhanced the expression of CYP1 isoforms and the associated formation of bulky DNA adducts (171). As expected a co-treatment with either α-naphthoflavone, an AHR antagonist, or the HSP90 blocker 17-AAG attenuated CYP1 induction and DNA adduct formation, thus confirming AHR dependency (171). Thierry Douki and co-workers, however, reported that a sequential treatment of keratinocytes and human skin explants with B[a]P or a mixture of PAHs and stimulated sunlight, reduced the expression of CYP1 isoforms, the generation of PAH metabolites and BPDE-DNA adduct formation (155, 170, 172). Given that the applied irradiation device emits UVB and UVA light (wavelengths from 290 nm - 400 nm), it is possible that the resulting generation of ROS is responsible for the observed downregulation of the expression of the CYP1 monooxygenases (173, 174). In addition, the spectrum of cytokines released by the irradiated epidermal cells may depend on the UV wavelength (175, 176). Tumor necrosis factor-α, for instance, is rather produced upon irradiation with UVA than with UVB light, and this cytokine is capable to suppress CYP1 gene expression via NF-κB-mediated trans-repression, i.e. a competition for common transcriptional co-activators (24, 177). However, as outlined in this article, PAHs and UV radiation may differently affect the expansion and function of immunosuppressive Tregs, which may be also an important factor that has to be considered in the context of a simultaneous exposure.
In summary, the current data concerning the simultaneous exposure of the skin to UV radiation and PAHs are not coherent and illustrate the need for further studies to ensure a proper risk assessment, in particular for roofers, roadmen, and other occupational groups that are frequently exposed to high doses of both, PAHs and sunlight. Chronic exposure studies on rodents, for instance, using the same irradiation device for single and simultaneous exposure to UVA and UVB light in combination with a pre- and post-treatment with environmentally-relevant PAH mixtures may shed light on a potential interaction of both risk factors in skin carcinogenesis.
Conclusion
Proper AHR signaling is indispensable for the development and physiology of the skin (178, 179). However, as outlined in this article, an overactivation of this signaling pathway in skin chronically exposed to one or more environmental stressors may have detrimental consequences (Figure 3). By modulating xenobiotic metabolism, different DNA repair systems, apoptosis, various functions of the immune system, and other processes, AHR-dependent signaling pathways may significantly contribute to the development of PAH- and UV radiation-induced skin carcinogenesis. Interestingly, both environmental factors seem to interact on the level of enzyme activity and DNA damage and repair, thus illustrating the critical role of the AHR in either restraining or facilitating the development of skin cancer. Even though, the published data on the potential co-carcinogenic action of UV radiation and PAHs do not produce a clear picture, a transient inhibition of cutaneous AHR signaling probably protects the skin of individuals exposed to UV radiation and PAHs alone or in combination. In the context of a co-exposure, it is tempting to speculate that the application of sunscreen does not only prevent the UV radiation-induced activation of AHR signaling but also the photoactivation of PAHs potentially present on the skin. Given that sun blockers do not protect against the genotoxicity of PAHs, the integration of transient AHR antagonists in sunscreens might be beneficial in order to protect the skin against both environmental/occupational stressors. Notably, it is widely accepted that the integration of antioxidants into sunscreens provides additional skin protection by neutralizing radiation- and pollution-induced ROS (180, 181) and, interestingly, several plant-derived polyphenols combine both properties, i.e. antagonizing AHR signaling and exhibiting antioxidative effects (182, 183). However, further research on the potential interaction of PAHs and UV radiation in the pathogenesis of SCCs is urgently needed in order to improve the risk assessment as well as the preventive strategies depending thereon.
Author Contributions
TH-S contributed to the conception and design of the review article. CV, KR, and TH-S wrote the manuscript. JK provided critical feedback during the preparation of the article and contributed to the revision of the manuscript. CV generated the figures. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Charlotte Esser for critical reading of the manuscript and helpful suggestions. The figures were created with BioRender software (www.biorender.com; agreement number JY23CK86BZ, SC23CK8D4Q and PB23CK8HNH). Research in the THS laboratory in supported by the Deutsche Forschungsgemeinschaft (HA 7346/2–2).
References
1. Rogers HW, Weinstock MA, Feldman SR, Coldiron BM. Incidence Estimate of Nonmelanoma Skin Cancer (Keratinocyte Carcinomas) in the U.S. Population, 2012. JAMA Dermatol (2015) 151:1081–6. doi: 10.1001/jamadermatol.2015.1187
2. Leiter U, Keim U, Eigentler T, Katalinic A, Holleczek B, Martus P, et al. Incidence, Mortality, and Trends of Nonmelanoma Skin Cancer in Germany. J Invest Dermatol (2017) 137:1860–7. doi: 10.1016/j.jid.2017.04.020
3. Green AC, Olsen CM. Cutaneous Squamous Cell Carcinoma: An Epidemiological Review. Br J Dermatol (2017) 177:373–81. doi: 10.1111/bjd.15324
4. Hiatt RA, Beyeler N. Cancer and Climate Change. Lancet Oncol (2020) 21:e519–27. doi: 10.1016/S1470-2045(20)30448-4
5. Lin MJ, Torbeck RL, Dubin DP, Lin CE, Khorasani H. Climate Change and Skin Cancer. J Eur Acad Dermatol Venereol (2019) 33:e324–5. doi: 10.1111/jdv.15622
6. Guy GP Jr, Machlin SR, Ekwueme DU, Yabroff KR. Prevalence and Costs of Skin Cancer Treatment in the U.S., 2002-2006 and 2007-2011. Am J Prev Med (2015) 48:183–7. doi: 10.1016/j.amepre.2014.08.036
7. Chahal HS, Lin Y, Ransohoff KJ, Hinds DA, Wu W, Dai HJ, et al. Genome-Wide Association Study Identifies Novel Susceptibility Loci for Cutaneous Squamous Cell Carcinoma. Nat Commun (2016) 7:12048. doi: 10.1038/ncomms12048
8. Berenblum I, Shubik P. The Role of Croton Oil Applications, Associated With a Single Painting of a Carcinogen, in Tumour Induction of the Mouse’s Skin. Br J Cancer (1947) 1:379–82. doi: 10.1038/bjc.1947.35
9. Berenblum I, Shubik P. A New, Quantitative, Approach to the Study of the Stages of Chemical Cartinogenesis in the Mouse’s Skin. Br J Cancer (1947) 1:383–91. doi: 10.1038/bjc.1947.36
10. Gelboin HV. Benzo[alpha]pyrene Metabolism, Activation and Carcinogenesis: Role and Regulation of Mixed-Function Oxidases and Related Enzymes. Physiol Rev (1980) 60:1107–66. doi: 10.1152/physrev.1980.60.4.1107
11. Luch A. Nature and Nurture - Lessons From Chemical Carcinogenesis. Nat Rev Cancer (2005) 5:113–25. doi: 10.1038/nrc1546
12. McCreery MQ, Balmain A. Chemical Carcinogenesis Models of Cancer: Back to the Future. Annu Rev Cancer Biol (2017) 1:295–312. doi: 10.1146/annurev-cancerbio-050216-122002
13. Bersten DC, Sullivan AE, Peet DJ, Whitelaw ML. bHLH-PAS Proteins in Cancer. Nat Rev Cancer (2013) 13:827–41. doi: 10.1038/nrc3621
14. Murray IA, Patterson AD, Perdew GH. Aryl Hydrocarbon Receptor Ligands in Cancer: Friend and Foe. Nat Rev Cancer (2014) 14:801–14. doi: 10.1038/nrc3846
15. Rothhammer V, Quintana FJ. The Aryl Hydrocarbon Receptor: An Environmental Sensor Integrating Immune Responses in Health and Disease. Nat Rev Immunol (2019) 19:184–97. doi: 10.1038/s41577-019-0125-8
16. Avilla MN, Malecki KMC, Hahn ME, Wilson RH, Bradfield CA. The Ah Receptor: Adaptive Metabolism, Ligand Diversity, and the Xenokine Model. Chem Res Toxicol (2020) 33:860–79. doi: 10.1021/acs.chemrestox.9b00476
17. Dvorak Z, Poulikova K, Mani S. Indole Scaffolds as a Promising Class of the Aryl Hydrocarbon Receptor Ligands. Eur J Med Chem (2021) 215:113231. doi: 10.1016/j.ejmech.2021.113231
18. Enan E, Matsumura F. Identification of C-Src as the Integral Component of the Cytosolic Ah Receptor Complex, Transducing the Signal of 2,3,7,8-Tetrachlorodibenzo-P-Dioxin (TCDD) Through the Protein Phosphorylation Pathway. Biochem Pharmacol (1996) 52:1599–612. doi: 10.1016/S0006-2952(96)00566-7
19. Mimura J, Ema M, Sogawa K, Fujii-Kuriyama Y. Identification of a Novel Mechanism of Regulation of Ah (Dioxin) Receptor Function. Genes Dev (1999) 13:20–5. doi: 10.1101/gad.13.1.20
20. Dong B, Cheng W, Li W, Zheng J, Wu D, Matsumura F, et al. FRET Analysis of Protein Tyrosine Kinase C-Src Activation Mediated via Aryl Hydrocarbon Receptor. Biochim Biophys Acta (2011) 1810:427–31. doi: 10.1016/j.bbagen.2010.11.007
21. Fritsche E, Schafer C, Calles C, Bernsmann T, Bernshausen T, Wurm M, et al. Lightening Up the UV Response by Identification of the Arylhydrocarbon Receptor as a Cytoplasmatic Target for Ultraviolet B Radiation. PNAS (2007) 104:8851–6. doi: 10.1073/pnas.0701764104
22. Vogeley C, Sondermann NC, Woeste S, Momin AA, Gilardino V, Hartung F, et al. Unraveling the Differential Impact of PAHs and Dioxin-Like Compounds on AKR1C3 Reveals the EGFR Extracellular Domain as a Critical Determinant of the AHR Response. Environ Int (2022) 158:106989. doi: 10.1016/j.envint.2021.106989
23. Vogel CF, Sciullo E, Li W, Wong P, Lazennec G, Matsumura F. RelB, a New Partner of Aryl Hydrocarbon Receptor-Mediated Transcription. Mol Endocrinol (2007) 21:2941–55. doi: 10.1210/me.2007-0211
24. Tian Y, Rabson AB, Gallo MA. Ah Receptor and NF-kappaB Interactions: Mechanisms and Physiological Implications. Chem Biol Interact (2002) 141:97–115. doi: 10.1016/S0009-2797(02)00068-6
25. Gradin K, McGuire J, Wenger RH, Kvietikova I, fhitelaw ML, Toftgard R, et al. Functional Interference Between Hypoxia and Dioxin Signal Transduction Pathways: Competition for Recruitment of the Arnt Transcription Factor. Mol Cell Biol (1996) 16:5221–31. doi: 10.1128/MCB.16.10.5221
26. Chan WK, Yao G, Gu YZ, Bradfield CA. Cross-Talk Between the Aryl Hydrocarbon Receptor and Hypoxia Inducible Factor Signaling Pathways. Demonstration of Competition and Compensation. J Biol Chem (1999) 274:12115–23. doi: 10.1074/jbc.274.17.12115
27. Wormke M, Stoner M, Saville B, Walker K, Abdelrahim M, Burghardt R, et al. The Aryl Hydrocarbon Receptor Mediates Degradation of Estrogen Receptor Alpha Through Activation of Proteasomes. Mol Cell Biol (2003) 23:1843–55. doi: 10.1128/MCB.23.6.1843-1855.2003
28. Ohtake F, Takeyama K, Matsumoto T, Kitagawa H, Yamamoto Y, Nohara K, et al. Modulation of Oestrogen Receptor Signalling by Association With the Activated Dioxin Receptor. Nature (2003) 423:545–50. doi: 10.1038/nature01606
29. Kalthoff S, Ehmer U, Freiberg N, Manns MP, Strassburg CP. Interaction Between Oxidative Stress Sensor Nrf2 and Xenobiotic-Activated Aryl Hydrocarbon Receptor in the Regulation of the Human Phase II Detoxifying UDP-Glucuronosyltransferase 1A10. J Biol Chem (2010) 285:5993–6002. doi: 10.1074/jbc.M109.075770
30. Tsuji G, Takahara M, Uchi H, Matsuda T, Chiba T, Takeuchi S, et al. Identification of Ketoconazole as an AhR-Nrf2 Activator in Cultured Human Keratinocytes: The Basis of Its Anti-Inflammatory Effect. J Invest Dermatol (2012) 132:59–68. doi: 10.1038/jid.2011.194
31. Boffetta P, Jourenkova N, Gustavsson P. Cancer Risk From Occupational and Environmental Exposure to Polycyclic Aromatic Hydrocarbons. Cancer Causes Control (1997) 8:444–72. doi: 10.1023/A:1018465507029
32. De Hertog SA, Wensveen CA, Bastiaens MT, Kielich CJ, Berkhout MJ, Westendorp RG, et al. Relation Between Smoking and Skin Cancer. J Clin Oncol (2001) 19:231–8. doi: 10.1200/JCO.2001.19.1.231
33. Dusingize JC, Olsen CM, Pandeya NP, Subramaniam P, Thompson BS, Neale RE, et al. Cigarette Smoking and the Risks of Basal Cell Carcinoma and Squamous Cell Carcinoma. J Invest Dermatol (2017) 137:1700–8. doi: 10.1016/j.jid.2017.03.027
34. Leonardi-Bee J, Ellison T, Bath-Hextall F. Smoking and the Risk of Nonmelanoma Skin Cancer: Systematic Review and Meta-Analysis. Arch Dermatol (2012) 148:939–46. doi: 10.1001/archdermatol.2012.1374
35. Shi S, Yoon DY, Hodge-Bell KC, Bebenek IG, Whitekus MJ, Zhang R, et al. The Aryl Hydrocarbon Receptor Nuclear Translocator (Arnt) Is Required for Tumor Initiation by Benzo[a]Pyrene. Carcinogenesis (2009) 30:1957–61. doi: 10.1093/carcin/bgp201
36. Shimizu Y, Nakatsuru Y, Ichinose M, Takahashi Y, Kume H, Mimura J, et al. Benzo[a]pyrene Carcinogenicity Is Lost in Mice Lacking the Aryl Hydrocarbon Receptor. PNAS (2000) 97:779–82. doi: 10.1073/pnas.97.2.779
37. Kleiner HE, Vulimiri SV, Reed MJ, Uberecken A, DiGiovanni J. Role of Cytochrome P450 1a1 and 1b1 in the Metabolic Activation of 7,12-Dimethylbenz[a]Anthracene and the Effects of Naturally Occurring Furanocoumarins on Skin Tumor Initiation. Chem Res Toxicol (2002) 15:226–35. doi: 10.1021/tx010151v
38. Modi BG, Neustadter J, Binda E, Lewis J, Filler RB, Roberts SJ, et al. Langerhans Cells Facilitate Epithelial DNA Damage and Squamous Cell Carcinoma. Science (2012) 335:104–8. doi: 10.1126/science.1211600
39. Buters JT, Mahadevan B, Quintanilla-Martinez L, Gonzalez FJ, Greim H, Baird WM, et al. Cytochrome P450 1B1 Determines Susceptibility to Dibenzo[a,L]Pyrene-Induced Tumor Formation. Chem Res Toxicol (2002) 15:1127–35. doi: 10.1021/tx020017q
40. Siddens LK, Bunde KL, Harper TA Jr., McQuistan TJ, Lohr CV, Bramer LM, et al. Cytochrome P450 1b1 in Polycyclic Aromatic Hydrocarbon (PAH)-Induced Skin Carcinogenesis: Tumorigenicity of Individual PAHs and Coal-Tar Extract, DNA Adduction and Expression of Select Genes in the Cyp1b1 Knockout Mouse. Toxicol Appl Pharmacol (2015) 287:149–60. doi: 10.1016/j.taap.2015.05.019
41. Zheng W, Jefcoate CR. Steroidogenic Factor-1 Interacts With cAMP Response Element-Binding Protein to Mediate cAMP Stimulation of CYP1B1 via a Far Upstream Enhancer. Mol Pharmacol (2005) 67:499–512. doi: 10.1124/mol.104.005504
42. Zheng W, Tong T, Lee J, Liu X, Marcus C, Jefcoate CR. Stimulation of Mouse Cyp1b1 During Adipogenesis: Characterization of Promoter Activation by the Transcription Factor Pax6. Arch Biochem Biophys (2013) 532:1–14. doi: 10.1016/j.abb.2013.01.007
43. Tsuchiya Y, Nakajima M, Kyo S, Kanaya T, Inoue M, Yokoi T. Human CYP1B1 Is Regulated by Estradiol via Estrogen Receptor. Cancer Res (2004) 64:3119–25. doi: 10.1158/0008-5472.CAN-04-0166
44. Ide F, Suka N, Kitada M, Sakashita H, Kusama K, Ishikawa T. Skin and Salivary Gland Carcinogenicity of 7,12-Dimethylbenz[a]Anthracene Is Equivalent in the Presence or Absence of Aryl Hydrocarbon Receptor. Cancer Lett (2004) 214:35–41. doi: 10.1016/j.canlet.2004.04.014
45. Matsumoto Y, Ide F, Kishi R, Akutagawa T, Sakai S, Nakamura M, et al. Aryl Hydrocarbon Receptor Plays a Significant Role in Mediating Airborne Particulate-Induced Carcinogenesis in Mice. Environ Sci Technol (2007) 41:3775–80. doi: 10.1021/es062793g
46. Marteijn JA, Lans H, Vermeulen W, Hoeijmakers JH. Understanding Nucleotide Excision Repair and Its Roles in Cancer and Ageing. Nat Rev Mol Cell Biol (2014) 15:465–81. doi: 10.1038/nrm3822
47. Roos WP, Thomas AD, Kaina B. DNA Damage and the Balance Between Survival and Death in Cancer Biology. Nat Rev Cancer (2016) 16:20–33. doi: 10.1038/nrc.2015.2
48. Penning TM. Human Aldo-Keto Reductases and the Metabolic Activation of Polycyclic Aromatic Hydrocarbons. Chem Res Toxicol (2014) 27:1901–17. doi: 10.1021/tx500298n
49. Birtwistle J, Hayden RE, Khanim FL, Green RM, Pearce C, Davies NJ, et al. The Aldo-Keto Reductase AKR1C3 Contributes to 7,12-Dimethylbenz(a)Anthracene-3,4-Dihydrodiol Mediated Oxidative DNA Damage in Myeloid Cells: Implications for Leukemogenesis. Mutat Res (2009) 662:67–74. doi: 10.1016/j.mrfmmm.2008.12.010
50. Park JH, Mangal D, Tacka KA, Quinn AM, Harvey RG, Blair IA, et al. Evidence for the Aldo-Keto Reductase Pathway of Polycyclic Aromatic Trans-Dihydrodiol Activation in Human Lung A549 Cells. Proc Natl Acad Sci USA (2008) 105:6846–51. doi: 10.1073/pnas.0802776105
51. Pettipher R, Hansel TT, Armer R. Antagonism of the Prostaglandin D2 Receptors DP1 and CRTH2 as an Approach to Treat Allergic Diseases. Nat Rev Drug Discov (2007) 6:313–25. doi: 10.1038/nrd2266
52. Massey WA, Hubbard WC, Liu MC, Kagey-Sobotka A, Cooper P, Lichtenstein LM. Profile of Prostanoid Release Following Antigen Challenge In Vivo in the Skin of Man. Br J Dermatol (1991) 125:529–34. doi: 10.1111/j.1365-2133.1991.tb14789.x
53. Mantel A, Carpenter-Mendini A, VanBuskirk J, Pentland AP. Aldo-Keto Reductase 1C3 Is Overexpressed in Skin Squamous Cell Carcinoma (SCC) and Affects SCC Growth via Prostaglandin Metabolism. Exp Dermatol (2014) 23:573–8. doi: 10.1111/exd.12468
54. Yusuf N, Nasti TH, Katiyar SK, Jacobs MK, Seibert MD, Ginsburg AC, et al. Antagonistic Roles of CD4+ and CD8+ T-Cells in 7,12-Dimethylbenz(a)Anthracene Cutaneous Carcinogenesis. Cancer Res (2008) 68:3924–30. doi: 10.1158/0008-5472.CAN-07-3059
55. Akiba H, Kehren J, Ducluzeau MT, Krasteva M, Horand F, Kaiserlian D, et al. Skin Inflammation During Contact Hypersensitivity Is Mediated by Early Recruitment of CD8+ T Cytotoxic 1 Cells Inducing Keratinocyte Apoptosis. J Immunol (2002) 168:3079–87. doi: 10.4049/jimmunol.168.6.3079
56. Anderson C, Hehr A, Robbins R, Hasan R, Athar M, Mukhtar H, et al. Metabolic Requirements for Induction of Contact Hypersensitivity to Immunotoxic Polyaromatic Hydrocarbons. J Immunol (1995) 155:3530–7.
57. Coussens LM, Zitvogel L, Palucka AK. Neutralizing Tumor-Promoting Chronic Inflammation: A Magic Bullet? Science (2013) 339:286–91. doi: 10.1126/science.1232227
58. He D, Li H, Yusuf N, Elmets CA, Athar M, Katiyar SK, et al. IL-17 Mediated Inflammation Promotes Tumor Growth and Progression in the Skin. PloS One (2012) 7:e32126. doi: 10.1371/journal.pone.0032126
59. Sun L, Fu J, Lin SH, Sun JL, Xia L, Lin CH, et al. Particulate Matter of 2.5 Mum or Less in Diameter Disturbs the Balance of TH17/regulatory T Cells by Targeting Glutamate Oxaloacetate Transaminase 1 and Hypoxia-Inducible Factor 1alpha in an Asthma Model. J Allergy Clin Immunol (2020) 145:402–14. doi: 10.1016/j.jaci.2019.10.008
60. Nadeau K, McDonald-Hyman C, Noth EM, Pratt B, Hammond SK, Balmes J, et al. Ambient Air Pollution Impairs Regulatory T-Cell Function in Asthma. J Allergy Clin Immunol (2010) 126:845–852 e810. doi: 10.1016/j.jaci.2010.08.008
61. O’Driscoll CA, Owens LA, Gallo ME, Hoffmann EJ, Afrazi A, Han M, et al. Differential Effects of Diesel Exhaust Particles on T Cell Differentiation and Autoimmune Disease. Part Fibre Toxicol (2018) 15:35. doi: 10.1186/s12989-018-0271-3
62. Castaneda AR, Vogel CFA, Bein KJ, Hughes HK, Smiley-Jewell S, Pinkerton KE. Ambient Particulate Matter Enhances the Pulmonary Allergic Immune Response to House Dust Mite in a BALB/c Mouse Model by Augmenting Th2- and Th17-Immune Responses. Physiol Rep (2018) 6:e13827. doi: 10.14814/phy2.13827
63. Wong TH, Lee CL, Su HH, Lee CL, Wu CC, Wang CC, et al. A Prominent Air Pollutant, Indeno[1,2,3-Cd]Pyrene, Enhances Allergic Lung Inflammation via Aryl Hydrocarbon Receptor. Sci Rep (2018) 8:5198. doi: 10.1038/s41598-018-23542-9
64. Weng CM, Wang CH, Lee MJ, He JR, Huang HY, Chao MW, et al. Aryl Hydrocarbon Receptor Activation by Diesel Exhaust Particles Mediates Epithelium-Derived Cytokines Expression in Severe Allergic Asthma. Allergy (2018) 73:2192–204. doi: 10.1111/all.13462
65. Hong CH, Lee CH, Yu HS, Huang SK. Benzopyrene, a Major Polyaromatic Hydrocarbon in Smoke Fume, Mobilizes Langerhans Cells and Polarizes Th2/17 Responses in Epicutaneous Protein Sensitization Through the Aryl Hydrocarbon Receptor. Int Immunopharmacol (2016) 36:111–7. doi: 10.1016/j.intimp.2016.04.017
66. Xia M, Viera-Hutchins L, Garcia-Lloret M, Noval Rivas M, Wise P, McGhee SA, et al. Vehicular Exhaust Particles Promote Allergic Airway Inflammation Through an Aryl Hydrocarbon Receptor-Notch Signaling Cascade. J Allergy Clin Immunol (2015) 136:441–53. doi: 10.1016/j.jaci.2015.02.014
67. Hidaka T, Ogawa E, Kobayashi EH, Suzuki T, Funayama R, Nagashima T, et al. The Aryl Hydrocarbon Receptor AhR Links Atopic Dermatitis and Air Pollution via Induction of the Neurotrophic Factor Artemin. Nat Immunol (2017) 18:64–73. doi: 10.1038/ni.3614
68. Krutmann J, Bouloc A, Sore G, Bernard BA, Passeron T. The Skin Aging Exposome. J Dermatol Sci (2017) 85:152–61. doi: 10.1016/j.jdermsci.2016.09.015
69. Young AR, Claveau J, Rossi AB. Ultraviolet Radiation and the Skin: Photobiology and Sunscreen Photoprotection. J Am Acad Dermatol (2017) 76:S100–9. doi: 10.1016/j.jaad.2016.09.038
70. Wondrak GT, Jacobson MK, Jacobson EL. Endogenous UVA-Photosensitizers: Mediators of Skin Photodamage and Novel Targets for Skin Photoprotection. Photochem Photobiol Sci (2006) 5:215–37. doi: 10.1039/B504573H
71. Ratushny V, Gober MD, Hick R, Ridky TW, Seykora JT. From Keratinocyte to Cancer: The Pathogenesis and Modeling of Cutaneous Squamous Cell Carcinoma. J Clin Invest (2012) 122:464–72. doi: 10.1172/JCI57415
72. Elmets CA, Athar M. Milestones in Photocarcinogenesis. J Invest Dermatol (2013) 133:E13–7. doi: 10.1038/skinbio.2013.179
73. Bernard JJ, Gallo RL, Krutmann J. Photoimmunology: How Ultraviolet Radiation Affects the Immune System. Nat Rev Immunol (2019) 19:688–701. doi: 10.1038/s41577-019-0185-9
74. Schwarz T, Beissert S. Milestones in Photoimmunology. J Invest Dermatol (2013) 133:E7–E10. doi: 10.1038/skinbio.2013.177
75. Cadet J, Grand A, Douki T. Solar UV Radiation-Induced DNA Bipyrimidine Photoproducts: Formation and Mechanistic Insights. Top Curr Chem (2015) 356:249–75. doi: 10.1007/128_2014_553
76. Rannug A, Rannug U, Rosenkranz HS, Winqvist L, Westerholm R, Agurell E, et al. Certain Photooxidized Derivatives of Tryptophan Bind With Very High Affinity to the Ah Receptor and Are Likely to be Endogenous Signal Substances. J Biol Chem (1987) 262:15422–7. doi: 10.1016/S0021-9258(18)47743-5
77. Herrlich P, Karin M, Weiss C. Supreme EnLIGHTenment: Damage Recognition and Signaling in the Mammalian UV Response. Mol Cell (2008) 29:279–90. doi: 10.1016/j.molcel.2008.01.001
78. Brash DE, Rudolph JA, Simon JA, Lin A, McKenna GJ, Baden HP, et al. A Role for Sunlight in Skin Cancer: UV-Induced P53 Mutations in Squamous Cell Carcinoma. Proc Natl Acad Sci USA (1991) 88:10124–8. doi: 10.1073/pnas.88.22.10124
79. Ziegler A, Jonason AS, Leffell DJ, Simon JA, Sharma HW, Kimmelman J, et al. Sunburn and P53 in the Onset of Skin Cancer. Nature (1994) 372:773–6. doi: 10.1038/372773a0
80. Jans J, Schul W, Sert YG, Rijksen Y, Rebel H, Eker AP, et al. Powerful Skin Cancer Protection by a CPD-Photolyase Transgene. Curr Biol (2005) 15:105–15. doi: 10.1016/j.cub.2005.01.001
81. Yarosh D, Alas LG, Yee V, Oberyszyn A, Kibitel JT, Mitchell D, et al. Pyrimidine Dimer Removal Enhanced by DNA Repair Liposomes Reduces the Incidence of UV Skin Cancer in Mice. Cancer Res (1992) 52:4227–31.
82. DiGiovanna JJ, Kraemer KH. Shining a Light on Xeroderma Pigmentosum. J Invest Dermatol (2012) 132:785–96. doi: 10.1038/jid.2011.426
83. Roos WP, Kaina B. DNA Damage-Induced Cell Death by Apoptosis. Trends Mol Med (2006) 12:440–50. doi: 10.1016/j.molmed.2006.07.007
84. Batista LF, Kaina B, Meneghini R, Menck CF. How DNA Lesions Are Turned Into Powerful Killing Structures: Insights From UV-Induced Apoptosis. Mutat Res (2009) 681:197–208. doi: 10.1016/j.mrrev.2008.09.001
85. Dunkern TR, Kaina B. Cell Proliferation and DNA Breaks Are Involved in Ultraviolet Light-Induced Apoptosis in Nucleotide Excision Repair-Deficient Chinese Hamster Cells. Mol Biol Cell (2002) 13:348–61. doi: 10.1091/mbc.01-05-0225
86. Garinis GA, Mitchell JR, Moorhouse MJ, Hanada K, de WH, Vandeputte D, et al. Transcriptome Analysis Reveals Cyclobutane Pyrimidine Dimers as a Major Source of UV-Induced DNA Breaks. EMBO J (2005) 24:3952–62. doi: 10.1038/sj.emboj.7600849
87. Sorensen CS, Hansen LT, Dziegielewski J, Syljuasen RG, Lundin C, Bartek J, et al. The Cell-Cycle Checkpoint Kinase Chk1 Is Required for Mammalian Homologous Recombination Repair. Nat Cell Biol (2005) 7:195–201. doi: 10.1038/ncb1212
88. Sanchez Y, Wong C, Thoma RS, Richman R, Wu Z, Piwnica-Worms H, et al. Conservation of the Chk1 Checkpoint Pathway in Mammals: Linkage of DNA Damage to Cdk Regulation Through Cdc25. Science (1997) 277:1497–501. doi: 10.1126/science.277.5331.1497
89. Helleday T. Homologous Recombination in Cancer Development, Treatment and Development of Drug Resistance. Carcinogenesis (2010) 31:955–60. doi: 10.1093/carcin/bgq064
90. Aziz MH, Reagan-Shaw S, Wu J, Longley BJ, Ahmad N. Chemoprevention of Skin Cancer by Grape Constituent Resveratrol: Relevance to Human Disease? FASEB J (2005) 19:1193–5. doi: 10.1096/fj.04-3582fje
91. Kawasumi M, Lemos B, Bradner JE, Thibodeau R, Kim YS, Schmidt M, et al. Protection From UV-Induced Skin Carcinogenesis by Genetic Inhibition of the Ataxia Telangiectasia and Rad3-Related (ATR) Kinase. PNAS (2011) 108:13716–21. doi: 10.1073/pnas.1111378108
92. Lu YP, Lou YR, Xie JG, Peng QY, Zhou S, Lin Y, et al. Caffeine and Caffeine Sodium Benzoate Have a Sunscreen Effect, Enhance UVB-Induced Apoptosis, and Inhibit UVB-Induced Skin Carcinogenesis in SKH-1 Mice. Carcinogenesis (2006) 28:199–206. doi: 10.1093/carcin/bgl112
93. Kim DJ, Kataoka K, Sano S, Connolly K, Kiguchi K, DiGiovanni J. Targeted Disruption of Bcl-xL in Mouse Keratinocytes Inhibits Both UVB- and Chemically Induced Skin Carcinogenesis. Mol Carcinog (2009) 48:873–85. doi: 10.1002/mc.20527
94. Applegate LA, Ley RD, Alcalay J, Kripke ML. Identification of the Molecular Target for the Suppression of Contact Hypersensitivity by Ultraviolet Radiation. J Exp Med (1989) 170:1117–31. doi: 10.1084/jem.170.4.1117
95. Kripke ML, Cox PA, Alas LG, Yarosh DB. Pyrimidine Dimers in DNA Initiate Systemic Immunosuppression in UV-Irradiated Mice. PNAS (1992) 89:7516–20. doi: 10.1073/pnas.89.16.7516
96. Stege H, Roza L, Vink AA, Grewe M, Ruzicka T, Grether-Beck S, et al. Enzyme Plus Light Therapy to Repair DNA Damage in Ultraviolet-B-Irradiated Human Skin. Proc Natl Acad Sci USA (2000) 97:1790–5. doi: 10.1073/pnas.030528897
97. Navid F, Bruhs A, Schuller W, Fritsche E, Krutmann J, Schwarz T, et al. The Aryl Hydrocarbon Receptor is Involved in UVR-Induced Immunosuppression. J Invest Dermatol (2013) 133:2763–70. doi: 10.1038/jid.2013.221
98. Veldhoen M, Hirota K, Westendorf AM, Buer J, Dumoutier L, Renauld JC, et al. The Aryl Hydrocarbon Receptor Links TH17-Cell-Mediated Autoimmunity to Environmental Toxins. Nature (2008) 453:106–9. doi: 10.1038/nature06881
99. Quintana FJ, Basso AS, Iglesias AH, Korn T, Farez MF, Bettelli E, et al. Control of T(reg) and T(H)17 Cell Differentiation by the Aryl Hydrocarbon Receptor. Nature (2008) 453:65–71. doi: 10.1038/nature06880
100. Rannug U, Rannug A, Sjoberg U, Li H, Westerholm R, Bergman J. Structure Elucidation of Two Tryptophan-Derived, High Affinity Ah Receptor Ligands. Chem Biol (1995) 2:841–5. doi: 10.1016/1074-5521(95)90090-X
101. Diani-Moore S, Ma Y, Labitzke E, Tao H, David Warren J, Anderson J, et al. Discovery and Biological Characterization of 1-(1H-Indol-3-Yl)-9H-Pyrido[3,4-B]Indole as an Aryl Hydrocarbon Receptor Activator Generated by Photoactivation of Tryptophan by Sunlight. Chem Biol Interact (2011) 193:119–28. doi: 10.1016/j.cbi.2011.05.010
102. Helferich WG, Denison MS. Ultraviolet Photoproducts of Tryptophan can Act as Dioxin Agonists. Mol Pharmacol (1991) 40:674–8.
103. Schallreuter KU, Salem MA, Gibbons NC, Maitland DJ, Marsch E, Elwary SM, et al. Blunted Epidermal L-Tryptophan Metabolism in Vitiligo Affects Immune Response and ROS Scavenging by Fenton Chemistry, Part 2: Epidermal H2O2/ONOO(-)-Mediated Stress in Vitiligo Hampers Indoleamine 2,3-Dioxygenase and Aryl Hydrocarbon Receptor-Mediated Immune Response Signaling. FASEB J (2012) 26:2471–85. doi: 10.1096/fj.11-201897
104. Goerz G, Barnstorf W, Winnekendonk G, Bolsen K, Fritsch C, Kalka K, et al. Influence of UVA and UVB Irradiation on Hepatic and Cutaneous P450 Isoenzymes. Arch Dermatol Res (1996) 289:46–51. doi: 10.1007/s004030050151
105. Mukhtar H, DelTito BJ Jr., Matgouranis PM, Das M, Asokan P, Bickers DR. Additive Effects of Ultraviolet B and Crude Coal Tar on Cutaneous Carcinogen Metabolism: Possible Relevance to the Tumorigenicity of the Goeckerman Regimen. J Invest Dermatol (1986) 87:348–53. doi: 10.1111/1523-1747.ep12524446
106. Tigges J, Haarmann-Stemmann T, Vogel CF, Grindel A, Hubenthal U, Brenden H, et al. The New Aryl Hydrocarbon Receptor Antagonist E/Z-2-Benzylindene-5,6-Dimethoxy-3,3-Dimethylindan-1-One Protects Against UVB-Induced Signal Transduction. J Invest Dermatol (2014) 134:556–9. doi: 10.1038/jid.2013.362
107. Katiyar SK, Matsui MS, Mukhtar H. Ultraviolet-B Exposure of Human Skin Induces Cytochromes P450 1A1 and 1B1. J Invest Dermatol (2000) 114:328–33. doi: 10.1046/j.1523-1747.2000.00876.x
108. Bergander L, Wincent E, Rannug A, Foroozesh M, Alworth W, Rannug U. Metabolic Fate of the Ah Receptor Ligand 6-Formylindolo[3,2-B]Carbazole. Chem Biol Interact (2004) 149:151–64. doi: 10.1016/j.cbi.2004.08.005
109. Wincent E, Bengtsson J, Mohammadi BA, Alsberg T, Luecke S, Rannug U, et al. Inhibition of Cytochrome P4501-Dependent Clearance of the Endogenous Agonist FICZ as a Mechanism for Activation of the Aryl Hydrocarbon Receptor. PNAS (2012) 109:4479–84. doi: 10.1073/pnas.1118467109
110. Walrant P, Santus R, Grossweiner LI. Photosensitizing Properties of N-Formylkynurenine. Photochem Photobiol (1975) 22:63–5. doi: 10.1111/j.1751-1097.1975.tb06723.x
111. Asquith RS, Rivett DE. Studies on the Photooxidation of Tryptophan. Biochim Biophys Acta (1971) 252:111–6. doi: 10.1016/0304-4165(71)90098-5
112. Youssef A, von Koschembahr A, Caillat S, Corre S, Galibert MD, Douki T. 6-Formylindolo[3,2-B]Carbazole (FICZ) Is a Very Minor Photoproduct of Tryptophan at Biologically Relevant Doses of UVB and Simulated Sunlight. Photochem Photobiol (2019) 95:237–43. doi: 10.1111/php.12950
113. DiNatale BC, Murray IA, Schroeder JC, Flaveny CA, Lahoti TS, Laurenzana EM, et al. Kynurenic Acid Is a Potent Endogenous Aryl Hydrocarbon Receptor Ligand That Synergistically Induces Interleukin-6 in the Presence of Inflammatory Signaling. Toxicol Sci (2010) 115:89–97. doi: 10.1093/toxsci/kfq024
114. Platten M, Nollen EAA, Rohrig UF, Fallarino F, Opitz CA. Tryptophan Metabolism as a Common Therapeutic Target in Cancer, Neurodegeneration and Beyond. Nat Rev Drug Discov (2019) 18:379–401. doi: 10.1038/s41573-019-0016-5
115. Pollet M, Shaik S, Mescher M, Frauenstein K, Tigges J, Braun SA, et al. The AHR Represses Nucleotide Excision Repair and Apoptosis and Contributes to UV-Induced Skin Carcinogenesis. Cell Death Differ (2018) 25:1823–36. doi: 10.1038/s41418-018-0160-1
116. Smith KJ, Murray IA, Boyer JA, Perdew GH. Allelic Variants of the Aryl Hydrocarbon Receptor Differentially Influence UVB-Mediated Skin Inflammatory Responses in SKH1 Mice. Toxicology (2018) 394:27–34. doi: 10.1016/j.tox.2017.11.020
117. Tauchi M, Hida A, Negishi T, Katsuoka F, Noda S, Mimura J, et al. Constitutive Expression of Aryl Hydrocarbon Receptor in Keratinocytes Causes Inflammatory Skin Lesions. Mol Cell Biol (2005) 25:9360–8. doi: 10.1128/MCB.25.21.9360-9368.2005
118. Bradford PT, Goldstein AM, Tamura D, Khan SG, Ueda T, Boyle J, et al. Cancer and Neurologic Degeneration in Xeroderma Pigmentosum: Long Term Follow-Up Characterises the Role of DNA Repair. J Med Genet (2011) 48:168–76. doi: 10.1136/jmg.2010.083022
119. Reid-Bayliss KS, Arron ST, Loeb LA, Bezrookove V, Cleaver JE. Why Cockayne Syndrome Patients do Not Get Cancer Despite Their DNA Repair Deficiency. Proc Natl Acad Sci USA (2016) 113:10151–6. doi: 10.1073/pnas.1610020113
120. Chu IM, Hengst L, Slingerland JM. The Cdk Inhibitor P27 in Human Cancer: Prognostic Potential and Relevance to Anticancer Therapy. Nat Rev Cancer (2008) 8:253–67. doi: 10.1038/nrc2347
121. Frauenstein K, Sydlik U, Tigges J, Majora M, Wiek C, Hanenberg H, et al. Evidence for a Novel Anti-Apoptotic Pathway in Human Keratinocytes Involving the Aryl Hydrocarbon Receptor, E2F1, and Checkpoint Kinase 1. Cell Death Differ (2013) 20:1425–34. doi: 10.1038/cdd.2013.102
122. Kalmes M, Hennen J, Clemens J, Blomeke B. Impact of Aryl Hydrocarbon Receptor (AhR) Knockdown on Cell Cycle Progression in Human HaCaT Keratinocytes. Biol Chem (2011) 392:643–51. doi: 10.1515/bc.2011.067
123. Dever DP, Opanashuk LA. The Aryl Hydrocarbon Receptor Contributes to the Proliferation of Human Medulloblastoma Cells. Mol Pharmacol (2012) 81:669–78. doi: 10.1124/mol.111.077305
124. Hsu HL, Chen HK, Tsai CH, Liao PL, Chan YJ, Lee YC, et al. Aryl Hydrocarbon Receptor Defect Attenuates Mitogen-Activated Signaling Through Leucine-Rich Repeats and Immunoglobulin-Like Domains 1 (LRIG1)-Dependent EGFR Degradation. Int J Mol Sci (2021) 22:9988. doi: 10.3390/ijms22189988
125. Compe E, Egly JM. TFIIH: When Transcription Met DNA Repair. Nat Rev Mol Cell Biol (2012) 13:343–54. doi: 10.1038/nrm3350
126. Coin F, Oksenych V, Mocquet V, Groh S, Blattner C, Egly JM. Nucleotide Excision Repair Driven by the Dissociation of CAK From TFIIH. Mol Cell (2008) 31:9–20. doi: 10.1016/j.molcel.2008.04.024
127. Gao S, Guo K, Chen Y, Zhao J, Jing R, Wang L, et al. Keratinocyte Growth Factor 2 Ameliorates UVB-Induced Skin Damage via Activating the AhR/Nrf2 Signaling Pathway. Front Pharmacol (2021) 12:655281. doi: 10.3389/fphar.2021.655281
128. Xie Y, Su N, Yang J, Tan Q, Huang S, Jin M, et al. FGF/FGFR Signaling in Health and Disease. Signal Transduct Target Ther (2020) 5:181. doi: 10.1038/s41392-020-00222-7
129. Schwarz A, Stander S, Berneburg M, Bohm M, Kulms D, van Steeg H, et al. Interleukin-12 Suppresses Ultraviolet Radiation-Induced Apoptosis by Inducing DNA Repair. Nat Cell Biol (2002) 4:26–31. doi: 10.1038/ncb717
130. Chan CY, Kim PM, Winn LM. TCDD-Induced Homologous Recombination: The Role of the Ah Receptor Versus Oxidative DNA Damage. Mutat Res (2004) 563:71–9. doi: 10.1016/j.mrgentox.2004.05.015
131. Chan CY, Kim PM, Winn LM. TCDD Affects DNA Double Strand-Break Repair. Toxicol Sci (2004) 81:133–8. doi: 10.1093/toxsci/kfh200
132. Stolpmann K, Brinkmann J, Salzmann S, Genkinger D, Fritsche E, Hutzler C, et al. Activation of the Aryl Hydrocarbon Receptor Sensitises Human Keratinocytes for CD95L- and TRAIL-Induced Apoptosis. Cell Death Dis (2012) 3:e388. doi: 10.1038/cddis.2012.127
133. Bruhs A, Haarmann-Stemmann T, Frauenstein K, Krutmann J, Schwarz T, Schwarz A. Activation of the Arylhydrocarbon Receptor Causes Immunosuppression Primarily by Modulating Dendritic Cells. J Invest Dermatol (2015) 135:435–44. doi: 10.1038/jid.2014.419
134. Jeon MS, Esser C. The Murine IL-2 Promoter Contains Distal Regulatory Elements Responsive to the Ah Receptor, a Member of the Evolutionarily Conserved bHLH-PAS Transcription Factor Family. J Immunol (2000) 165:6975–83. doi: 10.4049/jimmunol.165.12.6975
135. Nguyen NT, Kimura A, Nakahama T, Chinen I, Masuda K, Nohara K, et al. Aryl Hydrocarbon Receptor Negatively Regulates Dendritic Cell Immunogenicity via a Kynurenine-Dependent Mechanism. Proc Natl Acad Sci USA (2010) 107:19961–6. doi: 10.1073/pnas.1014465107
136. Opitz CA, Litzenburger UM, Sahm F, Ott M, Tritschler I, Trump S, et al. An Endogenous Tumour-Promoting Ligand of the Human Aryl Hydrocarbon Receptor. Nature (2011) 478:197–203. doi: 10.1038/nature10491
137. Munn DH, Mellor AL. Indoleamine 2,3 Dioxygenase and Metabolic Control of Immune Responses. Trends Immunol (2013) 34:137–43. doi: 10.1016/j.it.2012.10.001
138. Mezrich JD, Fechner JH, Zhang X, Johnson BP, Burlingham WJ, Bradfield CA. An Interaction Between Kynurenine and the Aryl Hydrocarbon Receptor Can Generate Regulatory T Cells. J Immunol (2010) 185:3190–8. doi: 10.4049/jimmunol.0903670
139. Litzenburger UM, Opitz CA, Sahm F, Rauschenbach KJ, Trump S, Winter M, et al. Constitutive IDO Expression in Human Cancer Is Sustained by an Autocrine Signaling Loop Involving IL-6, STAT3 and the AHR. Oncotarget (2014) 5:1038–51. doi: 10.18632/oncotarget.1637
140. D’Amato NC, Rogers TJ, Gordon MA, Greene LI, Cochrane DR, Spoelstra NS, et al. A TDO2-AhR Signaling Axis Facilitates Anoikis Resistance and Metastasis in Triple-Negative Breast Cancer. Cancer Res (2015) 75:4651–64. doi: 10.1158/0008-5472.CAN-15-2011
141. Kenison JE, Wang Z, Yang K, Snyder M, Quintana FJ, Sherr DH. The Aryl Hydrocarbon Receptor Suppresses Immunity to Oral Squamous Cell Carcinoma Through Immune Checkpoint Regulation. PNAS (2021) 118:e2012692118. doi: 10.1073/pnas.2012692118
142. Funatake CJ, Marshall NB, Steppan LB, Mourich DV, Kerkvliet NI. Cutting Edge: Activation of the Aryl Hydrocarbon Receptor by 2,3,7,8-Tetrachlorodibenzo-P-Dioxin Generates a Population of CD4+ CD25+ Cells With Characteristics of Regulatory T Cells. J Immunol (2005) 175:4184–8. doi: 10.4049/jimmunol.175.7.4184
143. Zhang L, Ma J, Takeuchi M, Usui Y, Hattori T, Okunuki Y, et al. Suppression of Experimental Autoimmune Uveoretinitis by Inducing Differentiation of Regulatory T Cells via Activation of Aryl Hydrocarbon Receptor. Invest Ophthalmol Vis Sci (2010) 51:2109–17. doi: 10.1167/iovs.09-3993
144. Ehrlich AK, Pennington JM, Bisson WH, Kolluri SK, Kerkvliet NI. TCDD, FICZ, and Other High Affinity AhR Ligands Dose-Dependently Determine the Fate of CD4+ T Cell Differentiation. Toxicol Sci (2018) 161:310–20. doi: 10.1093/toxsci/kfx215
145. Ronsein GE, Oliveira MCB, Miyamoto S, Medeiros MHG, Di Mascio P. Tryptophan Oxidation by Singlet Molecular Oxygen [O2(1Deltag)]: Mechanistic Studies Using 18O-Labeled Hydroperoxides, Mass Spectrometry, and Light Emission Measurements. Chem Res Toxicol (2008) 21:1271–83. doi: 10.1021/tx800026g
146. Van den Eynde BJ, Van Baren N, Baurain JF. Is There a Clinical Future for IDO1 Inhibitors After the Failure of Epacadostat in Melanoma? Annu Rev Cancer Biol (2020) 4:241–56. doi: 10.1146/annurev-cancerbio-030419-033635
147. Sadik A, Somarribas Patterson LF, Ozturk S, Mohapatra SR, Panitz V, Secker PF, et al. IL4I1 Is a Metabolic Immune Checkpoint That Activates the AHR and Promotes Tumor Progression. Cell (2020) 182:1252–1270 e1234. doi: 10.1016/j.cell.2020.07.038
148. Ramspott JP, Bekkat F, Bod L, Favier M, Terris B, Salomon A, et al. Emerging Role of IL-4-Induced Gene 1 as a Prognostic Biomarker Affecting the Local T-Cell Response in Human Cutaneous Melanoma. J Invest Dermatol (2018) 138:2625–34. doi: 10.1016/j.jid.2018.06.178
149. Wang GZ, Zhang L, Zhao XC, Gao SH, Qu LW, Yu H, et al. The Aryl Hydrocarbon Receptor Mediates Tobacco-Induced PD-L1 Expression and Is Associated With Response to Immunotherapy. Nat Commun (2019) 10:1125. doi: 10.1038/s41467-019-08887-7
150. Liu Y, Liang X, Dong W, Fang Y, Lv J, Zhang T, et al. Tumor-Repopulating Cells Induce PD-1 Expression in CD8(+) T Cells by Transferring Kynurenine and AhR Activation. Cancer Cell (2018) 33:480–94.e487. doi: 10.1016/j.ccell.2018.02.005
151. Labadie BW, Bao R, Luke JJ. Reimagining IDO Pathway Inhibition in Cancer Immunotherapy via Downstream Focus on the Tryptophan-Kynurenine-Aryl Hydrocarbon Axis. Clin Cancer Res (2019) 25:1462–71. doi: 10.1158/1078-0432.CCR-18-2882
152. Urbach F. Modification of Ultraviolet Carcinogenesis by Photoactive Agents; Preliminary Report. J Invest Dermatol (1959) 32:373–8. doi: 10.1038/jid.1959.63
153. Findlay GM. Ultra-Violet Light and Skin Cancer. Lancet (1928) 2:1070 – 1073. doi: 10.1016/S0140-6736(00)84845-X
154. Rusch HP, Kline BE, Baumann CA. The Nonadditive Effect of Ultraviolet Light and Other Carcinogenic Procedures. Cancer Res (1942) 2:183 – 188.
155. von Koschembahr A, Youssef A, Beal D, Gudimard L, Giot JP, Douki T. Toxicity and DNA Repair in Normal Human Keratinocytes Co-Exposed to Benzo[a]Pyrene and Sunlight. Toxicol In Vitro (2020) 63:104744. doi: 10.1016/j.tiv.2019.104744
156. Botta C, Di Giorgio C, Sabatier AS, De Meo M. Effects of UVA and Visible Light on the Photogenotoxicity of Benzo[a]Pyrene and Pyrene. Environ Toxicol (2009) 24:492–505. doi: 10.1002/tox.20455
157. Mauthe RJ, Cook VM, Coffing SL, Baird WM. Exposure of Mammalian Cell Cultures to Benzo[a]Pyrene and Light Results in Oxidative DNA Damage as Measured by 8-Hydroxydeoxyguanosine Formation. Carcinogenesis (1995) 16:133–7. doi: 10.1093/carcin/16.1.133
158. Wang S, Sheng Y, Feng M, Leszczynski J, Wang L, Tachikawa H, et al. Light-Induced Cytotoxicity of 16 Polycyclic Aromatic Hydrocarbons on the US EPA Priority Pollutant List in Human Skin HaCaT Keratinocytes: Relationship Between Phototoxicity and Excited State Properties. Environ Toxicol (2007) 22:318–27. doi: 10.1002/tox.20241
159. Shyong EQ, Lu Y, Goldstein A, Lebwohl M, Wei H. Synergistic Enhancement of H2O2 Production in Human Epidermoid Carcinoma Cells by Benzo[a]pyrene and Ultraviolet A Radiation. Toxicol Appl Pharmacol (2003) 188:104–9. doi: 10.1016/S0041-008X(03)00018-8
160. Soeur J, Belaidi JP, Chollet C, Denat L, Dimitrov A, Jones C, et al. Photo-Pollution Stress in Skin: Traces of Pollutants (PAH and Particulate Matter) Impair Redox Homeostasis in Keratinocytes Exposed to UVA1. J Dermatol Sci (2017) 86:162–9. doi: 10.1016/j.jdermsci.2017.01.007
161. Borska L, Andrys C, Krejsek J, Palicka V, Vorisek V, Hamakova K, et al. Influence of Dermal Exposure to Ultraviolet Radiation and Coal Tar (Polycyclic Aromatic Hydrocarbons) on the Skin Aging Process. J Dermatol Sci (2016) 81:192–202. doi: 10.1016/j.jdermsci.2015.12.010
162. Fu PP, Xia Q, Sun X, Yu H. Phototoxicity and Environmental Transformation of Polycyclic Aromatic Hydrocarbons (PAHs)-Light-Induced Reactive Oxygen Species, Lipid Peroxidation, and DNA Damage. J Environ Sci Health C Environ Carcinog Ecotoxicol Rev (2012) 30:1–41. doi: 10.1080/10590501.2012.653887
163. Park SL, Justiniano R, Williams JD, Cabello CM, Qiao S, Wondrak GT. The Tryptophan-Derived Endogenous Aryl Hydrocarbon Receptor Ligand 6-Formylindolo[3,2-B]Carbazole Is a Nanomolar UVA Photosensitizer in Epidermal Keratinocytes. J Invest Dermatol (2015) 135:1649–58. doi: 10.1038/jid.2014.503
164. Rolfes KM, Sondermann NC, Vogeley C, Dairou J, Gilardino V, Wirth R, et al. Inhibition of 6-Formylindolo[3,2-B]Carbazole Metabolism Sensitizes Keratinocytes to UVA-Induced Apoptosis: Implications for Vemurafenib-Induced Phototoxicity. Redox Biol (2021) 46:102110. doi: 10.1016/j.redox.2021.102110
165. Justiniano R, de Faria Lopes L, Perer J, Hua A, Park SL, Jandova J, et al. The Endogenous Tryptophan-Derived Photoproduct 6-Formylindolo[3,2-B]Carbazole (FICZ) Is a Nanomolar Photosensitizer That Can be Harnessed for the Photodynamic Elimination of Skin Cancer Cells in Vitro and in Vivo. Photochem Photobiol (2021) 97:180–91. doi: 10.1111/php.13321
166. Brem R, Macpherson P, Guven M, Karran P. Oxidative Stress Induced by UVA Photoactivation of the Tryptophan UVB Photoproduct 6-Formylindolo[3,2-B]Carbazole (FICZ) Inhibits Nucleotide Excision Repair in Human Cells. Sci Rep (2017) 7:4310. doi: 10.1038/s41598-017-04614-8
167. Dolmans DE, Fukumura D, Jain RK. Photodynamic Therapy for Cancer. Nat Rev Cancer (2003) 3:380–7. doi: 10.1038/nrc1071
168. Allegra A, Pioggia G, Tonacci A, Musolino C, Gangemi S. Oxidative Stress and Photodynamic Therapy of Skin Cancers: Mechanisms, Challenges and Promising Developments. Antioxid (Basel) (2020) 9:448. doi: 10.3390/antiox9050448
169. Hopf NB, Spring P, Hirt-Burri N, Jimenez S, Sutter B, Vernez D, et al. Polycyclic Aromatic Hydrocarbons (PAHs) Skin Permeation Rates Change With Simultaneous Exposures to Solar Ultraviolet Radiation (UV-S). Toxicol Lett (2018) 287:122–30. doi: 10.1016/j.toxlet.2018.01.024
170. Bourgart E, Persoons R, Marques M, Rivier A, Balducci F, von Koschembahr A, et al. Influence of Exposure Dose, Complex Mixture, and Ultraviolet Radiation on Skin Absorption and Bioactivation of Polycyclic Aromatic Hydrocarbons Ex Vivo. Arch Toxicol (2019) 93:2165–84. doi: 10.1007/s00204-019-02504-8
171. Nair S, Kekatpure VD, Judson BL, Rifkind AB, Granstein RD, Boyle JO, et al. UVR Exposure Sensitizes Keratinocytes to DNA Adduct Formation. Cancer Prev Res (Phila) (2009) 2:895–902. doi: 10.1158/1940-6207.CAPR-09-0125
172. von Koschembahr A, Youssef A, Beal D, Calissi C, Bourgart E, Marques M, et al. Solar Simulated Light Exposure Alters Metabolization and Genotoxicity Induced by Benzo[a]Pyrene in Human Skin. Sci Rep (2018) 8:14692. doi: 10.1038/s41598-018-33031-8
173. Barker CW, Fagan JB, Pasco DS. Down-Regulation of P4501A1 and P4501A2 mRNA Expression in Isolated Hepatocytes by Oxidative Stress. J Biol Chem (1994) 269:3985–90. doi: 10.1016/S0021-9258(17)41731-5
174. Morel Y, Barouki R. Down-Regulation of Cytochrome P450 1A1 Gene Promoter by Oxidative Stress. Critical Contribution of Nuclear Factor 1. J Biol Chem (1998) 273:26969–76. doi: 10.1074/jbc.273.41.26969
175. Vostalova J, Rajnochova Svobodova A, Galandakova A, Sianska J, Dolezal D, Ulrichova J. Differential Modulation of Inflammatory Markers in Plasma and Skin After Single Exposures to UVA or UVB Radiation In Vivo. BioMed Pap Med Fac Univ Palacky Olomouc Czech Repub (2013) 157:137–45. doi: 10.5507/bp.2013.036
176. Kim TY, Kripke ML, Ullrich SE. Immunosuppression by Factors Released From UV-Irradiated Epidermal Cells: Selective Effects on the Generation of Contact and Delayed Hypersensitivity After Exposure to UVA or UVB Radiation. J Invest Dermatol (1990) 94:26–32. doi: 10.1111/1523-1747.ep12873322
177. Ke S, Rabson AB, Germino JF, Gallo MA, Tian Y. Mechanism of Suppression of Cytochrome P-450 1A1 Expression by Tumor Necrosis Factor-Alpha and Lipopolysaccharide. J Biol Chem (2001) 276:39638–44. doi: 10.1074/jbc.M106286200
178. Vogeley C, Esser C, Tuting T, Krutmann J, Haarmann-Stemmann T. Role of the Aryl Hydrocarbon Receptor in Environmentally Induced Skin Aging and Skin Carcinogenesis. Int J Mol Sci (2019) 20:6005. doi: 10.3390/ijms20236005
179. Fernandez-Gallego N, Sanchez-Madrid F, Cibrian D. Role of AHR Ligands in Skin Homeostasis and Cutaneous Inflammation. Cells (2021) 10:3176. doi: 10.3390/cells10113176
180. Krutmann J, Schalka S, Watson REB, Wei L, Morita A. Daily Photoprotection to Prevent Photoaging. Photodermatol Photoimmunol Photomed (2021) 37:482–9. doi: 10.1111/phpp.12688
181. Burke KE. Mechanisms of Aging and Development-A New Understanding of Environmental Damage to the Skin and Prevention With Topical Antioxidants. Mech Ageing Dev (2018) 172:123–30. doi: 10.1016/j.mad.2017.12.003
182. Safe S, Jayaraman A, Chapkin RS, Howard M, Mohankumar K, Shrestha R. Flavonoids: Structure-Function and Mechanisms of Action and Opportunities for Drug Development. Toxicol Res (2021) 37:147–62. doi: 10.1007/s43188-020-00080-z
Keywords: aryl hydrocarbon receptor (AHR), apoptosis, DNA repair, immunosuppression, ultraviolet radiation, skin cancer, polycyclic aromatic hydrocarbons
Citation: Vogeley C, Rolfes KM, Krutmann J and Haarmann-Stemmann T (2022) The Aryl Hydrocarbon Receptor in the Pathogenesis of Environmentally-Induced Squamous Cell Carcinomas of the Skin. Front. Oncol. 12:841721. doi: 10.3389/fonc.2022.841721
Received: 22 December 2021; Accepted: 09 February 2022;
Published: 03 March 2022.
Edited by:
Suzie Chen, Rutgers, The State University of New Jersey, United StatesReviewed by:
Sheikh Umar Ahmad, Indian Institute of Integrative Medicine (CSIR), IndiaShobhan Gaddameedhi, North Carolina State University, United States
Copyright © 2022 Vogeley, Rolfes, Krutmann and Haarmann-Stemmann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thomas Haarmann-Stemmann, VGhvbWFzLmhhYXJtYW5uLXN0ZW1tYW5uQGl1Zi1kdWVzc2VsZG9yZi5kZQ==
†These authors have contributed equally to this work