 Kangzhe Xie
Kangzhe Xie Keely Tan
Keely Tan Matthew J. Naylor
Matthew J. Naylor- Charles Perkins Centre, School of Medical Sciences, Faculty of Medicine & Health, University of Sydney, Sydney, NSW, Australia
Prostate cancer is the second most diagnosed cancer among men worldwide. Androgen deprivation therapy, the most common targeted therapeutic option, is circumvented as prostate cancer progresses from androgen dependent to castrate-resistant disease. Whilst the nuclear receptor transcription factor, androgen receptor, drives the growth of prostate tumor during initial stage of the disease, androgen resistance is associated with poorly differentiated prostate cancer. In the recent years, increased research has highlighted the aberrant transcriptional activities of a small number of transcription factors. Along with androgen receptors, dysregulation of these transcription factors contributes to both the poorly differentiated phenotypes of prostate cancer cells and the initiation and progression of prostate carcinoma. As master regulators of cell fate decisions, these transcription factors may provide opportunity for the development of novel therapeutic targets for the management of prostate cancer. Whilst some transcriptional regulators have previously been notoriously difficult to directly target, technological advances offer potential for the indirect therapeutic targeting of these transcription factors and the capacity to reprogram cancer cell phenotype. This mini review will discuss how recent advances in our understanding of transcriptional regulators and material science pave the way to utilize these regulatory molecules as therapeutic targets in prostate cancer.
Introduction
Prostate cancer is the second most diagnosed cancer in men worldwide, with approximately 1.4 million cases in 2020 alone (1). Prostatic intraepithelial neoplasia (PIN) is a premalignant lesion characterized by the uncontrollable cell growth within the prostate gland (2). This unchecked proliferation precedes the development of localized prostate adenocarcinoma, whereby the tumor increases in volume and cells begin to infiltrate through the basement membrane. The initial pathogenesis of this disease is largely dependent on the activity of the transcriptional factor, androgen receptor (AR) (3). However, once the disease progresses to a more aggressive phenotype, the tumor becomes androgen resistant, evolving into castrate-resistant prostate carcinoma (CRPC) (4). Metastatic CRPC (mCRPC) is the advanced/final stage of the disease, with cancer cells undergoing metastasis to distal organs such as bone, liver and lungs (5). In addition to androgen resistance, phenotypic changes such as alteration to chromatin structure and nucleus enlargement also occur during the malignant transformation in prostate cells (6, 7).
AR is a ligand activated transcription factor and it functions through the binding of androgens, such as testosterone and 5-α-dihydrotestosterone, which releases AR from its chaperone heat shock protein (HSP) 90. Similar to many steroid hormone nuclear receptors, this results in translocation of AR to the nucleus to regulate the expression of genes associated with growth and maintenance of the prostate epithelium (8). AR, along with a small number of other transcription factors, have been well established as regulatory molecules that govern prostate cell phenotype and are implicated in the initiation and progression of prostate cancer (Table 1, also see reviews (2, 36). As the same transcription factor has the ability to bind to regulatory regions of different genes (37), targeting transcription factors provides a direct target in developing effective treatments for prostate cancer and allows for the coordinated inhibition of various oncogenic genes and signaling pathways. Whilst several direct approaches to alter transcription factor expression such as siRNA loaded nanoparticles and lentiviruses are under development, this mini review will focus on indirect approaches such as modulation of epigenetic mechanisms, manipulation of the ubiquitin-proteasome system, targeting the molecular chaperone network and exploitation of proteins in transcriptional complexes, with the discussion of some recent successful attempts (Figure 1).
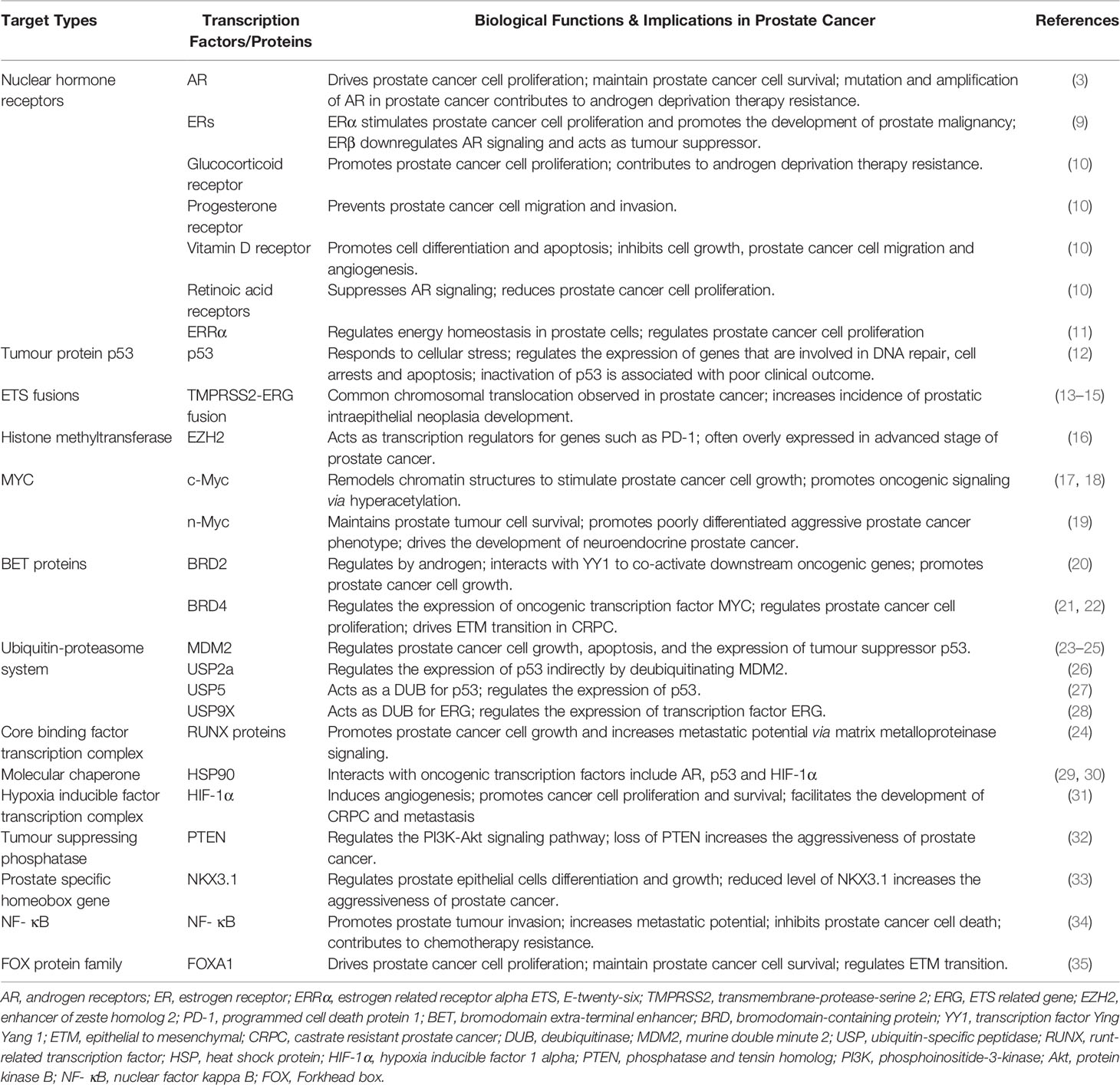
Table 1 Identified transcription factor targets and their implications in prostate cancer.
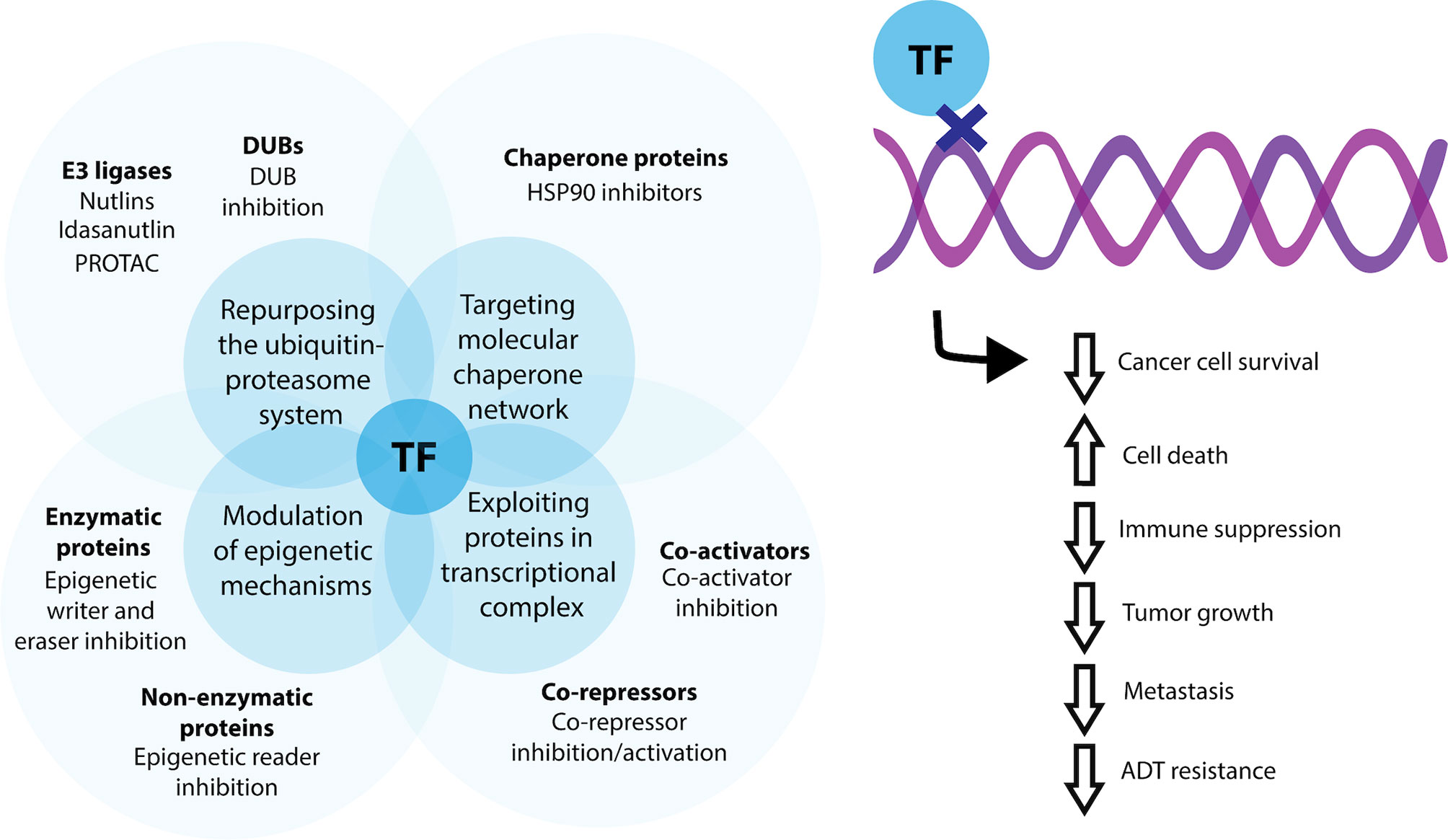
Figure 1 Current indirect methods for targeting transcription factors for prostate cancer therapy. Transcription factor (TF) activity may be indirectly modulated by targeting enzymatic and non-enzymatic proteins involved in epigenetic signaling, through repurposing and manipulating aspects of the ubiquitin-proteasomal system for control of transcription factor degradation, by targeting the molecular chaperone network and by exploiting co-activator and co-repressors associated with a transcriptional complex. Using these methods, transcription factor activity can be favorable modified to decrease cancer cell survival, overall tumour growth and the potential for metastatic dissemination.
Therapeutic Targeting of Transcription Factors
Historically, many transcription factors have been considered as ‘undruggable’ targets, owing to their ‘intrinsically disordered’ interaction network formed with their functional partners (38, 39). In cells, transcription factors regulate gene expression through protein-protein interactions (PPIs) with their co-activators and co-repressors as well as via direct sequence specific DNA binding (40). As a result, the lack of enzymatic activities and catalytic sites presents a major blockade in the development of transcription factor inhibitors and modulators (41). In addition, the transcription factor-DNA binding interfaces are often positively charged and structurally convex, whereas the sites for transcription factor-co-regulator interactions are much flatter than the typical enzyme ligand binding pockets (42, 43). Together, these properties further exacerbate the challenges in developing small molecule inhibitors and modulators with desirable ADME (Absorption, Distribution, Metabolism and Excretion) indices (42, 43). In recent years, a plethora of studies have demonstrated success in targeting transcription factors in prostate cancer, showcasing the feasibility of this approach, and challenging previous dogma. In particular, chemical inhibitors targeting the AR ligand binding domains such as bicalutamide and enzalutamide, have been developed (44, 45). Whilst the use of AR inhibitors are now amongst the primary options for androgen-targeted therapies in early-stage prostate cancers (45, 46), these options become ineffective once the tumor becomes castrate-resistant, as they are able to circumvent androgen targeted treatments via various mechanisms which include AR amplification, point mutation, splicing variants and replacing AR functions with glucocorticoid receptors (4, 47, 48). As there is a profound paucity of effective treatments for mCRPC patients (49), research into methods of targeting non-AR transcription factors in prostate cancer is critical.
Modulation of Epigenetic Mechanisms
Modulating epigenetic signaling pathways is one approach used to target oncogenic transcription factors within prostate cancer. As enzymes are druggable targets with relatively high tractability, epigenetic writers and erasers such as acetyl transferase, methyltransferase, deacetylases and demethylases provide a direct target for inhibitor development (50). Enhancer of zeste homolog 2 (EZH2) is an important epigenetic regulator and in prostate cancer, it was found that EZH2 negatively regulates the expression of interferon stimulated genes (ISGs) such as programmed cell death protein 1 (PD-1) and major histocompatibility complex, creating an immunosuppressive tumor microenvironment and increasing resistance to immune checkpoint blockade (ICB) therapies (16). In a recent study by Morel and colleagues, inhibition of EZH2 restored the expression of ISGs and reversed the resistance to ICB treatments, highlighting the therapeutic potential of EZH2 inhibitors in prostate cancer (51). The clinical applicability of EZH2 inhibitors was reinforced by Bai and colleagues, where EZH2 inhibition with GSK126 prevented prostate specific antigen expression and overcame enzalutamide resistance in CRPC (52). Furthermore, the development of neuroendocrine prostate cancer (NEPC), an aggressive subtype of CRPC, was also found to be associated with dysfunctional EZH2 activity (53). Using GSK126, Dardenne and colleagues showed that NEPC cells were more sensitive to EZH2 inhibition than androgen sensitive LNCaP cells (54), however, the clinical efficacy of EZH2 inhibitors against NEPC warrants further investigation. Whilst EZH2 represents a major target for prostate cancer, alternative targets include histone acetyl transferase E1A binding protein (p300) and CREB binding protein (CBP). In prostate cancer, p300 and CBP interact with numerous oncogenic transcription factors, including p53, MYC and AR, to drive tumour progression (55). These epigenetic enzymes can be inhibited by a CellCentric developed compound, CCS1477, where administration of CCS1477 was shown to downregulate the expression of AR and MYC, resulting in decreased tumor growth in a 22Rv1 xenograft model of CRPC (56, 57).
Alternatively, targeting regulators that do not possess enzymatic activities in the epigenetic signaling pathway have also proven successful. MYC is one of the most dysregulated transcription factors in human cancers (17). In prostate cancer, MYC remodels the chromatin structure to stimulate cell growth and promote oncogenic signaling via hyperacetylation (17, 18) and it has been shown that these oncogenic effects are partly mediated by the epigenetic reader protein, bromodomain extra-terminal (BET) (58). As a result, targeting the BET family of proteins provides a potential avenue to indirectly regulate the expression of MYC, ultimately regressing prostate tumor progression. Indeed, I-BET762, a BET inhibitor, has been shown to reduce MYC expression. This was associated with decreased prostate cancer cell proliferation, increased programmed cell death and reduced in vivo prostate tumour burden, highlighting the possibility of targeting BET proteins as a treatment for prostate cancer (59). The therapeutic potential of BET inhibition is further accentuated by JQ1, an inhibitor that targets bromodomain containing protein (BRD) 4 (60). BRD4 is a member of the BET family of proteins, and it has been shown to interact with transcription factors such as AR and MYC to mediate oncogenic effects (21). More recently, it has been suggested that BRD4 also plays a role in regulating tumor immune microenvironments. This is supported by Mao and colleagues, where BRD4 inhibition with JQ1 reduced PD-1 expression and promoted CD8-mediated lysis of prostate tumor cells both in vitro and in vivo (61). In addition, JQ1 was found to mediate anticancer effect by downregulating the expression of Achaete-scute homolog 1 (ASCL1) in small-cell lung cancer (62). With recent evidence suggesting that ASCL1 as a key driver for NEPC (63), JQ1 along with other BET inhibitors may have potential as NEPC therapeutics. The BET inhibitors, BMS-986158 and RO6870810 are now also in various phases of multicancer clinical trials, with the pan-BET inhibitor ZEN-3694, showcasing therapeutic efficacy in a Phase Ib/IIa mCRPC study (64–66).
Manipulation of the Ubiquitin-Proteasome System
Another way to target transcription factor in prostate cancer is utilizing the ubiquitin-proteasome system (UPS). Appropriate levels of transcription factor expression in cells is key in maintaining cellular homeostasis (67). Aberrant transcription factor expression or failure in the expression of regulatory circuits may lead to catastrophic effects and result in pathophysiological states. Ubiquitin ligases (E3 ligases) are enzymes in the UPS that catalyze the cellular process of ubiquitylation, in which ubiquitin covalently attaches to the substrate protein for proteasomal degradation as a method to regulate transcription factor expression (68, 69). This unique degradation pathway provides a potential platform for controlling transcription factor expression in prostate cancer.
In several human cancers, including prostate cancer, the expression of transcription factor and tumor suppressor p53, is known to be highly dysregulated (70). This dysregulation can arise from the increased activity of murine double minute 2 (MDM2), an E3 ligase, which decreases the expression of p53 and ultimately results in poor clinical outcomes for the patient (12, 23). Thus Nutlins, a novel class of MDM2 inhibitors, were developed by Vassilev and colleagues. The use of these inhibitors increased cellular expression of p53 and its target gene p21 (71), whilst further research using in vivo mouse xenograft models with androgen dependent LNCaP and androgen independent 22Rv1 cell lines, demonstrated increased apoptosis and reduced tumor burden in both cell types following Nutlins treatment (72, 73). To further improve the potency and selectivity of these Nutlins, a second-generation compound Idasanutlin (RG7738/RO5503781), was developed (74). Research shows that use of this compound induces cell death via a combined mechanism of cell cycle arrest and cytotoxic insult in LNCaP cells (75). The clinical applicability of E3 ligase inhibition has recently been assessed in a Phase I clinical trial for prostate cancer patients who have not received docetaxel treatment previously (76). Whilst the trial was terminated due to safety concerns and financial withdrawal from Roche, promising preliminary results highlight the potential for this class of compound, and E3 ligases in general, to be further investigated as a prostate cancer therapy.
The utilization of the UPS (more specifically E3 ligase) to target transcription factors within prostate cancer was taken further with the discovery of proteolysis targeting chimeras (PROTACs). PROTACs are bifunctional molecules comprised of a protein interacting ligand, as well as an E3 ligase recruiting ligand (77). The two ligands in PROTACs are linked in a covalent manner, with protein interacting ligand binding with the protein of interest such as a transcription factor, whereas E3 ligase recruiting ligand facilitates the process of ubiquitylation and subsequent protein degradation (78). PROTACs generate a ‘knocked-down’ effect in cells, abrogating the cellular function of the protein of interest (79). Furthermore, it was discovered that this process is highly catalytic, where a single PROTAC molecule can eliminate multiple protein of interest (80). As discussed above, BET proteins regulate the expression of many pro-oncogenic transcription factors such as AR and MYC, and pharmacological inhibition of these regulatory molecules results in an anticancer effect. Therefore, the use of PROTACs could represent another avenue of pharmacological modification on dysregulated transcription factor expression in prostate cancer. WWL0245 is a highly selective and potent PROTAC-based degrader of BRD4 and has been shown to function by inducing cell cycle arrest of the androgen sensitive prostate cancer cell lines, LNCaP and VCaP, in vitro. This was simultaneously associated with the downregulation of oncogenic transcription factors AR and c-Myc, which highlights the therapeutic potential and clinical feasibility of this approach in prostate cancer (81). Such notion was further supported by Raina and colleagues, where they demonstrated that pan-BET PROTAC, ARV-771, induced cell apoptosis and tumour regression in a mouse xenograft model of CRPC (82). Excluding BET, most of the current research is focused on the PROTAC-based approached on targeting AR (83–85), whereas attempts to target other dysregulated transcription factors in prostate cancer via PROTACs is limited. Thus, identifying a wider variety of protein interacting ligands should be the topic of prospective research.
Another way to alter transcription factor expression is to inhibit the activities of deubiquitinases (DUBs). DUBs are enzymes that remove ubiquitin proteins in the UPS, terminating the ubiquitin-mediated proteasomal degradation process (86). Ubiquitin-specific peptidase (USP) 2a act as a DUB for MDM2, in which it positively regulates the expression of MDM2 (26). Since MDM2 is an E3 ligase for transcription factor p53, inhibition of USP2a would promote the proteasomal degradation of MDM2 and indirectly regulate the expression of p53 (23, 86). This was supported by Stevenson and colleagues, where siRNA inhibition of USP2a led to the accumulation of p53 protein in vivo, highlighting the therapeutic potential to inhibit DUBs in prostate cancer (87). Inhibition of DUBs in the context of prostate cancer is not limited to the p53 signaling pathway. The gene fusion product transmembrane-protease-serine-2 (TMPRSS2)-ETS-related gene (ERG) as a result of chromosomal translocation is observed in 20 to 50% of prostate cancer patients with different ethnicities (13, 14). The TMPRSS2-ERG fusion protein was showed to mediate prostate cancer cell invasion and activation of transcriptional programs for invasion-associated genes (15). The expression of ERG is regulated by E3 ligase tripartite-motif-containing-25 (TRIM25), whereas USP9X acts as DUB for ERG deubiquitylation (28, 88). Thus, target inhibition of ERG or TMPRSS2 may be beneficial in prostate cancer. WP1130 is a small molecule inhibitor comprised of two protein reactive moieties, a 2-bromo-pyridine functional group as well as an α,β-unsaturated amide moiety that is able to undergo Michael addition reactions (89). This enables WP1130 to interact with proteins in a partly selective manner and exerts inhibitory effects on multiple DUBs such as USP5, USP9X and USP14 (90). It has been shown that WP1130 reduces the level of ERG in vitro by inhibiting the enzymatic function of USP9X. This was associated with a decrease in tumour volume in murine xenografts with VCaP cells, highlighting the clinical feasibility to target DUBs in prostate cancer (28).
Targeting the Molecular Chaperone Network
The molecular chaperone network is responsible for various biological processes such as appropriate protein folding, intracellular localization, and degradation, thus maintaining protein homeostasis in cells (29). Chaperone protein such as HSP90 exert these functions by interacting with a diverse range of client proteins, and amongst them, many are oncogenic transcription factors. They include AR, p53 and hypoxia inducible factor (HIF)-1α (29, 30). As a result, disrupting HSP90-transcription factor interactions via small molecule inhibitors provide a potential pathway to rectify the dysregulated mechanisms that cause prostate malignancy. 17-AAG is the first-in-class HSP90 inhibitor developed by Schnur and colleagues. However, the weak potency and poor bioavailability of this compound has sparked further optimization (30). Ganetespib is a second-generation HSP90 inhibitor with improved potency. It has been shown that Ganetespib induced cell cycle arrest in LNCaP and LAPC4 cells and resulted in tumor regression in a PDX model of CRPC (91), highlighting the feasibility and clinical applicability of HSP90 inhibition as an anticancer treatment. This notion was reinforced recently by SU086, another novel HSP90 inhibitor that was found to reduce the proliferation of PC3 and DU145 prostate cancer cells in vitro and inhibit tumor growth in a preclinical murine model of prostate cancer (92). In addition to HSP90, other emerging targets from the molecular chaperone network include HSP70 and HSP90 co-chaperone CDC37, however, drug-like inhibitors targeting these two proteins are yet to be developed (93). Whilst targeting chaperone proteins other than HSP90 in prostate cancer are not well understood and requires further investigation, it represents a novel strategy for prostate cancer treatment.
Exploiting Other Proteins in a Transcriptional Complex
Another approach to modulate transcription factor expressions can be achieved via the exploitation of proteins in a transcriptional complex. HIF is a transcriptional complex that plays a key role in inducing angiogenesis, an essential requirement for prostate tumour growth and the CRPC development. Prior to sufficient vascular development by the prostate tumour, cancer cells must adapt to the low oxygen concentration to fulfil their large energy expenditure (31). Estrogen related receptor alpha (ERRα) is involved in the regulation of prostate energy homeostasis (11). It has been shown that ERRα can interact with hypoxia inducible factor 1 (HIF) transcription factor complex to prevent HIF-1α from undergoing proteasomal degradation and augments the cellular adaptive response to hypoxia generated by the prostate tumour cells (94, 95). As a result, interference of this indispensable PPI is a lucrative approach to develop prostate cancer therapeutics. XCT790 is an inverse agonist of ERRα (94). It was demonstrated that administration of XCT790 attenuated ERRα-HIF-1 interactions and reduced the expressions of HIF-1 (95). This was associated with a decrease in LNCaP prostate cancer cell proliferation in vitro (95), outlining the clinical applicability of this approach to disrupt transcription factor interactions.
The heterodimeric transcription factor complex core binding factor (CBF) is another emerging target. CBF consists of two proteins: DNA binding runt-related transcription factor (RUNX) and its non-DNA binding beta subunit (CBFβ) (96). The CBFβ functions as a co-activator to RUNX, resulting in RUNX being relieved from its autoinhibited state, facilitating the CBF complex binding to DNA and regulation of target gene expression (97). In recent years, there has been growing recognition of RUNX transcription factors in promoting cell growth and metastatic potential of prostate cancer via matrix metalloproteinase signaling (24). Therefore, targeting such essential PPIs may disrupt the transcription process of oncogenic genes, resulting the anticarcinogenic effects. Successful targeting of this modality was achieved using a monovalent derivative of the AI-10-49 scaffolds, a bivalent inhibitor that was originally developed to target CBFβ-smooth muscle myosin heavy chain interactions (98). This novel monovalent inhibitor interferes the binding between wildtype CBFβ and the RUNX1 protein by altering their conformational dynamics (99). In a study on triple negative breast cancer, CBFβ-RUNX1 inhibition was shown to abolish colony formation and alter the expression of epithelial-mesenchymal transition genes, a characteristic cancer hallmark associated with metastasis (99). With regards to prostate cancer, this finding highlights the therapeutic potential to disrupt RUNX interaction circuity, which may be applicable for developing prostate malignancy therapeutics.
Conclusion
This mini review briefly summarized the recent success in targeting non-AR transcription factors. However, it is worth noting that possible approaches to modulate non-AR transcription factors are not restricted to the ones mentioned above, and these successful discoveries only mark the starting point of further transcription factor research. Nevertheless, the newly discovered inhibitors and modulators represent an encouraging potential to develop effective treatment options for mCRPC by targeting non-AR transcription factor.
Author Contributions
KX designed the study, was responsible for writing the article and the creation of the table. KT was responsible for writing the article and the creation of the figure. MN designed the study and was responsible for writing and revising the manuscript. All authors contributed to the generation of the concepts and ideas provided. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by Cancer Council NSW Research Project Grant (RG 20-08) and Priority-driven Collaborative Cancer Research Scheme (Grant #1130499), funded by the National Breast Cancer Foundation Australia with the assistance of Cancer Australia awarded to MN.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin (2021) 71(3):209–49. doi: 10.3322/caac.21660
2. Abate-Shen C, Shen MM. Molecular Genetics of Prostate Cancer. Genes Dev (2000) 14(19):2410–34. doi: 10.1101/gad.819500
3. Lonergan PE, Tindall DJ. Androgen Receptor Signaling in Prostate Cancer Development and Progression. J Carcinog (2011) 10:20. doi: 10.4103/1477-3163.83937
4. Sharma NL, Massie CE, Ramos-Montoya A, Zecchini V, Scott HE, Lamb AD, et al. The Androgen Receptor Induces a Distinct Transcriptional Program in Castration-Resistant Prostate Cancer in Man. Cancer Cell (2013) 23(1):35–47. doi: 10.1016/j.ccr.2012.11.010
5. Shen MM, Abate-Shen C. Molecular Genetics of Prostate Cancer: New Prospects for Old Challenges. Genes Dev (2010) 24(18):1967–2000. doi: 10.1101/gad.1965810
6. Epstein JI. Precursor Lesions to Prostatic Adenocarcinoma. Virchows Arch (2009) 454(1):1–16. doi: 10.1007/s00428-008-0707-5
7. Koh CM, Bieberich CJ, Dang CV, Nelson WG, Yegnasubramanian S, De Marzo AM. MYC and Prostate Cancer. Genes Cancer (2010) 1(6):617–28. doi: 10.1177/1947601910379132
8. Dai C, Heemers H, Sharifi N. Androgen Signaling in Prostate Cancer. Cold Spring Harb Perspect Med (2017) 7(9):a030452. doi: 10.1101/cshperspect.a030452
9. Yeh C-R, Da J, Song W, Fazili A, Yeh S. Estrogen Receptors in Prostate Development and Cancer. Am J Clin Exp urology (2014) 2(2):161.
10. Shiota M, Fujimoto N, Kashiwagi E, Eto M. The Role of Nuclear Receptors in Prostate Cancer. Cells (2019) 8(6):602. doi: 10.3390/cells8060602
11. Cheung CP, Yu S, Wong KB, Chan LW, Lai FM, Wang X, et al. Expression and Functional Study of Estrogen Receptor-Related Receptors in Human Prostatic Cells and Tissues. J Clin Endocrinol Metab (2005) 90(3):1830–44. doi: 10.1210/jc.2004-1421
12. Kluth M, Harasimowicz S, Burkhardt L, Grupp K, Krohn A, Prien K, et al. Clinical Significance of Different Types of P53 Gene Alteration in Surgically Treated Prostate Cancer. Int J Cancer (2014) 135(6):1369–80. doi: 10.1002/ijc.28784
13. Ren S, Peng Z, Mao JH, Yu Y, Yin C, Gao X, et al. RNA-Seq Analysis of Prostate Cancer in the Chinese Population Identifies Recurrent Gene Fusions, Cancer-Associated Long Noncoding RNAs and Aberrant Alternative Splicings. Cell Res (2012) 22(5):806–21. doi: 10.1038/cr.2012.30
14. Magi-Galluzzi C, Tsusuki T, Elson P, Simmerman K, LaFargue C, Esgueva R, et al. TMPRSS2-ERG Gene Fusion Prevalence and Class Are Significantly Different in Prostate Cancer of Caucasian, African-American and Japanese Patients. Prostate (2011) 71(5):489–97. doi: 10.1002/pros.21265
15. Tomlins SA, Laxman B, Varambally S, Cao X, Yu J, Helgeson BE, et al. Role of the TMPRSS2-ERG Gene Fusion in Prostate Cancer. Neoplasia (2008) 10(2):177–88. doi: 10.1593/neo.07822
16. Peng D, Kryczek I, Nagarsheth N, Zhao L, Wei S, Wang W, et al. Epigenetic Silencing of TH1-Type Chemokines Shapes Tumour Immunity and Immunotherapy. Nature (2015) 527(7577):249–53. doi: 10.1038/nature15520
17. Rebello RJ, Pearson RB, Hannan RD, Furic L. Therapeutic Approaches Targeting MYC-Driven Prostate Cancer. Genes (Basel) (2017) 8(2):71. doi: 10.3390/genes8020071
18. Grandori C, Gomez-Roman N, Felton-Edkins ZA, Ngouenet C, Galloway DA, Eisenman RN, et al. C-Myc Binds to Human Ribosomal DNA and Stimulates Transcription of rRNA Genes by RNA Polymerase I. Nat Cell Biol (2005) 7(3):311–8. doi: 10.1038/ncb1224
19. Lee JK, Phillips JW, Smith BA, Park JW, Stoyanova T, McCaffrey EF, et al. N-Myc Drives Neuroendocrine Prostate Cancer Initiated From Human Prostate Epithelial Cells. Cancer Cell (2016) 29(4):536–47. doi: 10.1016/j.ccell.2016.03.001
20. Xu C, Tsai YH, Galbo PM, Gong W, Storey AJ, Xu Y, et al. Cistrome Analysis of YY1 Uncovers a Regulatory Axis of YY1:BRD2/4-PFKP During Tumorigenesis of Advanced Prostate Cancer. Nucleic Acids Res (2021) 49(9):4971–88. doi: 10.1093/nar/gkab252
21. Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, et al. BET Bromodomain Inhibition as a Therapeutic Strategy to Target C-Myc. Cell (2011) 146(6):904–17. doi: 10.1016/j.cell.2011.08.017
22. Shafran JS, Jafari N, Casey AN, Gyorffy B, Denis GV. BRD4 Regulates Key Transcription Factors That Drive Epithelial-Mesenchymal Transition in Castration-Resistant Prostate Cancer. Prostate Cancer Prostatic Dis (2021) 24(1):268–77. doi: 10.1038/s41391-020-0246-y
23. Honda R, Tanaka H, Yasuda H. Oncoprotein MDM2 Is a Ubiquitin Ligase E3 for Tumor Suppressor P53. FEBS Lett (1997) 420(1):25–7. doi: 10.1016/S0014-5793(97)01480-4
24. Ashe H, Krakowiak P, Hasterok S, Sleppy R, Roller DG, Gioeli D. Role of the Runt-Related Transcription Factor (RUNX) Family in Prostate Cancer. FEBS J (2021) 288(21):6112–26. doi: 10.1111/febs.15804
25. Freedman DA, Wu L, Levine AJ. Functions of the MDM2 Oncoprotein. Cell Mol Life Sci (1999) 55(1):96–107. doi: 10.1007/s000180050273
26. Kwon SK, Saindane M, Baek KH. P53 Stability Is Regulated by Diverse Deubiquitinating Enzymes. Biochim Biophys Acta Rev Cancer (2017) 1868(2):404–11. doi: 10.1016/j.bbcan.2017.08.001
27. Dayal S, Sparks A, Jacob J, Allende-Vega N, Lane DP, Saville MK. Suppression of the Deubiquitinating Enzyme USP5 Causes the Accumulation of Unanchored Polyubiquitin and the Activation of P53. J Biol Chem (2009) 284(8):5030–41. doi: 10.1074/jbc.M805871200
28. Wang S, Kollipara RK, Srivastava N, Li R, Ravindranathan P, Hernandez E, et al. Ablation of the Oncogenic Transcription Factor ERG by Deubiquitinase Inhibition in Prostate Cancer. Proc Natl Acad Sci USA (2014) 111(11):4251–6. doi: 10.1073/pnas.1322198111
29. Hong DS, Banerji U, Tavana B, George GC, Aaron J, Kurzrock R. Targeting the Molecular Chaperone Heat Shock Protein 90 (HSP90): Lessons Learned and Future Directions. Cancer Treat Rev (2013) 39(4):375–87. doi: 10.1016/j.ctrv.2012.10.001
30. Powers MV, Workman P. Targeting of Multiple Signalling Pathways by Heat Shock Protein 90 Molecular Chaperone Inhibitors. Endocr Relat Cancer (2006) 13 Suppl 1:S125–35. doi: 10.1677/erc.1.01324
31. Ranasinghe WK, Sengupta S, Williams S, Chang M, Shulkes A, Bolton DM, et al. The Effects of Nonspecific HIF1alpha Inhibitors on Development of Castrate Resistance and Metastases in Prostate Cancer. Cancer Med (2014) 3(2):245–51. doi: 10.1002/cam4.189
32. Turnham DJ, Bullock N, Dass MS, Staffurth JN, Pearson HB. The PTEN Conundrum: How to Target PTEN-Deficient Prostate Cancer. Cells (2020) 9(11):2342. doi: 10.3390/cells9112342
33. Gurel B, Ali TZ, Montgomery EA, Begum S, Hicks J, Goggins M, et al. NKX3.1 as a Marker of Prostatic Origin in Metastatic Tumors. Am J Surg Pathol (2010) 34(8):1097–105. doi: 10.1097/PAS.0b013e3181e6cbf3
34. Verzella D, Fischietti M, Capece D, Vecchiotti D, Del Vecchio F, Cicciarelli G, et al. Targeting the NF-kappaB Pathway in Prostate Cancer: A Promising Therapeutic Approach? Curr Drug Targets (2016) 17(3):311–20. doi: 10.2174/1389450116666150907100715
35. Teng M, Zhou S, Cai C, Lupien M, He HH. Pioneer of Prostate Cancer: Past, Present and the Future of FOXA1. Protein Cell (2021) 12(1):29–38. doi: 10.1007/s13238-020-00786-8
36. Wang G, Zhao D, Spring DJ, DePinho RA. Genetics and Biology of Prostate Cancer. Genes Dev (2018) 32(17-18):1105–40. doi: 10.1101/gad.315739.118
37. Marco A, Konikoff C, Karr TL, Kumar S. Relationship Between Gene Co-Expression and Sharing of Transcription Factor Binding Sites in Drosophila Melanogaster. Bioinformatics (2009) 25(19):2473–7. doi: 10.1093/bioinformatics/btp462
38. Henley MJ, Koehler AN. Advances in Targeting 'Undruggable' Transcription Factors With Small Molecules. Nat Rev Drug Discov (2021) 20(9):669–88. doi: 10.1038/s41573-021-00199-0
39. Liu J, Perumal NB, Oldfield CJ, Su EW, Uversky VN, Dunker AK. Intrinsic Disorder in Transcription Factors. Biochemistry (2006) 45(22):6873–88. doi: 10.1021/bi0602718
40. Lambert SA, Jolma A, Campitelli LF, Das PK, Yin Y, Albu M, et al. The Human Transcription Factors. Cell (2018) 172(4):650–65. doi: 10.1016/j.cell.2018.01.029
41. Inamoto I, Shin JA. Peptide Therapeutics That Directly Target Transcription Factors. Pept Science (2019) 111(1):e24048. doi: 10.1002/pep2.24048
42. Arkin MR, Tang Y, Wells JA. Small-Molecule Inhibitors of Protein-Protein Interactions: Progressing Toward the Reality. Chem Biol (2014) 21(9):1102–14. doi: 10.1016/j.chembiol.2014.09.001
43. Bushweller JH. Targeting Transcription Factors in Cancer - From Undruggable to Reality. Nat Rev Cancer (2019) 19(11):611–24. doi: 10.1038/s41568-019-0196-7
44. Masiello D, Cheng S, Bubley GJ, Lu ML, Balk SP. Bicalutamide Functions as an Androgen Receptor Antagonist by Assembly of a Transcriptionally Inactive Receptor. J Biol Chem (2002) 277(29):26321–6. doi: 10.1074/jbc.M203310200
45. Ito Y, Sadar MD. Enzalutamide and Blocking Androgen Receptor in Advanced Prostate Cancer: Lessons Learnt From the History of Drug Development of Antiandrogens. Res Rep Urol (2018) 10:23–32. doi: 10.2147/RRU.S157116
46. Nevedomskaya E, Baumgart SJ, Haendler B. Recent Advances in Prostate Cancer Treatment and Drug Discovery. Int J Mol Sci (2018) 19(5):1359. doi: 10.3390/ijms19051359
47. Arora VK, Schenkein E, Murali R, Subudhi SK, Wongvipat J, Balbas MD, et al. Glucocorticoid Receptor Confers Resistance to Antiandrogens by Bypassing Androgen Receptor Blockade. Cell (2013) 155(6):1309–22. doi: 10.1016/j.cell.2013.11.012
48. Fujita K, Nonomura N. Role of Androgen Receptor in Prostate Cancer: A Review. World J Mens Health (2019) 37(3):288–95. doi: 10.5534/wjmh.180040
49. Moussa M, Papatsoris A, Abou Chakra M, Sryropoulou D, Dellis A. Pharmacotherapeutic Strategies for Castrate-Resistant Prostate Cancer. Expert Opin Pharmacother (2020) 21(12):1431–48. doi: 10.1080/14656566.2020.1767069
50. Mohammad HP, Barbash O, Creasy CL. Targeting Epigenetic Modifications in Cancer Therapy: Erasing the Roadmap to Cancer. Nat Med (2019) 25(3):403–18. doi: 10.1038/s41591-019-0376-8
51. Morel KL, Sheahan AV, Burkhart DL, Baca SC, Boufaied N, Liu Y, et al. EZH2 Inhibition Activates a dsRNA-STING-Interferon Stress Axis That Potentiates Response to PD-1 Checkpoint Blockade in Prostate Cancer. Nat Cancer (2021) 2(4):444–56. doi: 10.1038/s43018-021-00185-w
52. Bai Y, Zhang Z, Cheng L, Wang R, Chen X, Kong Y, et al. Inhibition of Enhancer of Zeste Homolog 2 (EZH2) Overcomes Enzalutamide Resistance in Castration-Resistant Prostate Cancer. J Biol Chem (2019) 294(25):9911–23. doi: 10.1074/jbc.RA119.008152
53. Luo J, Wang K, Yeh S, Sun Y, Liang L, Xiao Y, et al. LncRNA-P21 Alters the Antiandrogen Enzalutamide-Induced Prostate Cancer Neuroendocrine Differentiation via Modulating the EZH2/STAT3 Signaling. Nat Commun (2019) 10(1):2571. doi: 10.1038/s41467-019-09784-9
54. Dardenne E, Beltran H, Benelli M, Gayvert K, Berger A, Puca L, et al. N-Myc Induces an EZH2-Mediated Transcriptional Program Driving Neuroendocrine Prostate Cancer. Cancer Cell (2016) 30(4):563–77. doi: 10.1016/j.ccell.2016.09.005
55. Welti J, Sharp A, Brooks N, Yuan W, McNair C, Chand SN, et al. Targeting the P300/CBP Axis in Lethal Prostate Cancer. Cancer Discov (2021) 11(5):1118–37. doi: 10.1158/2159-8290.CD-20-0751
56. Pegg N, Brooks N, Worthington J, Young B, Prosser A, Lane J, et al. Characterisation of CCS1477: A Novel Small Molecule Inhibitor of P300/CBP for the Treatment of Castration Resistant Prostate Cancer. J Clin Oncol (2017) 35:11590. doi: 10.1200/JCO.2017.35.15_suppl.11590
57. Brooks N, Prosser A, Young B, Gaughan L, Elvin P, Pegg N. CCS1477, a Potent and Selective P300/CBP Bromodomain Inhibitor, Is Targeted & Differentiated From BET Inhibitors in Prostate Cancer Cell Lines In Vitro. AACR (2019) 79(13_Supplement):3826. doi: 10.1158/1538-7445.AM2019-3826
58. Mertz JA, Conery AR, Bryant BM, Sandy P, Balasubramanian S, Mele DA, et al. Targeting MYC Dependence in Cancer by Inhibiting BET Bromodomains. Proc Natl Acad Sci USA (2011) 108(40):16669–74. doi: 10.1073/pnas.1108190108
59. Wyce A, Degenhardt Y, Bai Y, Le B, Korenchuk S, Crouthame MC, et al. Inhibition of BET Bromodomain Proteins as a Therapeutic Approach in Prostate Cancer. Oncotarget (2013) 4(12):2419–29. doi: 10.18632/oncotarget.1572
60. Coleman DJ, Gao L, Schwartzman J, Korkola JE, Sampson D, Derrick DS, et al. Maintenance of MYC Expression Promotes De Novo Resistance to BET Bromodomain Inhibition in Castration-Resistant Prostate Cancer. Sci Rep (2019) 9(1):3823. doi: 10.1038/s41598-019-40518-5
61. Mao W, Ghasemzadeh A, Freeman ZT, Obradovic A, Chaimowitz MG, Nirschl TR, et al. Immunogenicity of Prostate Cancer Is Augmented by BET Bromodomain Inhibition. J Immunother Cancer (2019) 7(1):277. doi: 10.1186/s40425-019-0758-y
62. Lenhart R, Kirov S, Desilva H, Cao J, Lei M, Johnston K, et al. Sensitivity of Small Cell Lung Cancer to BET Inhibition Is Mediated by Regulation of ASCL1 Gene Expression. Mol Cancer Ther (2015) 14(10):2167–74. doi: 10.1158/1535-7163.MCT-15-0037
63. Fraser JA, Sutton JE, Tazayoni S, Bruce I, Poole AV. Hash1 Nuclear Localization Persists in Neuroendocrine Transdifferentiated Prostate Cancer Cells, Even Upon Reintroduction of Androgen. Sci Rep (2019) 9(1):19076. doi: 10.1038/s41598-019-55665-y
64. Shapiro GI, LoRusso P, Dowlati A, T Do K, Jacobson CA, Vaishampayan U, et al. A Phase 1 Study of RO6870810, a Novel Bromodomain and Extra-Terminal Protein Inhibitor, in Patients With NUT Carcinoma, Other Solid Tumours, or Diffuse Large B-Cell Lymphoma. Br J Cancer (2021) 124(4):744–53. doi: 10.1038/s41416-020-01180-1
65. US National Library of Medicine. Study of BMS-986158 in Subjects With Select Advanced Cancers (BET). ClinicalTrials.gov (2015). Available at: https://clinicaltrials.gov/ct2/show/NCT02419417.
66. Aggarwal RR, Schweizer MT, Nanus DM, Pantuck AJ, Heath EI, Campeau E, et al. A Phase Ib/IIa Study of the Pan-BET Inhibitor ZEN-3694 in Combination With Enzalutamide in Patients With Metastatic Castration-Resistant Prostate Cancer. Clin Cancer Res (2020) 26(20):5338–47. doi: 10.1158/1078-0432.CCR-20-1707
67. Seth A, Watson DK. ETS Transcription Factors and Their Emerging Roles in Human Cancer. Eur J Cancer (2005) 41(16):2462–78. doi: 10.1016/j.ejca.2005.08.013
68. Geng F, Wenzel S, Tansey WP. Ubiquitin and Proteasomes in Transcription. Annu Rev Biochem (2012) 81:177–201. doi: 10.1146/annurev-biochem-052110-120012
69. Yang Q, Zhao J, Chen D, Wang Y. E3 Ubiquitin Ligases: Styles, Structures and Functions. Mol Biomedicine (2021) 2(1):1–17. doi: 10.1186/s43556-021-00043-2
70. Meek DW. Tumour Suppression by P53: A Role for the DNA Damage Response? Nat Rev Cancer (2009) 9(10):714–23. doi: 10.1038/nrc2716
71. Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, et al. In Vivo Activation of the P53 Pathway by Small-Molecule Antagonists of MDM2. Science (2004) 303(5659):844–8. doi: 10.1126/science.1092472
72. Tovar C, Graves B, Packman K, Filipovic Z, Higgins B, Xia M, et al. MDM2 Small-Molecule Antagonist RG7112 Activates P53 Signaling and Regresses Human Tumors in Preclinical Cancer Models. Cancer Res (2013) 73(8):2587–97. doi: 10.1158/0008-5472.CAN-12-2807
73. Tovar C, Higgins B, Kolinsky K, Xia M, Packman K, Heimbrook DC, et al. MDM2 Antagonists Boost Antitumor Effect of Androgen Withdrawal: Implications for Therapy of Prostate Cancer. Mol Cancer (2011) 10:49. doi: 10.1186/1476-4598-10-49
74. Ding Q, Zhang Z, Liu JJ, Jiang N, Zhang J, Ross TM, et al. Discovery of RG7388, a Potent and Selective P53-MDM2 Inhibitor in Clinical Development. J Med Chem (2013) 56(14):5979–83. doi: 10.1021/jm400487c
75. Natarajan U, Venkatesan T, Radhakrishnan V, Samuel S, Rasappan P, Rathinavelu A. Cell Cycle Arrest and Cytotoxic Effects of SAHA and RG7388 Mediated Through P21(WAF1/CIP1) and P27(KIP1) in Cancer Cells. Medicina (Kaunas) (2019) 55(2):30. doi: 10.3390/medicina55020030
76. EU Clinical Trials Register. A Phase I/randomised Phase II Trial of Abiraterone Acetate or Enzalutamide With or Without Idasanutlin (RO5503781) in Patients With Metastatic Castration Resistant Prostate Cancer Who Have Not Previously Received Docetaxel. EU Clinical Trials Register (2013). Available at: https://www.clinicaltrialsregister.eu/ctr-search/trial/2013-002014-13/results.
77. Sun X, Gao H, Yang Y, He M, Wu Y, Song Y, et al. PROTACs: Great Opportunities for Academia and Industry. Signal Transduct Target Ther (2019) 4:64. doi: 10.1038/s41392-019-0101-6
78. Sakamoto KM, Kim KB, Kumagai A, Mercurio F, Crews CM, Deshaies RJ. Protacs: Chimeric Molecules That Target Proteins to the Skp1-Cullin-F Box Complex for Ubiquitination and Degradation. Proc Natl Acad Sci USA (2001) 98(15):8554–9. doi: 10.1073/pnas.141230798
79. Neklesa TK, Winkler JD, Crews CM. Targeted Protein Degradation by PROTACs. Pharmacol Ther (2017) 174:138–44. doi: 10.1016/j.pharmthera.2017.02.027
80. Bondeson DP, Mares A, Smith IE, Ko E, Campos S, Miah AH, et al. Catalytic In Vivo Protein Knockdown by Small-Molecule PROTACs. Nat Chem Biol (2015) 11(8):611–7. doi: 10.1038/nchembio.1858
81. Hu R, Wang WL, Yang YY, Hu XT, Wang QW, Zuo WQ, et al. Identification of a Selective BRD4 PROTAC With Potent Antiproliferative Effects in AR-Positive Prostate Cancer Based on a Dual BET/PLK1 Inhibitor. Eur J Med Chem (2022) 227:113922. doi: 10.1016/j.ejmech.2021.113922
82. Raina K, Lu J, Qian Y, Altieri M, Gordon D, Rossi AM, et al. PROTAC-Induced BET Protein Degradation as a Therapy for Castration-Resistant Prostate Cancer. Proc Natl Acad Sci USA (2016) 113(26):7124–9. doi: 10.1073/pnas.1521738113
83. Han X, Wang C, Qin C, Xiang W, Fernandez-Salas E, Yang CY, et al. Discovery of ARD-69 as a Highly Potent Proteolysis Targeting Chimera (PROTAC) Degrader of Androgen Receptor (AR) for the Treatment of Prostate Cancer. J Med Chem (2019) 62(2):941–64. doi: 10.1021/acs.jmedchem.8b01631
84. Liang JJ, Xie H, Yang RH, Wang N, Zheng ZJ, Zhou C, et al. Designed, Synthesized and Biological Evaluation of Proteolysis Targeting Chimeras (PROTACs) as AR Degraders for Prostate Cancer Treatment. Bioorg Med Chem (2021) 45:116331. doi: 10.1016/j.bmc.2021.116331
85. Neklesa T, Snyder LB, Willard RR, Vitale N, Pizzano J, Gordon DA, et al. ARV-110: An Oral Androgen Receptor PROTAC Degrader for Prostate Cancer. J Clin Oncol (2019) 37(259.10):1200. doi: 10.1200/JCO.2019.37.7_suppl.259
86. Antao AM, Tyagi A, Kim KS, Ramakrishna S. Advances in Deubiquitinating Enzyme Inhibition and Applications in Cancer Therapeutics. Cancers (Basel) (2020) 12(6):1579. doi: 10.3390/cancers12061579
87. Stevenson LF, Sparks A, Allende-Vega N, Xirodimas DP, Lane DP, Saville MK. The Deubiquitinating Enzyme USP2a Regulates the P53 Pathway by Targeting Mdm2. EMBO J (2007) 26(4):976–86. doi: 10.1038/sj.emboj.7601567
88. Wang S, Kollipara RK, Humphries CG, Ma SH, Hutchinson R, Li R, et al. The Ubiquitin Ligase TRIM25 Targets ERG for Degradation in Prostate Cancer. Oncotarget (2016) 7(40):64921–31. doi: 10.18632/oncotarget.11915
89. Kapuria V, Levitzki A, Bornmann WG, Maxwell D, Priebe W, Sorenson RJ, et al. A Novel Small Molecule Deubiquitinase Inhibitor Blocks Jak2 Signaling Through Jak2 Ubiquitination. Cell Signal (2011) 23(12):2076–85. doi: 10.1016/j.cellsig.2011.08.002
90. Kapuria V, Peterson LF, Fang D, Bornmann WG, Talpaz M, Donato NJ. Deubiquitinase Inhibition by Small-Molecule WP1130 Triggers Aggresome Formation and Tumor Cell Apoptosis. Cancer Res (2010) 70(22):9265–76. doi: 10.1158/0008-5472.CAN-10-1530
91. Jansson KH, Tucker JB, Stahl LE, Simmons JK, Fuller C, Beshiri ML, et al. High-Throughput Screens Identify HSP90 Inhibitors as Potent Therapeutics That Target Inter-Related Growth and Survival Pathways in Advanced Prostate Cancer. Sci Rep (2018) 8(1):17239. doi: 10.1038/s41598-018-35417-0
92. Rice MA, Kumar V, Tailor D, Garcia-Marques FJ, Hsu E-C, Liu S, et al. SU086, an Inhibitor of HSP90, Impairs Glycolysis and Represents a Treatment Strategy for Advanced Prostate Cancer. Cell Rep Med (2022) 3(2):100502. doi: 10.1016/j.xcrm.2021.100502
93. Wang L, Xu X, Jiang Z, You Q. Modulation of Protein Fate Decision by Small Molecules: Targeting Molecular Chaperone Machinery. Acta Pharm Sin B (2020) 10(10):1904–25. doi: 10.1016/j.apsb.2020.01.018
94. Matos B, Howl J, Jeronimo C, Fardilha M. The Disruption of Protein-Protein Interactions as a Therapeutic Strategy for Prostate Cancer. Pharmacol Res (2020) 161:105145. doi: 10.1016/j.phrs.2020.105145
95. Zou C, Yu S, Xu Z, Wu D, Ng CF, Yao X, et al. ERRalpha Augments HIF-1 Signalling by Directly Interacting With HIF-1alpha in Normoxic and Hypoxic Prostate Cancer Cells. J Pathol (2014) 233(1):61–73. doi: 10.1002/path.4329
96. Tahirov TH, Inoue-Bungo T, Morii H, Fujikawa A, Sasaki M, Kimura K, et al. Structural Analyses of DNA Recognition by the AML1/Runx-1 Runt Domain and Its Allosteric Control by CBFbeta. Cell (2001) 104(5):755–67. doi: 10.1016/S0092-8674(01)00271-9
97. Gu TL, Goetz TL, Graves BJ, Speck NA. Auto-Inhibition and Partner Proteins, Core-Binding Factor Beta (CBFbeta) and Ets-1, Modulate DNA Binding by CBFalpha2 (Aml1). Mol Cell Biol (2000) 20(1):91–103. doi: 10.1128/MCB.20.1.91-103.2000
98. Illendula A, Pulikkan JA, Zong H, Grembecka J, Xue L, Sen S, et al. Chemical Biology. A Small-Molecule Inhibitor of the Aberrant Transcription Factor CBFbeta-SMMHC Delays Leukemia in Mice. Science (2015) 347(6223):779–84. doi: 10.1126/science.aaa0314
Keywords: prostate cancer, transcription factor, epigenetic, ubiquitin-proteasome system, protein-protein interactions, targeting approaches
Citation: Xie K, Tan K and Naylor MJ (2022) Transcription Factors as Novel Therapeutic Targets and Drivers of Prostate Cancer Progression. Front. Oncol. 12:854151. doi: 10.3389/fonc.2022.854151
Received: 13 January 2022; Accepted: 23 March 2022;
Published: 25 April 2022.
Edited by:
Shashwat Sharad, Center for Prostate Disease Research (CPDR), United StatesReviewed by:
Andrew C. B. Cato, Karlsruhe Institute of Technology (KIT), GermanyRenea A Taylor, Monash University, Australia
Copyright © 2022 Xie, Tan and Naylor. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Matthew J. Naylor, bWF0dGhldy5uYXlsb3JAc3lkbmV5LmVkdS5hdQ==